 Katie J. Lee
Katie J. Lee Monika Janda
Monika Janda Mitchell S. Stark
Mitchell S. Stark Richard A. Sturm1
Richard A. Sturm1 H. Peter Soyer
H. Peter Soyer

95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Med. , 19 February 2021
Sec. Dermatology
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.635316
This article is part of the Research Topic The Genetics and Genomics of Melanoma and Benign Melanocytic Neoplasms View all 5 articles
Benign naevi are closely linked to melanoma, as risk factors, simulators, or sites of melanoma formation. There is a heavy genetic overlap between the two lesions, a shared environmental influence of ultraviolet radiation, and many similar cellular features, yet naevi remain locally situated while melanomas spread from their primary site and may progress systemically to distal organs. Untangling the overlapping contributors and predictors of naevi and melanoma is an ongoing area of research and should eventually lead to more personalized prevention and treatment strategies, through the development of melanoma risk stratification tools and early detection of evolving melanomas. This will be achieved through a range of complementary strategies: risk-adjusted primary prevention counseling; the use of lesion imaging technologies such as sequential 3D total body photography and consumer-performed lesion imaging; artificial intelligence deep phenotyping and clinical assistance; a better understanding of genetic drivers of malignancy, risk variants, clinical genetics, and polygenic effects; and the interplay between genetics, phenotype and the environment.
It is well-known that benign naevi and melanomas are closely linked. Many melanomas arise in or adjacent to otherwise benign naevi (1), and naevi are the most common simulators of melanoma. Number of naevi is the strongest phenotypic risk factor for melanoma, and high naevus count is associated with a younger age of melanoma onset (2, 3). A high total nevus count is also associated with a greater chance of having multiple primary melanomas, compared to single primary melanomas (4).
Not surprisingly, there is heavy genetic overlap between nevus- and melanoma-associated genes, and between the phenotypic and cellular features of naevi and melanoma, including activating mutations in oncogenes and increased proliferation rates (5). Indeed, histopathological diagnoses ranging from moderately dysplastic naevi to early stage invasive melanoma are not reproducible between different histopathologists, even though histopathological diagnosis remains the gold standard (6). Yet proliferating nevus cells migrate only locally, while melanoma cells have the capacity to spread systemically, and for any one individual nevus to transform into a melanoma is extremely rare (7).
This conundrum was the basis of our Center of Research Excellence for the Study of Naevi (CRE Naevi), a 5-year effort which aimed to advance our understanding of naevi and melanomas. The CRE teams study genetics of naevi formation, number and morphology, somatic mutations in naevi and environmental factors like UV exposure. The intention was to identify phenotypic and genetic markers that could lead to personalized genomic medicine via risk stratification, more appropriate surveillance practices, and improved health outcomes. Based in Queensland, Australia, the melanoma capital of the world (8), the CRE Naevi is in an ideal position to examine these questions, and this article will cover the state of this field at present.
Precancer (9) and preprocancer (10) concepts, which seek to determine which suspicious lesions and early cancers are likely to progress to invasive cancer, are increasing in importance. Despite the generally high sensitivity of Australian clinicians at detecting melanomas and the widespread use of dermoscopy, Australian primary care clinicians currently excise ~20 benign lesions for each melanoma, compared to 29 in other countries (11, 12). These currently-unavoidable excisions are perhaps unnecessary for cancer treatment, but provide peace of mind for clinicians and patients alike. However, being able to confidently exclude melanoma in cases where a benign nevus presents with suspicious features, and thus reduce excisions, would both benefit patients and reduce strain on the healthcare system.
On the other side of the coin, the incidence of in situ melanomas has also soared as detection methods become ever more refined, leading to overdiagnosis—defined as the diagnosis of lesions that are true cancers, but which would never have caused harm in the patient's lifetime (13, 14). As many as 58% of melanomas in Australia may be overdiagnosed, the majority of these being very thin in situ melanomas—currently indistinguishable from the very thin melanomas with invasive potential, where early excision is almost always curative (15); these figures are on top of benign naevi that are incorrectly diagnosed (or “overcalled”) by dermatopathologists as melanoma. The ability to distinguish indolent cancers from cancers with invasive potential would likewise reduce costs, adverse effects of treatments, and patient distress. Finally, studies of precursor lesions in other cancers have improved our knowledge of tumorigenesis (16), and there is every reason to suppose that nevus development can shed similar light on melanomagenesis.
In addition, technology advances in total body photography (TBP) (17) and artificial intelligence (18), and increasing access to large, high-quality, annotated image databases (19) in the last decade have opened the possibility of automated assessments of individual lesions and overall melanoma risk. Artificial intelligence-based support of clinical decision making is a particularly promising area of research (20).
Early diagnosis of melanoma is critical. People with a melanoma <1 mm thick at diagnosis have a 98.7% 5-year survival rate, but survival rates dive steeply with increasing thickness (21, 22). The basic detection method—clinician-led skin examinations, paying close attention to any suspicious lesion including naevi—has been greatly augmented by the widespread uptake of dermoscopy, and as reported by the Cochrane Collaboration, “visual inspection using the naked eye alone is not good enough and melanomas may be missed” (23). However, clinicians do require training in dermoscopy before they can expect to see improvements in diagnostic accuracy, and studies suggest that improvements in accuracy are more due to increases in specificity, rather than sensitivity; some featureless melanomas may still be missed (24).
However, new technologies emerging over the last decade are set to further improve both the accuracy of melanoma detection through 3D total body photography (3D TBP) and sequential dermoscopic imaging, and to extend the reach of specialist examinations to rural, remote, and underserved areas through mobile teledermatology and telehealth.
