 Shuang Bai
Shuang Bai Li Zhao
Li Zhao- Department of Pulmonary and Critical Care Medicine, Shengjing Hospital of China Medical University, Shenyang, China
The chronic obstructive pulmonary disease (COPD) emphysematous phenotype is characterized by destruction of lung tissue structure. Patients with this phenotype usually present with typical emphysema-like changes on chest computed Tomography CT, experience higher mortality and poorer prognosis, and are insensitive to routine pharmacological COPD therapy. However, the pathogenesis for the COPD emphysematous phenotype remains unclear, resulting in diagnostic and therapeutic challenges. The imbalance between injury and defense mechanisms is essential in the progression of many pulmonary diseases. Thus, in this review, we focus on the pathogenesis of the COPD emphysematous phenotype and discuss the pathophysiological processes involved in disease progression, from the perspective of injury and defense imbalance.
Introduction
Chronic obstructive pulmonary disease (COPD) is a chronic respiratory disease that generally is manifested as cough, sputum, and breathlessness (1) with structural lung tissue damage and airway reconstitution (2). The clinical manifestations, medical imaging examinations, and pathologies of patients with COPD are quite heterogeneous (3, 4). In recent years, the definition of the COPD phenotype was proposed, and one of the most typical phenotypes is the emphysematous phenotype.
COPD patients with the emphysematous phenotype present with severe structural damage of lung tissue but slight airway destruction (5, 6). Emphysema in these patients deteriorates with disease progression (7), which affects respiration and exercise tolerance of patients. Patients suffer from several complications and experience an extremely high mortality rate (8, 9). There are few COPD therapies that are effective for the emphysema lesions of these patients, and specific pharmacological therapy does not exist. Therefore, delineating the exact pathogenic mechanisms of the emphysematous phenotype can achieve earlier diagnosis, precise categorization, and personalized therapeutic strategies, all of which are required to improve patient prognosis.
The imbalance between injury and defense mechanisms is essential for many pulmonary diseases, such as COPD, asthma, and pulmonary fibrosis. However, the role of the injury and defense system imbalance in the COPD emphysematous phenotype is still not fully understood. Previously, alpha antitrypsin (AAT) deficiency was defined as an injury-induced factor causing emphysema. However, it was found that a substantial number of COPD patients with emphysema present with normal AAT levels. This review illustrates the recent progress in COPD emphysematous phenotype pathogenesis, from an injury and defense imbalance perspective, and summarizes diagnostics and available therapeutic options to help improve COPD emphysematous phenotype prognosis.
Manifestation and Pathophysiologic Processes of COPD Emphysematous Phenotype
The COPD phenotypes refer to disease characteristics that represent differences among COPD patients (10), including the chronic bronchitis phenotype, emphysematous phenotype, frequent exacerbators phenotype, and systemic inflammation phenotype. There are overlaps and interactions among these phenotypes (11). Patients with the emphysematous phenotype present a chronic pathophysiologic process with a decline of the elasticity of bronchioli terminals and distal airways and the destruction of lung tissue and alveolar walls, causing an overexpansion of distal airways and permanent enlargement in lung volume. As a result, COPD patients with an emphysematous phenotype present with reduced lung markings, lung volume enlargement, and decreased lung density (12).
Lung tissue damage and enhanced catabolism are the major pathophysiologic features of the COPD emphysematous phenotype. The restoring force of the lungs decreases as pulmonary elastic tissue is destroyed and insufficient pressure is applied to push air out during expiration. This results in the trapping of gas and excessive inflation. The motive range of the diaphragm narrows when the lungs expand, decreasing the blood volume returning to the heart. Thus, patients experience breathlessness and strain. Pulmonary ventilation dysfunction is also induced by the destruction of the air-blood barrier due to damaged alveoli and capillaries, resulting in air trapping and an imbalance of the ratio of ventilation and blood.
Clinical Characteristics of Patients with the COPD Emphysematous Phenotype
Patients with the emphysematous phenotype usually show common symptoms of COPD, such as cough, expectoration, breathlessness, and more severe dyspnea. Some patients suffer both obvious dyspnea and poor exercise tolerance without airway inflammation-related symptoms. We analyzed chest CT scans, which illustrated unambiguous differences, resulting in the diversity of respiratory manifestations across patients. Moreover, various positions of emphysematous lesions affect pulmonary ventilation to a different extent. The observed respiratory symptoms were inconsistent with the results of the pulmonary function tests—the gold standard of COPD (13, 14). In this light, it is not difficult to infer that when the emphysema lesion doesn't affect the large airway, the spirometry of patients is likely to be normal. In this condition, although some smokers suffer from symptoms such as expiratory dyspnea, breathlessness, limitation of motion, and their condition is diagnosed as emphysema based on CT images; their condition cannot be diagnosed as COPD owing to their ratio of forced expiratory volume in 1 s to forced vital capacity (FEV1/FVC) being >70% (15, 16). Therefore, more attention should be dedicated to the emphysematous phenotype, even to the cases without airflow limitation, and the development of a suitable treatment strategy that is effective during the earlier stages can improve patient prognosis.
