 Francesco Pesce1†
Francesco Pesce1† Emma D. Stea1†Michele Rossini1
Emma D. Stea1†Michele Rossini1 Marco Fiorentino1
Marco Fiorentino1 Fausta Piancone1Barbara Infante2Giovanni Stallone2Giuseppe Castellano2
Fausta Piancone1Barbara Infante2Giovanni Stallone2Giuseppe Castellano2 Loreto Gesualdo1*
Loreto Gesualdo1*- 1Nephrology, Dialysis and Transplantation Unit, Department of Emergency and Organ Transplantation, University of Bari, Bari, Italy
- 2Nephrology, Dialysis and Transplantation Unit, Department of Medical and Surgical Science, University of Foggia, Foggia, Italy
Acute kidney injury (AKI) is increasingly emerging as a global emergency. Sepsis, major surgery, and nephrotoxic drugs are the main causes of AKI in hospitalized patients. However, glomerulonephritis accounts for about 10% of AKI episodes in adults, mainly related to rapidly progressive glomerulonephritis resulting from granulomatous polyangiitis (GPA, Wegener granulomatosis), microscopic polyangiitis (MPA), and anti-glomerular basement membrane (GBM) disease. Also, diffuse proliferative lupus nephritis, immunoglobulin A nephropathy, post-streptococcal glomerulonephritis, mixed cryoglobulinemia, mesangiocapillary glomerulonephritis, membranous nephropathy, hemolytic uremic syndrome, thrombotic thrombocytopenic purpura, and scleroderma can induce acute renal failure. Early diagnosis of AKI due to glomerulonephritis is crucial for prompt, effective management to improve short- and long-term outcomes. Kidney biopsy is the gold standard for the diagnosis of glomerular disease, but it is not frequently performed in critically ill patients because of their clinical conditions. In this setting, a growing number of diagnostic assays can support the working hypothesis, including antineutrophil cytoplasmic antibodies (ANCAs), anti-double-stranded DNA antibodies, anti-GBM antibodies, antistreptolysin O and anti-DNase B antibodies, cryoglobulins, antiphospholipid antibodies, and complement levels. Therapeutic strategies in AKI patients with glomerulonephritis include high-dose corticosteroids, cyclophosphamide, and plasma exchange. This article reviews the wide spectrum of glomerulopathies associated with AKI, describing the immunological mechanisms underlying glomerular diseases and presenting an overview of the therapeutic options.
Introduction
Acute kidney injury (AKI) is a severe medical condition involving up to 10 million people worldwide (1), with an increasing global incidence especially in hospitalized patients (2). AKI affects ~10–15% of hospital inpatients and more than 50% of patients hospitalized in the intensive care units (ICUs) (2, 3). Renal replacement therapy (RRT) is necessary in 5–6% of critically ill patients and is characterized by an increased risk of progression to chronic kidney disease (CKD) and end-stage kidney disease (ESKD) (about 10% annually) (4–6). The definition of AKI is based on standard criteria such as serum creatinine and/or urine output (7, 8), although several biomarkers have recently been proposed in this clinical setting (9). Severe short- and long-term consequences are frequently associated with AKI, and the mortality rate in critically ill patients is still significant, ranging from 37% to 60% (4, 10–12). Moreover, patients who survive AKI present a major long-term mortality rate and an increased risk of developing CKD (13) and other chronic comorbidities (14–19).
Glomerulonephritis accounts for about 10% of AKI in adults (20). AKI episodes in glomerular disease are usually due to rapidly progressive glomerulonephritis (RPGN), in which the renal function declines over days or weeks. The most common causes are small-vessel vasculitis and anti-glomerular basement membrane (GBM) disease, although other glomerular diseases may manifest with acute renal impairment, including IgA nephropathy (IgAN), thrombotic microangiopathy (TMA), lupus nephritis, and post-streptococcal glomerulonephritis (16). Furthermore, acute renal failure in glomerulonephritis can also result from non-glomerular conditions such as acute tubular necrosis (ATN) from renal hypoperfusion or the nephrotic syndrome and drug- or radiocontrast agent-induced tubular epithelial cell injury.
Early diagnosis and prompt, effective treatment of glomerular disease may dramatically change the disease course and improve patient outcomes (21). In this scenario, kidney biopsy remains the gold standard for the diagnosis of kidney disease when the patient's clinical condition allows the performance of this procedure (22). This article reviews the main glomerular diseases manifesting with AKI (summarized in Table 1), describing the immunological mechanisms underlying glomerular diseases and the potential therapeutic strategies, summarizing the main features.
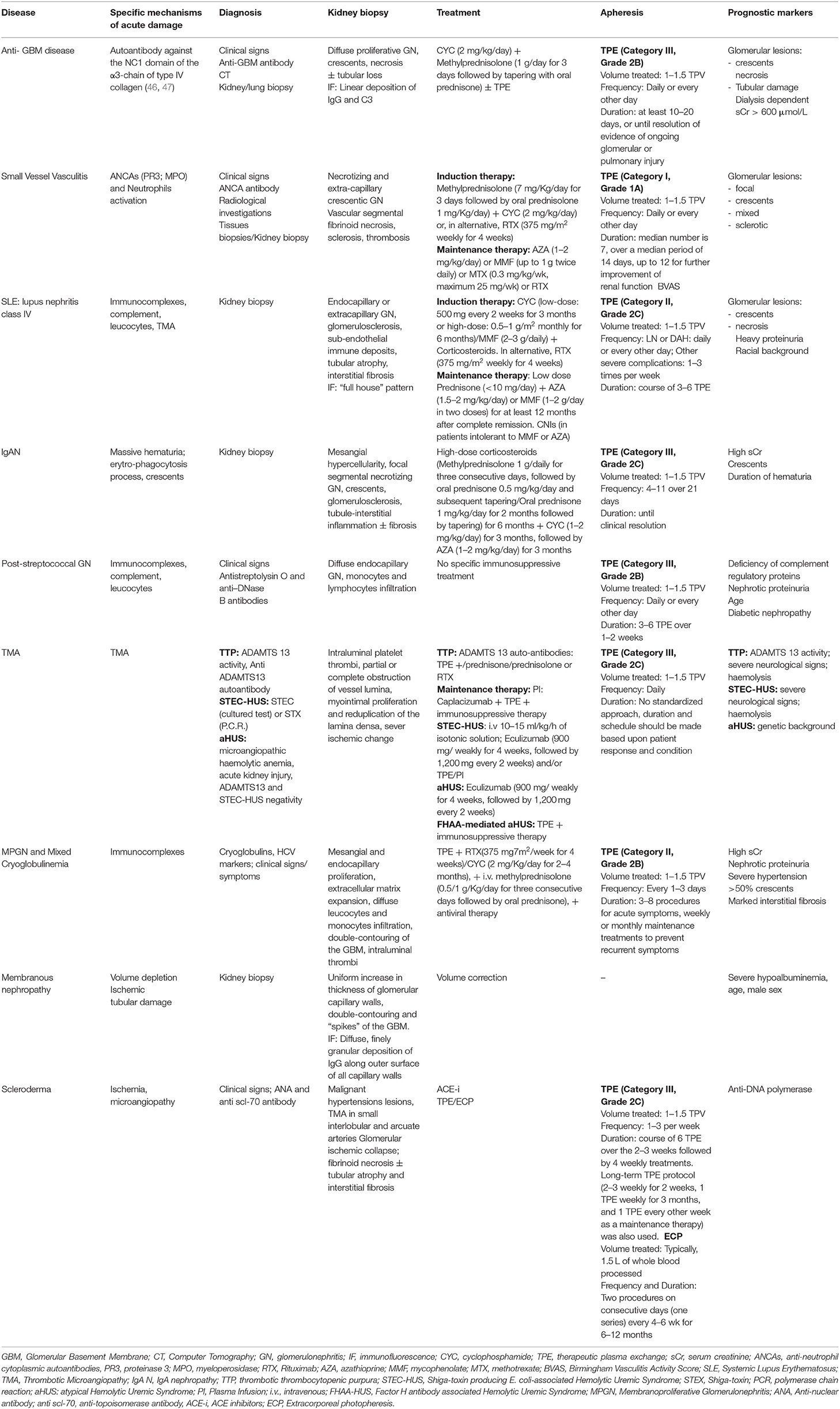
Table 1. Summary of key features for each disease.
