 Natalia Jarzebska1,2
Natalia Jarzebska1,2 Ekaterina S. Karetnikova3
Ekaterina S. Karetnikova3 Alexander G. Markov3
Alexander G. Markov3 Michael Kasper4
Michael Kasper4 Roman N. Rodionov2
Roman N. Rodionov2 Peter M. Spieth1*
Peter M. Spieth1*- 1Department of Anesthesiology and Critical Care Medicine, University Hospital Dresden, Technische Universität Dresden, Dresden, Germany
- 2Division of Angiology, Department of Internal Medicine III, University Center for Vascular Medicine, University Hospital Dresden, Technische Universität Dresden, Dresden, Germany
- 3Department of General Physiology, Saint-Petersburg State University, Saint Petersburg, Russia
- 4Institute of Anatomy, Technische Universität Dresden, Dresden, Germany
Radiation-induced pulmonary fibrosis is a common severe long-time complication of radiation therapy for tumors of the thorax. Current therapeutic options used in the clinic include only supportive managements strategies, such as anti-inflammatory treatment using steroids, their efficacy, however, is far from being satisfactory. Recent studies have demonstrated that the development of lung fibrosis is a dynamic and complex process, involving the release of reactive oxygen species, activation of Toll-like receptors, recruitment of inflammatory cells, excessive production of nitric oxide and production of collagen by activated myofibroblasts. In this review we summarized the current state of knowledge on the pathophysiological processes leading to the development of lung fibrosis and we also discussed the possible treatment options.
Introduction
Radiation therapy is an integral part of the treatment of various malignant neoplasms (1), including tumors of the thorax (2–4). Radiation induced pulmonary fibrosis is a common complication of this therapy affecting 5–50% of patients, which significantly limits available treatment options even after successful eradication of the tumor itself (5). Radiation-induced lung injury can be divided into two distinct forms: (1) classical radiation pneumonitis, which is restricted to the irradiated area and leads to fibrosis and (2) sporadic radiation pneumonitis, affecting much broader area than the tissue which was subjected to irradiation and resulting in bilateral lymphocytic alveolitis, mediated by various immunological processes (6, 7). Currently, corticosteroids, azathioprine, and cyclosporin A are used as first line treatment for post-radiation pneumonitis (8, 9). Ambroxol and angiotensin-converting enzyme inhibitors are suggested as additional drugs to prevent or attenuate the extent of pneumonitis (10, 11). Lung fibrosis, however, is an end stage of radiation-induced pneumonitis and in most of the cases it is resistant to all currently known pharmacological interventions. Previously only symptomatic treatment with antitussive agents, oxygen therapy, and mechanical ventilation was available (9). More recently, it was shown that nintedanib, a multiple tyrosine kinase inhibitor, and pirfenidone, a TGF-β inhibitor, exhibit anti-inflammatory and anti-fibrotic effects in experimental murine irradiation models (12, 13) and these drugs are now in clinical use for treatment of pulmonary fibrosis (13, 14).
The aim of this review is to summarize and analyse the currently available data on the pathophysiological mechanisms leading to lung fibrosis. Hopefully it will allow identifying promising directions for further research, which will lead to the development of effective preventive and/or treatment approaches to tackle post-radiation pulmonary fibrosis.
Clinical Perspective
The lung parenchyma is one of the most radiation-sensitive tissues with the alveolar-capillary unit being the most vulnerable part of the lung (15). The likelihood of developing side effects of radiation therapy, like difficulty swallowing, shortness of breath, breast or nipple soreness and shoulder stiffness depends on many factors like patient-related variables, the nature of the tumor itself and the parameters of radiation therapy (16–18). Specifically, it has been demonstrated that age above 65 years, smoking, presence of co-morbidities and poor pulmonary function tests (decreased forced expiratory volume and poor diffusing capacity of the lungs for carbon monoxide) have been associated with increased incidence of radiation pneumonitis (19, 20). Additionally, radiation therapy administered for mid and lower lobe lung tumors, breast cancer with tangential fields and esophageal cancer are also correlated with significantly increased incidence of radiation pneumonitis (20). Other risk factors include more than 30% of the planned target volume receiving 20 Gy or more, and the use of certain medications like bleomycin, cyclophosphamide, vincristine, taxanes, doxorubicin, dactiomycin, mitomycin, gemcitabine, erlotinib and bevacizumab (21–25). There are also certain molecular markers reported, which could be used to improve individualized treatment. For example, patients with elevated post treatment serum TGF-β1 (transforming growth factor beta 1) levels above baseline exhibit a significantly higher risk of radiation induced lung injury (26, 27). Furthermore, persistently elevated IL1a (interleukin 1 alpha) and IL6 (interleukin 6), ICAM 1 (intercellular adhesion molecule 1), SP-A (surfactant protein A) and SP-D (surfactant protein D) serum levels are predictive for the development of radiation pneumonitis (28–30). Furthermore, certain ATM (ataxia telangiectasia mutated) gene polymorphisms have been associated with increased risk of radiation pneumonitis (31). Finally, there are also studies showing that the risk of severe radiation pneumonitis is increased in patients with pre-existing idiopathic pulmonary fibrosis (IPF) (32–34). Nonetheless, there are no formal guidelines limiting the eligibility of IPF patients for thoracic radiotherapy, even though according to the recommendations of the European Organization for Research and Treatment of Cancer radiotherapy for lung cancer should be avoided in IPF patients (35). The decision of whether or not to treat these vulnerable patients with irradiation of the thorax should be made after a careful evaluation of a multidisciplinary team taking into account a comprehensive risk assessment. Clearly more research is needed to establish the safest treatment options. The group of Yoshitake and colleagues investigated the side effects of thoracic stereotactic body radiotherapy (SBRT) in patients with interstitial lung changes (ILC) and found that the risk of radiation pneumonitis grade 2 or more was increased in patients with subclinical ILC (36). Even though the topic still requires more research, dose limits for SBRT in patients with pre-existing ILC are recommended (37). There is an increasing body of evidence suggesting that proton beam therapy could be a safer option in patients with IPF than conventional radiotherapy or SBRT (38–41). However, prospective, multicentre studies with large number of patients enrolled are still needed. Nintedanib is a relatively new drug that can be used to slow the progression of IPF. It is a tyrosine kinase inhibitor affecting tyrosine phosphorylation on platelet-derived growth factor, vascular endothelial growth factor and fibroblast growth factor leading to suppression of inflammation, angiogenesis and fibroblast activation (42). The drug has been shown to prevent/retard acute exacerbations in patients with IPF (43, 44), however it was not effective in alleviating bleomycin-induced pulmonary fibrosis in an animal model (45).
Apart from causing pneumonitis and pulmonary fibrosis, radiotherapy is also thought to contribute to activation of cancer-associated fibroblasts (CAF), at least in some conditions. CAFs are a heterogeneous group of stromal cells inside a tumor, which differ epigenetically and phenotypically from normal fibroblasts. The major cellular origin of CAFs are normal fibroblasts that are transformed by the tumor microenvironment, however, smooth muscle cells, pericytes, adipocytes, mesenchymal stem cells and endothelial cells are also demonstrated to be the source of CAFs (46, 47). Those cells modulate the composition of the extracellular matrix through secretion of growth factors and cytokines which lead to regulation of tumor proliferation, invasion and the potential for metastasis. Interestingly, there are also studies demonstrating that CAFs can slow down tumor progression by formation of a physical barrier which limits tumor growth and the possibility to migrate (48). Radiotherapy affects the proliferation of CAFs at the genetic level, but does not limit the ability of these cells to sustain a microenvironment which supports tumor growth (49). The influence of radiotherapy on the CAFs is a matter of ongoing investigations. On one hand, there is data supporting the idea that irradiation can modify the CAFs leading to abrogation of tumor promoting ability (50), however, there are also studies showing that irradiated fibroblasts can induce epithelial-to-mesenchymal transition (EMT) in cancer cells and promote invasiveness through elevation of TGF-β levels (51, 52). In line with this data, the group of Ohuchida et al. showed that irradiated CAFs increase the invasiveness in pancreatic cancer cells (53). In another study it was demonstrated in the same type of cancer that CAFs after irradiation induce EMT and invasiveness by activation of the P38 pathway (54).
