
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Med. , 23 March 2021
Sec. Gastroenterology
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.643973
This article is part of the Research Topic Loss of Epithelial Barrier Integrity in Inflammatory Diseases: Cellular Mediators and Therapeutic Targets View all 13 articles
 Katrin Sommer1
Katrin Sommer1 Maximilian Wiendl1
Maximilian Wiendl1 Tanja M. Müller1
Tanja M. Müller1 Karin Heidbreder1
Karin Heidbreder1 Caroline Voskens2,3
Caroline Voskens2,3 Markus F. Neurath1,3
Markus F. Neurath1,3 Sebastian Zundler1,3*
Sebastian Zundler1,3*The intestinal epithelial barrier is carrying out two major functions: restricting the entry of potentially harmful substances while on the other hand allowing the selective passage of nutrients. Thus, an intact epithelial barrier is vital to preserve the integrity of the host and to prevent development of disease. Vice versa, an impaired intestinal epithelial barrier function is a hallmark in the development and perpetuation of inflammatory bowel disease (IBD). Besides a multitude of genetic, molecular and cellular alterations predisposing for or driving barrier dysintegrity in IBD, the appearance of intestinal mucosal wounds is a characteristic event of intestinal inflammation apparently inducing breakdown of the intestinal epithelial barrier. Upon injury, the intestinal mucosa undergoes a wound healing process counteracting this breakdown, which is controlled by complex mechanisms such as epithelial restitution, proliferation and differentiation, but also immune cells like macrophages, granulocytes and lymphocytes. Consequently, the repair of mucosal wounds is dependent on a series of events including coordinated trafficking of immune cells to dedicated sites and complex interactions among the cellular players and other mediators involved. Therefore, a better understanding of the crosstalk between epithelial and immune cells as well as cell trafficking during intestinal wound repair is necessary for the development of improved future therapies. In this review, we summarize current concepts on intestinal mucosal wound healing introducing the main cellular mediators and their interplay as well as their trafficking characteristics, before finally discussing the clinical relevance and translational approaches to therapeutically target this process in a clinical setting.
The intestinal mucosa forms a tight barrier with two opposing functions. While it is selectively permeable allowing the absorption of nutrients, it also separates the host from luminal toxins, antigens and microbes that potentially promote disease [reviewed in (1)]. Upon mucosal damage, the epithelial barrier gets leaky facilitating the translocation and therefore excessive exposure of deeper layers of the mucosa to intestinal microbial antigens. This may lead to the recruitment of immune cells releasing different cytokines and may result in disturbed homeostasis [further reviewed in (2, 3)]. Therefore, the regulation of the epithelial barrier function is essential to maintain mucosal homeostasis.
A variety of factors may potentially contribute to mucosal damage, including environmental factors, medication, diet, the host microbiota, infections like HIV as well as genetic factors such as polymorphisms in the CDH1 gene encoding E-Cadherin, which is associated with increased risk to develop ulcerative colitis (UC) [reviewed in (4, 5)]. In general, the pathogenesis of several chronic inflammatory diseases including the inflammatory bowel diseases (IBD) UC and Crohn's disease (CD) is associated with a dysfunctional intestinal epithelial barrier as well as insufficient and delayed mucosal wound healing (6–9). Particularly, wound repair as a pre-requisite to re-establish the mucosal epithelial barrier and intestinal homeostasis is crucial for efficient resolution of inflammation. Hence, mucosal healing (MH) is an increasingly acknowledged goal in IBD therapy in order to achieve and maintain long-term remission. However, mucosal repair and wound healing are complex processes coordinated by the dynamic crosstalk of different cellular players including epithelial cells and infiltrating immune cells as well as their mediators [reviewed in (10)] that are still incompletely understood. A better understanding of these interactions might therefore help to develop tissue-specific approaches to promote wound healing and to treat intestinal inflammation.
In the following paragraphs, we will review the current concepts of intestinal mucosal wound healing, shedding light on the contribution of infiltrating immune cells and their interaction with epithelial cells. Finally, we highlight the clinical relevance of MH and translational approaches to therapeutically target this process.
Intestinal epithelial wound healing is a complex process modulated by various regulatory peptides, including growth factors (GF), and cytokines. Three different phases can be distinguished: Restitution, proliferation, and differentiation and maturation. However, in vivo, these processes merge into each other and overlap [reviewed in (11)].
First, epithelial cells surrounding the wound migrate rapidly into the denuded area, form pseudopodia-like structures, re-organize themselves in order to extend into the wound and then re-differentiate after closing the wound defect. This process is termed epithelial restitution and occurs within minutes to hours [reviewed in (12)]. Interestingly, restitution is independent of cell proliferation and one of the most important stimulators of intestinal epithelial cell (IEC) restitution is transforming growth factor β (TGF-β) (13–15). Within the intestinal mucosa, TGF-β is produced by different cell types including epithelial cells, stromal cells, regulatory T cells (Tregs), dendritic cells (DC) and macrophages [reviewed in (16)]. Once TGF-β is activated, it enhances restitution by upregulating the expression of matrix metalloproteinase-1 (MMP-1), MMP-10 and a set of genes, including Slc28a2, Tubb2a, and Cpe that are preferentially expressed in fetal IECs (17, 18). Furthermore, mediators, such as vascular endothelial growth factor (VEGF), which are released from the inflamed mucosa, are involved in epithelial cell migration in a TGF-β-dependent manner (19). In addition, it was shown that amino acids like histidine and arginine play an important role in TGF-β-mediated IEC restitution probably via interaction with Smad signaling (20). Furthermore, Lopetuso et al. (21) showed that during acute resolving colitis, IL-33/ST2 promote epithelial repair and restitution by inducing miR-320. It had earlier been demonstrated that miR-320 is decreased in the context of intestinal inflammation, suggesting that this might lead to an inherent defect of epithelial repair (22). Recently, Desmocollin-2 (Dsc2), a desmosomal cadherin exclusively expressed on IECs, was identified as a further key contributor to IEC migration and restitution in vivo (23).
In order to increase the number of cells able to resurface the wound area, proliferation is necessary and occurs within hours or days [reviewed in (12)]. This phase is pre-dominantly promoted by various GFs, such as epidermal growth factor (EGF), keratinocyte growth factor (KGF), and fibroblast growth factor (FGF) (24–27), as well as different cytokines including IL-28, which was shown to control proliferation of IECs by activating STAT1 (28), and IL-22, which induces STAT3 signaling, an important regulator of immune homeostasis and mucosal wound healing in the gut (29). Moreover, TLR2 was shown to suppress apoptosis of IECs in vivo by selectively regulating trefoil factor 3 (TFF) expression and controlling intestinal epithelial wound repair by modulating epithelial connexin-43 (30, 31).
Finally, differentiation and maturation is needed to re-establish and maintain the mucosal barrier function. Under normal conditions, Lgr5+ intestinal stem cells (ISCs), which are located at the base of the crypts, differentiate into short-lived proliferating transit-amplifying progenitors, which further differentiate into absorptive (enterocyte) and secretory progenitors under the control of Wnt/Notch signaling [reviewed in (32, 33)]. Secretory precursors then develop into enteroendocrine cells in a Neurog3-dependent manner or into Goblet or Paneth cells following activation of Atoh1 also known as Math1. Later on, the different cell types acquire their lineage-specific expression of transcription factors (TFs), such as Sox9 for Paneth cells and Klf4 for Goblet cells (34–36). It is also worth mentioning, that there are two distinct ISC populations: Crypt base columnar (CBC) cells, which are actively proliferating and reserve intestinal stem cells (rISC) that are quiescent stem cells until activated upon injury. In line with this, Gonzalez et al. (37) showed that Hopx+ cells (rISC) are resistant to injury and are the likely source of epithelial renewal following prolonged ischemic injury (37).
Furthermore, host-microbiota interactions may substantially affect proliferation of epithelial cells and are implicated in intestinal barrier function. E.g., short chain fatty acids (SCFAs) produced by commensal bacteria promote proliferation and differentiation of cells along the crypt-villus axis and, thus, contribute to epithelial restitution (38). Moreover, they are also directly implicated in upholding epithelial integrity to counteract tissue damage (39). In addition to these direct effects on epithelial cells, SCFAs also profoundly impact on the differentiation of mucosal T cells and induce Tregs (40), which are involved in mucosal wound healing as described below. Further details on this emerging field are reviewed elsewhere [reviewed in (41, 42)].
Another important cellular mechanism that should be considered in the context of intestinal epithelial wound healing is epithelial-mesenchymal transition (EMT). During this process epithelial cells lose some of their epithelial characteristics, such as polarity and adhesiveness and acquire migratory functions and properties of mesenchymal cells. This transformation is characterized by the interplay of different mediators like TFs, RNAs, and TGF-β family proteins [reviewed in (43)]. In IBD patients, Leeb et al. (44) reported a reduced migratory ability of fibroblasts, which are normally essential in wound contraction during the initial phase of wound healing (44, 45). Based on these findings it is conceivable that epithelial cells are forced to undergo EMT in order to compensate fibroblast dysfunction and to rapidly restore the intestinal barrier function, which, in turn, might predispose for CD-associated fistulae formation (46).
