 Mazen Al-Qadi
Mazen Al-Qadi Barbara LeVarge
Barbara LeVarge H. James Ford
H. James Ford- Division of Pulmonary and Critical Care Medicine, Pulmonary Hypertension Program, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
Pulmonary hypertension (PH) is recognized to be associated with a number of comorbid conditions. Based on these associations, PH is classified into 5 groups, considering common pathophysiologic drivers of disease, histopathologic features, clinical manifestations and course, and response to PH therapy. However, in some of these associated conditions, these characteristics are less well-understood. These include, among others, conditions commonly encountered in clinical practice such as sarcoidosis, sickle cell disease, myeloproliferative disorders, and chronic kidney disease/end stage renal disease. PH in these contexts presents a significant challenge to clinicians with respect to disease management. The most recent updated clinical classification schemata from the 6th World Symposium on PH classifies such entities in Group 5, highlighting the often unclear and/or multifactorial nature of PH. An in-depth review of the state of the science of Group 5 PH with respect to epidemiology, pathogenesis, and management is provided. Where applicable, future directions with respect to research needed to enhance understanding of the clinical course of these entities is also discussed.
Introduction
Pulmonary hypertension (PH) is characterized by elevated mean pulmonary artery pressure (mPAP) of >20 mmHg as determined by right heart catheterization (RHC). PH is divided into 5 groups based on the underlying mechanism using the original World Health Organization classification system framework. In 2019, the proceedings of the World Symposium on Pulmonary Hypertension published the updated and revised classification based on the 6th World Symposium on PH (1). According to the new classification, causes of PH include pulmonary arterial hypertension (PAH, Group 1 PH), pulmonary venous hypertension due to elevated filling pressure of the left heart (Group 2 PH), PH due to chronic pulmonary disease or hypoxemia (Group 3 PH), chronic thromboembolic disease (Group 4), whereas PH that develops due to multiple or unclear mechanisms is referred to as Group 5 PH (Table 1). Within Group 5, several clinical disorders are implicated in association with the development of PH (Table 2). The most commonly seen clinical entities in Group 5 PH (Groups 5.1–5.3) are covered in detail: PH in the setting of hematologic disorders, sarcoidosis, and chronic renal failure. A brief overview of some of the remaining, less common but clinically relevant entities in Group 5 is also provided. Complex congenital heart disease (Group 5.4) is outside the scope of this manuscript.
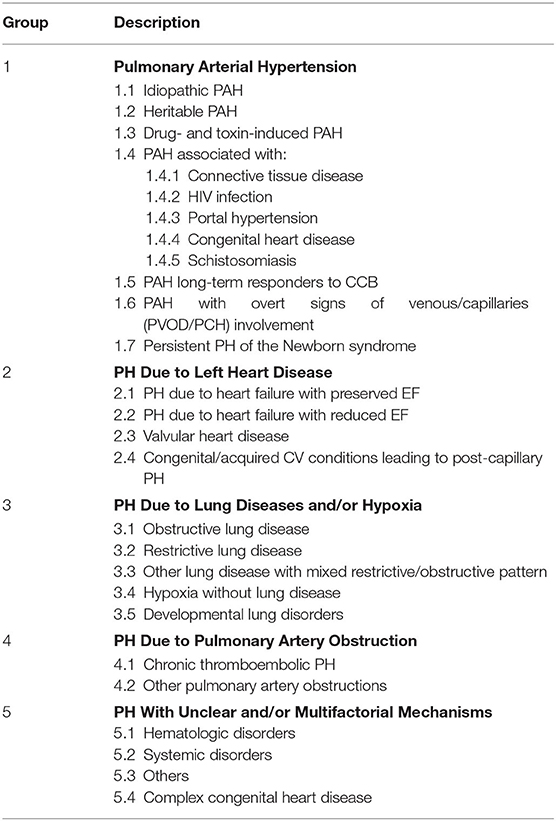
Table 1. Current clinical classification of pulmonary hypertension (1).
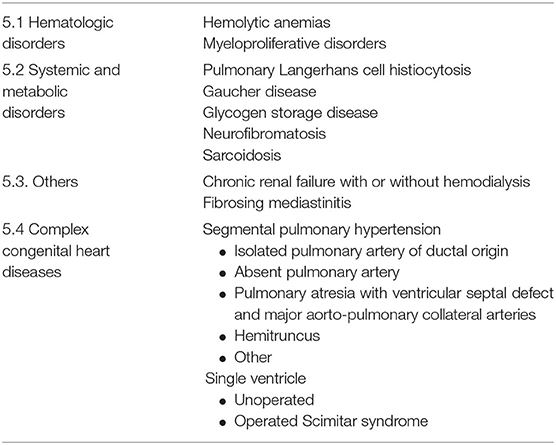
Table 2. Subclassifications of group 5 pulmonary hypertension: PH with unclear and/or multifactorial mechanisms (1).
Considering the true upper limits of normal mPAP, the 6th World Symposium on PH introduced revised hemodynamic definitions, affirming PH in those with mPAP >20 mmHg as opposed to mPAP ≥25 mmHg as previously defined. A pulmonary vascular resistance (PVR) of at least 3 Wood Units was further mandatory to define pre-capillary pulmonary hypertension. These changes impact identification and management of all groups of PH, including group 5 PH. With updated review and discussion of the literature, the proceedings outlined additional changes specifically impacting group 5 PH, including removal of both splenectomy and thyroid disease as specific subtypes of PH (asserting these conditions as risk factors) and reclassification of lymphangioleiomyomatosis into Group 3 PH (1).
Pulmonary Hypertension in Hematologic Disorders
Chronic Hemolytic Anemias
Prevalence
PH is increasingly recognized as a major source of morbidity and mortality in patients with chronic hemolysis, most notably in the context of sickle cell disease. The prevalence of PH in hemolytic anemia is variable depending upon the diagnostic criteria, methods used to diagnose PH, and population studied. In sickle cell disease (SCD, hemoglobin SS), 30–40% of patients have evidence of elevated pulmonary artery pressures based on tricuspid regurgitant jet velocity (TRV) ≥2.5 m/s on Doppler echocardiography. However, only 6–10.5% of patients have mPAP of ≥25 mmHg on invasive hemodynamic evaluation by RHC (2–5). This reflects the fact that estimation of pulmonary artery pressure is often inaccurate by echocardiogram. Additionally, elevated pulmonary artery pressures can be related to high cardiac output (due to anemia) and not always secondary to increased pulmonary vascular resistance (PVR). Using the new definition of PH (mPAP >20 mmHg), the prevalence of PH in SCD is probably higher than that reported in these studies.
PH has also been reported in association with other hemolytic anemias. In a study of 110 patients with β-thalassemia intermedia, 59% of patients had elevated peak systolic tricuspid gradient >30 mmHg on Doppler echocardiography, suggestive of PH. Only 6 patients had RHC and were found to have severe pre-capillary PH. All patients in this study had normal left ventricular function and high cardiac output (6). The prevalence of PH by echocardiography was even higher (75%) in a small study of 35 patients with homozygous β-thalassemia (7). More recently, Derchi et al. evaluated the prevalence of PH in a large multicenter cross-sectional study of 1,309 patients with β-thalassemia in Italy. Nine percent of patients had TRV >3 m/s; patients in the “PH likely” group with TRV ≥3.2 m/s underwent RHC. The prevalence of RHC-confirmed PH was 2.1%. Increased age and splenectomy were independent risk factors for PH in this study (8).
Notably, the prevalence of PH in patients with hereditary spherocytosis is much lower than reported with SCD and thalassemia. In two retrospective studies, no patients had a TRV of ≥2.8 m/s on echocardiography to suggest PH (9, 10). Nonetheless, PH has been described in hereditary spherocytosis and stomatocytosis, with most cases related to chronic thromboembolic pulmonary hypertension (CTEPH) following splenectomy (11).
Pathogenesis
The pathogenesis of PH in chronic hemolysis is multifaceted and is most extensively studied in SCD. The vasomotor tone of the pulmonary circulation is normally regulated by nitric oxide (NO), a powerful vasodilator and modulator of endothelial proliferation. NO is produced by the pulmonary endothelium in response to shear stress on the surface of endothelial cells (12). The enzyme NO synthase catalyzes cleavage of the terminal amino group from the amino acid L-arginine, producing NO. Hemolysis causes the release of arginase-1, which depletes L-arginine eliminating the NO precursor (13). Additionally, studies have shown that free hemoglobin produced by intravascular hemolysis in SCD consumes NO (14, 15). Impaired NO production is also related to hemolysis-induced generation of asymmetric dimethylarginine, an endogenous NO synthase inhibitor (16). Production of heme due to oxidation of free hemoglobin also activates Toll-like receptor 4 promoting vaso-occlusive events (17, 18). Interestingly, a growing body of literature indicates a possible role for free hemoglobin in the pathogenesis of PAH in patients with no known hemolysis. Rafikova et al. found that free hemoglobin levels were significantly higher in patients with known PAH compared to patients with no PAH. Furthermore, the severity of PAH correlated with higher cell-free hemoglobin levels. In an animal model of PAH, inhibition of heme translocation using sulfasalazine prevented the development of PH (19).
Endothelin-1 (ET-1), a potent vasoconstrictor of pulmonary vascular bed, plays a role in the imbalance between vasodilators and vasoconstrictors in SCD. Plasma and urine levels of ET-1 are elevated in patients with SCD compared with matched controls (20). Furthermore, Phelan et al. showed that exposure of cultured human endothelial cells to previously sickled erythrocytes results in enhanced gene expression of ET-1 (21).
Hypercoagulability is another important factor that adds to pulmonary endothelial dysfunction. A hypercoagulable state results from reduced NO, asplenia (functional or surgical), and activation of thrombotic factors (tissue factor, platelets, and thrombin) in SCD (22, 23). This is supported by findings of pulmonary thromboemboli in 38–80% of postmortem lung examinations of deceased SCD patients (24, 25).
Proliferative arteriopathy seen in SCD-PH implies possible contribution of angiogenic factors to the pathogenesis. A recent case-control study of children with β-thalassemia major demonstrated a significantly higher serum level of vascular endothelial growth factor in patients with PH than those without PH or matched healthy children (26).
Finally, a significant number of patients with SCD have been found to have restrictive cardiomyopathy and diastolic dysfunction (27). In fact, 50% of patients with SCD and PH have pulmonary artery wedge pressure (PAWP) of >15 mmHg, consistent with pulmonary venous hypertension (i.e., post-capillary PH) (3, 5, 28). Vaso-occlusive events, systemic hypertension, iron overload (due to transfusions) and end-organ damage (e.g., renal failure) in patients with chronic hemolysis contribute to diastolic left ventricular dysfunction as well (29). Collectively, all these factors increase the risk of developing PH in patients with SCD and other hemolytic anemias.
Evaluation
Pulmonary symptoms are common in patients with chronic hemolysis and often are multifactorial. Patients with SCD, for example, may have recurrent chest pains, fatigue, and chronic shortness of breath. These symptoms can be related to anemia, acute chest syndrome, obstructive airway disease, thromboembolism, and/or PH (30–32). Therefore, these should be systematically evaluated (Figure 1). Asymptomatic patients also remain at greater risk of developing PH; therefore, screening with Doppler echocardiography is recommended. The American Thoracic Society guidelines recommend screening patients with SCD with Doppler echocardiography every 1–3 years to assess mortality risk. Importantly, echocardiography should not be performed within 4 weeks of acute chest syndrome or within 2 weeks of acute vaso-occlusive crisis as these acutely increase the pulmonary artery systolic pressure (PASP) due to hypoxemia and worsening anemia (33). There are no screening guidelines for patients with other hemolytic diseases. Nonetheless, it is reasonable to obtain echocardiography on high-risk patients (e.g., severe hemolysis, asplenia, prior venous thromboembolism, and iron overload), especially those with unexplained pulmonary symptoms (34).
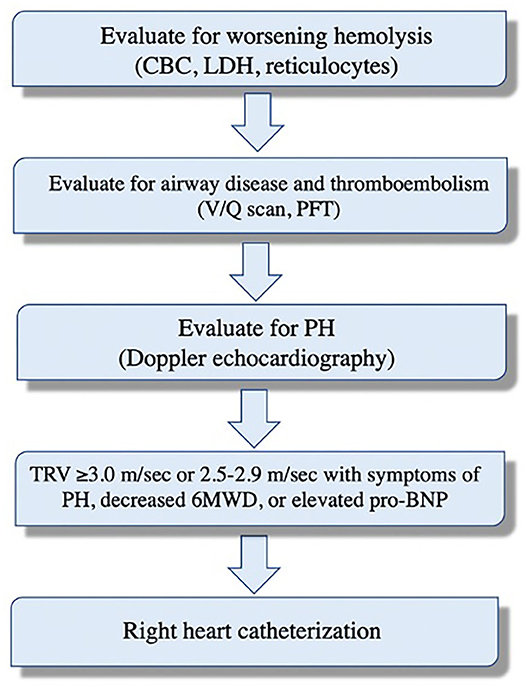
Figure 1. Systematic evaluation of dyspnea and potential pulmonary hypertension in patients with sickle cell disease.
PH is suspected if the TRV on echocardiography is >2.5 m/sec. However, TRV ≥2.5 m/s has a low positive predictive value as only 31% of such patients had mPAP ≥25 mmHg on RHC (27). A TRV ≥3 m/s is 3 standard deviations above the population mean and is more specific (PH confirmed in 66–77% of patients on RHC) (4, 5). A TRV between 2.5 and 3 m/s combined with either an NT-proBNP > 164.5 pg/ml or 6-min walk distance of < 333 m increases the likelihood of PH and warrants further evaluation and RHC (Figure 2) (3, 5). PASP is commonly estimated indirectly by measuring the right ventricular systolic pressure which (in the absence of pulmonic stenosis) are equivalent. Using the modified Bernoulli equation (P = 4V2), the PASP is calculated from the TRV. This means that minor variations in measurement of TRV can result in significant differences in estimated pulmonary pressures. Furthermore, even when accurate, elevated PASP can be related to high cardiac output often seen in patients with anemia. On the other hand, PH can be missed on echocardiography as the TRV is not always measurable (35). Thus, RHC remains the test of choice to confirm the presence of PH and assess the PAWP, cardiac output, and calculated PVR. It is worth mentioning that reduced blood viscosity and high cardiac output (due to anemia) result in a lower baseline PVR than healthy subjects. Thus, PVR of >2 Wood units is considered elevated in these patients (3–5).
