 Mary Stofan1
Mary Stofan1 Grace L. Guo1,2,3*
Grace L. Guo1,2,3*- 1Department of Pharmacology and Toxicology, Ernest Mario School of Pharmacy, Rutgers University, Piscataway, NJ, United States
- 2Environmental and Occupational Health Sciences Institute, Rutgers, The State University of New Jersey, Piscataway, NJ, United States
- 3VA New Jersey Health Care System, Veterans Administration Medical Center, East Orange, NJ, United States
Bile acids (BAs) are evolutionally conserved molecules synthesized in the liver from cholesterol and have been shown to be essential for lipid homeostasis. BAs regulate a variety of metabolic functions via modulating nuclear and membrane receptors. Farnesoid X receptor (FXR) is the most important nuclear receptor for maintaining BA homeostasis. FXR plays a tissue-specific role in suppressing BA synthesis and promoting BA enterohepatic circulation. Disruption of FXR in mice have been implicated in liver diseases commonly occurring in humans, including cholestasis, non-alcoholic fatty liver diseases, and hepatocellular carcinoma. Strategically targeting FXR activity has been rapidly used to develop novel therapies for the prevention and/or treatment of cholestasis and non-alcoholic steatohepatitis. This review provides an updated literature review on BA homeostasis and FXR modulator development.
Introduction
Bile acids (BAs) serve critical physiological functions, including elimination of cholesterol, absorption of fat and fat-soluble vitamins, regulation of the gut microbiome, and serving as important signaling molecules. BAs are endogenous ligands of farnesoid X receptor (FXR), Takeda G protein receptor 5 (TGR5), and sphingosine-1-phosphate receptor 2 (S1PR2). In the liver and intestine, BAs suppress their own synthesis, regulate glucose and lipid homeostasis, and inhibit inflammation and fibrogenesis. Disruption of BA homeostasis leads to severe pathological outcomes, including cholestasis, hepatic steatosis, fibrosis, and liver tumors. Regulating BA pathways has become a novel strategy to treat cholestasis and non-alcoholic steatohepatitis (NASH).
Overview of BAs
Synthesis
BAs are amphipathic molecules synthesized from cholesterol in the liver mainly through two pathways, the classical and the alternative pathway (1). In the classical pathway, the initial and rate-limiting step is the 7α-hydroxylation of cholesterol by a cytochrome P450 enzyme, cholesterol 7α-hydroxylase (CYP7A1) (2, 3). The crucial role of CYP7A1 has been demonstrated with Cyp7a1 knockout (KO) mice that have a high incidence of postnatal death due to abnormal neurological development following vitamin deficiencies (4). Afterwards, microsomal 3β-hydroxy-Δ5-C27-steroid dehydroxylase (3β-HSD) converts 7α-hydroxycholesterol to 7α-hydroxy-4-cholestene-3-one (C4) (5), which can be converted by sterol 12α-hydroxylase (CYP8B1) to cholic acid (CA) or alternatively catabolized by cytosolic Δ4-3-oxosteroid 5β-reductase (AKR1D1) and 3α-hydroxysteroid dehydrogenase (AKR1C4), yielding a sterol intermediate, 5β-cholestan-3α,7α-diol, which is further converted to chenodeoxycholic acid (CDCA) (5, 6). Cyp8b1 KO mice eliminated CA synthesis, suggesting that CYP8B1 is required for CA synthesis and is responsible for the CA-to-CDCA ratio in the classical pathway (7). Additionally, the C4 intermediate can be used as a serum marker for assessing BA synthesis levels in vivo (8).
In the alternative or acidic pathway, cholesterol is oxidized by mitochondrial sterol 27-hydroxylase (CYP27A1) to produce 27-hydroxycholesterol and 3β-hydroxy-5-cholestenoic acid, which is further hydroxylated by oxysterol 7α-hydroxylase (CYP7B1) to form the intermediate 3β, 7α-dihydroxy-5-cholestenoic acid (6, 9). Subsequent enzymatic conversions produce CDCA.
