 Yuliya Buinitskaya
Yuliya Buinitskaya Roman Gurinovich
Roman Gurinovich Clifford G. Wlodaver
Clifford G. Wlodaver Siarhei Kastsiuchenka
Siarhei Kastsiuchenka- 1Sci.AI, Tallinn, Estonia
- 2Oklahoma University Health Sciences Center, Oklahoma City, OK, United States
- 3Anesthesiology Institute, Cleveland Clinic Abu Dhabi, Abu Dhabi, United Arab Emirates
Introduction: COVID-19 is a novel and devastating disease. Its manifestations vary from asymptomatic to lethal. Moreover, mortality rates differ based on underlying health conditions and ethnicity. We investigated the biochemical rationale behind these observations using machine reasoning by the sci.AI system (https://sci.ai/). Facts were extracted and linked from publications available in nlm.nih.gov and Europe PMC to form the dataset which was validated by medical experts.
Results: Based on the analysis of experimental and clinical data, we synthesized detailed biochemical pathways of COVID-19 pathogenesis which were used to explain epidemiological and clinical observations. Clinical manifestations and biomarkers are highlighted to monitor the course of COVID-19 and navigate treatment. As depicted in the Graphical Abstract, SARS-CoV-2 triggers a pro-oxidant (PO) response leading to the production of reactive oxygen species (ROS) as a normal innate defense. However, SARS-CoV-2's unique interference with the antioxidant (AO) system, through suppression of nitric oxide (NO) production in the renin- angiotensin-aldosterone system (RAAS), leads to an excessive inflammatory PO response. The excessive PO response becomes critical in cohorts with a compromised AO system such as patients with glucose-6-phosphate dehydrogenase deficiency (G6PDd) where NO and glutathione (GSH) mechanisms are impaired. G6PDd develops in patients with metabolic syndrome. It is mediated by aldosterone (Ald) which also increases specifically in COVID-19.
Conclusion: G6PD is essential for an adequate immune response. Both G6PDd and SARS-CoV-2 compromise the AO system through the same pathways rendering G6PDd the Achilles' heel for COVID-19. Thus, the evolutionary antimalarial advantage of the G6PDd cohort can be a disadvantage against SARS-CoV-2.
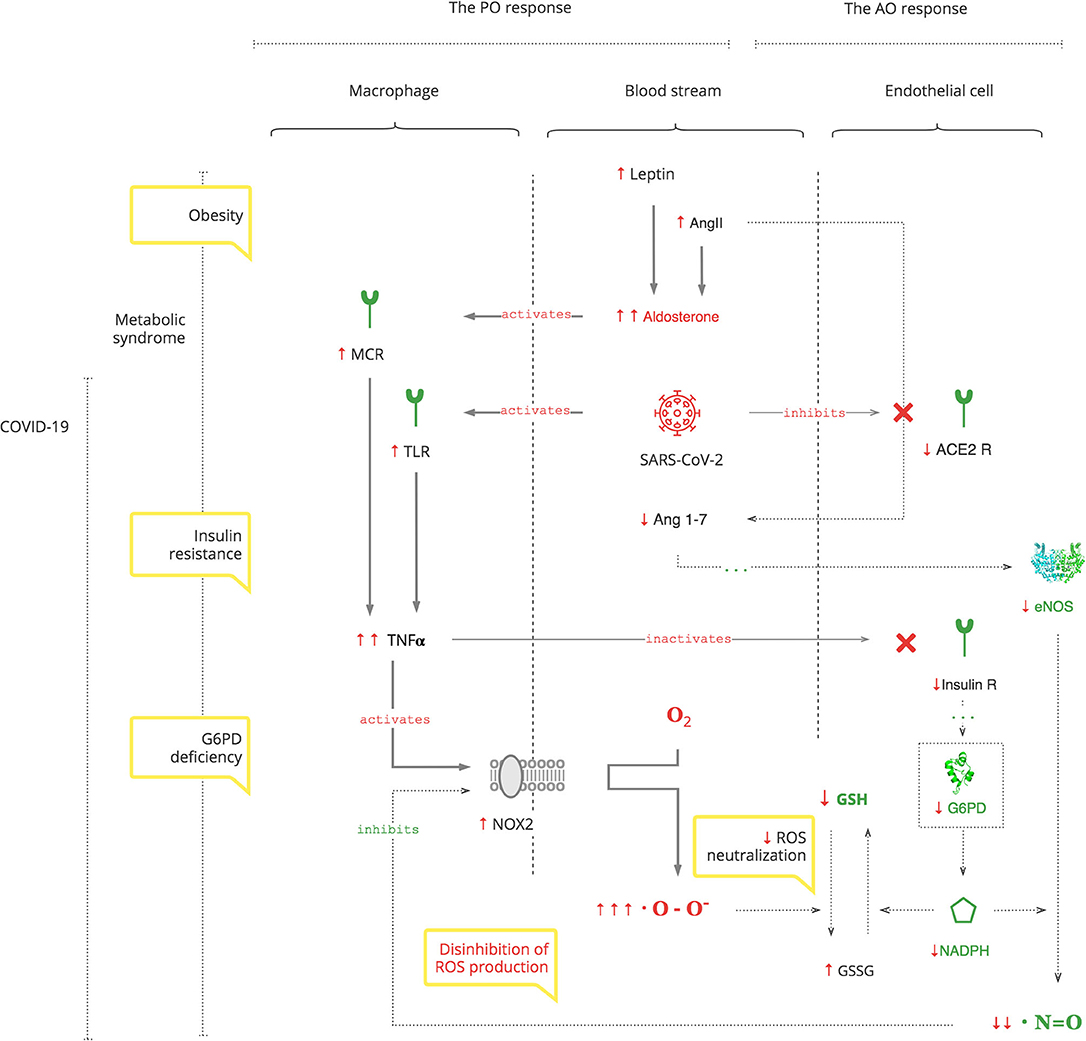
Graphical Abstract.
Introduction
Based on our previous discussion about the basic mechanisms of coronavirus disease 2019 (COVID-19) pathogenesis (1), in this paper, we will focus on particular parts of it to explain the present clinical and epidemiological observations.
The human host defends itself against infection through its immune response with two cooperative phases.
1. The first phase occurs early, is innate and nonspecific, and has two components.
(a) A pro-inflammatory pro-oxidant (PO) system mediates inflammation. It attacks pathogens with free radicals of the reactive oxygen species (ROS). However, when there is increased production and/or decreased neutralization, a heightened level of ROS occurs, and excessive levels cause collateral damage to normal cells and is referred to as “oxidative stress,” “cytokine storm,” and “systemic inflammatory response syndrome (SIRS).”
(b) An anti-inflammatory antioxidant (AO) system balances the PO response (2).
2. The second phase occurs with a delay and is adaptive and specific. It is mediated through antibody expression.
These two arms of immune response usually eradicate the pathogen (3). However, in COVID-19, both phases are delayed due to suppression of the host's gene expression by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)'s nsp1 protein (4).
Wu et al. (5) demonstrated, in vitro, that glucose-6-phosphate dehydrogenase deficiency (G6PDd) cell lines are vulnerable to coronavirus infection. There are two types of G6PDd: congenital and acquired. Congenital G6PDd is the most prevalent enzyme deficiency in the world, affecting 4.9% of the global population. It evolved against malaria and predominates in specific ethnic cohorts such as the Mediterranean, Asian, and African (6). Interestingly, these cohorts have been particularly affected by the COVID-19 pandemic (7–10). Acquired G6PDd develops in patients with underlying health conditions, especially the metabolic syndrome (11). The metabolic syndrome is prevalent and it spreads acquired G6PDd worldwide.