Cascade of Pathophysiological Processes Leading to Lung Fibrosis
Primary and Secondary Damage to Macromolecules
Exposure to radiation causes both direct and indirect macromolecular damage and also triggers the generation of various reactive oxygen species (ROS), including superoxide (O2−), hydrogen peroxide (H2O2) and hydroxyl radical (⦁OH) (55, 56). The combination of irradiation itself and the generation of ROS affect all types of macromolecules, including DNA, proteins, proteoglycans and lipids. However, double-strand breaks in DNA have most severe consequences for the cells (57). Primary and secondary damage triggers cascades and networks of biochemical reactions and the balance between them determines if normal lung tissue will be restored or if the fibrotic response will be initiated (58).
DAMPs-TLRs-Pro-Inflammatory Cytokines
Cell damage caused by irradiation leads to the accumulation of damage-associated molecular patterns (DAMPs) in the intercellular space of the lungs. The most prevalent types of DAMPs include extracellular DNA, extracellular ATP, high mobility group box chromosomal protein B1, heat-shock protein 70, uric acid and low-molecular hyaluronan (59). These DAMPs are generated in aseptic conditions and activate cell surface-bound TOLL-like receptors (TLR) 2 and 4 (18). TLRs are expressed in many cells, including alveolar epithelial cells of type II (60), endothelial cells, alveolar macrophages, fibroblasts, dendritic cells, monocytes, lymphocytes, neutrophils and natural killer cells (18, 61, 62) and their activation triggers sequential release of different mediators, including pro-IL-1β, pro-IL-18 and type I interferon (63). Interestingly, studies on the development of post-radiation pulmonary fibrosis in mice with global deficiency of TLR2 and TLR4, as well as with the knockout of MyD88 (Innate Immune Signal Transduction Adaptor) showed that these mice developed more severe pulmonary fibrosis compared to wild-type mice (60, 64, 65). This fact indirectly indicates that these signaling cascades promote regeneration of lung tissue rather than damage.
Pyroptosis Pathways of Inflammasome Activation
Simultaneously with the stimulation of synthesis of pro-IL1β, pro-IL18, and type I interferons, irradiation causes pyroptosis, a highly inflammatory form of programmed cell death (66). Specifically, irradiation can activate non-active multi-protein signaling complexes NLRP3 (NLR family, pyrin domain-containing 3) present in inflammasomes of various lung cells (18, 67, 68). The activation can occur directly through ROS (69) or indirectly through DAMPs like ATP, ADP, and adenosine which bind to P2XR, P2YR, and P1R (ATP-gated ion channels) (70) and through low molecular weight hyaluronan activating the surface receptors CD44 (69). Another possible pathway of activation is through the outflow of cell content, particularly uric acid, caused by irradiation-mediated damage to cell membranes (18, 68). Activated inflammasomes initiate activation of caspase-1 which cleaves pro-IL-1β and pro-IL-18 with the formation of active IL-1β and IL-18 (71–73) and also disrupts the integrity of the outer cytoplasmic membranes, which leads to osmotic lysis of cells and increases the amount of DAMPs in the intercellular milieu (73, 74). These processes lead to increased synthesis of proinflammatory cytokines and pyroptosis in lung tissue. IL-1β also plays an important role in initiation of the acute inflammatory responses by binding to its ubiquitously expressed receptor (IL-1R-1) and stimulation of the production of TGF-β, pro-IL-1β, TNF-α, and IL- 6. Together these cytokines promote the recruitment and activation of innate immune cells, as well as trigger and enhance cascades of aseptic inflammation in the damaged part of the lung (18, 71).
P2X Purinoreceptor 7
The NLRP3 inflammasome might be also involved in the pathogenesis of radiation-induced fibrosis by activating purinergic P2X and P2Y receptors (purinergic signaling). Purinergic signaling is involved in ionizing radiation-induced biological effects (75). An important protein in these pathways is the ATP-stimulated P2X purinoreceptor 7 (P2X7R), selectively present in alveolar macrophages and in alveolar epithelial type I cells in lungs and known to regulate the activation of the NLRP3 inflammasome. A rapid increase in the extracellular ATP concentration, for example after damage of a tissue or after cell death, causes an endogenous signal of danger and activates the NLRP3 inflammasome by binding of ATP to P2X7R which functions as ligand-controlled ion channel (76). The effect of ATP may be transmitted by P2X7R, which causes the formation of pannexin-1 pores. Their opening leads to a fast outflow of K+-ions from the cytosol (77). The decrease in the cytoplasmic K+ concentration causes assembly and activation of the inflammasome leading to an auto- catalytic cleavage of the inactive procaspase-1 to its active form. The inflammasome transmitted caspase-1-dependent proteolytic cleavage of inactive proforms of cytokines of the IL-1-family (pro-IL-1β, pro-IL-18) results in biological active forms, which are released from cells as a part of the inflammatory reaction. Further, the involvement of the connexin43 hemichannel in the ATP release downstream of the P2X7R in response to irradiation has to be taken into account (78). An upregulation of connexin43 in alveolar epithelial cells of rats with radiation-induced pulmonary fibrosis has been described earlier (79). Furthermore, P2X7R knockout mice exhibited dramatically reduced lung inflammation and fibrosis, underlining the important role of P2X7R in fibrotic diseases (80).
Inducible Nitric Oxide Synthase (iNOS), Asymmetric Dimethyarginine (ADMA) and Dimethyarginine Dimethyaminohydrolase (DDAH)
IL-1β, TNF-α, and other cytokines activate iNOS, which starts to produce large quantities of NO. When nitric oxide is produced, it can bind to O2− with the formation of peroxynitrite (ONOO−) causing secondary damage to macromolecules (DNA, proteins, glucosaminoglycan hyaluronan, etc.) (81). The importance of peroxynitrite in lung fibrosis development has been demonstrated in animal studies where is was shown that iNOS knockout mice had significantly less fibrotic changes in the lungs compared to wild-type mice after bleomycin treatment (81–83). The activity of iNOS in vivo is regulated by asymmetric dimethylarginine (ADMA), its endogenous inhibitor. It has been shown that at concentrations exceeding 10μM ADMA not only decreases NO production by iNOS but also uncouples it with the production of O2− (84). Therefore, it was speculated that the addition of ADMA to already fully active iNOS cannot reduce the damage to the lung tissue mediated by NO, but paradoxically can lead to further exacerbation of the damage due to the peroxynitrite formation. This assumption was confirmed by Wells and colleagues in in vitro and in vivo studies (84). They showed that ADMA elevates collagen production in primary mouse lung fibroblasts and that ADMA infusion via osmotic minipumps for 2 weeks caused collagen deposition in mice lungs (85). In the same manuscript the authors proposed an interesting pathway of pro-fibrotic ADMA activity where ADMA increases arginase activity leading to elevated levels of ornithine and urea; ornithine is a precursor of proline, an amino acid essential for collagen synthesis (85). In line with these observations, other study demonstrated that 21 days after bleomycin injury there was no difference in the fibrotic response in mice supplemented with ADMA and those given placebo (83). Concentration of ADMA is regulated by the activity of dimethylarginine dimethylaminohydrolase (DDAH), an enzyme which cleavages ADMA (86, 87). DDAH expression and activity increase in course of lung fibrosis development, through TGF-β and IL- 6 which increase mRNA levels of DDAH2 (one of two DDAH isoforms) (83); and through IL-1β which enhances DDAH activity and rises its intracellular concentrations (88). Increased DDAH expression and activity in turn reduces ADMA concentration in lung tissue, which results in greater amounts of NO produced by iNOS, contributing to further damage of lung tissue.