Anti-GBM Disease
Epidemiology and Disease Pathogenesis
Anti-GBM disease is a rare but severe immunological disorder manifesting with RPGN and pulmonary complications (lung hemorrhage). It accounts for about 5% of all cases of RPGN and affects especially young males and the elderly (23, 24). A genetic predisposition has been reported: anti-GBM disease is strongly associated with class II human leukocyte antigen (HLA), including DRB1*1501 and DR4 alleles, whereas DR1 and DR7 confer strong protection (25). Furthermore, several environmental triggers (smoking, hydrocarbon exposure, and drugs) appear to be important in the disease etiology (24, 26). The pathogenesis of the disease is immunological and related to the formation of antibodies specifically targeting the NC1 domain of the α3-chain of type IV collagen localized in the glomerular and alveolar basement membranes (24, 27, 28): complement activation, phagocyte accumulation, and T-cell recruitment after immunocomplex deposition contribute to the extent of the glomerular damage (29).
Clinical Presentations and Diagnosis
Patients typically present a mild prodromal phase followed by severe clinical features with macroscopic hematuria and/or AKI. Lung involvement is often characterized by hemoptysis and dyspnea that characterize the Goodpasture syndrome. Kidney and respiratory functions decline more rapidly than in any other form of RPGN, and mortality is often due to renal failure requiring RRT or to massive alveolar bleeding (30). The clinical suspicion, based on the simultaneous renal and pulmonary involvement, is a critical step to improve patient outcomes. The rapid renal function decline, the presence of a very active urinary sediment, and the scarce systemic involvement differentiate anti-GBM disease from vasculitis and lupus nephritis. An atypical variant of the disease, with no significant pulmonary involvement and undetectable serum anti-GBM antibodies, has also been described (30). Antineutrophil cytoplasmic antibody (ANCA) can be detected in rare cases showing double antibody positivity. This double-positive variant presents a bad prognosis requiring early and more aggressive treatment (30).
The diagnosis is confirmed by the detection of circulating anti-GBM antibodies, although their levels do not correlate with disease severity (27). Kidney biopsy is essential as it indicates the extent and severity of the renal lesions. The histological picture in anti-GBM disease is usually characterized by diffuse extracapillary proliferation with extensive crescent formation, often with associated fibrinoid necrosis of the glomerular tuft. Different degrees of glomerulosclerosis (as a result of previous proliferative lesions), tubular necrosis, and interstitial inflammation can also be observed. A pathognomonic sign of the disease is the linear deposition of IgG and, often, C3 (75%) along the glomerular and sometimes (10–67%) also distal tubular basement membranes and the alveolar basal membrane in the lung. The extent of glomerular involvement (percentage of crescents and fibrinoid necrosis) is correlated with the prognosis (29, 30).
Treatment
Since the clinical course is rapidly progressive, early treatment is needed, to remove circulating anti-GBM antibodies and reduce renal inflammation. Immunosuppressive therapy should be started immediately, except in patients with minimal renal involvement or irreversible kidney disease without lung involvement (31). The Kidney Disease Improving Global Outcomes (KDIGO) clinical practice guidelines for glomerulonephritis recommend the use of corticosteroids (usually intravenous pulses of methylprednisolone up to 1,000 mg/day for three consecutive days followed by oral prednisone of 1 mg/kg/day tapered to 20 mg/day for 6 weeks), oral cyclophosphamide (CYC, 2 mg/kg/day), and plasmapheresis (daily exchange of 1–1.5 volume of plasma against 5% human albumin for 14 days) in patients affected by anti-GBM disease (31–33). Lung hemorrhage is usually responsive to this treatment, and hemoptysis resolves within a few days. Renal recovery is more variable, and the clinical response to therapy is usually slower in patients requiring RRT and/or with serum creatinine values exceeding 600 μmol/L and in patients with histological evidence of a high proportion of crescents (34, 35). Oral prednisone and CYC are generally discontinued after 6 and 3 months, respectively, while plasmapheresis is continued for at least 14 days or until anti-GBM antibodies become undetectable.
Small-Vessel Vasculitis
Epidemiology and Disease Pathogenesis
Small-vessel vasculitis is a group of inflammatory systemic diseases characterized by a segmental necrotizing polyangiitis of small vessels, including GPA (Wegener granulomatosis), microscopic polyangiitis (MPA), eosinophilic granulomatosis with polyangiitis (Churg–Strauss syndrome), and renal-limited vasculitis (36). The first two diseases account for the majority of cases of RPGN, presenting as an emergency in the critical care setting (37, 38). The diseases can occur at any age, but the prevalence is greatest at ages 50–70 years, with a slight predominance in Caucasians and male gender (36). Small-vessel vasculitis is associated with the production of ANCAs, which have a central role in the disease pathogenesis, since they are directed against specific enzymes found within the cytoplasmic granules of neutrophils and monocytes, proteinase 3 (PR3), and myeloperoxidase (MPO) antigens (39, 40). Genetic background (specific α1-antitrypsin and neutrophil FcRγ111 receptor genotypes) and environmental triggers (in particular, infections and drugs) are reported as potential co-players in disease pathogenesis (41–45); specific HLA polymorphisms are associated with disease predisposition (46). After expression of PR3 and MPO on the surface following trigger conditions (infections), ANCAs bind these antigens, inducing neutrophil degranulation and the release of inflammatory mediators (cytokines, reactive oxygen species, and lytic enzymes), leading to endothelial damage (47, 48). Neutrophils also release PR3 and MPO, which adhere to the endothelium and induce in situ immunocomplex formation. Monocytes/macrophages, T cells, and complement system are also involved in the pathogenic process (49–52).
Clinical Presentations and Diagnosis
Wegener granulomatosis is characterized by a wide range of clinical manifestations, including RPGN with the formation of extracapillary crescents, alveolar hemorrhage, episcleritis, rhinitis, sinusitis, hearing loss, purpura, peripheral neuropathy, subglottic tracheal stenosis, and angina abdominis (53). The disease course is often recurrent, with relapses occurring within a few years after disease remission. In patients with MPA, renal involvement is always reported, while respiratory tract diseases are less common; moreover, the frequency of relapses is lower than that in Wegener granulomatosis.
The clinical manifestations drive the diagnosis, which is supported by the detection of circulating ANCA. Anti-PR3 autoantibodies [cytoplasmic ANCA (c-ANCA)] are positive in about 90% of patients with active Wegener granulomatosis, and anti-MPO autoantibodies [perinuclear ANCA (p-ANCA)] are typically detected in about 80% of patients with active MPA (54, 55). Typically, ANCA levels are higher at the onset, and their levels are directly correlated with disease activity; in fact, a significant increase in ANCA levels is reported in relapses. These antibodies are also found in other immunological disorders (inflammatory bowel disease, autoimmune liver disease, and rheumatoid arthritis). A limited percentage of patients (10–20%) affected by small-vessel vasculitis does not show ANCA positivity (56). Thus, ANCA negativity may not exclude the diagnosis in the presence of clinical symptoms. Radiological investigations and tissue biopsies can support the diagnosis.
Although kidney biopsy in these patients is considered to be at high risk, since bleeding complications are frequent due to vessel inflammation, histology can be very useful for predicting the response to treatment and clinical outcome.
ANCA-associated glomerulonephritis is histologically characterized by necrotizing and crescentic glomerulonephritis, with a variable degree of glomerular involvement (57). Granulomas are a specific feature of Wegener granulomatosis (58, 59). Immunofluorescence shows little or no glomerular staining for immunoglobulins or complement, and this feature is typically termed a “pauci-immune” staining pattern. Electron microscopy demonstrates subendothelial edema, microthrombosis, and degranulation of neutrophils, without immune deposits (60). Vascular lesions feature fibrinoid necrosis, sclerosis, and thrombosis. Apart from glomerular and vascular lesions, acute and chronic tubulointerstitial lesions are critical risk factors indicating disease severity and progression. A recent histological classification comprises four categories of glomerular lesions, which correlate the loss of function with increasing degrees of severity: focal, crescentic, mixed, and sclerotic. Kidney survival at 5 years is 93% in the focal, 76% in the crescentic, 61% the mixed, and 50% in the sclerotic class (55). A growing number of studies have investigated the utility of TIMP 1, C–X–C motif chemokine ligand 13, and matrix metalloproteinase-3 as biomarkers of ANCA-associated vasculitis and for treatment assessment (61, 62).