This paper presents a detailed description of how SARS-CoV-2 affects the innate PO and AO responses and how G6PDd potentiates COVID-19. In addition, we highlight accompanying clinical manifestations and biomarkers that are useful to monitor the clinical course and navigate treatment.
Methods
We used the sci.AI machine reasoning system (https://sci.ai/) to operate on publicly available datasets from nlm.nih.gov and Europe PMC. The process consisted of two stages: Representation and Reasoning.
Representation algorithms translate unstructured individual papers, documents, and files from heterogeneous sources into embeddings and graphs of entities relations. It goes beyond classic Named Entity Recognition (NER) and arbitrarily recognizes individual and composite biological entities and how they relate to each other. For example, in this sentence: “Obese patients with MetSyn had a significantly lower nitric oxide production rate (0.21 ± 0.13 μmol/h per kg; P = 0.009) than healthy normal-weight individuals (0.63 ± 0.30 μmol/h per kg), whereas nitric oxide (NO) production rate was intermediate in obese patients without MetSyn (0.49 ± 0.22 μmol/h per kg; P = 0.33)” (12); the machine recognizes the conditions “obesity” and “metabolic syndrome” and recognizes substance “nitric oxide” and links it to CHEBI:16480. Ultimately, “lower nitric oxide production rate” in the context of “metabolic syndrome” is recognized as a biomarker.
The second, Reasoning stage, synthesizes knowledge based on a subset of findings that appear to be relevant to COVID-19. The discovery process was triggered by textual queries “SARS” and “ARDS.” Traversing through the interlinked representations computed at the first stage produced multiple subgraphs. We progressively refined the generated knowledge and, in the last step, linked these excerpts to synthesize biochemical pathways to help explain the pathophysiology of COVID-19. We translated complex pathways into clinically relevant applications, conforming to our clinical observations.
Pathways were constructed iteratively; it is not a result of one time inference. Generally speaking, typical machine learning algorithms approximate previous data distributions. In contrast, our reasoning algorithm is based on graph traversing and utilizes biochemical properties in context. It allows to avoid bias caused by frequently mentioned terms, for example, angiotensin-converting enzyme 2 (ACE2). Subgraphs were interactively validated by a domain expert at every iteration. For instance, the term “SARS” mentioned together with “TLR” and “ACE2” led to the creation of two axes as described in our previous work (1):
– TLR/TNFα/NADPH oxidase (NOX2)/ROS, which is positively regulated, and
– ACE2/NOS3/NO, which is negatively regulated by SARS.
Both axes turn out to be composed mainly of canonical pathways: renin–angiotensin system, glutathione (GSH) metabolism, pentose phosphate pathway, aldosterone (Ald) synthesis and secretion, and NO production. When we placed all these pathways on the same canvas, reduced nicotinamide adenine dinucleotide phosphate (NADPH) appeared to be the cofactor of both axes and, in turn, is produced by glucose-6-phosphate dehydrogenase (G6PD). This biochemical rationale, together with the worldwide prevalence of congenital and acquired G6PDd, is consistent with COVID-19 outcomes at individual and epidemiological levels.
A limitation of our research is that it focuses on the centrality of G6PD. Yet, we acknowledge that there is certainly other biochemistry relevant to COVID-19 that remains open for investigation.
Results and Discussion
Based on machine reasoning of data from 30M papers, we demonstrate the results.
Severe Acute Respiratory Syndrome Coronavirus 2 Affects the Innate Immune Response
The PO System
The PO system is triggered by SARS-CoV-2, as for any pathogen, through toll-like receptors (TLRs) on macrophages, the first-line cell of innate defense (13). As depicted in Figure 1, this results in tumor necrosis factor-alpha (TNFα)-induced inflammation, which has two clinically relevant molecular effects: inactivation of insulin receptor signaling on endothelial cells (see Graphical Abstract) and activation of NOX2 on macrophages (14, 15). This response is acute and transient. Activated NOX2 produces ROS, particularly superoxide anion (O2*-) from oxygen (O2). Then a hydroxyl radical (OH*) is produced through the Fenton reaction (16). It destroys microorganisms (17). At the same time, it stresses the host's cells, especially platelets, lymphocytes, erythrocytes, and muscle cells (18–20). Muscle cell damage manifests with rhabdomyolysis (21). Damage of erythrocyte membranes results in latent hemolysis leaking lactate dehydrogenase (LDH), and hemoglobin (Hb) is oxidized to methemoglobin (MetHb) (22–24).
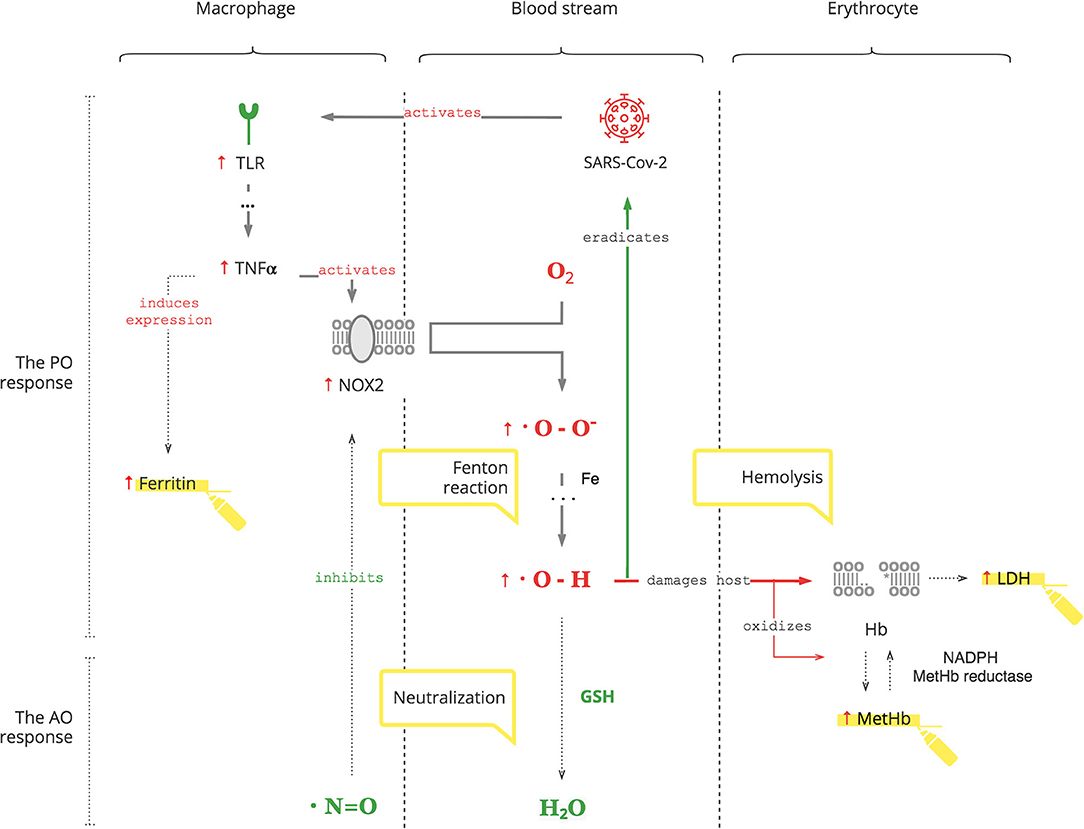
Figure 1. SARS-CoV-2 triggers the PO response resulting in ROS.