3D TBP differs from more standard 2D body photography by having the patient stand inside a matrix of cameras that all take photos at once, thus covering the body from all angles, which are then stitched together with a computer program to produce a full-body avatar on screen that can be turned around, zoomed in on, or other manipulations to allow a close look at almost any part of the skin surface. In contrast, 2D TBP requires the patient to adopt a series of anatomic poses for photography by a single camera, and these images are examined individually. While 2D TBP hardware is much cheaper than the 3D version, it is easier for individual lesions to be imaged twice or missed altogether due to the overlapping nature of the images. In both 2D and 3D imaging, there are still limitations on capturing the skin at the soles of the feet, scalp, genitals, and other folds of the body.
Sequential, 3D TBP is particularly useful for patients with many naevi or a personal or family history of melanoma, as it allows clinicians to monitor a large number of naevi for change, the number one marker of malignant transformation. Further refinement of these methods should reduce the benign to malignant excision ratio, by allowing clinicians to monitor lesions with more confidence. In addition, in our experience, study participants and patients are highly engaged by the 3D images and their use, and this may reinforce the importance of sun-protective behavior and skin self-examinations (25). TBP, including 2D photography, also seems to reduce patient anxiety about their skin in people with prior melanomas (26).
Consumer-facilitated nevus monitoring is an ongoing area of interest. More than half of all melanomas are first noticed by the patient themselves or a family member (27), and skin self-examinations are currently the recommended level of monitoring for many Australians (28). Mobile dermoscopes, partnered with apps for telehealth, may eventually be able to integrate regular skin self-examinations with teledermatology, especially for high-risk patients who need frequent monitoring; our CRE team found that such technology is feasible and acceptable to people, with further work needed to optimize lesions selection by consumers (29).
Earlier work shows that diagnostic accuracy for lesions selected by patients is similar between mobile dermoscopy and in-person examination, although further work is needed to guide people to patients reliably select concerning lesions (30, 31). Patients themselves nominate low cost, ease of access and convenience as the main factors inclining them to use mobile telehealth (32), and focus groups indicate that up to 95% of Australians would consider using an app to send images of skin lesions to a doctor, preferably for monitoring lesions between regular face-to-face appointments (33). Participant enthusiasm for mobile teledermatology decreases once they actually begin using it, but they still find it generally acceptable (34).
However, there are drawbacks to self-assessment to be addressed. For example, when asked whether they had few, some, or many naevi, 40% of participants misclassified themselves compared to a dermatologist's assessment, even with an error buffer of ±5 naevi (35). Self-imaging with a mobile device may also be limited by a participant's ability to image lesions on hard-to-reach body sites, or even to realize that those lesions might require imaging.
Nevus count is highly heritable [heritability (h2) = 60–70%] (36) and most nevus-associated genes are also associated with melanoma risk, including IRF4, MITF, MTAP, and PLA2G6 (37). However, not all melanoma genes affect nevus count (38). In addition, there is an overlap between somatic mutations found in naevi and melanomas, suggesting a procession of mutations necessary but not sufficient for malignant transformation that may in the future help diagnose lesions as needing monitoring but not yet excision.
Genome-wide association studies (GWAS), whole genome, and whole exome sequencing have identified many melanoma risk alleles, which have a low to moderate effect individually (39). However, their effects appear to be cumulative and may add up to a significant risk (40). A nevus GWAS meta-analysis shows that variants in MTAP, PLA2G6, IRF4, KITLG and the 9q32 region affect nevus count (38); combining this with a meta-analysis (41) of melanoma GWAS studies showed that GPRC5A, CYP1B1, PPARGC1B, HDAC4, FAM208B, DOCK8, and SYNE2 were associated with nevus count as well as melanoma risk. Recently, the number of genes identified by GWAS meta-analysis for melanoma susceptibility has expanded to 54 independent loci (42), reinforcing the importance of naevogenesis, pigmentation, telomere maintenance, and other potential pathways in the pathogenesis of melanoma (Table 1).
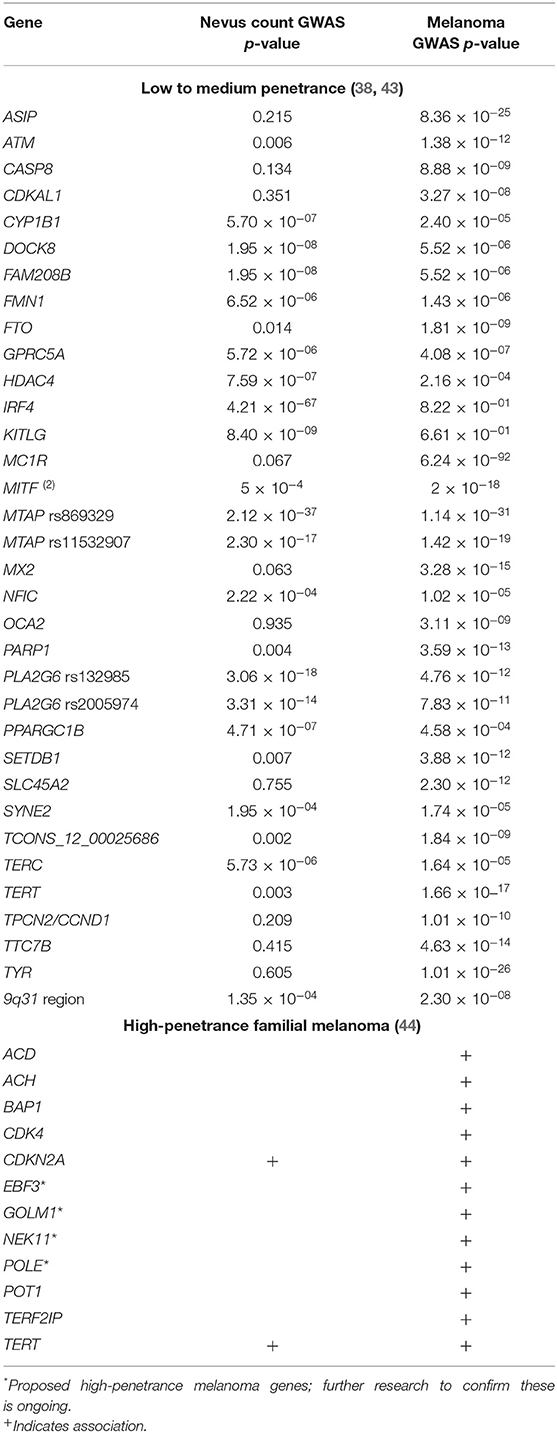
Table 1. Genes conferring susceptibility to naevi and/or melanoma.
