 Jiping Yao
Jiping Yao Min Zheng
Min Zheng- The State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, National Clinical Research Center for Infectious Diseases, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, China
Neddylation is a ubiquitin-like posttranslational modification that conjugates neural precursor cell expressed developmentally downregulated-8 (Nedd8) to specific substrates for regulation of protein activity. In light of current researches, the neddylation pathway is aberrant in the pathogenesis of many diseases. In our review, we summarize the versatile roles of neddylation in chronic liver diseases (CLDs). CLDs are one of the leading causes of chronic disease-associated deaths worldwide. There are diverse etiologic agents causing CLDs, mainly including hepatitis B virus (HBV) infection, nonalcoholic fatty liver disease (NAFLD), chronic exposure to alcohol or drugs, and autoimmune causes. So far, however, there remains a paucity of effective therapeutic approach to CLDs. In this review, we summarized the role of the neddylation pathway which runs through the chronic hepatitis B/NAFLD–liver fibrosis–cirrhosis–hepatocellular carcinoma (HCC) axis, a canonical pattern in the process of CLD development and progression. The dysregulation of neddylation may provide a better understanding of CLD pathology and even a novel therapeutic strategy. Correspondingly, inhibiting neddylation via MLN4924, a small molecule compound targeting NEDD8-activating enzyme (NAE), can potently alleviate CLD progression and improve the outcome. On this basis, profiling and characterization of the neddylation pathway can provide new insights into the CLD pathology as well as novel therapeutic strategies, independently of the etiology of CLD.
Introduction
Chronic liver disease (CLD) is posing a significant public health problem worldwide for it causes ~2 million deaths annually. It is desperately needed to pay high attention to it (1). There are diverse etiologic agents causing CLDs, primarily including chronic infection of hepatitis B or C viruses (HBV or HCV), chronic aberrant metabolic conditions [nonalcoholic fatty liver disease (NAFLD)], chronic consumption of alcohol or drugs, and abnormal autoimmunity (2, 3). Further, the onset, development, and deterioration of CLD are progressive processes. A variety of factors are involved in them, such as inflammatory cell infiltration, liver tissue reconstruction, fibrogenesis, and extracellular matrix (ECM) deposition (4, 5). Therefore, the pathological process of CLD is complicated, characterized by pan-cellular and pan-pathway mechanisms. Recently, more and more attention has been paid to the ubiquitination pathway due to its potential role in the therapy of CLD, particularly hepatocellular carcinoma (HCC), the advanced stage of CLD.
A new insight into the CLD field is the emerging role of the neddylation pathway. Neddylation is a ubiquitination-like modification. However, unlike ubiquitination that mainly targets proteins doomed to be degraded, neddylation regulates proteins' function and stability (6). The best-known physiological substrates of neddylation are the cullin family, the crucial component of cullin-RING ligases (CRLs). CRL is the largest family of ubiquitination E3 ligases (7) and responsible for about 20% of cellular protein degradation via proteasome (8). Neddylation of cullin activates CRLs (9) and promotes the ubiquitination of substrates (10). Hence, neddylation modulates the cellular function in some degree via manipulating ubiquitination E3 ligases behind the scene. Previous researches demonstrated that neddylation inhibition can inactivate CRLs (11) and then results in accumulation of their substrates, such candidates including chromatin licensing and DNA replication factor 1 (CDT1) (12), p21 (13), and nuclear factor erythroid 2-related factor 2 (NRF2) (14, 15). Most substrates of CRLs are tumor suppressors (16). The consequence of neddylation inhibition is cell apoptosis, cell autophagy, cell senescence, and ultimately cancer suppression (17) (Table 1). Current researches of noncullin substrates of neddylation uncover that neddylation might participate in additional biological process of cells (41) (Table 2). On this basis, recent studies have uncovered that neddylation inhibition can repress HBV survival (64), alleviate steatosis (65), reduce liver fibrosis (66), and restrain pro-tumor inflammation (67). Considering the progress that has been made to understand the role of neddylation, it is worthy to explore and conclude the relevance between neddylation and CLD. In this review, we particularly focus on the role of the neddylation pathway in CLD pathology and assess the therapeutic approach targeting neddylation in CLD.
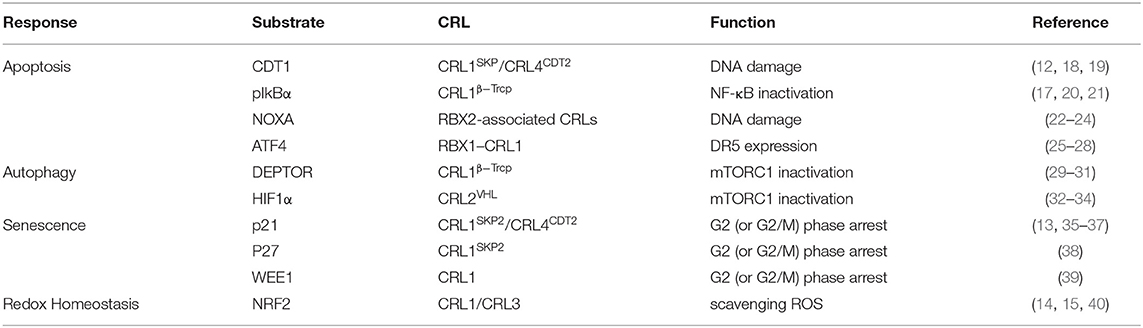
Table 1. Neddylation inhibition and CRL inactivation.
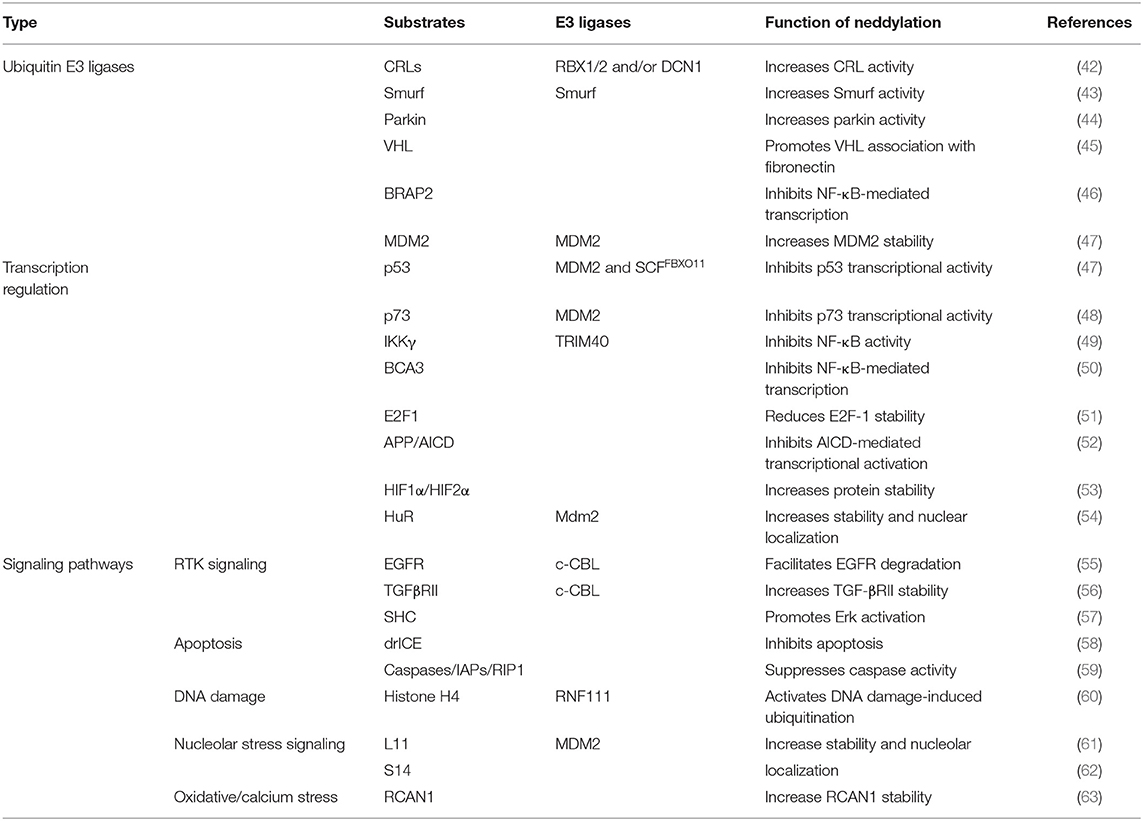
Table 2. Neddylation pathway substrates.