Clinical Pearls
• Hyperglycemia occurs during COVID-19. It is transient and reversible if there is no antecedent insulin resistance. Otherwise, underlying insulin resistance is aggravated by the stress of infection (25).
• Ferritin production is induced by TNFα and can be used to monitor the degree of the PO response (26).
• Thrombocytopenia and lymphopenia reflect the degree of oxidative stress and can be followed as biomarkers (27, 28).
• Since statins cause rhabdomyolysis as a complication, avoid these drugs in COVID-19 patients (29, 30).
• Erythrocytes are decreased due to latent hemolysis, which can be monitored by LDH levels (31).
• Increased MetHb makes SpO2 calculation inaccurate. It causes a low SpO2 by pulse oximetry in patients with a normal PaO2 (32, 33). This can be misleading and can result in an unnecessary administration of O2, the substrate of ROS.
Thus, SARS-CoV-2 interacts with the innate PO system, and adequate ROS levels are a first-line antimicrobial defense.
The AO System
The AO system balances the PO response through two central mechanisms: suppression of ROS production by NO and ROS neutralization by GSH (34, 35).
As depicted in Figure 2, SARS-CoV-2 binds to the ACE2 receptor in order to enter cells and, in turn, destroys this receptor. The ACE2 receptor is involved in the protective ACE2/endothelial nitric oxide synthase (eNOS)/NO pathway of the renin–angiotensin–aldosterone system (RAAS). It leads to the suppression of eNOS, the most prevalent isoform of NOS, and consequently decreased NO levels (36). In addition to its antioxidant property, NO is also essential for vasodilation, prevention of platelet aggregation, and inhibition of the replication of SARS-CoV (37, 38).
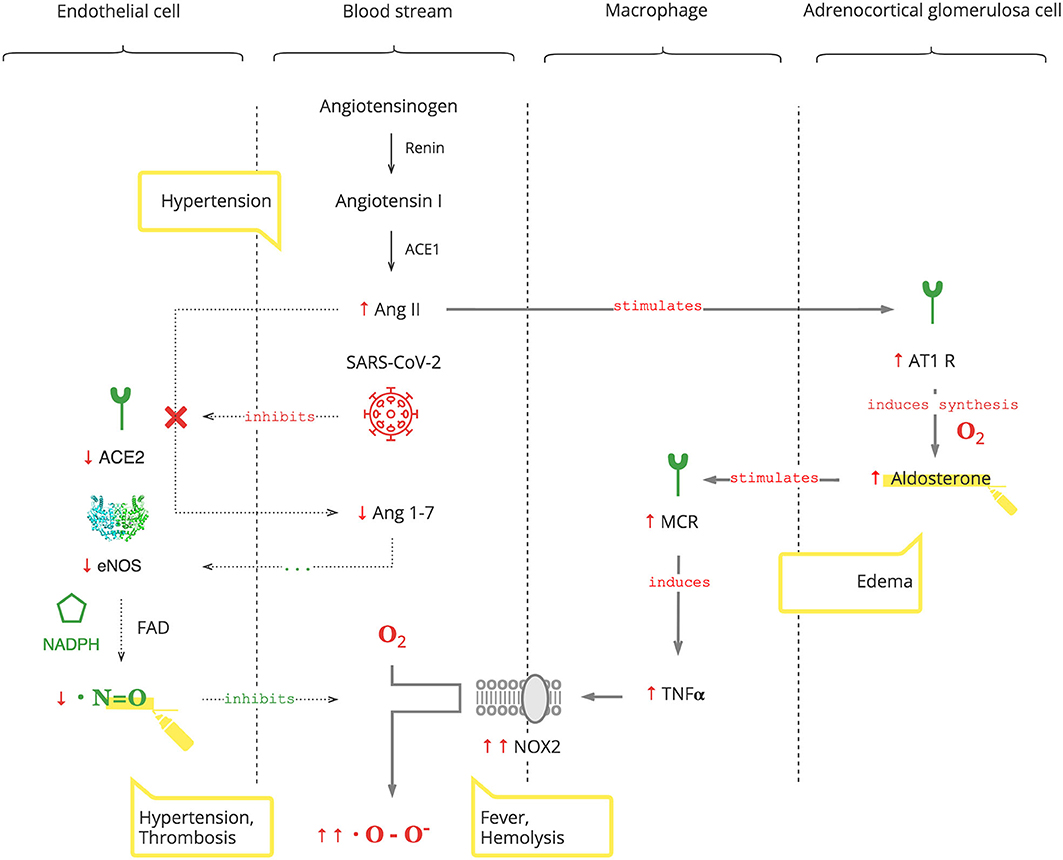
Figure 2. SARS-CoV-2 interferes with the AO response through RAAS resulting in excessive ROS.
Suppression of ACE2 activity also leads to an inability to convert angiotensin II (Ang II) to angiotensin 1-7 (Ang 1-7). Ang II is a potent vasoconstrictor and also stimulates Ald (39). This results in a transient increase in Ald that induces TNFα through mineralocorticoid receptor (MCR) on macrophages (40). NOX2 hyper-activation by TNFα, which is induced by the virus and Ald, and its disinhibition by virus-induced NO inhibition perpetuate ROS production, making it excessive.
It is also noteworthy that adrenocortical glomerulosa cells are extremely sensitive to dissolved O2 blood levels (41). And fever shifts the O2-Hb dissociation curve to the right, lowering the affinity of Hb to O2, further contributing to ROS production (42).
Clinical Pearls
• An increased Ald level is a specific biomarker of the SARS-CoV-2 infection. This is acute, transient, and Ang II-dependent.
• The virus-induced decrease in NO and increase in Ald (NO/RAAS dysbalance) render the immune response to COVID-19 excessive. This manifests with fever and hematological complications, especially progressive hemolysis, and thrombus formation (43).
• Vasoconstriction, mediated by NO/RAAS dysbalance, is a main pathophysiological component of COVID-19-associated acute respiratory distress syndrome (ARDS) and manifests as acute vascular distress syndrome (AVDS) (44).
• Pulmonary edema is potentiated by elevated levels of Ald and aggravates ARDS (45).
• Excessive O2 therapy can be deleterious.
Thus, SARS-CoV-2 interferes with the AO system, rendering the PO response excessive. Moreover, SARS-CoV-2-induced increases of Ald aggravate the condition, especially in patients with underlying health conditions.
The Role of Underlying Health Conditions
Individuals probably contract COVID-19 at similar rates. However, once infected, some persons do worse than others. The inoculum of infection may be an important variable (46) but will not be further discussed here. We will focus on the role of underlying health conditions. And we will relate these to the PO and AO immune responses that we discussed above.
As noted above, COVID-19 induces an excessive PO response. This needs to be balanced by AO mechanisms: NO and GSH. As depicted in Graphical Abstract, these two mechanisms are dependent on the cofactor NADPH (47, 48). It is produced mainly by G6PD in a rate-limiting manner in the pentose phosphate pathway (PPP) of glucose metabolism. In addition to NO and GSH, there are several other systems that require NADPH and compete for it: macrophage NADPH oxidase (NOX2) for antimicrobial defense, NADPH methemoglobin reductase for Hb recovery, and thyroid NADPH oxidase for triiodothyronine (T3) production (49). The inability of G6PD to supply enough NADPH for the excessive immune response, along with these other demands, aggravates G6PDd. Thus, G6PD is essential for both components of innate immune response and, particularly, for the AO system to balance the PO system (50).