Diagnostic Standards of COPD Emphysematous Phenotype
There is no uniform standard for the diagnosis of COPD patients with the emphysematous phenotype to date. The methods utilized in clinical studies can be divided into “spirometry-based classification” and “CT image-based classification.” The classifications of emphysematous phenotype analysis methods are shown in Table 1.

Table 1. Analysis methods of emphysematous phenotype judgement.
Spirometry-Based Classification
“Spirometry-based classification” is based on lung hyperinflation and impairment of lung diffusion capacity in COPD emphysematous phenotype. Cases of patients with total lung capacity (TLC) > 120% pred. (predicted value) and diffusion capacity for carbon monoxide in the lung (DLco) <80% pred. can be diagnosed as the emphysematous phenotype (17, 18). This classification method defines the COPD emphysematous phenotype from a qualitative perspective. However, the degree to which patients experience emphysema is ignored here. Diffusion measurements are taken into account, and this benefits the evaluation of the spirometry and respiratory symptoms.
CT Image-Based Classification
With the advent of high-resolution CT, the spatial resolution of CT scan has been improved greatly, and the consistency between CT images and pathological sections are verified (12, 19). Therefore, “CT image-based classification” is the predominant method in use currently. However, there was no unified criterion for CT image-based classification methods because of the difference between CT parameters and subjects in every study.
In the quantitative assessment method, emphysematous lesions are mainly estimated from the density, volume, and degree of destruction in the lung tissue of patients with COPD (20–24). Lung density was calculated using the “density masking” method to calculate the ratio of area change (25). The evaluation of lung volume is based on continuous scanning images. The region of lung tissue in each scanning image is indicated by image analysis software. Then lung volume is obtained combined with the thickness of layers by calculating the total area of lung tissue (26).
The evaluation of the degree of destruction can be divided into semi-quantitative evaluation based on subjective observation and quantitative evaluation based on software. In 1982, Goddard proposed a semi-quantitative evaluation method named the Goddard evaluation system, widely used to assess the degree of destruction from emphysema (26, 27). The evaluation method was subsequently improved to fit specific purposes, based on the Goddard evaluation system. Some select the representative layers to analyze the degree of emphysema instead of involving all layers (22, 28). Others change the emphysema threshold (21) and include the emphysema region evaluation in their system (29, 30). Compared with the semi-quantitative evaluation based on subjective observation, quantitative evaluation based on software determines the emphysema phenotype by analyzing the proportion of low-density regions in the lung field. However, the CT value threshold used in analysis remains variable across users.
Previously, the −950HU and −910HU in the inspiratory phase were set up as the CT value thresholds in emphysema. There was a definition of the low attenuation area (LAA) %, which referred to the proportion of regions with CT values below the CT value threshold in the lung field. Patients with a LAA% below 20% would be diagnosed as having the emphysematous phenotype. Though the CT value thresholds differ, the difference of the variable CT value threshold did not affect the conclusion of the studies (31). Subsequently, some studies classify the emphysema degree by CT value threshold in the inspiratory phase (20, 32). CT value threshold utilized in other studies include −910HU (33), −950HU (34), and −960HU (35).
In addition to the variance across subjects and equipment parameters, the limitation of sample size is the main reason the CT value threshold is unsuitable for all patients. From an analysis of the relations between CT value thresholds and the emphysema index in related studies (31), it was demonstrated that the CT value in a certain pixel point was affected greatly, but the emphysema indexes were not changed significantly. This result indicated a suitable CT value can be chosen as the CT value threshold according to the actual condition. However, it is still essential to obtain the CT value of emphysema for the target population according to pathological specimens.
Imbalance Between Injury and Defense Mechanism in the COPD Emphysematous Phenotype
The main pathological mechanism of emphysema includes protease activity enhancement, anti-protease activity reduction, oxidative stress, inflammation, apoptosis of alveolar epithelial and endothelial cells, tissue repair dysfunction, autophagy, the aging process, and necrosis (36), and most of these pathological processes were also related to the COPD emphysematous phenotype. We can summarize these pathological processes into two interactive aspects—injury factors and defense repairability of tissue (37). The schematic diagram is shown in Figure 1.
