
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Med. , 05 January 2021
Sec. Pulmonary Medicine
Volume 7 - 2020 | https://doi.org/10.3389/fmed.2020.588404
This article is part of the Research Topic Biomarkers in Pulmonary Diseases View all 19 articles
 Chintan K. Gandhi1
Chintan K. Gandhi1 Chixiang Chen2
Chixiang Chen2 Shaili Amatya1
Shaili Amatya1 Lili Yang3Chenqi Fu2Shouhao Zhou2Rongling Wu2
Lili Yang3Chenqi Fu2Shouhao Zhou2Rongling Wu2 Ivette Buendía-Roldan4
Ivette Buendía-Roldan4 Moisés Selman4Annie Pardo5
Moisés Selman4Annie Pardo5 Joanna Floros1,6*
Joanna Floros1,6*Background: Hypersensitivity pneumonitis (HP) is an interstitial lung disease caused by inhalation of common environmental organic particles. Surfactant proteins (SPs) play a role in innate immunity and surfactant function. We hypothesized that single nucleotide polymorphisms (SNPs) or haplotypes of the SP genes associate with HP.
Methods: Seventy-five HP patients caused by avian antigen and 258 controls, asymptomatic antigen exposed and non-exposed were enrolled. SNP association was performed using logistic regression analysis and SNP-SNP interaction models.
Results: Based on odds ratio, regression analyses showed association of (a) rs7316_G, 1A3 (protective) compared to antigen exposed; (b) male sex, smoking, rs721917_T and rs1130866_T (protective) compared to non-exposed controls with HP; (c) compared to antigen exposed, 25 interactions associated with HP in a three-SNP model; (d) compared to non-exposed, (i) rs1136451 associated with increased, whereas rs1136450 and rs1130866 associated with lower HP risk, (ii) 97 interactions associated with HP in a three-SNP model. The majority of SNP-SNP interactions associated with increased HP risk involved SNPs of the hydrophilic SPs, whereas, the majority of interactions associated with lower HP risk involved SNPs of both hydrophilic and hydrophobic SPs; (e) haplotypes of SP genes associated with HP risk.
Conclusions: The complexity of SNPs interactions of the SFTP genes observed indicate that the lung inflammatory response to avian antigens is modulated by a complex gene interplay rather than by single SNPs.
Hypersensitivity pneumonitis (HP) is an interstitial lung disease caused by an abnormal immune response to a wide variety of inhaled environmental antigens, mainly small organic particles (<5 μm) that reach the alveoli (1). These antigens provoke an exaggerated immune response in susceptible individuals (1, 2). HP is seen worldwide, and the most common implicated antigens are actinomyces species, fungi, and bird proteins (1). The pigeon breeder's disease caused by proteins from avian serum, feces, and feathers is the most common form of HP in Mexico (3). Given the universal and wide distribution of the offending antigens, it is unclear why only few individuals develop the disease, indicating the complex interaction between environment and genetic factors. Nonetheless, the host genetic factors that may play a role in HP susceptibility are understudied (1, 2).
Although pulmonary surfactant or its components have the potential to be contributors to the pathogenesis of HP, very little to no work has been done in this regard. Pulmonary surfactant is a complex mixture of 90% lipids and 10% proteins. The surfactant specific proteins are SP-A1, SP-A2, SP-B, and SP-C. Though SP-D is not part of the functional surfactant complex, it is grouped with the other surfactant proteins because it coisolates with them. SP-B and SP-C are hydrophobic proteins and play a role primarily in lowering the surface tension and stabilizing alveoli (4), whereas, SP-A and SP-D are hydrophilic proteins and play a role in innate immunity and host defense (5, 6), although SP-A also contributes to surfactant-related function (6). The human SP-A locus includes two functional genes, SFTPA1 and SFTPA2, in opposite transcriptional orientation (7), encoding SP-A1 and SP-A2, respectively. Several genetic polymorphisms for each SFTPA gene are found frequently in the general population (8). SP-B, SP-C, and SP-D are each encoded by a single gene, SFTPB, SFTPC, and SFTPD, respectively (9), and several polymorphisms have been described for each (10–12).
Single nucleotide polymorphisms (SNPs) of the SP genes have been shown to associate with various acute and chronic pulmonary diseases, such as idiopathic pulmonary fibrosis (IPF) (13), chronic obstructive pulmonary disease (COPD) (14, 15), acute respiratory distress syndrome (10), cystic fibrosis (16), and neonatal respiratory distress syndrome (RDS) (17–20). More importantly, we have shown an association of SP gene polymorphisms with IPF (13), COPD (14), and tuberculosis (21) in the Mexican population. Furthermore, some of the common SP-A variants have been shown to differentially affect function (22, 23) and regulation (24–27) of alveolar macrophages in an animal model, as well as SP-A variants have been associated with differential lung function mechanics and survival of mice (28–30). For the current study, we selected from physiologically and biologically relevant genes, 17 well-characterized SP SNPs that have been shown to associate with various acute and chronic pulmonary diseases (12, 31). Previous studies have shown increased SP-A in bronchoalveolar lavage (BAL) and alveolar macrophages, and elevated serum SP-A and SP-D concentration in HP patients compared to controls (32–34). Though the exact mechanisms are unknown, HP pathogenesis may include alveolar epithelial injury by altered immune response and increased leakage of SP-A and SP-D from the alveolar to the vascular compartment. Moreover, previous research showed altered concentration of SP-A, SP-B, and phospholipids in IPF, HP, and sarcoidosis patients (34, 35). Taken together, it is likely that SPs play a critical role in HP.
HP is characterized by an abnormal immune response leading to chronic inflammation and abnormal lung function and is probably the result of complex interactions of genetic and environmental factors. Due to the importance of SPs in normal lung function, innate immunity and host defense of the lung, we hypothesized that natural genetic variants of SPs and their interactions are associated with HP in Mexican population.
The study was conducted at the National Institute of Respiratory Diseases (INER) in Mexico City and the protocol was approved by the Ethics Committee. INER is a tertiary referral and one of the National Institutes of Health centers in Mexico. The study cohort compromised 3 groups (age > 18 years) and at least 3 generations of study participants were born in Mexico.
Group 1 (Cases) was composed of 75 consecutive unrelated patients 18 years of age or older with a diagnosis of HP (Table 1). HP was diagnosed as previously described using clinical characteristics, history of antecedent antigen exposure and specific antibodies against avian proteins, pulmonary function tests, high-resolution computed tomography, BAL findings (all 75 patients), and/or surgical biopsy when available (23 out of 75 patients) (1, 36). Attending clinicians systematically recorded the relevant data in every patient before making the final diagnosis and was confirmed by a multidisciplinary team. As per recently published official clinical practice guidelines from American Thoracic Society, all enrolled patients in the current study were classified to have a fibrotic HP (mixed inflammatory plus fibrosis) (37). Patients with other known interstitial lung diseases were excluded.

Table 1. Demographic and clinical characteristics of the study group.
Group 2 (Exposed controls) was composed of 64 healthy individuals with a history of avian antigen exposure but remained symptom-free. Some of these individuals were relatives of HP patients.
Group 3 (Non-exposed controls) was composed of 194 healthy individuals without a history of antigen exposure. Out of 194, 91 subjects were recruited randomly from the Smoking Cessation program with more than 5 years of smoking history and normal pulmonary function tests (smoker controls), and the other 103 unrelated healthy blood donors (non-smoker controls) were recruited randomly from the INER, as noted previously (13).
We collected blood samples of study participants after obtaining informed consent from subjects.
Genomic DNA was extracted from blood samples as described previously (8) and used as a template for polymerase chain reaction (PCR). A total of 17 targeted SNPs of surfactant protein genes, SFTPA1, SFTPA2, SFTPB, SFTPC, and SFTPD, were selected for the current study. The 17 SNPs include 5 SNPs from SFTPA1: rs1059047, rs1136450, rs1136451, rs1059057, and rs4253527; 4 SNPs from SFTPA2: rs1059046, rs17886395, rs1965707, and 1965708; 4 SNPs from SFTPB: rs2077079, rs3024798, rs1130866, and rs7316; 2 SNPs from SFTPC: rs4715 and rs1124; and 2 SNPs from SFTPD: rs721917 and rs2243639. The PCR-restriction fragment length polymorphism method was used to genotype the SFTPA1, SFTPA2, SFTPD (8, 10), SFTPB (10, 38), and SFTPC (13) gene polymorphisms, as described elsewhere in detail (8, 16). The PCR primer sequences used are reported in Supplementary Table 1. To avoid genotyping bias, each sample was given a sequential laboratory number without identifiers in the order they were received and all samples were genotyped together in a blinded fashion with those assigning genotype unaware of the group of subjects.
To determine the frequency of each SNP, we used the Chi-squared test, or Fisher's exact test when the expected frequency was too small (<5) and compared the allele distribution between the case and two control groups (avian antigen exposed and non-exposed). Dummy variables for each allele or SP-A genotype were created and applied for univariate logistic regression analysis, assuming no dose-effect for the alleles (13, 21). The selected markers, which were significantly associated with HP in unadjusted univariate analysis (p-value < 0.1), were passed to the multivariate logistic regression analysis after adjusting for smoking status and sex. Variable selection was performed using the backward elimination method with staying significance level less than 0.05.
