 Melise Chaves Silveira1
Melise Chaves Silveira1 Cláudio Marcos Rocha-de-Souza1
Cláudio Marcos Rocha-de-Souza1 Ivson Cassiano de Oliveira Santos1Leilane da Silva Pontes1Thamirys Rachel Tavares e Oliveira1Camila Bastos Tavares-Teixeira1Nataly de Almeida Cossatis1Natacha Ferreira Pereira1
Ivson Cassiano de Oliveira Santos1Leilane da Silva Pontes1Thamirys Rachel Tavares e Oliveira1Camila Bastos Tavares-Teixeira1Nataly de Almeida Cossatis1Natacha Ferreira Pereira1 Orlando Carlos da Conceição-Neto1
Orlando Carlos da Conceição-Neto1 Bianca Santos da Costa1
Bianca Santos da Costa1 Daiana Cristina Silva Rodrigues1Rodolpho Mattos Albano2
Daiana Cristina Silva Rodrigues1Rodolpho Mattos Albano2 Fabrício Alves Barbosa da Silva3Elizabeth Andrade Marques4
Fabrício Alves Barbosa da Silva3Elizabeth Andrade Marques4 Robson Souza Leão4
Robson Souza Leão4 Ana Paula D'Alincourt Carvalho-Assef1*
Ana Paula D'Alincourt Carvalho-Assef1*- 1Laboratório de Pesquisa em Infecção Hospitalar, Instituto Oswaldo Cruz - FIOCRUZ, Rio de Janeiro, Brazil
- 2Departamento de Bioquímica, Instituto de Biologia Roberto de Alcântara Gome, Universidade do Estado do Rio de Janeiro – UERJ, Rio de Janeiro, Brazil
- 3Programa de Computação Científica, Fundação Oswaldo Cruz - FIOCRUZ, Rio de Janeiro, Brazil
- 4Departamento de Microbiologia, Imunologia e Parasitologia, Faculdade de Ciências Médicas, Universidade do Estado do Rio de Janeiro – UERJ, Rio de Janeiro, Brazil
Multidrug-resistant microorganisms are a well-known global problem, and gram-negative bacilli are top-ranking. When these pathogens are associated with bloodstream infections (BSI), outcomes become even worse. Here we applied whole-genome sequencing to access information about clonal distribution, resistance mechanism diversity and other molecular aspects of gram-negative bacilli (GNB) isolated from bloodstream infections in Brazil. It was possible to highlight international high-risk clones circulating in the Brazilian territory, such as CC258 for Klebsiella pneumoniae, ST79 for Acinetobacter baumannii and ST233 for Pseudomonas aeruginosa. Important associations can be made such as a negative correlation between CRISPR-Cas and K. pneumoniae CC258, while the genes blaTEM, blaKPC and blaCTX−M are highly associated with this clone. Specific relationships between A. baumannii clones and blaOXA−51 variants were also observed. All P. aeruginosa ST233 isolates showed the genes blaVIM and blaOXA486. In addition, some trends could be identified, where a new P. aeruginosa MDR clone (ST3079), a novel A. baumannii clonal profile circulating in Brazil (ST848), and important resistance associations in the form of blaVIM−2 and blaIMP−56 being found together in one ST233 strain, stand out. Such findings may help to develop approaches to deal with BSI and even other nosocomial infections caused by these important GNB.
Introduction
Multidrug-resistant (MDR) microorganisms are spread worldwide, and global efforts have been made on different fronts to reduce the incidence of MDR infections. Among them, strengthening antimicrobial resistance knowledge through surveillance and research is a major action (1).
Bloodstream infections (BSI) are usually associated with poor outcomes especially when adequate antimicrobial therapy and source control are delayed. Carbapenems have been used for the treatment of severe infections caused by gram-negative bacilli (GNB). However, carbapenem-resistant GNB, such as Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacterales, are associated with decreased survival and have become a major challenge for therapeutics (2, 3). In this way, GNB bloodstream isolates are an important surveillance target for monitoring resistance and the World Health Organization (WHO) highlighted A. baumannii, P. aeruginosa, and Klebsiella pneumoniae as a critical priority to develop new antibiotics options (1).
Whole-genome sequencing (WGS) is a rapid molecular method for the search of molecular determinants of antimicrobial resistance (AMR). This tool has been proven to be fast and valuable to classify GNB in terms of resistance to the β-lactams and to guide antibiotic treatment decisions for BSI. WGS can detect not only important acquired genes but also chromosomal mutations, both of which contribute significantly to AMR (4). This approach improves our ability to analyze bacterial genomic content, and when these results are correlated with metadata and phylogenetic studies, we can understand with more precision the ways AMR bacteria multiply and spread (5).
Here, we conducted a surveillance about blood GNB isolates recovered from four of the five Brazilian regions using bioinformatic analysis of WGS data. The criteria and methods chosen are justified since as blood is a sterile site, these are more likely to represent true infections, and GNB is the group for which the highest resistance rates are currently found worldwide (2, 3). Phylogenetic analysis, clonal distribution, resistance mechanism diversity and other molecular aspects of nosocomial strains are described. Characteristics such as strain's national distribution, species diversity and the applied tools make this work the starting point for the construction of a new map of resistance in Brazil, contributing with global efforts to prevent the advance of AMR and assist in improving the therapeutic approach, especially for serious infections such as BSI.
Materials and Methods
Bacterial Strains
The Laboratório de Pesquisa em Infecção Hospitalar (LAPIH-FIOCRUZ) takes part in a National Bacterial Resistance Surveillance Network headed by The General Coordination of Public Health Laboratories (Brazilian Health Ministry). Our laboratory routinely receives clinical GNB isolates from hospitals located in different Brazilian states to confirm the mechanisms of drug resistance. The aim of this study was to identify the genetic basis AMR of selected bloodstream and catheter tip bacterial strains recovered from Brazilian states from January 2019 to September 2020.
All gram-negative isolates received by LAPIH in this period were submitted to carbapenemase genes detection by PCR (6) and colistin MICs by the broth microdilution method (http://www.eucast.org). The specie identification was confirmed by biochemical tests and for Complex A. baumannii isolates were performed the detection of blaOXA−51 gene by PCR.