NO and GSH are also dependent on flavin adenine dinucleotide (FAD). FAD production is catalyzed by T3, which requires NADPH for its synthesis by thyroid NADPH oxidase (51). Thus, G6PDd ultimately decreases T3, NO, and GSH, thereby compromising the body's defensive mechanisms.
Acquired G6PDd
While congenital G6PDd is well known, its acquired deficiency is less appreciated. It accompanies insulin resistance (52) and hypertension (53, 54), grouped together as the metabolic syndrome. In addition, advancing age also lowers it (55). We demonstrate the biochemical rationale of these findings and why these cohorts do worse with COVID-19.
As depicted in Figure 3, adipocytes secrete leptin. Obesity-induced hyper-leptinemia is chronic and progressive and directly stimulates Ald (56). Moreover, leptin suppresses atrial natriuretic peptide (ANP), which helps to “escape” Ald activity (57). Increased Ald, as discussed previously, results in increased TNFα (37). In addition to NOX2 activation, chronic TNFα stimulation also causes insulin resistance (14, 58). Under normal conditions, insulin receptor signaling is required for glucose entrance into cells. Glucose is phosphorylated to glucose-6- phosphate, which activates the carbohydrate response element-binding protein (ChREBP) (59). ChREBP regulates the expression of rate-limiting enzymes in glucose metabolism, in particular G6PD (60). Thus, decreased intracellular glucose results in decreased G6PD gene expression and, consequently, lower NADPH (61).
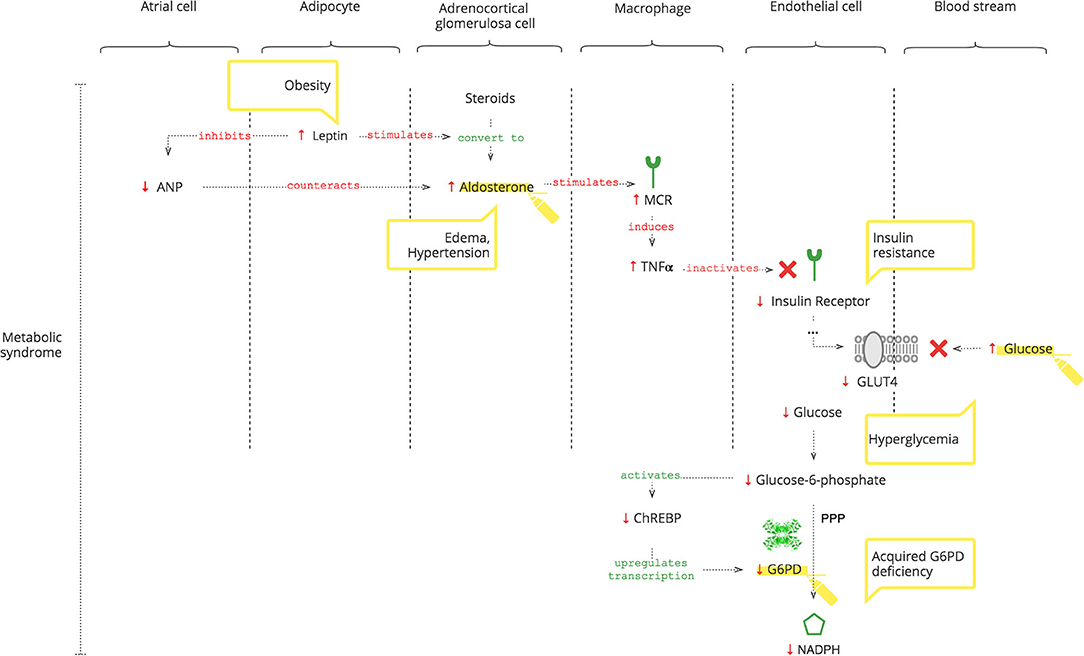
Figure 3. How the metabolic syndrome leads to acquired G6PDd.
Moreover, Liao et al. (62) showed that there is no significant difference in the expression of TNFα between G6PDd and normal patients.
Clinical Pearls
• Even when SARS-CoV-2 can no longer be detected and antibodies have formed, clinical manifestations of inflammation may ensue due to Ald-triggered TNFα.
• Leptin-induced increase of Ald is chronic, progressive, and AngII-independent, and it is not controlled by the RAAS, so angiotensin II receptor blockers (ARBs) and ACE inhibitors can be ineffective as antihypertensives (63).
• Decreased ANP levels in patients with metabolic syndrome render them vulnerable to COVID-19-induced acute increased Ald (64, 65).
• In COVID-19 patients with metabolic syndrome, hypertension, edema, and hyperglycemia accentuate.
• Chronic hyperglycemia can cause insulin resistance and can be a biomarker of developing G6PDd (66, 67).
• Laboratory values of G6PD levels and resulting NADPH activity can differ for several reasons: highly variable glucose level-dependent G6PD gene expression; the unique rate-limiting catalyzation of NADPH production; and the overload of immune mechanisms competing for NADPH, especially in patients with developing G6PDd.
• T3 levels reflect the NADPH activity but also can be involved in thyroid gland disorders.
• Metabolic syndrome-related chronic G6PDd can be aggravated by COVID-19-induced insulin resistance.
• As a consequence, patients with metabolic syndrome have a decreased level of NO and exogenous NO treatment can be considered (12, 68).
• Optimal control of underlying chronic diseases helps defend against COVID-19.
Thus, metabolic syndrome causes G6PDd. And G6PDd, by reducing NO, dysbalances the immune response to COVID-19. In addition, GSH plays a critical role as discussed below.
The Role of GSH System
GSH is an essential endogenous antioxidant. As depicted in Figure 4, it is composed of three amino acids: glycine, cysteine, and glutamate. The sulfhydryl (-SH) moiety of cysteine is responsible for the neutralization of toxic substances, both endogenous such as ROS and exogenous such as xenobiotics. During this reaction, GSH is oxidized to its inactive form, GSSG. The recycling requires NADPH and FAD (69).