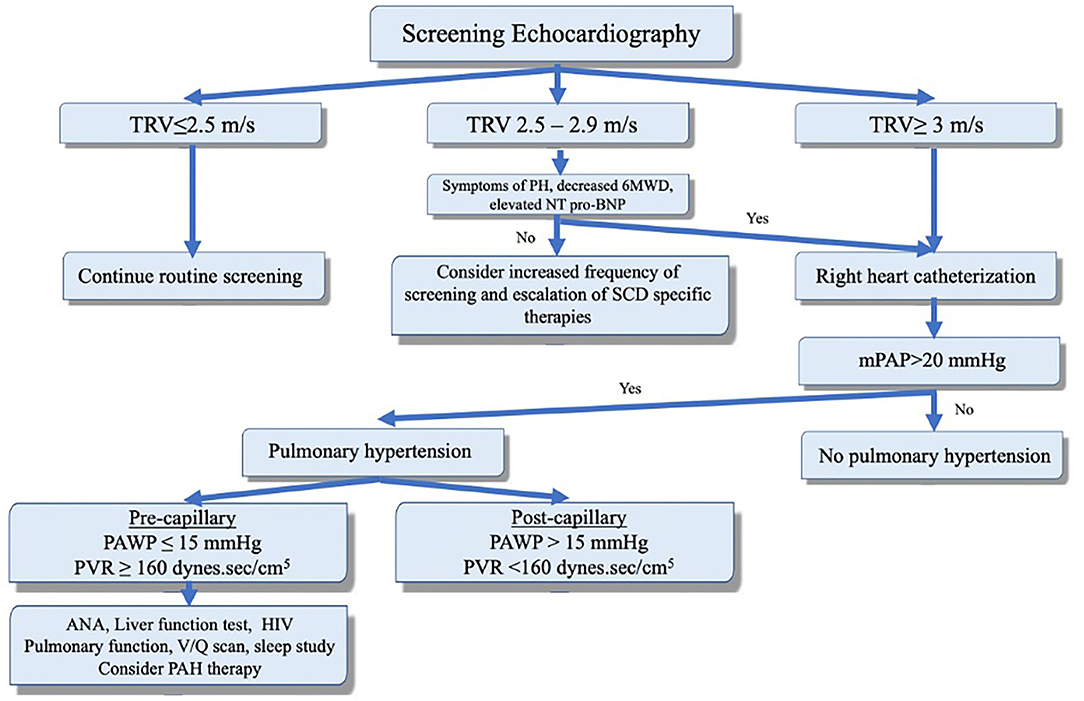
Figure 2. Screening, diagnosis, and clinical classification to guide management of pulmonary hypertension in the context of sickle cell disease. Adapted with permission of the American Thoracic Society. Copyright © 2020 American Thoracic Society. All rights reserved. Klings et al. (33). The American Journal of Respiratory and Critical Care Medicine is an official journal of the American Thoracic Society. Readers are encouraged to read the entire article for the correct context. The authors, editors, and The American Thoracic Society are not responsible for errors or omissions in adaptations.
Blood biomarkers can also be helpful in the evaluation of PH in chronic hemolytic anemia. Lactate dehydrogenase is a commonly used marker for hemolysis has been shown to correlate with NO resistance and severity of PH (36). NT-proBNP is another useful biomarker for PH and right ventricular dysfunction. In a study by Machado et al., elevated NT- proBNP (>160 pg/mL) in SCD patients correlated with TRV and was predictive of PH. Additionally, elevated NT- proBNP was an independent predictor of mortality (37).
Treatment
Management of PH in hemolytic anemias includes general measures such as supplemental oxygen to reverse hypoxemia and prevent its deleterious effect on the pulmonary vascular tone. Diuretics are used to treat volume overload but should be used cautiously due to risk of sickling in patients with SCD. Lifelong anticoagulation is indicated in SCD patients with PH secondary to CTEPH. Although surgical pulmonary thromboendarterectomy (PTE) can be curative in CTEPH, it is undoubtedly more challenging in SCD patients due to the increased risk of sickling and occlusive crises during cardiopulmonary bypass. The best perioperative approach in these patients remains controversial. Avoidance of hypoxemia, hypothermia, and acidosis reduces the risk of perioperative complications. Additionally, exchange transfusion to reduce hemoglobin S to <20% prior to surgery is advised (38, 39). This aggressive transfusion regimen, however, has not been shown to reduce perioperative complications in patients with SCD (40). Inoperable patients are considered for balloon pulmonary angioplasty (BPA). In a recent study of PH in SCD, three patients with CTEPH underwent BPA with significant reduction in PVR in 2 patients and normalization of mPAP in the third patient (41).
Optimizing treatment of the underlying hemolytic disease is of paramount importance. Hydroxyurea is a myelosuppressive agent that has been shown to decrease sickle cell hemoglobin polymerization, reducing hemolysis and frequency of acute chest syndrome, and vaso-occlusive crises. Furthermore, hydroxyurea decreases hospitalization and mortality in SCD patients with HbSS and is currently recommended for patients with more than one episode of acute chest syndrome or >3 vaso-occlusive crises per year (33, 42–44). The risk of developing PH is also reduced with hydroxyurea in patients with thalassemia (45). Chronic transfusion is recommended in SCD patients who fail to respond or cannot tolerate hydroxyurea. This strategy is associated with improvement in pulmonary vascular changes in SCD patients (46). Similarly, adherence to transfusion and iron chelation therapy in patients with thalassemia prevented PH (47, 48).
SCD patients who remain symptomatic despite optimization of their SCD therapy (i.e., hydroxyurea, transfusion) are considered for PH-targeted therapy. This is usually reserved for patients with moderate to severe (based on functional class) RHC-confirmed pre-capillary PH with relatively reduced cardiac output. In general, endothelin receptor antagonists (ERAs) are recommended in patients with moderate PH (Functional class 2-3). Bosentan, an endothelin receptor antagonist, was evaluated in two randomized placebo-controlled trials of SCD patients with RHC-proven pre- and post-capillary PH. Both studies were terminated early due to slow enrollment but modest improvement in hemodynamic parameters was observed. Efficacy endpoints were not analyzed to draw definitive conclusions but bosentan was well-tolerated (49). Patients with more severe PH (class IV functional status, evidence of right-sided heart failure, or reduced cardiac output) can be treated with prostanoids. The efficacy of prostacyclins has been evaluated in a small retrospective study of 11 patients who received treprostinil or epoprostenol and showed improvement in right ventricular systolic pressure on echocardiography (50). Patients who need such therapy should be referred to centers with expertise in managing PH in SCD patients due to concerns of complications (e.g., high-cardiac output pulmonary edema, line thrombosis). Phosphodiesterase type 5 inhibitors (PDE-5Is) should generally be avoided in this population due to risk of increased hospitalizations for painful crises noted in a study of sildenafil (51). Riociguat is a soluble guanylate cyclase stimulator that does not rely on NO and, therefore, is an attractive agent for SCD-PH. Riociguat is currently approved for treatment of Group 1 PAH and Group 4 CTEPH. Weir et al. recently reported in a small case series tolerability of riociguat in four of six patients with sickle related CTEPH. Additionally, riociguat was associated with significant improvement in exercise capacity (mean 6MWD improvement of 56.8 m), functional class, NT-proBNP, and RVSP in 3 patients. Vaso-occlusive crisis developed in only one patient who was not on hydroxyurea (52). A phase 2, multi-multi-center, randomized, double-blind, placebo-controlled, study is now underway to evaluate the effectiveness and safety of riociguat in patients with sickle cell disease.
Myeloproliferative Disorders
Prevalence
Myeloproliferative disorders (MPDs) are characterized by clonal expansion of the multipotent hematopoietic progenitor cell with overproduction of one of the blood elements (i.e., erythrocytes, leukocytes, or platelets). The prevalence of PH in patients with MPDs has been evaluated in multiple small studies. Most of these studies used Doppler echocardiography (PASP>35 mmHg) to diagnose PH, with an estimated prevalence of 13–48% of PH in MPDs (53–56). Interestingly, in a larger study of 103 patients with MPDs, the prevalence of PH was much lower with only 5 patients found to have PH (57). Among MPDs, patients with chronic myeloid leukemia, polycythemia vera, essential thrombocytosis, and myelofibrosis are particularly at higher risk of developing PH. The presence of PH is associated with poor prognosis in MPDs.
Pathogenesis
PH in MPDs has two main phenotypes: chronic thromboembolic (CTEPH) and proliferative pulmonary arteriopathy. Thrombotic events (both venous and arterial) and microcirculatory disturbances are common in polycythemia vera and essential thrombocytosis patients. In a large study of 1,213 patients with polycythemia vera, 41% developed thrombotic complications (58). This translates into higher risk of CTEPH. In a retrospective small study of 10 patients with MPDs and RHC-confirmed PH, six patients were diagnosed with CTEPH at the time of MPD diagnosis. CTEPH was strongly associated with elevated hematocrit (59). Presence of cardiovascular comorbidities, history of thrombosis or splenectomy and increased age further increase the risk of thrombosis in these patients (60–62).
High hematocrit in polycythemia vera is associated with hypercoagulability as demonstrated by the progressive increase in blood viscosity that parallels higher hematocrit values (63). The major impact of hematocrit on blood viscosity is in the venous (low-shear) circulation as it decreases blood flow and increases the risk of venous thrombosis. In the arterial circulation, under high-shear, platelets are displaced to the periphery by the large red cell mass and come in contact with endothelial cells, causing platelets activation (release of thromboxane A2, platelet-derived growth factor, vascular endothelial growth factor) and thrombosis (64, 65). The abnormalities of erythrocytes and platelets in MPDs also contribute to the aggregation of red blood cells and disturbed blood flow. Furthermore, chronically activated leukocytes (especially with JAK2 mutation) interact with platelets and endothelial cells and increase the risk of thrombosis (66–69). Splenectomy is used in the treatment of MPDs and can be associated with a hypercoagulable state as well (61). Notably, obstruction of pulmonary arteries in MPDs is not always related to thrombi but can be secondary to tumor emboli or circulating megakaryocytes with subsequent release of vasoactive substances that will further increase the tone in the pulmonary vascular bed (70, 71).
The second PH phenotype in MPDs is pulmonary arteriopathy which can result from different mechanisms. Portopulmonary hypertension may complicate chronic liver disease which is known to occur in MPDs (72). Medication-induced PH, fitting into Group 1 PH, is another potential cause of PH in these patients. Dasatinib, a tyrosine kinase inhibitor commonly used for treatment of chronic myeloid leukemia, has been associated with development of PH which seems to improve after discontinuation (73). Several TKIs other than dasatinib (e.g., bosutinib, ponatinib, lapatinib) have also been implicated in the development of PH. In a small study of 27 patients who received lapatinib, three patients were found to have PH by RHC with resolution of PH in all three after cessation of therapy (74).
Alkylating agents such as cyclophosphamide, melphalan, and busulfan (used for treatment of myelofibrosis) can cause PH with severely reduced diffusing capacity of the lung for carbon monoxide and abnormal findings on computed tomography of the chest (septal thickening, ground glass opacities, and lymph node enlargement) that are consistent with pulmonary veno-occlusive disease. Unlike dasatinib-induced PH, these patients have a poorer prognosis. Extramedullary hematopoiesis may complicate MPDs and result in infiltrative changes in the portal circulation with subsequent development of portal hypertension and portopulmonary hypertension. In cases of lung involvement, extramedullary hematopoiesis may lead to pulmonary vascular occlusion and PH. Proliferative changes in the pulmonary vessels may also occur due to increased circulating vascular endothelial growth factors and enhanced angiogenesis in MPDs (75). Finally, pre-capillary PH can be related to abnormal JAK2 signaling and depletion of NO. This is supported by improvement in PH and NO with JAK2 inhibitors.
Treatment
Since hematologic abnormalities contribute directly to the pulmonary vascular changes seen in MPDs, treatment of the underlying disease is a potential strategy. Indeed, allogeneic hematopoietic stem cell transplant and JAK inhibitors to treat MPDs have been shown to improve PH (76). For example, treatment with ruxolitinib (JAK1/JAK2 inhibitor) in 15 patients with myelofibrosis and PH resulted in improvement in right ventricular function and PASP by echocardiography in 66% of patients and was associated with significant reduction in NT-proBNP and increase in NO level (77). In cases of drug-induced PH, improvement in PH can be expected with discontinuation of the offending agent (especially in dasatinib-induced PH) although this alone may not be enough to reverse PH. When appropriate, CTEPH is treated with PTE or BPA. Riociguat, a guanylate cyclase stimulator, is associated with improved functional status and hemodynamics in patients with inoperable disease or suboptimal post-PTE hemodynamic outcomes (78, 79). External beam radiotherapy to the thorax has been used to treat extramedullary hematopoiesis in patients with MPDs and PH with improvement in pulmonary artery pressures (80). PAH-specific therapy has not been studied in MPD-associated PH.
Pulmonary Hypertension in Sarcoidosis
Prevalence
Sarcoidosis is characterized by granulomatous inflammation to an unknown trigger that primarily affects the lungs. The prevalence of sarcoidosis-associated PH (SAPH) is variable. This variation comes from inconsistency in methods used to diagnose SAPH. Autopsy studies have shown pulmonary vascular involvement in ~5% of patients with significant parenchymal lung disease (81). Similar prevalence has been found in studies that evaluated SAPH based on clinical evidence of right ventricular failure (i.e., cor pulmonale) (82). However, studies that used hemodynamic assessment using RHC to diagnose SAPH reported higher prevalence up to 23% (83, 84). The prevalence is substantially higher in patients with pulmonary symptoms and advanced lung disease due to sarcoidosis. In a study of 15 patients with advanced fibrotic sarcoidosis, Emirgil et al. found evidence of PH on RHC in 10 patients (67%) (85). Another study of 363 patients with advanced sarcoidosis (listed for lung transplantation) found evidence of PH on RHC in 74% of these patients (86). Racial differences may significantly impact the course of disease and risk of developing PH. For example, in a recent large prospective study of 512 patients, predominantly Caucasian, with sarcoidosis, SAPH was found in 2.9% of those with intermediate or high probability for PH (by echocardiography) (87). It is important to note that some patients with sarcoidosis may have normal pulmonary artery pressure at rest with evidence of exercise-induced PH (88).
Pathogenesis
There are several mechanisms of PH in sarcoidosis. Each one of these may have a different response and outcome to a specific therapy. Pre-capillary PH may result from hypoxic pulmonary vasoconstriction and destruction of the distal vascular bed with advanced interstitial lung disease. This is supported by the finding that the majority of SAPH patients have advanced fibrotic lung disease (89–91). This usually leads to mild to moderate PH (mPAP <35 mmHg). Nevertheless, at least one third of sarcoidosis patients have no significant fibrosis which indicates that hypoxemia and fibrosis are not the only explanation for SAPH.
Granulomatous vasculopathy and angiitis with obliteration of arterioles or venules has been reported in autopsies of patients with pulmonary sarcoidosis (92–94). Occlusion of pulmonary vessels by granulomas causes PH and sometimes can be mistaken with thromboembolic disease. This pathology is associated with more severe PH than that related to lung fibrosis. Granulomatous vasculitis can also affect the post-capillary vessels causing focal stenosis and a clinical pattern consistent with pulmonary veno-occlusive disease (95, 96).
Moreover, sarcoidosis is associated with increased risk of venous thromboembolism, which carries associated risk of developing CTEPH in some patients (97, 98). Sarcoidosis can be associated with mediastinal and hilar lymphadenopathy that can cause extrinsic compression of pulmonary arteries (and venous stenosis) or fibrosing mediastinitis, resulting in SAPH.