Only isolates of K. pneumoniae, A. baumannii, and P. aeruginosa, the three serious cause of healthcare-associated infections and an emerging health threat worldwide, were selected to evaluate the antimicrobial resistance profile and to be submitted to WGS. The selection criterion per batch of isolates received over the period studied included representative carbapenemase-producing and/or polymyxin-resistant strains from different states.
Antimicrobial Susceptibility
Antimicrobial susceptibility profiles were determined by an automated Vitek 2 system (bioMérieux-Vitek, Hazelwood, Mo).
Genome Sequencing and Bioinformatic Analysis
WGS was performed using the Illumina Miseq (Illumina, San Diego, California, USA). A genomic library was constructed by transposon tagmentation with the Nextera XT DNA Sample Prep kit (Illumina, Inc., USA). Kraken2 was used to classify the reads taxonomically (7). The paired-end reads were de novo assembled using SPAdes v3.13.1 (8). Contigs shorter than 500 bp were discarded. CheckM was applied to assess the quality of genome sequencing and assembly, estimating their completeness, contamination, size and contigs parameters (9). The genome coverage was calculated using: coverage = (forward reads count * 2) * read length/total genome size. Species from A. baumannii were confirmed using Kmer-db (10) and reference strains (A. baumannii KL810966.1, A. nosocomialis GCF_000368085.1, A. pittii CP002177, and A. calcoaceticus NZ_LS999521).
Draft genomes were then annotated using Prokka (11). The prediction of the resistome and plasmid incompatibility (Inc) group were made using the ABRicate (T. Seemann, https://github.com/tseemann/abricate) against ResFinder (12), and PlasmidFinder (13), respectively. Only the results above 90% of coverage and identity using ABRicate were considered. Besides that, BLAST (blastx) alignment results were analyzed to identify mutations related to resistance (14). The proteins analyzed were PhoP and PhoQ (polymyxin resistance), besides RamR and AdrR (tigecycline resistance) for K. pneumoniae, using MGH78578 as reference; ColR, ColS, and PhoQ (polymyxin resistance), besides OprD (carbapenem resistance), AmpC (cephalosporin resistance) and MexT (different classes) for P. aeruginosa using PA01 as reference (AAG07497.1, the functional variant CAA07694 being used for MexT); and LpxA, LpxC, and LpxD (polymyxin resistance), besides AdeN and AdeR (tigecycline resistance) for A. baumannii using ATCC 19606 as reference. Furthermore, proteins PmrA and PmrB (polymyxin resistance), and GyrA, GyrB, and ParC (fluoroquinolones resistance) were analyzed for all these three species. Deleterious mutations were checked using PROVEAN web server (15). We only considered mutations classified as deleterious in Results and Discussion section. Presence of a CRISPR-Cas system was assessed by the CRISPRCasFinder (16). MLST was performed with the MLST software (T. Seemann, https://github.com/tseemann/mlst) and the PubMLST database (17).
Phylogenetic analysis were performed using kSNP3.0 software, without core genome option (18). The program Kchooser, which is part of the kSNP package, was used to identify the optimal kmer length. iTOL was applied for visualizing and annotating the phylogenetic trees (19).
Genome Accession Number
All draft genomes are available at GenBank BioProject accession PRJNA677881.
Results
In the period of January 2019 to September 2020, 577 blood GNB isolates were received by LAPIH to characterize the resistance mechanisms (Supplementary Table 1). The isolates belonged to different species, the most frequent were K. pneumoniae (n = 200), A. baumannii (n = 192), and P. aeruginosa (n = 119). PCR analysis showed that the most prevalent carbapenemase gene among Enterobacterales isolates was blaKPC (69,6%, n = 176) and among Acinetobacter species was blaOXA−23like (89,6%, n = 172). Finally, Pseudomonas isolates were found to carry blaVIM (n = 20), blaKPC (n = 17), blaIMP (n = 8), and blaSPM (n = 4). However, 63.0% (n = 75) of P. aeruginosa isolates did not carry any of the investigated carbapenemases. Excluding species intrinsically resistant, polymyxin resistance was detected in 170 isolates (30.7%), belonging mainly to K. pneumoniae (n = 123/200, 61.5%). Furthermore, the plasmid-mediated mcr-1 gene was found in 2 Escherichia coli.
Based on the results obtained, WGS was performed on 84 selected isolates and included the following species: K. pneumoniae (n = 46/200, 23.5%, being one K. quasipneumoniae), A. baumannii (n = 23/192, 12%), and P. aeruginosa (n = 15/119, 12.6%). These isolates were representative of carrying carbapenemase genes and/or polymyxin resistance recovered from the 7 Brazilian states included in the study belonging to 4 geographic regions: Northeast (51.2%), Southeast (35.7%), Midwest (6%), and North (7.1%) (Supplementary Table 2).
Antimicrobial resistance levels for the sequenced organisms are shown in Figure 1. All strains from the three species analyzed displayed resistance to cefepime. Most significantly, the vast majority of the K. pneumoniae, A. baumannii, and P. aeruginosa isolates showed resistance to piperacillin-tazobactam, imipenem, and meropenem (±98.33%). Ampicillin-sulbactam, ceftazidime, and ceftriaxone were not active against all K. pneumoniae strains and the resistance rates for ertapenem, ciprofloxacin, gentamicin, tigecycline, and amikacin were 93.43, 89.13, 63.04, 26.08, and 15.22%, respectively. The P. aeruginosa isolates showed high resistance to amikacin, ceftazidime, gentamicin, and ciprofloxacin (93.33, 73.33, 66.66, and 66.66%, respectively). Furthermore, the antibiotics resistance pattern for A. baumannii isolates was: ciprofloxacin (100%), ceftriaxone (95.65%), ceftazidime (95.65%), gentamicin (43.48%), amikacin (39.13%), ampicillin-sulbactam (34.48%), and tigecycline (17.39%). Finally, 27/46 (56.46%) K. pneumoniae, 3/15 (20%) P. aeruginosa and 2/23 (8.69%) A. baumannii isolates showed polymyxin resistance (MICs >2mg/L) by broth microdilution.