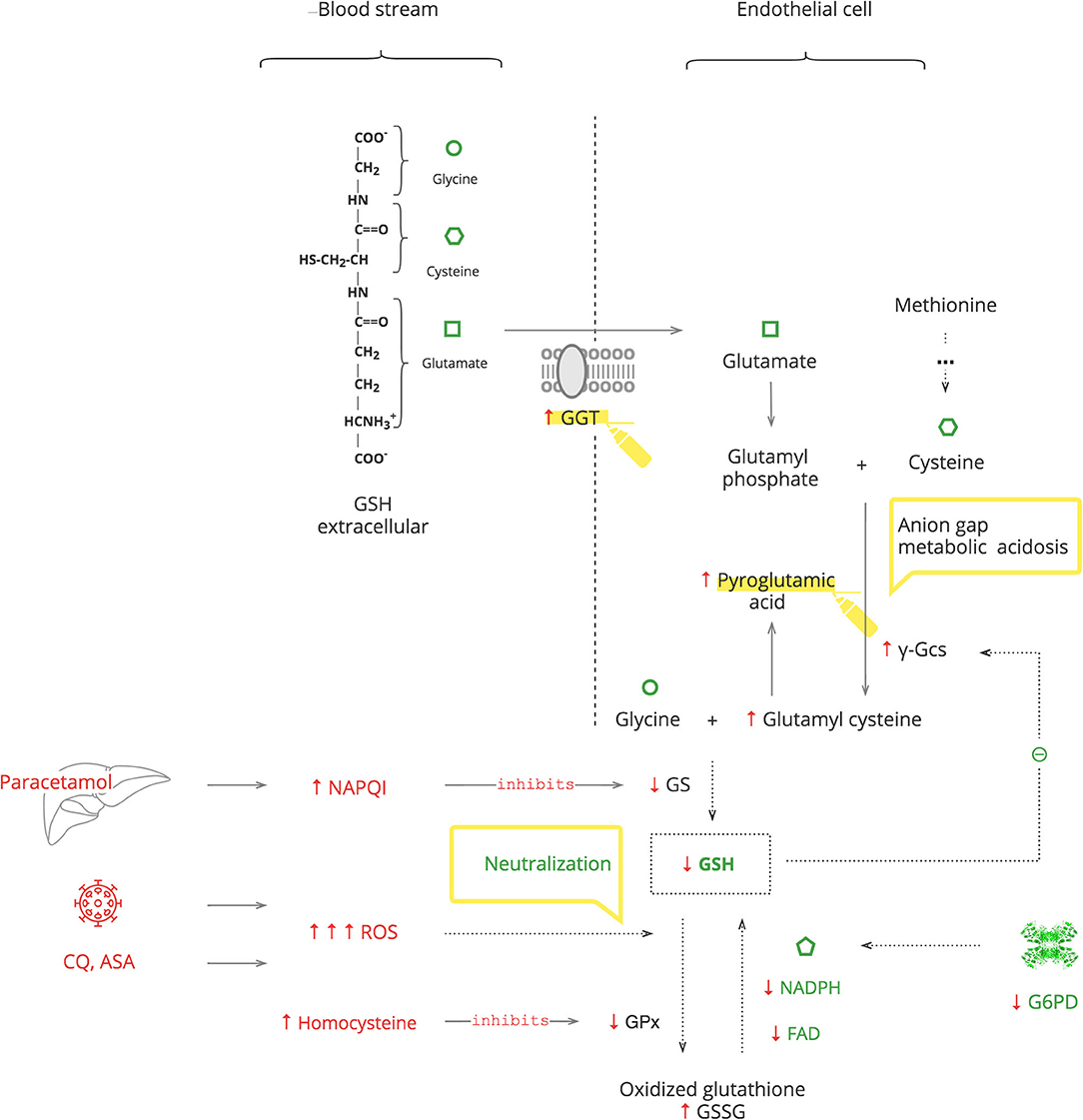
Figure 4. GSH depletion activates mechanisms of GSH repletion.
GSH depletion can be caused by G6PDd, which leads to an inability to recycle it (70, 71). It can also be caused by excessive levels of toxic substances that overload the capacity for its neutralization. Furthermore, the GSH system can be compromised by exogenous substances, e.g., the paracetamol metabolite, N-acetyl-p-benzoquinone imine (NAPQI), which inactivates glutathione synthetase (GS) of GSH production, and by endogenous substances, e.g., homocysteine, which inactivates glutathione peroxidase (GPx) of GSH function (72, 73). The body responds with γ-glutamyl transferase (GGT) upregulation to replete intracellular amino acids from extracellular GSH and also by de novo production of cysteine from methionine (74, 75). These amino acids then enter the γ-glutamyl cycle. When there is abundant GSH, it suppresses its own production by blocking γ-glutamyl cysteine synthase (γ-Gcs). Otherwise, GSH depletion results in increased γ-Gcs, leading to accumulation of pyroglutamic acid (76).
Clinical Pearls
• Exogenous stresses such as infection, medications, e.g., chloroquine (CQ), aspirin (ASA), and medical procedures, are accompanied by increased ROS production, which exacerbates GSH deficiency (77–80).
• In COVID-19, paracetamol is used as an antipyretic to avoid NSAIDs, and it accentuates GSH deficiency (69).
• Exacerbation of G6PDd manifests with fever and hematologic complications, especially hemolytic anemia. If a patient's Hb decreases after 2–3 days on certain treatments, e.g., CQ or O2 therapy, and the LDH level has increased, G6PDd should be considered.
• In critically ill patients, severe G6PDd manifests with transient hypothyroidism also known as the “low T3 syndrome” or the “euthyroid sick syndrome” (81).
• Patients with metabolic syndrome have increased levels of homocysteine. Consider folic acid and/or cyanocobalamin deficiency in these patients to prevent aggravation of GSH depletion (82, 83).
• Severe GSH deficiency clinically manifests with unexplained anion gap metabolic acidosis. This should be considered as pyroglutamic acidosis until proven otherwise. This acidemia, by itself, is not clinically important, but it is a sign of serious metabolic stress (73).
• An increased level of GGT can be used as a biomarker of GSH depletion.
Conclusion
G6PD activity is essential for the adequate functioning of both the PO and AO components of the innate immune response to counteract COVID-19-induced immune dysregulation. Therefore, in COVID-19 patients, inadequate G6PD activity should be considered and can be monitored with biomarkers. Recognizing these interactions is critical to avoid inappropriate treatment. “Primum non nocere.”
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
All authors contributed to the work equally. All authors read and approved the final manuscript.
Funding
This work was supported by Digital Science through their Catalyst Grant program. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of Interest
YB and RG were employed by sci.AI.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to express our deep gratitude to Mary J. Ruwart, Ph.D., research scientist and ethicist, and Nancy Lord, MD, independent researcher in age-reversal medicine and registered US patent attorney for their valuable and constructive suggestions during the planning and development of this research work. Secondly, we would also like to extend appreciation to our colleagues, anesthesiologists, and intensivists: Vasiliy Zaharevich, MD, State Institution Minsk Scientific, and Practical Center for Surgery, Transplantology and Hematology, Mikhail Stryzhak, MD, Minsk Children's Infectious Diseases Hospital, and Katsiaryna Liakhouskaya, Minsk City Emergency Hospital for their valuable comments, practical observations, and great support. Also, we wish to acknowledge the useful critics provided by Gennadiy Moiseyev, Ph.D., assistant professor of research at the Department of Physiology of the University of Oklahoma Health Sciences Center. Finally, we express special appreciation to Keely Harris, founder of g6pd Deficiency Foundation, Inc. (g6pdDF.org), to Niloofer Darbary, director–Transformation Management, Fiserv Inc., creator of the petition for the need for more awareness and research G6PD Deficiency and its role in the Corona Virus Pandemic, and to Terri Falbo, Non-Physician Member of Physicians for a National Health Program (USA), for their initiatives and enterprise in the global problem confronting the G6PDd cohort.
References
1. Buinitskaya Y, Gurinovich R, Wlodaver CG, Kastsiuchenka S. Highlights of COVID-19 pathogenesis. Insights into oxidative damage. (2020). doi: 10.6084/m9.figshare.12121575.v10
2. Rahal A, Kumar A, Singh V, Yadav B, Tiwari R, Chakraborty S, et al. Oxidative stress, prooxidants, and antioxidants: the interplay. BioMed Res Intern. (2014) 2014:761264. doi: 10.1155/2014/761264
3. Janeway CA Jr, Travers P, Walport M, Shlomchik MJ. Immunobiology: The Immune System in Health and Disease. 5th edition. New York, NY: Garland Science (2001).