Neddylation in CLD
Overactivation of Neddylation
Deregulation of the neddylation pathway has been described in various pathological conditions. Focusing on the CLD context like liver fibrosis and HCC, both NAE1 and global protein neddylation expressions are upregulated (66, 68). However, the initial triggering mechanism that overactivates neddylation pathways during CLD is not understood exactly. Significantly, a previous study demonstrated that diverse stress conditions such as heat shock and oxidative stress may lead to an entire increase of the neddylation pathway in vitro. Under the stress circumstance, the ubiquitin E1 enzyme Ube1, rather than NAE1, regulates the conjugation of the NEDD8 (69, 70). Further analysis reveals that the crosstalk between NEDD8 and ubiquitin causes a prompt and dramatic amplification of the NEDD8 proteome under stress conditions. In addition, neddylation of the substrate competing with its ubiquitination upon stress could stabilize its protein level and prevent its degradation (69). Subsequently, accumulated substrates mediate vital biological processes and trigger diverse cellular responses, which may result in multiple hepatic dysfunctions eventually. Besides, in HCC, a decrease in deneddylating enzyme NEDP1 with a concomitant increase of NEDD8 conjugates leads to the inhibition of ATPase activity of heat shock protein 70 (HSP70) (71). As a result, the formation of the apoptosome is disturbed, and subsequently, the apoptosis resistance of hepatoma cells is elevated (72).
Neddylation and HBV Infection
HBV chronic infection is a primary pathogeny leading to CLDs, especially cirrhosis and HBV-related HCC (64). HBV-encoded X protein (HBx) is a small regulatory protein that exhibits pleiotropic activities, including affecting transcription, DNA repair, cell growth, and apoptotic cell death (73). HBx can interact with various cellular proteins to influence its own activity. Among all these HBx-interacting proteins, the damage-specific DNA binding protein 1 (DDB1) is a well-characterized HBx binding partner (74). This interaction is worthy of attention because DDB1 is the adaptor protein of CRL4 ubiquitin E3 ligase. Previous researches reported that HBx–DDB1 interaction is indispensable in HBx-induced viral genome replication and affects cell viability. An interesting structural study revealed that HBx contains an α-helical motif termed the H-box, which is shared by several DDB1–CUL4-associated factor (DCAF) proteins (75). DCAF proteins act as well-known CRL4 substrate acceptors. According to the above, it is possible that HBx assembles an HBx–DDB1–CUL4–ROC1 E3 ligase complex (CRLHBx) as CRL4 and HBx plays the role of substrate receptor. HBx targets host proteins that suppress HBV genome replication, and CRLHBx promotes their ubiquitylation and degradation via the proteasome pathway (75–77). Neddylation activates CRL4HBx via conjugating Nedd8 to the cullin protein's conserved lysine residues to affect HBV replication indirectly.
Liu et al. also found that HBx can be neddylated by the Nedd8 E3 ligase human homolog of mouse double minute 2 (HDM2). Neddylation modification alters HBx's half-life and enhances its stability. Moreover, it also increases HBx's chromatin localization and the binding with DDB1 (64). Previous researches show that HBx is ubiquitylated by E3 ligase Siah-1 to induce HBx degradation (78). Interestingly, Liu and his colleagues found that the mechanism of HDM2-mediated neddylation modification increases HBx stability by preventing its ubiquitination-induced degradation (64). These processes ultimately favor HBx's activity of transcriptional regulation, cell proliferation, and HBV-driven tumor growth. In conclusion, it provides an insight into the neddylation's role in HBV invasion. Further study found that HDM2's expression is positively correlated with HBx expression in HBV-related HCC samples (Figure 1). HDM2 has the potential to act as a new prognostic marker for HBV-related HCC. Inhibition of the neddylation pathway may provide us with a novel therapeutic method for HBV-related HCC.
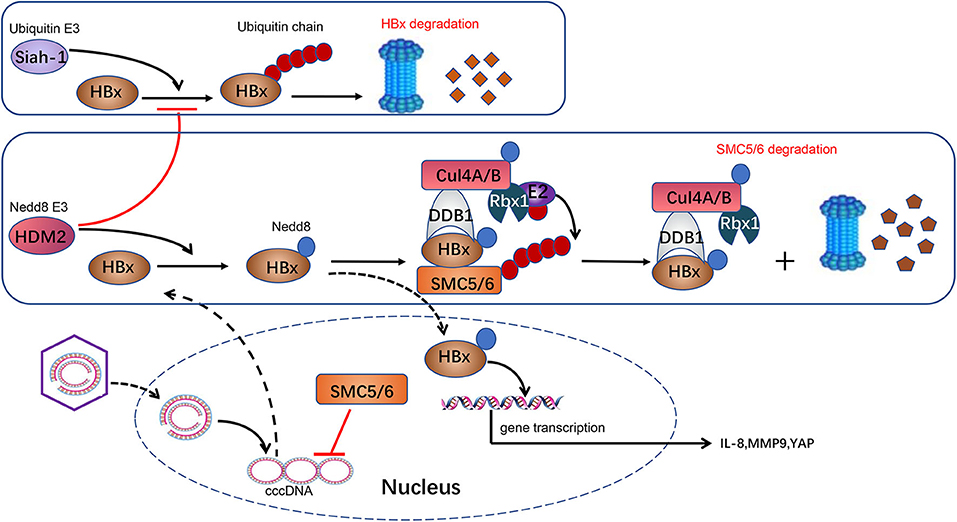
Figure 1. Neddylation and HBV infection. HBV is a DNA virus carrying a 3.2-kb genome. Following entry into the host cell, the 3.2-kb genome is transported into the nucleus and converted to a cccDNA, which serves as a crucial template for HBV transcription. HBx is transcripted from cccDNA and serves as a transcriptional activator to promote the expression of IL-8, MMP9, and YAP, which are implicated in HCC development. HBx in cytoplasm is ubiquitylated by E3 ligase Siah-1 to induce HBx degradation through proteasome. HBx can be neddylated by Nedd8 E3 ligase HDM2 in the cytoplasm. HDM2-mediated neddylation modification increases HBx stability by preventing its ubiquitination-induced degradation. Meanwhile, HBx plays a role as a substrate acceptor and assembles CRL4HBx ubiquitin E3 ligases. HBx targets SMC5/6, a host protein that suppresses HBV genome replication, and CRL4HBx promotes its ubiquitylation and degradation via proteasome. Besides, neddylation activates CRL4HBx via conjugating Nedd8 to cullin protein's conserved lysine residues to affect HBV replication indirectly.
Neddylation and NAFLD
NAFLD is an increasingly prevalent CLD and has become a prominent healthy concern globally due to dietary structure change and lifestyle change (79). NAFLD is characterized by steatosis, a pathologic phenomenon of excessive triglyceride accumulation in hepatocytes. NAFLD will frequently progress to its more severe form called nonalcoholic steatohepatitis (NASH), which consists of hepatic steatosis, inflammation, and fibrosis. NASH accompanied with advanced fibrosis may eventually lead to cirrhosis and even HCC (80, 81). Recently, more and more insight has been shed on the association between NAFLD and neddylation.
Neddylation plays a crucial physiological role in lipid metabolism. Recent research reported that liver-specific deficiency of NEDD8 or UBA3 causes neonatal death with spontaneous fatty liver in mouse models. Interestingly, electron transfer flavoproteins (ETFs), whose defects can lead to fatty acid oxidation disorder in glutaric aciduria type II (GA-II), are neddylation substrates. Hepatic neddylation modification can stabilize ETFs and even enhance ETF expression via suppressing their ubiquitination, which prevents fasting-induced steatosis (82).
Furthermore, another research of serine-rich splicing factor 3 (SRSF3) illuminates that neddylation is involved in the pathology of NAFLD and NASH. It suggested that the low expression of SRSF3 is correlated with an increased risk of NAFLD, NASH, or cirrhosis. In the condition of oxidative stress, SRSF3 could be modified by the NEDD8 protein at lysine 11, which results in the degradation of SRSF3 via proteasome (83). On this basis, the result above implies that intervening with the neddylation of SRSF3 contributes to its stability and accumulation, which is beneficial for preventing hepatic steatosis, fibrosis, and inflammation.
Recently, it was reported that sterol regulatory element-binding protein 1c (SREBP1c), a critical role in maintaining lipid homeostasis, is upregulated in liver to contribute to the progress of hepatic steatosis. However, unlike SRSF3, neddylation of SREBP1c competing with its ubiquitination facilitates its stability and, rather than promoting its degradation via proteasome, eventually contributes to hepatic steatosis. Further, SREBP1c can be neddylated by NEDD8 E3 ligase HDM2, which can also mediate the neddylation of HBx as described above (84).
Additionally, Dehnad et al. revealed that advanced glycation end product (AGE) clearance receptor AGER1 was decreased in NASH. Further analysis demonstrated that exposure to high AGEs promotes an AGER1/RAGE imbalance and subsequently promotes NRF2 degradation via neddylation of cullin3, which eventually causes downregulation of AGER1 (85).
Taken together, these studies come to the conclusion that a neddylation-dependent pathway is implicated in liver steatosis and fibrosis mainly via regulating the stability of its substrates, which function as critical regulators in the process of liver steatosis. Revealing the role of neddylation in hepatic lipid metabolism and fibrosis progression may pave the way for a novel therapeutic approach in NAFLD and NASH (65).
Neddylation and Liver Fibrosis
Liver fibrosis is a continuous wound-healing progress leading to sustained scarring response (86). Liver fibrosis can be triggered by underlying etiologies of CLDs, such as viral infection, alcohol, and NASH (87). The pathology progress is a dynamic and reversible response that can be regulated by halting or reversing the fibrosis to cirrhosis and HCC (3). To date, specific therapies validated as being effective for liver fibrosis have primarily been etiology induced. However, there is no thoroughly validated antifibrosis therapy that is independent of the underlying etiology in the clinic.