The effect of nevus-related genes on melanoma risk can be multiplied by polygenic effects for genes controlling other phenotypic risk factors. For example, MC1R “R” variants are best known for causing the red hair phenotype and have only a small effect on nevus count (43). However, R alleles and high nevus counts synergistically increase melanoma risk (45–47). People with MC1R wildtype (WT) genotype and 20+ naevi have a similar melanoma odds ratio (OR) to people homozygous for the MC1R R allele (R/R) with 0–4 naevi (OR 4.82 vs. 4.42, respectively.) In contrast, people with both risk factors—an R/R genotype and 20+ naevi—have a melanoma OR of 25.09 (47).
Genetic variations also influence the risk of having more than one primary melanoma. For example, the E318K variant in MITF, combined with MC1R R alleles and other haplotypes in e.g., ASIP, were all at elevated frequencies in people with multiple primary melanomas compared to people with a single primary melanoma (4). Polygenic risk scores, accounting for the cumulative risk of many low- to moderate-effect melanoma-associated alleles (40), are higher in multiple primary melanoma than single primary melanoma or no melanoma history participants (4, 48).
Various melanoma subtypes are also associated with particular genetic variants. For example, amelanotic and hypomelanotic melanomas are associated with the MTAP and PLA2G6 alleles associated with overall melanoma risk, nevus morphology and high nevus count; however, amelanotic/hypomelanotic melanoma patients do not have a significantly different total nevus count from those with the more common pigmented melanomas (49). This observation may be due to the older age at diagnosis of amelanotic/hypomelanotic melanoma compared to pigmented melanoma (50–52), as nevus count declines from the fourth decade onwards (53). Albinism-related genes such as TYR and OCA2 have deleterious variants that are also more common in patients with amelanotic/hypomelanotic melanoma (54).
Several melanoma/nevus genes have multiple associations to phenotypes. An example is the IRF4 intronic variant rs12203592*T, which is associated with increased melanoma risk, increased risk of nodular melanoma (55), increased Breslow thickness at diagnosis (56), high nevus count in childhood and low nevus count in adulthood (57), and globular dermoscopic subtype of naevi (43, 58). This SNP is associated with pigmentary phenotype of darker hair color, lighter eyes, and skin with a lower tanning response to sunlight (37). The different alleles exhibit differential expression levels that also influence a range of key immunomodulatory molecules and cytokines, and melanocyte growth and survival after UV exposure (59). Research continues on untangling which of these many effects contribute to nevus and/or melanoma formation and behavior (43, 60).
Approximately 5–10% of melanomas arise in a familial melanoma context. While there are a handful of well-established, rare, high penetrance mutations, such as deleterious variants of CDKN2A, CDK4, BAP1, TERT, POT1, TERF2IP, ACD, POLE, EBF3, GOLM1, and NEK11 associated with familial melanoma, these mutations account for only 30% of familial melanoma cases (44). Research is increasingly focusing on the 60% of familial melanoma that is not explained by these high penetrance mutations, and recent work shows that familial cluster without a high penetrance mutation are enriched for polygenic risk (48).
In the absence of an identifiable mendelian mutation, assessment of candidate risk genes and improvements in genotype-phenotype correlations, together with polygenic risk scores (4) will help identify people who are nevertheless at high risk. By assessing the cumulative risk of 21 gene regions associated with a low or moderate melanoma risk, Cust et al. (40) showed that, in a group of melanoma patients and control participants who all had a low phenotypic risk profile, 9–21% had a high polygenic risk.
The activation of the MAPK pathway is the first, essential step for melanocytic proliferation, but it is not sufficient for full malignant transformation (61). Either BRAF or NRAS, but rarely both, are mutated in almost all acquired melanocytic naevi (62, 63). In contrast, blue naevi and Spitz naevi often have GNAQ and HRAS mutations. GNAQ is also associated with uveal melanomas (64). This may indicate that GNAQ/HRAS are not key players in melanomagenesis, unlike BRAF and NRAS (64). BRAF mutations are also associated more strongly with the globular dermoscopic subtype than reticular naevi (92 vs. 67%) (63). Dermoscopic subtypes refer to the distribution of pigment visible in a dermoscopic image of a lesion, and correlated closely to histopathological structures, allowing a visual assessment of potentially pathological processes (64).
Most cutaneous melanomas fall into one of four categories based on their initial driver mutation: BRAF, NRAS, NF1 and triple wild-type, which lacks any of the three other drivers (65, 66). BRAF V600 mutations are found in 35–50% of melanomas, and V600 variants are also found in 67–100% of benign naevi (63).
TERT promotor mutations, found in 86% of cutaneous melanomas and also detected in intermediate melanocytic lesions, are an early secondary alteration necessary for malignant transformation leading to melanoma development and proliferation (64, 67). The aberrant TERT gene leads to lengthening of the telomeres, which is not recognized as abnormal by the cell and as such apoptosis or senescence does not occur. Now that the melanocyte has lost its ability to undergo apoptosis, the series of events that occur next are likely to be related to chromosomal instability and DNA copy number aberrations. The precise order of these events has not been determined for each aberrant melanocyte, but in a study by Shain et al. (68), it was found that UV-induced point mutations continued to increase, and invasive melanoma was associated with increased copy-number aberrations, particularly loss of CDKN2A. Further mutations and/or genomic aberrations (e.g., copy number gain/loss and promotor methylation) are necessary for the transition of locally invasive melanoma to metastatic disease and have been described in PTEN, AKT3, CDKN2A, CCND1, CDK4, RB1, TP53, MDM2, and ARID2 (Table 2) (65, 66, 70).