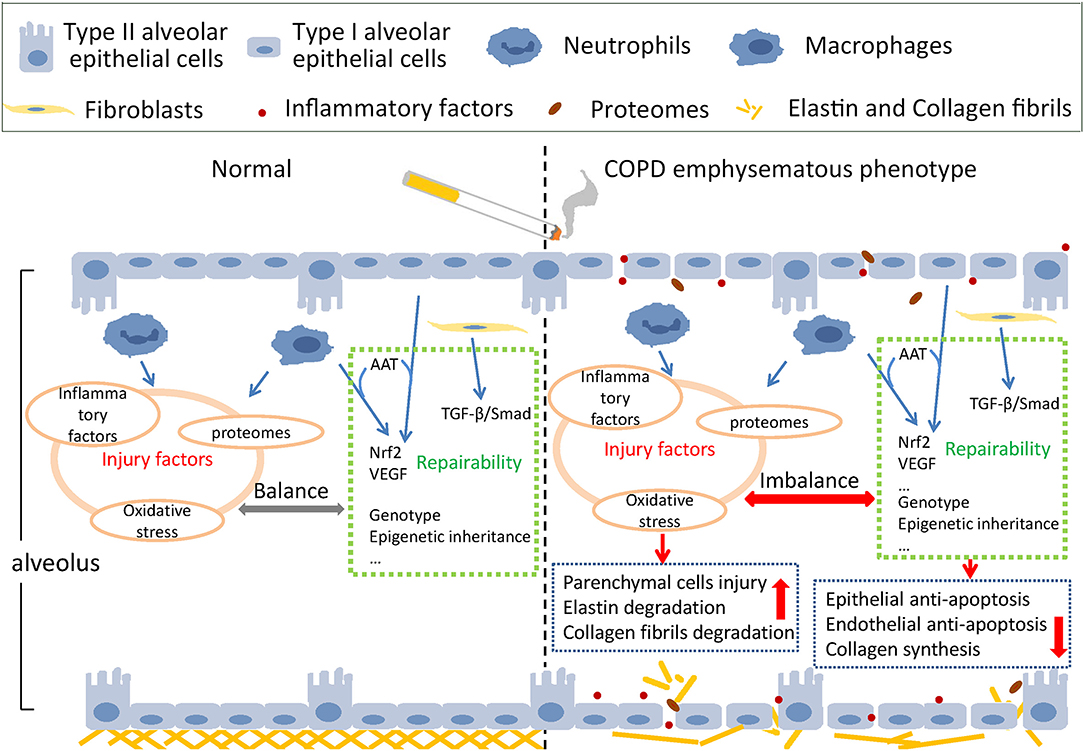
Figure 1. Schematic injury and defense imbalance in the COPD emphysematous phenotype. Cigarette smoke exposure induces injury enhancement and absence of defense repairability through various mechanisms and pathways, resulting in the destruction of alveolar structure and the development of emphysematous phenotype of chronic obstructive pulmonary disease.
Injury Factors
Usually, under the condition of stimulation, injury factors such as IL-6, IL-1β, and TNF-α are mainly produced by inflammatory cells and immune cells such as macrophages and neutrophils, which attack the lung parenchymal cells and mesenchymal components, including fibroblasts, elastin, and collagen fibrils. Many oxygen free radicals are produced, inducing severe inflammation and oxidative stress in alveolar epithelial cells and pulmonary vascular endothelial cells. Lung parenchymal cells experience uncontrolled apoptosis, and the pulmonary ventilation function is impaired (38, 39). At first, the AAT insufficiency was considered the main reason for emphysema occurrence in lungs (40). However, this hypothesis did not explain why smokers without AAT insufficiency still suffered from emphysema and the increase of neutrophil elastase (NE) was a typical feature for them (41). Accumulation of NE degrades elastin in lung mesenchyme causing irreversible damage to the mesenchymal structure.
The important role of matrix metalloproteinases (MMPs) was also identified in transgenic mice models. For example, knockout of MMP12 could rescue emphysema caused by smoking (42), even in mice with NE defect (43). Both inflammatory cells and immune cells, such as cytotoxicity T lymphocytes (CTLs) and natural killer (NK) cells, produce a type of proteinase—granzyme B (Gzm B)—under normal circumstances. Gzm B targets injury sites to clear abnormal cells, through perforin mediation. Once perforin deficient, excessive Gzm B degrades the extracellular matrix, destroys elastin and collagen, and causes severe lung damage (44).
All pathological processes mentioned above induce the imbalance of proteinase and antiprotease and degradation of elastin and collagen fibrils. Once the lung mesenchyme is destroyed by excessive injury factors, the lung tissue dynamic compliance is attenuated, and the alveolus inflates, eventually leading to emphysema.