To detect the effects of SNP-SNP interactions, we used Wang et al.'s case-control approach to study associations of SP genes polymorphisms with HP in cases vs. exposed controls and cases vs. non-exposed controls (39). Compared to traditional logistic methods, this approach is more efficient (40) in the detection of different genetic effects. It decomposes the overall genetic effect of each SNP into different components: the additive (a), recessive (r), and dominant (d) effect in a single SNP model. For two- and three-SNP models, this approach can also detect pairwise and triad-wise (high-order) epistasis, as described previously (41). If a statistically significant difference between the groups was observed, the p-value was corrected for variables such as sex and smoking status. The false discovery rate was controlled at 5% to account for multiple comparisons using the Benjamini- Hochberg method (42, 43). We used the Cochran's and Mantel–Haenszel test to adjust for variables (sex and smoking status) and calculated odds ratios (OR) with 95% of confidence interval (95% CI) (44). All possible SNP-SNP interactions were tested for single-, two-, and three-SNP interaction model and those with p-value < 0.05 are reported.
SP-A1 and SP-A2 haplotypes were assigned as described (8) and the frequency analysis was done similar to that of SNP frequencies. For haplotype estimation, we used the two-SNP haplotype estimation model to study association of haplotypes with HP patients (45). The effect of haplotype was studied in a similar way to that of the SNP-SNP interaction model in a case-control setting.
Table 1 shows the baseline characteristics of the HP patients, and exposed and non-exposed controls. Out of seventy-five HP patients, 92% (n = 70) were females compared to 30% (n = 19) of 64 exposed controls, and 36% (n = 70) of the 194 non-exposed controls (p < 0.00001). Only 19% (n = 14) of HP patients and 10% (n = 10) of exposed controls (p = NS) were current or former smokers compared to 47% (n = 91) of non-exposed controls (p = 0.00002). All HP patients demonstrated a significant reduction in FVC% and FEV1% on pulmonary function tests and hypoxemia at rest (Table 1).
The observed frequency distribution of the majority of SNPs did not deviate from Hardy-Weinberg equilibrium (data not shown). The univariate and multivariate logistic regression analysis was performed to study difference in frequencies of marker allele in HP patients compared to two control groups.
Marker allele that showed significant differences (p < 0.1) in the univariate analysis (Table 2) were included in the multivariate analysis. After multiple logistic regression analysis, the marker allele rs7316_G of the SFTPB is associated with increased risk for HP, p = 0.03, OR (95% CI) = 4.6 (1.3–22.0), whereas, male sex appears to be associated with decreased risk for HP, p < 0.01, OR = 0.02 (0.01–0.08).

Table 2. Hypersensitivity Pneumonitis (HP) vs. avian antigen exposed controls (univariate analysis).
Similarly, marker alleles that showed significant differences (p < 0.1) in the univariate analysis (Table 3) were included in the multivariate analysis. Based on the OR, marker allele (rs1130866_T of the SFTPB and rs721917_T of the SFTPD), as well as male sex and smoking appear to be associated with decreased risk of HP (Table 4).
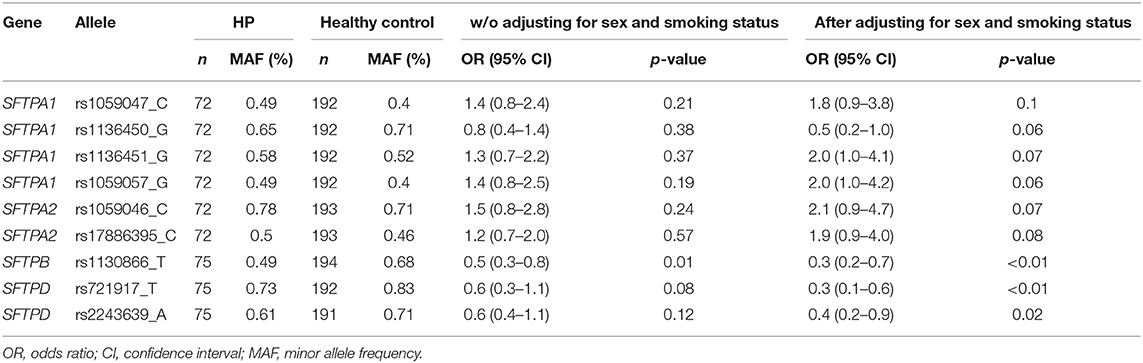
Table 3. Hypersensitivity Pneumonitis (HP) vs. non-exposed controls (univariate analysis).
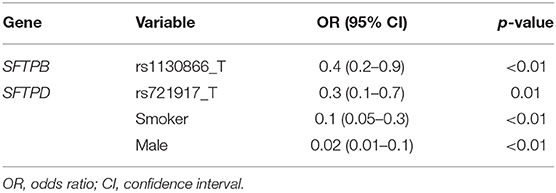
Table 4. Hypersensitivity Pneumonitis (HP) vs. non-exposed controls (multivariate analysis).
A particular SNP can have an additive (denoted as “a”), dominant (denoted as “d”) or recessive (not observed in our study) effect on the disease and the number that follows “a” or “d” indicates the position of the corresponding SNP. For example, a notation d1 x a2 x d3 means that SNPs in position 1 and 3 have dominant effect and SNP in position 2 has an additive effect on that particular interaction. These interactions could be intragenic, i.e., among SNPs of the same gene (shown in bold in the Tables), or intergenic, i.e., among SNPs of different genes. In general, each SNP exhibited either additive and/or dominant effect on HP in the single-, two-, and three-SNP model.
Since there was a statistically significant sex difference between the study groups, we adjusted for sex in the SNP analysis models. In the single- or two-SNP model, we did not observe significant association of SP genes SNPs with HP compared to exposed controls. Table 5 shows a total of 25 significant interactions associated with HP in the three-SNP model. Out of these 25 interactions, 16 were associated with increased risk for HP, p = 0.001–0.05, OR = 2.6–14.5, whereas 9 interactions were associated with lower risk for HP, p = 0.002–0.05, OR = 0.1–0.4. Out of those 25 interactions, 16 were with three dominant effects and 9 were with two dominant effects [d1 x a2 x d3 (n = 7), d1 x d2 x a3 (n = 2)]. We did not observe any significant interaction with additive effects only.
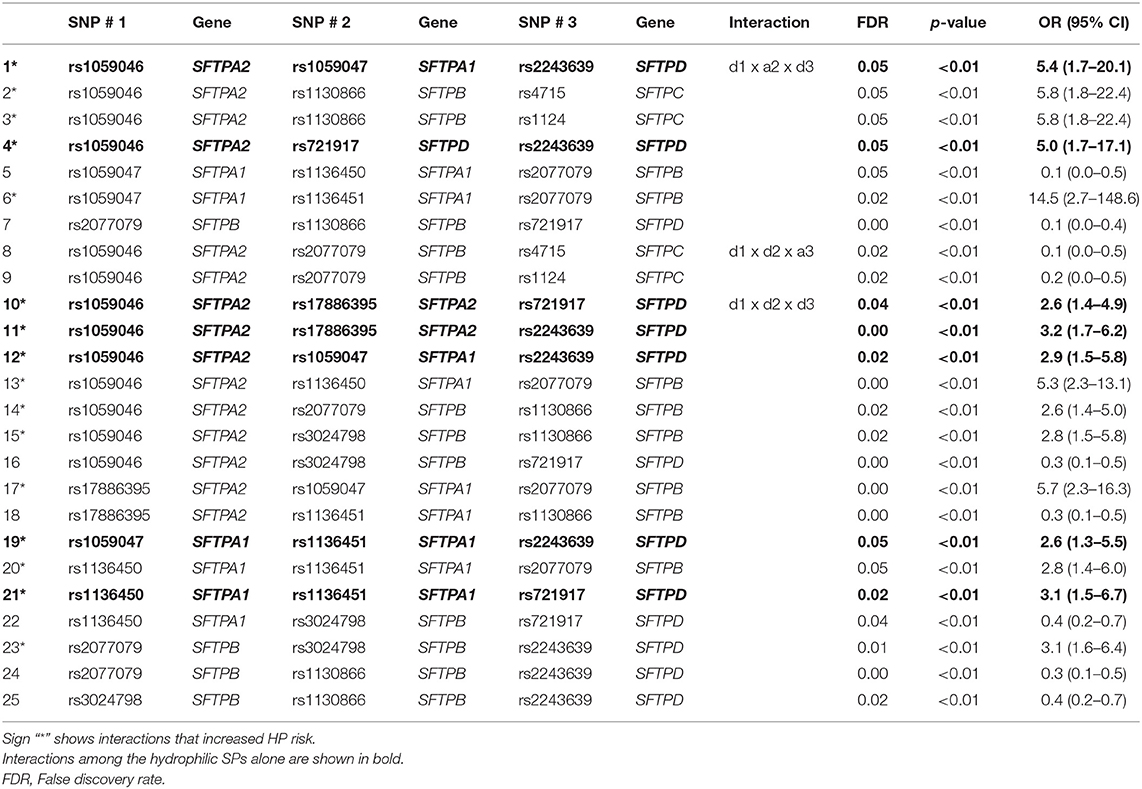
Table 5. Association of SP gene SNP interactions with HP compared to avian antigen exposed controls.
Out of the 16 interactions associated with increased HP risk, 7 were among SNPs of the hydrophilic SPs (SFTPA1, SFTPA2, and SFTPD) alone, the rest (n = 9) were among SNPs of both the hydrophilic and hydrophobic (SFTPB and SFTPC) SPs. There were no interactions among SNPs of the hydrophobic SP genes alone. Of note, all but one interaction had SNPs of either the SFTPA1 or SFTPA2 gene. The exception was the interaction among two SNPs of SFTPB (rs2077079 x rs3024798) and the rs2243639 of SFTPD, p = 0.01, OR (95% CI) = 3.1(1.6–6.4).