More recently, deregulated neddylation is founded in liver fibrosis (66). Firstly, neddylation inhibition decreases liver inflammation. It is reported that neddylation inhibition reduces the expression level of pro-inflammatory cytokines and chemokines, such as tumor necrosis factor α (TNFα), interleukin-6 (IL-6) and its receptor, the tumor necrosis factor alpha receptor (TNFR1) (88), the chemokine (C-X-C motif) ligand 1 (Cxcl1), the chemokine (C-X-C motif) ligand 2 (Cxcl2), the chemokine (C-C motif) ligand 2 (Ccl2), and the C-C chemokine receptors (Ccr) (Ccr1, Ccr2, and Ccr5) (89). Secondly, neddylation plays a role in hepatic stellate cell activation. In bile duct ligation (BDL) and carbon tetrachloride (CCl4)-induced injury rodents, neddylation inhibition decreases HSC activation and collagen accumulation, certified by reduced levels of the pro-fibrogenic factor, transforming growth factor (TGFβ), and the expression of collagen type I alpha 1 (ColIα1) (90). Thirdly, neddylation inhibition decreases fibrosis by inducing HSC apoptosis due to c-Jun accumulation. c-Jun, associated with apoptosis in several types of cells, such as HSC, could be targeted by neddylated cullins for degradation via the ubiquitin proteasome system. This provides a critical clue that neddylation inhibition could somehow modulate c-Jun levels and concomitant apoptosis (91).
Moreover, there are another two possible regulated mechanisms of neddylation in liver fibrogenesis. The transcription factor NF-κB mediates transcription of genes, such as pro-inflammation cytokines involved in the inflammation progress of fibrogenesis (92). Neddylation functions on NF-κB activation by promoting its nuclear translocation via activating SCFβTrCP's cullin. SCFβTrCP is a CRL that targets the NF-κB inhibitory protein IκBα for degradation via proteasome (93). It is reasonable to believe that neddylation inhibition could ameliorate inflammation and fibrosis via reducing specific NF-κB target genes' expressions. Another possible mechanism is associated with TGFβ. TGFβ is significantly important in HSC activation and involved in Smad2 signaling through transmembrane receptor serine/threonine kinases (94). An intriguing research shows that Casita B-lineage lymphoma (c-Cbl) can function as a Nedd8 E3 ligase of the type II receptor (TGFβ-RII) beyond CRL (56). The neddylation modification of TGFβ-RII interferes with its ubiquitination–proteasome degradation in blood cells. Therefore, we can speculate that the neddylation modification of TGFβ-RII stabilizes its function and thereby promotes TGFβ signaling, playing a key role in HSC activation (66, 95).
Neddylation and HCC
HCC, the most common and frequent primary liver cancer, is the advanced stage of CLD. Although some alteration of signal molecules involved in HCC progression is discovered, the high mortality and the poor prognosis make it the third malignancy that leads to cancer-related death globally (96). Recently, it was reported that the dysregulation of the neddylation pathway is associated with HCC. The whole neddylation pathway including NEDD8; NEDD8-specific E1, E2, and E3; and even deneddylation enzymes was upregulated in HCC (97).
Inhibiting the neddylation pathway can significantly suppress HCC cell proliferation and migration, induce apoptosis, and eventually inhibit HCC growth and metastasis (98). Nieves Embade and his colleagues found that Hu antigen R (HuR) was implicated in the above process. HuR plays a critical role in hepatocyte proliferation, survival, differentiation, and HCC transformation via enhancing the stability of target mRNAs (92). The stability of HuR itself in cells is attributed to Mdm2-mediated neddylation. The neddylation modification of HuR promotes its nuclear localization and reduces its degradation (54, 99). SREBP-1 is not merely a critical regulator of lipid metabolism but is also associated with cancer metabolism. In metastatic HCC samples, SREBP-1 is upregulated with a concomitant increase of UBC12. Recently, it was reported that SREBP-1 can be neddylated by NEDD8 E3 ligase UBC12. Consequently, neddylation of SREBP-1 competing with its ubiquitination promotes the stability of SREBP-1 (68). Otherwise, liver kinase B1 (LKB1) and Akt kinases, critical regulators in proliferative metabolism of the liver, could be neddylated to enhance their stability. Inhibition of neddylation leads to metabolic reprogramming and concomitant apoptosis of liver cancer cells via altering the stability of LKB1 and Akt (100).
Importantly, neddylation is closely related to regulation of autophagy in liver cancer cells (101). Autophagy is a cellular biological process in response to various stresses to maintain cellular homeostasis. Several researches have reported that autophagy could play a pro-survival role in cancer cells. Inhibition of neddylation would induce autophagy to promote survival of cancer cell and thus result in drug resistance. Mechanistically, suppressing neddylation causes inactivation of CRL1β−TrCP and CRL2VHL and subsequently contributes to the accumulation of their substrates: DEPTOR and HIF1α. DEPTOR and the HIF1–REDD1–TSC1 axis would induce mTOR inactivation, which partially leads to autophagy (34, 102, 103). Additionally, increased level of reactive oxygen species (ROS) and activating transcription factor 3 (ATF3) could also trigger autophagy in the circumstance of neddylation inhibition (104). Recently, it was reported that inactivation of CRL4 would block polyubiquitination and proteasomal degradation of the WD repeat domain, phosphoinositide interacting 2 (WIPI2) via inhibition of neddylation, which eventually induces autophagy during mitosis (105).
Moreover, neddylation also regulates pro-tumorigenic inflammation in liver cancer cells. Hypoxia-inducible factor-1 (HIF1), an oxygen homeostasis transcription factor, provides anti-inflammation activity under hypoxia conditions (106). The alpha subunit of HIF1 (HIF1α) can be targeted by the Von-Hippel-Lindau protein (pVHL), a substrate receptor of CRL2 (107). Under normoxic conditions, HIF1α prefers to be hydroxylated and then recognized by pVHL, which mediates the degradation of hydroxylated HIF1α by the UPS pathway (32, 33). Under hypoxic conditions, HIF1α tends to translocate to the nucleus and form a heterodimer with a HIF1β subunit and transcriptionally regulates a wide spectrum of genes significant for the anti-inflammatory response (108). Likewise, neddylation modification of cullin2 alters the activity of CRL2 and eventually influences the stability of HIF1α (109) (Figure 2). Recently, Cannito et al. (110) have suggested that SerpinB3, a serine protease inhibitor, can stimulate proliferation of hepatic tumor cells and subsequently facilitate HCC progression by enhancing the stabilization of HIF2α by promoting the direct and selective neddylation of HIF2α (111).
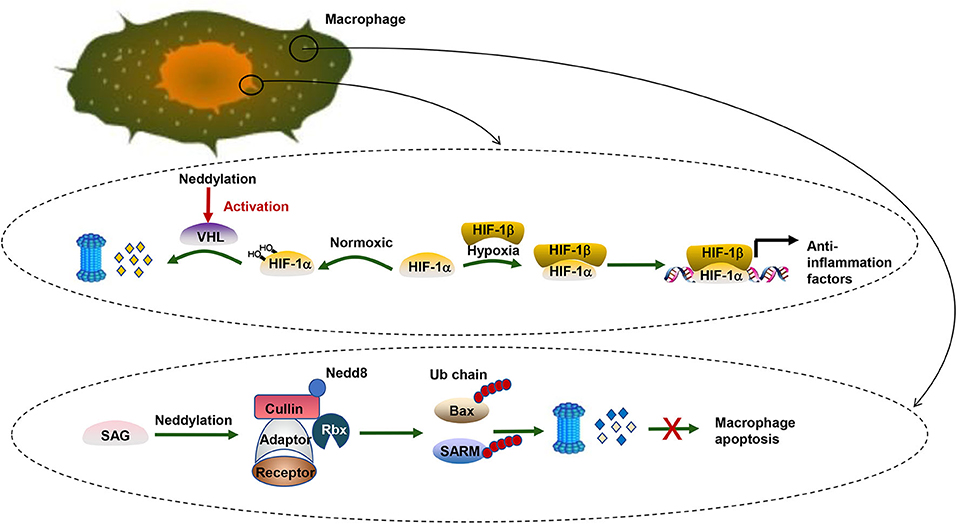
Figure 2. Neddylation and HCC. There are two biological processes taking place in pro-inflammation macrophages. One is related to the transcription factor HIF. HIF-1α can be targeted by pVHL, a substrate receptor of CRL2. Under normoxic conditions, HIF-1α prefers to be hydroxylated and then recognized by pVHL for degradation via the UPS pathway. Under hypoxic conditions, HIFα tends to translocate to the nucleus and form a heterodimer with a HIF1β subunit and transcriptionally regulates a wide spectrum of genes significant for the anti-inflammatory response. The other is about the apoptosis inhibition of pro-inflammation macrophages. SAG, a neddylation E3 ligase, collaborates with UPS to promote survival of infectious macrophages via ubiquitination of Bax and SARM. In contrast, SAG knockdown leads to the accumulation of proapoptotic Bax and SARM and breaks the balance between antiapoptotic Bcl-2 and Bax in the mitochondria, which induces the death of macrophages.