Cytokines and other mediators secreted by different T cell subsets play essential roles in wound healing (see Figure 1). Diverse injury models in mice (including models focusing on other organs than the gut, for which evidence is limited) show that depletion of Tregs during different phases of wound healing leads to a worse clinical outcome suggesting that they play an important role in the regulation of wound healing probably by counteracting pro-inflammatory stimuli (47–52). Nosbaum et al. (53) showed that Tregs in cutaneous wounds attenuated Interferon-γ (IFN-γ) production and reduced the accumulation of pro-inflammatory macrophages. Their elimination resulted in delayed wound re-epithelialization and wound closure. IFN-γ had previously been shown to affect epithelial intercellular junctions and to attenuate intestinal epithelial wound closure by inhibiting epithelial cell migration in a β1 integrin-dependent mechanism (54, 55). Nosbaum et al. (53) were also able to show that, mechanistically, Tregs induced the expression of EGFR early after wounding, and lineage-specific deletion of EGFR in Tregs resulted in a reduced accumulation and activation as well as increased accumulation of pro-inflammatory macrophages. Furthermore, there is evidence that FGF2 produced by Tregs together with IL-17 is involved in gene regulation to repair damaged cutaneous and intestinal epithelium (53, 56). Moreover, CD4+CD25+Foxp3+ Tregs isolated from peripheral blood of healthy individuals were reported to induce a phenotypical switch of human monocytes/macrophages to wound healing macrophages (57). Following IL-33 release from damaged epithelia, the GF amphiregulin is another mediator produced by Tregs, which is involved in limiting inflammation and promoting epithelial repair (47, 58).
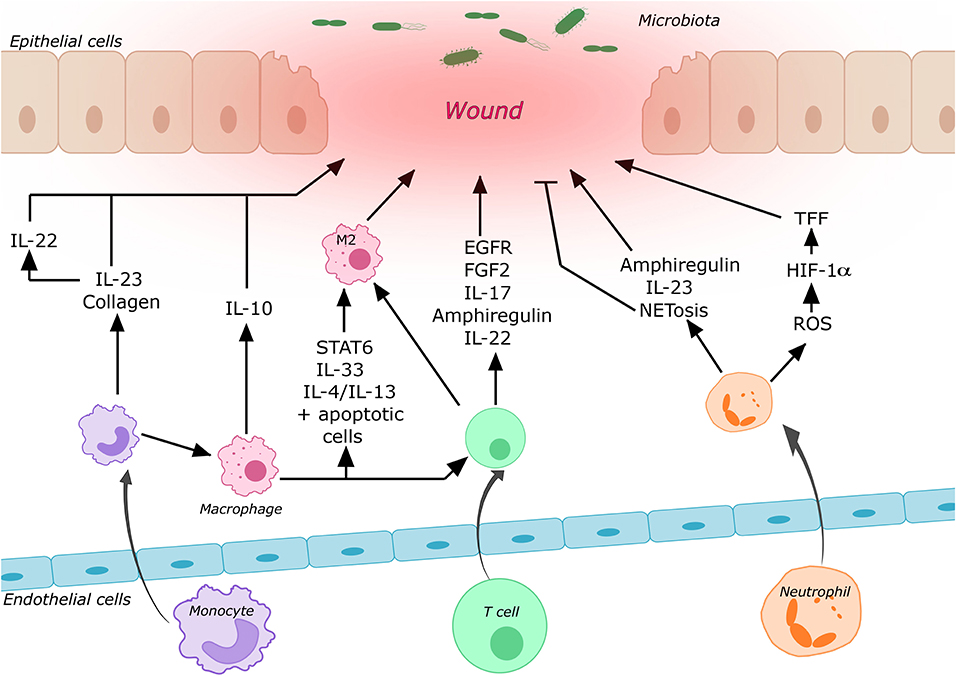
Figure 1. Contribution of some of the most important immune cells to intestinal wound healing. Circulating immune cells are recruited to the wound area by cell trafficking processes. After entering the tissue these cells may undergo differentiation processes and secrete various mediators, which promote or repress mucosal wound healing (for details cf. main text).
Other important cell types involved in intestinal mucosal wound healing are T helper cells (TH) and innate lymphoid cells (ILCs). IL-22 is produced by TH17 and TH22 cells as well as by group 3 ILCs (ILC3) at mucosal surfaces and is a key mediator of this process [reviewed in (59)]. By activating STAT3, IL-22 can not only accelerate proliferation of IECs, but also induce the expression of mucus-associated molecules and the restitution of mucus-producing cells (29, 60). Specifically, IL-22 produced by ILC3s after intestinal injury has been shown to activate intestinal stem cells to promote regeneration (61). Upstream, upon tissue damage, IL-23 may be released leading to the production of IL-22 by ILC3s (62). In line with this, mice deficient for IL-36γ, a potent inducer of IL-23, showed reduced levels of IL-22 and failed to recover from acute intestinal damage. This impaired recovery could be rescued by exogenous IL-23 application (63).
ILC1 show a similar cytokine expression pattern as TH1 cells and mainly exhibit their function by secreting tumor necrosis factor α (TNF-α) and IFN-γ to recruit and activate other inflammatory cells (64). As mentioned above, IFN-γ is also involved in the regulation of epithelial barrier integrity (54, 55). Thus, it is not surprising that depletion of intraepithelial ILC1s was associated with reduced proximal colon inflammation in a mouse model of colitis (65).
By contrast, ILC2s produce TH2-cell-associated cytokines including IL-4, IL-5, IL-9, and IL-13 [reviewed in (66, 67)]. Upon stimulation by IL-33 and similar to Tregs, ILC2s produce amphiregulin, which was shown to promote intestinal epithelial cell regeneration in dextran sodium sulfate (DSS)-treated mice (58).
Furthermore, γδ T cells need to be considered when talking about intestinal wound healing as they are the major source of KGF in the mucosa. KGF released from intraepithelial γδ T cells is important for maintaining intestinal epithelial cell proliferation and villus growth, for promoting the repair of epithelial lesions and is also involved in epithelial cell differentiation (68). It was shown that mice lacking γδ T cells have increased susceptibility to DSS-induced colitis and reduced ability to repair damaged epithelia (69). In line with this, Chen et al. (70) found that intraepithelial γδ T cells preserve the integrity of damaged epithelial surfaces by localized delivery of KGF (70, 71).
Neutrophils play a crucial role in the first line of defense against microbes. Their antimicrobial machineries include the formation of neutrophil extracellular traps called “NETs” (72) and the elimination of invading microbes through phagocytosis, degranulation and production of reactive oxygen species (ROS) [reviewed in (73)]. These mechanisms are essential for wound healing by on the one hand preventing infection through pathogen translocation, and on the other hand by mediating the early so-called inflammatory phase of wound healing. The recruitment of murine neutrophils to the site of cutaneous injury begins 4 h after the initial injury and peaks after 18 h (74). Depletion of neutrophils in damaged mucosa was shown to lead to a severer colitis as well as impaired recovery and restoration of epithelial integrity (75–77). Furthermore, it was shown that neutrophils enhance the production of amphiregulin by IECs promoting epithelial barrier function and tissue repair (75). Another mechanism contributing to the wound healing properties of neutrophils is their ability to generate a hypoxic microenvironment within the wounded tissue by producing ROS, which in turn leads to the stabilization of HIF-1α in the intestinal mucosa (78). HIF-1α was shown to enhance the epithelial expression of TFF3, which has a barrier-protective function (79). In addition, HIF-1α as a TF promotes the upregulation of genes involved in wound healing including adhesion proteins, different GFs and extracellular matrix components [reviewed in (80)]. Moreover, neutrophils produce IL-22 and IL-23, which are both essential mediators of wound healing as mentioned above (77, 81, 82).
However, neutrophils may also have a negative impact on wound healing. For instance, it was shown that counteracting the alarmin HMGB1 leads to reduced NET formation resulting in improved wound healing and inhibition of NETosis improves wound healing in diabetic mice (83). Furthermore, the accumulation of double strand breaks in the mucosa induced by neutrophils led to impaired wound healing and genomic instability (84). In summary, the effects of neutrophil in this process can be seen as a double-edged sword.
Circulating monocytes are rapidly recruited to sites of tissue damage or infection, where they further differentiate into inflammatory M1-like macrophages or wound healing M2-like macrophages. Although this classification has been used to explain many experimental observations, it is meanwhile regarded as oversimplification (85).
While the level of CD16 and CD14 expression can be used to differentiate three different monocyte subsets in humans, they are divided into two subpopulations based on their surface expression of Ly6C and/or CX3CR1 in mice (86, 87). Ly6Chi monocytes were shown to be more dominant in the early inflammatory phase exhibiting phagocytic and inflammatory functions, whereas Ly6Clow monocytes dominate the later phase displaying anti-inflammatory properties and promoting healing (88). The supportive role of macrophages for barrier function was shown by their ability to increase transepithelial electric resistance and cell height of enteroid monolayers (89). Depletion of macrophages in different mouse models led to severely altered wound morphology, delayed re-epithelialization, reduced collagen deposition, impaired angiogenesis, and decreased cell proliferation in the healing wounds (90, 91). Due to their heterogeneity, macrophages play essential roles in all phases of wound repair. More specifically, depletion after the inflammatory phase increased injury and delayed regeneration while depletion in the early inflammatory phase significantly reduced the formation of vascularized granulation tissue, impaired epithelialization, but also resulted in reduced scar formation in kidneys and skin (92, 93). As mentioned above, IL-23 is an important mediator of wound healing and macrophages were identified as a major source of this cytokine (94). Furthermore, the release of IL-10 by macrophages leads to endothelial cell proliferation and activation of epithelial pro-proliferative pathways in the intestine (95). Interestingly, monocytes and macrophages express virtually all known collagen and collagen-related mRNAs, which is essential for the remodeling phase of wound healing (96). Macrophages also have an impact on other immune cells, e.g., by inducing the differentiation of Foxp3+ Tregs in the lamina propria (97).
The polarization of macrophages to a wound healing phenotype is essential for repair processes and is regulated by different mediators. Blockade of IL-1β was shown to prime the generation of M2-like macrophages in diabetic mice and IL-33 significantly enhanced intestinal wound healing by promoting the M2 phenotype (98, 99). Moreover, STAT6-mediated M2 polarization promoted repair in 2,4,6-trinitrobenzenesulfonic acid (TNBS) treated mice through activation of the Wnt signaling pathway (100). In addition, IL-4 or IL-13 in combination with apoptotic cells are capable of activating wound healing macrophages. In the absence of apoptotic signals, the proliferation of tissue-resident macrophages, the induction of anti-inflammatory and tissue repair genes are impaired after induction of colitis (101). Recently, Fpr2/3, which is expressed by epithelial cells was shown to regulate the migration of monocytes to sites of mucosal injury, and CX3CR1 was important for the accumulation of macrophages in the wound (102).