While excessive UV exposure is well-known to promote melanomagenesis, a significant proportion of melanomas do form on sun-protected body sites. In a recent study, 75% of those diagnosed at ≤40 years old had one or more melanomas in a UV-protected site; in participants aged >40 years, only 18% of their melanomas were in UV-protected sites (4).
To explain this, the divergent pathway model of melanoma development posits that melanomas are caused either by a UV damage-related pathway or are secondary to inherent traits such as high nevus count (71). However, it appears that both pathways are at work in many melanoma cases, with genetic and phenotypic risk factors enhanced by a high-UV environment (72). For example, while nevus count is largely genetically controlled, increased UV exposure is associated with increased nevus count in those who are genetically predisposed (71). Usually only minimal or intermittent sun-exposure is required to activate the growth of aberrant melanocytes. It can be theorized that the BRAF mutation is already present in skin melanocytes scattered across the body surface (73) and that UV exposure contributes to the activation and increased proliferation of melanocytes harboring the BRAF mutation, leading to a nevus.
A study of genetic variations in melanoma patients who had all their melanomas in visibly sun-damaged sites found that these people were more likely to have variants at MC1R rs75570604 (OR 2·5), 9q31.2 rs10816595 (OR 1·4), and MTAP rs869329 (OR 1·4), compared to people with melanomas on both sun-exposed and sun-protected sites. Interestingly, these same variants were more common in people first diagnosed ≤40 years old than those diagnosed later in life (4).
Melanomas on sun-protected body sites are more likely to arise in an existing nevus (1), suggesting a link between defects in DNA repair and both nevus and melanoma formation. However, defects in DNA repair also appear to predispose to higher rates of UV damage on the sun-exposed sites of the body. A study of naevi on participants living in the high-UV environment of Queensland, Australia, found that sun-exposed lesions contained UV-related (signature 7) somatic mutations, while naevi on sun-protected sites had a higher proportion of mutation signatures associated with defective DNA repair, particularly indels associated with defective mismatch repair (74). This pattern was also observed in a lower-UV environment Spanish cohort (75). This suggests that defective DNA repair in melanocytes provides the necessary environment for both nevus formation and the accumulation of UV damage mutations.
Whole exome sequencing shows that benign naevi have a lower mutational burden than melanomas, particularly a lower level of UV-associated mutation signatures and melanoma driver mutations (76), although C>T transitions are still the most common SNV class (74). A WES study of 30 naevi and adjacent non-lesional skin identified UV-associated signatures in 97% of nevus samples, age-related signatures in 93%, and defective DNA repair signatures in 30%. This was reversed in the adjacent non-lesional skin, despite being exposed to the same level of UV as the naevi, with UV-related signatures in 10%, defective DNA repair signatures in 83% (signature 3) and 50% (signature 26), and age-related signatures in 100% of samples. In addition, two lesions in the study had low UV-associated and no age-related signatures, highlighting the importance of defective DNA mismatch repair in the development of at least some naevi (74).
Different dermoscopic patterns of naevi are also associated with different genomic signature; globular naevi have a higher proportion of C>T transitions, while reticular naevi have a higher proportion of copy number aberrations and indels. However, without longitudinal monitoring, it is difficult to know whether these differences reflect the patient's age at nevus onset, or reflect specific mutation types driving the development of specific dermoscopic patterns (74).
Even melanocytes in seemingly normal skin have numerous pathogenic somatic mutations, although the mutation load is even higher in melanocytes from skin adjacent to a skin cancer, where it is comparable to the mutational burden in melanoma cells. As might be expected, melanocytes from sun-protected sites have fewer somatic mutations than those from chronically sun-exposed sites, but melanocytes from intermittently sun-exposed sites, such as the thigh, have even more somatic mutations than either. Recent work has found that the majority of mutations are predicted to affect the MAPK pathway, including BRAF, NRAS, MAP2K1, NF1, CBL, and RASA2 (77). Other mutated genes included CDKN2A, ARID2, PTEN, and DDX3X.
Although recent advances have shed much light on the mechanisms linking naevi and melanoma, many questions remain. In particular, imaging, genetics and genomics, and cognitive computing have enormous potential to stratify risk categories and personalize skin cancer prevention and early detection, but protocols for integrating them into regular clinical care are still in the developmental stage.
TBP with sequential digital dermoscopy or pseudo-dermoscopy, supported by self-imaging with mobile devices during skin self-examinations, will likely become the new standard to facilitate early detection and prevent advanced and metastatic disease. Research will focus on designing and validating appropriate protocols and monitoring intervals, including tailored protocols for patients at different risk levels. This work will continue to investigate the natural history of naevi across the lifetime of individuals, an area still poorly understood, since most studies of naevi are cross-sectional rather than longitudinal (78).
TBP will be further augmented with cognitive computing (20), as artificial intelligence algorithms are developed to provide triaging, clinical decision support and risk assessment based not only on nevus imaging, but also deep phenotyping. Deep phenotyping is the process of assessing the whole skin surface for phenotypic signs of genetic susceptibility, namely freckling and nevus number, type and distribution, and signs of environmental risk, by assessing the amount of UV damage over large areas of skin. These should improve our overall assessment of melanoma risk by improving the objective assessment of accumulated sun damage and the visible effects of genetic risk factors.