Cardiac involvement occurs in 20–25% of patients with sarcoidosis (99). Thus, post-capillary PH can develop in sarcoidosis due systolic or diastolic dysfunction with left ventricular involvement. In a retrospective study of 130 sarcoidosis patients with persistent dyspnea, 29% of patients with SAPH had an elevated PAWP >15 mmHg (89). Because cardiac involvement can be subtle, screening for cardiac sarcoidosis using magnetic resonance imaging and 18 F-fluorodeoxyglucose positron emission tomography is recommended (100). Finally, post-capillary SAPH can be due to extrinsic compression of the pulmonary veins.
Treatment
As outlined earlier, SAPH can be related to multiple mechanisms (Figure 3). However, some patients may have a prominent mechanism. For example, in the presence of advanced parenchymal (fibrotic) lung disease, PH with mPAP of <35 mmHg is likely related to hypoxemia and destruction of the pulmonary vascular bed associated with fibrotic changes. Treatment in these patients should focus on treatment of parenchymal lung disease and correction of hypoxemia. PAH-specific therapy carries the risk of worsening ventilation-perfusion mismatch and, therefore, should be avoided.
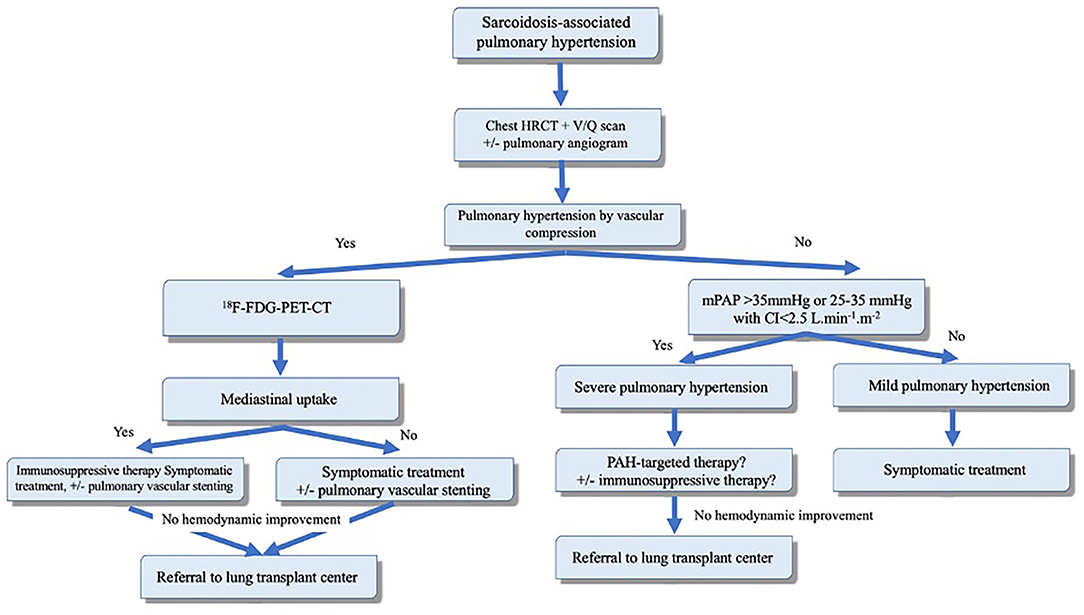
Figure 3. Proposed algorithm for evaluation and management of sarcoidosis-associated pulmonary hypertension (SAPH). Adapted from Boucly et al. (101). Reproduced with permission of the © ERS 2020.
PAH-specific therapy can be considered in carefully selected patients with moderate to severe SAPH that is “out-of-proportion” to the degree of parenchymal lung disease and hypoxia (i.e., mPAP >35 mmHg). Such degree of PH suggests a component of vasculopathy that may respond to pulmonary vasodilators. Vasoreactivity has been demonstrated in a small prospective observational study of 8 patients with moderate-to-severe SAPH with improvement in mPAP and PVR to inhaled NO (102). This highlights the potential role of pulmonary vasodilators in these patients. Such therapy has been evaluated in multiple studies and shown to improve the hemodynamic parameters with no significant change in functional status or 6-min walk distance (Table 3). Similarly, in a double-blind placebo-controlled trial, bosentan improved mPAP and PVR, but failed to demonstrate benefit in 6-min walk distance (110).

Table 3. Existing published studies evaluating treatment of sarcoid-associated pulmonary hypertension.
Immunosuppressive therapy alone may result in hemodynamic improvement in patients with granulomatous vasculopathy or compressive lymphadenopathy (101). SAPH due to extrinsic compression can be treated with pulmonary artery or vein angioplasty and stenting (114). Overall, patients with SAPH have poor prognosis and should be evaluated early for lung transplantation.
Pulmonary Hypertension in Chronic Kidney Disease and End Stage Renal Disease
The evaluation and management of PH in the context of chronic kidney disease (CKD), particularly end stage renal disease (ESRD), presents unique challenges to the clinician in terms of elucidating the main drivers of PH in this patient population. The clinical presentation of such patients is one in which PH is often multifactorial, hence the placement of this clinical entity into Group 5 of the PH classification schemata. As such, management relies in large part on attempts to correct and optimize multiple physiologic derangements often seen in CKD and ESRD.
Definition, Epidemiology, and Scope of the Problem
CKD is defined as abnormal renal function present for at least 3 months and is staged in severity based on the degree of reduction of glomerular filtration rate and the degree of albuminuria. ESRD is defined as renal failure requiring regular interval, long-term dialysis, or renal transplant to maintain survival. The true prevalence of PH amongst patients with CKD and ESRD is difficult to discern, due to the lack of robust data. Most of what is known is drawn from relatively small, retrospective, and almost exclusively echocardiography based studies. Amongst patients with CKD and not yet on dialysis, the prevalence of PH was observed to be upwards of 30% or more. Prevalence was highest among those with most advanced CKD (CKD 5) (115, 116). When considering those patients on hemodialysis (HD), a meta-analysis of known studies by Tang and colleagues (116) showed the prevalence of PH (based largely on echocardiography) between 19 and 56%. There is less data available with regard to the prevalence of PH in the setting of peritoneal dialysis (PD), but it appears to be lower than that of HD (117).
O'Leary et al. (118) recently published a retrospective analysis of a large cohort of patients with varying degrees of CKD that underwent RHC. PH was present in 1,267/1,873 (68%) of patients, using the accepted definition of PH at that time of mean PAP ≥ 25 mmHg. Of these, 76% had post-capillary PH and 24% had pre-capillary disease, using elevated PAWP (≥ 15 mmHg) as the differentiating factor. The authors also noted that mortality was higher among patients with advanced stages of CKD, and further compounded by the presence of PH. The distribution of hemodynamic profiles seen underscores the significant role of Group 2 PH phenomenon related to elevated left ventricular filling pressures in driving PH in an overwhelming majority of patient with CKD. Comorbid left ventricular systolic and/or diastolic dysfunction, as well as a propensity to volume overload at times are the main drivers of this. However, many of those with CKD/ESRD with and without elevated PAWP have other factors driving PH as well. Among these are alterations in circulating inflammatory and vasoactive mediators, endothelial dysfunction, alterations in cardiopulmonary flow related to anemia and arteriovenous fistula (AVF), factors related to dialysis itself, and comorbid Group 3 PH processes (see Figure 4).
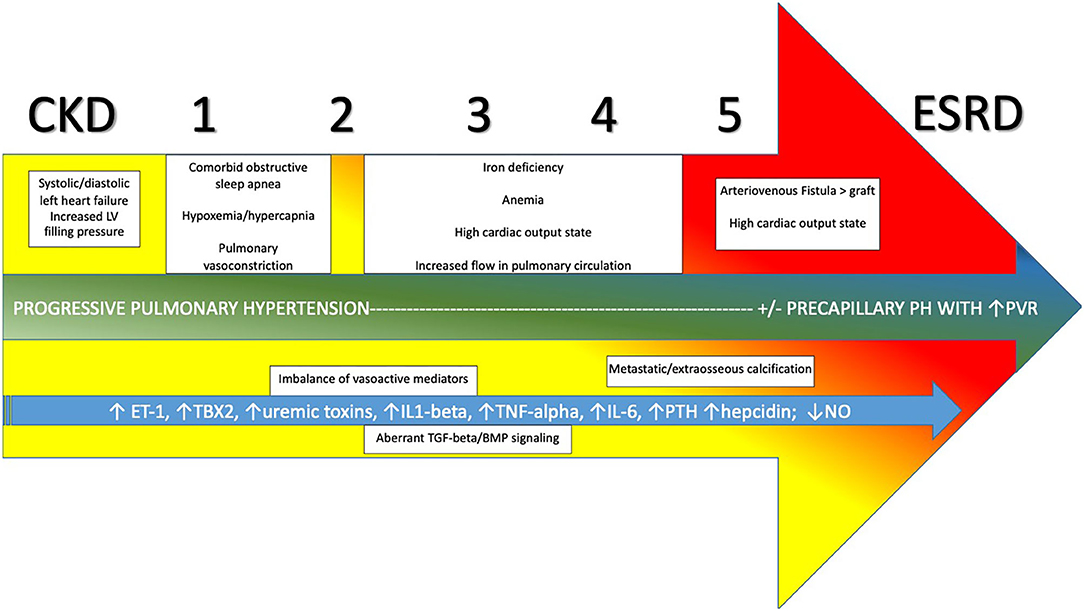
Figure 4. Evolution and development of pulmonary hypertension in CKD progressing through stages 1–5 to ESRD and relevant contributors. CKD, Chronic Kidney Disease; ESRD, End Stage Renal Disease; PVR, pulmonary vascular resistance; ET-1, endothelin-1; TBX2, thromboxane 2; IL1-beta, interleukin-1-beta; TNG-alpha, tumor necrosis factor-alpha; IL-6, interleukin-6; PTH, parathyroid hormone; NO, nitric oxide; TGF-beta, transforming growth factor-beta; BMP, bone morphogenetic protein.
Altered Inflammatory and Vasoactive Mediators and Related Endothelial Dysfunction
The presence of uremic toxins in patients with advanced CKD or ESRD can contribute to pulmonary vascular endothelial dysfunction. Specifically, mono-methyl L- Arginine, symmetric dimethyl arginine, and asymmetric dimethyl arginine are present in increased levels and inhibit NO synthase, impairing the intrinsic production and bioavailability of NO, the vasodilatory effects of which serve to maintain normal pulmonary vascular tone. There is also evidence that increased levels of uremic toxins are associated with increased inflammation, endothelial dysfunction, and oxidative stress (119). There is decreased availability of circulating NO in patients on HD with PH vs. those without PH. In addition, patients on HD with PH showed decreased ability to recover NO levels after HD, suggesting impairment in NO synthesis (120). As noted previously, ET-1 contributes to pulmonary vascular remodeling, is present in excess in patients with Group 1 PAH, and is also known to be elevated in patients with CKD and those on HD. Further, HD does not facilitate clearance of ET-1 and levels remain elevated after HD is performed (120). Angiopoietin 1 and 2 are additional mediators that modulate vascular smooth muscle growth via action on endothelial cells. There is evidence that both are important in the pathogenesis of CKD and PAH (121).
Cellular inflammation is a key driver of disease in both PH and CKD. In addition to the noted effects of uremic toxins, levels of circulating pro-inflammatory cytokines are increased in patients on HD with PH relative to those without, particularly IL-1beta, TNF-alpha, and IL-6. High-sensitivity c-reactive protein was also noted to be higher in the group with PH (122). The transforming growth factor beta (TGF-beta) and bone morphogenetic protein (BMP) signaling pathways modulate downstream gene expression that controls properties of vascular structure and tone. It has been demonstrated that this pathway is aberrant in CKD and PAH, leaning in a pro-inflammatory direction that favors vascular remodeling. Thus, there is a strong likelihood that the TGF-beta/BMP axis is relevant in patients with both PH and CKD/ESRD (123, 124).
Alterations in Cardiopulmonary Flow
Intentional arteriovenous shunting caused by the surgical creation of AVF or placement of a synthetic arteriovenous graft (AVG) contributes to a high cardiac output state. This leads initially to increased loading on the left ventricle. When this phenomenon is coupled with the increased output state, a syndrome of high-output heart failure may manifest. With time, persistence of this high flow is thought to lead to increased shear forces on the pulmonary vascular endothelium that can cause pulmonary vascular remodeling and an intrinsic arteriopathy in some patients (125). The propensity to develop PH is higher in patients with AVF relative to AVG, as AVF can enlarge with time and develop progressively higher flows, which in turn increases flow through the pulmonary circulation to increasing degrees. It has been noted when flow through the fistula exceeds 2 L/min or more than 20% of cardiac output, the risk of pulmonary hypertension is higher. Furthermore, presence of the AVF on the upper arm presents increased likelihood of PH as compared to AVF of the lower arm (126).
Practices regarding the placement and maintenance of AVF for HD have come under a bit more scrutiny in recent years as a result of noted effects on circulatory and pulmonary vascular physiology. There has been deliberate advocacy on the part of the Centers for Medicare and Medicaid Services to utilize AVF over catheter-based HD in most patients, driven largely by legitimate concern for the risk of catheter related bloodstream infection and thrombosis (127). Since the inception of this initiative, it has been increasingly recognized that some patients with CKD/ESRD may have subclinical or mild PH before arterio-venous access for dialysis is placed. Some advocate for more systematic echocardiographic screening for PH prior to placement of AVF. If evidence of PH is present, then PD and/or expedited/prioritized renal transplant evaluation may be more appropriate (128). Further study is needed in this regard.
Anemia due to reduced renal production of erythropoietin, as well as iron deficiency and anemia of chronic disease is very common in patients with CKD and ESRD. This contributes to compensatory increase in cardiac output (much like that seen in SCD) and thus exposes the pulmonary circulation to higher than usual flow and the risks of vascular remodeling noted above. Iron deficiency in both PAH and CKD appears to be modulated largely by increased levels of hepcidin, which inhibits enteric iron transport and uptake (129, 130). Iron deficiency thus may ultimately affect pulmonary vascular regulation through downstream effects on hypoxia inducible factors. Risk for development of PH in the context of CKD/ESRD and anemia is most significant when hemoglobin is <10 g/dl (131).
Factors Related to Dialysis
As noted, HD presents a higher likelihood of developing PH, due in part to the use of AVF or AVG in many patients. In addition, it appears that the type of dialysis membrane used may contribute to presence of PH as well. Thromboxane B2, a pro-proliferative, pro-thrombotic, and pro-inflammatory molecule, is known to be elevated in patients with PAH compared to controls. In addition, it has been shown that thromboxane B2 levels are higher in patients on HD and/or with PH as compared to those not on HD and/or with no evidence of PH (132). The reason for elevated thromboxane B2 in some patients on HD appears to be related to increased release from circulating monocytes when cellulose dialysis filter membranes are utilized as compared to polysulfone membranes (133, 134).