4. Huang C, Lokugamage KG, Rozovics JM, Narayanan K, Semler BL, Makino S. SARS coronavirus nsp1 protein induces template-dependent endonucleolytic cleavage of mRNAs: viral mRNAs are resistant to nsp1-induced RNA cleavage. PLoS Pathog. (2011) 7:e1002433. doi: 10.1371/journal.ppat.1002433
5. Wu YH, Tseng CP, Cheng ML, Ho HY, Shih SR, Chiu DT. Glucose-6-phosphate dehydrogenase deficiency enhances human coronavirus 229E infection. J Infect Dis. (2008) 197:812–6. doi: 10.1086/528377
6. Nkhoma ET, Poole C, Vannappagari V, Hall SA, Beutler E. The global prevalence of glucose-6-phosphate dehydrogenase deficiency: a systematic review and meta-analysis. Blood Cells Mol Dis. (2009) 42:267–78. doi: 10.1016/j.bcmd.2008.12.005
7. Vick DJ. Glucose-6-phosphate dehydrogenase deficiency and COVID-19 infection. Mayo Clinic Proc. (2020) 95:1803–4. doi: 10.1016/j.mayocp.2020.05.035
8. Al-Aamri M, Al-Khalifa F, Al-Nahwi F, Al-Ameer H, Al-Abdi S. G6PD deficiency overrepresented among pediatric COVID-19 cases in one saudi children hospital. MedRxiv. (2020) 2020:700. doi: 10.1101/2020.07.08.20148700
9. Al-Aamri MA, Al-Khars FT, Alkhwaitem SJ, AlHassan AK, Al Aithan AM, Alkhalifa FH, et al. A saudi G6PD deficient girl died with pediatric multisystem inflammatory syndrome-COVID-19. MedRxiv. (2020) 2020:497. doi: 10.1101/2020.07.08.20137497
10. Jamerson BD, Haryadi TH, Bohannon A. Glucose-6-phosphate dehydrogenase deficiency: an actionable risk factor for patients with COVID-19?. Arch Med Res. (2020). doi: 10.1016/j.arcmed.2020.06.006
11. Gheita TA, Kenawy SA, El Sisi RW, Gheita HA, Khalil H. Subclinical reduced G6PD activity in rheumatoid arthritis and Sjögren's Syndrome patients: relation to clinical characteristics, disease activity and metabolic syndrome. Mod Rheum. (2014) 24:612–7. doi: 10.3109/14397595.2013.851639
12. Siervo M, Jackson SJ, Bluck LJ. In-vivo nitric oxide synthesis is reduced in obese patients with metabolic syndrome: application of a novel stable isotopic method. J Hyperten. (2011) 29:1515–27. doi: 10.1097/HJH.0b013e3283487806
13. Totura AL, Whitmore A, Agnihothram S, Schäfer A, Katze MG, Heise MT, et al. Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. mBio. (2015) 6:e00638–15. doi: 10.1128/mBio.00638-15
14. Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci USA. (1994) 91:4854–8. doi: 10.1073/pnas.91.11.4854
15. Li JM, Mullen AM, Yun S, Wientjes F, Brouns GY, Thrasher AJ, et al. Essential role of the NADPH oxidase subunit p47(phox) in endothelial cell superoxide production in response to phorbol ester and tumor necrosis factor-alpha. Circul Res. (2002) 90:143–50. doi: 10.1161/hh0202.103615
16. Thomas C, Mackey MM, Diaz AA, Cox DP. Hydroxyl radical is produced via the Fenton reaction in submitochondrial particles under oxidative stress: implications for diseases associated with iron accumulation. Red Rep. (2009) 14:102–8. doi: 10.1179/135100009X392566
17. Kobayashi Y, Hayashi M, Yoshino F, Tamura M, Yoshida A, Ibi H, et al. Bactericidal effect of hydroxyl radicals generated from a low concentration hydrogen peroxide with ultrasound in endodontic treatment. J Clin Biochem Nutr. (2014) 54:161–5. doi: 10.3164/jcbn.13-86
18. Marx JJ, van Asbeck BS. Use of iron chelators in preventing hydroxyl radical damage: adult respiratory distress syndrome as an experimental model for the pathophysiology and treatment of oxygen-radical-mediated tissue damage. Acta Haematol. (1996) 95:49–62. doi: 10.1159/000203949
19. Allan IM, Vaughan AT, Milner AE, Lunec J, Bacon PA. Structural damage to lymphocyte nuclei by H2O2 or gamma irradiation is dependent on the mechanism of OH radical production. Br J Cancer. (1988) 58:34–7. doi: 10.1038/bjc.1988.156
20. Praticó D, Pasin M, Barry OP, Ghiselli A, Sabatino G, Iuliano L, et al. Iron-dependent human platelet activation and hydroxyl radical formation: involvement of protein kinase C. Circulation. (1999) 99:3118–24. doi: 10.1161/01.cir.99.24.3118
21. Zhang MH. Rhabdomyolosis and its pathogenesis. World J Emerg Med. (2012) 3:11–5. doi: 10.5847/wjem.j.issn.1920-8642.2012.01.002
22. Bhattacharyya J, Datta AG. Studies on the effects of lipopolysaccharide on lipid peroxidation of erythrocyte and its reversal by mannitol and glycerol. J Physiol Pharmacol. (2001) 52:145–52.
23. Antosik A, Czubak K, Cichon N, Nowak P, Zbikowska H. Vitamin E analogue protects red blood cells against storage-induced oxidative damage. Transfus Med Hemother. (2018) 45:347–54. doi: 10.1159/000486605
24. Stolze K, Nohl H. Formation of methemoglobin and phenoxyl radicals from p-hydroxyanisole and oxyhemoglobin. Free Rad Res Commun. (1991) 11:321–7. doi: 10.3109/10715769109088930
25. Wang A, Zhao W, Xu Z, Gu J. Timely blood glucose management for the outbreak of 2019 novel coronavirus disease (COVID-19) is urgently needed. Diabetes Res Clin Pract. (2020) 162:108118. doi: 10.1016/j.diabres.2020.108118
26. Miller LL, Miller SC, Torti SV, Tsuji Y, Torti FM. Iron-independent induction of ferritin H chain by tumor necrosis factor. Proc Natl Acad Sci USA. (1991) 88:4946–50. doi: 10.1073/pnas.88.11.4946
27. Erel O, Vural H, Aksoy N, Aslan G, Ulukanligil M. Oxidative stress of platelets and thrombocytopenia in patients with vivax malaria. Clin Biochem. (2001) 34:341–4. doi: 10.1016/s0009-9120(01)00221-1
28. Bermejo-Martin JF, Almansa R, Menéndez R, Mendez R, Kelvin DJ, Torres A. Lymphopenic community acquired pneumonia as signature of severe COVID-19 infection. J Infect. (2020) 80:e23–4. doi: 10.1016/j.jinf.2020.02.029
29. Torres PA, Helmstetter JA, Kaye AM, Kaye AD. Rhabdomyolysis: pathogenesis, diagnosis, and treatment. Ochsner J. (2015) 15:58–69.