Sources of Fibroblasts: Epithelial-Mesenchymal Transition (EMT), Endothelial-Mesenchymal Transition (EndMT) and Recruitment of Fibrocytes and Myofibroblasts
Increased concentrations of TGF-β1 in course of development of lung fibrosis induce EMT (epithelial-mesenchymal transition) of alveolar epithelial cells type II (AET II cells) (89) and EndMT (endothelial-mesenchymal transition) of microvascular endothelial cells (90). During EMT and EndMT, the polarized epithelial and endothelial cells loose their polarity and specific markers such as E- or VE-cadherin, tight junction proteins etc., increase expression of mesenchymal markers (vimentin, collagens I and III, α-SMA etc), move to the interstitium and gain phenotype like mesenchymal cells (89). A convincing proof of EMT in vivo was demonstrated in lineage-tracing studies (91–93) where mice with selective expression of β-galactosidase (β-gal) only in lung epithelial cells were used. After induction of lung fibrosis by TGF-β1 (92), thorax irradiation (91) or by bleomycin treatment (93) the authors reported the appearance of expression of different mesenchymal markers (α-SMA, vimentin, S100A4) in β-gal-positive cells and the co-expression of mesenchymal markers and pro-surfactant C (a marker of AET II cells) (91–94). There is a controversy about the contribution of epithelial cells to myofibroblast population and to the physiological role of EMT in vivo (95, 96). Moreover, recent findings suggest that AET II cells undergoing EMT promote a pro-fibrotic microenvironment through paracrine signaling activating local fibroblasts (97). An EMT of AET I cells is unknown. The various cytokines produced during aseptic inflammation also promote the recruitment of fibrocytes from the peripheral blood and their differentiation into fibroblasts in the lung tissue, where they produce extracellular matrix (93, 98). The last step in the development of radiation-induced pulmonary fibrosis is the formation of myofibroblasts from fibroblasts (99). The cellular source of myofibroblasts has been a subject of debate in recent years. It was suggested that the myofibroblasts pool is heterogeneous and derives from multiple sources, such as resident fibroblasts, circulating fibroblasts, perivascular mesenchymal cells, and alveolar epithelial cells (95, 100–102). However, recent studies using genetic lineage tracing identified the resident lipid-droplet-containing interstitial fibroblasts, as a precursor cell for the myofibroblast, at least in the bleomycin model of lung fibrosis in mice (103). Regardless of the source, the differentiation of fibroblast into myofibroblasts is driven by TGF-β1 (103). Upon activation these cells produce excessive amounts of extracellular matrix (ECM) (fibronectin, collagen etc), which causes contraction of the lung tissue (104, 105) and increase the thickness of the alveolar-capillary membrane together leading to irreversible compromised diffusing capacity of the lung (106) and restrictive ventilatory insufficiency.
The various processes leading to irradiation-induced lung fibrosis are summarized in Figure 1.
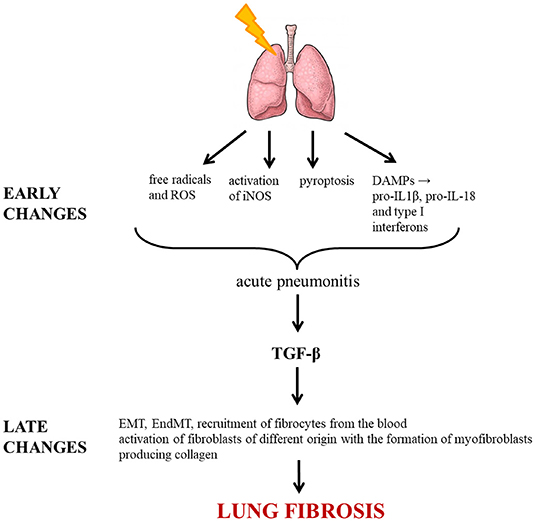
Figure 1. Molecular pathways triggered by irradiation leading to lung fibrosis. ROS, reactive oxygen species; DAMP, damage-associated molecular pattern; IL, interleukin; iNOS, inducible nitric oxide synthase; TGF-β, transforming growth factor beta; EMT, epithelial-mesenchymal transition; EndMT, endothelial-mesenchymal transition.
Research Targets Regarding Lung Fibrosis Prevention and Treatment
Minimizing Protein Damage
Since the primary and secondary damage to macromolecules is the starting point for the development of all pathophysiological mechanisms of lung fibrosis, one of the therapeutic approaches considered was the use of a non-toxic anti-ulcer drug geranylgeranlyacetone (GGA), which induces the expression of intracellular chaperone heat shock protein 70. The plausibility of this approach was shown by the group of Kim and colleagues in a study where they treated mice with GGA before and after irradiation and found out that after 6 months the animals had less pronounced lung fibrosis and had less signs of EMT in lung tissue compared with the placebo group (107). However, one possible drawback in using this drug in clinical practice is that it can also reduce the efficacy of irradiation as an anti-tumor treatment (108).
Influence on DDAH-ADMA-iNOS-NO Axis
The second possible therapeutic approach is based on the rationale that activated iNOS in the development of lung fibrosis is the source of excess of nitric oxide, which causes secondary injury to the lung. Studies in mice showed that at least in the bleomycin model the use of iNOS inhibitors resulted in significant reduction in the degree of lung fibrosis compared to animals from the placebo groups (81, 83). Pullamsetti and colleagues demonstrated a protective role of a selective DDAH inhibitor L-291 in the development of bleomycin-induced lung fibrosis. Interestingly, the inhibitor seemed to affect both ADMA-independent (antifibrotic) and ADMA-dependent (antiproliferative) pathways of lung fibrosis development (83).
Reducing TGF-β1-Mediated Effects
According to the current state of knowledge, TGF-β1 plays a key role in the development of lung fibrosis (108). Therefore, many efforts are concentrated on different ways to interfere with the pathways triggered by TGF-β1 activity. One possibility would be to block signal transduction mediated by TGF-β1 and the feasibility of this approach was demonstrated in rodents where the use of small molecules inhibiting TGF-β1 receptors resulted in decreased severity of lung fibrosis induced by irradiation (26, 109). Another approach to limit the activity of TGF-β1 is to use specific small interfering RNA (siRNA). In a study performed by Lu and co-workers it was demonstrated that in a mouse model the use of TGF-β1-siRNA significantly attenuated the increase in TGF-β1 serum levels after the entire thorax irradiation and that the treatment improved histological signs of inflammation and lung oedema (110). Another possibility considered is the use of a replication-defective adenoviral vector (AdTβ-ExR) that increases the levels of soluble TGF-β type II receptor (111). This approach has been tested in irradiated rats where it was shown that the adenoviral vector reduced TGF-β expression, myofibroblast proliferation, and macrophage infiltration in the lungs (111). Interestingly, there is already a drug – ambroxol – which affects TGF-β1 production. Clinical studies showed that oral treatment with ambroxol from the beginning of radiotherapy significantly attenuated the rise of TGF-β1 concentration in the blood, protected patients from declining lung diffusion capacity, and reduced the incidence of pneumonitis and lung fibrosis (11).
Another therapeutic option and promising approach is the application of pirfenidone, an oral synthetic molecule with antifibrotic, antioxidant and anti-inflammatory effects. Pirfenidone inhibits TGF-β1 (112) and has been successfully used in clinical studies (14, 113–115). Recently the use of this drug in a murine model of radiation-induced pulmonary fibrosis revealed an extended median survival time and decreased accumulation of collagen and fibrosis in lung tissues. Pirfenidone also reduced TGF-β1 levels and phosphorylation of Smad3 under experimental conditions (12). Further, pirfenidone has been efficiently used in combination with sunitinib and radiotherapy in Lewis lung carcinoma (116).
Regulation of Immune Response
One of main component in the development of lung fibrosis is the disbalance of the immune system. It can be speculated that a shift in the immune response from a pro-fibrotic pathway could attenuate and/or prevent lung fibrosis. There is a promising drug, cytosine-phosphate-guanine oligodeoxyribonucleotides (CpG-ODNs), which stimulates the production of pro-inflammatory cytokines and demonstrated a protective effect on post-radiation lung fibrosis development in a mouse model (117, 118). The attenuation of lung fibrosis was associated with reduction of serum concentrations of TGF-β1 and lower amount of hydroxyproline in the lung tissue (117, 118).
Preventing Alveolar Epithelial Injury
Earlier studies on the role of the alveolar epithelium in the pathogenesis of radiation-induced fibrosis concluded that the development of pneumonitis and pulmonary fibrosis is caused by the disruption of the balance between various cell populations of the pulmonary parenchyma (119). The myofibroblasts of the alveolar wall were formerly expected as the most active cell during fibrogenesis; however, AET I cells, endothelial cells, and the alveolar macrophages are the primary target of injury (108). Subsequently, products of all these cells, particularly of AET I cells, and signals from disturbed intercellular epithelial adhesion and other factors stimulate the AET II cells to proliferate faster, and to secrete many cytokines in autocrine and paracrine mode (119). This results in accelerated proliferation and differentiation of AET II cells into AET I cells (120). This presumably impaired transdifferentiation process includes an increased presence of an intermediate cell type that is cuboidal or flat and expresses both AET I and AET II cell markers (121). The underlying mechanisms which include apoptosis and senescence of alveolar epithelial cells are poorly understood. AET I cells seem to die rather by necrosis than by apoptosis as shown in ultrastructural studies (122, 123). Knockout of AET I-specific proteins (for example PAI-1, Cav-1, RAGE, P2X7R, ICAM-1) in mice show signs of resistance to lung injury and fibrosis (80, 124–127), thus indicating the importance and involvement of the alveolar epithelial cells, namely the AET I cells in fibrogenesis. Preventing alveolar epithelial injury implicates the maintenance of alveolar barrier function e.g., to stabilize epithelial tightness by proper tight junctions as predominant structures between alveolar epithelial cells (128). Disruption of TJs with subsequent loss of alveolar epithelial integrity plays an important role in the development of pulmonary fibrosis (129). Despite the alveolar epithelial barrier function being more resistant to radiation than that of the pulmonary capillary endothelium, intact alveolar epithelial permeability is of critical importance in keeping the alveolar space relatively free of fluid during acute radiation-induced lung injury (130). Promising tools for restoration of alveolar epithelial barrier function in radiation-induced pulmonary fibrosis, however, are missing.