All interactions (n = 9) associated with lower risk of HP were among SNPs of both hydrophilic and hydrophobic SPs. Moreover, SNPs of the SFTPB constituted the majority (~45%) of the SNPs in the significant interactions associated with lower risk of HP, whereas fewer than ~30% of the combined SFTPA1 and SFTPA2 SNPs in the interactions were associated with lower risk of HP.
We observed a statistically significant difference of sex and smoking status between the groups, therefore for SNP analysis, we adjusted for both of these covariates. In the single SNP model, we observed that rs1136451 of the SFTPA1 was associated with increased HP risk, p = 0.02, OR = 11.4 (2.3–57.9), whereas rs1136450 of SFTPA1 and rs1130866 of SFTPB were associated with lower risk of HP, p = 0.02, OR = 0.2 (0.0–0.6) compared to non-exposed controls. Each of these SNPs exhibited an additive effect on HP risk, e.g., in rs1136451 (A/G), risk allele “G” exhibited an additive (GG>GA>AA) effect on increased HP risk rather than the recessive (GG>GA=AA) or dominant (GG=GA>AA) effect. We did not observe any significant interactions associated with HP in the two-SNP model. We observed a total of 97 interactions associated with HP in the three-SNP model. Out of the 97 interactions, 29 were associated with increased HP risk, p = 0.00009–0.05, OR = 1.9–13.3 (1.2–128.8), and the remaining 68 were associated with lower risk for HP, p = 0.0001–0.05, OR = 0.1–0.6 (0.0–0.9). Of the 97 interactions, (a) 3 interactions with additive effects (a1 x a2 x a3, no dominant effect of any SNP), (b) 8 interactions had one dominant effect (a1 x a2 x d3), (c) 51 interactions had two dominant effects [a1 x d2 x d3 (n = 9), d1 x a2 x d3 (n = 12), d1x d2 x a3 (n = 30)], and (d) 35 interactions had three dominant effects (d1 x d2 x d3) as shown in Supplementary Tables 2, 3.
Of the 29 interactions associated with increased HP risk, we observed (a) one intragenic interaction among SNPs of the SFTPA1 (rs1059047 x rs1136450 x rs1136451), where each SNP exhibited a dominant effect, p = 0.03, OR = 1.9 (1.2–3) (shown in Figure 1), and the remaining 28 interactions were intergenic. (b) out of the 28 intergenic interactions, seven were among SNPs of the hydrophilic SPs (SFTPA1, SFTPA2, and SFTPD) alone, and the rest were among SNPs of both hydrophobic and hydrophilic SPs. (c) all but four of the significant intergenic interactions had SNPs of either SFTPA1 or SFTPA2.
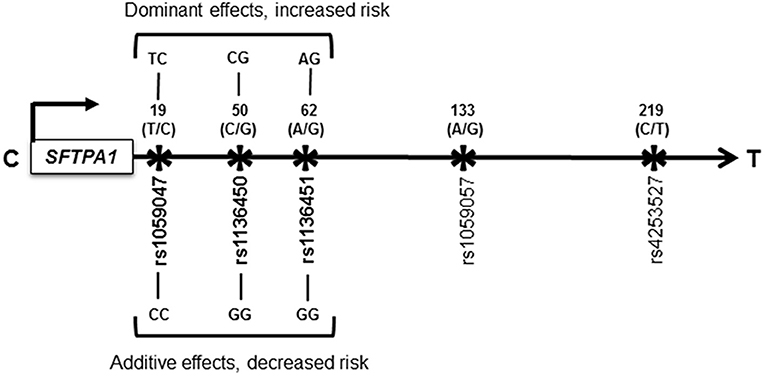
Figure 1. Schematic representation of the SFTPA1 single nucleotide polymorphisms (SNPs). All the studied SNPs of the SFTPA1 are shown. The relative location of the gene is shown from centromere (C) to telomere (T) and the arrow indicates transcriptional orientation. The number above the black arrow indicates the amino acid (AA) number of the precursor molecule and the corresponding nucleotide change shown in parenthesis. The SNP id is shown below the black arrow. The SNPs (rs1059047 x rs1136450 x rs1136451) of the SFTPA1 involved in an intragenic interaction in the three-SNP model are shown in bold font and are associated with HP risk compared to non-exposed controls. The change for AA at the codon 19 is Val/Ala corresponding to the T/C alleles, respectively; for AA50 is Val/Leu corresponding to C/G alleles. The SNP at the codon 62 does not change the encoded amino acid. Of note, the physical location of the SNPs in this interaction is very close to each other as shown in Figure. On the basis of odds ratio, the interaction (rs1059047 x rs1136450 x rs1136451) exhibiting a dominant effect is associated with increased risk of HP (d x d x d). The dominant genotype for each SNP is shown above the black arrow. On the other hand, the same interaction is associated with decreased risk of HP when each SNP exhibited an additive effect (a x a x a). The additive genotype for each SNP is shown below the black arrow.
Of the 68 interactions associated with lower risk of HP, we observed (a) two intragenic interactions: (i) among SNPs (rs1059047 x rs1136450 x rs1136451) of the SFTPA1, which were the same as the ones with increased HP risk. However, in this case, each SNP exhibited an additive effect, p = 0.05, OR = 0.1 (0–0.5) whereas when this interaction was associated with increased risk, each SNP exhibited a dominant effect (shown in Figure 1), and (ii) among SNPs of the SFTPB (rs2077079 x rs3024798 x rs1130866 exhibiting dominant x dominant x additive effect, respectively), p = 0.001, OR = 0.2 (0.1–0.5). (b) the remaining 66 intergenic interactions included; (i) 12 interactions among SNPs of the hydrophilic SPs alone, (ii) 3 interactions among SNPs of the hydrophobic SPs alone, and (iii) the rest (n = 51) were among SNPs of both hydrophobic and hydrophilic SPs.
We observed seven interactions associated with HP that were found to be in common in two separate comparisons, where cases were compared to either antigen exposed or to non-exposed controls (Figure 2). Out of the seven interactions: (a) five interactions were associated with lower risk of HP compared to antigen exposed and non-exposed controls (Panel A) and these interactions were among SNPs of both hydrophilic and hydrophobic SPs, p = 0.002–0.04, OR = 0.4–0.5 (0.3–0.8); (b) one was associated with increased risk of HP (Panel B – black dotted arrows) and this interaction was among SNPs of the hydrophilic SPs (rs1059046 x rs1059047 x rs2243639, SFTPA2 x SFTPA1 x SFTPD, d1 x a2 x d3), p = 0.009–0.05, OR = 3.5–5.4 (1.7–20.1); (c) In Panel B, one (red arrows) interaction (rs2077079 x rs3024798 x rs2243639, SFTPB x SFTPB x SFTPD, d1 x d2 x d3) was associated with increased risk of HP [p = 0.008, OR = 3.1 (1.6–6.4)] compared to antigen exposed controls, however, the same interaction was associated with lower risk of HP [p = 0.002, OR = 0.4 (0.3–0.7)] when compared to non-exposed controls.
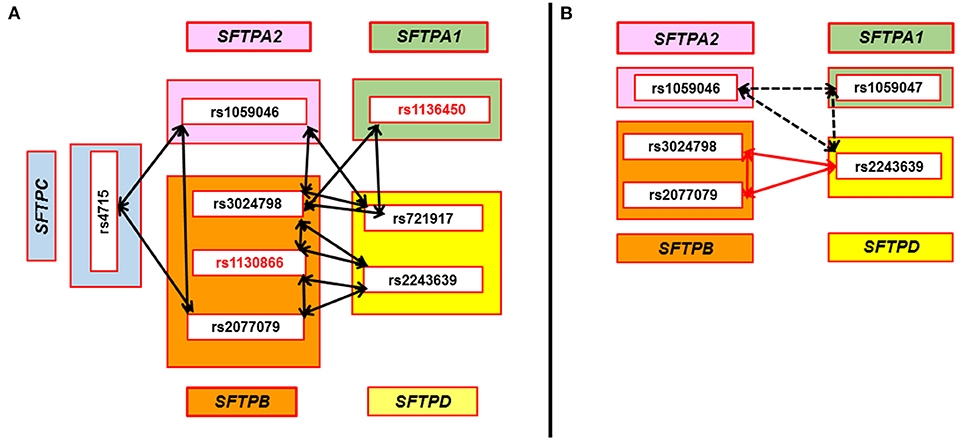
Figure 2. SNP-SNP interactions associated with HP compared to avian antigen exposed and non-exposed controls in the three-SNP model. Double head arrows represent common SNP-SNP interactions associated with HP and found to be significant in two sets of comparisons (HP vs. avian antigen exposed controls and HP vs. non-antigen exposed controls) in the three-SNP model. The SNPs of surfactant protein genes SFTPA1, SFTPA2, SFTPB, SFTPC, and SFTPD encoding SP-A1, SP-A2, SP-B, SP-C, and SP-D, respectively, are depicted within the green, pink, orange, blue, and yellow rectangles, respectively. (A) shows five significant interactions that were associated with lower risk of HP compared to antigen exposed and non-exposed controls. All these interactions were among SNPs of both hydrophobic and hydrophilic SPs and involve at least one SNP of SFTPB (n = 5) that interacts with SNPs of SFTPD (n = 4), SFTPA1 (n = 1), SFTPA2 (n = 2), and SFTPC (n = 1), p = 0.002–0.04, OR = 0.4–0.5 (0.3–0.8). Two cases of the three-SNP model involved two SFTPB SNPs interacting (rs3024798 and rs1130866, or rs1130866 and rs2077079) with the same SFTPD SNP (rs2243639). SNPs associated with lower risk of HP compared to non-exposed controls in the single-SNP model are shown in red bold font (rs1130866 of SFTPB and rs1136450 of SFTPA1). (B) shows two significant interactions associated with increased risk of HP compared to antigen exposed controls. One of the interactions, shown by the black dash double head arrow, is among SNPs of the hydrophilic SPs alone and was associated with increased risk of HP compared to antigen exposed and non-exposed controls, p = 0.009–0.05, OR = 3.5–5.4 (1.7–20.1). However, the other interaction, shown by red double head arrow among SNPs of the SFTPB and SFTPD, was associated with increased risk of HP compared to antigen exposed controls [p = 0.008, OR = 3.1 (1.6–6.4)]. Of interest, the same interaction was associated with lower risk of HP compared to non-exposed controls [p = 0.002, OR = 0.4 (0.3–0.7)].