There is clear species difference of the composition of BAs between humans and mice (Figure 1). Human primary BAs are CA and CDCA, that form a relatively hydrophobic BA pool consisting of 40% CA, 40% CDCA, and 20% deoxycholic acid (DCA) (9). Mouse primary BAs are CA and muricholic acid (MCA) that is from 6-hydroxylation of CDCA. Hydroxylation significantly changes the physicochemical properties of BAs, resulting in a BA pool that is more hydrophilic, less potent as detergents, and cytotoxic. More significantly, this additional conversion in mice markedly changes BA signaling properties, converting the most potent endogenous FXR agonist (CDCA) to antagonists (MCAs) (9). Three seminal studies discovered the mouse 6β-hydroxylase, CYP2C70, converting CDCA to MCA (10–12). Furthermore, the DCA levels are much higher in humans than in mice because humans are unable to rehydroxylate DCA and lithocholic acid (LCA) whereas mice can. A study by Honda et al. reported that mouse CYP2A12 is the enzyme responsible for 7α-rehydroxylation of taurodeoxycholic acid (TDCA) and taurolithocholic acid (TLCA), solving another unknown of the species difference between humans and mice (12).
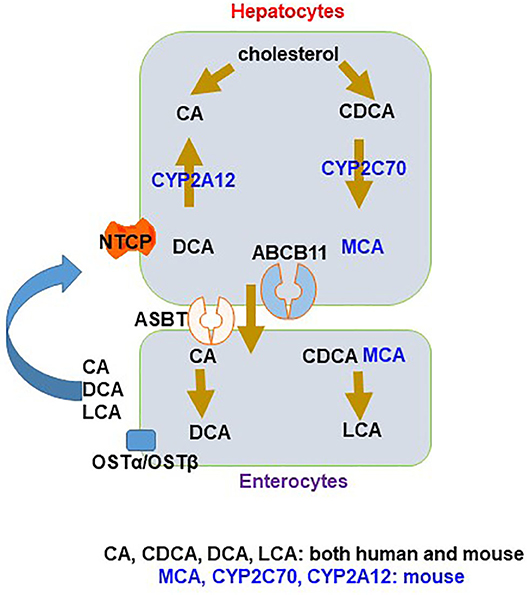
Figure 1. Species difference in bile acid (BA) synthesis and composition. In hepatocytes, primary BAs, cholic acid (CA) and chenodeoxycholic acid (CDCA), are made from cholesterol. In mice, CDCA is converted to muricholic acid (MCA) by CYP2C70. CA and CDCA are conjugated and then efflux via ABCB11 to the intestine through the uptake transporter ASBT, where they facilitate lipid absorption. Most BAs are transported back by effluxion out of enterocytes via the organic solute transporter (OST)α/OSTβ complex to the liver through portal circulation and taken up into hepatocytes mainly via sodium taurocholate co-transporting polypeptide (NTCP), with a small amount being converted to deoxycholic acid (DCA) and lithocholic acid (LCA), the secondary BAs in the large intestine. In mice, DCA can be transported back to the liver and converted to CA by CYP2A12.
Conjugation is considered to be the terminal step in BA synthesis and involves the addition of an amino acid, glycine or taurine, through an amide linkage at carbon 24 (13). Humans and rodents both utilize the enzyme bile acid-CoA:amino acid N-acyltransferase (BAAT) for conjugation; however, primary human BAs are mainly conjugated with glycine and, to a less extent, taurine, while rodent primary BAs are taurine conjugates (14, 15). Conjugation of BAs ultimately increases their solubility and amphipathicity (13).
Enterohepatic Circulation
BAs undergo constant enterohepatic circulation. Conjugated BAs are transported across the canalicular membrane into the bile and stored in the gallbladder in both humans and mice (9). Cholecystokinin, a hormone, is secreted by the duodenum following a meal to stimulate gallbladder contraction, leading to the release of BAs into the intestine (9), where BAs help absorb dietary lipids and fat-soluble vitamins. In the ileum, about 95% BAs are reabsorbed and transported back to the liver through portal circulation (9). Daily, ~0.5 g of BAs, or 5% of the total BA pool, is excreted in the feces, with BAs being recycled 4–12 times a day; this entire process comprises the enterohepatic circulation of BAs (9).