Defense Repairability
Defense repairability of lung parenchymal cells mainly involves the anti-apoptotic effects of alveolar epithelial cells and the “stem-like” repair function of type II alveolar epithelial cells. Type I and Type II alveolar epithelial cells, vascular endothelial cells, and myofibroblasts make up the alveolar interval. When cigarette smoke acts on the alveolar interval, cellular immunity is activated, and CD8+ T cells react immediately, resulting in epithelial cell apoptosis (45). Vascular endothelial growth factor (VEGF) is secreted by epithelial cells and macrophages and acts on pulmonary vascular endothelial cells to resist apoptosis. Nuclear Factor, Erythroid 2 Like 2 (NFE2L2/Nrf2), one of the most important transcription factors of antioxidant reaction, combines with AAT and VEGF to work together in the antiapoptotic process (41). AAT inhibits apoptosis of epithelial and vascular endothelial cells by interacting directly with cleaved-caspase3 (39, 46).
Under normal conditions, type II alveolar epithelial cells are responsible for secreting alveolar surfactant, sustaining tension and dynamic compliance, and regulating inflammation and cytokines (47). When alveolar epithelial cells are damaged, type II alveolar epithelial cells differentiate into type I alveolar epithelial cells to repair the alveolar structure. The destruction of type II alveolar epithelial cells affects lung parenchymal structure and pulmonary function. Mesenchymal cells, specifically fibroblasts, play an important role during the complicated process of lung mesenchyme repair (48–51). When lung mesenchyme hurts, elastases hydrolyze elastin, and collagen contraction is inhibited. Fibroblasts repair collagen through de novo collagen synthesis (52–54). The dysfunction-related pathways of fibroblasts were also examined through bioinformatic analysis of emphysema transcriptomics (55). As macrophages and other inflammatory cells accumulate around emphysema lesions and small airways, inflammatory factors and proteases are secreted, resulting in inflammation and mesenchymal degradation. TGF-β combines with its receptors and phosphorylates Smad2/3 by cascade reactions to activate the repairing-related pathways and improve the synthesis of fibrin in fibroblasts (56). The activity of TGF-β pathways is spatially different in the lungs. Smad2 phosphorylation decreased in emphysematous lesions, whereas it increased around small airways, consistent with the pathological manifestation of COPD patients with emphysema—alveolar destruction and thickening of the small airway mesenchyme (57).
Members of the lysamyl oxidase (LOX) superfamily also play an important role in the repair mechanism of emphysema. Specific amino acid residues on collagen and elastin can be oxidized by LOX in the extracellular matrix, forming covalent bonds between collagen and elastin, with the participation of Cu2+ (58–62). Single-nucleotide polymorphisms (SNP), such as rs1828591 and rs13118928 on the gene of Hedgehog Interacting Protein (HHIP), are intensively related to COPD (63). HHIP negatively regulates cell proliferation and matrix repair under normal conditions (64). By comparing mesenchymal cells in the lung tissue of emphysema and normal subjects through single-cell sequencing, it was found that the Hh signaling pathways were atopically activated in the alveolar matrix, impairing alveolar stem cells. Therefore, it is speculated that the SNP of HHIP facilitates the formation of emphysema by functional incapacitation (65). In addition to the influence of genotype on COPD emphysema phenotype, epigenetic inheritance also plays an important role. Some studies have documented that cigarette smoke can change the expression of COPD-related genes by DNA methylation and posttranslational modification of histones.
In brief, inflammatory factors and proteinases multiply and damage pulmonary structural cells; matrix components are degraded, and alveolar cell apoptosis is induced. The repair and regeneration of parenchymal and interstitial cells do not compensate for the impaired pulmonary tissue. Eventually, all the pulmonary tissue gets structurally damaged, enabling emphysema formation (38, 65, 66).
Conclusion
The COPD emphysematous phenotype is characterized by specific manifestations and has a high mortality rate. The injury and defense mechanisms play important roles in the occurrence and development of this phenotype. Thus, a more in-depth understanding of the related radiological and mechanistic features to define a suitable CT value threshold and create personalized therapies is warranted. This can enable improved diagnostic and treatment options that improve patient prognosis in those affected with the COPD emphysematous phenotype.