The likelihood of PH is also directly proportional to older age and number of years on HD (131). The reason for this is likely simply related to the duration of exposure of the pulmonary circulation to the aforementioned pathophysiologic effects of HD. An additional phenomenon that may contribute to pulmonary vascular changes and PH is the showering of micro-bubbles from the dialysis machine with each HD session. These micro-bubbles have the potential to obstruct small pulmonary capillaries, leading to local inflammatory changes and microthrombosis, setting the stage for progressive pre-capillary arteriopathy to progress as HD continues over time (135, 136).
Comorbid Group 3 PH Processes
As noted previously, Group 3 PH is often driven by abnormal oxygenation and ventilation, commonly seen during sleep in patients with obstructive sleep apnea (OSA). There is a high prevalence of OSA amongst patients with CKD, independent of the need for HD (137, 138). The associated hypoxemia, and in some cases hypercapnia, contributes to pulmonary vasoconstriction and some degree of PH in many patients with CKD. Asymmetric dimethyl arginine is also noted to be elevated in patients with sleep disordered breathing, implicating impairment of NO production as at least one plausible molecular mechanism for the development of PH attributable to OSA (139). This pathway may be of particular relevance to patients with CKD. As such, screening for nocturnal hypoxemia and OSA with formal polysomnography is of paramount importance. Those found to have sleep disordered breathing should be titrated for and treated with nocturnal non-invasive positive airway pressure therapy.
In addition to OSA, it has been noted that secondary hyperparathyroidism seen in CKD/ESRD can lead to extraosseous calcium deposition in the interalveolar septae and pulmonary arterial system. Alveolar septal deposition can contribute to restrictive physiology on pulmonary function testing, as well as reduction in diffusing capacity (140). The overall clinical impact of these phenomenon with respect to the development of PH is not as clear compared to that of OSA. Additionally, it has been shown that parathyroid hormone levels do not differ between patients with and without PH in the context of CKD/ESRD (141).
PH Diagnosis and Invasive Hemodynamic Evaluation in Patients With CKD/ESRD
As with all forms of pulmonary hypertension, the initial preferred tool for screening is transthoracic echocardiography. This is especially important in CKD/ESRD patients with unexplained dyspnea. Timing of echocardiography is most optimal soon after the completion of a session of dialysis, with the patient at or very near their established dry weight. The presence of estimated PASP ≥ 50 mmHg and or significant right ventricular dilatation or dysfunction should prompt invasive hemodynamic measurement by RHC. As with echocardiography, RHC should be performed in the relative immediate time period after a session of dialysis, such that the left and right sided circulation are as volume and pressure off-loaded as possible, ensuring that hemodynamic measurements are reflective largely of alterations in flow and/or PVR that may be present.
The PEPPER study (142) was an important endeavor that helped to understand the frequency of PH in patients with advanced CKD (stage 4 or 5) and unexplained dyspnea, as well as to understand the effect of HD on pulmonary hemodynamics. In the study, 31 CKD patients not on dialysis and 31 patients on HD with dyspnea underwent RHC. In the group on HD, 78% were found to have PH (13% pre-capillary PH, 65% post-capillary PH). Among those not on HD, 77% had PH (6% pre-capillary PH, 71% post-capillary PH). Further, in the group on HD, 25/31 underwent RHC pre and post dialysis. It was noted that mean PAP and PAWP were both significantly lower post-dialysis, underscoring the importance of timing of RHC relative to HD.
In patients that are on HD and have an AVF, it may be useful to study the effects of fistula occlusion during RHC, however there is scant literature to guide this practice. This must be done cautiously and for a relatively short time, as there is some risk of associated thrombosis of the AVF. Published studies looking at the hemodynamic effects of fistula compression are echocardiography based. Yigla et al. found that after compression of AVF for 1 min in four patients on HD with echocardiographic evidence of PH resulted in a 10% reduction in mean estimated PA systolic pressure and a 15% reduction in estimated cardiac output (reduction from supranormal values) (143).
Treatment of PH in the Context of CKD and ESRD
Given that the majority of PH in the context of ESRD/HD is pulmonary venous hypertension, the mainstay of treatment is optimizing volume status with diuretics or dialysis, controlling systemic blood pressure, and medical therapies that optimize systolic and diastolic function of the left ventricle. The importance of correcting hypoxemia and sleep disordered breathing when relevant was previously discussed. If clinical history or risk factors suggest that CTEPH may be a possibility, then consideration must be given to screening ventilation-perfusion lung scan. If there is evidence of CTEPH, then additional imaging and hemodynamic workup is indicated to determine if the patient is a candidate for PTE or BPA. For patients in whom there is exceedingly high AVF flow and elevation in cardiac output driving PH, consideration should be given to revision of the AVF via banding or ligation (144, 145).
In general, trials of off label use of PAH-specific therapies in group 5 PH related to CKD/ESRD should be reserved for a very select few patients for whom all other contributing factors to PH have been optimized but have persistent and significant RHC-proven PVR elevation, particularly in the context of reduced cardiac output reflective of RV failure. The presence of clear Group 1 PAH comorbidities such as connective tissue disease, HIV, or portal hypertension, may also make a case for treatment with PAH therapies more compelling in the setting of a hemodynamic profile consistent with clear pre-capillary disease. As with all group 5 PH, there is no randomized, controlled data regarding the use of PAH-specific therapy in CKD/ESRD associated PH. Though not absolutely contraindicated in renal failure, PDE-5 inhibitors and riociguat should be used with caution in patients with advanced CKD/ESRD due to potential alterations in pharmacokinetics. As such, dose adjustments may be needed in this context. (146, 147).
Renal transplant surgery is generally regarded as not presenting prohibitive levels of operative risk to patients with mild to moderate degrees of PH and RV dysfunction, particularly considering the procedure is not usually associated with large intravascular volume and hemodynamic shifts as seen for example in liver transplantation. Patients with significant PH, and particularly severe right ventricular dysfunction, are considered with an abundance of caution for renal transplantation, especially if interventions to attempt to improve pulmonary hemodynamics have failed. There is no clear evidence based data available to systematically risk stratify patients with ESRD and PH for renal transplant surgery (148). Independent of immediate perioperative surgical risk however, it is noted that pre-transplant PH (based on echocardiographic studies) is associated with a significant increased risk of graft failure (149). Furthermore, post-transplant survival is decreased in patients with elevated estimated PASP on echocardiogram > 50 mmHg and in those with elevated PVR > 3 Wood units (150, 151). In general, approach to renal transplantation candidacy in the setting of PH should be done in an individualized, multidisciplinary fashion, especially given the lack of robust studies to reliably predict outcomes.
Pulmonary Hypertension Associated With Less Common Comorbidities
Pulmonary Langerhans Cell Histiocytosis
Pulmonary Langerhans cell histiocytosis (PLCH) is an uncommon cystic lung disease characterized by pathologic dendritic cell infiltration of the walls of the distal bronchiole. The involved dendritic cell subset, the Langerhans cell, localizes within the airway epithelium and processes encountered inhaled antigens, becoming activated when additional molecular signals indicate a state of danger (152). Activated dendritic cells migrate to lymphoid tissue to induce the adaptive immune response. PLCH occurs almost exclusively in smokers (153), and smokers with or without pulmonary diseases have significantly greater presence of Langerhans cells in the lung (154, 155). Cigarette smoking further induces production of molecules such as TNF-alpha, TGF-beta, GM-CSF, and osteopontin; these factors promote development, activation, and survival of Langerhans cells, though migration out of the airway tissue may be impaired (156–158). The bronchiolocentric nodules characteristic of PLCH contain a mixed population of Langerhans and other inflammatory cell types (159). Factors predisposing the small population of smokers developing PLCH remain incompletely understood.
Relative to other forms of chronic lung disease, PLCH is associated with higher prevalence of PH, and the vascular component of disease can be great (160, 161). PH has been found in 41–100% of PLCH patients, with prevalence particularly high (>90%) in those undergoing lung transplant evaluation (160, 162–164). Presence of PH has been consistently associated with increased mortality in PLCH (163, 165). While inverse correlation between pulmonary artery pressure elevation and spirometry has been noted in some studies (163, 166), others have not demonstrated this finding (160, 161). Examinations of tissue specimens have demonstrated typical vascular changes of PAH, including intimal fibrosis and medial hypertrophy of small and medium-sized pulmonary arteries (160). Observation of Langerhans cells with vessels is rare, and vascular involvement can also be noted in regions without parenchymal infiltration of Langerhans cells (160, 161). In contrast to most forms of PAH, capillary and venular pathology appears to be common in PLCH-PH (160, 165–168).
Management of PLCH, for those with or without associated PH, should most importantly center around smoking cessation. Stabilization and even substantial disease regression has been observed in those successful at quitting (169–172). While most case reports and series have focused on improvements in radiology or pulmonary function following smoking cessation, benefits on the pulmonary vascular component of disease should also be presumed. One case report of a patient with PLCH and associated PH has described PH resolution following smoking cessation (173). For patients with symptomatic PLCH refractory to smoking cessation, steroids, and chemotherapeutic agents have been used with mixed results (174–178). Use of PAH-specific therapy in those with PLCH-PH has been best described in a series by Le Pavec et al., where 14 patients were treated with an ERA, PDE-5I, and/or inhaled iloprost. Use of PAH-specific therapy was associated with improvements in mPAP and PVR on follow-up hemodynamic reassessments; a trend toward improved survival was also observed (167). With histologic observation of pulmonary venous involvement in PLCH-PH, the potential for development of pulmonary edema in response to PAH-specific therapy must be acknowledged, and cases consistent with this outcome have been documented after initiation of intravenous epoprostenol (160). Encouragingly, oxygenation of treated patients did not worsen in the series by Le Pavec (167). Given vascular complexity, lung transplant should be considered in those with associated PH and in others with severe PLCH. PLCH patients make up <1 percent of lung transplant recipients in the United States, with overall similar post-transplant outcomes to other patient groups (162, 164).
Neurofibromatosis Type 1
Neurofibromatosis type 1 (NF1) is an autosomal dominant genetic disorder caused by mutation in NF1, the tumor suppressor gene encoding neurofibromin. Incidence is 1 in 3,000 births, with near complete penetrance; skin manifestations are usually prominent and early in onset. Major manifestations leading to diagnosis include café au lait macules, cutaneous and subcutaneous neurofibromas, axillary and inguinal freckling, and Lisch nodules of the iris (179). NF1-related parenchymal lung disease affects 10–20% of adults and is characterized by cysts, micronodules, and interstitial abnormalities (179–181). Arterial and venous vascular lesions, including arterio-venous malformations, arterial aneurysms, coarctation of the abdominal aorta, and renal artery stenosis, are important but under recognized sources of morbidity in NF1 (182, 183). Neurofibromin is expressed in endothelial and vascular smooth muscle cells, and it is thus suspected that absence of functional protein leads to dysregulation of growth and proliferation of intimal and medial layer cells, in systemic and pulmonary circulations (182, 184). Patients with NF1 also have increased malignancy risk, and cancer is the leading cause of death with life expectancy reduced by 10–15 years (185, 186).
Pulmonary hypertension in NF1 patients has been described only in case reports and series, thus prevalence of NF1-PH is difficult to estimate but thought to be rare. The largest series of PH in NF1 recently characterized 49 patients from the French PH National Referral Center. Despite NF1 affecting males and female equally, 80% of NF1-PH patients were female, and median age at diagnosis of PH was 62. No specific NF1 mutations were associated with PH, and others with NF1-PH were not observed in the families of most patients. Hemodynamics at diagnosis were consistent with severe pre-capillary PH (mean mPAP 45 with PVR 10.7 WU). The majority of patients had normal or mildly abnormal spirometry, with decreased diffusion capacity for carbon monoxide (median 30% predicted) and resting hypoxemia. Imaging was most frequently notable for diffuse lung cysts, ground glass opacities, and emphysema (187). Importantly, this series and another literature review have identified normal imaging or single radiologic abnormalities in nearly 30% of NF1-PH patients (184, 187). Lung tissue has been infrequently available; the few histologic studies have demonstrated arterial remodeling with muscularization of small arterioles as well as particular increase in intimal layer thickness (187, 188). Findings of thrombosis have not been noted, and presence of plexiform lesions has been variable (187, 189). Three patients undergoing transplant all had pulmonary venous wall thickening (187), and a patient with pulmonary capillary hemangiomatosis associated with NF1 has also been reported (190).
Prognosis of NF1-PH is poor, with transplant free survival of 87, 54, and 42% at 1, 3, and 5 years, respectively (187). Of patients in the large series by Jutant et al., 45/49 were started on PAH-specific therapy; ERAs, PDE-5Is, and prostacyclin derivatives were all used alone or in combination. Treated patients demonstrated improvement in cardiac index and PVR; however, oxygenation worsened slightly and clinical improvements were modest and not sustained (187). Lung transplantation has been successfully performed for NF1 patients (187, 188); however, concern for malignancy risk following transplant and immunosuppression can be a barrier (191). The tyrosine kinase inhibitor sorafenib, which is capable of suppressing kinases operating in pathways also shared by targets of neurofibromin, was used in a single patient with refractory NF1-PH. Hemodynamic and clinical improvements resulted after 3 months of use (192). Given neurofibromin's role in containment of cell proliferation, investigation of drugs impacting cell cycle regulation are particularly of interest in NF1-PH.
Gaucher Disease
Gaucher disease (GD) is an autosomal recessive lysosomal storage disease with an incidence of 1 in 50,000 births. GD results from inadequate function of the lysosomal enzyme glucocerebrosidase, leading to accumulation of its substrate in macrophages, described as Gaucher cells. Accumulation of macrophages is particularly prominent in the bone marrow, liver, and spleen, with resulting hepatosplenomegaly, thrombocytopenia, anemia, osteopenia, and painful bone crises. GD is classified into three subtypes (GD 1, 2, and 3), with GD type 1, the non-neuronopathic form of GD, being the most common. Pulmonary involvement can be seen in all GD types, though those with GD1 are at most risk for developing pulmonary vascular complications of the disease (193). Enzyme replacement therapy for GD was first approved by the United States Food and Drug Administration in 1991 and has provided significant improvements in hematologic counts, spleen and liver volumes, and other clinical parameters for GD type 1 (194, 195). Importantly, use of enzyme replacement therapy has dramatically decreased need for splenectomy, previously performed for GD due to lack of other management options (195–197).