Treatment
A prompt diagnosis and early, appropriate treatment are essential to prevent loss of renal function and progression to CKD. Kidney and patient survival rates range between 65 and 75% at 5 years after appropriate treatment (63). KDIGO guidelines recommend the use of high-dose corticosteroids (intravenous methylprednisolone ar 7 mg/kg/day for 3 days followed by oral prednisone at 1 or 1 mg/kg/day of oral prednisone tapered to 0.25 mg/day for 3 months) plus oral or intravenous CYC (2 mg/kg/day or 0.5 g/m2 monthly, respectively. Combined treatment induces remission in about 90% of patients at 6 months. When CYC is contraindicated or the disease is resistant to this regime, the anti-CD20 monoclonal antibody rituximab (RTX) (375 mg/m2 weekly for 4 weeks) plus steroids is recommended (27–29). In this context, the RAVE and RITUXIVAS studies have shown that RTX is equivalent to CYC in terms of efficacy, with a similar risk profile (64, 65).
The addition of plasma exchange is recommended for patients requiring dialysis, with rapid increases of serum creatinine, diffuse pulmonary hemorrhage, and/or with an overlap syndrome of ANCA vasculitis and anti-GBM disease (31–33). Therapeutic plasma exchange (TPE) reduces the development of ESKD by ~40% (66), but the true efficacy is still questioned (67).
During maintenance therapy, CYC is replaced by azathioprine (AZA, 1–2 mg/kg/day) or alternatively mycophenolate (MMF, up to 1 g twice daily) or methotrexate (initially 0.3 mg/kg/week, maximum 25 mg/week). An alternative regimen includes the use of CYC for the induction treatment and perhaps RTX for maintenance (32, 33). Patients should be monitored for general symptoms and laboratory data, as well as urinary RBC counts, C-reactive protein, renal function, and ANCA levels. A rapid response to treatment is typical, with a significant improvement of the clinical condition within few weeks. High ANCA levels may persist for several months. The maintenance treatment is carried out for at least 18 months up to 3 years in Wegener granulomatosis and 2 years in MPA if no relapses occur (68), which can often happen during immunosuppression tapering and may require a further short course of high-dose corticosteroids or CYC.
In ANCA-associated pauci-immune glomerulonephritis, the mortality rate within the first year of diagnosis is reported to be about 20% of cases: ESKD occurs in up to 25% of the patients within 4 years after diagnosis (63).
Other Glomerulonephritis Forms That Cause AKI
Systemic Lupus Erythematosus (SLE)
Renal involvement in patients with SLE disease is associated with a worse outcome and higher mortality. Kidney damage is characterized by the deposition of immunocomplexes, complement activation, leukocyte infiltrations, and microangiopathic thrombosis probably driven by the type I interferon signature, involving all renal compartments (glomeruli, vessels, and tubule interstitium) (69). The histological findings in patients with lupus nephritis (LN) may range from initial mesangial immune deposits [class I in the World Health Organization (WHO) classification] to diffuse global glomerulosclerosis (class VI) (70); however, <10% of all patients with LN develop RPGN, which is associated with the diffuse proliferative glomerulonephritis form (Class IV).
Clinically, LN should be suspected in the presence of urinary abnormalities (hematuria) and nephrotic proteinuria; in addition, patients with Class IV LN typically exhibit acute impairment of renal function and hypertension. Low complement levels and positivity for anti-dsDNA antibodies may confirm the clinical suspicion. However, renal biopsy is crucial to assess the extent of renal lesions and to tailor appropriate treatment (21, 71, 72). In class IV LN, more than 50% of the glomeruli exhibit endocapillary hypercellularity and/or cellular crescents associated with other active (i.e., fibrinoid necrosis, karyorrhexis, presence of neutrophils, abundant subendothelial immunocomplex deposits, and interstitial inflammation) and chronic lesions (global and segmental glomerulosclerosis, adhesions, fibrous crescents, interstitial fibrosis, and tubular atrophy). Immunofluorescence shows a typical “full house” pattern, characterized by the evidence of IgG, IgM, IgA, C3, and C1q immune deposits. Electron microscopy reveals abundant electron-dense subendothelial deposits conferring a “wire loop” profile to the capillary walls (70, 73).
In patients with class IV LN, induction therapy can be based on intravenous CYC (low dose: 500 mg every 2 weeks for 3 months or high dose: 0.5–1 g/m2 monthly for 6 months) or MMF (2–3 g/day) combined with oral glucocorticoids (0.5–1.0 mg/kg/day) with or without three pulses of intravenous methylprednisolone (32, 33). In addition, RTX and other biological agents may be useful in class IV LN and in refractory LN as induction therapy (71). The induction therapy may be associated with TPE, as reported in Table 1 (67). After the induction treatment, maintenance therapy is required; oral AZA (1.5–2.5 mg/kg/day) or MMF (1–2 g/day in divided doses) is recommended and effective, combined with low-dose oral prednisone (≤10 mg/day) (31–33). The maintenance therapy should be continued for at least 1 year after complete remission is achieved, while a repeat kidney biopsy is required when complete remission has not been achieved. The maintenance phase of the Aspreva Lupus Management Study (ALMS) showed that regardless of the induction therapy, MMF was superior to AZA in maintaining the renal response and preventing relapse of proliferative LN: the overall treatment failure rate in the MMF group was half that observed in the AZA group (16.4 vs. 32.4%), and renal flares were significantly higher in patients treated with AZA (12.9 vs. 23.4%) (74). The superiority of MMF demonstrated in the ALMS was not apparent in the MAINTAIN nephritis trial, published in 2010 (probably due to differences in the study design): however, renal flares occurred in 19% of the MMF group, compared with 25% of the AZA group, suggesting that MMF should be considered the drug of choice (75). Finally, calcineurin inhibitors (CNIs) should be used for maintenance therapy in patients intolerant to MMF and AZA.
IgAN
IgAN is the most common primary glomerulonephritis worldwide; it is characterized by persistent microscopic hematuria, mild proteinuria, and episodes of gross hematuria concurrently with upper respiratory tract infections (76). However, RPGN has been described in one third of patients with IgAN where histology reveals >50% crescents. Crescentic IgAN is a critical condition, leading to CKD and ESKD in a few years and so requiring prompt, efficacious treatment (77, 78). High-dose corticosteroids and CYC are recommended (79, 80). Corticosteroid treatment can be based on methylprednisolone (1 g/day for three consecutive days), followed by oral prednisone 0.5 mg/kg/day and subsequent tapering or, alternatively, on oral prednisone 1 mg/kg/day for 2 months followed by tapering (0.2 mg/kg monthly) within 6 months. CYC [1–2 mg/kg/day, based on the patient's glomerular filtration rate (GFR)] should be administered for 3 months, followed by AZA (1–2 mg/kg/day) for the following 3 months. TPE can be combined with immunosuppressive treatments until clinical improvement is achieved (67).
In addition, AKI episodes in IgAN patients can be due to massive hematuria of glomerular origin (81). In cases of massive hematuria, tubular cell damage has been described as the consequence of red blood cell cast formation and intratubular obstructions, combined with erythrophagocytosis processes (81). The hemoglobin products released by red blood cells induce oxidative stress, inflammation, podocytes, and tubular cell apoptosis and consequently cell detachment and fibrosis (82). In this setting, adequate and sustained hydration is crucial to prevent renal damage and enhance renal recovery. Renal biopsy should be performed in cases of persistent kidney function impairment despite supportive care and massive hydration.
Post-streptococcal Glomerulonephritis
Post-streptococcal glomerulonephritis typically occurs in children in developing countries at 1–3 weeks after upper respiratory tract infections (83). Clinically, 20% of affected patients develop a classic nephritic syndrome characterized by hematuria, hypertension, oliguria, and worsening renal function. The diagnosis is based on the presence of these symptoms associated with the detection of antistreptolysin O and anti-DNase B antibodies and low C3 (83). In this setting, renal biopsy is not mandatory since the prognosis is usually excellent and no specific treatments are needed (21). However, 1% of patients develop crescentic glomerulonephritis, which is sometimes associated with ANCA (83). The role of immunosuppression in these patients is debated. Although there is no evidence yet from randomized controlled trials (RCTs), the use of high-dose corticosteroids can be considered in patients with extensive glomerular crescents and RPGN.