Further analysis elucidates that the role of neddylation poses a profound effect not only on liver cancer cells but also on immune cells. In macrophages, blocking neddylation regulates NF-κB signaling and eventually causes the downregulation of proinflammatory cytokines. Besides, further research reveals that the sensitive to apoptosis gene (SAG), a neddylation E3 ligase, collaborates with UPS to promote survival of infectious macrophages via degrading proapoptotic Bax and sterile α and HEAT/armadillo-motif-containing protein (SARM) (112). SAG also affects cytokine secretion of macrophages (113). Similar to innate immune cells, SAG-deficient T cells also show decreased proliferation, reduced production of cytokines, and diminished release of the T-cell lineage. Besides, knockdown of Ubc12 in CD4+ T cells caused impaired T-cell receptor/CD28-induced proliferation because T cells were arrested in the G0/G1 phase of the cell cycle (113). Moreover, cytokine production like IL-2 and the differentiation of CD4+ T cells into effector Th-cell subsets are decreased when the expression of Ubc12 is reduced. The neddylation pathway regulates various aspects of CD4+ T-cell function. However, the exact mechanism remains to be investigated. A similar result was found in B cells (114–116).
Therapeutic Strategies Targeting Neddylation
MLN4924 (pevonedistat), a small molecule inhibitor of NAE, disrupts CRL-mediated protein turnover; causes restraining tumor cell growth by inducing apoptosis, senescence, and autophagy; and causes sensitization to chemoradiation therapies in a cellular context-dependent manner (8). Currently, MLN4924 is being evaluated in several phase Ib/II/III clinical trials (https://www.clinicaltrials.gov/). Among these clinical trials, five completed phase I clinical trials in solid tumors and hematological malignancies have verified that MLN4924 is safe and feasible. Several phase II clinical trials are under way. Particularly, a phase III clinical trial of MLN4924 combined with azacytidine is currently recruiting volunteers with hematological malignancies. Previous researches suggested that MLN4924 is a potent and selective inhibitor in hematological neoplasms as well as many solid neoplasms, including HCC. Given that the summaries about previous studies of MLN4924 in HCC are relatively sufficient, we primarily demonstrate the use of MLN4924 in HBV infectious, NAFLD, and liver fibrosis.
Currently, although nucleoside or nucleoside analogs can suppress new viral DNA replication, they are unable to eradicate the cccDNA from infected hepatocytes thoroughly (117). This is the most critical reason resulting in HBV rebound and obstinacy. Recently, it is reported that MLN4924 can suppress HBV transcription and protein expression significantly via restoring the structural maintenance of chromosome (SMC) complex protein (SMC5/6) levels (118). Murphy et al. (119) identified that SMC5/6, a host restriction that restricts HBV replication by inhibiting HBV gene expression, is the substrate of CRLHBx (120). HBx targets SMC5/6 for degradation by the ubiquitin–proteasome system, and subsequently, the transcription from cccDNA is elevated (Figure 1). Neddylation of cullin is necessary for activation of CRLHBx. MLN4924 inhibits the neddylation process, promotes SMC5/6 accumulation, and ultimately leads to restriction of viral transcription and HBV DNA level, particularly cccDNA (118). Thereby, the neddylation pathway is a potential target for HBV treatment. MLN4924 may become a novel anti-HBV agent, though evidence is far from being enough.
In view of the role of neddylation in regulating stabilization of SRSF3, MLN4924 can repress the degradation of SRSF3 and reduce the accumulation of SREBP1c, which alleviate the steatosis and prevent the progression of NAFLD (65). Serrano-Macia et al. have found that β-oxidation activity and ketone body levels were enhanced after treatment. On the contrary, the levels of lipid peroxidation and ROS are significantly reduced after MLN4924 treatment. Further study suggests that in a NASH mouse model, MLN4924 treatment can reverse steatosis, inflammation, and fibrosis. Hence, inhibition of neddylation via MLN4924 is a potent therapeutic option because it can ameliorate fatty acid metabolism (121).
Similarly, using the pharmacological inhibitor MLN4924 could protect liver from injury, inflammation, and fibrosis via regulating the function of hepatocytes. To be more specific, MLN4924 treatment reduces the expression of pro-inflammatory cytokines previously associated with liver damage and, therefore, ameliorate the inflammation after liver injury (122). In agreement, pro-fibrogenic factors implicated in liver fibrosis, such as TGFβ, COL1α1, matrix metalloproteinase-9 (MMP9), and interferon-α (IFNα), are consistently decreased after using MLN4924. More importantly, MLN4924 diminishes the activation of HSC (66). Together, these results highlight that MLN4924 treatment is pointed out as a potential antifibrosis therapy that is independent of the underlying etiology in the clinic.
In this review, we conclude that inhibition of neddylation pharmacologically via MLN4924 can significantly alleviate CLD exacerbation and progression in view of recent researches (8). Given its well-tolerated toxicity and potent antitumor activity in preclinical trails, MLN4924 is anticipated to be a promising therapeutic approach for CLD. However, there still remains some challenges for final application of MLN4924 in the clinic. Due to the treatment-emergent NAEb mutations, resistance to MLN4924 during therapy will appear, which subsequently reduces the effectiveness of this medicine (123). Therefore, it is indispensable to design next-generation NAE inhibitors that can overcome treatment-emergent resistance.
Conclusions and Perspectives
Chronic hepatitis/NAFLD–liver fibrosis–cirrhosis–HCC axis is a canonical pattern in the process of CLD progression (124). Recent studies in the neddylation pathway provides us with crucial clues that neddylation is a versatile pathway that takes on various aspects and phases of CLD (Table 3). Although the complicated mechanism underlying the overactivation of the neddylation pathway during CLD still remains elusive, there are several notable features of neddylation in CLD. First, the NEDD8 and NAE1 expressions are upregulated significantly in NAFLD, liver fibrosis, and HCC, and the components of the neddylation pathway like Nedd8, E1 (NAE), E2, and E3 may become novel biomarkers for CLD diagnosis. Second, under stress conditions, neddylation modification directly or by activating CRL indirectly promotes the stability of the substrate (promotes degradation in rare cases), and the accumulated substrate functions as a crucial molecule to facilitate the development of CLD. Third, MLN4924 could inhibit the overactivation of neddylation during CLD and thus alleviates the pathological process.
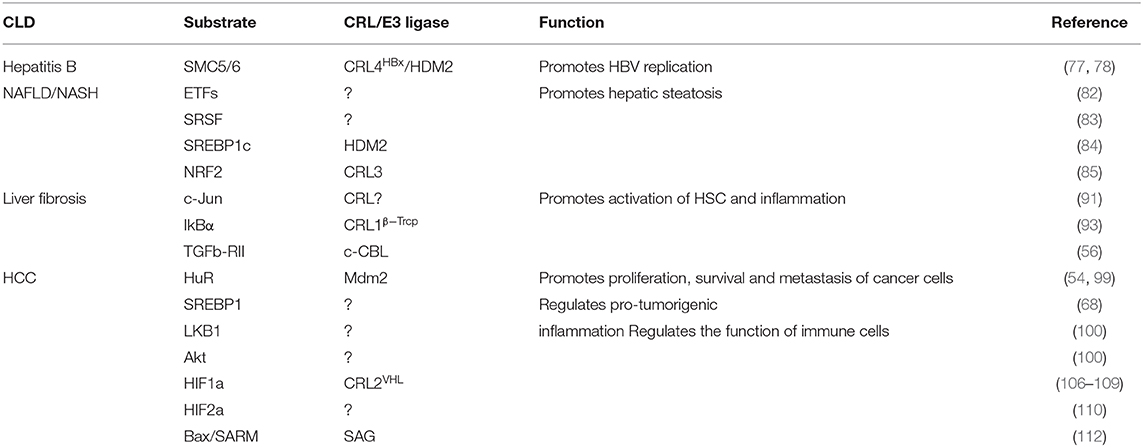
Table 3. Neddylation in CLDs.
However, there remain several issues that need to be further explained correspondingly. First, the underlying mechanism by which the neddylation pathway is overactivated in CLD has not been demonstrated clearly so far. Second, besides HSC and cancer cells, whether and how the upregulated neddylation pathway plays a significant role in hepatic parenchyma, Kupffer cells, and tumor microenvironment require further exploration. Third, MLN4924 suppresses the whole neddylation pathway and may cause potential unforeseeable secondary effects. Moreover, MLN4924 would induce autophagy that acts as a pro-survival signal in cancer cells. Given this situation, it is probably sensible to be cautious about using MLN4924 for CLD therapy.
Notably, sumoylation, another important ubiquitin-like posttranslational modification, is identified as a double-edged sword in CLD while neddylation generally causes pathological consequences. Inhibition of the global sumoylation pathway might not always be an optimal therapeutic strategy due to its “two faces” in CLD (125–128). Therefore, targeting the neddylation pathway holds promise for the therapy of CLD. On the context of drug discovery, it is highly anticipated that more specific and safer small molecule inhibitors aiming at special targets such as specific E3 Nedd8 ligases or deneddylase enzymes should be discovered as novel therapeutic approaches for CLD.