BA transporters are responsible for dynamically moving BAs during the enterohepatic circulation. Efflux of BAs from the hepatocytes into canaliculi is mainly mediated by the bile salt export pump (BSEP; ABCB11/Abcb11) (16). The multidrug resistance-associated protein (MRP2; ABCC2/Abcc2) effluxes divalent BAs along with other organic substrates, bilirubin conjugates, glutathione, and drugs (17). Like BSEP, MRP2 is an ATP-binding cassette transporter localized to the canalicular membrane of hepatocytes (17). There seems to be a species difference between humans and mice regarding the roles of BSEP and MRP2. Mice use mainly BSEP and, to a smaller extent, MRP2, to efflux BAs into the bile, whereas humans mainly rely on BSEP to efflux BAs into the bile, which could at least partially explain the more severe cholestasis development in human patients with BSEP mutation compared to mice with BSEP deficiency (18). Mutation of the ABCB11 gene causes BSEP deficiency and progressive familial intrahepatic cholestasis type 2 (PFIC2) (19). PFIC2 is an inherited disorder characterized by severe cholestasis beginning at infancy that can progress to cirrhosis, hepatic failure, hepatocellular carcinoma (HCC), and death (20, 21). Due to the species differences mentioned above, the PFIC2 phenotype cannot be achieved in Abcb11 KO mice. This leaves a void for a translational model for PFIC2 to study potential therapies as the standard treatment remains to be liver transplantation.
Reabsorption of BAs in the terminal ileum mainly occurs through the uptake mediated by the apical sodium-dependent bile salt transporter (ASBT; SLC10A2/Slc10a2) (22), intracellular binding to intestinal bile acid-binding protein (IBABP) (23), and basolateral BA efflux into the portal circulation by the organic solute transporters OSTα and OSTβ heterodimer (24).
At the basolateral (sinusoidal) membrane of hepatocytes, the major BA uptake transporter is the sodium taurocholate co-transporting polypeptide (NTCP; SLC10A1/Slc10a1) (25). Interestingly, human NTCP seems to have higher affinity than does the rat transporter, allowing more efficient BA extraction at low plasma levels (25). Sodium-independent basolateral BA uptake into hepatocytes is mediated by organic anion transporting polypeptides (OATPs) (16). Only 25% of the hepatic BA uptake is estimated to be mediated by Na+-independent mechanism and responsible for mainly unconjugated BA uptake (16).
Although not directly involved in enterohepatic circulation, an important canalicular membrane flippase encoded by the multidrug resistance gene (MDR3;ABCB4 in humans and Mdr2;Abcb4 in mice) is responsible for phospholipid secretion into the bile (26, 27). Disruption of Mdr2 prevents the secretion of phospholipids, a component of BA mixed micelles, thus increasing the concentration of free BAs that can damage the biliary epithelium (21). Defects in ABCB4 are associated with progressive familial intrahepatic cholestasis type 3 (PFIC3), intrahepatic cholestasis of pregnancy, and adult biliary cirrhosis (28, 29). Mdr2 KO mice develop severe biliary fibrosis and are a well-established model for primary sclerosing cholangitis (PSC) (30, 31).
The gut microbiota play an important role in BA biotransformation and are responsible for secondary BA formation. Conjugated BAs that remain in the intestine are deconjugated by bacterial bile salt hydrolases (BSHs) (32). In the large intestine, bacterial 7α-dehydroxylase converts CA to DCA and CDCA to LCA through the removal of the hydroxyl group at the C-7 position (32). These secondary BAs are more cytotoxic. While LCA is highly insoluble and mostly excreted by fecal excretion, DCA can be reabsorbed through passive diffusion (33). As mentioned above, mouse hepatocytes can rehydroxylate DCA to CA by CYP2A12 (12). Species differences in the gut microbiota may affect the generation of secondary BAs and should be considered when using animal models to study human BA signaling (21).
BAs are important for the intestinal absorption of lipids and lipid-soluble nutrients, removal of excess cholesterol, regulating bile flow, modulating the gut microbiome, and modulating energy homeostasis. Many of these functions are performed by modulating a nuclear receptor (NR) FXR in a tissue-specific manner. Additional NRs and membrane-bound receptors that have been identified to be activated by BAs include pregnane X receptor (PXR), vitamin D receptor (VDR), Takeda G protein-coupled receptor (TGR5), and sphingosine-1-phosphate receptor 2 (S1PR2) (34–36).