Author Contributions
LZ substantially contributed to the conception and design of the manuscript. SB and LZ contributed to the acquisition, analysis, interpretation of data, drafted the article, revised it critically and substantially, have approved the final version, and accepted accountability for all aspects of the study. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Vogelmeier CF, Criner GJ, Martinez FJ, Anzueto A, Barnes PJ, Bourbeau J, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report. GOLD executive summary. Am J Respir Crit Care Med. (2017) 195:557–82. doi: 10.1164/rccm.201701-0218PP
2. Mohamed Hoesein FA, de Hoop B, Zanen P, Gietema H, Kruitwagen CL, van Ginneken B, et al. CT-quantified emphysema in male heavy smokers: association with lung function decline. Thorax. (2011) 66:782–7. doi: 10.1136/thx.2010.145995
3. Ostridge K, Williams NP, Kim V, Harden S, Bourne S, Clarke SC, et al. Relationship of CT-quantified emphysema, small airways disease and bronchial wall dimensions with physiological, inflammatory and infective measures in COPD. Respir Res. (2018) 19:31. doi: 10.1186/s12931-018-0734-y
4. Agusti A, Calverley PM, Celli B, Coxson HO, Edwards LD, Lomas DA, et al. Characterisation of COPD heterogeneity in the ECLIPSE cohort. Respir Res. (2010) 11:122. doi: 10.1186/1465-9921-11-122
5. Castaldi PJ, Dy J, Ross J, Chang Y, Washko GR, Curran-Everett D, et al. Cluster analysis in the COPDGene study identifies subtypes of smokers with distinct patterns of airway disease and emphysema. Thorax. (2014) 69:415–22. doi: 10.1136/thoraxjnl-2013-203601
6. Ziegler-Heitbrock L, Frankenberger M, Heimbeck I, Burggraf D, Wjst M, Häussinger K, et al. The EvA study: aims and strategy. Eur Respir J. (2012) 40:823–9. doi: 10.1183/09031936.00142811
7. Wille MM, Thomsen LH, Dirksen A, Petersen J, Pedersen JH, Shaker SB. Emphysema progression is visually detectable in low-dose CT in continuous but not in former smokers. Eur Radiol. (2014) 24:2692–9. doi: 10.1007/s00330-014-3294-7
8. Kamio K, Yoshida T, Gao C, Ishii T, Ota F, Motegi T, et al. α1,6-Fucosyltransferase (Fut8) is implicated in vulnerability to elastase-induced emphysema in mice and a possible non-invasive predictive marker for disease progression and exacerbations in chronic obstructive pulmonary disease (COPD). Biochem Biophys Res Commun. (2012) 424:112–7. doi: 10.1016/j.bbrc.2012.06.081
9. Fan L, Xia Y, Guan Y, Zhang TF, Liu SY. Characteristic features of pulmonary function test, CT volume analysis and MR perfusion imaging in COPD patients with different HRCT phenotypes. Clin Respir J. (2014) 8:45–54. doi: 10.1111/crj.12033
10. Han MK, Agusti A, Calverley PM, Celli BR, Criner G, Curtis JL, et al. Chronic obstructive pulmonary disease phenotypes: the future of COPD. Am J Respir Crit Care Med. (2010) 182:598–604. doi: 10.1164/rccm.200912-1843CC
11. Mirza S, Benzo R. Chronic obstructive pulmonary disease phenotypes: implications for care. Mayo Clin Proc. (2017) 92:1104–12. doi: 10.1016/j.mayocp.2017.03.020
12. Smith BM, Austin JH, Newell JD Jr., D'Souza BM, Rozenshtein A, Hoffman EA, et al. Pulmonary emphysema subtypes on computed tomography: the MESA COPD study. Am J Med. (2014) 127:94.e7–23. doi: 10.1016/j.amjmed.2013.09.020
13. Sverzellati N, Calabrò E, Randi G, La Vecchia C, Marchianò A, Kuhnigk JM, et al. Sex differences in emphysema phenotype in smokers without airflow obstruction. Eur Respir J. (2009) 33:1320–8. doi: 10.1183/09031936.00109808
14. DeMeo DL, Hersh CP, Hoffman EA, Litonjua AA, Lazarus R, Sparrow D, et al. Genetic determinants of emphysema distribution in the national emphysema treatment trial. Am J Respir Crit Care Med. (2007) 176:42–8. doi: 10.1164/rccm.200612-1797OC
15. Castaldi PJ, San José Estépar R, Mendoza CS, Hersh CP, Laird N, Crapo JD, et al. Distinct quantitative computed tomography emphysema patterns are associated with physiology and function in smokers. Am J Respir Crit Care Med. (2013) 188:1083–90. doi: 10.1164/rccm.201305-0873OC
16. Sheen S, Sun JS, Park JH, Oh YM, Ki SK, Kim K, et al. Unique features of non-obstructive emphysema and pure airway obstruction. Int J Tuberc Lung Dis. (2014) 18:109–16. doi: 10.5588/ijtld.13.0258