However, monocytes and macrophages may also have negative effects on the epithelial barrier. Mononuclear phagocytes interact with IECs by E-Cadherin leading to dysregulated epithelial cell differentiation and intestinal inflammation by disrupting mucosal homeostasis (103, 104). In line with this, a combination of paracrine and hetero-cellular communication between IECs and macrophages was suggested to play a pivotal role in regulating epithelial cell function and dysregulation of intestinal epithelial barrier (105). Sablet et al. demonstrated that inflammatory monocytes contribute to the loss of intestinal barrier function during cryptosporidiosis by producing TNF-α and IL-1β (106).
Taken together, macrophages are crucially involved in many aspects of intestinal wound healing. Depending on their polarization and the phase of wound healing, they may either promote wound closure or predispose for dysregulation of MH.
As all of the immune cells discussed in the scope of this review are circulating cells or descendants from such cells, there is an obvious need of trafficking for these effectors to reach the site of insult. Thus, cell trafficking should be considered as an integral part of wound healing processes and will shortly be reviewed here.
Described in greater detail elsewhere, cell trafficking describes all processes that are involved in the localization of cells and therefore comprises cellular influx to, retention in and egress from effector tissues [as reviewed in (3, 107)]. Influx from the circulation is regulated by a tightly controlled multistep adhesion cascade. As a prerequisite for transmigration through the endothelium, interaction of selectins and their respective ligands on endothelial cells recruit circulating cells to the vessel walls of high endothelial venules (HEVs) leading to rolling and reduced velocity (108). This slow-down increases the availability of circulating cells to chemotactic stimuli, especially to chemokines, thereby enabling chemokine-induced conformational changes of heterodimeric integrins. Activated integrins are able to firmly bind to endothelial cell adhesion molecules, leading to the arrest of circulating cells on the vessel wall and subsequent para- or intracellular transmigration and target tissue invasion (109).
With regard to gut homing, the α4β7 integrin-mucosal vascular addressin cell adhesion molecule 1 (MAdCAM-1) axis was identified as important due to the virtually exclusive expression of MAdCAM-1 on HEVs of the intestinal tract (110). The relevance of this axis in intestinal wound healing was recently demonstrated, as anti-α4β7 antibody treatment of mice in a colon wound model led to impaired intestinal wound closure, most likely due to reduced homing of non-classical monocytes (NCMs) and a reduction of NCM-derived wound healing macrophages (111). Further, gut specificity in trafficking processes may be provided by the exclusive expression of chemokines in the intestine, as for instance of the CCR9 ligand CCL25 and the GPR15 ligand in the small and large intestine, respectively (112, 113). Their participation in cell recruitment to intestinal wounds has not been studied so far and needs to be further elucidated. Interestingly, both α4β7 and CCR9 are induced on gut-homing T cells through retinoic acid (RA) produced by dendritic cells in the gut-associated lymphoid tissue. With regard to ILCs, it has been shown that this is the case only for ILC1s and ILC3s, while α4β7 expression on ILC2s occurs independent of RA and is already induced in the bone marrow (114). In connection with the above-mentioned roles of ILCs in wound healing, it is tempting to speculate that this might lead to continuous gut homing of amphiregulin-secreting ILC2s promoting homeostasis, while ILC3 recruitment might be regulated by the level of inflammation present. However, it is difficult to envision the consequences for wound healing, since ILC3s not only promote mucosal repair through IL-22, but may also promote inflammation and, thus, secondary tissue injury (115).
Retention of homed cells within the target tissues is either controlled indirectly by the regulation of egress signal receptors or by direct anchoring to tissue structures. A key example of indirect retention is the interaction of CD69 with sphingosine 1-phosophate receptor-1 (S1PR1), leading to degradation of the latter and inhibition of extravasation along the S1P gradient into the bloodstream (116, 117). Further extravasation signals might be provided by the interaction of CCR7 and CCL19 or CCL21, facilitating the recruitment of receptor-bearing cells to the lymphatic system (118, 119). Direct anchoring of recruited cells can be provided by the interaction of integrins with cell adhesion molecules in the tissue. E.g., αE-integrin (CD103) dimerizes with β7-integrin and mediates tissue retention through interaction with E-Cadherin (120, 121). Although the retention of cells in the wound area or their recirculation to the blood will certainly be of relevance in the spatiotemporal orchestration of the wound healing process, since it might lead to the accumulation or reduction of repair-promoting or -impeding cell populations, these mechanisms have not been specifically investigated in this context in the gut. However, and interestingly, there is evidence from skin models, that tissue resident memory T cells (TRM cells), which play important roles in the pathogenesis of IBD (122), promote epithelial wound healing (123, 124).
It is also worth mentioning that mucosal cytokine profiles differ between CD and UC. While UC is dominated by TH2-associated cytokines like IL-5, IL-13, IL-9, and IL-4 (125–129), CD is marked by cytokines, such as IFN-γ and IL-2, associated with a TH1 phenotype (125, 130). TH17 cells and cytokines seem to be involved in both entities (131). At the same time, macroscopic differences in ulcerations between CD and UC exist (132) and inflammation patterns between CD and UC differ with immune cell infiltration restricted to the mucosa of the colon in UC, but being transmural and potentially occurring in the whole gastrointestinal tract in CD [reviewed in (133)]. This strongly suggests that different homing mechanisms apply for immune cells in CD and UC that might also impact wound healing. Interestingly, differences in the expression of gut homing markers on different T cell subsets and differential usage of gut homing pathways in ileal CD as compared to colonic UC have been observed [reviewed in (134, 135)]. However, further dedicated studies are needed to explore this assumption in depth.
Taken together, the implications of immune trafficking for intestinal wound healing are obvious. Particularly, they need to be considered in a therapeutic context, especially when trafficking mechanisms are directly manipulated by antibodies. This also highlights the need for further investigation of the trafficking mechanisms participating in intestinal wound healing.
Mucosal healing (MH), a term coined by Truelove and Witts in 1955 (136), is nowadays considered an important study endpoint and increasingly important treatment goal in IBD. Several clinical trials showed the importance and improved clinical outcomes after achieving MH, defined as absent or low signs of mucosal injury on endoscopy (137–141). In UC and CD, it is associated with long-term remission and reduced need for surgery (142, 143). On tissue level and mechanistically, it is obvious that wound healing and restitution of the intestinal epithelial barrier function are major steps in achieving MH. Consequently, the promotion of wound healing has been suggested as a potential therapeutic tool (144). Calprotectin is a soluble protein in the cytosol of neutrophils and known to be elevated in both the intestinal mucosa and feces of IBD patients (145). Several studies have shown a correlation between low fecal calprotectin (FC) concentration and histological remission as well as MH in UC and CD patients. Therefore, low calprotectin levels might be an early predictor of therapeutic success in terms of MH (146, 147).
One experimental approach to achieve wound healing that was addressed by several studies, but not in the gut, was the promotion of recruitment and polarization of monocytes and wound healing macrophages (148, 149). Maruyama and colleagues (150) showed that upon injection of IL-1β-activated macrophages in mice, the production of VEGF-C was increased and cutaneous wound healing improved. Interestingly, one mechanism of action of corticosteroids is M1 macrophage suppression in response to LPS stimulation, which involves the miR-155 (151). Moreover, neutrophils as cellular mediators can be targeted. In the context of peritonitis, Norling et al. (152) showed that nanoparticles containing aspirin-triggered resolvin D1 or a lipoxin A4 analog reduced polymorphonuclear cell influx and enhanced wound healing. As different GFs like EGF, VEGF, and KGF mediate epithelial repair, they might also be interesting candidates [reviewed in (153)]. Another promising therapeutic approach is targeting IL-22, which is considered to promote epithelial integrity via STAT3. Consequently, an IL-22 IgG4 Fc fusion protein (UTTR1147A) is currently tested in patients with moderate-to-severe UC and CD (ClinicalTrials.gov Identifier: NCT03558152, NCT03650413).
In addition to these experimental concepts, several current IBD treatments were shown to have a protective or regenerative effect on the damaged epithelium and to promote MH [reviewed in (154)]. Aminosalicylates not only affect intestinal inflammation via various signaling pathways such as NF-κB, but also directly stimulate epithelial wound healing by enhancing epithelial cell restitution and proliferation (155–157). Anti-TNF-α antibodies such as infliximab and adalimumab are able to induce and maintain MH (144, 158–160) by restricting the inflammatory infiltrate and T cell proliferation within the lamina propria and by downregulating the expression of metalloproteinases and pro-inflammatory molecules (161). For infliximab, a single nucleotide polymorphism in the TRAP1 gene has been described to be associated with MH in CD patients (162). Moreover, anti-TNF-α antibodies support regenerative processes by reducing inflammation, restoring gut barrier function, mucosal secretion and by activating fibroblasts (163). In addition, it has been suggested that these antibodies mediate Fc region-dependent induction of wound healing macrophages. It was shown that infliximab as well as adalimumab can induce wound healing macrophages in vitro and in vivo (164, 165). Similarly, ustekinumab, a monoclonal antibody directed against IL-12 and IL-23, successfully induced MH in CD patients (166).
As the JAK/STAT pathway seems to play an important role in the interaction of lymphocytes and IECs through a variety of cytokines, it is not surprising that tofacitinib, a JAK inhibitor routinely used in UC treatment, is able to induce and maintain MH (167). Lechner et al. (168) recently demonstrated that tofacitinib specifically reduces pro-inflammatory cytokines that are produced by lamina propria T cells and affects their homing potential by suppressing the surface integrin expression on T cells. However, in an experimental model of intestinal mucosal wounding, high concentrations of tofacitinib rather prolonged wound healing (168), an observation that requires further translational studies to reconcile it with the clinical outcomes.