Farnesoid X Receptor
FXR is the most important NR to regulate BA homeostasis. NRs are ligand-activated transcription factors that regulate the expression of genes involved in various processes, including cell growth, differentiation, and metabolism (37). The general structure of NRs consists of an N-terminal DNA-binding domain (DBD) and a C-terminal ligand-binding domain (LBD), with the DBD being the most conserved area that contains two zinc finger motifs (9). These zinc fingers allow the NR to bind to DNA elements, known as hormone response elements (HREs), composed of direct, inverted, or everted repeats of the sequence AGGTCA and separated by a variable number of nucleotides (38). NR activation also requires either homodimerization or heterodimerization with retinoid X receptor (RXR) (39).
FXR was originally labeled as an orphan NR (38). After multiple groups demonstrated that physiological concentrations of free or conjugated BAs could activate FXR, with CDCA being the most potent, followed by DCA, CA, and LCA, BAs were recognized to be the endogenous ligands of FXR and FXR is now considered an “adopted” NR (40–42). FXR is highly expressed in the liver, ileum, kidneys, and adrenal glands (40). The most common FXR response element (FXRE) consists of an inverted AGGTCA repeat separated by one nucleotide (IR1); FXR could also bind to an everted repeat separated by two nucleotides (ER2) (43). Both steroidal and non-steroidal FXR agonists are being developed in the treatment of various liver diseases and include semi-synthetic BA obeticholic acid (OCA), cilofexor, and tropifexor, with OCA being used clinically to treat primary biliary cholangitis (PBC).
Regulation of BA Homeostasis
It has been well-established that FXR is involved in the regulation of BA homeostasis. As shown in Figure 2, there is a clear tissue-specific role of FXR in the liver and intestine to regulate BA synthesis (44). Activation of intestinal FXR plays a major role and activation of liver FXR serves a minor role in suppressing CYP7A1/Cyp7a1 gene expression through the induction of the ileal hormone fibroblast growth factor 19 (FGF19) in humans and FGF15 in mice and hepatic small heterodimer partner 1 (SHP-1), respectively (42, 44–47). In contrast, Cyp8b1 gene repression via FXR is almost equally dependent on both intestinal and liver FXR (44). Furthermore, FXR is critical in regulating the enterohepatic circulation of BAs by inducing the expression of BSEP, IBABP, and OSTα/β and suppressing those of NTCP and ASBT (48–52).
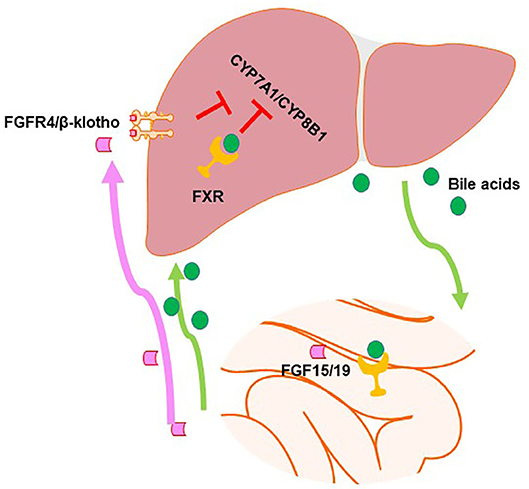
Figure 2. Farnesoid X receptor (FXR) regulates bile acid (BA) synthesis in a tissue-specific manner. In the intestine, FXR activation induces fibroblast growth factor (FGF)15/19, which can go to the liver and activate the FGFR4/β-klotho dimer to activate signaling pathways in order to inhibit the expression of genes in the classical BA synthesis pathway. Hepatic FXR activation also inhibits BA synthesis, albeit to a smaller degree.