Genomics remains a fruitful area of research, with polygenic risk scores (40) and the identification and characterization of rare candidate risk genes (38) a particularly promising avenue of research. A combination of further discoveries of nevus- and melanoma-associated genes and regulatory regions with polygenic scores incorporating many small-to-moderate effect risk variants, as well as the well-known larger-effect variants, will eventually allow for risk stratification that directs more appropriate surveillance protocols for people with different levels of melanoma risk.
Finally, it will be critical to develop algorithms able to capture all the relevant phenotypic and genotypic factors and synthesize them into a complete risk-stratification tool to allow risk-adjusted surveillance that detects melanoma early while minimizing unintended harms of surveillance.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
KL wrote the first draft of the manuscript. MJ, MS, RS, and HS wrote sections of the manuscript. All authors contributed to manuscript revision and read and approved the submitted version.
This work was funded by the Australian National Health and Medical Research Council (NHMRC) Centre of Research Excellence for the Study of Naevi (APP1099021). HS holds an NHMRC Medical Research Future Fund Next Generation Clinical Researchers Program Practitioner Fellowship (APP1137127). MJ was funded by a National Health and Medical Research Council (NHMRC) TRIP Fellowship (APP1151021).
HS is a shareholder of MoleMap NZ Limited and e-derm consult GmbH, and undertakes regular teledermatological reporting for both companies. HS has NHMRC partnership grants with Canfield Scientific Inc. (APP1153046) and Fotofinder Systems Inc. (APP1113962). HS provides medical consultant services for Canfield Scientific Inc., First Derm by iDoc24 Inc, and Revenio Research Oy. HS is a board member of Melanoma and Skin Cancer Trials Limited and the QLD Skin & Cancer Foundation. HS is a member of the Australian Academy of Health and Medical Sciences Reports committee.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Pampena R, Kyrgidis A, Lallas A, Moscarella E, Argenziano G, Longo C. A meta-analysis of nevus-associated melanoma: prevalence and practical implications. J Am Acad Dermatol. (2017) 77:938–45.e4. doi: 10.1016/j.jaad.2017.06.149
2. Chang YM, Newton-Bishop JA, Bishop DT, Armstrong BK, Bataille V, Bergman W, et al. A pooled analysis of melanocytic nevus phenotype and the risk of cutaneous melanoma at different latitudes. Int J Cancer. (2009) 124:420–8. doi: 10.1002/ijc.23869
3. Taylor NJ, Thomas NE, Anton-Culver H, Armstrong BK, Begg CB, Busam KJ, et al. Nevus count associations with pigmentary phenotype, histopathological melanoma characteristics and survival from melanoma. Int J Cancer. (2016) 139:1217–22. doi: 10.1002/ijc.30157
4. McMeniman EK, Duffy DL, Jagirdar K, Lee KJ, Peach E, McInerney-Leo AM, et al. The interplay of sun damage and genetic risk in Australian multiple and single primary melanoma cases and controls. Br J Dermatol. (2019) 183:357–66. doi: 10.1111/bjd.18777
5. Bastian BC. The molecular pathology of melanoma: an integrated taxonomy of melanocytic neoplasia. Annu Rev Pathol. (2014) 9:239–71. doi: 10.1146/annurev-pathol-012513-104658
6. Elmore JG, Barnhill RL, Elder DE, Longton GM, Pepe MS, Reisch LM, et al. Pathologists' diagnosis of invasive melanoma and melanocytic proliferations: observer accuracy and reproducibility study. BMJ. (2017) 357:j2813. doi: 10.1136/bmj.j2813
7. Tsao H, Bevona C, Goggins W, Quinn T. The transformation rate of moles (melanocytic nevi) into cutaneous melanoma: a population-based estimate. Arch Dermatol. (2003) 139:282–8. doi: 10.1001/archderm.139.3.282
9. Srivastava S, Ghosh S, Kagan J, Mazurchuk R. The PreCancer Atlas (PCA). Trends Cancer. (2018) 4:513–4. doi: 10.1016/j.trecan.2018.06.003
11. Youl PH, Baade PD, Janda M, Del Mar CB, Whiteman DC, Aitken JF. Diagnosing skin cancer in primary care: how do mainstream general practitioners compare with primary care skin cancer clinic doctors? Med J Austral. (2007) 187:215–20. doi: 10.5694/j.1326-5377.2007.tb01202.x
12. Argenziano G, Cerroni L, Zalaudek I, Staibano S, Hofmann-Wellenhof R, Arpaia N, et al. Accuracy in melanoma detection: a 10-year multicenter survey. J Am Acad Dermatol. (2012) 67:54–9. doi: 10.1016/j.jaad.2011.07.019
13. Welch HG, Black WC. Overdiagnosis in cancer. J Natl Cancer Inst. (2010) 102:605–13. doi: 10.1093/jnci/djq099
14. Welch HG, Mazer BL, Adamson AS. The rapid rise in cutaneous melanoma diagnoses. N Engl J Med. (2021) 384:72–9. doi: 10.1056/NEJMsb2019760
15. Glasziou PP, Jones MA, Pathirana T, Barratt AL, Bell KJ. Estimating the magnitude of cancer overdiagnosis in Australia. Med J Austral. (2020) 212:163–8. doi: 10.5694/mja2.50455
16. Bettington M, Walker N, Rosty C, Brown I, Clouston A, Wockner L, et al. Critical appraisal of the diagnosis of the sessile serrated adenoma. Am J Surg Pathol. (2014) 38:158–66. doi: 10.1097/PAS.0000000000000103
17. Fried L, Tan A, Bajaj S, Liebman TN, Polsky D, Stein JA. Technological advances for the detection of melanoma: Part I. Advances in diagnostic techniques. J Am Acad Dermatol. (2020) 83:983–92. doi: 10.1016/j.jaad.2020.03.121
18. Puri P, Comfere N, Drage LA, Shamim H, Bezalel SA, Pittelkow MR, et al. Deep learning for dermatologists: Part II. Current applications. J Am Acad Dermatol. (2020). doi: 10.1016/j.jaad.2020.05.053. [Epub ahead of print].