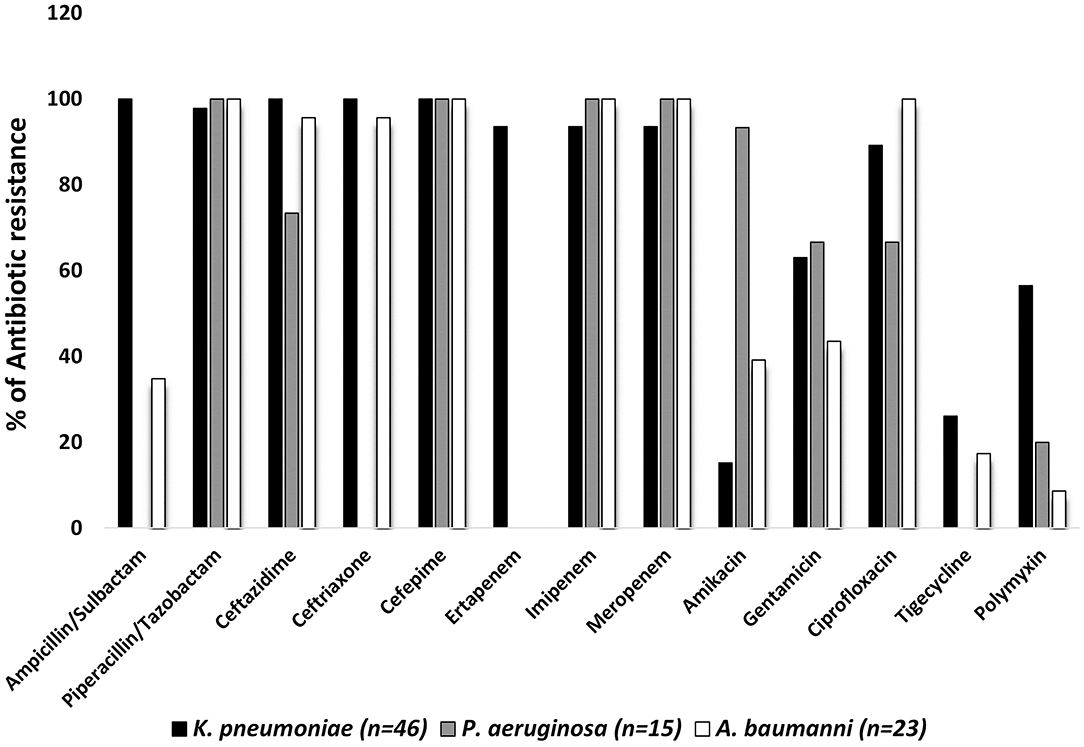
Figure 1. Antibiotics resistance patterns in percentages from selected bloodstream GNB isolated from January 2019 to September 2020. Antimicrobial susceptibility tests were interpreted according to the European/Brazilian Committee on Antimicrobial Susceptibility Testing guidelines. The minimum inhibitory concentration for colistin was obtained using the broth microdilution test, according to the European/Brazilian Committee on Antimicrobial Susceptibility Testing guidelines.
The bioinformatic analysis results are summarized in Figure 2. The acquired genes found are related to resistance to 11 different classes, including beta-lactams, aminoglycosides, sulfonamides, fluoroquinolone, macrolide, trimethoprim, tetracycline, fosfomycin, chloramphenicol, rifamycin, and macrolide/streptogramin/lincosamide (MLS phenotype). Most of the sequenced strains were positive for gene families sul (76.2%, sulfonamides resistance), aph(3) (73.8%, aminoglycosides resistance), and fosA (71.4%, fosfomycin resistance). Fifteen families of β-lactamase genes were found (blaKPC, blaNDM, blaSPM, blaIMP, blaVIM, blaCTX−M, blaGES, blaTEM, blaSHV, blaSCO, blaCARB, blaADC, blaOKP, blaPAO, and blaOXA).
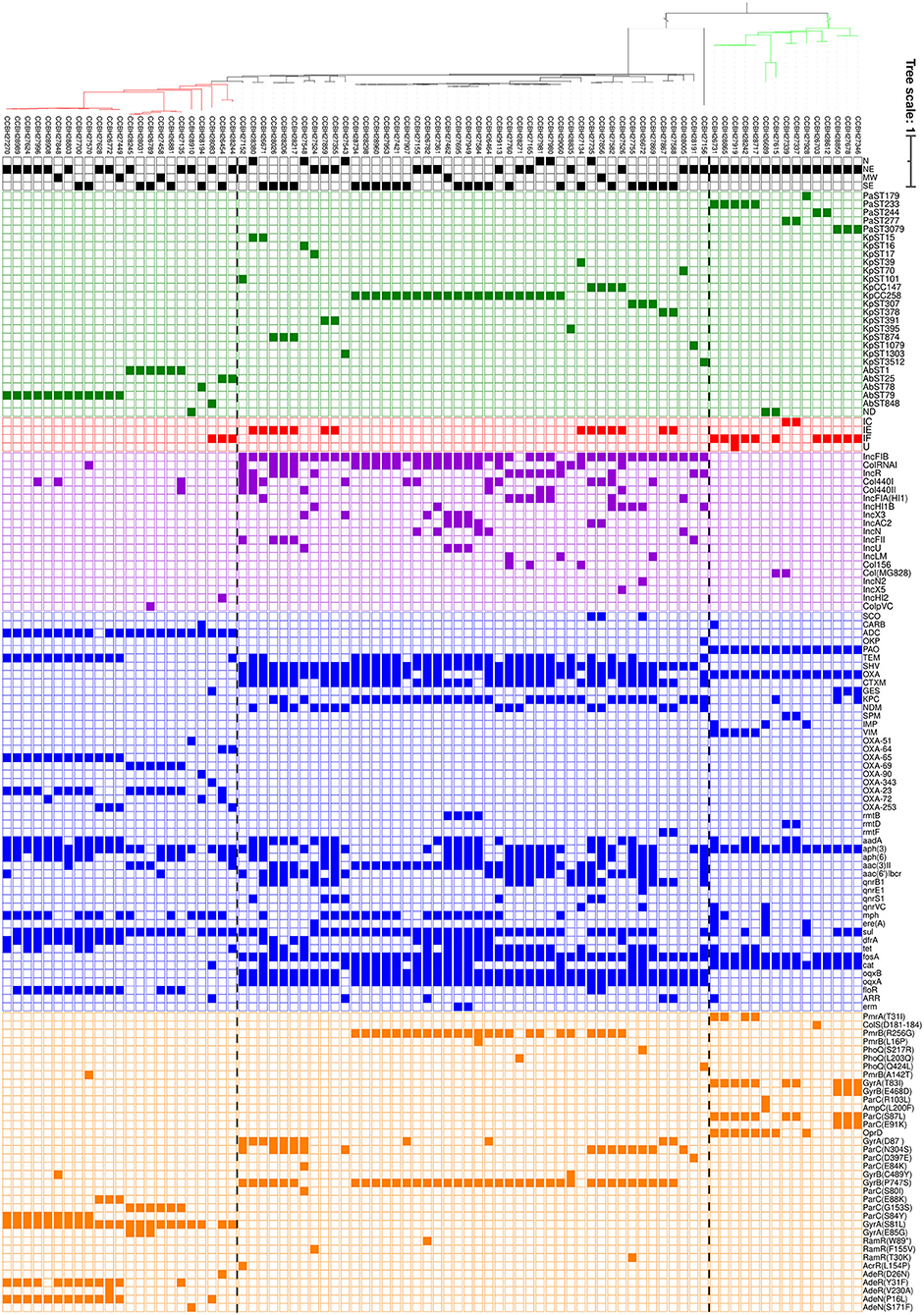
Figure 2. Whole-genome SNP-based parsimony tree including isolates of K. pneumoniae (46 isolates, black clade), A. baumannii (23 isolates, red clade), and P. aeruginosa (15 isolates, green clade) generated by kSNP3.0. The branch lengths are expressed in terms of changes per number of SNPs. The tree was visualized using iTol (19). The panel shows the presence (filled squares) and absence (empty squares) of the characteristics analyzed. Each color corresponds to a characteristic: black (Brazilian regions), green (Sequence Types), red (Cas type), purple (Inc groups), blue (resistance acquired genes), and orange (deleterious mutations in proteins related to antimicrobial resistance). N, north; NE, northeast; MW, Midwest; SE, southeast; ND, not determined.