The univariate analysis (Supplementary Table 4) showed that the frequency of several haplotypes of the SFTPA1 and SFTPA2 differed between HP patients vs. each of the two control groups (p < 0.1). In the multivariate analysis, the 1A3 of the SFTPA2 and male sex were associated with decreased risk of HP compared to avian antigen control, p < 0.05, OR = 0.1 (1A3) and OR = 0.03 (male), whereas smoking and male sex, but none of the SFTPA1 and SFTPA2 haplotypes, appeared to decrease risk of HP compared to non-exposed healthy control, p < 0.01, OR = 0.13 (smoking) and OR = 0.03 (male).
Using the two-SNP haplotype model, Figure 3 shows associations of haplotypes with HP compared to avian antigen exposed and non-exposed controls. Compared to avian antigen exposed controls, six haplotypes of the SFTPA1, SFTPA2, and SFTPB were associated with HP. Of the six haplotypes, four were associated with decreased risk of HP (OR = 0.02–0.3) and the remaining were associated with increased risk of HP (OR = 4.26–13.19), Supplementary Table 5. As shown in Figure 3, the haplotype “CC” of the SFTPB (rs2077079 (A/C) x rs3024798 (C/A)) is associated with decreased risk of HP and exhibited an additive, OR = 0.02 (0.002–0.2) as well as a dominant effect, OR = 0.12 (0.07–0.39). In this example, each parent can transmit the risk haplotype in four forms: AC, AA, CC, and CA. The additive effect of the risk haplotype “CC” means that the presence of two copies of “CC/CC” decreases the risk of HP compared to the combination of two copies of any non-risk haplotypes (AC/AC, AA/AA, and CA/CA). Whereas the dominant effect of the same haplotype “CC” means that the presence of the risk haplotype “CC” in combination with any other haplotype (AC, AA, and CA) decreases the risk of HP compared to the presence of two copies of CC or any other combination of the non-risk haplotypes (AC/AC, AC/AA, AC/CA, AA/AA, AA/CA, and CA/CA). The dominant effect of a risk haplotype may have a non-linear interaction effect in which the combination of the risk haplotype “CC” with other non-risk haplotype (AC, AA, and CA) may produce a larger effect on the disease risk than the sum of two copies of the risk haplotype “CC.” This complexity (non-linear) of interactions is our challenge for the study of human disease. Unexpected and not readily understood phenomena may occur, and this finding is one of the examples of this phenomenon. In other words, 1 + 1 > 2 in this risk haplotype. The haplotypes “CC” of the SFTPA2 (rs1059046 x rs17886395), OR = 0.07 (0.02–0.26) and “AA” of the SFTPA1 (rs1136451 x rs1059057), OR = 0.30 (0.12–0.78), exhibited a dominant effect on decreased HP risk. The haplotypes “AC” of the SFTPA1 (rs1059057 x rs4253527), OR = 13.19 (4.44–39.17) and “CA” of the SFTPB (rs1130866 x rs7316), OR = 13.19 (4.44–39.17), exhibited a dominant effect on increased HP risk (Figure 3, red arrows).
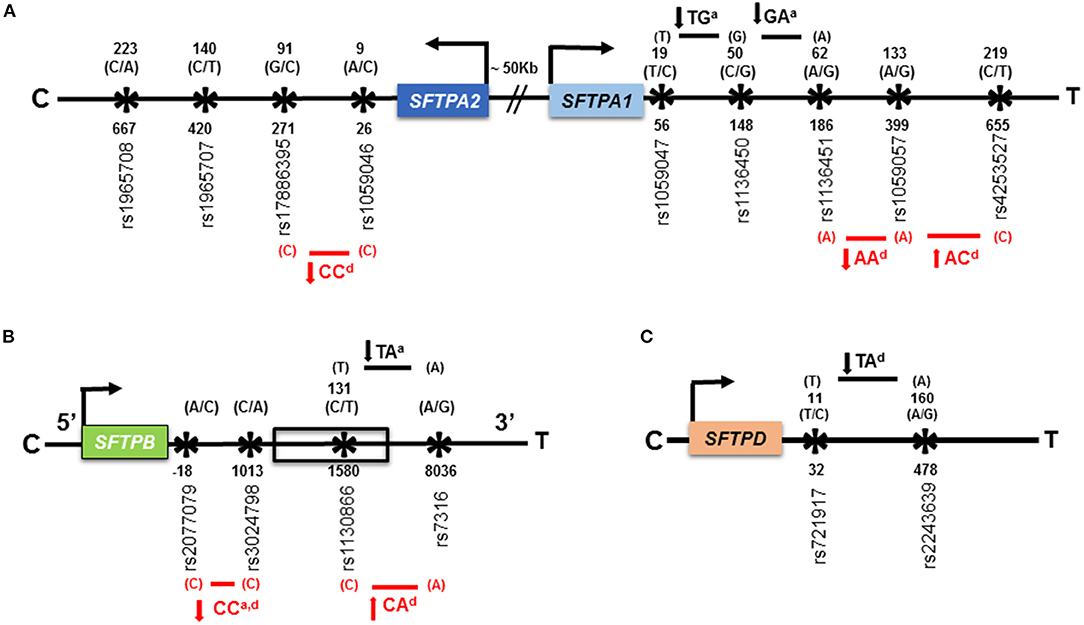
Figure 3. Association of haplotypes of the SFTPA1, SFTPA2, SFTPB, and SFTPD genes with HP compared to avian antigen exposed and non-exposed controls in the two-SNP haplotype estimation model. (A) is a schematic representation of SNPs of the SFTPA1 and SFTPA2 genes. The distance between the genes is ~50 kb shown with sign “//.” The studied SNPs are located within exons. (B) is a schematic representation of the SFTPB gene shown in 5' to 3' UTR direction. Only rs1130866 of the SFTPB shown in box, is located within an exon and the corresponding amino acid (AA) is shown above that. The SFTPB (1) rs2077079 is located 10 nucleotides downstream of TATAA box, in the 5′ regulatory region; (2) rs3024798 is located in the intron; and (3) rs7316 is located in the 3′UTR. Thus, no AA is shown for these SNPs. (C) is a schematic representation of SNPs of the SFTPD gene. The studied SNPs are located within exons. The gene direction is shown from centromere (C) to telomere (T) and the arrow above the color box indicates transcriptional orientation. The number above the black arrow indicates the AA number and the corresponding nucleotide change for that particular SNP is shown in parenthesis. The numbering of AAs in SFTPA2, SFTPA1, and SFTPB is based on the sequence of the precursor molecule, whereas, it is based on the mature protein (i.e., minus the signal peptide) in SFTPD. The numbers below the black arrow indicate the nucleotide number and the corresponding SNP ids. The transmitted haplotypes and nucleotide changes are shown in bold. The haplotypes associated with HP risk compared to avian antigen exposed and non-exposed controls are shown in red font with red line and black font with black line, respectively. The direction of the solid arrows besides haplotypes indicates increased or decreased HP risk compared to control groups. The superscript “a” and “d” after a given haplotype indicates additive and dominant effect, respectively, of that particular haplotype.
Compared to non-exposed controls, all four significant susceptibility haplotypes of the SFTPA1, SFTPB, and SFTPD were associated with decreased risk of HP, OR = 0.15–0.35, Supplementary Table 6 and Figure 3 (black arrows). The haplotypes “TG” and “GA” of the SFTPA1, rs1059047 x rs1136450 and rs1136450 x rs1136451, respectively, exhibited an additive effect, OR = 0.15 (0.04–0.54). The haplotype “TA” of the SFTPB (rs1130866 x rs7316) exhibited an additive effect, OR = 0.17 (0.05–0.64), whereas, the haplotype “TA” of the SFTPD (rs721917 x rs2243639) exhibited a dominant effect, OR = 0.35 (0.18–0.66) on decreasing the risk of HP.