19. Rotemberg V, Kurtansky N, Betz-Stablein B, Caffery L, Chousakos E, Codella N, et al. A patient-centric dataset of images and metadata for identifying melanomas using clinical context. arXiv. (2020). doi: 10.1038/s41597-021-00815-z
20. Tschandl P, Rinner C, Apalla Z, Argenziano G, Codella N, Halpern A, et al. Human-computer collaboration for skin cancer recognition. Nat Med. (2020) 26:1229–34. doi: 10.1038/s41591-020-0942-0
21. Rigel DS, Carucci JA. Malignant melanoma: prevention, early detection, and treatment in the 21st century. CA Cancer J Clin. (2000) 50:215–36; quiz 37–40. doi: 10.3322/canjclin.50.4.215
22. Green AC, Baade P, Coory M, Aitken JF, Smithers M. Population-based 20-year survival among people diagnosed with thin melanomas in Queensland, Australia. J Clin Oncol. (2012) 30:1462–7. doi: 10.1200/JCO.2011.38.8561
23. Dinnes J, Deeks JJ, Grainge MJ, Chuchu N, Ferrante di Ruffano L, Matin RN, et al. Visual inspection for diagnosing cutaneous melanoma in adults. Cochrane Database Systematic Rev. (2018) 12:Cd013194. doi: 10.1002/14651858.CD013194
24. Lee KJ, di Meo N, Yelamos O, Malvehy J, Zalaudek I, Soyer HP. Dermoscopy/confocal microscopy. In: Balch C, Gershenwald J, Thompson J, Atkins M, Kirkwood J, Kefford R, et al., editors. Cutaneous Melanoma. 6th ed. Cham: Springer (2019) 1–50.
25. Rayner JE, Laino AM, Nufer KL, Adams L, Raphael AP, Menzies SW, et al. Clinical perspective of 3D total body photography for early detection and screening of melanoma. Front Med. (2018) 5:152. doi: 10.3389/fmed.2018.00152
26. Moye MS, King SM, Rice ZP, DeLong LK, Seidler AM, Veledar E, et al. Effects of total-body digital photography on cancer worry in patients with atypical mole syndrome. JAMA Dermatol. (2015) 151:137–43. doi: 10.1001/jamadermatol.2014.2229
27. Hamidi R, Peng D, Cockburn M. Efficacy of skin self-examination for the early detection of melanoma. Int J Dermatol. (2010) 49:126–34. doi: 10.1111/j.1365-4632.2009.04268.x
28. Cancer Council Australia. Position Statement - Early Detection of Skin Cancer 2019. Available online at: https://wiki.cancer.org.au/policy/Position_statement_-_Screening_and_early_detection_of_skin_cancer (accessed September 24, 2020).
29. Janda M, Horsham C, Koh U, Gillespie N, Loescher LJ, Vagenas D, et al. Redesigning skin cancer early detection and care using a new mobile health application: protocol of the SKIN research project, a randomised controlled trial. Dermatology. (2019) 235:11–8. doi: 10.1159/000493729
30. Manahan MN, Soyer HP, Loescher LJ, Horsham C, Vagenas D, Whiteman DC, et al. A pilot trial of mobile, patient-performed teledermoscopy. Br J Dermatol. (2015) 172:1072–80. doi: 10.1111/bjd.13550
31. Wu X, Oliveria SA, Yagerman S, Chen L, DeFazio J, Braun R, et al. Feasibility and efficacy of patient-initiated mobile teledermoscopy for short-term monitoring of clinically atypical nevi. JAMA Dermatol. (2015) 151:489–96. doi: 10.1001/jamadermatol.2014.3837
32. Kong F, Horsham C, Rayner J, Simunovic M, O'Hara M, Soyer HP, et al. Consumer preferences for skin cancer screening using mobile teledermoscopy: a qualitative study. Dermatology. (2020) 236:97–104. doi: 10.1159/000505620
33. Koh U, Horsham C, Soyer HP, Loescher LJ, Gillespie N, Vagenas D, et al. Consumer acceptance and expectations of a mobile health application to photograph skin lesions for early detection of melanoma. Dermatology. (2019) 235:4–10. doi: 10.1159/000493728
34. Horsham C, Snoswell C, Vagenas D, Loescher LJ, Gillespie N, Soyer HP, et al. Is teledermoscopy ready to replace face-to-face examinations for the early detection of skin cancer? Consumer views, technology acceptance, and satisfaction with care. Dermatology. (2020) 236:90–6. doi: 10.1159/000506154
35. Betz-Stablein B, Koh U, Plasmeijer EI, Janda M, Aitken JF, Soyer HP, et al. Self-reported naevus density may lead to misclassification of melanoma risk. Br J Dermatol. (2020) 182:1488–90. doi: 10.1111/bjd.18802
36. Lee S, Duffy DL, McClenahan P, Lee KJ, McEniery E, Burke B, et al. Heritability of naevus patterns in an adult twin cohort from the Brisbane Twin Registry: a cross-sectional study. Br J Dermatol. (2016) 174:356–63. doi: 10.1111/bjd.14291
37. Ainger SA, Jagirdar K, Lee KJ, Soyer HP, Sturm RA. Skin pigmentation genetics for the clinic. Dermatology. (2017) 233:1–15. doi: 10.1159/000468538
38. Duffy DL, Zhu G, Li X, Sanna M, Iles MM, Jacobs LC, et al. Novel pleiotropic risk loci for melanoma and nevus density implicate multiple biological pathways. Nat Commun. (2018) 9:4774. doi: 10.1038/s41467-018-06649-5
39. Aoude LG, Wadt KA, Pritchard AL, Hayward NK. Genetics of familial melanoma: 20 years after CDKN2A. Pigment Cell Melanoma Res. (2015) 28:148–60. doi: 10.1111/pcmr.12333
40. Cust AE, Drummond M, Kanetsky PA, Goldstein AM, Barrett JH, MacGregor S, et al. Assessing the incremental contribution of common genomic variants to melanoma risk prediction in two population-based studies. J Invest Dermatol. (2018) 138:2617–24. doi: 10.1016/j.jid.2018.05.023
41. Law MH, Bishop DT, Lee JE, Brossard M, Martin NG, Moses EK, et al. Genome-wide meta-analysis identifies five new susceptibility loci for cutaneous malignant melanoma. Nat Genet. (2015) 47:987–95.