The prevalence of PH in adult GD type 1 patients, as estimated by echocardiography, ranges from 7 to 30% (198, 199). Mild PASP elevation (35–50 mmHg) is noted in the majority, with 0.76% documented to have PASP >50 (198). Treatment with enzyme replacement therapy is associated with lower prevalence of PH (7.4% in treated patients vs. 30% in untreated patients) (198). Though enzyme replacement therapy has been questioned to be causative of PH (200, 201), most current evidence suggests protection, and multiple examples of PH-related clinical improvements after initiation exist (198). In contrast, splenectomy has strong association with development of GD-PH. Severe PH complicating GD occurs predominantly in females who have undergone splenectomy as part of GD management (198, 199). The largest published series of GD-PH patients included 14 adult patients from a single referral center. Females made up 71% of the population, with a mean age of 36 at PH diagnosis, and all patients had previously undergone splenectomy at mean age 12. Other pertinent findings from this series includes lack of correlation between development of PH and GD severity by other metrics. Heteroallelic mutations (with one copy of the most common mutation N370S) were more prevalent than N370S homozygotes (199). Of interest, this report and others provide examples of patients also meeting criteria for hepatopulmonary syndrome; in these cases PH was either coexisting with hepatopulmonary syndrome or was unmasked after initiation of enzyme replacement therapy and resulting resolution of hepatopulmonary syndrome (199, 202). When tissue has been available, observed arterial changes have resembled those of PAH, including medial hypertrophy and intimal fibrosis, as well as plexiform lesions (203–205). In addition, intravascular Gaucher cells have been observed, though are not uniformly present (205–207).
If not previously initiated, most GD-PH patients will start on enzyme replacement therapy, with case reports of this alone leading to significant improvement (198, 199). ERAs, PDE-5Is, and prostanoids have all been used in individual patients with GD-PH, generally with positive outcomes (198, 199, 208). Given shared risk factor (splenectomy) with CTEPH, use of anticoagulation should be discussed particularly in splenectomized patients, though data about its role is lacking. Coumadin has been used in several patients in combination with enzyme replacement therapy and PAH-specific therapy (198). In cases of refractory GD-PH, imatinib has been used successfully in a single patient (209). Finally, though other comorbidities can pose a challenge to candidacy, lung transplant has been used as a treatment modality for a few cases of severe GD-PH (203, 205).
Discussion
Group 5 pulmonary hypertension remains a varied and challenging group of clinical entities. Understanding of the pathogenetic factors underlying many of the clinical conditions in the group has advanced considerably, however the relative rarity and/or variable clinical presentation of many of these disease processes has made it challenging to identify unifying treatment principles that can be applied broadly. Greater understanding may be gained by enhanced prospective registries of the more common conditions associated with PH, particularly sarcoidosis, sickle cell disease, myeloproliferative disorders, and chronic kidney disease/ESRD. More robust randomized trials of PAH therapies will be needed to understand if any of the ever-increasing number of PAH treatments may be of benefit in any of these populations.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
HF has received institutional research grant support from United Therapeutics/Lung Biotechnology, Actelion/Jannsen, Liquidia, and Eiger. He has received consulting honoraria from Actelion/Jannsen, United Therapeutics/Lung Biotechnology, Liquidia, Bayer, and Altavant. BL has received institutional research grant support from United Therapeutics and Actelion/Jannsen. MA-Q has received institutional research grant support from Bellerophon and Actelion/Jannsen.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. (2019) 53:1801913. doi: 10.1183/13993003.01913-2018
2. Rosenzweig EB, Abman SH, Adatia I, Beghetti M, Bonnet D, Haworth S, et al. Paediatric pulmonary arterial hypertension: updates on definition, classification, diagnostics and management. Eur Respir J. (2019) 53:1801916. doi: 10.1183/13993003.01916-2018
3. Fonseca GH, Souza R, Salemi VM, Jardim CV, Gualandro SF. Pulmonary hypertension diagnosed by right heart catheterisation in sickle cell disease. Eur Respir J. (2012) 39:112–8. doi: 10.1183/09031936.00134410
4. Mehari A, Gladwin MT, Tian X, Machado RF, Kato GJ. Mortality in adults with sickle cell disease and pulmonary hypertension. JAMA. (2012) 307:1254–6. doi: 10.1001/jama.2012.358
5. Parent F, Bachir D, Inamo J, Lionnet F, Driss F, Loko G, et al. A hemodynamic study of pulmonary hypertension in sickle cell disease. N Engl J Med. (2011) 365:44–53. doi: 10.1056/NEJMoa1005565
6. Aessopos A, Farmakis D, Karagiorga M, Voskaridou E, Loutradi A, Hatziliami A, et al. Cardiac involvement in thalassemia intermedia: a multicenter study. Blood. (2001) 97:3411–6. doi: 10.1182/blood.V97.11.3411
7. Grisaru D, Rachmilewitz EA, Mosseri M, Gotsman M, Lafair JS, Okon E, et al. Cardiopulmonary assessment in beta-thalassemia major. Chest. (1990). 98:1138–42. doi: 10.1378/chest.98.5.1138
8. Derchi G, Galanello R, Bina P, Cappellini MD, Piga A, Lai ME, et al. Prevalence and risk factors for pulmonary arterial hypertension in a large group of β-thalassemia patients using right heart catheterization: a Webthal study. Circulation. (2014) 129:338–45. doi: 10.1161/CIRCULATIONAHA.113.002124
9. Crary SE, Ramaciotti C, Buchanan GR. Prevalence of pulmonary hypertension in hereditary spherocytosis. Am J Hematol. (2011) 86:E73–6. doi: 10.1002/ajh.22182
10. Das A, Bansal D, Ahluwalia J, Das R, Rohit MK, Attri SV, et al. Risk factors for thromboembolism and pulmonary artery hypertension following splenectomy in children with hereditary spherocytosis. Pediatr Blood Cancer. (2014) 61:29–33. doi: 10.1002/pbc.24766
11. Jaïs X, Ioos V, Jardim C, Sitbon O, Parent F, Hamid A, et al. Splenectomy and chronic thromboembolic pulmonary hypertension. Thorax. (2005) 60:1031–4. doi: 10.1136/thx.2004.038083
12. Joannides R, Haefeli WE, Linder L, Richard V, Bakkali EH, Thuillez C, et al. Nitric oxide is responsible for flow-dependent dilatation of human peripheral conduit arteries in vivo. Circulation. (1995) 91:1314–9. doi: 10.1161/01.CIR.91.5.1314
13. Morris CR, Kato GJ, Poljakovic M, Wang X, Blackwelder WC, Sachdev V, et al. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA. (2005) 294:81–90. doi: 10.1001/jama.294.1.81
14. Liu X, Miller MJ, Joshi MS, Sadowska-Krowicka H, Clark DA, Lancaster JR Jr. Diffusion-limited reaction of free nitric oxide with erythrocytes. J Biol Chem. (1998) 273:18709–13. doi: 10.1074/jbc.273.30.18709
15. Reiter CD, Wang X, Tanus-Santos JE, Hogg N, Cannon RO III, Schechter AN, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. (2002). 8:1383–9. doi: 10.1038/nm1202-799
16. Kato GJ, Wang Z, Machado RF, Blackwelder WC, Taylor JG VI, Hazen SL. Endogenous nitric oxide synthase inhibitors in sickle cell disease: abnormal levels and correlations with pulmonary hypertension, desaturation, haemolysis, organ dysfunction and death. Br J Haematol. (2009) 145:506–13. doi: 10.1111/j.1365-2141.2009.07658.x
17. Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood. (2014) 123:377–90. doi: 10.1182/blood-2013-04-495887
18. Ghosh S, Adisa OA, Chappa P, Tan F, Jackson KA, Archer DR, et al. Extracellular hemin crisis triggers acute chest syndrome in sickle mice. J Clin Invest. (2013) 123:4809–20. doi: 10.1172/JCI64578
19. Rafikova O, Williams ER, McBride ML, Zemskova M, Srivastava A, Nair V, et al. Hemolysis-induced lung vascular leakage contributes to the development of pulmonary hypertension. Am J Respir Cell Mol Biol. (2018) 59:334–45. doi: 10.1165/rcmb.2017-0308OC
20. Tharaux PL, Hagège I, Placier S, Vayssairat M, Kanfer A, Girot R, et al. Urinary endothelin-1 as a marker of renal damage in sickle cell disease. Nephrol Dial Transplant. (2005) 20:2408–13. doi: 10.1093/ndt/gfi111
21. Phelan M, Perrine SP, Brauer M, Faller DV. Sickle erythrocytes, after sickling, regulate the expression of the endothelin-1 gene and protein in human endothelial cells in culture. J Clin Invest. (1995) 96:1145–51. doi: 10.1172/JCI118102
22. Berney SI, Ridler CD, Stephens AD, Thomas AE, Kovacs IB. Enhanced platelet reactivity and hypercoagulability in the steady state of sickle cell anaemia. Am J Hematol. (1992) 40:290–4. doi: 10.1002/ajh.2830400409
23. Solovey A, Gui L, Key NS, Hebbel RP. Tissue factor expression by endothelial cells in sickle cell anemia. J Clin Invest. (1998) 101:1899–904. doi: 10.1172/JCI1932
24. Carstens GR, Paulino BBA, Katayama EH, Amato-Lourenço LF, Fonseca GH, Souza R, et al. Clinical relevance of pulmonary vasculature involvement in sickle cell disease. Br J Haematol. (2019) 185:317–26. doi: 10.1111/bjh.15795
25. Graham JK, Mosunjac M, Hanzlick RL, Mosunjac M. Sickle cell lung disease and sudden death: a retrospective/prospective study of 21 autopsy cases and literature review. Am J Forensic Med Pathol. (2007) 28:168–72. doi: 10.1097/01.paf.0000257397.92466.50
26. Alkholy UM, Mohamed SA, Elhady M, Attar SE, Abdalmonem N, Zaki A. Vascular endothelial growth factor and pulmonary hypertension in children with beta thalassemia major. J Pediatr. (2019) 95:593–9. doi: 10.1016/j.jpedp.2018.06.015
27. Niss O, Quinn CT, Lane A, Daily J, Khoury PR, Bakeer N, et al. Cardiomyopathy with restrictive physiology in sickle cell disease. JACC Cardiovasc Imaging. (2016) 9:243–52. doi: 10.1016/j.jcmg.2015.05.013
28. Castro O, Hoque M, Brown BD. Pulmonary hypertension in sickle cell disease: cardiac catheterization results and survival. Blood. (2003) 101:1257–61. doi: 10.1182/blood-2002-03-0948
29. Sachdev V, Machado RF, Shizukuda Y, Rao YN, Sidenko S, Ernst I, et al. Diastolic dysfunction is an independent risk factor for death in patients with sickle cell disease. J Am Coll Cardiol. (2007) 49:472–9. doi: 10.1016/j.jacc.2006.09.038
30. Lisbona R, Derbekyan V, Novales-Diaz JA. Scintigraphic evidence of pulmonary vascular occlusion in sickle cell disease. J Nucl Med. (1997) 38:1151–3.
31. Santoli F, Zerah F, Vasile N, Bachir D, Galacteros F, Atlan G. Pulmonary function in sickle cell disease with or without acute chest syndrome. Eur Respir J. (1998) 12:1124–9. doi: 10.1183/09031936.98.12051124
32. Vichinsky EP, Styles LA, Colangelo LH, Wright EC, Castro O, Nickerson B. Acute chest syndrome in sickle cell disease: clinical presentation and course. cooperative study of sickle cell disease. Blood. (1997) 89:1787–92. doi: 10.1182/blood.V89.5.1787.1787_1787_1792
33. Klings ES, Machado RF, Barst RJ, Morris CR, Mubarak KK, Gordeuk VR, et al. An official American Thoracic Society clinical practice guideline: diagnosis, risk stratification and management of pulmonary hypertension of sickle cell disease. Am J Respir Crit Care Med. (2014) 189:727–40. doi: 10.1164/rccm.201401-0065ST
34. Morris CR, Kim HY, Trachtenberg F, Wood J, Quinn CT, Sweeters N, et al. Risk factors and mortality associated with an elevated tricuspid regurgitant jet velocity measured by Doppler-echocardiography in thalassemia: a Thalassemia Clinical Research Network report. Blood. (2011) 118:3794–802. doi: 10.1182/blood-2010-11-319152
35. Ristow B, Ali S, Ren X, Whooley MA, Schiller NB. Elevated pulmonary artery pressure by Doppler echocardiography predicts hospitalization for heart failure and mortality in ambulatory stable coronary artery disease: the Heart and Soul Study. J Am Coll Cardiol. (2007) 49:43–9. doi: 10.1016/j.jacc.2006.04.108
36. Kato GJ, McGowan V, Machado RF, Little JA, Taylor J VI, Morris CR, et al. Lactate dehydrogenase as a biomarker of hemolysis-associated nitric oxide resistance, priapism, leg ulceration, pulmonary hypertension, and death in patients with sickle cell disease. Blood. (2006) 107:2279–85. doi: 10.1182/blood-2005-06-2373
37. Machado RF, Anthi A, Steinberg MH, Bonds D, Sachdev V, Kato GJ, et al. N-terminal pro-brain natriuretic peptide levels and risk of death in sickle cell disease. JAMA. (2006). 296:310–8. doi: 10.1001/jama.296.3.310
38. Mahesh B, Besser M, Ravaglioli A, Pepke-Zaba J, Martinez G, Klein A, et al. Pulmonary endarterectomy is effective and safe in patients with haemoglobinopathies and abnormal red blood cells: the Papworth experience. Eur J Cardiothorac Surg. (2016) 50:537–41. doi: 10.1093/ejcts/ezw062
39. Yung GL, Channick RN, Fedullo PF, Auger WR, Kerr KM, Jamieson SW, et al. Successful pulmonary thromboendarterectomy in two patients with sickle cell disease. Am J Respir Crit Care Med. (1998) 157:1690–3. doi: 10.1164/ajrccm.157.5.9710032
40. Vichinsky EP, Haberkern CM, Neumayr L, Earles AN, Black D, Koshy M, et al. A comparison of conservative and aggressive transfusion regimens in the perioperative management of sickle cell disease. the preoperative transfusion in sickle cell disease study group. N Engl J Med. (1995) 333:206–13. doi: 10.1056/NEJM199512143332415
41. Savale L, Habibi A, Lionnet F, Maitre B, Cottin V, Jais X, et al. Clinical phenotypes and outcomes of precapillary pulmonary hypertension of sickle cell disease. Eur Respir J. (2019) 54:1900585. doi: 10.1183/13993003.00585-2019
42. Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. investigators of the multicenter study of hydroxyurea in sickle cell anemia. N Engl J Med. (1995) 332:1317–22. doi: 10.1056/NEJM199505183322001
43. Ferster A, Vermylen C, Cornu G, Buyse M, Corazza F, Devalck C, et al. Hydroxyurea for treatment of severe sickle cell anemia: a pediatric clinical trial. Blood. (1996) 88:1960–4. doi: 10.1182/blood.V88.6.1960.bloodjournal8861960
44. Steinberg MH, Barton F, Castro O, Pegelow CH, Ballas SK, Kutlar A, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA. (2003) 289:1645–51. doi: 10.1001/jama.289.13.1645
45. Karimi, Borzouee M, Mehrabani A, Cohan N. Echocardiographic finding in beta-thalassemia intermedia and major: absence of pulmonary hypertension following hydroxyurea treatment in beta-thalassemia intermedia. Eur J Haematol. (2009) 82:213–8. doi: 10.1111/j.1600-0609.2008.01192.x
46. Detterich JA, Kato RM, Rabai M, Meiselman HJ, Coates TD, Wood JC. Chronic transfusion therapy improves but does not normalize systemic and pulmonary vasculopathy in sickle cell disease. Blood. (2015) 126:703–10. doi: 10.1182/blood-2014-12-614370
47. Aessopos A, Farmakis D, Hatziliami A, Fragodimitri C, Karabatsos F, Joussef J, Mitilineou E, et al. Cardiac status in well-treated patients with thalassemia major. Eur J Haematol. (2004) 73:359–66. doi: 10.1111/j.1600-0609.2004.00304.x
48. Taher AT, Musallam KM, Karimi M, El-Beshlawy A, Belhoul K, Daar S, et al. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the OPTIMAL CARE study. Blood. (2010) 115:1886–92. doi: 10.1182/blood-2009-09-243154
49. Barst RJ, Mubarak KK, Machado RF, Ataga KI, Benza RL, Castro O, et al. Exercise capacity and haemodynamics in patients with sickle cell disease with pulmonary hypertension treated with bosentan: results of the ASSET studies. Br J Haematol. (2010) 149:426–35. doi: 10.1111/j.1365-2141.2010.08097.x
50. Weir NA, Saiyed R, Alam S, Conrey A, Desai HD, George MP, et al. Prostacyclin-analog therapy in sickle cell pulmonary hypertension. Haematologica. (2017) 102:e163–5. doi: 10.3324/haematol.2015.131227
51. Machado RF, Barst RJ, Yovetich NA, Hassell KL, Kato GJ, Gordeuk VR, et al. Hospitalization for pain in patients with sickle cell disease treated with sildenafil for elevated TRV and low exercise capacity. Blood. (2011) 118:855–64. doi: 10.1182/blood-2010-09-306167
52. Weir NA, Conrey A, Lewis D, Mehari A. Riociguat use in sickle cell related chronic thromboembolic pulmonary hypertension: a case series. Pulm Circ. (2018) 8:2045894018791802. doi: 10.1177/2045894018791802
53. Altintas A, Karahan Z, Pasa S, Cil T, Boyraz T, Iltumur K, et al. Pulmonary hypertension in patients with essential thrombocythemia and reactive thrombocytosis. Leuk Lymphoma. (2007) 48:1981–7. doi: 10.1080/10428190701493928
54. Garypidou V, Vakalopoulou S, Dimitriadis D, Tziomalos K, Sfikas G, Perifanis V. Incidence of pulmonary hypertension in patients with chronic myeloproliferative disorders. Haematologica. (2004) 89:245–6.