Regulation of Lipid and Glucose Homeostasis
FXR also shows critical effects in regulating lipid and glucose homeostasis. In general, FXR activation leads to lower lipid levels in the circulation as it suppresses de novo fatty acid synthesis (53, 54), decreases very low-density lipoprotein (VLDL) hepatic secretion (55), and increases triglyceride hydrolysis and clearance as well as fatty acid oxidation (56–60). Activation of FXR may reduce glucose intolerance by reducing hepatic gluconeogenesis and glycolysis and increasing glycogen synthesis (61). FXR activation may decrease gluconeogenesis via SHP-mediated suppression of the critical transcription factors involved in gluconeogenesis (62). In contrast, a different study utilizing human and rat hepatocytes and mouse livers showed that FXR agonism induced phosphoenolpyruvate carboxykinase (PEPCK) expression and glucose levels (63). Our genome-wide ChIP-seq analysis also suggests that FXR could regulate glucose homeostasis, but there may be species differences among humans and mice (43, 64). Despite conflicting evidence, it is apparent that FXR may play important roles in glucose homeostasis as FXR KO mice develop fatty livers, elevate circulating free fatty acids (FFAs) and serum glucose levels, and present insulin resistance (65). In both diabetic db/db and wild-type mice, FXR activation or hepatic overexpression significantly lowered the blood glucose levels, decreased the FFA levels, and increased the insulin sensitivity (66), suggesting FXR activation may improve metabolic syndrome.
Role in Inflammation and Fibrosis
During liver injury, FXR has been shown to play an anti-inflammatory role (67, 68). Monocyte chemoattractant protein-1 (MCP-1/CCL2) is a key chemokine that regulates the migration and infiltration of monocytes/macrophages (69). In the methionine/choline-deficient (MCD) diet-induced NASH model, the synthetic FXR agonist WAY-362450 decreased MCP-1 expression and significantly decreased inflammatory cell infiltration in the liver (68). Nuclear factor kappa-light-chain enhancer of activated B cell (NF-κB) is a transcription factor that induces the expression of various pro-inflammatory genes (70). FXR KO mice displayed strong hepatic inflammation after treatment with lipopolysaccharide (LPS), confirmed by massive liver necrosis and the significant increase in the hepatic cytokine signaling molecules inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), and interferon-γ (IFN-γ) (67). Ultimately, the pretreatment of HepG2 cells and mouse primary hepatocytes with FXR agonists suppressed the NF-κB-mediated inflammation in an FXR-dependent manner (67). FXR could suppress inflammation via an indirect mechanism by reducing cholestasis and the levels of toxic BA production and accumulation in the liver, as described above.
FXR activation suppresses the development of hepatic fibrosis. In addition to regulating hepatic lipid metabolism and reducing hepatic fibrosis, FXR seems to directly inactivate hepatic fibrosis by inducing anti-fibrotic gene expression in hepatic stellate cells (HSCs). Activation of FXR induces SHP to increase the peroxisomal proliferator-activated receptor γ (PPARγ) expression in HSCs, and PPARγ is well-known to inactivate HSCs (71, 72). Recently, we have shown that FGF15 deficiency reduces hepatic fibrosis through increasing FXR activation following loss of FGF15-mediated suppression of BA synthesis (73, 74). Interestingly, in a human HSC cell line, LX2, FGF19 does not suppress fibrogenic gene expression, but suppresses inflammation, likely through modulating the inhibitor of nuclear factor kappa B (IκB) activity (74). These studies provide another group of evidence to support the role of FXR as a homeostatic regulator to suppress liver inflammation and fibrosis.
Role in Cholestasis
There is conflicting evidence regarding the role of FXR in cholestatic diseases. In an early study, the synthetic FXR agonist GW4064 was investigated in rat models of extrahepatic and intrahepatic cholestasis through bile duct ligation (BDL) and α-naphthylisothiocyanate (ANIT) administration, respectively (75). Significant reductions in liver injury were observed in GW4064-treated animals in both cholestatic models, revealed by the reduced alanine aminotransferase (ALT) and aspartate transaminase (AST), necrosis, inflammation, and bile duct proliferation (75). The observed protective effects of GW4064 suggest that FXR agonists may be helpful in treating cholestatic diseases (75).
However, another group found that FXR KO mice were protected from obstructive cholestasis achieved through BDL (76). In FXR KO mice after BDL, mortality and liver injury were reduced as serum bilirubin was not significantly elevated (76). FXR KO mice had reduced serum total BA concentrations and had a marked induction of the basolateral transporter multidrug resistance-associated protein 4 (Mrp4), suggesting that these animals had a greater capacity to export BAs back into circulation and reduce hepatoxicity (76). This study supports the potential clinical use of FXR antagonists in the treatment of obstructive cholestatic diseases (76).