Another important class of IBD therapeutics are anti-trafficking agents [reviewed in (3)]. Vedolizumab, a humanized monoclonal anti-α4β7 antibody, inhibits the binding and subsequent migration of lymphocytes into the gut (169). The GEMINI I trial showed that significantly more UC patients treated with vedolizumab than with placebo achieved MH (140, 170). However, mechanistic data explaining the impact of vedolizumab on trafficking of cells implicated in wound healing in inflammation are so far missing. Thus, it is not clear, whether this is a direct effect or secondarily resulting from reduced inflammation and associated changes in the balance of cells promoting and counteracting mucosal repair. In seeming contrast to data on MH as a study endpoint assessing control of inflammation, several (but not all) studies reported that patients treated with vedolizumab are more vulnerable to post-operative complications (171–176). A potential explanation might be that, according to a recent study from our group, blocking α4β7 impaired gut homing of NCMs, which was associated with delayed wound healing and reduced perilesional presence of wound healing macrophages (111). It is important to mention that this is not necessarily contradicting the mentioned MH data, since this study exclusively addressed exogenous tissue injury in the absence of inflammation and it is likely that ongoing inflammation will substantially modulate trafficking, communication and signaling pathways.
Collectively, almost all available therapies for the treatment of IBD have demonstrated their potential to induce MH, although it is not clear to what extent this is a result from direct impact on wound healing processes or a secondary effect of the reduction of inflammation. Thus, further mechanistic data and additional efforts to directly promote wound healing and barrier integrity in the context of IBD are necessary.
Intestinal mucosal wound repair are key steps for achieving and maintaining MH, which is associated with beneficial clinical outcomes. However, the interplay as well as the trafficking characteristics of the most important cellular mediators like lymphocytes, neutrophils and monocytes/macrophages are not sufficiently characterized. Further research is necessary in order to better understand the contribution of cell trafficking to mucosal wound repair and to base targeted therapeutic approaches on this process.
KS, MW, TM, KH, and SZ wrote the manuscript. All authors critically revised the manuscript and approved the final version.
The research of MN and SZ was supported by the Interdisciplinary Center for Clinical Research (IZKF) and the ELAN program of the University Erlangen-Nuremberg, the Else Kröner-Fresenius-Stiftung, the Fritz Bender-Stiftung, the Dr. Robert Pfleger Stiftung, the Litwin IBD Pioneers Initiative of the Crohn's and Colitis Foundation of America (CCFA), the Kenneth Rainin Foundation, the Ernst Jung-Stiftung for Science and Research, the German Crohn's and Colitis Foundation (DCCV) and the German Research Foundation (DFG) through individual grants (ZU 377/4-1) and the Collaborative Research Centers TRR241, 643, 796 and 1181.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We acknowledge support by Deutsche Forschungsgemeinschaft and Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) within the funding program Open Access Publishing.
1. Allaire JM, Crowley SM, Law HT, Chang SY, Ko HJ, Vallance BA. The intestinal epithelium: central coordinator of mucosal immunity. Trends Immunol. (2018) 39:677–96. doi: 10.1016/j.it.2018.04.002
2. Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. (2014) 14:329–42. doi: 10.1038/nri3661
3. Zundler S, Becker E, Schulze LL, Neurath MF. Immune cell trafficking and retention in inflammatory bowel disease: mechanistic insights and therapeutic advances. Gut. (2019) 68:1688–700. doi: 10.1136/gutjnl-2018-317977
4. Guan Q. A comprehensive review and update on the pathogenesis of inflammatory bowel disease. J Immunol Res. (2019) 2019:7247238. doi: 10.1155/2019/7247238
5. Crakes KR, Jiang G. Gut microbiome alterations during HIV/SIV infection: implications for HIV cure. Front Microbiol. (2019) 10:1104: doi: 10.3389/fmicb.2019.01104
6. Pearson AD, Eastham EJ, Laker MF, Craft AW, Nelson R. Intestinal permeability in children with Crohn's disease and coeliac disease. Br Med J (Clin Res Ed). (1982) 285:20–1. doi: 10.1136/bmj.285.6334.20
7. Irvine EJ, Marshall JK. Increased intestinal permeability precedes the onset of Crohn's disease in a subject with familial risk. Gastroenterology. (2000) 119:1740–4. doi: 10.1053/gast.2000.20231
8. Hollander D, Vadheim CM, Brettholz E, Petersen GM, Delahunty T, Rotter JI. Increased intestinal permeability in patients with Crohn's disease and their relatives. A possible etiologic factor. Ann Intern Med. (1986) 105:883–5. doi: 10.7326/0003-4819-105-6-883
9. Münch A, Söderholm JD, Ost A, Ström M. Increased transmucosal uptake of E. coli K12 in collagenous colitis persists after budesonide treatment. Am J Gastroenterol. (2009) 104:679–85. doi: 10.14309/00000434-200903000-00028
10. Brazil JC, Quiros M, Nusrat A, Parkos CA. Innate immune cell-epithelial crosstalk during wound repair. J Clin Invest. (2019) 129:2983–93. doi: 10.1172/JCI124618
11. Dignass AU, Baumgart DC, Sturm A. Review article: the aetiopathogenesis of inflammatory bowel disease–immunology and repair mechanisms. Aliment Pharmacol Ther. (2004) 4(Suppl. 20):9–17. doi: 10.1111/j.1365-2036.2004.02047.x
12. Iizuka M, Konno S. Wound healing of intestinal epithelial cells. World J Gastroenterol. (2011) 17:2161–71. doi: 10.3748/wjg.v17.i17.2161
13. Basson MD, Modlin IM, Flynn SD, Jena BP, Madri JA. Independent modulation of enterocyte migration and proliferation by growth factors, matrix proteins, and pharmacologic agents in an in vitro model of mucosal healing. Surgery. (1992) 112:299–307.
14. Dignass AU, Podolsky DK. Cytokine modulation of intestinal epithelial cell restitution: central role of transforming growth factor beta. Gastroenterology. (1993) 105:1323–32. doi: 10.1016/0016-5085(93)90136-Z
15. Beck PL, Rosenberg IM, Xavier RJ, Koh T, Wong JF, Podolsky DK. Transforming growth factor-beta mediates intestinal healing and susceptibility to injury in vitro and in vivo through epithelial cells. Am J Pathol. (2003) 162:597–608. doi: 10.1016/S0002-9440(10)63853-9
16. Shen Y, Zhang C, Chen Y. TGF-β in inflammatory bowel diseases: a tale of the janus-like cytokine. Crit Rev Eukaryot Gene Expr. (2015) 25:335–47. doi: 10.1615/CritRevEukaryotGeneExpr.2015013974
17. Salmela MT, Pender SL, Karjalainen-Lindsberg ML, Puolakkainen P, Macdonald TT, Saarialho-Kere U. Collagenase-1 (MMP-1), matrilysin-1 (MMP-7), and stromelysin-2 (MMP-10) are expressed by migrating enterocytes during intestinal wound healing. Scand J Gastroenterol. (2004) 39:1095–104. doi: 10.1080/00365520410003470
18. Anzai S, Kawamoto A, Nagata S, Takahashi J, Kawai M, Kuno R, et al. TGF-β promotes fetal gene expression and cell migration velocity in a wound repair model of untransformed intestinal epithelial cells. Biochem Biophys Res Commun. (2020) 524:533–41. doi: 10.1016/j.bbrc.2020.01.108
19. Bulut K, Pennartz C, Felderbauer P, Ansorge N, Banasch M, Schmitz F, et al. Vascular endothelial growth factor (VEGF164) ameliorates intestinal epithelial injury in vitro in IEC-18 and Caco-2 monolayers via induction of TGF-beta release from epithelial cells. Scand J Gastroenterol. (2006) 41:687–92. doi: 10.1080/00365520500408634
20. Matsui T, Ichikawa H, Fujita T, Takemura S, Takagi T, Osada-Oka M, et al. Histidine and arginine modulate intestinal cell restitution via transforming growth factor-β(1). Eur J Pharmacol. (2019) 850:35–42. doi: 10.1016/j.ejphar.2019.02.006
21. Lopetuso LR, De Salvo C, Pastorelli L, Rana N, Senkfor HN, Petito V, et al. IL-33 promotes recovery from acute colitis by inducing miR-320 to stimulate epithelial restitution and repair. Proc Natl Acad Sci U S A. (2018) 115:E9362–e9370. doi: 10.1073/pnas.1803613115
22. Pierdomenico M, Cesi V, Cucchiara S, Vitali R, Prete E, Costanzo M, et al. NOD2 is regulated by Mir-320 in physiological conditions but this control is altered in inflamed tissues of patients with inflammatory bowel disease. Inflamm Bowel Dis. (2016) 22:315–26. doi: 10.1097/MIB.0000000000000659
23. Flemming S, Luissint AC, D.Kusters HM, Raya-Sandino A, Fan S, Zhou DW, et al. Desmocollin-2 promotes intestinal mucosal repair by controlling integrin-dependent cell adhesion and migration. Mol Biol Cell. (2020) 31:407–18. doi: 10.1091/mbc.E19-12-0692
24. Dise RS, Frey MR, Whitehead RH, Polk DB. Epidermal growth factor stimulates Rac activation through Src and phosphatidylinositol 3-kinase to promote colonic epithelial cell migration. Am J Physiol Gastrointest Liver Physiol. (2008) 294:G276–85. doi: 10.1152/ajpgi.00340.2007
25. Grotendorst GR, Soma Y, Takehara K, Charette M. EGF and TGF-alpha are potent chemoattractants for endothelial cells and EGF-like peptides are present at sites of tissue regeneration. J Cell Physiol. (1989) 139:617–23. doi: 10.1002/jcp.1041390323
26. Brown GL, Curtsinger L 3rd, Brightwell JR, Ackerman DM, Tobin GR, Polk HC Jr, et al. Enhancement of epidermal regeneration by biosynthetic epidermal growth factor. J Exp Med. (1986) 163:1319–24. doi: 10.1084/jem.163.5.1319
27. Zheng Z, Kang HY, Lee S, Kang SW, Goo B, Cho SB. Up-regulation of fibroblast growth factor (FGF) 9 expression and FGF-WNT/β-catenin signaling in laser-induced wound healing. Wound Repair Regen. (2014) 22:660–5. doi: 10.1111/wrr.12212
28. Chiriac MT, Buchen B, Wandersee A, Hundorfean G, Günther C, Bourjau Y, et al. Activation of epithelial signal transducer and activator of transcription 1 by interleukin 28 controls mucosal healing in mice with colitis and is increased in mucosa of patients with inflammatory bowel disease. Gastroenterology. (2017) 153:123–38.e8. doi: 10.1053/j.gastro.2017.03.015
29. Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. (2009) 206:1465–72. doi: 10.1084/jem.20082683
30. Ey B, Eyking A, Gerken G, Podolsky DK, Cario E. TLR2 mediates gap junctional intercellular communication through connexin-43 in intestinal epithelial barrier injury. J Biol Chem. (2009) 284:22332–43. doi: 10.1074/jbc.M901619200
31. Podolsky DK, Gerken G, Eyking A, Cario E. Colitis-associated variant of TLR2 causes impaired mucosal repair because of TFF3 deficiency. Gastroenterology. (2009) 137:209–20. doi: 10.1053/j.gastro.2009.03.007
32. Lueschow SR, McElroy SJ. The paneth cell: the curator and defender of the immature small intestine. Front Immunol. (2020) 11:587. doi: 10.3389/fimmu.2020.00587
33. Sanman LE, Chen IW, Bieber JM, Steri V, Trentesaux C, Hann B, et al. Transit-amplifying cells coordinate changes in intestinal epithelial cell-type composition. Dev Cell. (2021) 56:356–65.e9. doi: 10.1016/j.devcel.2020.12.020
34. Jenny M, Uhl C, Roche C, Duluc I, Guillermin V, Guillemot F, et al. Neurogenin3 is differentially required for endocrine cell fate specification in the intestinal and gastric epithelium. Embo J. (2002) 21:6338–47. doi: 10.1093/emboj/cdf649
35. Mori-Akiyama Y, van den Born M, van Es JH, Hamilton SR, Adams HP, Zhang J, et al. SOX9 is required for the differentiation of paneth cells in the intestinal epithelium. Gastroenterology. (2007) 133:539–46. doi: 10.1053/j.gastro.2007.05.020
36. Katz JP, Perreault N, Goldstein BG, Lee CS, Labosky PA, Yang VW, et al. The zinc-finger transcription factor Klf4 is required for terminal differentiation of goblet cells in the colon. Development. (2002) 129:2619–28.