55. Gupta R, Perumandla S, Patsiornik Y, Niranjan S, Ohri A. Incidence of pulmonary hypertension in patients with chronic myeloproliferative disorders. J Natl Med Assoc. (2006) 98:1779–82.
56. Reisner SA, Rinkevich D, Markiewicz W, Tatarsky I, Brenner B. Cardiac involvement in patients with myeloproliferative disorders. Am J Med. (1992) 93:498–504. doi: 10.1016/0002-9343(92)90576-W
57. Chebrek S, Aïssi K, Francès Y, Mercier C, Farnault L, Sébahoun G, et al. Pulmonary hypertension in patients with chronic myeloproliferative neoplasms. Leuk Lymphoma. (2014) 55:223–5. doi: 10.3109/10428194.2013.797083
58. Polycythemia vera: the natural history of 1213 patients followed for 20 years. Gruppo Italiano Studio Policitemia. Ann Intern Med. (1995) 123:656–64. doi: 10.7326/0003-4819-123-9-199511010-00003
59. Guilpain P, Montani D, Damaj G, Achouh L, Lefrère F, Le Pavec J, et al. Pulmonary hypertension associated with myeloproliferative disorders: a retrospective study of ten cases. Respiration. (2008) 76:295–302. doi: 10.1159/000112822
60. Marchioli R, Finazzi G, Landolfi R, Kutti J, Gisslinger H, Patrono C, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol. (2005) 23:2224–32. doi: 10.1200/JCO.2005.07.062
61. Mohren M, Markmann I, Dworschak U, Franke A, Maas C, Mewes S, et al. Thromboembolic complications after splenectomy for hematologic diseases. Am J Hematol. (2004) 76:143–7. doi: 10.1002/ajh.20018
62. Wolanskyj AP, Schwager SM, McClure RF, Larson DR, Tefferi A, et al. Essential thrombocythemia beyond the first decade: life expectancy, long-term complication rates, and prognostic factors. Mayo Clin Proc. (2006) 81:159–66. doi: 10.4065/81.2.159
63. Pearson TC, Wetherley-Mein G, Vascular occlusive episodes and venous haematocrit in primary proliferative polycythaemia. Lancet. (1978) 2:1219–22. doi: 10.1016/S0140-6736(78)92098-6
64. Huang PY, Hellums JD. Aggregation and disaggregation kinetics of human blood platelets: Part III. the disaggregation under shear stress of platelet aggregates. Biophys J. (1993) 65:354–61. doi: 10.1016/S0006-3495(93)81080-4
65. Turitto VT, Weiss HJ. Platelet and red cell involvement in mural thrombogenesis. Ann N Y Acad Sci. (1983) 416:363–76. doi: 10.1111/j.1749-6632.1983.tb35199.x
66. Arellano-Rodrigo E, Alvarez-Larrán A, Reverter JC, Villamor N, Colomer D, Cervantes F, et al. Increased platelet and leukocyte activation as contributing mechanisms for thrombosis in essential thrombocythemia and correlation with the JAK2 mutational status. Haematologica. (2006) 91:169–75.
67. Carobbio A, Finazzi G, Guerini V, Spinelli O, Delaini F, Marchioli R, et al. Leukocytosis is a risk factor for thrombosis in essential thrombocythemia: interaction with treatment, standard risk factors, and Jak2 mutation status. Blood. (2007) 109:2310–3. doi: 10.1182/blood-2006-09-046342
68. Falanga A, Marchetti M, Evangelista V, Vignoli A, Licini M, Balicco M, et al. Polymorphonuclear leukocyte activation and hemostasis in patients with essential thrombocythemia and polycythemia vera. Blood. (2000) 96:4261–6. doi: 10.1182/blood.V96.13.4261.h8004261_4261_4266
69. Jensen MK, de Nully Brown P, Lund BV, Nielsen OJ, Hasselbalch HC. Increased circulating platelet-leukocyte aggregates in myeloproliferative disorders is correlated to previous thrombosis, platelet activation and platelet count. Eur J Haematol. (2001) 66:143–51. doi: 10.1034/j.1600-0609.2001.00359.x
70. Dot JM, Sztrymf B, Yaïci A, Dorfmüller P, Capron F, Parent F, et al. Pulmonary arterial hypertension due to tumor emboli. Rev Mal Respir. (2007) 24:359–66. doi: 10.1016/S0761-8425(07)91070-0
71. Marvin KS, Spellberg RD. Pulmonary hypertension secondary to thrombocytosis in a patient with myeloid metaplasia. Chest. (1993) 103:642–4. doi: 10.1378/chest.103.2.642
72. Toros AB, Gokcay S, Cetin G, Ar MC, Karagoz Y, Kesici B, et al. Portal hypertension and myeloproliferative neoplasms: a relationship revealed. ISRN Hematol. (2013) 2013:673781. doi: 10.1155/2013/673781
73. Montani D, Bergot E, Günther S, Savale L, Bergeron A, Bourdin A, et al. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation. (2012) 125:2128–37. doi: 10.1161/CIRCULATIONAHA.111.079921
74. Alkhatib Y, Albashaireh D, Al-Aqtash T, Awdish R. The role of tyrosine kinase inhibitor “Lapatinib” in pulmonary hypertension. Pulm Pharmacol Ther. (2016) 37:81–4. doi: 10.1016/j.pupt.2016.03.002
75. Cortelezzi A, Gritti G, Del Papa N, Pasquini MC, Calori R, Gianelli U, et al. Pulmonary arterial hypertension in primary myelofibrosis is common and associated with an altered angiogenic status. Leukemia. (2008) 22:646–9. doi: 10.1038/sj.leu.2404943
76. Faiz SA, Iliescu C, Lopez-Mattei J, Patel B, Bashoura L, Popat U. Resolution of myelofibrosis-associated pulmonary arterial hypertension following allogeneic hematopoietic stem cell transplantation. Pulm Circ. (2016) 6:611–13. doi: 10.1086/687291
77. Tabarroki A, Lindner DJ, Visconte V, Zhang L, Rogers HJ, Parker Y, et al. Ruxolitinib leads to improvement of pulmonary hypertension in patients with myelofibrosis. Leukemia. (2014) 28:1486–93. doi: 10.1038/leu.2014.5
78. Ghofrani HA, D'Armini AM, Grimminger F, Hoeper MM, Jansa P, Kim NH, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med. (2013) 369:319–29. doi: 10.1056/NEJMoa1209657
79. Ghofrani HA, Hoeper MM, Halank M, Meyer FJ, Staehler G, Behr J, et al. Riociguat for chronic thromboembolic pulmonary hypertension and pulmonary arterial hypertension: a phase II study. Eur Respir J. (2010) 36:792–9. doi: 10.1183/09031936.00182909
80. Steensma DP, Hook CC, Stafford SL, Tefferi A. Low-dose, single-fraction, whole-lung radiotherapy for pulmonary hypertension associated with myelofibrosis with myeloid metaplasia. Br J Haematol. (2002) 118:813–6. doi: 10.1046/j.1365-2141.2002.03695.x
81. Iwai K, Tachibana T, Takemura T, Matsui Y, Kitaichi M, Kawabata Y. Pathological studies on sarcoidosis autopsy. I. epidemiological features of 320 cases in Japan. Acta Pathol Jpn. (1993) 43:372–6. doi: 10.1111/j.1440-1827.1993.tb01148.x
82. Mayock RL, Bertrand P, Morrison CE, Scott JH. Manifestations of sarcoidosis. analysis of 145 patients, with a review of nine series selected from the literature. Am J Med. (1963) 35:67–89. doi: 10.1016/0002-9343(63)90165-7
83. Gluskowski J, Hawrylkiewicz I, Zych D, Zieliński J. Effects of corticosteroid treatment on pulmonary haemodynamics in patients with sarcoidosis. Eur Respir J. (1990) 3:403–7.
84. Rizzato G, Pezzano A, Sala G, Merlini R, Ladelli L, Tansini G, et al. Right heart impairment in sarcoidosis: haemodynamic and echocardiographic study. Eur J Respir Dis. (1983) 64:121–8.
85. Emirgil C, Sobol BJ, Herbert WH, Trout K. The lesser circulation in pulmonary fibrosis secondary to sarcoidosis and its relationship to respiratory function. Chest. (1971) 60:371–8. doi: 10.1378/chest.60.4.371
86. Shorr AF, Helman DL, Davies DB, Nathan SD. Pulmonary hypertension in advanced sarcoidosis: epidemiology and clinical characteristics. Eur Respir J. (2005) 25:783–8. doi: 10.1183/09031936.05.00083404
87. Huitema MP, Bakker ALM, Mager JJ, Rensing BJWM, Smits F, Snijder RJ, et al. Prevalence of pulmonary hypertension in pulmonary sarcoidosis: the first large European prospective study. Eur Respir J. (2019) 54:1900897. doi: 10.1183/13993003.00897-2019
88. Głuskowski J, Hawryłkiewicz I, Zych D, Wojtczak A, Zieliński J. Pulmonary haemodynamics at rest and during exercise in patients with sarcoidosis. Respiration. (1984) 46:26–32. doi: 10.1159/000194667
89. Baughman RP, Engel PJ, Taylor L, Lower EE. Survival in sarcoidosis-associated pulmonary hypertension: the importance of hemodynamic evaluation. Chest. (2010) 138:1078–85. doi: 10.1378/chest.09-2002
90. Bourbonnais JM, Samavati L. Clinical predictors of pulmonary hypertension in sarcoidosis. Eur Respir J. (2008) 32:296–302. doi: 10.1183/09031936.00175907
91. Handa T, Nagai S, Miki S, Fushimi Y, Ohta K, Mishima M, et al. Incidence of pulmonary hypertension and its clinical relevance in patients with sarcoidosis. Chest. (2006) 129:1246–52. doi: 10.1378/chest.129.5.1246
92. Rosen Y, Moon S, Huang CT, Gourin A, Lyons HA. Granulomatous pulmonary angiitis in sarcoidosis. Arch Pathol Lab Med. (1977) 101:170–4.
93. Smith LJ, Lawrence JB, Katzenstein AA. Vascular sarcoidosis: a rare cause of pulmonary hypertension. Am J Med Sci. (1983) 285:38–44. doi: 10.1097/00000441-198301000-00004
94. Takemura T, Matsui Y, Saiki S, Mikami R. Pulmonary vascular involvement in sarcoidosis: a report of 40 autopsy cases. Hum Pathol. (1992) 23:1216–23. doi: 10.1016/0046-8177(92)90288-E
95. Hoffstein V, Ranganathan N, Mullen JB. Sarcoidosis simulating pulmonary veno-occlusive disease. Am Rev Respir Dis. (1986) 134:809–11.
96. Jones RM, Dawson A, Jenkins GH, Nicholson AG, Hansell DM, Harrison NK. Sarcoidosis-related pulmonary veno-occlusive disease presenting with recurrent haemoptysis. Eur Respir J. (2009) 34:517–20. doi: 10.1183/09031936.00044609
97. Labyk A, Wretowski D, Zybińska-Oksiutowicz S, Furdyna A, Ciesielska K, Piotrowska-Kownacka D, et al. Balloon pulmonary angioplasty - efficient therapy of chronic thromboembolic pulmonary hypertension in the patient with advanced sarcoidosis - a case report. BMC Pulm Med. (2018) 18:139. doi: 10.1186/s12890-018-0695-4
98. Ungprasert P, Crowson CS, Matteson EL. Association of sarcoidosis with increased risk of VTE: a population-based study, 1976 to 2013. Chest. (2017) 151:425–30. doi: 10.1016/j.chest.2016.09.009
99. Birnie D, Ha AC, Gula LJ, Chakrabarti S, Beanlands RS, Nery P. Cardiac Sarcoidosis. Clin Chest Med. (2015) 36:657–8. doi: 10.1016/j.ccm.2015.08.008
100. Birnie DH, Sauer WH, Bogun F, Cooper JM, Culver DA, Duvernoy CS, et al. HRS expert consensus statement on the diagnosis and management of arrhythmias associated with cardiac sarcoidosis. Heart Rhythm. (2014) 11:1305–23. doi: 10.1016/j.hrthm.2014.03.043
101. Boucly A, Cottin V, Nunes H, Jaïs X, Tazi A, Prévôt G, et al. Management and long-term outcomes of sarcoidosis-associated pulmonary hypertension. Eur Respir J. (2017) 50:1700465. doi: 10.1183/13993003.00465-2017
102. Preston IR, Klinger JR, Landzberg MJ, Houtchens J, Nelson D, Hill NS. Vasoresponsiveness of sarcoidosis-associated pulmonary hypertension al. Chest. (2001) 120:866–72. doi: 10.1378/chest.120.3.866
103. Culver DA. Treatment of pulmonary hypertension in sarcoidosis. Proc Am Thorac Soc. (2005) 2:A862.
104. Fisher KA, Serlin DM, Wilson KC, Walter RE, Berman JS, Farber HW. Sarcoidosis-associated pulmonary hypertension: outcome with long-term epoprostenol treatment. Chest. (2006) 130:1481–8. doi: 10.1378/chest.130.5.1481
105. Milman N, Burton CM, Iversen M, Videbaek R, Jensen CV, Carlsen J. Pulmonary hypertension in end-stage pulmonary sarcoidosis: therapeutic effect of sildenafil? J Heart Lung Transplant. (2008) 27:329–34. doi: 10.1016/j.healun.2007.11.576
106. Barnett CF, Bonura EJ, Nathan SD, Ahmad S, Shlobin OA, Osei K, et al. Treatment of sarcoidosis-associated pulmonary hypertension. a two-center experience. Chest. (2009) 135:1455–61. doi: 10.1378/chest.08-1881
107. Baughman RP, Judson MA, Lower EE, Highland K, Kwon S, Craft N, et al. Inhaled iloprost for sarcoidosis associated pulmonary hypertension. Sarcoidosis Vasc Diffuse Lung Dis. (2009) 26:110–20.