New Techniques of Radiotherapy
Radiotherapy techniques have changed significantly over the past few decades due to improvements in engineering and computing. The use of state-of-the art equipment employing the recent advances in radiation oncology has a huge potential to limit the risk of radiation induced pulmonary damage. By the end of the 90s, 3-dimentional conformal radiotherapy (3DCRT) was developed, where 3D imaging data is used before the irradiation to design a minimum number of radiation beams with a predefined fixed shape and uniform dose distribution matching the shape of the tumor mass (131). Later this approach was improved into IMRT (intensity-modulated radiotherapy), which allows variation of dose within each beam and typically uses more beams than 3DCRT which leads to more conformal dose distribution (132). This technique was made possible by the use of computer-controlled multi-leaf collimators and advanced treatment planning algorithms that are capable of creating the desired dose variation inside the radiation field (133). In the past decade this approach was further developed into techniques like VMAT (volumetric modulated arc therapy) where the irradiation is carried out while rotating the irradiator around the patient and the radiation dose is accurately shaped to the tumor while minimizing the dose received by the surrounding tissues (134). Further advances include the stereotactic body radiotherapy (SBRT), where immobilization devices and improved real-time imaging have allowed clinicians to administer high ablative doses to precisely target the tumor. The effectiveness of SBRT arises from the cumulative biologically effective dose that can be achieved while maintaining a sharp dose gradient fall off outside the target, preventing dose to critical structures (135). However, this technique is not free of side effects, including airway toxicity and consequent atelectasis, stenosis/stricture, airway necrosis and/or fistula formation (more prominent in patients with centrally located tumors) (136–138), spontaneous pneumothorax (139, 140), pneumonitis (141), chest wall pain and rib fracture (142, 143). Parallel to the development of these photon-based irradiation techniques, research is also focused on particle therapy, which is based on the use of protons and carbon ions to further reduce the radiation dose received by the healthy tissue. In contrast to photons, particle therapy aims to achieve proper radiation dose concentrated predominantly at a precise depth, which allows additional protection to the normal tissue (144, 145).
Conclusion
Post-radiation lung fibrosis is a common and currently untreatable adverse event of radiation therapy. To date, much is known about the pathophysiological processes of the development of lung fibrosis and pathways supporting the spread of pathological processes in the lungs. This knowledge led to recognition of the key points of fibrotic reaction and hopefully in the future will result in a development of highly effective therapeutic approaches which could be used to improve the duration and quality of life of patients after radiotherapy while at the same time will not limit the effectiveness of the applied anti-neoplastic therapy.
Author Contributions
NJ, EK, AM, MK, RR, and PS: drafted the manuscript and approved the final version.
Funding
This review was supported by the Partnership Program Technische Universität Dresden – Saint Petersburg State University. Open Access Funding by the Publication Fund of the TU Dresden.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. eviCore Radiation Therapy Clinical Guidelines. Available online at: https://www.evicore.com/ReferenceGuidelines/eviCore%20Radiation%20Therapy%20Criteria_V1.0.2018_Eff04.01.2018.pdf (accessed April 1, 2018).
2. Tyldesley S, Delaney G, Foroudi F, Barbera L, Kerba M, Mackillop W. Estimating the need for radiotherapy for patients with prostate, breast, and lung cancers: verification of model estimates of need with radiotherapy utilization data from British Columbia. Int J Radiat Oncol Biol Phys. (2011) 79:1507–15. doi: 10.1016/j.ijrobp.2009.12.070
3. Chen MJ, Novaes PE, Gadia R, Motta R. Guidelines for the treatment of lung cancer using radiotherapy. Rev Assoc Med Bras. (2017) 63:729–32. doi: 10.1590/1806-9282.63.09.729
4. Lim E, Baldwin D, Beckles M, Duffy J, Entwisle J, Faivre-Finn C, et al. Guidelines on the radical management of patients with lung cancer. Thorax. (2010) 65(Suppl. 3):iii1–27. doi: 10.1136/thx.2010.145938
5. Marks LB, Bentzen SM, Deasy JO, Kong FM, Bradley JD, Vogelius IS. Radiation dose-volume effects in the lung. Int J Radiat Oncol Biol Phys. (2010) 76:S70–6. doi: 10.1016/j.ijrobp.2009.06.091
6. Morgan GW, Breit SN. Radiation and the lung: a reevaluation of the mechanisms mediating pulmonary injury. Int J Radiat Oncol Biol Phys. (1995) 31:361–9. doi: 10.1016/0360-3016(94)00477-3
7. Makimoto T, Tsuchiya S, Hayakawa K, Saitoh R, Mori M. Risk factors for severe radiation pneumonitis in lung cancer. Jpn J Clin Oncol. (1999) 29:192–7. doi: 10.1093/jjco/29.4.192
8. Weytjens R, Erven K, De Ruysscher D. Radiation pneumonitis: occurrence, prediction, prevention and treatment. Belg J Med Oncol. (2013) 7:105–110.
9. Williams JP, Johnston CJ, Finkelstein JN. Treatment for radiation-induced pulmonary late effects: spoiled for choice or looking in the wrong direction? Curr Drug Targets. (2010) 11:1386–94. doi: 10.2174/1389450111009011386
10. Medhora M, Gao F, Jacobs ER, Moulder JE. Radiation damage to the lung: mitigation by angiotensin-converting enzyme (ACE) inhibitors. Respirology. (2012) 17:66–71. doi: 10.1111/j.1440-1843.2011.02092.x
11. Xia DH, Xi L, Xv C, Mao WD, Shen WS, Shu ZQ, et al. The protective effects of ambroxol on radiation lung injury and influence on production of transforming growth factor beta1 and tumor necrosis factor alpha. Med Oncol. (2010) 27:697–701. doi: 10.1007/s12032-009-9271-3
12. Qin W, Liu B, Yi M, Li L, Tang Y, Wu B, et al. Antifibrotic agent pirfenidone protects against development of radiation-induced pulmonary fibrosis in a murine model. Radiat Res. (2018) 190:396–403. doi: 10.1667/RR15017.1
13. De Ruysscher D, Granton PV, Lieuwes NG, van Hoof S, Wollin L, Weynand B, et al. Nintedanib reduces radiation-induced microscopic lung fibrosis but this cannot be monitored by CT imaging: a preclinical study with a high precision image-guided irradiator. Radiother Oncol. (2017) 124:482–7. doi: 10.1016/j.radonc.2017.07.014
14. Simone NL, Soule BP, Gerber L, Augustine E, Smith S, Altemus RM, et al. Oral pirfenidone in patients with chronic fibrosis resulting from radiotherapy: a pilot study. Radiat Oncol. (2007) 2:19. doi: 10.1186/1748-717X-2-19
15. Ghafoori P, Marks LB, Vujaskovic Z, Kelsey CR. Radiation-induced lung injury. Assessment, management, and prevention. Oncology (Williston Park). (2008) 22:37–47.