30. Jin M, Tong Q. Rhabdomyolysis as potential late complication associated with COVID-19. Emerg Infect Dis. (2020) 26:1618–20. doi: 10.3201/eid2607.200445
31. Lu G, Wang J. Dynamic changes in routine blood parameters of a severe COVID-19 case. Clin Chim Acta. (2020) 508:98–102. doi: 10.1016/j.cca.2020.04.034
32. Naymagon L, Berwick S, Kessler A, Lancman G, Gidwani U, Troy K. The emergence of methemoglobinemia amidst the COVID-19 pandemic. Am J Hematol. (2020) 95:E196–7. doi: 10.1002/ajh.25868
33. Rizvi I, Zaman S, Zaidi N, Asif MS, Abdali N. Acute life-threatening methaemoglobinaemia following ingestion of chloroquine. BMJ Case Rep. (2012). doi: 10.1136/bcr.12.2011.5383
34. Yang T, Peleli M, Zollbrecht C, Giulietti A, Terrando N, Lundberg JO, et al. Inorganic nitrite attenuates NADPH oxidase-derived superoxide generation in activated macrophages via a nitric oxide-dependent mechanism. Free Rad Biol Med. (2015) 83:159–66. doi: 10.1016/j.freeradbiomed.2015.02.016
35. Stańczyk M, Gromadzińska J, Wasowicz W. Roles of reactive oxygen species and selected antioxidants in regulation of cellular metabolism. Int J Occup Med Environ Health. (2005) 18:15–26.
36. Green SJ. Covid-19 accelerates endothelial dysfunction and nitric oxide deficiency. Microb Infect. (2020) 22:149–50. doi: 10.1016/j.micinf.2020.05.006
37. Chirkov YY, Belushkina NN, Tyshchuk IA, Severina IS, Horowitz JD. Increase in reactivity of human platelet guanylate cyclase during aggregation potentiates the disaggregating capacity of sodium nitroprusside. Clin Exp Pharmacol. (1991) 18:517–24. doi: 10.1111/j.1440-1681.1991.tb01486.x
38. Akerström S, Gunalan V, Keng CT, Tan YJ, Mirazimi A. Dual effect of nitric oxide on SARS-CoV replication: viral RNA production and palmitoylation of the S protein are affected. Virology. (2009) 395:1–9. doi: 10.1016/j.virol.2009.09.007
39. Tanabe A, Naruse M, Arai K, Naruse K, Yoshimoto T, Seki T, et al. Angiotensin II stimulates both aldosterone secretion and DNA synthesis via type 1 but not type 2 receptors in bovine adrenocortical cells. J Endocrinol Invest. (1998) 21:668–72. doi: 10.1007/BF03350796
40. Chantong B, Kratschmar DV, Nashev LG, Balazs Z, Odermatt A. Mineralocorticoid and glucocorticoid receptors differentially regulate NF-kappaB activity and pro-inflammatory cytokine production in murine BV-2 microglial cells. J Neuroinflamm. (2012) 9:260. doi: 10.1186/1742-2094-9-260
41. Brickner RC, Raff H. Oxygen sensitivity of potassium- and angiotensin II-stimulated aldosterone release by bovine adrenal cells. J Endocrinol. (1991) 129:43–8. doi: 10.1677/joe.0.1290043
42. Kaufman DP, Kandle PF, Murray I, Dhamoon AS. Physiology, Oxyhemoglobin Dissociation Curve. In: StatPearls. Treasure Island, FL: StatPearls Publishing (2020).
43. Terpos E, Ntanasis-Stathopoulos I, Elalamy I, Kastritis E, Sergentanis TN, Politou M, et al. Hematological findings and complications of COVID-19. Am J Hematol. (2020) 95:834–47. doi: 10.1002/ajh.25829
44. Mahjoub Y, Rodenstein DO, Jounieaux V. Severe covid-19 disease: rather AVDS than ARDS?. Critical Care. (2020) 24:327. doi: 10.1186/s13054-020-02972-w
45. Semler MW, Marney AM, Rice TW, Nian H, Yu C, Wheeler AP, et al. B-type natriuretic peptide, aldosterone, and fluid management in ARDS. Chest. (2016) 150:102–11. doi: 10.1016/j.chest.2016.03.017
46. Guallar MP, Meiriño R, Donat-Vargas C, Corral O, Jouvé N, Soriano V. Inoculum at the time of SARS-CoV-2 exposure and risk of disease severity. Int J Infect Dis. (2020) 97:290–2. doi: 10.1016/j.ijid.2020.06.035
47. Leopold JA, Cap A, Scribner AW, Stanton RC, Loscalzo J. Glucose-6-phosphate dehydrogenase deficiency promotes endothelial oxidant stress and decreases endothelial nitric oxide bioavailability. FASEB J. (2001) 15:1771–3. doi: 10.1096/fj.00-0893fje
48. Parsanathan R, Jain SK. Glucose-6-phosphate dehydrogenase deficiency increases cell adhesion molecules and activates human monocyte-endothelial cell adhesion: Protective role of l-cysteine. Arch Biochem Biophys. (2019) 663:11–21. doi: 10.1016/j.abb.2018.12.023
49. Kuhn V, Diederich L, Keller T, Kramer CM, Lückstädt W, Panknin C, et al. Red blood cell function and dysfunction: redox regulation, nitric oxide metabolism, anemia. Antioxid Redox Signal. (2017) 26:718–42. doi: 10.1089/ars.2016.6954
50. Yang HC, Wu YH, Yen WC, Liu HY, Hwang TL, Stern A, et al. The redox role of G6PD in cell growth, cell death, and cancer. Cells. (2019) 8:1055. doi: 10.3390/cells8091055
51. Lee SS, McCormick DB. Thyroid hormone regulation of flavocoenzyme biosynthesis. Arch Biochem Biophys. (1985) 237:197–201. doi: 10.1016/0003-9861(85)90269-3
52. Sun Q, Zhang BY, Zhang PA, Hu J, Zhang HH, Xu GY. Downregulation of glucose-6-phosphate dehydrogenase contributes to diabetic neuropathic pain through upregulation of toll-like receptor 4 in rats. Mol Pain. (2019) 15:1744806919838659. doi: 10.1177/1744806919838659
53. Zhao J, Zhang X, Guan T, Wang X, Zhang H, Zeng X, et al. The association between glucose-6-phosphate dehydrogenase deficiency and abnormal blood pressure among prepregnant reproductive-age Chinese females. Hypertension research: official journal of the Japanese Society of Hypertension. (2019) 42:75–84. doi: 10.1038/s41440-018-0118-1
54. Moss ME, Jaffe IZ. Mineralocorticoid receptors in the pathophysiology of vascular inflammation and atherosclerosis. Front Endocrinol. (2015) 6:153. doi: 10.3389/fendo.2015.00153
55. Maurya PK, Kumar P, Chandra P. Age-dependent detection of erythrocytes glucose-6-phosphate dehydrogenase and its correlation with oxidative stress. Arch Physiol Biochem. (2016) 122:61–6. doi: 10.3109/13813455.2015.1136648
56. Belin de Chantemèle EJ, Mintz JD, Rainey WE, Stepp DW. Impact of leptin-mediated sympatho-activation on cardiovascular function in obese mice. Hypertension. (2011) 58:271–9. doi: 10.1161/HYPERTENSIONAHA.110.168427
57. Yuan K, Yu J, Shah A, Gao S, Kim SY, Kim SZ, et al. Leptin reduces plasma ANP level via nitric oxide-dependent mechanism. Am J Physiol Regul Integr Comp Physiol. (2010) 298:R1007–16. doi: 10.1152/ajpregu.00598.2009
58. Sowers JR. Obesity as a cardiovascular risk factor. Am J Med. (2003) 115: Suppl 8A, 37S−41S. doi: 10.1016/j.amjmed.2003.08.012