17. Camiciottoli G, Diciotti S, Bigazzi F, Lombardo S, Bartolucci M, Paoletti M, et al. Is intrathoracic tracheal collapsibility correlated to clinical phenotypes and sex in patients with COPD? Int J Chron Obstruct Pulmon Dis. (2015) 10:843–52. doi: 10.2147/COPD.S80558
18. Paige M, Burdick MD, Kim S, Xu J, Lee JK, Shim YM. Pilot analysis of the plasma metabolite profiles associated with emphysematous Chronic Obstructive Pulmonary Disease phenotype. Biochem Biophys Res Commun. (2011) 413:588–93. doi: 10.1016/j.bbrc.2011.09.006
19. Park HJ, Jung SM, Song JJ, Park YB, Song JS, Lee SW. Comparison of radiological and histological findings of lung parenchyma in patients with antineutrophil cytoplasmic antibody-associated vasculitis. Yonsei Med J. (2019) 60:454–60. doi: 10.3349/ymj.2019.60.5.454
20. Izquierdo-Alonso JL, Rodriguez-Gonzálezmoro JM, de Lucas-Ramos P, Unzueta I, Ribera X, Antón E, et al. Prevalence and characteristics of three clinical phenotypes of chronic obstructive pulmonary disease (COPD). Respir Med. (2013) 107:724–31. doi: 10.1016/j.rmed.2013.01.001
21. Papaioannou AI, Kostikas K, Papaporfyriou A, Angelakis L, Papathanasiou E, Hillas G, et al. Emphysematous phenotype is characterized by low blood eosinophils: a cross-sectional study. COPD. (2017) 14:635–40. doi: 10.1080/15412555.2017.1386644
22. Márquez-Martín E, Ramos PC, López-Campos JL, Serrano Gotarredona MP, Herrero SN, Aguilar RT, et al. Components of physical capacity in patients with chronic obstructive pulmonary disease: relationship with phenotypic expression. Int J Chron Obstruct Pulmon Dis. (2011) 6:105–12.
23. Tam A, Churg A, Wright JL, Zhou S, Kirby M, Coxson HO, et al. Sex differences in airway remodeling in a mouse model of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2016) 193:825–34. doi: 10.1164/rccm.201503-0487OC
24. Han MK, Wise R, Mumford J, Sciurba F, Criner GJ, Curtis JL, et al. Prevalence and clinical correlates of bronchoreversibility in severe emphysema. Eur Respir J. (2010) 35:1048–56. doi: 10.1183/09031936.00052509
25. Schiska A. The role of computed tomography in the diagnosis and treatment of emphysema. Radiol Technol. (2016) 87:340–3.
26. de Torres JP, Bastarrika G, Zagaceta J, Sáiz-Mendiguren R, Alcaide AB, Seijo LM, et al. Emphysema presence, severity, and distribution has little impact on the clinical presentation of a cohort of patients with mild to moderate COPD. Chest. (2011) 139:36–42. doi: 10.1378/chest.10-0984
27. Goddard PR, Nicholson EM, Laszlo G, Watt I. Computed tomography in pulmonary emphysema. Clin Radiol. (1982) 33:379–87. doi: 10.1016/S0009-9260(82)80301-2
28. Fouda MA, Alhamad EH, Al-Hajjaj MS, Shaik SA, Alboukai AA, Al-Kassimi FA. A study of chronic obstructive pulmonary disease-specific causes of osteoporosis with emphasis on the emphysema phenotype. Ann Thorac Med. (2017) 12:101–6. doi: 10.4103/atm.ATM_357_16
29. Wakayama K, Kurihara N, Fujimoto S, Hata M, Takeda T. Relationship between exercise capacity and the severity of emphysema as determined by high resolution CT. Eur Respir J. (1993) 6:1362–7.
30. Lamers RJ, Thelissen GR, Kessels AG, Wouters EF, van Engelshoven JM. Chronic obstructive pulmonary disease: evaluation with spirometrically controlled CT lung densitometry. Radiology. (1994) 193:109–13. doi: 10.1148/radiology.193.1.8090878
31. Camp PG, Coxson HO, Levy RD, Pillai SG, Anderson W, Vestbo J, et al. Sex differences in emphysema and airway disease in smokers. Chest. (2009) 136:1480–8. doi: 10.1378/chest.09-0676
32. Yamada M, Ishii T, Ikeda S, Naka-Mieno M, Tanaka N, Arai T, et al. Association of fucosyltransferase 8 (FUT8) polymorphism Thr267Lys with pulmonary emphysema. J Hum Genet. (2011) 56:857–60. doi: 10.1038/jhg.2011.118
33. Ley-Zaporozhan J, van Beek EJ. Imaging phenotypes of chronic obstructive pulmonary disease. J Magn Reson Imaging. (2010) 32:1340–52. doi: 10.1002/jmri.22376
34. Oh YM, Sheen SS, Park JH, Jin UR, Yoo JW, Seo JB, et al. Emphysematous phenotype is an independent predictor for frequent exacerbation of COPD. Int J Tuberc Lung Dis. (2014) 18:1407–14. doi: 10.5588/ijtld.14.0205
35. Mishima M, Hirai T, Jin Z, Oku Y, Sakai N, Nakano Y, et al. Standardization of low attenuation area versus total lung area in chest X-ray CT as an indicator of chronic pulmonary emphysema. Front Med Biol Eng. (1997) 8:79–86.