16. Emami B. Tolerance of normal tissue to therapeutic radiation. Rep Radiother Oncol. (2013) 1:35–48. doi: 10.1016/0360-3016(91)90171-y
17. Giridhar P, Mallick S, Rath GK, Julka PK. Radiation induced lung injury: prediction, assessment and management. Asian Pac J Cancer Prev. (2015) 16:2613–7. doi: 10.7314/APJCP.2015.16.7.2613
18. Wirsdorfer F, Jendrossek V. Modeling DNA damage-induced pneumopathy in mice: insight from danger signaling cascades. Radiat Oncol. (2017) 12:142. doi: 10.1186/s13014-017-0865-1
19. Palma DA, Senan S, Tsujino K, Barriger RB, Rengan R, Moreno M, et al. Predicting radiation pneumonitis after chemoradiation therapy for lung cancer: an international individual patient data meta-analysis. Int J Radiat Oncol Biol Phys. (2013) 85:444–50. doi: 10.1016/j.ijrobp.2012.04.043
20. Vogelius IR, Bentzen SM. A literature-based meta-analysis of clinical risk factors for development of radiation induced pneumonitis. Acta Oncol. (2012) 51:975–83. doi: 10.3109/0284186X.2012.718093
21. McDonald S, Rubin P, Phillips TL, Marks LB. Injury to the lung from cancer therapy: clinical syndromes, measurable endpoints, and potential scoring systems. Int J Radiat Oncol Biol Phys. (1995) 31:1187–203. doi: 10.1016/0360-3016(94)00429-O
22. Taghian AG, Assaad SI, Niemierko A, Kuter I, Younger J, Schoenthaler R, et al. Risk of pneumonitis in breast cancer patients treated with radiation therapy and combination chemotherapy with paclitaxel. J Natl Cancer Inst. (2001) 93:1806–11. doi: 10.1093/jnci/93.23.1806
23. Rancati T, Ceresoli GL, Gagliardi G, Schipani S, Cattaneo GM. Factors predicting radiation pneumonitis in lung cancer patients: a retrospective study. Radiother Oncol. (2003) 67:275–83. doi: 10.1016/S0167-8140(03)00119-1
24. Urbanic JJ, Lally B, Blackstock AW. The best-laid plans … often go awry …. J Thorac Oncol. (2009) 4:783–4. doi: 10.1097/JTO.0b013e3181a99bf0
25. Lind JS, Senan S, Smit EF. Pulmonary toxicity after bevacizumab and concurrent thoracic radiotherapy observed in a phase I study for inoperable stage III non-small-cell lung cancer. J Clin Oncol. (2012) 30:e104–8. doi: 10.1200/JCO.2011.38.4552
26. Anscher MS, Thrasher B, Zgonjanin L, Rabbani ZN, Corbley MJ, Fu K, et al. Small molecular inhibitor of transforming growth factor-beta protects against development of radiation-induced lung injury. Int J Radiat Oncol Biol Phys. (2008) 71:829–37. doi: 10.1016/j.ijrobp.2008.02.046
27. Zhao L, Sheldon K, Chen M, Yin MS, Hayman JA, Kalemkerian GP, et al. The predictive role of plasma TGF-beta1 during radiation therapy for radiation-induced lung toxicity deserves further study in patients with non-small cell lung cancer. Lung Cancer. (2008) 59:232–9. doi: 10.1016/j.lungcan.2007.08.010
28. Chen Y, Hyrien O, Williams J, Okunieff P, Smudzin T, Rubin P. Interleukin (IL)-1A and IL-6: applications to the predictive diagnostic testing of radiation pneumonitis. Int J Radiat Oncol Biol Phys. (2005) 62:260–6. doi: 10.1016/j.ijrobp.2005.01.041
29. Tsujisaki M, Imai K, Hirata H, Hanzawa Y, Masuya J, Nakano T, et al. Detection of circulating intercellular adhesion molecule-1 antigen in malignant diseases. Clin Exp Immunol. (1991) 85:3–8. doi: 10.1111/j.1365-2249.1991.tb05673.x
30. Sasaki R, Soejima T, Matsumoto A, Maruta T, Yamada K, Ota Y, et al. Clinical significance of serum pulmonary surfactant proteins a and d for the early detection of radiation pneumonitis. Int J Radiat Oncol Biol Phys. (2001) 50:301–7. doi: 10.1016/S0360-3016(00)01591-1
31. Xiong H, Liao Z, Liu Z, Xu T, Wang Q, Liu H, et al. ATM polymorphisms predict severe radiation pneumonitis in patients with non-small cell lung cancer treated with definitive radiation therapy. Int J Radiat Oncol Biol Phys. (2013) 85:1066–73. doi: 10.1016/j.ijrobp.2012.09.024
32. Turkkan G, Willems Y, Hendriks LEL, Mostard R, Conemans L, Gietema HA, et al. Idiopathic pulmonary fibrosis: current knowledge, future perspectives and its importance in radiation oncology. Radiother Oncol. (2020) 155:269–77. doi: 10.1016/j.radonc.2020.11.020
33. Lee YH, Kim YS, Lee SN, Lee HC, Oh SJ, Kim SJ, et al. Interstitial lung change in pre-radiation therapy computed tomography is a risk factor for severe radiation pneumonitis. Cancer Res Treat. (2015) 47:676–86. doi: 10.4143/crt.2014.180
34. Ueki N, Matsuo Y, Togashi Y, Kubo T, Shibuya K, Iizuka Y, et al. Impact of pretreatment interstitial lung disease on radiation pneumonitis and survival after stereotactic body radiation therapy for lung cancer. J Thorac Oncol. (2015) 10:116–25. doi: 10.1097/JTO.0000000000000359
35. De Ruysscher D, Faivre-Finn C, Moeller D, Nestle U, Hurkmans CW, Le Pechoux C, et al. European Organization for Research and Treatment of Cancer (EORTC) recommendations for planning and delivery of high-dose, high precision radiotherapy for lung cancer. Radiother Oncol. (2017) 124:1–10. doi: 10.1016/j.radonc.2017.06.003
36. Yoshitake T, Shioyama Y, Asai K, Nakamura K, Sasaki T, Ohga S, et al. Impact of interstitial changes on radiation pneumonitis after stereotactic body radiation therapy for lung cancer. Anticancer Res. (2015) 35:4909–13. doi: 10.1016/j.ijrobp.2015.07.1608
37. Chen H, Senan S, Nossent EJ, Boldt RG, Warner A, Palma DA, et al. Treatment-related toxicity in patients with early-stage non-small cell lung cancer and coexisting interstitial lung disease: a systematic review. Int J Radiat Oncol Biol Phys. (2017) 98:622–31. doi: 10.1016/j.ijrobp.2017.03.010
38. Harada H, Murayama S. Proton beam therapy in non-small cell lung cancer: state of the art. Lung Cancer (Auckl). (2017) 8:141–5. doi: 10.2147/LCTT.S117647
39. Ono T, Hareyama M, Nakamura T, Kimura K, Hayashi Y, Azami Y, et al. The clinical results of proton beam therapy in patients with idiopathic pulmonary fibrosis: a single center experience. Radiat Oncol. (2016) 11:56. doi: 10.1186/s13014-016-0637-3
40. Yamaguchi S, Ohguri T, Ide S, Aoki T, Imada H, Yahara K, et al. Stereotactic body radiotherapy for lung tumors in patients with subclinical interstitial lung disease: the potential risk of extensive radiation pneumonitis. Lung Cancer. (2013) 82:260–5. doi: 10.1016/j.lungcan.2013.08.024
41. Kim H, Pyo H, Noh JM, Lee W, Park B, Park HY, et al. Preliminary result of definitive radiotherapy in patients with non-small cell lung cancer who have underlying idiopathic pulmonary fibrosis: comparison between X-ray and proton therapy. Radiat Oncol. (2019) 14:19. doi: 10.1186/s13014-019-1221-4
42. Chaudhary NI, Roth GJ, Hilberg F, Muller-Quernheim J, Prasse A, Zissel G, et al. Inhibition of PDGF, VEGF and FGF signalling attenuates fibrosis. Eur Respir J. (2007) 29:976–85. doi: 10.1183/09031936.00152106
43. Inomata M, Nishioka Y, Azuma A. Nintedanib: evidence for its therapeutic potential in idiopathic pulmonary fibrosis. Core Evid. (2015) 10:89–98. doi: 10.2147/CE.S82905
44. Richeldi L, Kolb M, Jouneau S, Wuyts WA, Schinzel B, Stowasser S, et al. Efficacy and safety of nintedanib in patients with advanced idiopathic pulmonary fibrosis. BMC Pulm Med. (2020) 20:3. doi: 10.1186/s12890-019-1030-4
45. Chu KA, Yeh CC, Kuo FH, Lin WR, Hsu CW, Chen TH, et al. Comparison of reversal of rat pulmonary fibrosis of nintedanib, pirfenidone, and human umbilical mesenchymal stem cells from Wharton's jelly. Stem Cell Res Ther. (2020) 11:513. doi: 10.1186/s13287-020-02012-y
46. Ishii G, Ochiai A, Neri S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv Drug Deliv Rev. (2016) 99:186–96. doi: 10.1016/j.addr.2015.07.007
47. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. (2016) 16:582–98. doi: 10.1038/nrc.2016.73
48. Barbazan J, Matic Vignjevic D. Cancer associated fibroblasts: is the force the path to the dark side? Curr Opin Cell Biol. (2019) 56:71–9. doi: 10.1016/j.ceb.2018.09.002
49. Hellevik T, Pettersen I, Berg V, Winberg JO, Moe BT, Bartnes K, et al. Cancer-associated fibroblasts from human NSCLC survive ablative doses of radiation but their invasive capacity is reduced. Radiat Oncol. (2012) 7:59. doi: 10.1186/1748-717X-7-59
50. Grinde MT, Vik J, Camilio KA, Martinez-Zubiaurre I, Hellevik T. Ionizing radiation abrogates the pro-tumorigenic capacity of cancer-associated fibroblasts co-implanted in xenografts. Sci Rep. (2017) 7:46714. doi: 10.1038/srep46714
51. Barker HE, Paget JT, Khan AA, Harrington KJ. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer. (2015) 15:409–25. doi: 10.1038/nrc3958
52. Arshad A, Deutsch E, Vozenin MC. Simultaneous irradiation of fibroblasts and carcinoma cells repress the secretion of soluble factors able to stimulate carcinoma cell migration. PLoS ONE. (2015) 10:e0115447. doi: 10.1371/journal.pone.0115447
53. Ohuchida K, Mizumoto K, Murakami M, Qian LW, Sato N, Nagai E, et al. Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor-stromal interactions. Cancer Res. (2004) 64:3215–22. doi: 10.1158/0008-5472.CAN-03-2464
54. Li D, Qu C, Ning Z, Wang H, Zang K, Zhuang L, et al. Radiation promotes epithelial-to-mesenchymal transition and invasion of pancreatic cancer cell by activating carcinoma-associated fibroblasts. Am J Cancer Res. (2016) 6:2192–206.