Author Contributions
JY wrote the manuscript and prepared figures: MZ, YL, and XL provided expert comments and edits. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by Grants from the National Nature Science Foundation of China, No. 81871646; the State S & T Project of 13th Five Year, No. 2018ZX10302206.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
CLD, chronic liver diseases; MDM2, murine double minute-2; NEDD8, neural precursor cell expressed developmentally downregulated-8; HBV, hepatitis B virus; HCV, hepatitis C virus; HCC, hepatocellular carcinoma; HBx, HBV-encoded X protein; HSC, hepatic stellate cell; NAE, NEDD8-activating enzyme; E2F-1, E2F transcription factor 1; ECM, extracellular matrix; CRL, cullin-RING ligases; VHL, von Hippel-Lindau; RBX, RING box protein-1; SKP, S-phase kinase-associated protein; CDT1, chromatin licensing and DNA replication factor; CDT2, chromatin licensing and DNA replication factor 2; NRF2, nuclear factor erythroid 2-related factor 2; NF-κB, the nuclear factor kappa-light-chain-enhancer of activated B cells; ATF4, activating transcription factor 4; βTrCP, beta-transducin repeat containing protein; DCN1, defective in cullin neddylation 1; ROS, reactive oxygen species; mTORC, mammalian target of rapamycin complex; NASH, nonalcoholic steatohepatitis; NAFLD, nonalcoholic fatty liver disease; TRIM40, tripartite motif containing 40; BCA3, breast cancer-associated protein 3; FBXO11, F-box protein 11; HuR, Hu antigen R; TGFβ-RII, transforming growth factor β type II receptor; AICD, APP intracellular domain; EGFR, epidermal growth factor receptor; BRAP2, BRCA1-associated protein 2; SCF, Skp1, cullin, and F-box protein; RTK, receptor tyrosine kinase; cccDNA, covalently closed circular DNA; IL-8, interleukin-8; MMP9, matrix metalloproteinase-9; DEPTOR, DEP domain containing mTOR-interacting protein; HIFα, hypoxia-inducible factor-α; DCAF, DDB1–CUL4-associated factor; IFNα, interferon-α; ColIα1, collagen type I alpha 1; TGFβ, transforming growth factor; TNFα, tumor necrosis factor α; IL-6, interleukin-6; Cxcl, the chemokine (C-X-C motif) ligand; Ccl, the chemokine (C-C motif) ligand; Ccr, the C-C chemokine receptors; c-Cbl, Casita B-lineage lymphoma; pVHL, Von-Hippel-Lindau protein; DDB1, the damage-specific DNA binding protein 1; SARM, sterile α and HEAT/armadillo-motif-containing protein; Bax, Bcl-2 associated protein X; CUL, cullin; KC, Kupffer cells; CCl4, carbon tetrachloride; JNK, c-Jun N-terminal kinase; Bcl-2, β-cell lymphoma 2; SMC, the structural maintenance of chromosomes; ETFs, electron transfer flavoproteins; SRSF3, serine-rich splicing factor 3; BDL, bile duct ligation; CCl4, carbon tetrachloride; HSP70, heat shock protein 70; SREBP1c, sterol regulatory element-binding protein 1c; HDM2, human homolog of mouse double minute 2; LKB1, liver kinase B1; AGEs, advanced glycation end products; WIPI2, WD repeat domain, phosphoinositide interacting 2.
References
1. Global regional and national age-sex specific all-cause and cause-specific mortality for 240 causes of death 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. (2015) 385:117–71. doi: 10.1016/S0140-6736(14)61682-2
2. Parola M, Pinzani M. Liver fibrosis: pathophysiology, pathogenetic targets and clinical issues. Mol Aspects Med. (2019) 65:37–55. doi: 10.1016/j.mam.2018.09.002
3. Trautwein C, Friedman SL, Schuppan D, Pinzani M. Hepatic fibrosis: concept to treatment. J Hepatol. (2015) 62:S15–S24. doi: 10.1016/j.jhep.2015.02.039
4. Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut. (2015) 64:830–41. doi: 10.1136/gutjnl-2014-306842
5. Rosselli M, MacNaughtan J, Jalan R, Pinzani M. Beyond scoring: a modern interpretation of disease progression in chronic liver disease. Gut. (2013) 62:1234–41. doi: 10.1136/gutjnl-2012-302826
6. Kamitani T, Kito K, Nguyen HP, Yeh ETH. Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J Biol Chem. (1997) 272:28557–62. doi: 10.1074/jbc.272.45.28557
7. Pan ZQ, Kentsis A, Dias DC, Yamoah K, Wu K. Nedd8 on cullin: building an expressway to protein destruction. Oncogene. (2004) 23:1985–97. doi: 10.1038/sj.onc.1207414
8. Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. (2009) 458:732–6. doi: 10.1038/nature07884
9. Huang DT, Ayrault O, Hunt HW, Taherbhoy AM, Duda DM, Scott DC, et al. E2-RING expansion of the NEDD8 cascade confers specificity to cullin modification. Mol Cell. (2009) 33:483–95. doi: 10.1016/j.molcel.2009.01.011
10. Saha A, Deshaies RJ. Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Mol Cell. (2008) 32:21–31. doi: 10.1016/j.molcel.2008.08.021
11. Brownell JE, Sintchak MD, Gavin JM, Liao H, Bruzzese FJ, Bump NJ, et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell. (2010) 37:102–11. doi: 10.1016/j.molcel.2009.12.024
12. Hu J, McCall CM, Ohta T, Xiong Y. Targeted ubiquitination of CDT1 by the DDB1-CUL4A-ROC1 ligase in response to DNA damage. Nat Cell Biol. (2004) 6:1003–9. doi: 10.1038/ncb1172
13. Abbas T, Sivaprasad U, Terai K, Amador V, Pagano M, Dutta A. PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev. (2008) 22:2496–506. doi: 10.1101/gad.1676108
14. Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol. (2011) 31:1121–33. doi: 10.1128/MCB.01204-10
15. Rada P, Rojo AI, Evrard-Todeschi N, Innamorato NG, Cotte A, Jaworski T, et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta-TrCP axis. Mol Cell Biol. (2012) 32:3486–99. doi: 10.1128/MCB.00180-12
16. Welcker M, Orian A, Jin JP, Grim JE, Harper JW, Eisenman RN, et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation (vol 101, pg 9085, 2004). Proc Natl Acad Sci U S A. (2006) 103:504. doi: 10.1073/pnas.0402770101
17. Milhollen MA, Traore T, Adams-Duffy J, Thomas MP, Berger AJ, Dang L, et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-{kappa}B-dependent lymphoma. Blood. (2010) 116:1515–23. doi: 10.1182/blood-2010-03-272567
18. Li X, Zhao Q, Liao R, Sun P, Wu X. The SCF(Skp2) ubiquitin ligase complex interacts with the human replication licensing factor Cdt1 and regulates Cdt1 degradation. J Biol Chem. (2003) 278:30854–8. doi: 10.1074/jbc.C300251200
19. Blank JL, Liu XJ, Cosmopoulos K, Bouck DC, Garcia K, Bernard H, et al. Novel DNA damage checkpoints mediating cell death induced by the NEDD8-activating enzyme inhibitor MLN4924. Cancer Res. (2013) 73:225–34. doi: 10.1158/0008-5472.CAN-12-1729
20. Godbersen JC, Humphries LA, Danilova OV, Kebbekus PE, Brown JR, Eastman A, et al. The Nedd8-activating enzyme inhibitor MLN4924 thwarts microenvironment-driven NF-κB activation and induces apoptosis in chronic lymphocytic leukemia B cells. Clin Cancer Res. (2014) 20:1576–89. doi: 10.1158/1078-0432.CCR-13-0987
21. Swords RT, Kelly KR, Smith PG, Garnsey JJ, Mahalingam D, Medina E, et al. Inhibition of NEDD8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood. (2010) 115:3796–800. doi: 10.1182/blood-2009-11-254862