37. Gonzalez LM, Stewart AS, Freund J, Kucera CR, Dekaney CM, Magness ST, et al. Preservation of reserve intestinal epithelial stem cells following severe ischemic injury. Am J Physiol Gastrointest Liver Physiol. (2019) 316:G482–g494. doi: 10.1152/ajpgi.00262.2018
38. Park JH, Kotani T, Konno T, Setiawan J, Kitamura Y, Imada S, et al. Promotion of intestinal epithelial cell turnover by commensal bacteria: role of short-chain fatty acids. PLoS ONE. (2016) 11:e0156334. doi: 10.1371/journal.pone.0156334
39. Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, et al. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe. (2015) 17:662–71. doi: 10.1016/j.chom.2015.03.005
40. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. (2013) 504:451–5. doi: 10.1038/nature12726
41. Alam A, Neish A. Role of gut microbiota in intestinal wound healing and barrier function. Tissue Barriers. (2018) 6:1539595. doi: 10.1080/21688370.2018.1539595
42. Kayama H, Okumura R, Takeda K. Interaction between the microbiota, epithelia, and immune cells in the intestine. Annu Rev Immunol. (2020) 38:23–48. doi: 10.1146/annurev-immunol-070119-115104
43. Jiang H, Shen J, Ran Z. Epithelial-mesenchymal transition in Crohn's disease. Mucosal Immunol. (2018) 11:294–303. doi: 10.1038/mi.2017.107
44. Leeb SN, Vogl D, Gunckel M, Kiessling S, Falk W, Göke M, et al. Reduced migration of fibroblasts in inflammatory bowel disease: role of inflammatory mediators and focal adhesion kinase. Gastroenterology. (2003) 125:1341–54. doi: 10.1016/j.gastro.2003.07.004
45. Bainbridge P. Wound healing and the role of fibroblasts. J Wound Care. (2013) 22:407–8, 410–12. doi: 10.12968/jowc.2013.22.8.407
46. Bataille F, Rohrmeier C, Bates R, Weber A, Rieder F, Brenmoehl J, et al. Evidence for a role of epithelial mesenchymal transition during pathogenesis of fistulae in Crohn's disease. Inflamm Bowel Dis. (2008) 14:1514–27. doi: 10.1002/ibd.20590
47. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A special population of regulatory T cells potentiates muscle repair. Cell. (2013) 155:1282–95. doi: 10.1016/j.cell.2013.10.054
48. Ito M, Komai K, Mise-Omata S, Iizuka-Koga M, Noguchi Y, Kondo T, et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature. (2019) 565:246–50. doi: 10.1038/s41586-018-0824-5
49. Kinsey GR, Sharma R, Huang L, Li L, Vergis AL, Ye H, et al. Regulatory T cells suppress innate immunity in kidney ischemia-reperfusion injury. J Am Soc Nephrol. (2009) 20:1744–53. doi: 10.1681/ASN.2008111160
50. Weirather J, Hofmann UD, Beyersdorf N, Ramos GC, Vogel B, Frey A, et al. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res. (2014) 115:55–67. doi: 10.1161/CIRCRESAHA.115.303895
51. Sharir R, Semo J, Shimoni S, Ben-Mordechai T, Landa-Rouben N, Maysel-Auslender S, et al. Experimental myocardial infarction induces altered regulatory T cell hemostasis, and adoptive transfer attenuates subsequent remodeling. PLoS ONE. (2014) 9:e113653. doi: 10.1371/journal.pone.0113653
52. Villalta SA, Rosenthal W, Martinez L, Kaur A, Sparwasser T, Tidball JG, et al. Regulatory T cells suppress muscle inflammation and injury in muscular dystrophy. Sci Transl Med. (2014) 6:258ra142. doi: 10.1126/scitranslmed.3009925
53. Nosbaum A, Prevel N, Truong HA, Mehta P, Ettinger M, Scharschmidt TC, et al. Cutting edge: regulatory T cells facilitate cutaneous wound healing. J Immunol. (2016) 196:2010–4. doi: 10.4049/jimmunol.1502139
54. Tong Q, Vassilieva EV, Ivanov AI, Wang Z, Brown GT, Parkos CA, et al. Interferon-gamma inhibits T84 epithelial cell migration by redirecting transcytosis of beta1 integrin from the migrating leading edge. J Immunol. (2005) 175:4030–8. doi: 10.4049/jimmunol.175.6.4030
55. Bruewer M, Luegering A, Kucharzik T, Parkos CA, Madara JL, Hopkins AM, et al. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol. (2003) 171:6164–72. doi: 10.4049/jimmunol.171.11.6164
56. Song X, Dai D, He X, Zhu S, Yao Y, Gao H, et al. Growth factor FGF2 cooperates with interleukin-17 to repair intestinal epithelial damage. Immunity. (2015) 43:488–501. doi: 10.1016/j.immuni.2015.06.024
57. Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci U S A. (2007) 104:19446–51. doi: 10.1073/pnas.0706832104
58. Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DM, Artis D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci U S A. (2015) 112:10762–7. doi: 10.1073/pnas.1509070112
59. Rutz S, Eidenschenk C, Ouyang W. IL-22, not simply a Th17 cytokine. Immunol Rev. (2013) 252:116–32. doi: 10.1111/imr.12027
60. Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. (2008) 118:534–44. doi: 10.1172/JCI33194
61. Lindemans CA, Calafiore M, Mertelsmann AM, O'Connor MH, Dudakov JA, Jenq RR, et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature. (2015) 528:560–4. doi: 10.1038/nature16460
62. Hanash AM, Dudakov JA, Hua G, O'Connor MH, Young LF, Singer NV, et al. Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity. (2012) 37:339–50. doi: 10.1016/j.immuni.2012.05.028
63. Ngo VL, Abo H, Maxim E, Harusato A, Geem D, Medina-Contreras O, et al. A cytokine network involving IL-36γ, IL-23, and IL-22 promotes antimicrobial defense and recovery from intestinal barrier damage. Proc Natl Acad Sci U S A. (2018) 115:E5076–e5085. doi: 10.1073/pnas.1718902115
64. Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol. (2013) 14:221–9. doi: 10.1038/ni.2534
65. Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-γ-producing cells. Immunity. (2013) 38:769–81. doi: 10.1016/j.immuni.2013.02.010
66. Xue X, Falcon DM. The role of immune cells and cytokines in intestinal wound healing. Int J Mol Sci. (2019) 20:6097: doi: 10.3390/ijms20236097
67. Gurram RK, Zhu J. Orchestration between ILC2s and Th2 cells in shaping type 2 immune responses. Cell Mol Immunol. (2019) 16:225–35. doi: 10.1038/s41423-019-0210-8
68. Yang H, Antony PA, Wildhaber BE, Teitelbaum DH. Intestinal intraepithelial lymphocyte gamma delta-T cell-derived keratinocyte growth factor modulates epithelial growth in the mouse. J Immunol. (2004) 172:4151–8. doi: 10.4049/jimmunol.172.7.4151
69. Kober OI, Ahl D, Pin C, Holm L, Carding SR, Juge N. γδ T-cell-deficient mice show alterations in mucin expression, glycosylation, and goblet cells but maintain an intact mucus layer. Am J Physiol Gastrointest Liver Physiol. (2014) 306:G582–93. doi: 10.1152/ajpgi.00218.2013
70. Chen Y, Chou K, Fuchs E, Havran WL, Boismenu R. Protection of the intestinal mucosa by intraepithelial gamma delta T cells. Proc Natl Acad Sci U S A. (2002) 99:14338–43. doi: 10.1073/pnas.212290499
71. Boismenu R, Feng L, Xia YY, Chang JC, Havran WL. Chemokine expression by intraepithelial gamma delta T cells. Implications for the recruitment of inflammatory cells to damaged epithelia. J Immunol. (1996) 157:985–92.
72. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
73. Liew PX, Kubes P. The neutrophil's role during health and disease. Physiol Rev. (2019) 99:1223–48. doi: 10.1152/physrev.00012.2018
74. Kim MH, Liu W, Borjesson DL, Curry FR, Miller LS, Cheung AL, et al. Dynamics of neutrophil infiltration during cutaneous wound healing and infection using fluorescence imaging. J Invest Dermatol. (2008) 128:1812–20. doi: 10.1038/sj.jid.5701223
75. Chen F, Yang W, Huang X, Cao AT, Bilotta AJ, Xiao Y, et al. Neutrophils promote amphiregulin production in intestinal epithelial cells through TGF-β and contribute to intestinal homeostasis. J Immunol. (2018) 201:2492–501. doi: 10.4049/jimmunol.1800003
76. Kühl AA, Kakirman H, Janotta M, Dreher S, Cremer P, Pawlowski NN, et al. Aggravation of different types of experimental colitis by depletion or adhesion blockade of neutrophils. Gastroenterology. (2007) 133:1882–92. doi: 10.1053/j.gastro.2007.08.073
77. Zindl CL, Lai JF, Lee YK, Maynard CL, Harbour SN, Ouyang W, et al. IL-22-producing neutrophils contribute to antimicrobial defense and restitution of colonic epithelial integrity during colitis. Proc Natl Acad Sci U S A. (2013) 110:12768–73. doi: 10.1073/pnas.1300318110
78. Campbell EL, Bruyninckx WJ, Kelly CJ, Glover LE, McNamee EN, Bowers BE, et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity. (2014) 40:66–77. doi: 10.1016/j.immuni.2013.11.020
79. Furuta GT, Turner JR, Taylor CT, Hershberg RM, Comerford K, Narravula S, et al. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J Exp Med. (2001) 193:1027–34. doi: 10.1084/jem.193.9.1027
80. Hong WX, Hu MS, Esquivel M, Liang GY, Rennert RC, McArdle A, et al. The role of hypoxia-inducible factor in wound healing. Adv Wound Care (New Rochelle). (2014) 3:390–9. doi: 10.1089/wound.2013.0520
81. Zhou G, Yu L, Fang L, Yang W, Yu T, Miao Y, et al. CD177(+) neutrophils as functionally activated neutrophils negatively regulate IBD. Gut. (2018) 67:1052–63. doi: 10.1136/gutjnl-2016-313535
82. Kvedaraite E, Lourda M, Ideström M, Chen P, Olsson-Åkefeldt S, Forkel M, et al. Tissue-infiltrating neutrophils represent the main source of IL-23 in the colon of patients with IBD. Gut. (2016) 65:1632–41. doi: 10.1136/gutjnl-2014-309014
83. Lee YS, Kang SU, Lee MH, Kim HJ, Han CH, Won HR, et al. GnRH impairs diabetic wound healing through enhanced NETosis. Cell Mol Immunol. (2020) 17:856–64. doi: 10.1038/s41423-019-0252-y
84. Butin-Israeli V, Bui TM, Wiesolek HL, Mascarenhas L, Lee JJ, Mehl LC, et al. Neutrophil-induced genomic instability impedes resolution of inflammation and wound healing. J Clin Invest. (2019) 129:712–26. doi: 10.1172/JCI122085
85. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. (2010) 11:889–96. doi: 10.1038/ni.1937
86. Shaw TN, Houston SA, Wemyss K, Bridgeman HM, Barbera TA, Zangerle-Murray T, et al. Tissue-resident macrophages in the intestine are long lived and defined by Tim-4 and CD4 expression. J Exp Med. (2018) 215:1507–18. doi: 10.1084/jem.20180019
87. Bain CC, Bravo-Blas A, Scott CL, Perdiguero EG, Geissmann F, Henri S, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol. (2014) 15:929–37. doi: 10.1038/ni.2967
88. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. (2007) 204:3037–47. doi: 10.1084/jem.20070885
89. Noel G, Baetz NW, Staab JF, Donowitz M, Kovbasnjuk O, Pasetti MF, et al. A primary human macrophage-enteroid co-culture model to investigate mucosal gut physiology and host-pathogen interactions. Sci Rep. (2017) 7:45270. doi: 10.1038/srep45270
90. Goren I, Allmann N, Yogev N, Schürmann C, Linke A, Holdener M, et al. A transgenic mouse model of inducible macrophage depletion: effects of diphtheria toxin-driven lysozyme M-specific cell lineage ablation on wound inflammatory, angiogenic, contractive processes. Am J Pathol. (2009) 175:132–47. doi: 10.2353/ajpath.2009.081002
91. Mirza R, DiPietro LA, Koh TJ. Selective and specific macrophage ablation is detrimental to wound healing in mice. Am J Pathol. (2009) 175:2454–62. doi: 10.2353/ajpath.2009.090248
92. Zhang MZ, Yao B, Yang S, Jiang L, Wang S, Fan X, et al. CSF-1 signaling mediates recovery from acute kidney injury. J Clin Invest. (2012) 122:4519–32. doi: 10.1172/JCI60363
93. Lucas T, Waisman A, Ranjan R, Roes J, Krieg T, Müller W, et al. Differential roles of macrophages in diverse phases of skin repair. J Immunol. (2010) 184:3964–77. doi: 10.4049/jimmunol.0903356
94. Arnold IC, Mathisen S, Schulthess J, Danne C, Hegazy AN, Powrie F. CD11c(+) monocyte/macrophages promote chronic Helicobacter hepaticus-induced intestinal inflammation through the production of IL-23. Mucosal Immunol. (2016) 9:352–63. doi: 10.1038/mi.2015.65
95. Quiros M, Nishio H, Neumann PA, Siuda D, Brazil JC, Azcutia V, et al. Macrophage-derived IL-10 mediates mucosal repair by epithelial WISP-1 signaling. J Clin Invest. (2017) 127:3510–20. doi: 10.1172/JCI90229
96. Schnoor M, Cullen P, Lorkowski J, Stolle K, Robenek H, Troyer D, et al. Production of type VI collagen by human macrophages: a new dimension in macrophage functional heterogeneity. J Immunol. (2008) 180:5707–19. doi: 10.4049/jimmunol.180.8.5707
97. Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. (2007) 8:1086–94. doi: 10.1038/ni1511
98. Mirza RE, Fang MM, Ennis WJ, Koh TJ. Blocking interleukin-1β induces a healing-associated wound macrophage phenotype and improves healing in type 2 diabetes. Diabetes. (2013) 62:2579–87. doi: 10.2337/db12-1450
99. Seo DH, Che X, Kwak MS, Kim S, Kim JH, Ma HW, et al. Interleukin-33 regulates intestinal inflammation by modulating macrophages in inflammatory bowel disease. Sci Rep. (2017) 7:851. doi: 10.1038/s41598-017-00840-2
100. Cosin-Roger J, Ortiz-Masià MD, Barrachina MD. Macrophages as an emerging source of Wnt ligands: relevance in mucosal integrity. Front Immunol. (2019) 10:2297. doi: 10.3389/fimmu.2019.02297
101. Bosurgi L, Cao YG, Cabeza-Cabrerizo M, Tucci A, Hughes LD, Kong Y, et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science. (2017) 356:1072–6. doi: 10.1126/science.aai8132
102. Birkl D, O'Leary MN, Quiros M, Azcutia V, Schaller M, Reed M, et al. Formyl peptide receptor 2 regulates monocyte recruitment to promote intestinal mucosal wound repair. FASEB J. (2019) 33:13632–43. doi: 10.1096/fj.201901163R
103. Ihara S, Hirata Y, Hikiba Y, Yamashita A, Tsuboi M, Hata M, et al. Adhesive interactions between mononuclear phagocytes and intestinal epithelium perturb normal epithelial differentiation and serve as a therapeutic target in inflammatory bowel disease. J Crohns Colitis. (2018) 12:1219–31. doi: 10.1093/ecco-jcc/jjy088
104. Farro G, Stakenborg M, Gomez-Pinilla PJ, Labeeuw E, Goverse G, Di Giovangiulio M, et al. CCR2-dependent monocyte-derived macrophages resolve inflammation and restore gut motility in postoperative ileus. Gut. (2017) 66:2098–109. doi: 10.1136/gutjnl-2016-313144
105. Al-Ghadban S, Kaissi S, Homaidan FR, Naim HY, El-Sabban ME. Cross-talk between intestinal epithelial cells and immune cells in inflammatory bowel disease. Sci Rep. (2016) 6:29783. doi: 10.1038/srep29783
106. de Sablet T, Potiron L, Marquis M, Bussière FI, Lacroix-Lamandé S, Laurent F. Cryptosporidium parvum increases intestinal permeability through interaction with epithelial cells and IL-1β and TNFα released by inflammatory monocytes. Cell Microbiol. (2016) 18:1871–80. doi: 10.1111/cmi.12632
107. Habtezion A, Nguyen LP, Hadeiba H, Butcher EC. Leukocyte trafficking to the small intestine and colon. Gastroenterology. (2016) 150:340–54. doi: 10.1053/j.gastro.2015.10.046
108. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. (2007) 7:678–89. doi: 10.1038/nri2156
109. Kinashi T. Intracellular signalling controlling integrin activation in lymphocytes. Nat Rev Immunol. (2005) 5:546–59. doi: 10.1038/nri1646
110. Berlin C, Berg EL, Briskin MJ, Andrew DP, Kilshaw PJ, Holzmann B, et al. Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell. (1993) 74:185–95. doi: 10.1016/0092-8674(93)90305-A