Looking further into the role of FXR during intrahepatic cholestasis, ANIT-induced injury was utilized in wild-type (WT), FXR KO, and PXR KO mice (77). Serum ALT, alkaline phosphatase (ALP), and bilirubin were elevated in all genotypes after ANIT administration, with the highest ALP levels seen in FXR KO mice (77). ANIT-treated FXR KO mice had higher concentrations of serum and liver unconjugated BAs across all genotypes (77). While ANIT treatment induced the messenger RNA (mRNA) expressions of Mdr2, Bsep, and ATPase, class I, type 8B and member 1 (Atp8b1) in WT and PXR KO mice, no upregulation was observed in FXR KOs (77). It was concluded that FXR deficiency, not PXR deficiency, was responsible for the increased susceptibility to injury in the ANIT-induced intrahepatic cholestasis model due to the reduction of hepatobiliary efflux transporters and the accumulation of unconjugated BAs (77). Furthermore, pretreatment of the FXR agonist GW4064 was also investigated in ANIT-treated WT mice (77). GW4064 treatment was shown to be protective as it reduced necrosis compared to ANIT treatment alone (77). This reproduces what Liu et al. had found in BDL and ANIT-induced injury in rats and further supports FXR as a therapeutic target for intrahepatic cholestasis (77).
Through the use of reversible BDL (rBDL) in the rat to model cholestasis, FXR activation by OCA worsened the biliary injury, shown by a considerable increase in ALT and ALP compared to the controls (78). OCA treatment in rBDL rats upregulated Bsep, multidrug resistance-associated protein 3 (Mrp3), Mrp4, and Ostβ transporters (78). The 8-fold induction of the FXR target gene Bsep was suggested to be the cause of biliary injury as BAs would be pumped via BSEP into an already obstructed biliary tree (78).
Cholangiocytes
Cholangiocytes are epithelial cells which line the bile ducts of the biliary tree (79). Through absorptive and secretory transport systems in cholangiocytes, bile is modified to become more fluid and alkaline (80). Bile then enters the gallbladder for concentration and storage or delivered to the intestinal lumen (80). Cholangiocytes have also been shown to be actively involved in bile homeostasis (81). Compared to hepatocytes, cholangiocytes have no or low expressions of Cyp7a1 and Cyp8b1, but considerable expression of Cyp27a1, suggesting that cholangiocytes are involved in cholesterol metabolism (81). Measurement of the mRNA levels revealed that Fgf15 was expressed at higher levels in cholangiocytes compared to hepatocytes, while the fibroblast growth factor receptor 4 (Fgfr4) expression was lower (81). As FXR is known to regulate Fgf15/FGF19 levels, investigation of a similar regulation in cholangiocytes was achieved through treatment of rat cholangiocytes with CDCA and the FXR agonist GW4064, with both treatments inducing the expression of Fgf15 (81). Additionally, cultured human cholangiocytes treated with CDCA induced the secretion of FGF19 in the medium (81). FGF15/19-mediated repression of Cyp27a1 in cholangiocytes was found to differ from hepatocytes and is mediated through p38 kinase (81). Ultimately, understanding BA metabolism in cholangiocytes may help provide therapeutic pathways for cholangiopathy treatments (81).
One of the most common biliary complications after liver transplantation (LT) is non-anastomotic strictures that develop after biliary epithelial damage and can result from BA toxicity (82). To investigate the mechanism of cholangiocyte BA transport following LT, a rat LT model was utilized. After transplantation, a prolonged biliary transport time of BAs was observed, while the expression of FXR was dramatically decreased and was related to cold ischemic time of the donor liver. Furthermore, in vitro-cultured human biliary epithelial cells under hypoxic conditions exhibited a repression of FXR expression and DNA binding activities (82). Hypoxic conditions also altered the expressions of BA transporters as hypoxia slightly induced Asbt expression and repressed both Ostα and Ostβ (82). This led to the intracellular accumulation of BAs, increased cell apoptosis, and increased expression of profibrotic factors in cholangiocytes (82). It was concluded that, after LT, repression of FXR under ischemic/hypoxic conditions led to the disruption of BA transport of cholangiocytes and, thus, biliary damage (82).