55. Renschler MF. The emerging role of reactive oxygen species in cancer therapy. Eur J Cancer. (2004) 40:1934–40. doi: 10.1016/j.ejca.2004.02.031
56. Jeong H, Bok S, Hong BJ, Choi HS, Ahn GO. Radiation-induced immune responses: mechanisms and therapeutic perspectives. Blood Res. (2016) 51:157–63. doi: 10.5045/br.2016.51.3.157
57. Mladenov E, Magin S, Soni A, Iliakis G. DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Front Oncol. (2013) 3:113. doi: 10.3389/fonc.2013.00113
58. Park B, Yee C, Lee KM. The effect of radiation on the immune response to cancers. Int J Mol Sci. (2014) 15:927–43. doi: 10.3390/ijms15010927
59. Portella L, Scala S. Ionizing radiation effects on the tumor microenvironment. Semin Oncol. (2019) 46:254–60. doi: 10.1053/j.seminoncol.2019.07.003
60. Liang J, Zhang Y, Xie T, Liu N, Chen H, Geng Y, et al. Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. Nat Med. (2016) 22:1285–93. doi: 10.1038/nm.4192
61. Huebener P, Schwabe RF. Regulation of wound healing and organ fibrosis by toll-like receptors. Biochim Biophys Acta. (2013) 1832:1005–17. doi: 10.1016/j.bbadis.2012.11.017
62. Wirsdorfer F, Jendrossek V. The role of lymphocytes in radiotherapy-induced adverse late effects in the lung. Front Immunol. (2016) 7:591. doi: 10.3389/fimmu.2016.00591
63. Takeda K, Kaisho T, Akira S. Toll-like receptors. Ann Rev Immunol. (2003) 21:335–76. doi: 10.1146/annurev.immunol.21.120601.141126
64. Paun A, Fox J, Balloy V, Chignard M, Qureshi ST, Haston CK. Combined Tlr2 and Tlr4 deficiency increases radiation-induced pulmonary fibrosis in mice. Int J Radiat Oncol Biol Phys. (2010) 77:1198–205. doi: 10.1016/j.ijrobp.2009.12.065
65. Brickey WJ, Neuringer IP, Walton W, Hua X, Wang EY, Jha S, et al. MyD88 provides a protective role in long-term radiation-induced lung injury. Int J Radiat Biol. (2012) 88:335–47. doi: 10.3109/09553002.2012.652723
66. Tabraue C, Lara PC, De Mirecki-Garrido M, De La Rosa JV, Lopez-Blanco F, Fernandez-Perez L, et al. LXR signaling regulates macrophage survival and inflammation in response to ionizing radiation. Int J Radiat Oncol Biol Phys. (2019) 104:913–23. doi: 10.1016/j.ijrobp.2019.03.028
67. Hirota JA, Knight DA. Human airway epithelial cell innate immunity: relevance to asthma. Curr Opin Immunol. (2012) 24:740–6. doi: 10.1016/j.coi.2012.08.012
68. Jo EK, Kim JK, Shin DM, Sasakawa C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol. (2016) 13:148–59. doi: 10.1038/cmi.2015.95
69. Wei J, Wang H, Wang H, Wang B, Meng L, Xin Y, et al. The role of NLRP3 inflammasome activation in radiation damage. Biomed Pharmacother. (2019) 118:109217. doi: 10.1016/j.biopha.2019.109217
70. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 Inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. (2019) 20:3328. doi: 10.3390/ijms20133328
71. Abderrazak A, Syrovets T, Couchie D, El Hadri K, Friguet B, Simmet T, et al. NLRP3 inflammasome: from a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. (2015) 4:296–307. doi: 10.1016/j.redox.2015.01.008
72. He X, Qian Y, Li Z, Fan EK, Li Y, Wu L, et al. TLR4-Upregulated IL-1beta and IL-1RI promote alveolar macrophage pyroptosis and lung inflammation through an autocrine mechanism. Sci Rep. (2016) 6:31663. doi: 10.1038/srep31663
73. Sohn SH, Lee JM, Park S, Yoo H, Kang JW, Shin D, et al. The inflammasome accelerates radiation-induced lung inflammation and fibrosis in mice. Environ Toxicol Pharmacol. (2015) 39:917–26. doi: 10.1016/j.etap.2015.02.019
74. Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. (2012) 19:107–20. doi: 10.1038/cdd.2011.96
75. Tsukimoto M. Purinergic signaling is a novel mechanism of the cellular response to ionizing radiation. Biol Pharm Bull. (2015) 38:951–9. doi: 10.1248/bpb.b15-00062
76. Pelegrin P, Surprenant A. Pannexin-1 couples to maitotoxin- and nigericin-induced interleukin-1beta release through a dye uptake-independent pathway. J Biol Chem. (2007) 282:2386–94. doi: 10.1074/jbc.M610351200
77. Kanneganti TD, Lamkanfi M, Kim YG, Chen G, Park JH, Franchi L, et al. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity. (2007) 26:433–43. doi: 10.1016/j.immuni.2007.03.008
78. Ohshima Y, Tsukimoto M, Harada H, Kojima S. Involvement of connexin43 hemichannel in ATP release after gamma-irradiation. J Radiat Res. (2012) 53:551–7. doi: 10.1093/jrr/rrs014
79. Kasper M, Traub O, Reimann T, Bjermer L, Grossmann H, Muller M, et al. Upregulation of gap junction protein connexin43 in alveolar epithelial cells of rats with radiation-induced pulmonary fibrosis. Histochem Cell Biol. (1996) 106:419–24. doi: 10.1007/BF02473301
80. Riteau N, Gasse P, Fauconnier L, Gombault A, Couegnat M, Fick L, et al. Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. Am J Respir Crit Care Med. (2010) 182:774–83. doi: 10.1164/rccm.201003-0359OC
81. Genovese T, Cuzzocrea S, Di Paola R, Failla M, Mazzon E, Sortino MA, et al. Inhibition or knock out of inducible nitric oxide synthase result in resistance to bleomycin-induced lung injury. Res Res. (2005) 6:58. doi: 10.1186/1465-9921-6-58
82. Janssen W, Pullamsetti SS, Cooke J, Weissmann N, Guenther A, Schermuly RT. The role of dimethylarginine dimethylaminohydrolase (DDAH) in pulmonary fibrosis. J Pathol. (2013) 229:242–9. doi: 10.1002/path.4127
83. Pullamsetti SS, Savai R, Dumitrascu R, Dahal BK, Wilhelm J, Konigshoff M, et al. The role of dimethylarginine dimethylaminohydrolase in idiopathic pulmonary fibrosis. Sci Transl Med. (2011) 3:87ra53. doi: 10.1126/scitranslmed.3001725
84. Wells SM, Holian A. Asymmetric dimethylarginine induces oxidative and nitrosative stress in murine lung epithelial cells. Am J Respir Cell Mol Biol. (2007) 36:520–8. doi: 10.1165/rcmb.2006-0302SM
85. Wells SM, Buford MC, Migliaccio CT, Holian A. Elevated asymmetric dimethylarginine alters lung function and induces collagen deposition in mice. Am J Respir Cell Mol Biol. (2009) 40:179–88. doi: 10.1165/rcmb.2008-0148OC
86. Murray-Rust J, Leiper J, McAlister M, Phelan J, Tilley S, Santa Maria J, et al. Structural insights into the hydrolysis of cellular nitric oxide synthase inhibitors by dimethylarginine dimethylaminohydrolase. Nat Struct Biol. (2001) 8:679–83. doi: 10.1038/90387
87. Cooke JP, Ghebremariam YT. DDAH says NO to ADMA. Arterioscler Thromb Vasc Biol. (2011) 31:1462–4. doi: 10.1161/ATVBAHA.111.228833
88. Ueda S, Kato S, Matsuoka H, Kimoto M, Okuda S, Morimatsu M, et al. Regulation of cytokine-induced nitric oxide synthesis by asymmetric dimethylarginine: role of dimethylarginine dimethylaminohydrolase. Circ Res. (2003) 92:226–33. doi: 10.1161/01.RES.0000052990.68216.EF
89. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Investig. (2009) 119:1420–8. doi: 10.1172/JCI39104
90. Choi SH, Hong ZY, Nam JK, Lee HJ, Jang J, Yoo RJ, et al. A hypoxia-induced vascular endothelial-to-mesenchymal transition in development of radiation-induced pulmonary fibrosis. Clin Cancer Res. (2015) 21:3716–26. doi: 10.1158/1078-0432.CCR-14-3193
91. Balli D, Ustiyan V, Zhang Y, Wang IC, Masino AJ, Ren X, et al. Foxm1 transcription factor is required for lung fibrosis and epithelial-to-mesenchymal transition. EMBO J. (2013) 32:231–44. doi: 10.1038/emboj.2012.336
92. Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Nat Acad Sci U S A. (2006) 103:13180–5. doi: 10.1073/pnas.0605669103
93. Tanjore H, Xu XC, Polosukhin VV, Degryse AL, Li B, Han W, et al. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med. (2009) 180:657–65. doi: 10.1164/rccm.200903-0322OC
94. Almeida C, Nagarajan D, Tian J, Leal SW, Wheeler K, Munley M, et al. The role of alveolar epithelium in radiation-induced lung injury. PLoS ONE. (2013) 8:e53628. doi: 10.1371/journal.pone.0053628
95. Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A. (2011) 108:E1475–83. doi: 10.1073/pnas.1117988108
96. Yamada M, Kuwano K, Maeyama T, Hamada N, Yoshimi M, Nakanishi Y, et al. Dual-immunohistochemistry provides little evidence for epithelial-mesenchymal transition in pulmonary fibrosis. Histochem Cell Biol. (2008) 129:453–62. doi: 10.1007/s00418-008-0388-9
97. Hill C, Jones MG, Davies DE, Wang Y. Epithelial-mesenchymal transition contributes to pulmonary fibrosis via aberrant epithelial/fibroblastic cross-talk. J Lung Health Dis. (2019) 3:31–5. doi: 10.29245/2689-999X/2019/2.1149
98. Garibaldi BT, D'Alessio FR, Mock JR, Files DC, Chau E, Eto Y, et al. Regulatory T cells reduce acute lung injury fibroproliferation by decreasing fibrocyte recruitment. Am J Respir Cell Mol Biol. (2013) 48:35–43. doi: 10.1165/rcmb.2012-0198OC
99. Todd NW, Luzina IG, Atamas SP. Molecular and cellular mechanisms of pulmonary fibrosis. Fibrogenesis Tissue Repair. (2012) 5:11. doi: 10.1186/1755-1536-5-11
100. El Agha E, Kramann R, Schneider RK, Li X, Seeger W, Humphreys BD, et al. Mesenchymal stem cells in fibrotic disease. Cell Stem Cell. (2017) 21:166–77. doi: 10.1016/j.stem.2017.07.011
101. Xie T, Liang J, Liu N, Huan C, Zhang Y, Liu W, et al. Transcription factor TBX4 regulates myofibroblast accumulation and lung fibrosis. J Clin Invest. (2016) 126:3626. doi: 10.1172/JCI89968
102. Kramann R, Schneider RK, DiRocco DP, Machado F, Fleig S, Bondzie PA, et al. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell. (2015) 16:51–66. doi: 10.1016/j.stem.2014.11.004
103. El Agha E, Moiseenko A, Kheirollahi V, De Langhe S, Crnkovic S, Kwapiszewska G, et al. Two-way conversion between lipogenic and myogenic fibroblastic phenotypes marks the progression and resolution of lung fibrosis. Cell Stem Cell. (2017) 20:571. doi: 10.1016/j.stem.2017.03.011
104. Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmouliere A, Varga J, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. (2012) 180:1340–55. doi: 10.1016/j.ajpath.2012.02.004
105. Sisson TH, Ajayi IO, Subbotina N, Dodi AE, Rodansky ES, Chibucos LN, et al. Inhibition of myocardin-related transcription factor/serum response factor signaling decreases lung fibrosis and promotes mesenchymal cell apoptosis. Am J Pathol. (2015) 185:969–86. doi: 10.1016/j.ajpath.2014.12.005
106. Hanania AN, Mainwaring W, Ghebre YT, Hanania NA, Ludwig M. Radiation-induced lung injury: assessment and management. Chest. (2019) 156:150–62. doi: 10.1016/j.chest.2019.03.033
107. Kim JS, Son Y, Jung MG, Jeong YJ, Kim SH, Lee SJ, et al. Geranylgeranylacetone alleviates radiation-induced lung injury by inhibiting epithelial-to-mesenchymal transition signaling. Mol Med Rep. (2016) 13:4666–70. doi: 10.3892/mmr.2016.5121
108. Bartholomew JR. Update on the management of venous thromboembolism. Cleve Clin J Med. (2017) 84:39–46. doi: 10.3949/ccjm.84.s3.04
109. Flechsig P, Dadrich M, Bickelhaupt S, Jenne J, Hauser K, Timke C, et al. LY2109761 attenuates radiation-induced pulmonary murine fibrosis via reversal of TGF-beta and BMP-associated proinflammatory and proangiogenic signals. Clin Cancer Res. (2012) 18:3616–27. doi: 10.1158/1078-0432.CCR-11-2855
110. Lu Z, Ma Y, Zhang S, Liu F, Wan M, Luo J. Transforming growth factor-beta1 small interfering RNA inhibits growth of human embryonic lung fibroblast HFL-I cells in vitro and defends against radiation-induced lung injury in vivo. Mol Med Rep. (2015) 11:2055–61. doi: 10.3892/mmr.2014.2923
111. Nishioka A, Ogawa Y, Kariya S, Hamada N, Nogami M, Inomata T, et al. Reduction of fibroproliferative changes in irradiated rat lung with soluble transforming growth factor-beta receptor. Mol Med Rep. (2015) 11:2659–63. doi: 10.3892/mmr.2014.3064
112. Nakayama S, Mukae H, Sakamoto N, Kakugawa T, Yoshioka S, Soda H, et al. Pirfenidone inhibits the expression of HSP47 in TGF-beta1-stimulated human lung fibroblasts. Life Sci. (2008) 82:210–7. doi: 10.1016/j.lfs.2007.11.003
113. Li SM, Lin Y, Liang SS. Efficacy of pirfenidone for the treatment of pulmonary fibrosis: an updated systematic review protocol of randomized controlled trial. Med (Baltimore). (2019) 98:e15407. doi: 10.1097/MD.0000000000015407
114. Margaritopoulos GA, Trachalaki A, Wells AU, Vasarmidi E, Bibaki E, Papastratigakis G, et al. Pirfenidone improves survival in IPF: results from a real-life study. BMC Pulm Med. (2018) 18:177. doi: 10.1186/s12890-018-0736-z
115. Costabel U, Albera C, Glassberg MK, Lancaster LH, Wuyts WA, Petzinger U, et al. Effect of pirfenidone in patients with more advanced idiopathic pulmonary fibrosis. Respir Res. (2019) 20:55. doi: 10.1186/s12931-019-1021-2
116. Choi SH, Nam JK, Jang J, Lee HJ, Lee YJ. Pirfenidone enhances the efficacy of combined radiation and sunitinib therapy. Biochem Biophys Res Commun. (2015) 462:138–43. doi: 10.1016/j.bbrc.2015.04.107
117. Li X, Xu G, Qiao T, Yuan S, Zhuang X, Zhang J, et al. Effects of CpG Oligodeoxynucleotide 1826 on transforming growth factor-beta 1 and radiation-induced pulmonary fibrosis in mice. J Inflamm. (2016) 13:16. doi: 10.1186/s12950-016-0125-4
118. Zhang C, Zhao H, Li BL, Fu G, Liu H, Cai JM, et al. CpG-oligodeoxynucleotides may be effective for preventing ionizing radiation induced pulmonary fibrosis. Toxicol Lett. (2018) 292:181–9. doi: 10.1016/j.toxlet.2018.04.009
119. Kasper M, Haroske G. Alterations in the alveolar epithelium after injury leading to pulmonary fibrosis. Histol Histopathol. (1996) 11:463–83.