The most prevalent resistance genes among K. pneumoniae and K. quasipneumoniae isolates were fosA (100%), blaSHV (97.8%), and oqxAB (97.8%, fluoroquinolones resistance). Plasmids from the incompatibility group IncFIB were found in 93.5% of all isolates. The strains belonged to 20 STs. The CC258 (ST11, n = 16, ST258, n = 4, and ST437, n = 1) was the most prevalent clonal complex (45.7%) found in four Brazilian regions, followed by CC147 (ST147, n = 1, ST392, n = 2, and ST273, n = 1) (8.7%). All CC258 strains analyzed showed absence of CRISPR-Cas and 85.7% were found to carry the genes blaKPC, blaCTX−M, and blaTEM. The rmtB gene (aminoglycosides resistance) was detected solely in ST258, in 4 strains. Of these, three are the only amikacin resistant strains from CC258. IncA/C2 was found only in ST258 strains. The CC258 showed a high Inc groups diversity, being IncFIB the most prevalent (90.5%), followed by ColRNAI (66.7%) and IncR (28.6%). In addition, deleterious mutations were identified in ParC (S80I) for all CC258 strains and in PmrB (R256G) for 90.5% of them. ParC could interfere in fluoroquinolones resistance, and indeed all CC258 strains were ciprofloxacin resistant. On the other hand, 5 K. pneumoniae strains from CC258 were polymyxin-susceptible despite the presence of mutation in PmrB (R256G). The CC258 strains with PhoQ (L203Q) or PmrB (L16P) deleterious mutations were resistant to polymyxin. All CC147 strains showed the adaptative immune system CRISPR-Cas type IE and were found to carry the resistance genes blaOXA−1, blaSHV, oqxAB, and sul1. IncFIB and deleterious mutations in PmrB (R256G), ParC (S80I), and ParC (N304S) were also found in all strains. Despite PmrB mutation, 75% of CC147 strains were polymyxin susceptible. In contrast, all of them were ciprofloxacin resistant and it could be related to ParC mutations. The ST3512 K. quasipneumoniae strain (CCBH27156), presented some unique characteristics, such as the simultaneous presence of the blaKPC and blaNDM, absence of the blaSHV, and mutation in PhoQ (Q424L), however, it was not associated with polymyxin resistance. Among all strains with PmrB R256G mutation, 33.3% were susceptible to polymyxin. The deleterious (T30K) and non-sense (W89*) mutations on RamR were only present in tigecycline resistant strains. Another 10 strains proved to be tigecycline resistant without any characteristic mutation identified. The tet(x) gene, related to tigecycline resistance, was not found.
The 23 A. baumannii strains analyzed belonged to 6 STs and were recovered from three Brazilian regions. ST79 was the most prevalent ST found (52.2%), followed by ST1 (26.1%) and ST25 (8.7%). The most common characteristics were the deleterious mutation S81L in GyrA (95.7%), and the resistance genes blaADC (95.7%), aph(3) (91.3%), and sul (91.3%). The resistance genes blaOXA−65 (blaOXA−51 allelic variant), blaTEM−1A, and dfrA (trimethoprim resistance) were only detected in all ST79 strains. The blaOXA−23 was the most prevalent carbapenemase gene among ST79 strains (66.7%). The carbapenemase gene blaOXA−253 (blaOXA−143 allelic variant), was also detected in three strains from this ST. The resistance gene blaOXA−69 (blaOXA−51 allelic variant), and the deleterious mutation G153S in ParC were only found in all ST1 strains. The carbapemase gene blaOXA−23 was also found in all ST1 strains. A few A. baumannii strains presented the Inc groups investigated (26.1%). A IncHI2 plasmid and the carbapenemase genes blaOXA−23 and blaOXA−72 (blaOXA−24 allelic variant), were found in one strain belonging to ST25. The CRISPR-Cas system type IF was detected in only three strains, assigned to ST25 and ST848. Interestingly, the only ST848 strain presented blaGES−11 and arr genes (rifamycin resistance) and the absence of S81L mutation in GyrA, differing from the other A. baumannii isolates. The only strain with a PmrB deleterious mutation showed polymyxin susceptibility (A142T in CCBH27570). We did not identify deleterious mutation associated to the two polymyxin resistant strains. The mutations AdeR(D26N), AdeR(V230A), and AdeN(S171F) were found in tigecycline-susceptible strains. Two mutations were majority related to ST79 (AdeR, Y31F and AdeN, P16L), present in both susceptible and resistant strains. The tet(x) gene was not found.