Fibroblast Growth Factors 15/19
Fibroblast growth factors (FGFs) make up a family of at least 22 proteins that regulate various biological processes including growth, development, and differentiation (83, 84). FGF15 and its human ortholog FGF19 belong to the subfamily of endocrine FGFs that act as hormones due to their low or no affinity for heparin sulfate, which allows them to enter systemic circulation (84, 85). FGF19 was originally identified in the fetal brain during a screen for novel FGFs (86). Although FGF15 and FGF19 are orthologs, they interestingly only share ~50% amino acid identity (86). For high-affinity receptor binding, the endocrine FGFs require klotho proteins that interact with fibroblast growth factor receptors (FGFRs) (87). β-Klotho specifically binds to FGF15/19 which has high affinity for fibroblast growth factor receptor 4 (FGFR4) and less for fibroblast growth factor receptor 1 (FGFR1) that are highly expressed in hepatocytes and white adipose tissue (WAT), respectively (88, 89). Low levels of FGFR4 expression are also detected in other cell types, including HSCs, macrophages, and some central neurons (90). FGF15/19 is expressed in ileal enterocytes, where it is strongly induced by FXR activation (84). Once released into blood circulation, FGF15/19 acts on the liver to repress BA synthesis, as described above.
However, mouse FGFR4 does not recognized human FGF19 (91). Therefore, when using high dosage of FGF19 in mice, the observed effects may be due to the activation of FGFR1 or other FGFRs, but not FGFR4 by FGF19.
Role in Energy Expenditure
To investigate the role of FGF19 in physiological homeostasis, transgenic mice expressing human FGF19 were utilized (92). FGF19 transgenic mice had a significant reduction in fat mass arising from an increase in energy expenditure (92). When fed a high-fat diet, FGF19 transgenic mice did not become obese or diabetic (92). The results suggest two mechanisms by which FGF19 may increase energy expenditure through an increase in brown adipose tissue (BAT) and through a decrease in liver enzyme acetyl CoA carboxylase 2 (ACC2) (92). Reduction in ACC2, the rate-limiting enzyme for fatty acid entry into the mitochondria, also resulted in reduced liver triglyceride levels (92). In an additional study, FGF19 increased the metabolic rate in mice fed a high-fat diet while reducing body weight and diabetes in leptin-deficient mice (93). FGF19 also acts in the central nervous system to improve insulin sensitivity by reducing hypothalamic agouti-related peptide (AGRP)/neuropeptide Y (NPY) neuron activity (94). In summary, FGF15/19 increases insulin sensitivity, thermogenesis, and weight loss and decreases serum cholesterol and triglyceride levels.
Protein and Glycogen Synthesis
FGF15/19 also regulates hepatic protein and glycogen synthesis (95). Fgf15 KO mice were shown to be glucose-intolerant and store half as much hepatic glycogen compared to control wild-type mice (95). In diabetic mice lacking insulin, FGF19 treatment restored the hepatic glycogen concentrations to normal levels, indicating that FGF19 activates an insulin-independent pathway to regulate glycogen metabolism (95). It was determined that FGF15/19 uses a RAS/extracellular signal-regulated protein kinase (ERK)/p90RSK pathway to induce hepatic glycogen and protein synthesis in vivo (95). FGF19 also shows a positive effect on muscle weight, revealed by a study showing that FGF19 stimulates the phosphorylation of the ERK1/2 and the ribosomal protein S6 kinase (S6K1), an mTOR-dependent master regulator of muscle cell growth (96).
Gluconeogenesis
Energy homeostasis is additionally regulated through FGF15/19 repressing gluconeogenesis, like insulin (97). While insulin peaks in serum 15 min after feeding, FGF15/19 peaks ~45 min later due to the increase of BAs in the small intestine (97). In vivo, FGF15/19 blocks the expression of gluconeogenesis genes through the dephosphorylation and inactivation of the transcription factor cAMP regulatory element-binding protein (CREB) (97). This then inhibits the expression of peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) and other downstream hepatic metabolism genes (97).