42. Landi MT, Bishop DT, MacGregor S, Machiela MJ, Stratigos AJ, Ghiorzo P, et al. Genome-wide association meta-analyses combining multiple risk phenotypes provide insights into the genetic architecture of cutaneous melanoma susceptibility. Nat Genet. (2020) 52:494–504. doi: 10.1038/s41588-020-0611-8
43. Duffy DL, Jagirdar K, Lee KJ, McWhirter SR, McMeniman EK, De'Ambrosis B, et al. Genes determining nevus count and dermoscopic appearance in Australian melanoma cases and controls. J Investig Dermatol. (2020) 140:498–501.e17. doi: 10.1016/j.jid.2019.05.032
44. Dalmasso B, Ghiorzo P. Evolution of approaches to identify melanoma missing heritability. Expert Rev Mol Diagn. (2020) 20:523–31. doi: 10.1080/14737159.2020.1738221
45. Veierod MB, Adami HO, Lund E, Armstrong BK, Weiderpass E. Sun and solarium exposure and melanoma risk: effects of age, pigmentary characteristics, and nevi. Cancer Epidemiol Biomarkers Prev. (2010) 19:111–20. doi: 10.1158/1055-9965.EPI-09-0567
46. Olsen CM, Zens MS, Stukel TA, Sacerdote C, Chang YM, Armstrong BK, et al. Nevus density and melanoma risk in women: a pooled analysis to test the divergent pathway hypothesis. Int J Cancer. (2009) 124:937–44. doi: 10.1002/ijc.24011
47. Duffy DL, Lee KJ, Jagirdar K, Pflugfelder A, Stark MS, McMeniman EK, et al. High naevus count and MC1R red hair alleles contribute synergistically to increased melanoma risk. Brit J Dermatol. (2019) 181:1009–16. doi: 10.1111/bjd.17833
48. Law MH, Aoude LG, Duffy DL, Long GV, Johansson PA, Pritchard AL, et al. Multiplex melanoma families are enriched for polygenic risk. Hum Mol Genet. (2020) 29:2976–85. doi: 10.1093/hmg/ddaa156
49. Rayner JE, McMeniman EK, Duffy DL, De'Ambrosis B, Smithers BM, Jagirdar K, et al. Phenotypic and genotypic analysis of amelanotic and hypomelanotic melanoma patients. J Eur Acad Dermatol Venereol. (2019) 33:1076–83. doi: 10.1111/jdv.15446
50. McClain SE, Mayo KB, Shada AL, Smolkin ME, Patterson JW, Slingluff CL Jr. Amelanotic melanomas presenting as red skin lesions: a diagnostic challenge with potentially lethal consequences. Int J Dermatol. (2012) 51:420–6. doi: 10.1111/j.1365-4632.2011.05066.x
51. Cheung WL, Patel RR, Leonard A, Firoz B, Meehan SA. Amelanotic melanoma: a detailed morphologic analysis with clinicopathologic correlation of 75 cases. J Cutaneous Pathol. (2012) 39:33–9. doi: 10.1111/j.1600-0560.2011.01808.x
52. Moreau JF, Weissfeld JL, Ferris LK. Characteristics and survival of patients with invasive amelanotic melanoma in the USA. Melanoma Res. (2013) 23:408–13. doi: 10.1097/CMR.0b013e32836410fe
53. Piliouras P, Gilmore S, Wurm EM, Soyer HP, Zalaudek I. New insights in naevogenesis: number, distribution and dermoscopic patterns of naevi in the elderly. Australas J Dermatol. (2011) 52:254–8. doi: 10.1111/j.1440-0960.2011.00794.x
54. Rayner JE, Duffy DL, Smit DJ, Jagirdar K, Lee KJ, De'Ambrosis B, et al. Albinism variants in individuals with amelanotic/hypomelanotic melanoma: increased carriage of TYR and OCA2 variants. PLoS ONE. (2020) 15:e0238529. doi: 10.1371/journal.pone.0238529
55. Rayner JE, McMeniman EK, Duffy DL, De'Ambrosis B, Smithers BM, Jagirdar K, et al. IRF4 rs12203592*T/T genotype is associated with nodular melanoma. Melanoma Res. (2019) 29:445–6. doi: 10.1097/CMR.0000000000000596
56. Gibbs DC, Ward SV, Orlow I, Cadby G, Kanetsky PA, Luo L, et al. Functional melanoma-risk variant IRF4 rs12203592 associated with Breslow thickness: a pooled international study of primary melanomas. Br J Dermatol. (2017) 117:e180–2. doi: 10.1111/bjd.15784
57. Duffy DL, Iles MM, Glass D, Zhu G, Barrett JH, Hoiom V, et al. IRF4 variants have age-specific effects on nevus count and predispose to melanoma. Am J Hum Genet. (2010) 87:6–16. doi: 10.1016/j.ajhg.2010.05.017
58. Orlow I, Satagopan JM, Berwick M, Enriquez HL, White KA, Cheung K, et al. Genetic factors associated with naevus count and dermoscopic patterns: preliminary results from the Study of Nevi in Children (SONIC). Br J Dermatol. (2015) 172:1081–9. doi: 10.1111/bjd.13467
59. Chhabra Y, Yong HXL, Fane ME, Soogrim A, Lim W, Mahiuddin DN, et al. Genetic variation in IRF4 expression modulates growth characteristics, tyrosinase expression and interferon-gamma response in melanocytic cells. Pigm Cell Melanoma R. (2018) 31:51–63. doi: 10.1111/pcmr.12620