22. Tan M, Li Y, Yang R, Xi N, Sun Y. Inactivation of SAG E3 ubiquitin ligase blocks embryonic stem cell differentiation and sensitizes leukemia cells to retinoid acid. PLoS One. (2011) 6:e27726. doi: 10.1371/journal.pone.0027726
23. Jia L, Yang J, Hao X, Zheng M, He H, Xiong X, et al. Validation of SAG/RBX2/ROC2 E3 ubiquitin ligase as an anticancer and radiosensitizing target. Clin Cancer Res. (2010) 16:814–24. doi: 10.1158/1078-0432.CCR-09-1592
24. Wei D, Li H, Yu J, Sebolt JT, Zhao L, Lawrence TS, et al. Radiosensitization of human pancreatic cancer cells by MLN4924, an investigational NEDD8-activating enzyme inhibitor. Cancer Res. (2012) 72:282–93. doi: 10.1158/0008-5472.CAN-11-2866
25. Chen P, Hu T, Liang Y, Li P, Chen X, Zhang J, et al. Neddylation inhibition activates the extrinsic apoptosis pathway through ATF4-CHOP-DR5 axis in human esophageal cancer cells. Clin Cancer Res. (2016) 22:4145–57. doi: 10.1158/1078-0432.CCR-15-2254
26. Knorr KL, Schneider PA, Meng XW, Dai H, Smith BD, Hess AD, et al. MLN4924 induces Noxa upregulation in acute myelogenous leukemia and synergizes with Bcl-2 inhibitors. Cell Death Differ. (2015) 22:2133–42. doi: 10.1038/cdd.2015.74
27. Pike LR, Phadwal K, Simon AK, Harris AL. ATF4 orchestrates a program of BH3-only protein expression in severe hypoxia. Mol Biol Rep. (2012) 39:10811–22. doi: 10.1007/s11033-012-1975-3
28. Liu X, Jiang Y, Wu J, Zhang W, Liang Y, Jia L, et al. NEDD8-activating enzyme inhibitor, MLN4924 (Pevonedistat) induces NOXA-dependent apoptosis through up-regulation of ATF-4. Biochem Biophys Res Commun. (2017) 488:1–5. doi: 10.1016/j.bbrc.2017.04.122
29. Duan S, Skaar JR, Kuchay S, Toschi A, Kanarek N, Ben-Neriah Y, et al. mTOR generates an auto-amplification loop by triggering the βTrCP- and CK1α-dependent degradation of DEPTOR. Mol Cell. (2011) 44:317–24. doi: 10.1016/j.molcel.2011.09.005
30. Gao D, Inuzuka H, Tan MK, Fukushima H, Locasale JW, Liu P, et al. mTOR drives its own activation via SCF(betaTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Mol Cell. (2011) 44:290–303. doi: 10.1016/j.molcel.2011.08.030
31. Zhao Y, Xiong X, Sun Y. DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(betaTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol Cell. (2011) 44:304–16. doi: 10.1016/j.molcel.2011.08.029
32. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. (2001) 292:464–8. doi: 10.1126/science.1059817
33. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. (2001) 292:468–72. doi: 10.1126/science.1059796
34. Zhao Y, Xiong X, Jia L, Sun Y. Targeting Cullin-RING ligases by MLN4924 induces autophagy via modulating the HIF1-REDD1-TSC1-mTORC1-DEPTOR axis. Cell Death Dis. (2012) 3:e386. doi: 10.1038/cddis.2012.125
35. Jia L, Li H, Sun Y. Induction of p21-dependent senescence by an NAE inhibitor, MLN4924, as a mechanism of growth suppression. Neoplasia. (2011) 13:561–9. doi: 10.1593/neo.11420
36. Kim Y, Starostina NG, Kipreos ET. The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev. (2008) 22:2507–19. doi: 10.1101/gad.1703708
37. Yu ZK, Gervais JL, Zhang H. Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21(CIP1/WAF1) and cyclin D proteins. Proc Natl Acad Sci U S A. (1998) 95:11324–9. doi: 10.1073/pnas.95.19.11324
38. Zhao Y, Morgan MA, Sun Y. Targeting Neddylation pathways to inactivate cullin-RING ligases for anticancer therapy. Antioxid Redox Signal. (2014) 21:2383–400. doi: 10.1089/ars.2013.5795
39. Mackintosh C, Garcia-Dominguez DJ, Ordonez JL, Ginel-Picardo A, Smith PG, Sacristan MP, et al. WEE1 accumulation and deregulation of S-phase proteins mediate MLN4924 potent inhibitory effect on Ewing sarcoma cells. Oncogene. (2013) 32:1441–51. doi: 10.1038/onc.2012.153
40. Chowdhry S, Zhang Y, McMahon M, Sutherland C, Cuadrado A, Hayes JD. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene. (2013) 32:3765–81. doi: 10.1038/onc.2012.388
41. Batuello CN, Hauck PM, Gendron JM, Lehman JA, Mayo LD. Src phosphorylation converts Mdm2 from a ubiquitinating to a neddylating E3 ligase. Proc Natl Acad Sci U S A. (2015) 112:1749–54. doi: 10.1073/pnas.1416656112
42. Abidi N, Xirodimas DP. Regulation of cancer-related pathways by protein NEDDylation and strategies for the use of NEDD8 inhibitors in the clinic. Endocr Relat Cancer. (2015) 22:T55–70. doi: 10.1530/ERC-14-0315
43. Xie P, Zhang M, He S, Lu K, Chen Y, Xing G, et al. The covalent modifier Nedd8 is critical for the activation of Smurf1 ubiquitin ligase in tumorigenesis. Nat Commun. (2014) 5:3733. doi: 10.1038/ncomms4733
44. Choo YS, Vogler G, Wang D, Kalvakuri S, Iliuk A, Tao WA, et al. Regulation of parkin and PINK1 by neddylation. Hum Mol Genet. (2012) 21:2514–23. doi: 10.1093/hmg/dds070
45. Russell RC, Ohh M. NEDD8 acts as a ‘molecular switch' defining the functional selectivity of VHL. EMBO Rep. (2008) 9:486–91. doi: 10.1038/embor.2008.19
46. Takashima O, Tsuruta F, Kigoshi Y, Nakamura S, Kim J, Katoh MC, et al. Brap2 regulates temporal control of NF-kappaB localization mediated by inflammatory response. PLoS One. (2013) 8:e58911. doi: 10.1371/journal.pone.0058911
47. Xirodimas DP, Saville MK, Bourdon JC, Hay RT, Lane DP. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell. (2004) 118:83–97. doi: 10.1016/j.cell.2004.06.016
48. Watson IR, Blanch A, Lin DC, Ohh M, Irwin MS. Mdm2-mediated NEDD8 modification of TAp73 regulates its transactivation function. J Biol Chem. (2006) 281:34096–103. doi: 10.1074/jbc.M603654200
49. Noguchi K, Okumura F, Takahashi N, Kataoka A, Kamiyama T, Todo S, et al. TRIM40 promotes neddylation of IKKgamma and is downregulated in gastrointestinal cancers. Carcinogenesis. (2011) 32:995–1004. doi: 10.1093/carcin/bgr068
50. Gao F, Cheng J, Shi T, Yeh ET. Neddylation of a breast cancer-associated protein recruits a class III histone deacetylase that represses NFκB-dependent transcription. Nat Cell Biol. (2006) 8:1171–7. doi: 10.1038/ncb1483
51. Loftus SJ, Liu G, Carr SM, Munro S, La Thangue NB. NEDDylation regulates E2F-1-dependent transcription. EMBO Rep. (2012) 13:811–8. doi: 10.1038/embor.2012.113
52. Lee MR, Lee D, Shin SK, Kim YH, Choi CY. Inhibition of APP intracellular domain (AICD) transcriptional activity via covalent conjugation with Nedd8. Biochem Biophys Res Commun. (2008) 366:976–81. doi: 10.1016/j.bbrc.2007.12.066
53. Ryu JH, Li SH, Park HS, Park JW, Lee B, Chun YS. Hypoxia-inducible factor alpha subunit stabilization by NEDD8 conjugation is reactive oxygen species-dependent. J Biol Chem. (2011) 286:6963–70. doi: 10.1074/jbc.M110.188706
54. Embade N, Fernandez-Ramos D, Varela-Rey M, Beraza N, Sini M, Gutierrez de Juan V, et al. Murine double minute 2 regulates Hu antigen R stability in human liver and colon cancer through NEDDylation. Hepatology. (2012) 55:1237–48. doi: 10.1002/hep.24795