The 15 P. aeruginosa strains analyzed belonged to 5 STs and were from 1 Brazilian region. ST233 (33.3%) was the most prevalent ST found, followed by ST3079 (20%). All strains showed the resistance genes blaPAO, aph(3), and catB7 (chloramphenicol resistance). The CRISPR-Cas system was detected in 86.6% of the strains, being type I-F the most frequent. Deleterious mutations T83I and S87L in GyrA and ParC, respectively, were the most frequently found (66.7%), and were distributed in different clones. These two mutations were present in all ciprofloxacin resistant strains and absent in strains susceptible to this antibiotic. All 5 ST233 strains showed the genes blaVIM and blaOXA486. The PmrA deleterious mutation T31I was found in four strains belonging to this ST, and two of them are resistant to polymyxin according to MIC. Furthermore, one ST233 isolate carried the carbapenemase genes blaVIM−2 and blaIMP−56 together. The three ST3079 strains were the only ones to show the gene blaGES−1 and the deleterious mutations in Parc (E91K) and GyrB (E468D). The genes blaSPM, blaOXA−56, and rmtD were found only in all ST277 isolates. A strain from ST244 with ColS amino acids deletion (D181–184) was polymyxin susceptible. The plasmid incompatibility group Col(MG828) was found in two P. aeruginosa isolates from different STs.
Discussion
In the present study we analyzed gram-negative pathogens causing BSI and K. pneumoniae was the major species among MDR strains. A national study about nosocomial BSI in Brazil reveled that monomicrobial episodes of BSI were mostly caused by gram-negative bacteria, highlighting K. pneumoniae (20). Other Brazilian data about BSI, from a single hospital, showed that K. pneumoniae infections had a positive association with the MDR phenotype (21). Both studies corroborate the high prevalence of this specie among the isolates selected for the present work.
Antibiotic resistance within the group of gram-negative bacteria related to BSI is worrisome in Brazil, and the pathogens analyzed here are among the most outstanding (20, 21). According to our results, amikacin was the most effective antimicrobial against K. pneumoniae, and, A. baumannii and P. aeruginosa showed a great sensibility to polymyxin.
A predominance of worldwide distributed clones among K. pneumoniae (CC258), A. baumannii (ST79) and P. aeruginosa (ST233) strains was observed. Isolates from international high-risk CC258 have been found in all Brazilian states and are highly associated with the dissemination of carbapenemase KPC (6, 22). The ST1 is designed as the second major A. baumannii clone and is globally associated to OXA-23 (23), however in South America this carbapenemase has been most commonly related with ST79, the third major international distribution clone, as noted here (23, 24). The most prevalent ST in P. aeruginosa, ST233, is a worldwide disseminated MDR clone particularly linked to VIM-2, as was corroborated by our results (25).
The prevalence of gram-negative BSI has practical importance, especially when dealing with the issue of treatment, creating a vicious cycle of antimicrobial heavy use and resistance development (20). A great variety of resistance genes was found, highlighting sul and fosA. High prevalence rates of sul variants have been observed mainly in gram-negative bacteria isolated all over the world (26). Meanwhile, fosA is commonly found in K. pneumoniae and P. aeruginosa contributing to intrinsic fosfomycin resistance, but is largely absent in A. baumannii (27), as confirmed here. The resistance rates to carbapenems equal or higher than 93,48% can be associated to specific carbapenemases genes found more often. The carbapenemase blaKPC−2 was very prevalent in K. pneumoniae, contrasting with blaNDM−1. This difference is expected once blaKPC−2 circulates in Brazil since 2009 (28), and blaNDM−1 was detected only in 2013 in this country (29). Furthermore, most of the analyzed strains are CC258 K. pneumoniae, which is spread in Brazil mainly carrying blaKPC (22). The most prevalent carbapenemase in A. baumannii strains, OXA-23, was detected here in already associated clones as ST1, ST25 and ST79 (23, 30). Although this gene was not detected in some ST79 strains from this surveillance, all of them have OXA-65 carbapenemase, as previously reported in a Brazilian surveillance (31). The single strain from A. baumannii ST848 calls to attention since the occurrence of blaGES−11, which mediates resistance to β-lactam and reduced susceptibility to carbapenem, has been reported in other continents and clones but neither in America nor in ST848 (32). Furthermore, this clone is not clearly associated with A. baumannii isolates from Brazil. The carbapenemases genes found in P. aeruginosa are highly associated with a specific clone. The association of blaVIM−2 to ST233 (25) and blaSPM−1 to ST277 (33) have been reported. On the other hand, the association of ESBL blaGES−1 to ST3079 has not yet been described. Carbapenemase KPC-2 was also present in 2 of the 3 strains analyzed from this clone. It is important that the dispersion of P. aeruginosa ST3079 be monitored and the prevalence of these important genes confirmed. The coexistence of genes blaVIM and blaIMP in P. aeruginosa found here for a single strain was described before in India, associated with a MIC for meropenem higher than 32 μg/ml (34), so this association also demands attention.
Since resistance to carbapenems is usually associated with multidrug resistance, polymyxins became the last alternative (20). Here we detected different deleterious mutations in PhoQ for K. pneumoniae, PmrA for P. aeruginosa and PmrB for K. pneumoniae and A. baumannii. It is difficult to extrapolate whether some substitutions identified in proteins, mainly in P. aeruginosa and A. baumannii, leads to polymyxin resistance, and the levels of gene expression may vary and consequently influence the level of resistance (35). The deleterious mutation R256G in PmrB which seems common in K. pneumoniae CC258 and CC147, has been found in polymyxin-susceptible isolates (36), and the same was observed here. Other studies will be necessary to verify if PhoQ (L203Q) and PmrB (L16P) deleterious mutations are indeed associated to polymyxin resistant in K. pneumoniae. As far as we know, the PmrA deleterious mutation shown in P. aeruginosa (T31I) has not yet been linked to polymyxin resistance, but among the 4 strains that showed this mutation, 2 were resistant according to MIC. The PmrB A142T mutation in A. baumannii is present in a susceptible strain, and has not yet been linked to polymyxin resistance.