TMA
TMA is a clinical phenotype which includes multiple diseases leading to thrombosis of arterioles and capillary vessels. Laboratory findings (thrombocytopenia with microangiopathic hemolytic anemia) lead to a suspicion of TMAs and reflect the mechanical disruption of red blood cells and platelets in the microvasculature (84). Thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS) are the two classic TMA disorders (85). TTP is mainly characterized by neurological involvement, while only a limited proportion of patients (25%) show renal involvement; TTP patients usually exhibit congenital or acquired ADAMTS 13 deficiency leading to the accumulation of the ultra-large multimers of von Willebrand factors (vWFs) that cause platelet aggregation and consequently microvascular thrombosis (86). Conversely, kidney injury typically occurs in all patients with HUS, as a consequence of the massive thrombotic capillaries' occlusion (87). The histological features comprise microvascular thrombosis, swelling, and detachment of the endothelial cells from the GBM; patchy cortical necrosis; and focal-segmental sclerosis, which is a long-term sequela of acute HUS (88, 89).
HUS includes STEC-HUS due to Shiga toxin-producing Escherichia coli infection (90–92), pneumococcal HUS caused by infections with Streptococcus pneumoniae, and genetic HUS, also called aHUS (93). STEC-HUS occurs most frequently in pediatric patients (the incidence is five to six children per 10,000 children population per year). Patients typically present bloody diarrhea and gastroenteritis 6–10 days before the development of TMA. STX closely adheres to gut epithelial cells, causing apoptosis and destruction of the brush border of the villi. Then, the toxin enters the circulation: in the kidney, STX is internalized via the Gb3 receptor and releases a protease into the cytoplasm. This protease inhibits protein synthesis and activates inflammatory pathways, inducing cell death (92). The diagnosis is driven by the clinical suspicion and the detection of STEC (cultured test) or STEX (polymerase chain reaction). Prompt intravenous hydration (10–15 ml/kg/h of isotonic solution) is recommended as soon as E. coli infection is suspected, in order to reduce the systemic effects of STX (94, 95). The use of antibiotics must be avoided, because they increase STX gene expression and induce massive STX release. Only azithromycin can be administered to a very limited number of children presenting with bacteremia (94). Supportive management of anemia, renal failure, and hypertension is required in these patients. In cases of severe clinical manifestations and neurological involvement, eculizumab and/or plasma exchange can be used as rescue therapy (88). The prognosis in children is generally favorable, and up to 70–85% of patients recover renal function.
aHUS is a multifactorial rare disease linked to an uncontrolled activation of the alternative complement pathway. About 60% of aHUS patients carry genetic variants or risk haplotypes in genes of the alternative pathway (CFH, CFI, CFB, MCP, C3, CFHR1-5, and THBD) and/or in diacylglycerol kinase E (DGKE). Genetic background accounts for different outcomes, as response to therapy and risk of relapses may vary depending on the underlying mutation; in addition, specific environmental factors (pregnancy, drugs, infections, etc.) have been reported as triggers in disease pathogenesis (93). aHUS is a life-threatening disorder requiring immediate diagnosis and treatment, often in the ICU. In addition, 50% of aHUS patients progress to ESKD, and the mortality rate during the acute phase of the disease is significant. The diagnosis of aHUS is made on exclusion of STEC infection and ADAMTS 13 deficiency; moreover, a genetic screening should be recommended, particularly in patients who are candidates for kidney transplantation. Treatment should be promptly started. The use of eculizumab in the aHUS treatment has dramatically changed short- and long-term outcomes in this setting and is currently the first-line treatment. The attack dose is 900 mg weekly for 4 weeks, followed by 1,200 mg at week 5 and then every 2 weeks: however, the duration of treatment is not yet well established. The Food and Drug Administration and the European Medicines Agency have recently approved a long-acting anti-C5 antibody (ravulizumab) for the treatment of aHUS, to be administered every 8 weeks (half-life of approximately 51 days).
It is important to remember that patients with DGKE variants do not respond to eculizumab therapy and thus pose an open challenge for scientists and physicians (96). Finally, 3–8% of aHUS patients present antibodies to complement factor H protein (FHAA), which are associated with a homozygous deficiency of FHR1 (85%) and homozygous deletion of CFHR3-CFHR1 (97). In this setting, plasma exchange alone was associated with disease recurrence (58%), CKD (39%), ESKD (27%), and death (9%) (98). A combination of TPE and immunosuppression reduces the antibody production and improves patient outcomes (99, 100).
Membranoproliferative Glomerulonephritis (MPGN) and Mixed Cryoglobulinemia
In MPGN, the histological pattern includes a group of chronic immune-mediated diseases characterized by GBM thickening and proliferative changes (101). On the basis of the pathophysiological processes, MPG is currently classified as immunocomplex-mediated MPGN and complement-mediated GNs (DDD and C3GN) (102). Among these disorders, mixed cryoglobulinemia is the most prevalent disorder in older adults (103). Mixed cryoglobulinemia is a small-vessel vasculitis characterized by the precipitation of circulating immunocomplexes after cold exposure (polyclonal IgG with or without monoclonal IgM with rheumatoid factor activity), involving several organs such as the skin, the joints, the peripheral nervous system, and the kidneys (103). Hepatitis C virus (HCV) disease is the main cause of mixed cryoglobulinemia. In HCV-positive patients, renal involvement accounts for about 30–40% of all cases (104). Clinical symptoms are weakness, arthralgias, and purpura, followed by a nephritic/nephrotic syndrome and rapidly deteriorating kidney function (104, 105). Kidney histology is characterized by mesangial and endocapillary proliferation, extracellular matrix expansion, diffuse leukocyte and monocyte infiltration, double contouring of the GBM, and the presence of intraluminal PAS-positive thrombi. On electron microscopy, electron-dense deposits (cryo-immunocomplexes) can be detected in subendothelial and mesangial regions (104, 106). The diagnosis is based on specific laboratory findings (detection of serum cryoglobulins and serologic hepatitis C markers) combined with the clinical signs and symptoms (104). In this scenario, KDIGO guidelines suggest the use of TPE combined with immunosuppressive treatment, such as RTX (375 mg/m2/week for 4 weeks) or CYC (2 mg/kg/day for 2–4 months), combined with intravenous methylprednisolone (0.5/1 g/kg/day for three consecutive days followed by oral prednisone). Importantly, concomitant antiviral therapy should be performed in HCV-positive patients (31–33).
Glomerulonephritis Manifesting With Nephrotic Syndrome
Several forms of glomerulonephritis are associated with a nephrotic syndrome, characterized by heavy proteinuria, severe hypoalbuminemia, edema, weight gain due to fluid retention, and dyslipidemia. Membranous nephropathy is the main cause of nephrotic syndrome in adults (107); in addition, diabetic nephropathy, minimal change disease, focal-segmental glomerulosclerosis, SLE, and amyloidosis usually manifest with nephrotic syndrome. AKI episodes have been in rare cases described as a potential complication of nephrotic syndrome, independently of the underlying pathogenesis (108, 109). The AKI pathogenesis in this setting is multifactorial. Some clinical observations supported the hypothesis that the massive urinary albumin loss induces microvascular injury due to volume depletion (109), leading to a massive expression of endothelin 1, inducing a decrease in blood flow and GFR (110). In addition, ischemic tubular damage and cell necrosis have been reported in some cases. Recent studies have shown that some urine biomarkers (NGAL and alpha 1-microglobulin) are elevated in patients with AKI and nephrotic syndrome (111). Although the pathogenesis still needs to be clarified, AKI in NS is generally reversible. The majority of patients with idiopathic nephrotic syndrome have a complete recovery of kidney function after volume depletion correction. Severe hypoalbuminemia, older age, and male sex are risk factors (109).