59. Li MV, Chen W, Harmancey RN, Nuotio-Antar AM, Imamura M, Saha P, et al. Glucose-6-phosphate mediates activation of the carbohydrate responsive binding protein (ChREBP). Biochem Biophys Res Commun. (2010) 395:395–400. doi: 10.1016/j.bbrc.2010.04.028
60. Vijayakumar A, Aryal P, Wen J, Syed I, Vazirani RP, Moraes-Vieira PM, et al. Absence of carbohydrate response element binding protein in adipocytes causes systemic insulin resistance and impairs glucose transport. Cell Rep. (2017) 21:1021–35. doi: 10.1016/j.celrep.2017.09.091
61. Leopold JA, Dam A, Maron BA, Scribner AW, Liao R, Handy DE, et al. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med. (2007) 13:189–97. doi: 10.1038/nm1545
62. Liao SL, Lai SH, Tsai MH, Weng YH. Cytokine responses of TNF-α, IL-6, and IL-10 in G6PD-deficient infants. Pedi Hematol Oncol. (2014) 31:87–94. doi: 10.3109/08880018.2013.865821
63. Dudenbostel T, Ghazi L, Liu M, Li P, Oparil S, Calhoun DA. Body mass index predicts 24-hour urinary aldosterone levels in patients with resistant hypertension. Hypertension. (2016) 68:995–1003. doi: 10.1161/HYPERTENSIONAHA.116.07806
64. Wang JH, Lee CJ, Hsieh JC, Chen YC, Hsu BG. Serum atrial natriuretic peptide level inversely associates with metabolic syndrome in older adults. Geri Gerontol Int. (2014) 14:640–6. doi: 10.1111/ggi.12151
65. Yokota N, Bruneau BG, Kuroski de Bold ML, de Bold AJ. Atrial natriuretic factor significantly contributes to the mineralocorticoid escape phenomenon. Evidence for a guanylate cyclase-mediated pathway. J Clin Invest. (1994) 94:1938–46. doi: 10.1172/JCI117544
66. Zhang Z, Liew CW, Handy DE, Zhang Y, Leopold JA, Hu J, et al. High glucose inhibits glucose-6-phosphate dehydrogenase, leading to increased oxidative stress and beta-cell apoptosis. FASEB J. (2010) 24:1497–505. doi: 10.1096/fj.09-136572
67. Zhang Z, Apse K, Pang J, Stanton RC. High glucose inhibits glucose-6-phosphate dehydrogenase via cAMP in aortic endothelial cells. J Biol Chem. (2000) 275:40042–7. doi: 10.1074/jbc.M007505200
68. Chen L, Liu P, Gao H, Sun B, Chao D, Wang F, et al. Inhalation of nitric oxide in the treatment of severe acute respiratory syndrome: a rescue trial in Beijing. Clin Infect Dis. (2004) 39:1531–5. doi: 10.1086/425357
69. Lushchak VI. Glutathione homeostasis and functions: potential targets for medical interventions. J Amino Acids. (2012) 2012:736837. doi: 10.1155/2012/736837
70. Rajasekaran NS, Connell P, Christians ES, Yan LJ, Taylor RP, Orosz A, et al. Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell. (2007) 130:427–39. doi: 10.1016/j.cell.2007.06.044
71. Jain M, Cui L, Brenner DA, Wang B, Handy DE, Leopold JA, et al. Increased myocardial dysfunction after ischemia-reperfusion in mice lacking glucose-6-phosphate dehydrogenase. Circulation. (2004) 109:898–903. doi: 10.1161/01.CIR.0000112605.43318.CA
72. Walker V, Mills GA, Anderson ME, Ingle BL, Jackson JM, Moss CL, et al. The acetaminophen metabolite N-acetyl-p-benzoquinone imine (NAPQI) inhibits glutathione synthetase in vitro; a clue to the mechanism of 5-oxoprolinuric acidosis?. Xenobiotica. (2017) 47:164–75. doi: 10.3109/00498254.2016.1166533
73. Handy DE, Zhang Y, Loscalzo J. Homocysteine down-regulates cellular glutathione peroxidase (GPx1) by decreasing translation. J Biol Chem. (2005) 280:15518–25. doi: 10.1074/jbc.M501452200
74. Zhang H, Forman HJ. Redox regulation of gamma-glutamyl transpeptidase. Am J Resp Cell Mol Biol. (2009) 41:509–15. doi: 10.1165/rcmb.2009-0169TR
75. Vitvitsky V, Mosharov E, Tritt M, Ataullakhanov F, Banerjee R. Redox regulation of homocysteine-dependent glutathione synthesis. Redox Report. (2003) 8:57–63. doi: 10.1179/135100003125001260
76. Gamarra Y, Santiago FC, Molina-López J, Castaño J, Herrera-Quintana L, Domínguez Á, et al. Pyroglutamic acidosis by glutathione regeneration blockage in critical patients with septic shock. Critical Care. (2019) 23:162. doi: 10.1186/s13054-019-2450-5
77. Nasi A, McArdle S, Gaudernack G, Westman G, Melief C, Rockberg J, et al. Reactive oxygen species as an initiator of toxic innate immune responses in retort to SARS-CoV-2 in an ageing population, consider N-acetylcysteine as early therapeutic intervention. Toxicol Rep. (2020) 7:768–71. doi: 10.1016/j.toxrep.2020.06.003
78. Park J, Choi K, Jeong E, Kwon D, Benveniste EN, Choi C. Reactive oxygen species mediate chloroquine-induced expression of chemokines by human astroglial cells. Glia. (2004) 47:9–20. doi: 10.1002/glia.20017
79. Raza H, John A, Shafarin J. Potentiation of LPS-induced apoptotic cell death in human hepatoma HepG2 cells by aspirin via ROS and mitochondrial dysfunction: protection by N-acetyl cysteine. PLoS ONE. (2016) 11:e0159750. doi: 10.1371/journal.pone.0159750
80. Kavazis AN, Talbert EE, Smuder AJ, Hudson MB, Nelson WB, Powers SK. Mechanical ventilation induces diaphragmatic mitochondrial dysfunction and increased oxidant production. Free Rad Biol Med. (2009) 46:842–50. doi: 10.1016/j.freeradbiomed.2009.01.002
81. Economidou F, Douka E, Tzanela M, Nanas S, Kotanidou A. Thyroid function during critical illness. Hormones. (2011) 10:117–24. doi: 10.14310/horm.2002.1301
82. Sreckovic B, Sreckovic VD, Soldatovic I, Colak E, Sumarac-Dumanovic M, Janeski H, et al. Homocysteine is a marker for metabolic syndrome and atherosclerosis. Diab Metab Syndr. (2017) 11:179–82. doi: 10.1016/j.dsx.2016.08.026
Keywords: COVID-19, glucose-6-phosphate dehydrogenase (G6PD), reactive oxygen species, nitric oxide - NO, glutathione, aldosterone (Ald), Metabolic syndrome
Citation: Buinitskaya Y, Gurinovich R, Wlodaver CG and Kastsiuchenka S (2020) Centrality of G6PD in COVID-19: The Biochemical Rationale and Clinical Implications. Front. Med. 7:584112. doi: 10.3389/fmed.2020.584112
Received: 16 July 2020; Accepted: 27 August 2020;
Published: 22 October 2020.
Edited by:
John Hay, University at Buffalo, United StatesReviewed by:
Jane A. Leopold, Massachusetts General Hospital and Harvard Medical School, United StatesGiovanni Mario Pes, University of Sassari, Italy
Copyright © 2020 Buinitskaya, Gurinovich, Wlodaver and Kastsiuchenka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuliya Buinitskaya, anVsaWFAc2NpLmFp
†These authors have contributed equally to this work