Although ParC deleterious mutations occurred in different K. pneumoniae clones and may cause ciprofloxacin resistance, we could not ignore the high frequency of oqxAB genes, which encode a efflux pump that confer resistance to multiple agents including fluoroquinolones. Some mutations observed for P. aeruginosa are relevant examples of gain-of-function and are associated with resistance to fluoroquinolones [GyrA (T83I), GyrB (E468D) and ParC (S87L)] (37). Indeed, we showed that the GyrA and ParC deleterious mutations were strict associated to ciprofloxacin resistance. For A. baumannii, the simultaneous presence of mutations in GyrA (S81L) and in ParC (S84L) have been associated to resistance to ciprofloxacin and nalidixic acid (38). All A. baumannii strains analyzed here are ciprofloxacin resistant and most of them have the GyrA S81L mutation (95.8%). For ST1, the deleterious mutations ParC (S84L e G153S) could be noted simultaneously to GyrA(S81L), and for ST79 the concomitant mutation was ParC(S84Y). These associations could explain ciprofloxacin resistance for the major A. baumannii STs in this study.
Plasmid-mediated transfer of resistance has led to widespread dissemination, outbreaks, and untreatable infections. Molecular identification of plasmid and strain genotypes can distinguish whether the spread of AMR genes is driven by epidemic plasmids to different hosts or by clonal spread of bacterial organisms harboring these plasmids with AMR genes (39). Here, the diversity and abundance of Inc groups found in K. pneumoniae strains contrast with P. aeruginosa and A. baumannii results. The majority of K. pneumoniae strains analyzed have the IncFIB plasmid, a group known for their MDR characteristic (39, 40). In Brazil, the IncFIB plasmid has already been described as carrying blaKPC−2 and blaNDM−1 in K. pneumoniae (40, 41). A recent manuscript reported that plasmids with IncR, ColRNAI and IncF co-exists in KPC-2-producing K. pneumoniae strains from ST11 (42). Interestingly, we detected IncR in most ST11 isolates, but not in other CC258 clones. IncN was detected mainly in CC258, which is expected as in Brazil the spread of blaKPC−2 has already been related to dispersion of Tn4401 'b', carried by IncN plasmids mainly disseminated by this clonal complex (22). IncA/C2 was only detected in ST258 strains, the same strains which carry rmtB and tet(G) genes. This profile is very similar to plasmid pMTY16641, identified in the first KPC-producing K. pneumoniae ST258 isolated from a Japanese patient without a history of foreign travel (43). The incompatibility group IncHI2 was found in a single A. baumannii strain, the only one positive both for blaOXA−23 and blaOXA−72, and as far as we know, this group has not yet been associated with such genes.
The only species for which almost all strains present CRISPR-Cas systems was P. aeruginosa. Types I-F and I-C identified here were already described in P. aeruginosa strains. Type I-F is the most widely distributed system among P. aeruginosa, while most type I-C-positive strains seem restricted to MDR clones, like ST277 (44), and a similar scenario was observed here. Only CRIPR-Cas type I-E is already described in the literature for K. pneumoniae, as was seen in this surveillance (45). It was also confirmed that this type is extremely rare in CC258 (45). However, other linages positive for blaKPC, like CC147 and ST874, presented half or more positive strains for type I-E CRISPR-Cas system, so the negative association between CRISPR-Cas system and blaKPC seems to be specific to CC258. In A. baumannii, only the CRISPR type I–F system has been found so far (46), and we detected it in 12.5% of the strains. One of these was also positive for IncHI2 contradicting the recently reported negative association between A. baumannii strains with CRISPR system and plasmid detection (46).
WGS is a powerful tool to provide reliable data for monitoring clonal dispersion, antimicrobial resistance and other related characteristics. In the presented study, it was possible to highlight high-risk clones circulating in Brazilian territory. Some important associations could be made between these clones, important resistance genes and specific deleterious mutations in genes also related to antibiotic resistance. In addition, some trends could be identified such as new MDR clones, and novel important resistance genes and clonal profiles that are circulating. Such findings may help to develop approaches to deal with BSI and even other nosocomial infections caused by these important GNB, mainly in Brazil but also in other countries, since most clones are globally dispersed.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA677881.
Author Contributions
AC-A conceived the research. IO, LP, TO, CT-T, NC, NP, OC-N, BC, and DR were responsible for the execution of phenotypic and molecular tests to identify and detect resistance of Gram-negative bacteria. AC-A and CR participated in the selection of sequenced strains. MS, CR, and RL participated in method design, whole genome sequencing, and data handling. MS performed the data analysis. MS and CR made the figures. AC-A, MS, CR, RA, FS, EM, and RL wrote parts and edited the complete manuscript. All authors have read and approved the manuscript.
Funding
This manuscript received specific funding from Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro - FAPERJ (E-26/202.554.2019) and Conselho Nacional de Desenvolvimento Científico e Tecnológico - CNPq (402524/2018-7).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2021.635206/full#supplementary-material
Supplementary Table 1. PCR and MIC results about the 577 blood GNB isolates received between January 2019 to September 2020.
Supplementary Table 2. General information about 84 blood GNB isolates submitted to whole genome sequencing.
References
1. World Health Organization. Global Action Plan on Antimicrobial Resistance. (2015). Available online at: https://apps.who.int/iris/bitstream/handle/10665/193736/9789241509763_eng.pdf?sequence=1 (accessed February 7, 2021).