120. Trott KR, Herrmann T, Kasper M. Target cells in radiation pneumopathy. Int J Radiat Oncol Biol Phys. (2004) 58:463–9. doi: 10.1016/j.ijrobp.2003.09.045
121. Kasper M, Barth K. Potential contribution of alveolar epithelial type I cells to pulmonary fibrosis. Biosci Rep. (2017) 37:BSR20171301. doi: 10.1042/BSR20171301
122. Adamson IY, Bowden DH. The pathogenesis of bleomycin-induced pulmonary fibrosis in mice. Am J Pathol. (1974) 77:185–97.
123. Jones AW, Reeve NL. Ultrastructural study of bleomycin-induced pulmonary changes in mice. J Pathol. (1978) 124:227–33. doi: 10.1002/path.1711240407
124. Chuang-Tsai S, Sisson TH, Hattori N, Tsai CG, Subbotina NM, Hanson KE, et al. Reduction in fibrotic tissue formation in mice genetically deficient in plasminogen activator inhibitor-1. Am J Pathol. (2003) 163:445–52. doi: 10.1016/S0002-9440(10)63674-7
125. Shivshankar P, Brampton C, Miyasato S, Kasper M, Thannickal VJ, Le Saux CJ. Caveolin-1 deficiency protects from pulmonary fibrosis by modulating epithelial cell senescence in mice. Am J Respir Cell Mol Biol. (2012) 47:28–36. doi: 10.1165/rcmb.2011-0349OC
126. Englert JM, Kliment CR, Ramsgaard L, Milutinovic PS, Crum L, Tobolewski JM, et al. Paradoxical function for the receptor for advanced glycation end products in mouse models of pulmonary fibrosis. Int J Clin Exp Pathol. (2011) 4:241–54.
127. Hamaguchi Y, Nishizawa Y, Yasui M, Hasegawa M, Kaburagi Y, Komura K, et al. Intercellular adhesion molecule-1 and L-selectin regulate bleomycin-induced lung fibrosis. Am J Pathol. (2002) 161:1607–18. doi: 10.1016/S0002-9440(10)64439-2
128. Wittekindt OH. Tight junctions in pulmonary epithelia during lung inflammation. Pflugers Arch. (2017) 469:135–47. doi: 10.1007/s00424-016-1917-3
129. Kulkarni T, de Andrade J, Zhou Y, Luckhardt T, Thannickal VJ. Alveolar epithelial disintegrity in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. (2016) 311:L185–91. doi: 10.1152/ajplung.00115.2016
130. Suzuki S, Noda M, Akaizawa T, Song C, Tsubochi H, Suzuki T, et al. Intact alveolar epithelial permeability and transalveolar fluid absorption after thoracic irradiation in rats. Radiat Res. (1999) 152:517–22. doi: 10.2307/3580148
131. Barazzuol L, Coppes RP, van Luijk P. Prevention and treatment of radiotherapy-induced side effects. Mol Oncol. (2020) 14:1538–54. doi: 10.1002/1878-0261.12750
132. Veldeman L, Madani I, Hulstaert F, De Meerleer G, Mareel M, De Neve W. Evidence behind use of intensity-modulated radiotherapy: a systematic review of comparative clinical studies. Lancet Oncol. (2008) 9:367–75. doi: 10.1016/S1470-2045(08)70098-6
133. Garibaldi C, Jereczek-Fossa BA, Marvaso G, Dicuonzo S, Rojas DP, Cattani F, et al. Recent advances in radiation oncology. Ecancermedicalscience. (2017) 11:785. doi: 10.3332/ecancer.2017.785
134. Nicosia L, Sicignano G, Rigo M, Figlia V, Cuccia F, De Simone A, et al. Daily dosimetric variation between image-guided volumetric modulated arc radiotherapy and MR-guided daily adaptive radiotherapy for prostate cancer stereotactic body radiotherapy. Acta Oncol. (2020). doi: 10.1080/0284186X.2020.1821090. [Epub ahead of print].
135. Amini A, Yeh N, Gaspar LE, Kavanagh B, Karam SD. Stereotactic body radiation therapy (SBRT) for lung cancer patients previously treated with conventional radiotherapy: a review. Radiat Oncol. (2014) 9:210. doi: 10.1186/1748-717X-9-210
136. Karlsson K, Nyman J, Baumann P, Wersall P, Drugge N, Gagliardi G, et al. Retrospective cohort study of bronchial doses and radiation-induced atelectasis after stereotactic body radiation therapy of lung tumors located close to the bronchial tree. Int J Radiat Oncol Biol Phys. (2013) 87:590–5. doi: 10.1016/j.ijrobp.2013.06.2055
137. Speiser BL, Spratling L. Radiation bronchitis and stenosis secondary to high dose rate endobronchial irradiation. Int J Radiat Oncol Biol Phys. (1993) 25:589–97. doi: 10.1016/0360-3016(93)90003-E
138. Kang KH, Okoye CC, Patel RB, Siva S, Biswas T, Ellis RJ, et al. Complications from stereotactic body radiotherapy for lung cancer. Cancers (Basel). (2015) 7:981–1004. doi: 10.3390/cancers7020820
139. Ohnishi K, Shioyama Y, Nomoto S, Sasaki T, Ohga S, Yoshitake T, et al. Spontaneous pneumothorax after stereotactic radiotherapy for non-small-cell lung cancer. Jpn J Radiol. (2009) 27:269–74. doi: 10.1007/s11604-009-0333-4
140. Seppenwoolde Y, De Jaeger K, Boersma LJ, Belderbos JS, Lebesque JV. Regional differences in lung radiosensitivity after radiotherapy for non-small-cell lung cancer. Int J Radiat Oncol Biol Phys. (2004) 60:748–58. doi: 10.1016/j.ijrobp.2004.04.037
141. Macfadyen RJ, Lees KR, Reid JL. Perindopril. A review of its pharmacokinetics and clinical pharmacology. Drugs. (1990) 39(Suppl. 1):49–63. doi: 10.2165/00003495-199000391-00009
142. Andolino DL, Forquer JA, Henderson MA, Barriger RB, Shapiro RH, Brabham JG, et al. Chest wall toxicity after stereotactic body radiotherapy for malignant lesions of the lung and liver. Int J Radiat Oncol Biol Phys. (2011) 80:692–7. doi: 10.1016/j.ijrobp.2010.03.020
143. Voroney JP, Hope A, Dahele MR, Purdie TG, Franks KN, Pearson S, et al. Chest wall pain and rib fracture after stereotactic radiotherapy for peripheral non-small cell lung cancer. J Thorac Oncol. (2009) 4:1035–7. doi: 10.1097/JTO.0b013e3181ae2962
144. Durante M, Orecchia R, Loeffler JS. Charged-particle therapy in cancer: clinical uses and future perspectives. Nat Rev Clin Oncol. (2017) 14:483–95. doi: 10.1038/nrclinonc.2017.30
Keywords: radiation, pulmonary fibrosis, macromolecular damage, nitric oxide, dimethylarginine dimethylaminohydrolase
Citation: Jarzebska N, Karetnikova ES, Markov AG, Kasper M, Rodionov RN and Spieth PM (2021) Scarred Lung. An Update on Radiation-Induced Pulmonary Fibrosis. Front. Med. 7:585756. doi: 10.3389/fmed.2020.585756
Received: 13 August 2020; Accepted: 17 December 2020;
Published: 15 January 2021.
Edited by:
Olivier Sitbon, Université Paris-Saclay, FranceReviewed by:
Hilario Nunes, Hôpital Avicenne, FranceVera Luiza Capelozzi, University of São Paulo, Brazil
Copyright © 2021 Jarzebska, Karetnikova, Markov, Kasper, Rodionov and Spieth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter M. Spieth, UGV0ZXIuU3BpZXRoQHVuaWtsaW5pa3VtLWRyZXNkZW4uZGU=