36. Mizumura K, Maruoka S, Shimizu T, Gon Y. Autophagy, selective autophagy, and necroptosis in COPD. Int J Chron Obstruct Pulmon Dis. (2018) 13:3165–72. doi: 10.2147/COPD.S175830
37. Taraseviciene-Stewart L, Voelkel NF. Molecular pathogenesis of emphysema. J Clin Invest. (2008) 118:394–402. doi: 10.1172/JCI31811
38. Nakanishi K, Takeda Y, Tetsumoto S, Iwasaki T, Tsujino K, Kuhara H, et al. Involvement of endothelial apoptosis underlying chronic obstructive pulmonary disease-like phenotype in adiponectin-null mice: implications for therapy. Am J Respir Crit Care Med. (2011) 183:1164–75. doi: 10.1164/rccm.201007-1091OC
39. Tuder RM, Yoshida T, Fijalkowka I, Biswal S, Petrache I. Role of lung maintenance program in the heterogeneity of lung destruction in emphysema. Proc Am Thorac Soc. (2006) 3:673–9. doi: 10.1513/pats.200605-124SF
40. Janssen R, Piscaer I, Franssen FME, Wouters EFM. Emphysema: looking beyond alpha-1 antitrypsin deficiency. Expert Rev Respir Med. (2019) 13:381–97. doi: 10.1080/17476348.2019.1580575
41. Lee KH, Lee CH, Jeong J, Jang AH, Yoo CG. Neutrophil elastase differentially regulates Interleukin 8 (IL-8) and Vascular Endothelial Growth Factor (VEGF) production by cigarette smoke extract. J Biol Chem. (2015) 290:28438–45. doi: 10.1074/jbc.M115.663567
42. Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. (1997) 277:2002–4. doi: 10.1126/science.277.5334.2002
43. Shapiro SD, Goldstein NM, Houghton AM, Kobayashi DK, Kelley D, Belaaouaj A. Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am J Pathol. (2003) 163:2329–35. doi: 10.1016/S0002-9440(10)63589-4
44. Ngan DA, Vickerman SV, Granville DJ, Man SF, Sin DD. The possible role of granzyme B in the pathogenesis of chronic obstructive pulmonary disease. Ther Adv Respir Dis. (2009) 3:113–29. doi: 10.1177/1753465809341965
45. Borchers MT, Wesselkamper SC, Curull V, Ramirez-Sarmiento A, Sánchez-Font A, Garcia-Aymerich J, et al. Sustained CTL activation by murine pulmonary epithelial cells promotes the development of COPD-like disease. J Clin Invest. (2009) 119:636–49. doi: 10.1172/JCI34462
46. Stewart JI, Criner GJ. The small airways in chronic obstructive pulmonary disease: pathology and effects on disease progression and survival. Curr Opin Pulm Med. (2013) 19:109–15. doi: 10.1097/MCP.0b013e32835ceefc
47. Jin S, Zhao G, Li Z, Nishimoto Y, Isohama Y, Shen J, et al. Age-related pulmonary emphysema in mice lacking alpha/beta hydrolase domain containing 2 gene. Biochem Biophys Res Commun. (2009) 380:419–24. doi: 10.1016/j.bbrc.2009.01.098
48. Rennard SI, Wachenfeldt K. Rationale and emerging approaches for targeting lung repair and regeneration in the treatment of chronic obstructive pulmonary disease. Proc Am Thorac Soc. (2011) 8:368–75. doi: 10.1513/pats.201102-019RM
49. Kanaji N, Basma H, Nelson A, Farid M, Sato T, Nakanishi M, et al. Fibroblasts that resist cigarette smoke-induced senescence acquire profibrotic phenotypes. Am J Physiol Lung Cell Mol Physiol. (2014) 307:L364–73. doi: 10.1152/ajplung.00041.2014
50. Sirianni FE, Milaninezhad A, Chu FS, Walker DC. Alteration of fibroblast architecture and loss of Basal lamina apertures in human emphysematous lung. Am J Respir Crit Care Med. (2006) 173:632–8. doi: 10.1164/rccm.200509-1434OC
51. Togo S, Holz O, Liu X, Sugiura H, Kamio K, Wang X, et al. Lung fibroblast repair functions in patients with chronic obstructive pulmonary disease are altered by multiple mechanisms. Am J Respir Crit Care Med. (2008) 178:248–60. doi: 10.1164/rccm.200706-929OC