2. Kadri SS, Adjemian J, Lai YL, Spaulding AB, Ricotta E, Rebecca Prevots D, et al. Difficult-to-treat resistance in gram-negative bacteremia at 173 US hospitals: retrospective cohort analysis of prevalence, predictors, and outcome of resistance to all first-line agents. Clin Infect Dis. (2018) 67:1803–14. doi: 10.1093/cid/ciy378
3. Diekema DJ, Hsueh P-R, Mendes RE, Pfaller MA, Rolston KV, Sader HS, et al. The microbiology of bloodstream infection: 20-year trends from the SENTRY antimicrobial Surveillance Program. Antimicrob Agents Chemother. (2019) 63:e00355-19. doi: 10.1128/AAC.00355-19
4. Shelburne SA, Kim J, Munita JM, Sahasrabhojane P, Shields RK, Press EG, et al. Whole-genome sequencing accurately identifies resistance to extended-spectrum β-lactams for major gram-negative bacterial pathogens. Clin Infect Dis. (2017) 65:738–45. doi: 10.1093/cid/cix417
5. McEwen SA, Collignon PJ. Antimicrobial resistance: a one health colloquium. Microbiol Spectr. (2018) 6:1–26. doi: 10.1128/microbiolspec.ARBA-0009-2017
6. Aires CAM, Pereira PS, Rocha-De-Souza CM, Silveira MC, Carvalho-Assef APDA, Asensi MD. Population structure of KPC-2-producing Klebsiella pneumoniae isolated from surveillance rectal swabs in Brazil. Microb Drug Resist. (2020) 26:652–60. doi: 10.1089/mdr.2019.0166
7. Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. (2019) 20:257. doi: 10.1186/s13059-019-1891-0
8. Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. (2012) 19:455–77. doi: 10.1089/cmb.2012.0021
9. Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. (2015) 25:1043–55. doi: 10.1101/gr.186072.114
10. Deorowicz S, Gudys A, Dlugosz M, Kokot M, Danek A. Kmer-db: instant evolutionary distance estimation. Bioinformatics. (2019) 35:133–6. doi: 10.1101/263590
11. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. (2014) 30:2068–9. doi: 10.1093/bioinformatics/btu153
12. Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. (2012) 67:2640–4. doi: 10.1093/jac/dks261
13. Carattoli A, Zankari E, Garciá-Fernández A, Larsen MV, Lund O, Villa L, et al. In Silico detection and typing of plasmids using plasmidfinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother. (2014) 58:3895–903. doi: 10.1128/AAC.02412-14
14. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. (1990) 215:403–10. doi: 10.1016/S0022-2836(05)80360-2
15. Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. (2015) 31:2745–7. doi: 10.1093/bioinformatics/btv195
16. Couvin D, Bernheim A, Toffano-Nioche C, Touchon M, Michalik J, Néron B, et al. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. (2018) 46:W246–W251. doi: 10.1093/nar/gky425
17. Jolley KA, Bray JE, Maiden MCJ. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications [version 1; referees: 2 approved]. Wellcome Open Res. (2018) 3:124. doi: 10.12688/wellcomeopenres.14826.1
18. Gardner SN, Slezak T, Hall BG. kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics. (2015) 31:2877–8. doi: 10.1093/bioinformatics/btv271
19. Letunic I, Bork P. Interactive Tree of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. (2019) 47:W256–9. doi: 10.1093/nar/gkz239
20. Marra AR, Camargo LFA, Pignatari ACC, Sukiennik T, Behar PRP, Medeiros EAS, et al. Nosocomial bloodstream infections in Brazilian hospitals: analysis of 2,563 cases from a prospective nationwide surveillance study. J Clin Microbiol. (2011) 49:1866–71. doi: 10.1128/JCM.00376-11
21. Ferreira Leal H, Azevedo J, Evelyn Oliveira Silva G, Maria Lima Amorim A, Rangel Cabral de Roma L, Carolina Palmeira Arraes A, et al. Bloodstream infections caused by multidrug-resistant gram-negative bacteria: epidemiological, clinical and microbiological features. BMC Infect. Dis. (2019) 19:609. doi: 10.1186/s12879-019-4265-z
22. Pereira PS, De araujo CFM, Seki LM, Zahner V, Carvalho-Assef APDA, Asensi MD. Update of the molecular epidemiology of KPC-2-producing Klebsiella pneumoniae in Brazil: spread of clonal complex 11 (ST11, ST437 and ST340). J Antimicrob Chemother. (2013) 68:312–6. doi: 10.1093/jac/dks396
23. Hamidian M, Nigro SJ. Emergence, molecular mechanisms and global spread of carbapenem-resistant acinetobacter baumannii. Microb Genomics. (2019) 5:e000306. doi: 10.1099/mgen.0.000306
24. Rodríguez CH, Nastro M, Famiglietti A. Carbapenemases in Acinetobacter baumannii. Review of their dissemination in Latin America. Rev Argent Microbiol. (2018) 50:327–33. doi: 10.1016/j.ram.2017.10.006
25. del Barrio-Tofiño E, Causapé CL, Oliver A. Pseudomonas aeruginosa epidemic high-risk clones and their association with horizontally-acquired β-lactamases: 2020 update. Int J Antimicrob Agents. (2020) 2020:106196. doi: 10.1016/j.ijantimicag.2020.106196
26. Jiang H, Cheng H, Liang Y, Yu S, Yu T, Fang J, et al. Diverse mobile genetic elements and conjugal transferability of sulfonamide resistance genes (sul1, sul2, and sul3) in Escherichia coli isolates from Penaeus vannamei and pork from large markets in Zhejiang, China. Front Microbiol. (2019) 10:1787. doi: 10.3389/fmicb.2019.01787
27. Ito R, Mustapha MM, Tomich AD, Callaghan JD, McElheny CL, Mettus RT, et al. Widespread fosfomycin resistance in gram-negative bacteria attributable to the chromosomal fosA gene. MBio. (2017) 8:e00749-17. doi: 10.1128/mBio.00749-17
28. Monteiro J, Santos AF, Asensi MD, Peirano G, Gales AC. First report of KPC-2-producing Klebsiella pneumoniae strains in Brazil. Antimicrob Agents Chemother. (2009) 53:333–4. doi: 10.1128/AAC.00736-08
29. Carvalho-Assef AP, Pereira PS, Albano RM, Berião GC, Chagas TP, Timm LN, et al. (2013). Isolation of NDM-producing providencia rettgeri in brazil. J Antimicrob Chemother. (2013) 68:2956–7. doi: 10.1093/jac/dkt298