Scleroderma
Scleroderma is an autoimmune systemic disorder characterized by uncontrolled expansion of connective tissue in skins and other visceral organs. Kidney involvement is usually characterized by the presence of low-grade proteinuria, but AKI episodes can occur (112, 113). The so-called scleroderma renal crisis is a life-threatening complication of scleroderma and typically presents with the abrupt onset of hypertension and rapid progressive renal insufficiency, followed by encephalopathy, heart failure, and signs and symptoms of TMA (113, 114). The typical histological features are ischemic glomerular changes, malignant hypertension lesions, and TMA in small interlobular and arcuate arteries: glomeruli may show ischemic collapse and fibrin thrombi; various degrees of tubular atrophy and interstitial fibrosis have also been reported (115). Clinical presentation, along with the detection of speckled ANA (positive in 90% of cases) and anti-topoisomerase I antibodies (scl-70) (positive in 20% of cases), supports the diagnosis (113). Renal biopsy should be performed in patients with atypical presentations (increased serum creatinine in normotensive patients or in the presence of active urine sediment or nephrotic range proteinuria). Activation of the renin–angiotensin–aldosterone system (RAAS) is crucial in the pathogenesis of scleroderma renal crisis. Thus, the introduction of angiotensin-converting enzyme (ACE) inhibitors has substantially improved the prognosis of patients with scleroderma crisis, reducing the mortality associated with scleroderma crisis to <10% (113). Calcium channel blockers and, as third line, diuretics and alpha-blockers could be used as additional therapy if blood pressure control remains suboptimal despite maximum doses of ACE inhibitors (116). In addition, recent evidence suggests a role of complement activation and endothelin-1 in the pathogenesis of scleroderma crisis, thus suggesting the use of C5 inhibitors and endothelin receptor antagonists, particularly in refractory cases. Finally, TPE seems to confer some benefits in patients with scleroderma crisis with evidence of TMA or in patients with ACE inhibitor intolerance (116): a typical course of six TPE over 2–3 weeks followed by 4-weekly treatments is a reasonable therapeutic approach, resulting in long-lasting improvements in symptoms (67).
Conclusions
Several glomerular diseases have been reported to manifest with AKI episodes: early diagnosis is crucial since different conditions have a similar clinical profile but require different, often aggressive, treatment in order to preserve renal function and delay the onset of ESKD. In this scenario, renal biopsy is essential for an accurate diagnosis and to describe the extent of reversible/irreversible renal lesions.
Author Contributions
FPe and ES wrote the manuscript. MR provided histological images. MF, FPi, GS, GC, and LG revised the paper. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Mehta RL, Burdmann EA, Cerdá J, Feehally J, Finkelstein F, García-García G, et al. Recognition and management of acute kidney injury in the International Society of Nephrology 0by25 Global Snapshot: a multinational cross-sectional study. Lancet. (2016) 387:2017–25. doi: 10.1016/S0140-6736(16)30240-9
2. Al-Jaghbeer M, Dealmeida D, Bilderback A, Ambrosino R, Kellum JA. Clinical decision support for In-Hospital AKI. J Am Soc Nephrol. (2018) 29:654–60. doi: 10.1681/ASN.2017070765
3. Hoste EA, Bagshaw SM, Bellomo R, Cely CM, Colman R, Cruz DN, et al. Epidemiology of acute kidney injury in critically ill patients: the multinational AKI-EPI study. Intens Care Med. (2015) 41:1411–23. doi: 10.1007/s00134-015-3934-7
4. Uchino S, Kellum JA, Bellomo R, Doig GS, Morimatsu H, Morgera S, et al. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA. (2005) 294:813–8. doi: 10.1001/jama.294.7.813
5. Hsu RK, McCulloch CE, Dudley RA, Lo LJ, Hsu CY. Temporal changes in incidence of dialysis-requiring AKI. J Am Soc Nephrol. (2013) 24:37–42. doi: 10.1681/ASN.2012080800
6. Fiaccadori E, Regolisti G, Cademartiri C, Cabassi A, Picetti E, Barbagallo M, et al. Efficacy and safety of a Citrate-based protocol for sustained low-efficiency dialysis in AKI using standard dialysis equipment. Clin J Am Soc Nephrol. (2013) 8:1670–8. doi: 10.2215/CJN.00510113
7. Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, et al. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. (2007) 11:R31. doi: 10.1186/cc5713
8. Kellum JA, Lameire N, Aspelin P, Barsoum RS, Burdmann EA, Goldstein SL, et al. Kidney disease: improving global outcomes (KDIGO) acute kidney injury work group. KDIGO clinical practice guideline for acute kidney injury. Kidney Int Suppl. (2012) 2:1–138. doi: 10.1038/kisup.2012.1
9. Fiorentino M, Castellano G, Kellum JA. Differences in acute kidney injury ascertainment for clinical and preclinical studies. Nephrol Dial Transplant. (2017) 32:1789–805. doi: 10.1093/ndt/gfx002
10. Hsu CY, Chertow GM, McCulloch CE, Fan D, Ordonez JD, Go AS. Nonrecovery of kidney function and death after acute on chronic renal failure. Clin J Am Soc Nephrol. (2009) 4:891–8. doi: 10.2215/CJN.05571008
11. Kistler BM, Khubchandani J, Wiblishauser M, Wilund KR, Sosnoff JJ. Epidemiology of falls and fall-related injuries among middle-aged adults with kidney disease. Int Urol Nephrol. (2019) 51:1613–21. doi: 10.1007/s11255-019-02148-8
12. Stasi A, Intini A, Divella C, Franzin R, Montemurno E, Grandaliano G, et al. Emerging role of lipopolysaccharide binding protein insepsis-induced acute kidney injury. Nephrol Dial Transplant. (2017) 32:24–31. doi: 10.1093/ndt/gfw250
13. Fiorentino M, Grandaliano G, Gesualdo L, Castellano G. Acute kidney injury to chronic kidney disease transition. Cont Nephrol. (2018) 193:45–54. doi: 10.1159/000484962
14. Fiorentino M, Tohme FA, Wang S, Murugan R, Angus DC, Kellum JA. Long-term survival in patients with septic acute kidney injury is strongly influenced by renal recovery. PLoS ONE. (2018) 13:e0198269. doi: 10.1371/journal.pone.0198269
15. Moore PK, Hsu RK, Liu KD. Management of acute kidney injury: Core Curriculum 2018. Am J Kidney Dis. (2018) 72:136–48. doi: 10.1053/j.ajkd.2017.11.021
16. Lafrance J-P, Miller DR. Acute kidney injury associates with increased long-term mortality. J Am Soc Nephrol. (2010) 21:345–52. doi: 10.1681/ASN.2009060636
17. Coca SG, Yusuf B, Shlipak MG, Garg AX, Parikh CR. Long-term risk of mortality and other adverse outcomes after acute kidney injury: a systematic review and meta-analysis. Am J Kidney Dis. (2009) 53:961–73. doi: 10.1053/j.ajkd.2008.11.034
18. Fani F, Regolisti G, Delsante M, Cantaluppi V, Castellano G, Gesualdo L, et al. Recent advances in the pathogenetic mechanisms of sepsis-associated acute kidney injury. J Nephrol. (2018) 31:351–9. doi: 10.1007/s40620-017-0452-4
19. Regolisti G, Maggiore U, Cademartiri C, Belli L, Gherli T, Cabassi A, et al. Renal resistive index by transesophageal and transparietal echo-doppler imaging for the prediction of acute kidney injury in patients undergoing major heart surgery. J Nephrol. (2017) 30:243–53. doi: 10.1007/s40620-016-0289-2
20. Thervet É. Insuffisance rénale aiguë d'origine glomérulaire [Acute renal failure of glomerular origin]. Rev Prat. (2018) 68:160–5.