108. Judson MA, Highland KB, Kwon S, Donohue JF, Aris R, Craft N, et al. Ambrisentan for sarcoidosis associated pulmonary hypertension. Sarcoidosis Vasc Diffuse Lung Dis. (2011) 28:139–45. doi: 10.1164/ajrccm-conference.2010.181.1_MeetingAbstracts.A2368
109. Dobarro D, Schreiber BE, Handler C, Beynon H, Denton CP, Coghlan JG. Clinical characteristics, haemodynamics and treatment of pulmonary hypertension in sarcoidosis in a single centre, and meta-analysis of the published data. Am J Cardiol. (2013) 111:278–85. doi: 10.1016/j.amjcard.2012.09.031
110. Baughman RP, Culver DA, Cordova FC, Padilla M, Gibson KF, Lower EE, et al. Bosentan for sarcoidosis-associated pulmonary hypertension: a double-blind placebo controlled randomized trial. Chest. (2014) 145:810–7. doi: 10.1378/chest.13-1766
111. Keir GJ, Walsh SL, Gatzoulis MA, Marino PS, Dimopoulos K, Alonso R, et al. Treatment of sarcoidosis-associated pulmonary hypertension: a single centre retrospective experience using targeted therapies. Sarcoidosis Vasc Diffuse Lung Dis. (2014) 31:82–90.
112. Bonham CA, Oldham JM, Gomberg-Maitland M, Vij R. Prostacyclin and oral vasodilator therapy in sarcoidosis-associated pulmonary hypertension: a retrospective case series. Chest. (2015) 148:1055–62. doi: 10.1378/chest.14-2546
113. Ford HJ, Baughman RP, Aris R, Engel P, Donohue JF. Tadalafil therapy for sarcoidosis-associated pulmonary hypertension. Pulm Circ. (2016) 6:557–62. doi: 10.1086/688775
114. Condado JF, Babaliaros V, Henry TS, Kaebnick B, Kim D, Staton GW Jr. Pulmonary stenting for the treatment of sarcoid induced pulmonary vascular stenosis. Sarcoidosis Vasc Diffuse Lung Dis. (2016) 33:281–7.
115. Shang W, Li Y, Ren Y, Li W, Wei HL, Dong J. Prevalence of pulmonary hypertension in patients with chronic kidney disease without dialysis: a meta-analysis. Int Urol Nephrol. (2018) 50:1497–504. doi: 10.1007/s11255-018-1853-6
116. Tang M, Batty JA, Lin C, Fan X, Chan KE, Kalim S. Pulmonary hypertension, mortality, and cardiovascular disease in CKD and ESRD patients: a systematic review and meta-analysis. Am J Kidney Dis. (2018) 72:75–83. doi: 10.1053/j.ajkd.2017.11.018
117. Zhang Q, Wang L, Zeng H, Lv Y, Huang Y. Epidemiology and risk factors in CKD patients with pulmonary hypertension: a retrospective study. BMC Nephrol. (2018) 19:1–8. doi: 10.1186/s12882-018-0866-9
118. O'Leary JM, Assad TR, Xu M, Birdwell KA, Farber-Eger E, Wells QS, et al. Pulmonary hypertension in patients with chronic kidney disease: invasive hemodynamic etiology and outcomes. Pulm Circ. (2017) 7:674–83 doi: 10.1177/2045893217716108
119. Böger RH. Asymmetric dimethylarginine (ADMA) and cardiovascular disease: insights from prospective clinical trials. Vasc Med. (2005) 10(Suppl. 1):S19–25 doi: 10.1177/1358836X0501000104
120. Nakhoul F, Yigla M, Gilman R, Reisner SA, Abassi Z. The pathogenesis of pulmonary hypertension in haemodialysis patients via arterio-venous access. Nephrol Dial Transplant. (2005) 20:1686–92. doi: 10.1093/ndt/gfh840
121. Nickel NP, O'Leary JM, Brittain EL, Fessel JP, Zamanian RT, West JD, et al. Kidney dysfunction in patients with pulmonary arterial hypertension. Pulm Circ. (2017) 7:38–54 doi: 10.1086/690018
122. Yu TM, Chen YH, Hsu JY, Sun CS, Chuang YW, Chen CH, et al. Systemic inflammation is associated with pulmonary hypertension in patients undergoing haemodialysis. Nephrol Dial Transplant. (2009) 24:1946–51. doi: 10.1093/ndt/gfn751
123. Rol N, Kurakula KB, Happé C, Bogaard HJ, Goumans MJ. TGF-β and BMPR2 signaling in PAH: Two black sheep in one family. Int J Mol Sci. (2018) 19:1–17. doi: 10.3390/ijms19092585
124. Chen L, Yang T, Lu DW, Zhao H, Feng YL, Chen H, et al. Central role of dysregulation of TGF-β/Smad in CKD progression and potential targets of its treatment. Biomed Pharmacother. (2018) 101:670–81. doi: 10.1016/j.biopha.2018.02.090
125. Alkhouli M, Sandhu P, Boobes K, Hatahet K, Raza F, Boobes Y. Cardiac complications of arteriovenous fistulas in patients with end-stage renal disease. Nefrologia. (2015) 35:234–45. doi: 10.1016/j.nefroe.2015.07.005
126. Basile C, Lomonte C, Vernaglione L, Casucci F, Antonelli M, Losurdo N. The relationship between the flow of arteriovenous fistula and cardiac output in haemodialysis patients. Nephrol Dial Transplant. (2008) 23:282–7. doi: 10.1093/ndt/gfm549
127. ESRD NCC (2019). Available from: https://esrdncc.org/en/fistula-first-catheter-last/ (accessed June 13, 2019).
128. Yigla M, Banderski R, Azzam ZS, Reisner SA, Nakhoul F. Arterio-venous access in end-stage renal disease patients and pulmonary hypertension. Ther Adv Respir Dis. (2008) 2:49–53. doi: 10.1177/1753465808089456
129. Rhodes CJ, Wharton J, Howard L, Gibbs JS, Vonk-Noordegraaf A, Wilkins MR. Iron deficiency in pulmonary arterial hypertension: a potential therapeutic target. Eur Respir J. (2011) 38:1453–60. doi: 10.1183/09031936.00037711
130. Ganz T. Iron balance and the role of hepcidin in chronic kidney disease. Semin Nephrol. (2016) 36:87–93. doi: 10.1016/j.semnephrol.2016.02.001
131. Navaneethan SD, Roy J, Tao K, Brecklin CS, Chen J, Deo R, et al. Chronic renal insufficiency cohort investigators. prevalence, predictors, and outcomes of pulmonary hypertension in CKD. J Am Soc Nephrol. (2016) 27:877–86. doi: 10.1681/ASN.2014111111
132. Abdelwhab S, Elshinnawy S. Pulmonary hypertension in chronic renal failure patients. Am J Nephrol. (2008) 28:990–7. doi: 10.1159/000146076
133. Mahiout A, Jörres A, Hiss R, Meinhold H, Kessel M. Effects of blood-dialyser interaction on prostaglandins in uraemic patients and in healthy man. Nephrol Dial Transplant. (1987) 2:546–50.
134. Kiykim AA, Horoz M, Ozcan T, Yildiz I, Sari S, Genctoy G. Pulmonary hypertension in hemodialysis patients without arteriovenous fistula: the effect of dialyzer composition. Ren Fail. (2010) 32:1148–52. doi: 10.3109/0886022X.2010.516854
135. Barak M, Nakhoul F, Katz Y. Pathophysiology and clinical implications of microbubbles during hemodialysis. Semin Dial. (2008) 21:232–8. doi: 10.1111/j.1525-139X.2008.00424.x
136. Perkett EA, Brigham KL, Meyrick B. Continuous air embolization into sheep causes sustained pulmonary hypertension and increased pulmonary vasoreactivity. Am J Pathol. (1988) 132:444–54.
137. Sakaguchi Y, Shoji T, Kawabata H, Niihata K, Suzuki A, Kaneko T, et al. High prevalence of obstructive sleep apnea and its association with renal function among nondialysis chronic kidney disease patients in Japan: a cross-sectional study. Clin J Am Soc Nephrol. (2011) 6:995–1000. doi: 10.2215/CJN.08670910
138. Zoccali C, Mallamaci F, Tripepi G. Nocturnal hypoxemia predicts incident cardiovascular complications in dialysis patients. J Am Soc Nephrol. (2002) 13:729–33.
139. Barceló A, de la Peña M, Ayllón O, Vega-Agapito MV, Piérola J, Pérez G, et al. Increased plasma levels of asymmetric dimethylarginine and soluble CD40 ligand in patients with sleep apnea. Respiration. (2009) 77:85–90. doi: 10.1159/000165630
140. Conger JD, Hammond WS, Alfrey AC, Contiguglia SR, Stanford RE, Huffer WE. Pulmonary calcification in chronic dialysis patients. clinical and pathologic studies. Ann Intern Med. (1975) 83:330–6. doi: 10.7326/0003-4819-83-3-330
141. Amin M, Fawzy A, Hamid MA, Elhendy A. Pulmonary hypertension in patients with chronic renal failure: role of parathyroid hormone and pulmonary artery calcifications. Chest. (2003) 124:2093–7. doi: 10.1378/chest.124.6.2093
142. Pabst S, Hammerstingl C, Hundt F, Gerhardt T, Grohé C, Nickenig G, et al. Pulmonary hypertension in patients with chronic kidney disease on dialysis and without dialysis: results of the PEPPER-study. PLoS ONE. (2012) 7:e35310. doi: 10.1371/journal.pone.0035310
143. Yigla M, Nakhoul F, Sabag A, Tov N, Gorevich B, Abassi Z, et al. Pulmonary hypertension in patients with end-stage renal disease. Chest. (2003) 123:1577–82. doi: 10.1378/chest.123.5.1577
144. Vaes RHD, Wouda R, Van Loon M, Van Hoek F, Tordoir JH, Scheltinga MR. Effectiveness of surgical banding for high flow in brachial artery-based hemodialysis vascular access. J Vasc Surg. (2015) 61:762–6. doi: 10.1016/j.jvs.2014.09.034
145. Rao NN, Stokes MB, Rajwani A, Ullah S, Williams K, King D, et al. Effects of arteriovenous fistula ligation on cardiac structure and function in kidney transplant recipients. Circulation. (2019) 139:2809–18 doi: 10.1161/CIRCULATIONAHA.118.038505
146. Frey R, Becker C, Saleh S, Unger S, van der Mey D, Mück W. Clinical pharmacokinetic and pharmacodynamic profile of riociguat. Clin Pharmacokinet. (2018) 57:647–661. doi: 10.1007/s40262-017-0604-7
147. Montani D, Chaumais MC, Savale L, Natali D, Price LC, Jaïs X, et al. Phosphodiesterase type 5 inhibitors in pulmonary arterial hypertension. Adv Ther. (2009) 26:813–25. doi: 10.1007/s12325-009-0064-z
148. Lentine KL, Villines TC, Axelrod D, Kaviratne S, Weir MR, Costa SP. Evaluation and management of pulmonary hypertension in kidney transplant candidates and recipients: Concepts and controversies. Transplantation. (2017) 101:166–81 doi: 10.1097/TP.0000000000001043
149. Foderaro AE, Baird GL, Bazargan-Lari A, Morrissey PE, Gohh RY, Poppas A, et al. Echocardiographic pulmonary hypertension predicts post-transplantation renal allograft failure. Transplant Proc. (2017) 49:1256–61. doi: 10.1016/j.transproceed.2017.01.085
150. Issa N, Krowka MJ, Griffin MD, Hickson LJ, Stegall MD, Cosio FG. Pulmonary hypertension is associated with reduced patient survival after kidney transplantation. Transplantation. (2008) 86:1384–8 doi: 10.1097/TP.0b013e318188d640
151. Wolfe JD, Hickey GW, Althouse AD, Sharbaugh MS, Kliner DE, Mathier MA, et al. Pulmonary vascular resistance determines mortality in end-stage renal disease patients with pulmonary hypertension. Clin Transplant. (2018) 32. doi: 10.1111/ctr.13270
152. Vermaelen K, Pauwels R. Pulmonary dendritic cells. Am J Respir Crit Care Med. (2005) 172:530–51. doi: 10.1164/rccm.200410-1384SO
153. Vassallo R, Ryu JH, Schroeder DR, Decker PA, Limper AH. Clinical outcomes of pulmonary Langerhans'-cell histiocytosis in adults. N Engl J Med. (2002) 346:484–90. doi: 10.1056/NEJMoa012087
154. Casolaro MA, Bernaudin JF, Saltini C, Ferrans VJ, Crystal RG. Accumulation of Langerhans' cells on the epithelial surface of the lower respiratory tract in normal subjects in association with cigarette smoking. Am Rev Respir Dis. (1988) 137:406–11. doi: 10.1164/ajrccm/137.2.406
155. Vassallo R, Walters PR, Lamont J, Kottom TJ, Yi ES, Limper AH. Cigarette smoke promotes dendritic cell accumulation in COPD; a Lung Tissue Research Consortium study. Respir Res. (2010) 11:45. doi: 10.1186/1465–9921-11-45
156. Suri HS, Yi ES, Nowakowski GS, Vassallo R. Pulmonary langerhans cell histiocytosis. Orphanet J Rare Dis. (2012) 7:16. doi: 10.1186/1750–1172-7-16
157. Prasse A, Stahl M, Schulz G, Kayser G, Wang L, Ask K, et al. Essential role of osteopontin in smoking-related interstitial lung diseases. Am J Pathol. (2009) 174:1683–91. doi: 10.2353/ajpath.2009.080689
158. Tazi A, Bonay M, Bergeron A, Grandsaigne M, Hance AJ, Soler P. Role of granulocyte-macrophage colony stimulating factor (GM-CSF) in the pathogenesis of adult pulmonary histiocytosis X. Thorax. (1996) 51:611–4. doi: 10.1136/thx.51.6.611
159. Travis WD, Borok Z, Roum JH, Zhang J, Feuerstein I, Ferrans VJ, et al. Pulmonary langerhans cell granulomatosis (histiocytosis X). a clinicopathologic study of 48 cases. Am J Surg Pathol. (1993) 17:971–86. doi: 10.1097/00000478-199310000-00002
160. Fartoukh M, Humbert M, Capron F, Maître S, Parent F, Le Gall C, et al. Severe pulmonary hypertension in histiocytosis X. Am J Respir Crit Care Med. (2000) 161:216–23. doi: 10.1164/ajrccm.161.1.9807024