30. Chagas TPG, Carvalho KR, de Oliveira Santos IC, Carvalho-Assef APDA, Asensi MD. Characterization of carbapenem-resistant Acinetobacter baumannii in Brazil (2008-2011): countrywide spread of OXA-23-producing clones (CC15 and CC79). Diagn Microbiol Infect Dis. (2014) 79:468–72. doi: 10.1016/j.diagmicrobio.2014.03.006
31. Leal NC, Campos TL, Rezende AM, Docena C, Mendes-Marques CL, de Sá Cavalcanti FL, et al. Comparative genomics of Acinetobacter baumannii clinical strains from Brazil reveals polyclonal dissemination and selective exchange of mobile genetic elements associated with resistance genes. Front Microbiol. (2020) 11:1176. doi: 10.3389/fmicb.2020.01176
32. Wibberg D, Salto IP, Eikmeyer FG, Maus I, Winkler A, Nordmann P, et al. Complete genome sequencing of acinetobacter baumannii strain K50 discloses the large conjugative plasmid pK50a encoding carbapenemase OXA-23 and extended-spectrum β-Lactamase GES-11. Antimicrob Agents Chemother. (2018) 62:e00212-18. doi: 10.1128/AAC.00212-18
33. Silveira MC, Rocha-De-Souza CM, Albano RM, De Oliveira Santos IC, Carvalho-Assef APDA. Exploring the success of Brazilian endemic clone Pseudomonas aeruginosa ST277 and its association with the CRISPR-Cas system type I-C. BMC Genomics. (2020) 21:255. doi: 10.1186/s12864-020-6650-9
34. Mohanam L, Menon T. Coexistence of metallo-beta-lactamase-encoding genes in Pseudomonas aeruginosa. Indian J Med Res Suppl. (2017) 146:46–52. doi: 10.4103/ijmr.IJMR_29_16
35. Poirel L, Jayol A, Nordmanna P. Polymyxins: antibacterial activity, susceptibility testing, and resistance mechanisms encoded by plasmids or chromosomes. Clin Microbiol Rev. (2017) 30:557–96. doi: 10.1128/CMR.00064-16
36. Pitt ME, Elliot AG, Cao MD, Ganesamoorthy D, Karaiskos I, Giamarellou H, et al. Multifactorial chromosomal variants regulate polymyxin resistance in extensively drug-resistant Klebsiella pneumoniae. Microb Genomics. (2018) 4:e000158. doi: 10.1099/mgen.0.000158
37. López-Causapé C, Cabot G, del Barrio-Tofiño E, Oliver A. The versatile mutational resistome of Pseudomonas aeruginosa. Front Microbiol. (2018) 9:1–9. doi: 10.3389/fmicb.2018.00685
38. Kumburu HH, Sonda T, Van Zwetselaar M, Leekitcharoenphon P, Lukjancenko O, Mmbaga BT, et al. Using WGS to identify antibiotic resistance genes and predict antimicrobial resistance phenotypes in MDR Acinetobacter baumannii in Tanzania. J Antimicrob Chemother. (2019) 74:1484–93. doi: 10.1093/jac/dkz055
39. Rozwandowicz M, Brouwer MSM, Fischer J, Wagenaar JA, Gonzalez-Zorn B, Guerra B, et al. Plasmids carrying antimicrobial resistance genes in Enterobacteriaceae. J Antimicrob Chemother. (2018) 73:1121–37. doi: 10.1093/jac/dkx488
40. Raro OHF, da Silva RMC, Filho EMR, Sukiennik TCT, Stadnik C, Dias CAG, et al. Carbapenemase-Producing Klebsiella pneumoniae from transplanted patients in Brazil: phylogeny, resistome, virulome and mobile genetic elements harboring blaKPC−2 or blaNDM−1. Front Microbiol. (2020) 11:1563. doi: 10.3389/fmicb.2020.01563
41. Dalmolin TV, Martins AF, Zavascki AP, de Lima-Morales D, Barth AL. Acquisition of the mcr-1 gene by a high-risk clone of KPC-2-producing Klebsiella pneumoniae ST437/CC258, Brazil. Diagn Microbiol Infect Dis. (2018) 90:132–3. doi: 10.1016/j.diagmicrobio.2017.09.016
42. Yu X, Zhang W, Zhao Z, Ye C, Zhou S, Wu S, et al. Molecular characterization of carbapenem-resistant Klebsiella pneumoniae isolates with focus on antimicrobial resistance. BMC Genomics. (2019) 20:822. doi: 10.1186/s12864-019-6225-9
43. Ainoda Y, Aoki K, Ishii Y, Okuda K, Furukawa H, Manabe R, et al. Klebsiella pneumoniae carbapenemase (KPC)-producing Klebsiella pneumoniae ST258 isolated from a Japanese patient without a history of foreign travel - a new public health concern in Japan: a case report. BMC Infect Dis. (2019) 19:1–20. doi: 10.1186/s12879-018-3649-9
44. Belkum A, Van Soriaga LB, Lafave MC, Akella S, Veyrieras J, Barbu EM, et al. Phylogenetic distribution of CRISPR-cas systems in antibiotic- resistant Pseudomonas aeruginosa. MBio. (2015) 6:1–13. doi: 10.1128/mBio.01796-15
45. Zhou Y, Tang Y, Fu P, Tian D, Yu L, Huang Y, et al. The type I-E CRISPR-Cas system influences the acquisition of blaKPC-IncF plasmid in Klebsiella pneumonia. Emerg Microbes Infect. (2020) 9:1011–22. doi: 10.1080/22221751.2020.1763209
46. Mangas EL, Rubio A, Álvarez-Marín R, Labrador-Herrera G, Pachón J, Pachón-Ibáñez ME, et al. Pangenome of acinetobacter baumannii uncovers two groups of genomes, one of them with genes involved in CRISPR/Cas defence systems associated with the absence of plasmids and exclusive genes for biofilm formation. Microb Genomics. (2019) 5:e000309. doi: 10.1099/mgen.0.000309
Keywords: multidrug-resistance, bloodstream infections, gram-negative bacilli, whole-genome sequencing, Brazil, surveillance
Citation: Silveira MC, Rocha-de-Souza CM, de Oliveira Santos IC, Pontes LdS, Oliveira TRTe, Tavares-Teixeira CB, Cossatis NdA, Pereira NF, Conceição-Neto OC, da Costa BS, Rodrigues DCS, Albano RM, da Silva FAB, Marques EA, Leão RS and Carvalho-Assef APD (2021) Genetic Basis of Antimicrobial Resistant Gram-Negative Bacteria Isolated From Bloodstream in Brazil. Front. Med. 8:635206. doi: 10.3389/fmed.2021.635206
Received: 30 November 2020; Accepted: 22 February 2021;
Published: 15 March 2021.
Edited by:
Alessandro Russo, University of Pisa, ItalyReviewed by:
Andrei Nicoli Gebieluca Dabul, University of São Paulo, BrazilHui Wang, Peking University People's Hospital, China
Copyright © 2021 Silveira, Rocha-de-Souza, de Oliveira Santos, Pontes, Oliveira, Tavares-Teixeira, Cossatis, Pereira, da Conceição-Neto, da Costa, Rodrigues, Albano, da Silva, Marques, Leão and Carvalho-Assef. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana Paula D'Alincourt Carvalho-Assef, YW5hcGRjYUBpb2MuZmlvY3J1ei5icg==