60. Pozzobon FC, Tell-Marti G, Calbet-Llopart N, Barreiro A, Espinosa N, Potrony M, et al. Influence of germline genetic variants on dermoscopic features of melanoma. Pigment Cell Melanoma Res. (2020) 181:1009–16. doi: 10.1111/pcmr.12954
61. Pollock PM, Harper UL, Hansen KS, Yudt LM, Stark M, Robbins CM, et al. High frequency of BRAF mutations in nevi. Nat Genet. (2003) 33:19–20. doi: 10.1038/ng1054
62. Kumar R, Angelini S, Snellman E, Hemminki K. BRAF mutations are common somatic events in melanocytic nevi. J Investig Dermatol. (2004) 122:342–8. doi: 10.1046/j.0022-202X.2004.22225.x
63. Tan JM, Tom LN, Jagirdar K, Lambie D, Schaider H, Sturm RA, et al. The BRAF and NRAS mutation prevalence in dermoscopic subtypes of acquired naevi reveals constitutive mitogen-activated protein kinase pathway activation. Br J Dermatol. (2018) 178:191–7. doi: 10.1111/bjd.15809
64. Tan JM, Tom LN, Soyer HP, Stark MS. Defining the molecular genetics of dermoscopic naevus patterns. Dermatology. (2019) 235:19–34. doi: 10.1159/000493892
65. Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell. (2015) 161:1681–96. doi: 10.1016/j.cell.2015.05.044
66. Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, et al. Whole-genome landscapes of major melanoma subtypes. Nature. (2017) 545:175–80. doi: 10.1038/nature22071
67. Shain AH, Yeh I, Kovalyshyn I, Sriharan A, Talevich E, Gagnon A, et al. The genetic evolution of melanoma from precursor lesions. N Engl J Med. (2015) 373:1926–36. doi: 10.1056/NEJMoa1502583
68. Shain AH, Joseph NM, Yu R, Benhamida J, Liu S, Prow T, et al. Genomic and transcriptomic analysis reveals incremental disruption of key signaling pathways during melanoma evolution. Cancer Cell. (2018) 34:45–55.e4. doi: 10.1016/j.ccell.2018.06.005
69. Griewank KG, Murali R, Wiesner T. Molecular pathology and genomics of melanoma. In: Balch CM, Atkins MB, Garbe C, Gershenwald JE, Halpern AC, Kirkwood JM, et al., editors. Cutaneous Melanoma. Cham: Springer International Publishing (2020). p. 381–422.
70. Griewank KG, Murali R, Wiesner T. Molecular pathology and genomics of melanoma. In: Balch C, editor. Cutaneous Melanoma. 6 ed. Charm: Springer (2019) 1–42.
71. Whiteman DC, Pavan WJ, Bastian BC. The melanomas: a synthesis of epidemiological, clinical, histopathological, genetic, and biological aspects, supporting distinct subtypes, causal pathways, and cells of origin. Pigment Cell Melanoma Res. (2011) 24:879–97. doi: 10.1111/j.1755-148X.2011.00880.x
72. Bauer J, Buttner P, Wiecker TS, Luther H, Garbe C. Risk factors of incident melanocytic nevi: a longitudinal study in a cohort of 1,232 young German children. Int J Cancer. (2005) 115:121–6. doi: 10.1002/ijc.20812
73. Kittler H, Tschandl P. Driver mutations in the mitogen-activated protein kinase pathway: the seeds of good and evil. Br J Dermatol. (2018) 178:26–7. doi: 10.1111/bjd.16119
74. Stark MS, Tan JM, Tom L, Jagirdar K, Lambie D, Schaider H, et al. Whole-exome sequencing of acquired nevi identifies mechanisms for development and maintenance of benign neoplasms. J Investig Dermatol. (2018) 138:1636–44. doi: 10.1016/j.jid.2018.02.012
75. Stark MS, Denisova E, Kays TA, Heidenreich B, Rachakonda S, Requena C, et al. Mutation signatures in melanocytic nevi reveal characteristics of defective DNA repair. J Investig Dermatol. (2020) 140:2093–6.e2. doi: 10.1016/j.jid.2020.02.021
76. Melamed RD, Aydin IT, Rajan GS, Phelps R, Silvers DN, Emmett KJ, et al. Genomic characterization of dysplastic nevi unveils implications for diagnosis of melanoma. J Investig Dermatol. (2017) 137:905–9. doi: 10.1016/j.jid.2016.11.017
77. Tang J, Fewings E, Chang D, Zeng H, Liu S, Jorapur A, et al. The genomic landscapes of individual melanocytes from human skin. Nature. (2020) 586:600–5. doi: 10.1038/s41586-020-2785-8
Keywords: precancer, precursor lesion, genetics and genomics, artificial intelligence, risk stratification, melanoma, naevi
Citation: Lee KJ, Janda M, Stark MS, Sturm RA and Soyer HP (2021) On Naevi and Melanomas: Two Sides of the Same Coin? Front. Med. 8:635316. doi: 10.3389/fmed.2021.635316
Received: 30 November 2020; Accepted: 01 February 2021;
Published: 19 February 2021.
Edited by:
Youwen Zhou, University of British Columbia, CanadaReviewed by:
Remco Van Doorn, Leiden University Medical Center, NetherlandsCopyright © 2021 Lee, Janda, Stark, Sturm and Soyer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: H. Peter Soyer, cC5zb3llckB1cS5lZHUuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.