52. Stone PJ, Morris SM, Thomas KM, Schuhwerk K, Mitchelson A. Repair of elastase-digested elastic fibers in acellular matrices by replating with neonatal rat-lung lipid interstitial fibroblasts or other elastogenic cell types. Am J Respir Cell Mol Biol. (1997) 17:289–301. doi: 10.1165/ajrcmb.17.3.2597
53. Zhu YK, Liu XD, Sköld CM, Umino T, Wang HJ, Spurzem JR, et al. Synergistic neutrophil elastase-cytokine interaction degrades collagen in three-dimensional culture. Am J Physiol Lung Cell Mol Physiol. (2001) 281:L868–78. doi: 10.1152/ajplung.2001.281.4.L868
54. Sköld CM, Liu X, Umino T, Zhu Y, Ohkuni Y, Romberger DJ, et al. Human neutrophil elastase augments fibroblast-mediated contraction of released collagen gels. Am J Respir Crit Care Med. (1999) 159(Pt. 1):1138–46. doi: 10.1164/ajrccm.159.4.9805033
55. Campbell JD, McDonough JE, Zeskind JE, Hackett TL, Pechkovsky DV, Brandsma CA, et al. A gene expression signature of emphysema-related lung destruction and its reversal by the tripeptide GHK. Genome Med. (2012) 4:67. doi: 10.1186/gm367
56. Nakao A, Imamura T, Souchelnytskyi S, Kawabata M, Ishisaki A, Oeda E, et al. TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. Embo J. (1997) 16:5353–62. doi: 10.1093/emboj/16.17.5353
57. Leppäranta O, Myllärniemi M, Salmenkivi K, Kinnula VL, Keski-Oja J, Koli K. Reduced phosphorylation of the TGF-Beta signal transducer Smad2 in emphysematous human lung. Copd. (2009) 6:234–41. doi: 10.1080/15412550903049173
58. Niewoehner DE, Hoidal JR. Lung fibrosis and emphysema: divergent responses to a common injury? Science. (1982) 217:359–60. doi: 10.1126/science.7089570
59. Mizuno S, Yasuo M, Bogaard HJ, Kraskauskas D, Alhussaini A, Gomez-Arroyo J, et al. Copper deficiency induced emphysema is associated with focal adhesion kinase inactivation. PLoS ONE. (2012) 7:e30678. doi: 10.1371/journal.pone.0030678
60. Besiktepe N, Kayalar O, Ersen E, Oztay F. The copper dependent-lysyl oxidases contribute to the pathogenesis of pulmonary emphysema in chronic obstructive pulmonary disease patients. J Trace Elem Med Biol. (2017) 44:247–55. doi: 10.1016/j.jtemb.2017.08.011
61. Soskel NT, Watanabe S, Sandberg LB. Mechanisms of lung injury in the copper-deficient hamster model of emphysema. Chest. (1984) 85(Suppl. 6):70–3s. doi: 10.1378/chest.85.6_Supplement.70S
62. Liu L, Geng X, McDermott J, Shen J, Corbin C, Xuan S, et al. Copper deficiency in the lungs of TNF-α transgenic mice. Front Physiol. (2016) 7:234. doi: 10.3389/fphys.2016.00234
63. Pillai SG, Ge D, Zhu G, Kong X, Shianna KV, Need AC, et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet. (2009) 5:e1000421. doi: 10.1371/journal.pgen.1000421
64. Zhou X, Qiu W, Sathirapongsasuti JF, Cho MH, Mancini JD, Lao T, et al. Gene expression analysis uncovers novel hedgehog interacting protein (HHIP) effects in human bronchial epithelial cells. Genomics. (2013) 101:263–72. doi: 10.1016/j.ygeno.2013.02.010
65. Yao H, Rahman I. Current concepts on oxidative/carbonyl stress, inflammation and epigenetics in pathogenesis of chronic obstructive pulmonary disease. Toxicol Appl Pharmacol. (2011) 254:72–85. doi: 10.1016/j.taap.2009.10.022
Keywords: chronic obstructive pulmonary disease, emphysematous phenotype, pathogenesis, diagnosis, wound healing
Citation: Bai S and Zhao L (2021) Imbalance Between Injury and Defense in the COPD Emphysematous Phenotype. Front. Med. 8:653332. doi: 10.3389/fmed.2021.653332
Received: 27 January 2021; Accepted: 10 March 2021;
Published: 05 May 2021.
Edited by:
Konstantinos Kostikas, University of Ioannina, GreeceReviewed by:
Chris Kyriakopoulos, University Hospital of Ioannina, GreeceKonstantinos Bartziokas, University Hospital of Ioannina, Greece
Copyright © 2021 Bai and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Zhao, bHpoYW9saUAxNjMuY29t