21. Fiorentino M, Bolignano D, Tesar V, Pisano A, Van Biesen W, D'Arrigo G, et al. Renal biopsy in 2015–From epidemiology to evidence-based indications. Am J Nephrol. (2016) 43:1–19. doi: 10.1159/000444026
22. Fiorentino M, Bolignano D, Tesar V, Pisano A, Biesen W Van, Tripepi G, et al. Renal biopsy in patients with diabetes: a pooled meta-analysis of 48 studies. Nephrol Dial Transplant. (2017) 32:97–110. doi: 10.1093/ndt/gfw070
23. McAdoo SP, Pusey CD. Anti-glomerular basement membrane disease. Clin J Am Soc Nephrol. (2017) 12:1162–72. doi: 10.2215/CJN.01380217
24. Cui Z, Zhao M-H. Advances in human antiglomerular basement membrane disease. Nat Rev Nephrol. (2011) 7:697–705. doi: 10.1038/nrneph.2011.89
25. Rees AJ, Peters DK, Compston DA, Batchelor JR. Strong association between HLA-DRW2 and antibody-mediated Goodpasture's syndrome. Lancet. (1978) 1:966–8. doi: 10.1016/S0140-6736(78)90252-0
26. Donaghy M, Rees AJ. Cigarette smoking and lung haemorrhage in glomerulonephritis caused by autoantibodies to glomerular basement membrane. Lancet. (1983) 2:1390–3. doi: 10.1016/S0140-6736(83)90923-6
27. Jayne DR, Marshall PD, Jones SJ, Lockwood CM. Autoantibodies to GBM and neutrophil cytoplasm in rapidly progressive glomerulonephritis. Kidney Int. (1990) 37:965–70. doi: 10.1038/ki.1990.72
28. Saus J, Wieslander J, Langeveld JP, Quinones S, Hudson BG. Identification of the Goodpasture antigen as the alpha 3(IV) chain of collagen IV. J Biol Chem. (1988) 263:13374–80.
29. Lahmer T, Heemann U. Anti-glomerular basement membrane antibody disease: a rare autoimmune disorder affecting the kidney and the lung. Autoimmunity Rev. (2012) 12:169–73. doi: 10.1016/j.autrev.2012.04.002
30. McAdoo SP, Tanna A, Hrušková Z, Holm L, Weiner M, Arulkumaran N, et al. Patients double-seropositive for ANCA and anti-GBM antibodies have varied renal survival, frequency of relapse, and outcomes compared to single-seropositive patients. Kidney Int. (2017) 92:693–702. doi: 10.1016/j.kint.2017.03.014
31. Beck L, Bomback AS, Choi MJ, Holzman LB, Langford C, Mariani LH, et al. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for glomerulonephritis. Am J Kidney Dis. (2013) 62:403–41. doi: 10.1053/j.ajkd.2013.06.002
32. Floege J, Barbour SJ, Cattran DC, Hogan JJ, Nachman PH, Tang SCW, et al. Management and treatment of glomerular diseases (part 1): conclusions from a kidney disease: improving global outcomes (KDIGO) Controversies Conference. Kidney Int. (2019) 95:268–80. doi: 10.1016/j.kint.2018.10.018
33. Rovin BH, Caster DJ, Cattran DC, Gibson KL, Hogan JJ, Moeller MJ, et al. Management and treatment of glomerular diseases (part 2): conclusions from a Kidney Disease: improving global outcomes (KDIGO) Controversies Conference. Kidney Int. (2019) 95:281–95. doi: 10.1016/j.kint.2018.11.008
34. Lockwood CM, Rees AJ, Pearson TA, Evans DJ, Peters DK, Wilson CB. Immunosuppression and plasma-exchange in the treatment of Goodpasture's syndrome. Lancet. (1976) 1:711–5. doi: 10.1016/S0140-6736(76)93089-0
35. Savige JA, Dowling J, Kincaid-Smith P. Superimposed glomerular immune complexes in anti-glomerular basement membrane disease. Am J Kidney Dis. (1989) 14:145–53. doi: 10.1016/S0272-6386(89)80190-8
36. Villacorta J, Diaz-Crespo F, Acevedo M, Cavero T, Guerrero C, Praga M, et al. Renal vasculitis presenting with acute kidney injury. Rheumatol Int. (2017) 37:1035–41. doi: 10.1007/s00296-017-3697-2
37. Semple D, Keogh J, Forni L, Venn R. Clinical review: vasculitis on the intensive care unit–part 1: diagnosis. Crit Care. (2005) 9:92–7. doi: 10.1186/cc2936
38. Griffith M, Brett S. The pulmonary physician in critical care * illustrative case 3: pulmonary vasculitis. Thorax. (2003) 58:543–6. doi: 10.1136/thorax.58.6.543
39. van der Woude FJ, Rasmussen N, Lobatto S, Wiik A, Permin H, van Es LA, et al. Autoantibodies against neutrophils and monocytes: tool for diagnosis and marker of disease activity in Wegener's granulomatosis. Lancet. (1985) 1:425–9. doi: 10.1016/S0140-6736(85)91147-X
40. Savage CO, Winearls CG, Jones S, Marshall PD, Lockwood CM. Prospective study of radioimmunoassay for antibodies against neutrophil cytoplasm in diagnosis of systemic vasculitis. Lancet. (1987) 1:1389–93. doi: 10.1016/S0140-6736(87)90591-5
41. Falk RJ, Hogan S, Carey TS, Jennette JC. Clinical course of anti-neutrophil cytoplasmic autoantibody-associated glomerulonephritis and systemic vasculitis. The Glomerular Disease Collaborative Network. Ann Int Med. (1990) 113:656–63. doi: 10.7326/0003-4819-113-9-656
42. Choi HK, Merkel PA, Walker AM, Niles JL. Drug-associated antineutrophil cytoplasmic antibody-positive vasculitis: prevalence among patients with high titers of antimyeloperoxidase antibodies. Arthrit Rheumatol. (2000) 43:405–13. doi: 10.1002/1529-0131(200002)43:2<405::AID-ANR22>3.0.CO;2-5
43. Tse WY, Abadeh S, Jefferis R, Savage CO, Adu D. Neutrophil FcgammaRIIIb allelic polymorphism in anti-neutrophil cytoplasmic antibody (ANCA)-positive systemic vasculitis. Clin Exp Immunol. (2000) 119:574–7. doi: 10.1046/j.1365-2249.2000.01182.x
44. Liu M, Chen X, Sun X, Zhou J, Zhang X, Zhu H, et al. Validation of a differential diagnostic model of diabetic nephropathy and non-diabetic renal diseases and the establishment of a new diagnostic model. J Diabetes. (2014) 6:519–26. doi: 10.1111/1753-0407.12150
45. Esnault VL, Testa A, Audrain M, Rogé C, Hamidou M, Barrier JH, et al. Alpha 1-antitrypsin genetic polymorphism in ANCA-positive systemic vasculitis. Kidney Int. (1993) 43:1329–32. doi: 10.1038/ki.1993.186
46. Merkel PA, Xie G, Monach PA, Ji X, Ciavatta DJ, Byun J, et al. Identification of functional and expression polymorphisms associated with risk for antineutrophil cytoplasmic autoantibody-associated vasculitis. Arthrit Rheumatol. (2017) 69:1054–66. doi: 10.1002/art.40034
47. Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, Aratani Y, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Investig. (2002) 110:955–63. doi: 10.1172/JCI0215918
48. Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci USA. (1990) 87:4115–9. doi: 10.1073/pnas.87.11.4115
49. Jennette JC, Xiao H, Falk RJ. Pathogenesis of vascular inflammation by anti-neutrophil cytoplasmic antibodies. J Am Soc Nephrol. (2006) 17:1235–42. doi: 10.1681/ASN.2005101048
50. Kallenberg CGM, Heeringa P, Stegeman CA. Mechanisms of Disease: pathogenesis and treatment of ANCA-associated vasculitides. Nat Clin Pract Rheumatol. (2006) 2:661–70. doi: 10.1038/ncprheum0355
51. Couser WG, Johnson RJ. What is myeloperoxidase doing in ANCA-associated glomerulonephritis? Kidney Int. (2015) 88:938–40. doi: 10.1038/ki.2015.259
52. O'Sullivan KM, Lo CY, Summers SA, Elgass KD, McMillan PJ, Longano A, et al. Renal participation of myeloperoxidase in antineutrophil cytoplasmic antibody (ANCA)-associated glomerulonephritis. Kidney Int. (2015) 88:1030–46. doi: 10.1038/ki.2015.202