Hypersensitivity Pneumonitis is an interstitial lung disease caused by an abnormal immune response to antigen exposure and most likely due to complex interactions between environmental and genetic factors (1, 2). Surfactant proteins play an important role in normal lung function as well as innate immunity and host defense (6, 46), and alterations in their function is central to several pulmonary diseases. Several genetic variants have been identified for SFTPA1, SFTPA2, SFTPB, SFTPC, and SFTPD that are associated with pulmonary diseases (12, 47). Taking advantage of the homogeneity of the Mexican population, in the current study, we tested the hypothesis that SP genetic variants are associated with susceptibility of HP. Our results showed that (a) in the multivariate analysis, the rs7316_G of the SFTPB and male sex were associated with increased and decreased risk of HP, respectively, compared to antigen exposed controls, whereas the rs1130866_T of the SFTPB, rs721917_T of the SFTPD, male sex, and smoking were associated with decreased risk of HP compared to non-exposed controls; (b) in the single-SNP model, the rs1136451 of SFTPA1 was associated with increased HP risk whereas the rs1136450 of the SFTPA1 and the rs1130866 of the SFTPB, each associated with lower risk and exhibited an additive effect on HP compared to non-exposed controls; (c) in the three-SNP model, when HP patients were compared to antigen exposed and non-exposed controls, the majority of SNP-SNP interactions associated with increased risk of HP involved SNPs of the hydrophilic SPs alone, whereas, the majority of the interactions with hydrophobic SPs were associated with lower risk of HP; (d) based on OR, the 1A3 and “CC” haplotypes of the SFTPA2, each associated with decreased risk and certain haplotypes of the SFTPA1 and SFTPB were associated with increased or decreased risk to HP compared to antigen exposed controls, whereas, certain haplotypes of SFTPA1, SFTPB, and SFTPD, each associated with decreased risk of HP compared to non-exposed controls. Thus, the SP genetic marker alleles, SNPs and haplotypes, either alone and/or via their interactions may contribute to the development of HP. These results also indicate that the use of multiple markers may better predict the risk of disease and be used as diagnostic and/or prognostic markers of HP.
For the current study, we used two statistical approaches: (1) a traditional logistic regression analysis, and (2) a newer SNP-SNP interaction models in case-control settings. Moreover, to avoid overestimation of an association of SNP and SNP-SNP interactions for HP patients and also to account for the heterogeneity secondary to genetic differences and environmental factors that could affect the case-control study, we used two distinct controls: (1) asymptomatic antigen exposed and (2) non-exposed healthy controls. The antigen exposed controls who did not develop disease can be classified as a resistant population and non-exposed controls represent the general population where the history of exposure is not known. Of note, 30–40% of confirmed HP patients do not have a history of antigen exposure (1) making this study clinically useful.
The regression analysis revealed a protective association of male sex and smoking with HP, although the duration and/or amount of smoking was unknown. Previous human and animal studies have shown a protective role of smoking in HP under similar antigen risk exposure (48, 49). Smoking has a paradoxical role in HP in the sense that HP develops more frequently in non-smokers than in smokers but when HP occurs in smokers, the outcome is poor (50). Similar to our study, a higher incidence of HP has been reported in females in a recent epidemiological study (49). Thus, modifiable and non-modifiable environmental factors may change the susceptibility of complex diseases. Adjusting for these significant variables and the use of two different statistical approaches could provide confidence in the observations made.
The rs7316_G of the SFTPB is shown to associate with increase HP risk compared to antigen exposed controls only in the multivariate regression analysis. Although the rs7316 is located in the 3′ untranslated region that frequently acts as a regulatory region affecting mRNA stability, the functional significance of this variant is unclear (51). In the single-SNP model and the univariate analysis, we found three SNPS, two of the SFTPA1 and one of the SFTPB, each associated with HP compared to non-exposed controls. The rs1136451_G of the SFTPA1 associated with increased risk of HP. This SNP at codon 62 of SP-A1 does not result in an amino acid change (proline). Moreover, the C allele of this SNP is previously shown to associate with increased risk of COPD (14) and decreased risk of TB (21) in Mexicans. Of note, the G allele of the same SNP was found to associate with decreased risk of COPD in a Chinese population (52). Though the biological effect of the variant is not known, collectively these data indicate that this variant is differentially associated with pulmonary diseases in various ethnic groups. Moreover, we observed that the rs1136450_G allele of the SFTPA1 is associated with decreased risk of HP compared to general controls. This SNP is known to change the encoded amino acid Leucine to Valine at codon 50, but its effect on structure, function and/or stability of the SP-A is not known. Though there are some similarities in the pathogenesis of the two types of interstitial lung diseases (IPF and HP), the C allele of rs1136450 is shown to associate with six-times higher odds of developing IPF in the Mexican population compared to general controls (13), but the same allele is associated with a decreased risk of HP. Whether this along with other markers could be used to differentiate between interstitial lung diseases in Mexicans remains to be determined. A previous study has shown a 3-fold increase in SP-A in BAL of HP patients (34), however, the question remains whether increased SP-A causes or contributes to HP or it is the result of the disease process. The level of SP-A1 and SP-A2 differs among individual as a function of age and lung health status (e.g., heathy vs. cystic fibrosis, culture positive vs. culture negative), as shown by differences in the protein ration of SP-A1 to total SP-A in human BAL samples (53). In addition, several studies have shown that SPs levels are influence by age, health, smoking status, lung health disease as well as by genetic factors (54–56), however, very few studies have correlated genetic polymorphisms with serum levels (57). For the current study, we did not measure the level of SPs in BAL; therefore, the effect of SP SNPs on surfactant protein concentration is unknown in healthy and/or in HP subjects. The N-terminal segment, the collagen-like region and the neck domain but not the carbohydrate recognition domain participate in SP-A oligomerization (6). Of interest, all the significant SNPs of the SFTPA1 and SFTPA2 associated with HP are located within regions that participate in oligomerization, whereas SNPs (rs1965707, rs1965708, and rs4253527) located in the carbohydrate recognition domain are not associated with HP. Currently it is unknown if and how any of the significant SNPs alone or in combination contribute to SP-A oligomerization.
The rs1130866_T allele of the SFTPB was also associated with lower risk of HP. This SNP is a missense mutation that changes the encoded amino acid Threonine to Isoleucine and eliminates an N-linked glycosylation site (58). The T allele of rs1130866 is protective against neonatal RDS (20) and systemic sclerosis associated interstitial lung disease in a Japanese population (59). On the other hand, the C allele that has the N-linked glycosylation site is associated with increased risk of COPD (15), acute respiratory distress syndrome (10), and IPF in Mexicans (13). The presence of N-linked glycosylation may interfere with SP-B processing and protein folding in disease conditions, hence the T allele, without the N-linked glycosylation somehow protects against HP. This has been shown in a transgenic mouse model of pneumonia and sepsis, where the C allele of rs1130866 of human SP-B resulted in a decreased number of lamellar bodies, SP-B concentration, and increase surface tension compared to the T allele of rs1130866 and compared to the wild type mice (60).
In summary, the fact that the marker alleles (rs1130866_T, rs1136450_G, and rs1136451_G) identified by logistic regression analysis and SNPs identified (see below) by the single-SNP model (i.e., the additive effect of the T, G, and G alleles of the rs1130866, rs1136450, and rs1136451, respectively) are identical, based on the calculated OR (regardless of the statistical approaches used), associate with HP provide confidence that these associations are true rather than spurious.
It is known that a genetic variant in the presence of another variant can alter the susceptibility of an individual to certain diseases (61). By studying SNP-SNP interactions, we can better understand the role of genetics in complex diseases such as HP. It is possible that networks of additive and/or epistatic interactions among surfactant protein genetic variants may alter functional capabilities of certain SPs, more importantly, alveolar integrity and/or host defense at the cellular, molecular or tissue level (61). In the three-SNP model, we observed 96 interactions (HP vs. non-exposed) compared to 25 interactions (HP vs. avian antigen exposed), with only 7 interactions being in common between them. The observed difference in the number of interactions could be due to the difference in the patient population, statistical approach and/or sample size. For the current study, we enrolled HP patients, exposed and non-exposed controls from a homogeneous Mexican population and used similar statistical approach to compare HP patients with two different control groups. Therefore, the observed difference in the number of interactions is likely due to the sample size difference.
We observed 44 interactions associated with avian-antigen controls compared to non-exposed controls in a three-SNP model after adjusting for smoking status (data not shown). This indicates that although the demographics of the two control groups (antigen-exposed and non-exposed) appear identical except for the smoking status (Table 1), genetically the two control groups differ. This difference between the two control groups may be in part due to the fact that 25% of the antigen exposed controls were relatives to the cases or other. However, despite genetic differences between the two control groups, we observed seven interactions associated with HP that were found to be in common in the two separate comparisons, where HP cases were compared to either antigen exposed or to non-exposed controls (Figure 2). The association of these interactions with HP is likely robust given the two different controls and considering the baseline genetic differences between the two control groups and should be investigated further in the future.
The complex three SNP model identified various significant interactions among the hydrophilic and hydrophobic SPs associated with HP after adjusting for sex and smoking status of study participants. Interestingly, we noted that one of the common interactions, involving the SFTPB (rs3024798 and rs2077079) and the rs2243639 of the SFTPD, was associated with increased risk of HP compared to the homogeneous antigen exposed control group, however, the same interaction was associated with decreased risk of HP compared to the heterogeneous non-exposed control group, in which the history of exposure and living conditions were unknown. Heterogeneity due to differences in genetic background and/or environmental conditions is probably one of the sources of apparent discrepancies in case-control study results. Therefore, the differences indicate that caution should be exercised in the definition of cases and control groups and that findings should be interpreted within the context of experimental design.