111. Schleier L, Wiendl M, Heidbreder K, Binder MT, Atreya R, Rath T, et al. Non-classical monocyte homing to the gut via α4β7 integrin mediates macrophage-dependent intestinal wound healing. Gut. (2020) 69:252–63. doi: 10.1136/gutjnl-2018-316772
112. Nguyen LP, Pan J, Dinh TT, Hadeiba H, O'Hara E II, Ebtikar A, et al. Role and species-specific expression of colon T cell homing receptor GPR15 in colitis. Nat Immunol. (2015) 16:207–13. doi: 10.1038/ni.3079
113. Kunkel EJ, Butcher EC. Chemokines and the tissue-specific migration of lymphocytes. Immunity. (2002) 16:1–4. doi: 10.1016/S1074-7613(01)00261-8
114. Kim MH, Taparowsky EJ, Kim CH. Retinoic acid differentially regulates the migration of innate lymphoid cell subsets to the gut. Immunity. (2015) 43:107–19. doi: 10.1016/j.immuni.2015.06.009
115. Zeng X, Xu WK, Lok TM, Ma HT, Poon RYC. Imbalance of the spindle-assembly checkpoint promotes spindle poison-mediated cytotoxicity with distinct kinetics. Cell Death Dis. (2019) 10:314. doi: 10.1038/s41419-019-1539-8
116. Bankovich AJ, Shiow LR, Cyster JG. CD69 suppresses sphingosine 1-phosophate receptor-1 (S1P1) function through interaction with membrane helix 4. J Biol Chem. (2010) 285:22328–37. doi: 10.1074/jbc.M110.123299
117. Hla T, Venkataraman K, Michaud J. The vascular S1P gradient-cellular sources and biological significance. Biochim Biophys Acta. (2008) 1781:477–82. doi: 10.1016/j.bbalip.2008.07.003
118. Randolph GJ. CCR7: Unifying disparate journeys to the lymph node. J Immunol. (2016) 196:3–4. doi: 10.4049/jimmunol.1502372
119. Martens R, Permanyer M, Werth K, Yu K, Braun A, Halle O, et al. Efficient homing of T cells via afferent lymphatics requires mechanical arrest and integrin-supported chemokine guidance. Nat Commun. (2020) 11:1114. doi: 10.1038/s41467-020-14921-w
120. Mokrani M, Klibi J, Bluteau D, Bismuth G, Mami-Chouaib F. Smad and NFAT pathways cooperate to induce CD103 expression in human CD8 T lymphocytes. J Immunol. (2014) 192:2471–9. doi: 10.4049/jimmunol.1302192
121. Cepek KL, Shaw SK, Parker CM, Russell GJ, Morrow JS, Rimm DL, et al. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the alpha E beta 7 integrin. Nature. (1994) 372:190–3. doi: 10.1038/372190a0
122. Zundler S, Becker E, Spocinska M, Slawik M, Parga-Vidal L, Stark R, et al. Hobit- and Blimp-1-driven CD4(+) tissue-resident memory T cells control chronic intestinal inflammation. Nat Immunol. (2019) 20:288–300. doi: 10.1038/s41590-018-0298-5
123. Toulon A, Breton L, Taylor KR, Tenenhaus M, Bhavsar D, Lanigan C, et al. A role for human skin-resident T cells in wound healing. J Exp Med. (2009) 206:743–50. doi: 10.1084/jem.20081787
124. Zaid A, Mackay LK, Rahimpour A, Braun A, Veldhoen M, Carbone FR, et al. Persistence of skin-resident memory T cells within an epidermal niche. Proc Natl Acad Sci U S A. (2014) 111:5307–12. doi: 10.1073/pnas.1322292111
125. Fuss IJ, Neurath M, Boirivant M, Klein JS, de la Motte C, Strong SA, et al. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn's disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol. (1996) 157:1261–70.
126. Berrebi D, Languepin J, Ferkdadji L, Foussat A, De Lagausie P, Paris R, et al. Cytokines, chemokine receptors, and homing molecule distribution in the rectum and stomach of pediatric patients with ulcerative colitis. J Pediatr Gastroenterol Nutr. (2003) 37:300–8. doi: 10.1097/00005176-200309000-00018
127. Heller F, Fuss IJ, Nieuwenhuis EE, Blumberg RS, Strober W. Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity. (2002) 17:629–38. doi: 10.1016/S1074-7613(02)00453-3
128. Gerlach K, Hwang Y, Nikolaev A, Atreya R, Dornhoff H, Steiner S, et al. TH9 cells that express the transcription factor PU.1 drive T cell-mediated colitis via IL-9 receptor signaling in intestinal epithelial cells. Nat Immunol. (2014) 15:676–86. doi: 10.1038/ni.2920
129. Popp V, Gerlach K, Mott S, Turowska A, Garn H, Atreya R, et al. Rectal delivery of a DNAzyme that specifically blocks the transcription factor GATA3 and reduces colitis in mice. Gastroenterology. (2017) 152:176–92.e5. doi: 10.1053/j.gastro.2016.09.005
130. Mullin GE, Maycon ZR, Braun-Elwert L, Cerchia R, James SP, Katz S, et al. Inflammatory bowel disease mucosal biopsies have specialized lymphokine mRNA profiles. Inflamm Bowel Dis. (1996) 2:16–26. doi: 10.1002/ibd.3780020105
131. Zundler S, Neurath MF. Pathogenic T cell subsets in allergic and chronic inflammatory bowel disorders. Immunol Rev. (2017) 278:263–76. doi: 10.1111/imr.12544
132. Lee JY, Min BH, Lee JG, Noh D, Lee JH, Rhee PL, et al. Endoscopic submucosal dissection for early gastric neoplasia occurring in the remnant stomach after distal gastrectomy. Clin Endosc. (2016) 49:182–6. doi: 10.5946/ce.2015.049
133. Neurath MF. Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nat Immunol. (2019) 20:970–9. doi: 10.1038/s41590-019-0415-0
134. Zundler S, Schillinger D, Fischer A, Atreya R, López-Posadas R, Watson A, et al. Blockade of αEβ7 integrin suppresses accumulation of CD8(+) and Th9 lymphocytes from patients with IBD in the inflamed gut in vivo. Gut. (2017) 66:1936–48. doi: 10.1136/gutjnl-2016-312439
135. Zundler S, Neurath MF. Novel insights into the mechanisms of gut homing and antiadhesion therapies in inflammatory bowel diseases. Inflamm Bowel Dis. (2017) 23:617–27. doi: 10.1097/MIB.0000000000001067
136. Truelove SC, Witts LJ. Cortisone in ulcerative colitis; final report on a therapeutic trial. Br Med J. (1955) 2:1041–8. doi: 10.1136/bmj.2.4947.1041
137. Hanauer SB, Feagan BG, Lichtenstein GR, Mayer LF, Schreiber S, Colombel JF, et al. Maintenance infliximab for Crohn's disease: the ACCENT I randomised trial. Lancet. (2002) 359:1541–9. doi: 10.1016/S0140-6736(02)08512-4
138. Colombel JF, Sandborn WJ, Reinisch W, Mantzaris GJ, Kornbluth A, Rachmilewitz D, et al. Infliximab, azathioprine, or combination therapy for Crohn's disease. N Engl J Med. (2010) 362:1383–95. doi: 10.1056/NEJMoa0904492
139. Adedokun OJ, Sandborn WJ, Feagan BG, Rutgeerts P, Xu Z, Marano CW, et al. Association between serum concentration of infliximab and efficacy in adult patients with ulcerative colitis. Gastroenterology. (2014) 147:1296–307.e5. doi: 10.1053/j.gastro.2014.08.035
140. Feagan BG, Rutgeerts P, Sands BE, Hanauer S, Colombel JF, Sandborn WJ, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. (2013) 369:699–710. doi: 10.1056/NEJMoa1215734
141. Sandborn WJ, Feagan BG, Rutgeerts P, Hanauer S, Colombel JF, Sands BE, et al. Vedolizumab as induction and maintenance therapy for Crohn's disease. N Engl J Med. (2013) 369:711–21. doi: 10.1056/NEJMoa1215739
142. Frøslie KF, Jahnsen J, Moum BA, Vatn MH. Mucosal healing in inflammatory bowel disease: results from a Norwegian population-based cohort. Gastroenterology. (2007) 133:412–22. doi: 10.1053/j.gastro.2007.05.051
143. Reinink AR, Lee TC, Higgins PD. Endoscopic mucosal healing predicts favorable clinical outcomes in inflammatory bowel disease: a meta-analysis. Inflamm Bowel Dis. (2016) 22:1859–69. doi: 10.1097/MIB.0000000000000816
144. Colombel JF, Rutgeerts P, Reinisch W, Esser D, Wang Y, Lang Y, et al. Early mucosal healing with infliximab is associated with improved long-term clinical outcomes in ulcerative colitis. Gastroenterology. (2011) 141:1194–201. doi: 10.1053/j.gastro.2011.06.054
145. Berstad A, Arslan G, Folvik G. Relationship between intestinal permeability and calprotectin concentration in gut lavage fluid. Scand J Gastroenterol. (2000) 35:64–9. doi: 10.1080/003655200750024551
146. Zittan E, Kelly OB, Kirsch R, Milgrom R, Burns J, Nguyen GC, et al. Low fecal calprotectin correlates with histological remission and mucosal healing in ulcerative colitis and colonic Crohn's disease. Inflamm Bowel Dis. (2016) 22:623–30. doi: 10.1097/MIB.0000000000000652
147. Bertani L, Blandizzi C, Mumolo MG, Ceccarelli L, Albano E, Tapete G, et al. Fecal calprotectin predicts mucosal healing in patients with ulcerative colitis treated with biological therapies: a prospective study. Clin Transl Gastroenterol. (2020) 11:e00174. doi: 10.14309/ctg.0000000000000174