53. DeRemee RA. Antineutrophil cytoplasmic autoantibody-associated diseases: a pulmonologist's perspective. Am J Kidney Dis. (1991) 18:180–3. doi: 10.1016/S0272-6386(12)80877-8
54. Savige J, Gillis D, Benson E, Davies D, Esnault V, Falk RJ, et al. International Consensus statement on testing and reporting of Antineutrophil Cytoplasmic Antibodies (ANCA). Am J Clin Pathol. (1999) 111:507–13. doi: 10.1093/ajcp/111.4.507
55. Savige J, Davies D, Falk RJ, Jennette JC, Wiik A. Antineutrophil cytoplasmic antibodies and associated diseases: a review of the clinical and laboratory features. Kidney Int. (2000) 57:846–62. doi: 10.1046/j.1523-1755.2000.057003846.x
56. Eisenberger U, Fakhouri F, Vanhille P, Beaufils H, Mahr A, Guillevin L, et al. ANCA-negative pauci-immune renal vasculitis: histology and outcome. Nephrol Dial Transplant. (2005) 20:1392–9. doi: 10.1093/ndt/gfh830
57. Aasarød K, Bostad L, Hammerstrøm J, Jørstad S, Iversen BM. Renal histopathology and clinical course in 94 patients with Wegener's granulomatosis. Nephrol Dial Transplant. (2001) 16:953–60. doi: 10.1093/ndt/16.5.953
58. Hauer HA, Bajema IM, Hagen EC, Noël L-H, Ferrario F, Waldherr R, et al. Long-term renal injury in ANCA-associated vasculitis: an analysis of 31 patients with follow-up biopsies. Nephrol Dial Transplant. (2002) 17:587–96. doi: 10.1093/ndt/17.4.587
59. Berden AE, Ferrario F, Hagen EC, Jayne DR, Jennette JC, Joh K, et al. Histopathologic classification of ANCA-associated glomerulonephritis. J Am Soc Nephrol. (2010) 21:1628–36. doi: 10.1681/ASN.2010050477
60. Joh K, Muso E, Shigematsu H, Nose M, Nagata M, Arimura Y, et al. Renal pathology of ANCA-related vasculitis: proposal for standardization of pathological diagnosis in Japan. Clin Exp Nephrol. (2008) 12:277–91. doi: 10.1007/s10157-008-0052-7
61. Monach PA, Warner RL, Tomasson G, Specks U, Stone JH, Ding L, et al. Serum proteins reflecting inflammation, injury and repair as biomarkers of disease activity in ANCA-associated vasculitis. Ann Rheumat Dis. (2013) 72:1342–50. doi: 10.1136/annrheumdis-2012-201981
62. Ishizaki J, Takemori A, Suemori K, Matsumoto T, Akita Y, Sada K-E, et al. Targeted proteomics reveals promising biomarkers of disease activity and organ involvement in antineutrophil cytoplasmic antibody-associated vasculitis. Arthrit Res Therapy. (2017) 19:218. doi: 10.1186/s13075-017-1429-3
63. Little MA, Nightingale P, Verburgh CA, Hauser T, De Groot K, Savage C, et al. Early mortality in systemic vasculitis: relative contribution of adverse events and active vasculitis. Ann Rheumat Dis. (2010) 69:1036–43. doi: 10.1136/ard.2009.109389
64. Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, Hoffman GS, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. New Engl J Med. (2010) 363:221–32. doi: 10.1056/NEJMoa0909905
65. Jones RB, Tervaert JWC, Hauser T, Luqmani R, Morgan MD, Peh CA, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. New Engl J Med. (2010) 363:211–20. doi: 10.1056/NEJMoa0909169
66. Walters G. Role of therapeutic plasmapheresis in ANCA-associated vasculitis. Pediatr Nephrol. (2016) 31:217–25. doi: 10.1007/s00467-014-3038-6
67. Padmanabhan A, Connelly-Smith L, Aqui N, Balogun RA, Klingel R, Meyer E, et al. Guidelines on the use of therapeutic apheresis in clinical practice - evidence-based approach from the writing committee of the american society for apheresis: the eighth special issue. J Clin Apheresis. (2019) 34:171–354. doi: 10.1002/jca.21705
68. Hruskova Z, Stel VS, Jayne D, Aasarød K, De Meester J, Ekstrand A, et al. Characteristics and outcomes of Granulomatosis with polyangiitis (Wegener) and Microscopic polyangiitis requiring renal replacement therapy: results from the European Renal Association-European Dialysis and Transplant Association Registry. Am J Kidney Dis. (2015) 66:613–20. doi: 10.1053/j.ajkd.2015.03.025
69. Castellano G, Cafiero C, Divella C, Sallustio F, Gigante M, Pontrelli P, et al. Local synthesis of interferon-alpha in lupus nephritis is associated with type I interferons signature and LMP7 induction in renal tubular epithelial cells. Arthrit Res Therapy. (2015) 17:72. doi: 10.1186/s13075-015-0588-3
70. Almaani S, Meara A, Rovin BH. Update on lupus nephritis. Clin J Am Soc Nephrol. (2017) 12:825–35. doi: 10.2215/CJN.05780616
71. Chan TM. Treatment of severe lupus nephritis: the new horizon. Nat Rev Nephrol. (2015) 11:46–61. doi: 10.1038/nrneph.2014.215
72. Mok CC. Towards new avenues in the management of lupus glomerulonephritis. Nat Rev Rheumatol. (2016) 12:221–34. doi: 10.1038/nrrheum.2015.174
73. Wilhelmus S, Bajema IM, Bertsias GK, Boumpas DT, Gordon C, Lightstone L, et al. Lupus nephritis management guidelines compared. Nephroogyl Dial Transplanat. (2016) 31:904–13. doi: 10.1093/ndt/gfv102
74. Dooley MA, Jayne D, Ginzler EM, Isenberg D, Olsen NJ, Wofsy D, et al. Mycophenolate versus azathioprine as maintenance therapy for lupus nephritis. New Engl J Med. (2011) 365:1886–95. doi: 10.1056/NEJMoa1014460
75. Houssiau FA, D'Cruz D, Sangle S, Remy P, Vasconcelos C, Petrovic R, et al. Azathioprine versus mycophenolate mofetil for long-term immunosuppression in lupus nephritis: results from the MAINTAIN Nephritis Trial. Ann Rheumat Dis. (2010) 69:2083–9. doi: 10.1136/ard.2010.131995
76. Wyatt RJ, Julian BA. IgA nephropathy. New Engl J Med. (2013) 368:2402–14. doi: 10.1056/NEJMra1206793
77. Roberts ISD, Cook HT, Troyanov S, Alpers CE, Amore A, Barratt J, et al. The Oxford classification of IgA nephropathy: pathology definitions, correlations, and reproducibility. Kidney Int. (2009) 76:546–56. doi: 10.1038/ki.2009.168
78. Lv J, Yang Y, Zhang H, Chen W, Pan X, Guo Z, et al. Prediction of outcomes in crescentic IgA nephropathy in a multicenter cohort study. J Am Soc Nephrol. (2013) 24:2118–25. doi: 10.1681/ASN.2012101017
79. Natale P, Palmer SC, Ruospo M, Saglimbene VM, Craig JC, Vecchio M, et al. Immunosuppressive agents for treating IgA nephropathy. Cochr Database Syst Rev. (2020) CD003965. doi: 10.1002/14651858.CD003965.pub3
80. Rauen T, Eitner F, Fitzner C, Sommerer C, Zeier M, Otte B, et al. Intensive supportive care plus immunosuppression in IgA nephropathy. New Engl J Med. (2015) 373:2225–36. doi: 10.1056/NEJMoa1415463
81. Schulman G, Berl T, Beck GJ, Remuzzi G, Ritz E, Shimizu M, et al. Risk factors for progression of chronic kidney disease in the EPPIC trials and the effect of AST-120. Clin Exp Nephrol. (2018) 22:299–308. doi: 10.1007/s10157-017-1447-0
82. Rubio-Navarro A, Sanchez-Niño MD, Guerrero-Hue M, García-Caballero C, Gutiérrez E, Yuste C, et al. Podocytes are new cellular targets of haemoglobin-mediated renal damage. J Pathol. (2018) 244:296–310. doi: 10.1002/path.5011
83. Eison TM, Ault BH, Jones DP, Chesney RW, Wyatt RJ. Post-streptococcal acute glomerulonephritis in children: clinical features and pathogenesis. Pediatr Nephrol. (2011) 26:165–80. doi: 10.1007/s00467-010-1554-6