The intragenic interaction (rs1059047 x rs1136450 x rs1136451) of the SFTPA1 is associated with increased or decreased risk of HP depending on dominant or additive effects of each SNP, respectively, in the three-SNP model compared to non-exposed controls (Figure 1). Although the study population remained the same, the susceptibility to disease changes based on the effect the particular SNP exhibits in a particular interaction. It is possible that either too much or too little of a gene product and their interactions could lead to either over or under function, of genes in a disease state (62). Of note, the concentration and biochemical properties of surfactant proteins are altered in HP patients compared to controls (32–34). This may explain the change in HP risk based on the effect of a particular SNP in that interaction. Moreover, the majority of previous associations studies have assumed only additive effects of SNPs, without considering non-additive effects (63). By studying non-additive effects (i.e., dominant or recessive) of SNPs and their interactions in the present study, we identified associations that could change the disease risk as shown in this SFTPA1 intragenic SNP interaction. Conversely, the same SFTPA1 intragenic SNP interaction (rs1059047 x rs1136450 x rs1136451) with dominant effects of each SNP was associated with decreased risk of pediatric acute respiratory failure (41). The clinical outcome of a quantitative or qualitative imbalance of a given gene product in a given microenvironment, may differ among individuals. The fact that enrolled subjects in that study (41) were predominantly white children aged less than 2 years and admitted with viral infections, the contrasting effect of the same interaction on disease risk is not surprising.
Interactions of the SFTPB and SFTPD SNPs had a variable susceptibility to HP (Figure 2). For example, the rs1130866 of the SFTPB is associated with lower risk of HP in the single-SNP model. Interactions of that particular SNP with other SFTPB SNPs (rs3024798 and rs2077079); and with the rs2243639 of the SFTPD associated with a decreased risk of HP. These findings likely highlight the protective role of rs1130866 of the SFTPB in HP. However, the interaction of two other SNPs of the SFTPB (rs3024798 and rs2077079) with the rs2243639 of the SFTPD was associated with increased risk of HP. In addition, the rs1136451 of the SFTPA1 is associated with increased risk of HP in the single-SNP model but interactions of the same SNP with other hydrophobic SPs (SFTPB and SFTPC) SNPs were associated with decreased risk of HP in the three-SNP model. As shown in the current study, a given SNP could change the susceptibility of a disease depending on its interactions with other SNPs, and this highlights the importance of studying SNP-SNP interactions rather than a single SNP association to fully understand the role of genetics in complex diseases such as HP.
In general, the majority of interactions associated with increased HP risk involved SNPs of either the SFTPA1 and/or SFTPA2, whereas their interactions with the hydrophobic SPs (SFTPB and SFTPC) were associated with a decreased risk of HP. The significance for the SFTPAs association may be due to the differential effect of SFTPA gene variants in lung function parameters (29), which, in the case of HP, is also altered due to antigen-induced lung inflammation. Based on these, we speculate that the presence of the hydrophilic SP genetic variants, particularly of SFTPA1 and SFTPA2, in a susceptible population contribute to a dysfunction/poor functioning of the innate immune response to avian antigen exposure, alters lung function and this in turn may contribute to the pathogenesis of HP. However, interactions of these SNPs with the hydrophobic SP gene variants, particularly with SFTPB, may confer protection against HP. SP-B profoundly influences intracellular processing, secretion, and the pool size of surfactant (46). SP-A and SP-B have an interactive role in maintaining surface activity in vitro, and both are essential components of tubular myelin, an extracellular form of surfactant (64, 65). Together, these interactions among SNPs of SFTPA and SFTPB may alter the level and/or properties of SPs in HP patients that may provide protection against dysregulated inflammation. Moreover, interactions of the SFTPA with SFTPB have been previously shown to change susceptibility to neonatal RDS based on ethnic background, where certain variants increased risk of RDS in white neonates compared to black neonates (20). Nonetheless, the impact of these actual gene-gene interactions on levels and biophysical/biochemical properties of SPs need to be studied in biological experiments.
The study of haplotypes (SNPs that are inherited together) is shown to be a powerful tool to identify associations in complex diseases such as HP (66). The 1A3 and “CC” haplotypes of the SFTPA2 were associated with decreased risk of HP compared to avian antigen controls. Moreover, the 1A3 haplotype is shown to associate with increased risk of TB in a Mexican population compared to healthy controls. Of note, the 1A3 differs from most other SP-A2 haplotypes at amino acid 223 (Lys instead of Gly) located within carbohydrate recognition domain that is responsible for recognizing, binding and clearing foreign materials entering into the lungs (9). Previous studies of SP-A1 and SP-A2 variants using the transgenic mouse model showed that the 1A3 of the SFTPA2 was associated with better survival (28) and exhibited significantly higher lung function compared to other SP-A1 variants (29). Thus, findings of the current study, although it is an association study, are consistent with our previous animal data where a functional outcome was measured. How this haplotype alters the functional capabilities of SP-A remains to be determined, particularly in response to a potentially dysregulated inflammation and infection. It would be interesting, guided by the SNP-SNP interaction data, to generate additional SP-A cDNAs to use them to either generate stably transfected cell lines or transgenic mice for functional and regulatory studies as done with some of the common SP-A variants (65, 67). Similar to the SNP-SNP interaction model, a haplotype estimation model showed increased or decreased risk of HP based on the effect (i.e., dominant and recessive) of that particular haplotype. It is important to note that the significant haplotypes associated with HP are located very close to each other on the gene as shown in Figure 3 and biologically have a higher chance of transmitted together.
The strengths of the study includes, (1) some of the susceptibility SNPs and SNP-SNP interactions associated with HP were the same by two different statistical approaches as well as by multiple comparisons adjusting for variables; (2) use of physiologically and biologically relevant SP SNPs, which is prerequisite for the SNP-SNP interaction model to study complex diseases (39). The limitations of the present study are the moderate sample size and the homogeneous patient population. A previous simulation study of SNP-SNP interaction indicated that a sample size of at least 100 in both case and control groups is needed to detect all the possible interactions. The moderate sample size of the current study may have resulted in under-reporting of causative SNP-SNP interactions and haplotypes, since the power to detect small differences is limited by the sample size, particularly for comparison of HP patients (n = 75) with antigen-exposed controls (n = 64). Furthermore, the findings of the present study may not be generalized in heterogeneous non-Hispanic patients. These associations should be strengthened and validated by increasing the sample size and replicating the findings in other groups of heterogeneous non-Hispanic HP patients. In addition, we did not measure SP expression/level in BAL of HP patients, therefore, the impact of these SNPs on SP expression, and in turn, on HP is unknown.
In summary, this is the first study showing association of SP SNPs and haplotypes with HP using two different statistical approaches in a Mexican population. The rs1136451 of the SFTPA1 is associated with increased risk, whereas, the rs1136450 and the rs1130866 of the SFTPA1 and SFTPB, respectively, are associated with decreased risk of HP compared to non-exposed controls using logistic regression analysis and a single-SNP model. Moreover, SNPs that are significantly associated with HP in the multivariate analysis also remained significant in the three-SNP interaction model after adjusting for smoking and sex. The rs1965707, rs1965708, and rs4253527 located within the carbohydrate recognition domain of the SFTPA1 and SFTPA2 were not associated with HP. SNPs of the SFTPA1 and SFTPA2 were overrepresented in interactions associated with increased HP risk, and their interactions with SNPs of the hydrophobic SPs for the most part associated with decreased HP risk. These observations indicate that specific SP genetic variants play role in the susceptibility of Mexicans to HP. This study focuses on complex and unique interactions of the SP SNPs with HP and gives valuable information of possible functional role of surfactant proteins in innate immunity against antigens as well as in the pathogenesis of HP. This knowledge may be useful in specific marker development for diagnosis of HP.
The data that support the findings of this study are available from the corresponding author, Chintan K. Gandhi, upon reasonable request.
The studies involving human participants were reviewed and approved by National Institute of Respiratory Diseases (INER). The patients/participants provided their written informed consent to participate in this study.
CG: data curation. LY, CC, CF, SZ, RW, and CG: formal analysis. JF: funding acquisition. MS, IB-R, AP, and JF: resources. RW, MS, AP, and JF: supervision and writing – review & editing. CG, SA, and JF: writing – original draft. All authors read and approved the final manuscript.
This work was supported by NIH HL34788 to JF.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank the study participants. We thank Susan DiAngelo for her technical assistance in genotyping samples.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2020.588404/full#supplementary-material
HP, Hypersensitivity Pneumonitis; SFTPA1, gene encoding SP-A1; SFTPA2, gene encoding SP-A2; SFTPB, gene encoding SP-B; SFTPC, gene encoding SP-C; SFTPD, gene encoding SP-D; SNPs, Single nucleotide polymorphisms; IPF, Interstitial pulmonary fibrosis; COPD, Chronic obstructive pulmonary disease; RDS, Respiratory distress syndrome; OR, Odds ratio.