55. Oved S, Mosesson Y, Zwang Y, Santonico E, Shtiegman K, Marmor MD, et al. Conjugation to Nedd8 instigates ubiquitylation and down-regulation of activated receptor tyrosine kinases. J Biol Chem. (2006) 281:21640–51. doi: 10.1074/jbc.M513034200
56. Zuo W, Huang F, Chiang YJ, Li M, Du J, Ding Y, et al. c-Cbl-mediated neddylation antagonizes ubiquitination and degradation of the TGF-β type II receptor. Mol Cell. (2013) 49:499–510. doi: 10.1016/j.molcel.2012.12.002
57. Jin HS, Liao L, Park Y, Liu YC. Neddylation pathway regulates T-cell function by targeting an adaptor protein Shc and a protein kinase Erk signaling. Proc Natl Acad Sci U S A. (2013) 110:624–9. doi: 10.1073/pnas.1213819110
58. Nagano T, Hashimoto T, Nakashima A, Kikkawa U, Kamada S. X-linked inhibitor of apoptosis protein mediates neddylation by itself but does not function as a NEDD8-E3 ligase for caspase-7. FEBS Lett. (2012) 586:1612–6. doi: 10.1016/j.febslet.2012.04.056
59. Broemer M, Tenev T, Rigbolt KT, Hempel S, Blagoev B, Silke J, et al. Systematic in vivo RNAi analysis identifies IAPs as NEDD8-E3 ligases. Mol Cell. (2010) 40:810–22. doi: 10.1016/j.molcel.2010.11.011
60. Ma T, Chen Y, Zhang F, Yang CY, Wang S, Yu X. RNF111-dependent neddylation activates DNA damage-induced ubiquitination. Mol Cell. (2013) 49:897–907. doi: 10.1016/j.molcel.2013.01.006
61. Sundqvist A, Liu G, Mirsaliotis A, Xirodimas DP. Regulation of nucleolar signalling to p53 through NEDDylation of L11. EMBO Rep. (2009) 10:1132–9. doi: 10.1038/embor.2009.178
62. Zhang J, Bai D, Ma X, Guan J, Zheng X. hCINAP is a novel regulator of ribosomal protein-HDM2-p53 pathway by controlling NEDDylation of ribosomal protein S14. Oncogene. (2014) 33:246–54. doi: 10.1038/onc.2012.560
63. Noh EH, Hwang HS, Hwang HS, Min B, Im E, Chung KC. Covalent NEDD8 conjugation increases RCAN1 protein stability and potentiates its inhibitory action on calcineurin. PLos One. (2012) 7:e0048315. doi: 10.1371/journal.pone.0048315
64. Liu N, Zhang J, Yang X, Jiao T, Zhao X, Li W, et al. HDM2 promotes NEDDylation of Hepatitis B virus HBx to enhance its stability and function. J Virol. (2017) 91:e00340–17. doi: 10.1128/JVI.00340-17
65. Kumar D, Das M, Sauceda C, Ellies LG, Kuo K, Parwal P, et al. Degradation of splicing factor SRSF3 contributes to progressive liver disease. J Clin Invest. (2019) 130:4477–91. doi: 10.1172/JCI127374
66. Zubiete-Franco I, Fernandez-Tussy P, Barbier-Torres L, Simon J, Fernandez-Ramos D, Lopitz-Otsoa F, et al. Deregulated neddylation in liver fibrosis. Hepatology. (2017) 65:694–709. doi: 10.1002/hep.28933
67. Mathewson N, Toubai T, Kapeles S, Sun Y, Oravecz-Wilson K, Tamaki H, et al. Neddylation plays an important role in the regulation of murine and human dendritic cell function. Blood. (2013) 122:2062–73. doi: 10.1182/blood-2013-02-486373
68. Heo MJ, Kang SH, Kim YS, Lee JM, Yu J, Kim HR, et al. UBC12-mediated SREBP-1 neddylation worsens metastatic tumor prognosis. Int J Cancer. (2020) 147:2550–63. doi: 10.1002/ijc.33113
69. Leidecker O, Matic I, Mahata B, Pion E, Xirodimas DP. The ubiquitin E1 enzyme Ube1 mediates NEDD8 activation under diverse stress conditions. Cell Cycle. (2012) 11:1142–50. doi: 10.4161/cc.11.6.19559
70. Schmidt MHH, Dikic I. Ubiquitin and NEDD8: brothers in arms. Sci STKE. (2006) 2006:.pe50. doi: 10.1126/stke.3622006pe50
71. Richter K, Haslbeck M, Buchner J. The heat shock response: life on the verge of death. Mol Cell. (2010) 40:253–66. doi: 10.1016/j.molcel.2010.10.006
72. Bailly AP, Perrin A, Serrano-Macia M, Maghames C, Leidecker O, Trauchessec H, et al. The balance between mono- and NEDD8-chains controlled by NEDP1 upon DNA damage is a regulatory module of the HSP70 ATPase activity. Cell Rep. (2019) 29:212–24.e8. doi: 10.1016/j.celrep.2019.08.070
73. Slagle BL, Bouchard MJ. Hepatitis B virus X and regulation of viral gene expression. Cold Spring Harb Perspect Med. (2016) 6:a021402. doi: 10.1101/cshperspect.a021402
74. Guo L, Wang X, Ren L, Zeng M, Wang S, Weng Y, et al. HBx affects CUL4-DDB1 function in both positive and negative manners. Biochem Biophys Res Commun. (2014) 450:1492–7. doi: 10.1016/j.bbrc.2014.07.019
75. Li T, Robert EI, van Breugel PC, Strubin M, Zheng N. A promiscuous alpha-helical motif anchors viral hijackers and substrate receptors to the CUL4-DDB1 ubiquitin ligase machinery. Nat Struct Mol Biol. (2010) 17:105–11. doi: 10.1038/nsmb.1719
76. Hodgson AJ, Hyser JM, Keasler VV, Cang Y, Slagle BL. Hepatitis B virus regulatory HBx protein binding to DDB1 is required but is not sufficient for maximal HBV replication. Virology. (2012) 426:73–82. doi: 10.1016/j.virol.2012.01.021
77. Leupin O, Bontron S, Schaeffer C, Strubin M. Hepatitis B virus X protein stimulates viral genome replication via a DDB1-dependent pathway distinct from that leading to cell death. J Virol. (2005) 79:4238–45. doi: 10.1128/JVI.79.7.4238-4245.2005
78. Zhao J, Wu J, Cai H, Wang D, Yu L, Zhang WH. E3 ubiquitin ligase siah-1 is down-regulated and fails to target natural HBx truncates for degradation in hepatocellular carcinoma. J Cancer. (2016) 7:418–26. doi: 10.7150/jca.13019
79. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. (2018) 15:11–20. doi: 10.1038/nrgastro.2017.109
80. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology. (2018) 67:328–57. doi: 10.1002/hep.29367
81. Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology. (2010) 51:679–89. doi: 10.1002/hep.23280
82. Zhang X, Zhang Y-L, Qiu G, Pian L, Guo L, Cao H, et al. Hepatic neddylation targets and stabilizes electron transfer flavoproteins to facilitate fatty acid beta-oxidation. Proc Natl Acad Sci U S A. (2020) 117:2473–83. doi: 10.1073/pnas.1910765117
83. Sen S, Langiewicz M, Jumaa H, Webster NJG. Deletion of serine/arginine-rich splicing factor 3 in hepatocytes predisposes to hepatocellular carcinoma in mice. Hepatology. (2015) 61:171–83. doi: 10.1002/hep.27380
84. Ju UI, Jeong DW, Seo J, Park JB, Park JW, Suh KS, et al. Neddylation of sterol regulatory element-binding protein 1c is a potential therapeutic target for nonalcoholic fatty liver treatment. Cell Death Dis. (2020) 11:283. doi: 10.1038/s41419-020-2472-6
85. Dehnad A, Fan W, Jiang JX, Fish SR, Li Y, Das S, et al. AGER1 downregulation associates with fibrosis in nonalcoholic steatohepatitis and type 2 diabetes. J Clin Invest. (2020) 130:4320–30. doi: 10.1172/JCI133051
86. Pinzani M, Rombouts K. Liver fibrosis: from the bench to clinical targets. Dig Liver Dis. (2004) 36:231–42. doi: 10.1016/j.dld.2004.01.003
87. Schuppan D, Kim YO. Evolving therapies for liver fibrosis. J Clin Invest. (2013) 123:1887–901. doi: 10.1172/JCI66028
88. Leist M, Gantner F, Bohlinger I, Tiegs G, Germann PG, Wendel A. Tumor necrosis factor-induced hepatocyte apoptosis precedes liver failure in experimental murine shock models. Am J Pathol. (1995) 146:1220–34.