84. Remuzzi G. HUS and TTP: variable expression of a single entity. Kidney Int. (1987) 32:292–308. doi: 10.1038/ki.1987.206
85. Noris M, Mescia F, Remuzzi G. STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol. (2012) 8:622–33. doi: 10.1038/nrneph.2012.195
86. Moake JL. Moschcowitz, multimers, and metalloprotease. New Engl J Med. (1998) 339:1629–31. doi: 10.1056/NEJM199811263392210
87. Eknoyan G, Riggs SA. Renal involvement in patients with thrombotic thrombocytopenic purpura. Am J Nephrol. (1986) 6:117–31. doi: 10.1159/000167066
88. Goodship THJ, Cook HT, Fakhouri F, Fervenza FC, Frémeaux-Bacchi V, Kavanagh D, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. (2017) 91:539–51. doi: 10.1016/j.kint.2016.10.005
89. Brocklebank V, Wood KM, Kavanagh D. Thrombotic microangiopathy and the kidney. Clin J Am Soc Nephrol. (2018) 13:300–17. doi: 10.2215/CJN.00620117
90. Bruyand M, Mariani-Kurkdjian P, Gouali M, de Valk H, King LA, Le Hello S, et al. Hemolytic uremic syndrome due to Shiga toxin-producing Escherichia coli infection. Méd Maladies Infect. (2018) 48:167–74. doi: 10.1016/j.medmal.2017.09.012
91. Giordano M, Castellano G, Messina G, Divella C, Bellantuono R, Puteo F, et al. Preservation of renal function in atypical hemolytic uremic syndrome by Eculizumab: a case report. Pediatrics. (2012) 130:e1385–e8. doi: 10.1542/peds.2011-1685
92. Obrig TG. Escherichia coli Shiga toxin mechanisms of action in renal disease. Toxins. (2010) 2:2769–94. doi: 10.3390/toxins2122769
93. Zipfel PF, Wiech T, Stea ED, Skerka C. CFHR gene variations provide insights in the pathogenesis of the kidney diseases atypical hemolytic uremic syndrome and C3 glomerulopathy. J Am Soc Nephrol. (2020) 31:241–56. doi: 10.1681/ASN.2019050515
94. Loconsole D, Giordano M, Laforgia N, Torres D, Santangelo L, Carbone V, et al. Case-management protocol for bloody diarrhea as a model to reduce the clinical impact of Shiga toxin-producing Escherichia coli infections. Experience from Southern Italy. Eur J Clin Microbiol Infect Dis. (2020) 39:539–47. doi: 10.1007/s10096-019-03755-0
95. Corogeanu D, Willmes R, Wolke M, Plum G, Utermöhlen O, Krönke M. Therapeutic concentrations of antibiotics inhibit Shiga toxin release from enterohemorrhagic E. coli O104:H4 from the 2011 German outbreak. BMC Microbiol. (2012) 12:160. doi: 10.1186/1471-2180-12-160
96. Nester CM, Barbour T, de Cordoba SR, Dragon-Durey MA, Fremeaux-Bacchi V, Goodship THJ, et al. Atypical aHUS: state of the art. Mol Immunol. (2015) 67:31–42. doi: 10.1016/j.molimm.2015.03.246
97. Zipfel PF, Mache C, Müller D, Licht C, Wigger M, Skerka C. DEAP-HUS: deficiency of CFHR plasma proteins and autoantibody-positive form of hemolytic uremic syndrome. Pediatr Nephrol. (2010) 25:2009–19. doi: 10.1007/s00467-010-1446-9
98. Dragon-Durey M-A, Sethi SK, Bagga A, Blanc C, Blouin J, Ranchin B, et al. Clinical features of anti-factor H autoantibody-associated hemolytic uremic syndrome. J Am Soc Nephrol. (2010) 21:2180–7. doi: 10.1681/ASN.2010030315
99. Fremeaux-Bacchi V, Fakhouri F, Garnier A, Bienaimé F, Dragon-Durey M-A, Ngo S, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol. (2013) 8:554–62. doi: 10.2215/CJN.04760512
100. Sinha A, Gulati A, Saini S, Blanc C, Gupta A, Gurjar BS, et al. Prompt plasma exchanges and immunosuppressive treatment improves the outcomes of anti-factor H autoantibody-associated hemolytic uremic syndrome in children. Kidney Int. (2014) 85:1151–60. doi: 10.1038/ki.2013.373
101. Bomback AS, Appel GB. Pathogenesis of the C3 glomerulopathies and reclassification of MPGN. Nat Rev Nephrol. (2012) 8:634–42. doi: 10.1038/nrneph.2012.213
102. Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis–a new look at an old entity. New Engl J Med. (2012) 366:1119–31. doi: 10.1056/NEJMra1108178
103. Terrier B, Cacoub P. Cryoglobulinemia vasculitis: an update. Curr Opin Rheumatol. (2013) 25:10–8. doi: 10.1097/BOR.0b013e32835b15f7
104. Roccatello D, Saadoun D, Ramos-Casals M, Tzioufas AG, Fervenza FC, Cacoub P, et al. Cryoglobulinaemia. Nat Rev Dis Primers. (2018) 4:11. doi: 10.1038/s41572-018-0009-4
105. Spatola L, Generali E, Angelini C, Badalamenti S, Selmi C. HCV-negative mixed cryoglobulinemia and kidney involvement: in-depth review on physiopathological and histological bases. Clin Exp Med. (2018) 18:465–471. doi: 10.1007/s10238-018-0514-5
106. D'Amico G, Colasanti G, Ferrario F, Sinico RA. Renal involvement in essential mixed cryoglobulinemia. Kidney Int. (1989) 35:1004–1014. doi: 10.1038/ki.1989.84
107. Glassock RJ. The pathogenesis of idiopathic membranous nephropathy: a 50-year odyssey. Am J Kidney Dis. (2010) 56:157–67. doi: 10.1053/j.ajkd.2010.01.008
108. Meyrier A, Niaudet P. Acute kidney injury complicating nephrotic syndrome of minimal change disease. Kidney Int. (2018) 94:861–9. doi: 10.1016/j.kint.2018.04.024
109. Chen T, Lv Y, Lin F, Zhu J. Acute kidney injury in adult idiopathic nephrotic syndrome. Renal Fail J. (2011) 33:144–9. doi: 10.3109/0886022X.2011.553301
110. Koomans HA. Pathophysiology of acute renal failure in idiopatic nephrotic syndrome. Nephrol Dial Transplant. (2001) 16:221–4. doi: 10.1093/ndt/16.2.221
111. Fujigaki Y, Tamura Y, Nagura M, Arai S, Ota T, Shibata S, et al. Unique proximal tubular cell injury and the development of acute kidney injury in adult patients with minimal change nephrotic syndrome. BMC Nephrol. (2017) 18:339. doi: 10.1186/s12882-017-0756-6
112. Steen VD. Kidney involvement in systemic sclerosis. La Presse Méd. (2014) 43:e305–14. doi: 10.1016/j.lpm.2014.02.031
113. Bose N, Chiesa-Vottero A, Chatterjee S. Scleroderma renal crisis. Semin Arthrit Rheumat. (2015) 44:687–94. doi: 10.1016/j.semarthrit.2014.12.001
114. Ghossein C, Varga J, Fenves AZ. Recent developments in the classification, evaluation, pathophysiology, and management of Scleroderma renal crisis. Curr Rheumatol Rep. (2016) 18:5. doi: 10.1007/s11926-015-0551-y
115. Guillevin L, Mouthon L. Scleroderma renal crisis. Rheumat Dis Clin N Am. (2015) 41:475–88. doi: 10.1016/j.rdc.2015.04.008
Keywords: AKI, glomerulonephritis, antibodies, complement, immunosuppression
Citation: Pesce F, Stea ED, Rossini M, Fiorentino M, Piancone F, Infante B, Stallone G, Castellano G and Gesualdo L (2021) Glomerulonephritis in AKI: From Pathogenesis to Therapeutic Intervention. Front. Med. 7:582272. doi: 10.3389/fmed.2020.582272
Received: 11 July 2020; Accepted: 25 November 2020;
Published: 02 March 2021.
Edited by:
Vincenzo Cantaluppi, Università del Piemonte Orientale, ItalyReviewed by:
Bassam G. Abu Jawdeh, University of Cincinnati, United StatesKirk Campbell, Icahn School of Medicine at Mount Sinai, United States
Copyright © 2021 Pesce, Stea, Rossini, Fiorentino, Piancone, Infante, Stallone, Castellano and Gesualdo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Loreto Gesualdo, bG9yZXRvLmdlc3VhbGRvQHVuaWJhLml0
†These authors have contributed equally to this work