1. Selman M, Pardo A, Talmadge E, King J. Hypersensitivity Pneumonitis. Am J Respir Crit Care Med. (2012) 186:314–24. doi: 10.1164/rccm.201203-0513CI
2. Vasakova M, Selman M, Morell F, Sterclova M, Molina-Molina M, Raghu G. Hypersensitivity pneumonitis: current concepts of pathogenesis and potential targets for treatment. Am J Respir Crit Care Med. (2019) 200:301–8. doi: 10.1164/rccm.201903-0541PP
3. Selman M, Lacasse Y, Pardo A, Cormier Y. Hypersensitivity pneumonitis caused by fungi. Proc Am Thorac Soc. (2010) 7:229–36. doi: 10.1513/pats.200906-041AL
4. Serrano AG, Perez-Gil J. Protein-lipid interactions and surface activity in the pulmonary surfactant system. Chem Phys Lipids. (2006) 141:105–18. doi: 10.1016/j.chemphyslip.2006.02.017
5. Wright JR. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol. (2005) 5:58–68. doi: 10.1038/nri1528
6. Kishore U, Greenhough TJ, Waters P, Shrive AK, Ghai R, Kamran MF, et al. Surfactant proteins SP-A and SP-D: structure, function and receptors. Mol Immunol. (2006) 43:1293–315. doi: 10.1016/j.molimm.2005.08.004
7. Hoover RR, Floros J. Organization of the human SP-A and SP-D loci at 10q22-q23. Physical and radiation hybrid mapping reveal gene order and orientation. Am J Respir Cell Mol Biol. (1998) 18:353–62. doi: 10.1165/ajrcmb.18.3.3035
8. DiAngelo S, Lin Z, Wang G, Phillips S, Ramet M, Luo J, et al. Novel, non-radioactive, simple and multiplex PCR-cRFLP methods for genotyping human SP-A and SP-D marker alleles. Dis Markers. (1999) 15:269–81.
9. Floros J, Phelps D. Pulmonary surfactant. In: Yaksh TL, Lynch III C, Zapol WM, Maze M, Biebuyck JF, Saidman LJ, editors. Anesthesia: Biologic Foundations. Philadelphia, PA: Lippincott-Raven (1997). p. 1259–79.
10. Lin Z, Pearson C, Chinchilli V, Pietschmann SM, Luo J, Pison U, et al. Polymorphisms of human SP-A, SP-B, and SP-D genes: association of SP-B Thr131Ile with ARDS. Clin Genet. (2000) 58:181–91. doi: 10.1034/j.1399-0004.2000.580305.x
11. Wert SE, Whitsett JA, Nogee LM. Genetic disorders of surfactant dysfunction. Pediatr Dev Pathol. (2009) 12:253–74. doi: 10.2350/09-01-0586.1
12. Silveyra P, Floros J. Genetic variant associations of human SP-A and SP-D with acute and chronic lung injury. Front Biosci.(2012) 17:407–29. doi: 10.2741/3935
13. Selman M, Lin HM, Montano M, Jenkins AL, Estrada A, Lin Z, et al. Surfactant protein A and B genetic variants predispose to idiopathic pulmonary fibrosis. Hum Genet. (2003) 113:542–50. doi: 10.1007/s00439-003-1015-4
14. Guo X, Lin HM, Lin Z, Montano M, Sansores R, Wang G, et al. Surfactant protein gene A, B, and D marker alleles in chronic obstructive pulmonary disease of a Mexican population. Eur Respir J. (2001) 18:482–90. doi: 10.1183/09031936.01.00043401
15. Seifart C, Plagens A, Brodje D, Muller B, von Wichert P, Floros J. Surfactant protein B intron 4 variation in German patients with COPD and acute respiratory failure. Dis Markers. (2002) 18:129–36. doi: 10.1155/2002/194075
16. Lin Z, Thorenoor N, Wu R, DiAngelo SL, Ye M, Thomas NJ, et al. Genetic association of pulmonary surfactant protein genes, SFTPA1, SFTPA2, SFTPB, SFTPC, and SFTPD with cystic fibrosis. Front Immunol. (2018) 9:2256. doi: 10.3389/fimmu.2018.02256
17. Kala P, Ten Have T, Nielsen H, Dunn M, Floros J. Association of pulmonary surfactant protein A (SP-A) gene and respiratory distress syndrome: interaction with SP-B. Pediatr Res. (1998) 43:169–77. doi: 10.1203/00006450-199804001-01004
18. Nogee LM, Wert SE, Proffit SA, Hull WM, Whitsett JA. Allelic heterogeneity in hereditary surfactant protein B (SP-B) deficiency. Am J Respir Crit Care Med. (2000) 161(3 Pt 1):973–81. doi: 10.1164/ajrccm.161.3.9903153
19. Ramet M, Haataja R, Marttila R, Floros J, Hallman M. Association between the surfactant protein A (SP-A) gene locus and respiratory-distress syndrome in the Finnish population. Am J Hum Genet. (2000) 66:1569–79. doi: 10.1086/302906
20. Floros J, Fan R, Diangelo S, Guo X, Wert J, Luo J. Surfactant protein (SP) B associations and interactions with SP-A in white and black subjects with respiratory distress syndrome. Pediatr Int. (2001) 43:567–76. doi: 10.1046/j.1442-200x.2001.01474.x
21. Floros J, Lin HM, Garcia A, Salazar MA, Guo X, DiAngelo S, et al. Surfactant protein genetic marker alleles identify a subgroup of tuberculosis in a Mexican population. J Infect Dis. (2000) 182:1473–8. doi: 10.1086/315866
22. Mikerov AN, Umstead TM, Huang W, Liu W, Phelps DS, Floros J. SP-A1 and SP-A2 variants differentially enhance association of Pseudomonas aeruginosa with rat alveolar macrophages. Am J Physiol Lung Cell Mol Physiol. (2005) 288:L150–8. doi: 10.1152/ajplung.00135.2004
23. Mikerov AN, Wang G, Umstead TM, Zacharatos M, Thomas NJ, Phelps DS, et al. Surfactant protein A2 (SP-A2) variants expressed in CHO cells stimulate phagocytosis of Pseudomonas aeruginosa more than do SP-A1 variants. Infect Immun. (2007) 75:1403–12. doi: 10.1128/iai.01341-06
24. Phelps DS, Umstead TM, Floros J. Sex differences in the acute in vivo effects of different human SP-A variants on the mouse alveolar macrophage proteome. J Proteomics. (2014) 108:427–44. doi: 10.1016/j.jprot.2014.06.007
25. Thorenoor N, Kawasawa YI, Gandhi CK, Zhang X, Floros J. Differential impact of co-expressed SP-A1/SP-A2 protein on AM miRNome; sex differences. Front Immunol. (2019) 10:1960. doi: 10.3389/fimmu.2019.01960
26. Phelps DS, Chinchilli VM, Weisz J, Yang L, Shearer D, Zhang X, et al. Differences in the alveolar macrophage toponome in humanized SP-A1 and SP-A2 transgenic mice. JCI Insight. (2020) 3:141410. doi: 10.1172/jci.insight.141410. [Epub ahead of print].
27. Thorenoor N, Kawasawa YI, Gandhi CK, Floros J. Sex-specific regulation of gene expression networks by surfactant protein A (SP-A) variants in alveolar macrophages in response to Klebsiella pneumoniae. Front Immuno. (2020) 11:1290. doi: 10.3389/fimmu.2020.01290
28. Thorenoor N, Umstead TM, Zhang X, Phelps DS, Floros J. Survival of surfactant protein-A1 and SP-A2 transgenic mice after Klebsiella pneumoniae infection, exhibits sex-, gene-, and variant specific differences; treatment with surfactant protein improves survival. Front Immunol. (2018) 9:2404. doi: 10.3389/fimmu.2018.02404
29. Thorenoor N, Zhang X, Umstead TM, Scott Halstead E, Phelps DS, Floros J. Differential effects of innate immune variants of surfactant protein-A1 (SFTPA1) and SP-A2 (SFTPA2) in airway function after Klebsiella pneumoniae infection and sex differences. Respir Res. (2018) 19:23. doi: 10.1186/s12931-018-0723-1
30. Thorenoor NS, Phelps D, Kala P, Ravi R, Floros Phelps A, Umstead T, et al. Impact of surfactant protein-A variants on survival in aged mice in response to Klebsiella pneumoniae infection and ozone: serendipity in action. Microorganisms. (2020) 8:1276. doi: 10.3390/microorganisms8091276
31. Floros J, Thomas N. Genetic variations of surfactant proteins and lung injury. In: Nakos G, Papathanasiou A, editors. Surfactant Pathogenesis and Treatment of Lung Diasease (2009). pp. 25–48.
32. Guzman J, Wang YM, Kalaycioglu O, Schoenfeld B, Hamm H, Bartsch W, et al. Increased surfactant protein A content in human alveolar macrophages in hypersensitivity pneumonitis. Acta Cytol. (1992) 36:668–73.
33. Hamm H, Lührs J, Rotaeche JG, Costabel U, Fabel H, Bartsch W. Elevated surfactant protein A in bronchoalveolar lavage fluids from sarcoidosis and hypersensitivity pneumonitis patients. Chest. (1994) 106:1766–70. doi: 10.1378/chest.106.6.1766
34. Phelps DS, Umstead TM, Mejia M, Carrillo G, Pardo A, Selman M. Increased surfactant protein-A levels in patients with newly diagnosed idiopathic pulmonary fibrosis. Chest. (2004) 125:617–25. doi: 10.1378/chest.125.2.617
35. Gunther A, Schmidt R, Nix F, Yabut-Perez M, Guth C, Rosseau S, et al. Surfactant abnormalities in idiopathic pulmonary fibrosis, hypersensitivity pneumonitis and sarcoidosis. Eur Respir J. (1999) 14:565.