161. Harari S, Brenot F, Barberis M, Simmoneau G. Advanced pulmonary histiocytosis X is associated with severe pulmonary hypertension. Chest. (1997) 111:1142–4. doi: 10.1378/chest.111.4.1142-a
162. Wajda N, Zhu Z, Jandarov R, Dilling DF, Gupta N. Clinical outcomes and survival following lung transplantation in patients with pulmonary Langerhans cell histiocytosis. Respirology. (2020) 25:644–50. doi: 10.1111/resp.13671
163. Chaowalit N, Pellikka PA, Decker PA, Aubry MC, Krowka MJ, Ryu JH, et al. Echocardiographic and clinical characteristics of pulmonary hypertension complicating pulmonary Langerhans cell histiocytosis. Mayo Clin Proc. (2004) 79:1269–75. doi: 10.4065/79.10.1269
164. Dauriat G, Mal H, Thabut G, Mornex JF, Bertocchi M, Tronc F, et al. Lung transplantation for pulmonary langerhans' cell histiocytosis: a multicenter analysis. Transplantation. (2006) 81:746–50. doi: 10.1097/01.tp.0000200304.64613.af
165. Bois MC, May AM, Vassallo R, Jenkins SM, Yi ES, Roden AC. Morphometric study of pulmonary arterial changes in pulmonary langerhans cell histiocytosis. Arch Pathol Lab Med. (2018) 142:929–37. doi: 10.5858/arpa.2017-0463-OA
166. Heiden GI, Sobral JB, Freitas CSG, Pereira de Albuquerque AL, Salge JM, Kairalla RA, et al. Mechanisms of exercise limitation and prevalence of pulmonary hypertension in pulmonary langerhans cell histiocytosis. Chest. (2020) 158:2440–8. doi: 10.1016/j.chest.2020.05.609
167. Le Pavec J, Lorillon G, Jaïs X, Tcherakian C, Feuillet S, Dorfmüller P, et al. Pulmonary Langerhans cell histiocytosis-associated pulmonary hypertension: clinical characteristics and impact of pulmonary arterial hypertension therapies. Chest. (2012) 142:1150–7. doi: 10.1378/chest.11-2490
168. Hamada K, Teramoto S, Narita N, Yamada E, Teramoto K, Kobzik L. Pulmonary veno-occlusive disease in pulmonary Langerhans' cell granulomatosis. Eur Respir J. (2000) 15:421–3. doi: 10.1034/j.1399-3003.2000.15b33.x
169. Tazi A, de Margerie C, Naccache JM, Fry S, Dominique S, Jouneau S, et al. The natural history of adult pulmonary Langerhans cell histiocytosis: a prospective multicentre study. Orphanet J Rare Dis. (2015) 10:30. doi: 10.1186/s13023-015-0249-2
170. Mogulkoc N, Veral A, Bishop PW, Bayindir U, Pickering CA, Egan JJ. Pulmonary Langerhans' cell histiocytosis: radiologic resolution following smoking cessation. Chest. (1999) 115:1452–5. doi: 10.1378/chest.115.5.1452
171. Wolters PJ, Elicker BM. Subacute onset of pulmonary langerhans cell histiocytosis with resolution after smoking cessation. Am J Respir Crit Care Med. (2014) 190:e64. doi: 10.1164/rccm.201405-0974IM
172. Elia D, Torre O, Cassandro R, Caminati A, Harari S. Pulmonary Langerhans cell histiocytosis: a comprehensive analysis of 40 patients and literature review. Eur J Intern Med. (2015) 26:351–6. doi: 10.1016/j.ejim.2015.04.001
173. Kinoshita Y, Watanabe K, Sakamoto A, Hidaka K. Pulmonary Langerhans cell histiocytosis-associated pulmonary hypertension showing a drastic improvement following smoking cessation. Intern Med. (2016) 55:491–5. doi: 10.2169/internalmedicine.55.5152
174. Delobbe A, Durieu J, Duhamel A, Wallaert B. Determinants of survival in pulmonary Langerhans' cell granulomatosis (histiocytosis X). Groupe d'Etude en Pathologie Interstitielle de la Société de Pathologie Thoracique du Nord. Eur Respir J. (1996) 9:2002–6. doi: 10.1183/09031936.96.09102002
175. Schönfeld N, Frank W, Wenig S, Uhrmeister P, Allica E, Preussler H, et al. Clinical and radiologic features, lung function and therapeutic results in pulmonary histiocytosis X. Respiration. (1993) 60:38–44. doi: 10.1159/000196171
176. Grobost V, Khouatra C, Lazor R, Cordier JF, Cottin V. Effectiveness of cladribine therapy in patients with pulmonary Langerhans cell histiocytosis. Orphanet J Rare Dis. (2014) 9:191. doi: 10.1186/s13023-014-0191-8
177. Lorillon G, Bergeron A, Detourmignies L, Jouneau S, Wallaert B, Frija J, et al. Cladribine is effective against cystic pulmonary Langerhans cell histiocytosis. Am J Respir Crit Care Med. (2012) 186:930–2. doi: 10.1164/ajrccm.186.9.930
178. Tazi A, Lorillon G, Haroche J, Neel A, Dominique S, Aouba A, et al. Vinblastine chemotherapy in adult patients with langerhans cell histiocytosis: a multicenter retrospective study. Orphanet J Rare Dis. (2017) 12:95. doi: 10.1186/s13023-017-0651-z
179. Alves Júnior SF, Zanetti G, Alves de Melo AS, Souza AS Jr, Souza LS, de Souza Portes Meirelles G, et al. Neurofibromatosis type 1: state-of-the-art review with emphasis on pulmonary involvement. Respir Med. (2019) 149:9–15. doi: 10.1016/j.rmed.2019.01.002
180. Oikonomou A, Vadikolias K, Birbilis T, Bouros D, Prassopoulos P. HRCT findings in the lungs of non-smokers with neurofibromatosis. Eur J Radiol. (2011) 80:e520–3. doi: 10.1016/j.ejrad.2010.11.033
181. Zamora AC, Collard HR, Wolters PJ, Webb WR, King TE. Neurofibromatosis-associated lung disease: a case series and literature review. Eur Respir J. (2007) 29:210–4. doi: 10.1183/09031936.06.00044006
182. Lie JT. Vasculopathies of neurofibromatosis Type 1 (von Recklinghausen Disease). Cardiovasc Pathol. (1998) 7:97–108. doi: 10.1016/S1054-8807(97)00081-1
183. Oderich GS, Sullivan TM, Bower TC, Gloviczki P, Miller DV, Babovic-Vuksanovic D, et al. Vascular abnormalities in patients with neurofibromatosis syndrome type I: clinical spectrum, management, and results. J Vasc Surg. (2007) 46:475–84. doi: 10.1016/j.jvs.2007.03.055
184. Jutant EM, Girerd B, Jaïs X, Savale L, O'Connell C, Perros F, et al. Pulmonary hypertension associated with neurofibromatosis type 1. Eur Respir Rev. (2018) 27:180053. doi: 10.1183/16000617.0053-2018
185. Rasmussen SA, Yang Q, Friedman JM. Mortality in neurofibromatosis 1: an analysis using U.S. death certificates. Am J Hum Genet. (2001) 68:1110–8. doi: 10.1086/320121
186. Zöller M, Rembeck B, Akesson HO, Angervall L. Life expectancy, mortality and prognostic factors in neurofibromatosis type 1. a twelve-year follow-up of an epidemiological study in Göteborg, Sweden. Acta Derm Venereol. (1995) 75:136–40.
187. Jutant EM, Jaïs X, Girerd B, Savale L, Ghigna MR, Perros F, et al. Phenotype and outcomes of pulmonary hypertension associated with neurofibromatosis Type 1. Am J Respir Crit Care Med. (2020) 202:843–52. doi: 10.1164/rccm.202001-0105OC
188. Montani D, Coulet F, Girerd B, Eyries M, Bergot E, Mal H, et al. Pulmonary hypertension in patients with neurofibromatosis type I. Medicine. (2011) 90:201–11. doi: 10.1097/MD.0b013e31821be2b7
189. Aoki Y, Kodama M, Mezaki T, Ogawa R, Sato M, Okabe M, et al. von Recklinghausen disease complicated by pulmonary hypertension. Chest. (2001) 119:1606–8. doi: 10.1378/chest.119.5.1606
190. Chaddha U, Puscas I, Prosper A, Ganesh S, Yaghmour B. A 63-year-old woman with neurofibromatosis Type 1 and pulmonary hypertension with worsening hypoxemia. Chest. (2017) 152:e89–93. doi: 10.1016/j.chest.2017.05.014
191. Merlo CA, Studer SM, Conte JV, Yang SC, Sonnett J, Orens JB. The course of neurofibromatosis type 1 on immunosuppression after lung transplantation: report of 2 cases. J Heart Lung Transplant. (2004) 23:774–6. doi: 10.1016/S1053-2498(03)00265-1
192. Tamura Y, Ono T, Sano M, Fukuda K, Kataoka M, Satoh T. Favorable effect of sorafenib in a patient with neurofibromatosis-associated pulmonary hypertension. Am J Respir Crit Care Med. (2012) 186:291–2. doi: 10.1164/ajrccm.186.3.291
193. Baris HN, Cohen IJ, Mistry PK. Gaucher disease: the metabolic defect, pathophysiology, phenotypes and natural history. Pediatr Endocrinol Rev. (2014) 12(Suppl. 1):72–81.
194. Weinreb NJ, Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, et al. Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am J Med. (2002) 113:112–9. doi: 10.1016/S0002-9343(02)01150-6
195. Charrow J, Scott CR. Long-term treatment outcomes in Gaucher disease. Am J Hematol. (2015) 90(Suppl. 1):S19–24. doi: 10.1002/ajh.24056
196. Jmoudiak M, Futerman AH. Gaucher disease: pathological mechanisms and modern management. Br J Haematol. (2005) 129:178–88. doi: 10.1111/j.1365-2141.2004.05351.x
197. van Dussen L, Biegstraaten M, Dijkgraaf MG, Hollak CE. Modelling Gaucher disease progression: long-term enzyme replacement therapy reduces the incidence of splenectomy and bone complications. Orphanet J Rare Dis. (2014) 9:112. doi: 10.1186/s13023-014-0112-x
198. Mistry PK, Sirrs S, Chan A, Pritzker MR, Duffy TP, Grace ME, et al. Pulmonary hypertension in type 1 Gaucher's disease: genetic and epigenetic determinants of phenotype and response to therapy. Mol Genet Metab. (2002) 77:91–8. doi: 10.1016/S1096-7192(02)00122-1
199. Lo SM, Liu J, Chen F, Pastores GM, Knowles J, Boxer M, et al. Pulmonary vascular disease in Gaucher disease: clinical spectrum, determinants of phenotype and long-term outcomes of therapy. J Inherit Metab Dis. (2011) 34:643–50. doi: 10.1007/s10545-011-9313-9
200. Elstein D, Klutstein MW, Lahad A, Abrahamov A, Hadas-Halpern I, Zimran A. Echocardiographic assessment of pulmonary hypertension in Gaucher's disease. Lancet. (1998) 351:1544–6. doi: 10.1016/S0140-6736(98)10194-0
201. Harats D, Pauzner R, Elstein D, Many A, Klutstein MW, Kramer MR, et al. Pulmonary hypertension in two patients with type I Gaucher disease while on alglucerase therapy. Acta Haematol. (1997) 98:47–50. doi: 10.1159/000203562
202. Dawson A, Elias DJ, Rubenson D, Bartz SH, Garver PR, Kay AC, et al. Pulmonary hypertension developing after alglucerase therapy in two patients with type 1 Gaucher disease complicated by the hepatopulmonary syndrome. Ann Intern Med. (1996) 125:901–4. doi: 10.7326/0003-4819-125-11-199612010-00005
203. de Boer GM, van Dussen L, van den Toorn LM, den Bakker MA, Hoek RA, Hesselink DA, et al. Lung transplantation in Gaucher disease: a learning lesson in trying to avoid both scylla and charybdis. Chest. (2016) 149:e1–5. doi: 10.1016/j.chest.2015.09.011
204. den Bakker MA, Grünberg K, Boonstra A, van Hal PT, Hollak CE. Pulmonary arterial hypertension with plexogenic arteriopathy in enzyme-substituted Gaucher disease. Histopathology. (2012) 61:324–6. doi: 10.1111/j.1365-2559.2012.04261.x
205. Goobie GC, Sirrs SM, Yee J, English JC, Bergeron C, Nador R, et al. Lessons from lung transplantation: cause for redefining the pathophysiology of pulmonary hypertension in gaucher disease. Respir Med Case Rep. (2019) 28:100893. doi: 10.1016/j.rmcr.2019.100893
206. Roberts WC, Fredrickson DS. Gaucher's disease of the lung causing severe pulmonary hypertension with associated acute recurrent pericarditis. Circulation. (1967) 35:783–9. doi: 10.1161/01.CIR.35.4.783
207. Ross DJ, Spira S, Buchbinder NA. Gaucher cells in pulmonary-capillary blood in association with pulmonary hypertension. N Engl J Med. (1997) 336:379–81. doi: 10.1056/NEJM199701303360516
208. Fernandes CJ, Jardim C, Carvalho LA, Farias AQ, Filho MT, Souza R. Clinical response to sildenafil in pulmonary hypertension associated with Gaucher disease. J Inherit Metab Dis. (2005) 28:603–5. doi: 10.1007/s10545-005-0603-y
Keywords: multifactorial, pulmonary hypertension, Group 5, hematologic, sickle cell, sarcoidosis, metabolic, CKD - chronic kidney disease
Citation: Al-Qadi M, LeVarge B and Ford HJ (2021) Epidemiology, Pathogenesis, and Clinical Approach in Group 5 Pulmonary Hypertension. Front. Med. 7:616720. doi: 10.3389/fmed.2020.616720
Received: 13 October 2020; Accepted: 17 December 2020;
Published: 25 March 2021.
Edited by:
Vinicio De Jesus Perez, Stanford University, United StatesReviewed by:
Roberto J. Bernardo, University of Oklahoma, United StatesJeremy Mazurek, University of Pennsylvania, United States
Nicholas Kolaitis, University of California, San Francisco, United States
Copyright © 2021 Al-Qadi, LeVarge and Ford. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: H. James Ford, aGpmb3JkQG1lZC51bmMuZWR1