89. Saiman Y, Friedman SL. The role of chemokines in acute liver injury. Front Physiol. (2012) 3:213. doi: 10.3389/fphys.2012.00213
90. Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. (2013) 4:2823. doi: 10.1038/ncomms3823
91. Wertz IE, O'Rourke KM, Zhang Z, Dornan D, Arnott D, Deshaies RJ, et al. Human De-etiolated-1 regulates c-Jun by assembling a CUL4A ubiquitin ligase. Science. (2004) 303:1371–4. doi: 10.1126/science.1093549
92. Collins PE, Mitxitorena I, Carmody RJ. The ubiquitination of NF-κB subunits in the control of transcription. Cells. (2016) 5:23. doi: 10.3390/cells5020023
93. Sasaki R, Kanda T, Nakamura M, Nakamoto S, Haga Y, Wu S, et al. Possible involvement of Hepatitis B virus infection of hepatocytes in the attenuation of apoptosis in hepatic stellate cells. PLoS One. (2016) 11:e0146314. doi: 10.1371/journal.pone.0146314
94. Hellerbrand C, Stefanovic B, Giordano F, Burchardt ER, Brenner DA. The role of TGFβ1 in initiating hepatic stellate cell activation in vivo. J Hepatol. (1999) 30:77–87. doi: 10.1016/S0168-8278(99)80010-5
95. Troeger JS, Mederacke I, Gwak G-Y, Dapito DH, Mu X, Hsu CC, et al. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology. (2012) 143:1073–83.e22. doi: 10.1053/j.gastro.2012.06.036
96. Tapper EB, Parikh ND. Mortality due to cirrhosis and liver cancer in the United States, 1999-2016: observational study. BMJ Br Med J. (2018) 362:k2817. doi: 10.1136/bmj.k2817
97. Yu J, Huang WL, Xu QG, Zhang L, Sun SH, Zhou WP, et al. Overactivated neddylation pathway in human hepatocellular carcinoma. Cancer Med. (2018) 7:3363–72. doi: 10.1002/cam4.1578
98. Yang Z, Zhang J, Lin X, Wu D, Li G, Zhong C, et al. Inhibition of neddylation modification by MLN4924 sensitizes hepatocellular carcinoma cells to sorafenib. Oncol Rep. (2019) 41:3257–69. doi: 10.3892/or.2019.7098
99. McLarnon A. Cancer: Mdm2-regulated stabilization of HuR by neddylation in HCC and colon cancer–a possible target for therapy. Nat Rev Gastroenterol Hepatol. (2011) 9:4. doi: 10.1038/nrgastro.2011.241
100. Barbier-Torres L, Delgado TC, García-Rodríguez JL, Zubiete-Franco I, Fernández-Ramos D, Buqué X, et al. Stabilization of LKB1 and Akt by neddylation regulates energy metabolism in liver cancer. Oncotarget. (2015) 6:2509–23. doi: 10.18632/oncotarget.3191
101. Zhou L, Zhang W, Sun Y, Jia L. Protein neddylation and its alterations in human cancers for targeted therapy. Cell Signal. (2018) 44:92–102. doi: 10.1016/j.cellsig.2018.01.009
102. Luo Z, Yu G, Lee HW, Li L, Wang L, Yang D, et al. The Nedd8-activating enzyme inhibitor MLN4924 induces autophagy and apoptosis to suppress liver cancer cell growth. Cancer Res. (2012) 72:3360–71. doi: 10.1158/0008-5472.CAN-12-0388
103. Zhao Y, Sun Y. Targeting the mTOR-DEPTOR pathway by CRL E3 ubiquitin ligases: therapeutic application. Neoplasia. (2012) 14:360–7. doi: 10.1593/neo.12532
104. Liang Y, Jiang Y, Jin X, Chen P, Heng Y, Cai L, et al. Neddylation inhibition activates the protective autophagy through NF-κB-catalase-ATF3 Axis in human esophageal cancer cells. Cell Commun Signal. (2020) 18:72. doi: 10.1186/s12964-020-00576-z
105. Lu G, Yi J, Gubas A, Wang YT, Wu Y, Ren Y, et al. Suppression of autophagy during mitosis via CUL4-RING ubiquitin ligases-mediated WIPI2 polyubiquitination and proteasomal degradation. Autophagy. (2019) 15:1917–34. doi: 10.1080/15548627.2019.1596484
106. Gorlach A, Bonello S. The cross-talk between NF-kappaB and HIF-1: further evidence for a significant liaison. Biochem J. (2008) 412:e17–9. doi: 10.1042/BJ20080920
107. Hon WC, Wilson MI, Harlos K, Claridge TDW, Schofield CJ, Pugh CW, et al. Structural basis for the recognition of hydroxyproline in alpha IF-1 alpha by pVHL. Nature. (2002) 417:975–8. doi: 10.1038/nature00767
108. Zhang W, Shi X, Peng Y, Wu M, Zhang P, Xie R, et al. HIF-1α promotes epithelial-mesenchymal transition and metastasis through direct regulation of ZEB1 in colorectal cancer. PLoS One. (2015) 10:e0129603. doi: 10.1371/journal.pone.0129603
109. Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, et al. Hypoxia-induced neutrophil survival is mediated by HIF-1α-dependent NF-κB activity. J Exp Med. (2005) 201:105–15. doi: 10.1084/jem.20040624
110. Cannito S, Foglia B, Villano G, Turato C, Delgado TC, Morello E, et al. SerpinB3 differently up-regulates hypoxia inducible factors-1 alpha and-2 alpha in hepatocellular carcinoma: mechanisms revealing novel potential therapeutic targets. Cancers. (2019) 11:1933. doi: 10.3390/cancers11121933
111. Belaiba RS, Bonello S, Zahringer C, Schmidt S, Hess J, Kietzmann T, et al. Hypoxia up-regulates hypoxia-inducible factor-1alpha transcription by involving phosphatidylinositol 3-kinase and nuclear factor κB in pulmonary artery smooth muscle cells. Mol Biol Cell. (2007) 18:4691–7. doi: 10.1091/mbc.e07-04-0391
112. Chang SC, Ding JL. Ubiquitination by SAG regulates macrophage survival/death and immune response during infection. Cell Death Differ. (2014) 21:1388–98. doi: 10.1038/cdd.2014.54
113. Mathewson ND, Fujiwara H, Wu SR, Toubai T, Oravecz-Wilson K, Sun Y, et al. SAG/Rbx2-dependent neddylation regulates T-cell responses. Am J Pathol. (2016) 186:2679–91. doi: 10.1016/j.ajpath.2016.06.014
114. Wang Y, Luo Z, Pan Y, Wang W, Zhou X, Jeong LS, et al. Targeting protein neddylation with an NEDD8-activating enzyme inhibitor MLN4924 induced apoptosis or senescence in human lymphoma cells. Cancer Biol Ther. (2015) 16:420–9. doi: 10.1080/15384047.2014.1003003
115. Godbersen JC, Paiva C, Danilova OV, Berger A, Brown JR, Danilov AV. Targeting neddylation effectively antagonizes nuclear factor-κB in chronic lymphocytic leukemia B-cells. Leuk Lymphoma. (2015) 56:1566–9. doi: 10.3109/10428194.2014.990901
116. Paiva C, Godbersen JC, Berger A, Brown JR, Danilov AV. Targeting neddylation induces DNA damage and checkpoint activation and sensitizes chronic lymphocytic leukemia B cells to alkylating agents. Cell Death Dis. (2015) 6:e1807. doi: 10.1038/cddis.2015.161
117. Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol. (2013) 59:583–94. doi: 10.1016/j.jhep.2013.03.033
118. Sekiba K, Otsuka M, Ohno M, Yamagami M, Kishikawa T, Seimiya T, et al. Pevonedistat, a neuronal precursor cell-expressed developmentally down-regulated protein 8–activating enzyme inhibitor of hepatitis B virus. Hepatology. (2019) 69, 1903–15. doi: 10.1002/hep.30491
119. Murphy CM, Xu Y, Li F, Nio K, Reszka-Blanco N, Li X, et al. Hepatitis B virus X protein promotes degradation of SMC5/6 to enhance HBV replication. Cell Rep. (2016) 16:2846–54. doi: 10.1016/j.celrep.2016.08.026
120. Decorsiere A, Mueller H, Van Breugel PC, Abdul F, Gerossier L, Beran RK, et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature. (2016) 531:386–9. doi: 10.1038/nature17170
121. Serrano-Macia M, Azkargorta M, Simon J, Lopitz Otsoa F, Cardoso Delgado T, Fernandez Ramos D, et al. NEDDylation inhibition as a new potential therapy of nonalcoholic fatty liver disease. J Hepatol. (2019) 70:E546. doi: 10.1016/S0618-8278(19)31082-5
122. Chang F-M, Reyna SM, Granados JC, Wei S-J, Innis-Whitehouse W, Maffi SK, et al. Inhibition of neddylation represses lipopolysaccharide-induced proinflammatory cytokine production in macrophage cells. J Biol Chem. (2012) 287:35756–67. doi: 10.1074/jbc.M112.397703
123. Milhollen MA, Thomas MP, Narayanan U, Traore T, Riceberg J, Amidon BS, et al. Treatment-emergent mutations in NAE beta confer resistance to the NEDD8-activating enzyme inhibitor MLN4924. Cancer Cell. (2012) 21:388–401. doi: 10.1016/j.ccr.2012.02.009
124. Dawkins J, Webster RM. The hepatocellular carcinoma market. Nat Rev Drug Discov. (2019) 18:13–4. doi: 10.1038/nrd.2018.146
125. Bu FT, Chen Y, Yu HX, Chen X, Yang Y, Pan XY, et al. SENP2 alleviates CCl(4)-induced liver fibrosis by promoting activated hepatic stellate cell apoptosis and reversion. Toxicol Lett. (2018) 289:86–98. doi: 10.1016/j.toxlet.2018.03.010
126. Ramani K, Tomasi ML, Yang H, Ko K, Lu SC. Mechanism and significance of changes in glutamate-cysteine ligase expression during hepatic fibrogenesis. J Biol Chem. (2012) 287:36341–55. doi: 10.1074/jbc.M112.370775
127. Lachiondo-Ortega S, Mercado-Gómez M, Serrano-Maciá M, Lopitz-Otsoa F, Salas-Villalobos TB, Varela-Rey M, et al. Ubiquitin-like post-translational modifications (Ubl-PTMs): small peptides with huge impact in liver fibrosis. Cells. (2019) 8:1575. doi: 10.3390/cells8121575
Keywords: neddylation, HBV, NAFLD, liver fibrosis, HCC, therapy, MLN4924
Citation: Yao J, Liang X, Liu Y and Zheng M (2020) Neddylation: A Versatile Pathway Takes on Chronic Liver Diseases. Front. Med. 7:586881. doi: 10.3389/fmed.2020.586881
Received: 24 July 2020; Accepted: 31 August 2020;
Published: 19 October 2020.
Edited by:
Gianfranco Danilo Alpini, Indiana University, United StatesReviewed by:
Maria L. Martinez Chantar, CIC bioGUNE, SpainKeisaku Sato, Indiana University, United States
Copyright © 2020 Yao, Liang, Liu and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Zheng, bWluemhlbmdAemp1LmVkdS5jbg==