148. Hu MS, Walmsley GG, Barnes LA, Weiskopf K, Rennert RC, Duscher D, et al. Delivery of monocyte lineage cells in a biomimetic scaffold enhances tissue repair. JCI Insight. (2017) 2:e96260: doi: 10.1172/jci.insight.96260
149. Fang Y, Shen J, Yao M, Beagley KW, Hambly BD, Bao S. Granulocyte-macrophage colony-stimulating factor enhances wound healing in diabetes via upregulation of proinflammatory cytokines. Br J Dermatol. (2010) 162:478–86. doi: 10.1111/j.1365-2133.2009.09528.x
150. Maruyama K, Asai J, Ii M, Thorne T, Losordo DW, D'Amore PA. Decreased macrophage number and activation lead to reduced lymphatic vessel formation and contribute to impaired diabetic wound healing. Am J Pathol. (2007) 170:1178–91. doi: 10.2353/ajpath.2007.060018
151. Zheng Y, Xiong S, Jiang P, Liu R, Liu X, Qian J, et al. Glucocorticoids inhibit lipopolysaccharide-mediated inflammatory response by downregulating microRNA-155: a novel anti-inflammation mechanism. Free Radic Biol Med. (2012) 52:1307–17. doi: 10.1016/j.freeradbiomed.2012.01.031
152. Norling LV, Spite M, Yang R, Flower RJ, Perretti M, Serhan CN. Cutting edge: humanized nano-proresolving medicines mimic inflammation-resolution and enhance wound healing. J Immunol. (2011) 186:5543–7. doi: 10.4049/jimmunol.1003865
153. Krishnan K, Arnone B, Buchman A. Intestinal growth factors: potential use in the treatment of inflammatory bowel disease and their role in mucosal healing. Inflamm Bowel Dis. (2011) 17:410–22. doi: 10.1002/ibd.21316
154. Neurath MF, Travis SP. Mucosal healing in inflammatory bowel diseases: a systematic review. Gut. (2012) 61:1619–35. doi: 10.1136/gutjnl-2012-302830
155. Baumgart DC, Vierziger K, Sturm A, Wiedenmann B, Dignass AU. Mesalamine promotes intestinal epithelial wound healing in vitro through a TGF-beta-independent mechanism. Scand J Gastroenterol. (2005) 40:958–64. doi: 10.1080/00365520510015854
156. Nakase H. Optimizing the use of current treatments and emerging therapeutic approaches to achieve therapeutic success in patients with inflammatory bowel disease. Gut Liver. (2020) 14:7–19. doi: 10.5009/gnl18203
157. Bokemeyer B, Hommes D, Gill I, Broberg P, Dignass A. Mesalazine in left-sided ulcerative colitis: efficacy analyses from the PODIUM trial on maintenance of remission and mucosal healing. J Crohns Colitis. (2012) 6:476–82. doi: 10.1016/j.crohns.2011.10.006
158. D'Haens G, Van Deventer S, Van Hogezand R, Chalmers D, Kothe C, Baert F, et al. Endoscopic and histological healing with infliximab anti-tumor necrosis factor antibodies in Crohn's disease: a European multicenter trial. Gastroenterology. (1999) 116:1029–34. doi: 10.1016/S0016-5085(99)70005-3
159. Rutgeerts P, Van Assche G, Sandborn WJ, Wolf DC, Geboes K, Colombel JF, et al. Adalimumab induces and maintains mucosal healing in patients with Crohn's disease: data from the EXTEND trial. Gastroenterology. (2012) 142:1102–11.e2. doi: 10.1053/j.gastro.2012.01.035
160. Cholapranee A, Hazlewood GS, Kaplan GG, Peyrin-Biroulet L, Ananthakrishnan AN. Systematic review with meta-analysis: comparative efficacy of biologics for induction and maintenance of mucosal healing in Crohn's disease and ulcerative colitis controlled trials. Aliment Pharmacol Ther. (2017) 45:1291–302. doi: 10.1111/apt.14030
161. Baert FJ, D'Haens GR, Peeters M, Hiele MI, Schaible TF, Shealy D, et al. Tumor necrosis factor alpha antibody (infliximab) therapy profoundly down-regulates the inflammation in Crohn's ileocolitis. Gastroenterology. (1999) 116:22–8. doi: 10.1016/S0016-5085(99)70224-6
162. Park SH, Hong M, Lee HS, Ye BD, Hwang SW, Jung S, et al. Association of TRAP1 with infliximab-induced mucosal healing in Crohn's disease. J Gastroenterol Hepatol. (2019) 34:2118–25. doi: 10.1111/jgh.14696
163. Suenaert P, Bulteel V, Lemmens L, Noman M, Geypens B, Van Assche G, et al. Anti-tumor necrosis factor treatment restores the gut barrier in Crohn's disease. Am J Gastroenterol. (2002) 97:2000–4. doi: 10.1111/j.1572-0241.2002.05914.x
164. Vos AC, Wildenberg ME, Arijs I, Duijvestein M, Verhaar AP, de Hertogh G, et al. Regulatory macrophages induced by infliximab are involved in healing in vivo and in vitro. Inflamm Bowel Dis. (2012) 18:401–8. doi: 10.1002/ibd.21818
165. Vos AC, Wildenberg ME, Duijvestein M, Verhaar AP, van den Brink GR, Hommes DW. Anti-tumor necrosis factor-α antibodies induce regulatory macrophages in an Fc region-dependent manner. Gastroenterology. (2011) 140:221–30. doi: 10.1053/j.gastro.2010.10.008
166. Rutgeerts P, Gasink C, Chan D, Lang Y, Pollack P, Colombel JF, et al. Efficacy of ustekinumab for inducing endoscopic healing in patients with Crohn's disease. Gastroenterology. (2018) 155:1045–58. doi: 10.1053/j.gastro.2018.06.035
167. Sandborn WJ, Su C, Sands BE, D'Haens GR, Vermeire S, Schreiber S, et al. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med. (2017) 376:1723–36. doi: 10.1056/NEJMoa1606910
168. Gerlach K, Lechner K, Popp V, Offensperger L, Zundler S, Wiendl M, et al. The JAK1/3 inhibitor to tofacitinib suppresses T cell homing and activation in chronic intestinal inflammation. J Crohns Colitis. (2021) 15:244–57. doi: 10.1093/ecco-jcc/jjaa162
169. Binder MT, Becker E, Wiendl M, Schleier L, Fuchs F, Leppkes M, et al. Similar inhibition of dynamic adhesion of lymphocytes from IBD patients to MAdCAM-1 by vedolizumab and etrolizumab-s. Inflamm Bowel Dis. (2018) 24:1237–50. doi: 10.1093/ibd/izy077
170. Colombel JF, Sands BE, Rutgeerts P, Sandborn W, Danese S, D'Haens G, et al. The safety of vedolizumab for ulcerative colitis and Crohn's disease. Gut. (2017) 66:839–51. doi: 10.1136/gutjnl-2015-311079
171. Lightner AL, Raffals LE, Mathis KL, Cima RR, Tse CS, Pemberton JH, et al. Postoperative outcomes in vedolizumab-treated patients undergoing abdominal operations for inflammatory bowel disease. J Crohns Colitis. (2017) 11:185–90. doi: 10.1093/ecco-jcc/jjw147
172. C.Law CY, Narula A, Lightner AL, McKenna NP, Colombel JF, Narula N. Systematic review and meta-analysis: preoperative vedolizumab treatment and postoperative complications in patients with inflammatory bowel disease. J Crohns Colitis. (2018) 12:538–45. doi: 10.1093/ecco-jcc/jjy022
173. Lightner AL, McKenna NP, Tse CS, Raffals LE, Loftus EV Jr, Mathis KL. Postoperative outcomes in vedolizumab-treated Crohn's disease patients undergoing major abdominal operations. Aliment Pharmacol Ther. (2018) 47:573–80. doi: 10.1111/apt.14459
174. Lightner AL, Mathis KL, Tse CS, Pemberton JH, Shen B, Kochhar G, et al. Postoperative outcomes in vedolizumab-treated patients undergoing major abdominal operations for inflammatory bowel disease: retrospective multicenter cohort study. Inflamm Bowel Dis. (2018) 24:871–6. doi: 10.1093/ibd/izx076
175. Yung DE, Horesh N, Lightner AL, Ben-Horin S, Eliakim R, Koulaouzidis A, et al. Systematic review and meta-analysis: vedolizumab and postoperative complications in inflammatory bowel disease. Inflamm Bowel Dis. (2018) 24:2327–38. doi: 10.1093/ibd/izy156
Keywords: wound healing, intestinal epithelial cells, mucosal healing, IBD, intestinal epithelial barrier function
Citation: Sommer K, Wiendl M, Müller TM, Heidbreder K, Voskens C, Neurath MF and Zundler S (2021) Intestinal Mucosal Wound Healing and Barrier Integrity in IBD–Crosstalk and Trafficking of Cellular Players. Front. Med. 8:643973. doi: 10.3389/fmed.2021.643973
Received: 19 December 2020; Accepted: 24 February 2021;
Published: 23 March 2021.
Edited by:
Levinus Albert Dieleman, University of Alberta, CanadaReviewed by:
Matthew Richard Olson, Purdue University, United StatesCopyright © 2021 Sommer, Wiendl, Müller, Heidbreder, Voskens, Neurath and Zundler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sebastian Zundler, c2ViYXN0aWFuLnp1bmRsZXJAdWstZXJsYW5nZW4uZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.