36. Buendía-Roldán I, Santiago-Ruiz L, Pérez-Rubio G, Mejía M, Rojas-Serrano J, Ambrocio-Ortiz E, et al. A major genetic determinant of autoimmune diseases is associated with the presence of autoantibodies in Hypersensitivity Pneumonitis. Eur Respir J. (2020) 56:1901380. doi: 10.1183/13993003.01380-2019
37. Raghu G, Remy-Jardin M, Ryerson CJ, Myers JL, Kreuter M, Vasakova M, et al. Diagnosis of hypersensitivity pneumonitis in adults. Am J Respir Crit Care Med. (2020) 202:e36–69. doi: 10.1164/rccm.202005-2032ST
38. Lin Z, deMello DE, Batanian JR, Khammash HM, DiAngelo S, Luo J, et al. Aberrant SP-B mRNA in lung tissue of patients with congenital alveolar proteinosis (CAP). Clin Genet. (2000) 57:359–69. doi: 10.1034/j.1399-0004.2000.570506.x
39. Wang Z, Liu T, Lin Z, Hegarty J, Koltun WA, Wu R. A general model for multilocus epistatic interactions in case-control studies. PLoS ONE. (2010) 5:e11384. doi: 10.1371/journal.pone.0011384
40. Liu T, Thalamuthu A, Liu JJ, Chen C, Wang Z, Wu R. Asymptotic distribution for epistatic tests in case-control studies. Genomics. (2011) 98:145–51. doi: 10.1016/j.ygeno.2011.05.001
41. Gandhi CK, Chen C, Wu R, Yang L, Thorenoor N, Thomas NJ, et al. Association of SNP–SNP interactions of surfactant protein genes with pediatric acute respiratory failure. J Clin Med. (2020) 9:1183. doi: 10.3390/jcm9041183
42. Hope ACA. A simplified Monte Carlo significance test procedure. J R Stat Soc Ser B. (1968) 30:582–98. doi: 10.1111/j.2517-6161.1968.tb00759.x
43. Hochberg YBY. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc. Ser B. (1995) 57:289–300.
44. Day NE, Byar DP. Testing hypotheses in case-control studies-equivalence of mantel-haenszel statistics and logit score tests. Biometrics. (1979) 35:623–30. doi: 10.2307/2530253
45. Zhang L, Liu R, Wang Z, Culver D, Wu R. Modeling haplotype-haplotype interactions in case-control genetic association studies. Front Genet. (2012) 3:2. doi: 10.3389/fgene.2012.00002
46. Cañadas O, Olmeda B, Alonso A, Pérez-Gil J. Lipid–protein and protein–protein interactions in the pulmonary surfactant system and their role in lung homeostasis. Int J Mol Sci. (2020) 21:3708. doi: 10.3390/ijms21103708
47. Floros J, Thomas NJ. Surfactant protein genetics in community-acquired pneumonia: balancing the host inflammatory state. Crit Care. (2011) 15:156. doi: 10.1186/cc10115
48. Blanchet M-R, Israël-Assayag E, Cormier Y. Inhibitory effect of nicotine on experimental hypersensitivity pneumonitis in vivo and in vitro. Am J Respir Crit Care Med. (2004) 169:903–9. doi: 10.1164/rccm.200210-1154OC
49. Pérez ERF, Kong AM, Raimundo K, Koelsch TL, Kulkarni R, Cole AL. Epidemiology of hypersensitivity pneumonitis among an insured population in the United States: a claims-based cohort analysis. Ann Am Thorac Soc. (2018) 15:460–9. doi: 10.1513/AnnalsATS.201704-288OC
50. Ohtsuka Y, Munakata M, Tanimura K, Ukita H, Kusaka H, Masaki Y, et al. Smoking promotes insidious and chronic farmer's lung disease, and deteriorates the clinical outcome. Intern Med. (1995) 34:966–71. doi: 10.2169/internalmedicine.34.966
51. Fatahi N, Niknafs N, Kalani M, Dalili H, Shariat M, Amini E, et al. Association of SP-B gene 9306 A/G polymorphism (rs7316) and risk of RDS. J Matern Fetal Neonat Med. (2018) 31:2965–70. doi: 10.1080/14767058.2017.1359829
52. Guan J, Liu X, Xie J, Xu X, Luo S, Wang R, et al. Surfactant protein a polymorphism is associated with susceptibility to chronic obstructive pulmonary disease in Chinese Uighur population. J Huazhong Univ Sci Technol. (2012) 32:186–9. doi: 10.1007/s11596-012-0033-7
53. Tagaram HR, Wang G, Umstead TM, Mikerov AN, Thomas NJ, Graff GR, et al. Characterization of a human surfactant protein A1 (SP-A1) gene-specific antibody; SP-A1 content variation among individuals of varying age and pulmonary health. Am J Physiol Lung Cell Mol Physiol. (2007) 292:L1052–1063. doi: 10.1152/ajplung.00249.2006
54. Betsuyaku T, Kuroki Y, Nagai K, Nasuhara Y, Nishimura M. Effects of ageing and smoking on SP-A and SP-D levels in bronchoalveolar lavage fluid. Eur. Respir J. (2004) 24:964. doi: 10.1183/09031936.04.00064004
55. Sørensen GL, Hjelmborg JB, Kyvik KO, Fenger M, Høj A, Bendixen C, et al. Genetic and environmental influences of surfactant protein D serum levels. Am J Physiol Lung Cell Mol Physiol. (2006) 290:L1010–7. doi: 10.1152/ajplung.00487.2005
56. Sorensen GL, Husby S, Holmskov U. Surfactant protein A and surfactant protein D variation in pulmonary disease. Immunobiology. (2007) 212:381–416. doi: 10.1016/j.imbio.2007.01.003
57. Heidinger K, König IR, Bohnert A, Kleinsteiber A, Hilgendorff A, Gortner L, et al. Polymorphisms in the human surfactant protein-D (SFTPD) gene: strong evidence that serum levels of surfactant protein-D (SP-D) are genetically influenced. Immunogenetics. (2005) 57:1–7. doi: 10.1007/s00251-005-0775-5
58. Wang G, Christensen ND, Wigdahl B, Guttentag SH, Floros J. Differences in N-linked glycosylation between human surfactant protein-B variants of the C or T allele at the single-nucleotide polymorphism at position 1580: implications for disease. Biochem J. (2003) 369(Pt 1):179–84. doi: 10.1042/bj20021376
59. Sumita Y, Sugiura T, Kawaguchi Y, Baba S, Soejima M, Murakawa Y, et al. Genetic polymorphisms in the surfactant proteins in systemic sclerosis in Japanese: T/T genotype at 1580 C/T (Thr131Ile) in the SP-B gene reduces the risk of interstitial lung disease. Rheumatology. (2008) 47:289–91. doi: 10.1093/rheumatology/kem355
60. Yang F, Zhang J, Yang Y, Ruan F, Chen X, Guo J, et al. Regulatory roles of human surfactant protein B variants on genetic susceptibility to Pseudomonas aeruginosa Pneumonia-Induced sepsis. Shock. 54:507–19. (2019). doi: 10.1097/shk.0000000000001494
61. Cordell HJ. Detecting gene-gene interactions that underlie human diseases. Nat Rev Genet. (2009) 10:392–404. doi: 10.1038/nrg2579
62. Floros J, Wang G. A point of view: quantitative and qualitative imbalance in disease pathogenesis; pulmonary surfactant protein A genetic variants as a model. Comp Biochem Physiol A Mol Integr Physiol. (2001) 129:295–303. doi: 10.1016/s1095-6433(01)00325-7
63. Sabourin J, Nobel AB, Valdar W. Fine-mapping additive and dominant SNP effects using group-LASSO and fractional resample model averaging. Genet Epidemiol. (2015) 39:77–88. doi: 10.1002/gepi.21869
64. Poulain FR, Nir S, Hawgood S. Kinetics of phospholipid membrane fusion induced by surfactant apoproteins A and B. Biochim Biophys Acta. (1996) 1278:169–75. doi: 10.1016/0005-2736(95)00212-X
65. Wang G, Guo X, Diangelo S, Thomas NJ, Floros J. Humanized SFTPA1 and SFTPA2 transgenic mice reveal functional divergence of SP-A1 and SP-A2: formation of tubular myelin in vivo requires both gene products. J Biol Chem. (2010) 285:11998–2010. doi: 10.1074/jbc.M109.046243
66. Stram DO. Multi-SNP haplotype analysis methods for association analysis. In: Elston RC, editor. Statistical Human Genetics: Methods and Protocols. New York, NY: Springer (2017). p. 485–504.
Keywords: SNP-SNP interaction, surfactant protein gene polymorphism, SFTPA1, SFTPA2, SFTPB, SFTPC, SFTPD, genetic susceptibility
Citation: Gandhi CK, Chen C, Amatya S, Yang L, Fu C, Zhou S, Wu R, Buendía-Roldan I, Selman M, Pardo A and Floros J (2021) SNP and Haplotype Interaction Models Reveal Association of Surfactant Protein Gene Polymorphisms With Hypersensitivity Pneumonitis of Mexican Population. Front. Med. 7:588404. doi: 10.3389/fmed.2020.588404
Received: 28 July 2020; Accepted: 07 December 2020;
Published: 05 January 2021.
Edited by:
Bassam Mahboub, Rashid Hospital, United Arab EmiratesReviewed by:
Haruhiko Furusawa, University of Colorado, United StatesCopyright © 2021 Gandhi, Chen, Amatya, Yang, Fu, Zhou, Wu, Buendía-Roldan, Selman, Pardo and Floros. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joanna Floros, amZsb3Jvc0BwZW5uc3RhdGVoZWFsdGgucHN1LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.