Fatty Acid Synthesis
Lastly, FGF19 inhibits hepatic fatty acid synthesis. Primary hepatocytes incubated with recombinant FGF19 protein in the presence or absence of insulin showed that FGF19 suppressed the insulin-dependent stimulation of fatty acid synthesis (98). Similar to the SHP-mediated suppression of sterol regulatory element-binding protein 1c (SREBP1c) following FXR activation, FGF19 was shown to decrease SREBP1c through increasing the signal transducer and activator of transcription 3 (STAT3) and decreasing the peroxisome proliferator-activated receptor-γ coactivator-1β (PGC-1β), while also increasing the expression of SHP (98). This favorable inhibition of hepatic fatty acid synthesis, along with the promotion of protein and glycogen synthesis and the repression of gluconeogenesis, supports the beneficial effects of FGF15/19 on metabolic syndrome and warrants further investigation of FGF15/19 in the prevention and treatment of NASH. Indeed, modified FGF19 has been shown to be beneficial in mouse models of NASH and cholestasis (99, 100).
FXR as Drug Targets—FXR Agonists
There are many FXR modulators that have undergone clinical trials for the treatment of chronic liver diseases. The focus for most of these trials is the efficacy of FXR activation on cholestasis, NASH, and obesity; however, there are some studies focused on minor indications, including bile acid diarrhea or association with reactivation of latent pro-virus (clinical trials.gov). Currently, two types of FXR agonists—steroidal represented by OCA vs. non-steroidal represented by tropifexor—are front-runners for obtaining U.S. Food and Drug Administration (FDA) approval for the treatment of NASH.
The first FDA-approved FXR agonist for the treatment of PBC is OCA, which is a steroidal FXR agonist modified from CDCA (101). When compared to CDCA, OCA was shown to be ~100 times more potent (101). In a model of cholestasis, male Wistar rats were administered LCA through an intravenous infusion to impair bile flow. Administration of OCA alone did not induce cholestasis, while co-infusion of LCA and OCA fully reversed bile flow impairment and protected hepatocytes from necrosis (101). This initial study confirmed OCA as a selective, potent FXR agonist and warranted further investigation of additional therapeutic uses.
The traditional first-line treatment for PBC is ursodeoxycholic acid (UDCA) as it has been shown to improve liver tests and transplant-free survival with minimal side effects (102). However, not all patients respond to UDCA (102). In a randomized, double-blinded, 12-week, phase II clinical trial, the efficacy of OCA in PBC patients who did not respond favorably to UDCA was evaluated (103). Patients (n = 165) were randomly assigned to receive 10, 25, or 50 mg of OCA or placebo once daily in addition to an existing dose of UDCA (103). The primary endpoint was level change of ALP from baseline until the conclusion of the study (103). All three doses significantly reduced the levels of ALP, γ-glutamyltransferase (GGT), and ALT compared to placebo. However, pruritus was reported in all groups, with severity correlating to the dose of OCA (103). Based on the efficacy and tolerability, the once daily dose of 10 mg OCA was determined to be the most effective (103).
In the randomized, double-blinded, phase III POISE trial, 217 PBC patients who had an inadequate response to UDCA were assigned to receive 10 mg OCA, 5–10 mg OCA, or placebo once daily for 12 months (104). Patients still received UCDA as a background therapy (104). The primary endpoint was a reduction in ALP from baseline and a normal total bilirubin level, which was reached in more patients in both OCA groups compared to placebo (104). As seen previously in the phase II trial, an OCA dose-dependent increase in the incidence of pruritus was reported (104). Based on the favorable effects of OCA on important biochemical markers, the FDA approved OCA for the treatment of PBC patients with an inadequate response of intolerance to UDCA in 2016 (105).
Another phase II study investigated OCA as a monotherapy in PBC patients (106). Patients received 10 mg OCA, 50 mg OCA, or placebo once daily for 3 months and then followed up for up to 6 years (106). OCA treatment as a monotherapy significantly improved ALP and other biochemical markers associated with improved clinical outcomes (106). However, severe pruritus was reported in almost all patients who received 50 mg OCA (106). Compared to UDCA co-therapy, no additional benefits for OCA as a monotherapy were reported (107).
A multiyear study (COBALT) to determine the effects of OCA in PBC patients with more advanced liver disease is ongoing (104). In 2017, after 11 cases of serious liver injury and 19 cases of death associated with OCA were reported, the FDA released a black box warning for the use of OCA in patients with decompensated cirrhosis (105). Many of the cases of increased liver injury appeared to be due to inappropriate high dosing of OCA (105).
Due to arising side effects including pruritus and increased risk of liver decompensation in cirrhotic PBC patients administered OCA, a study to determine whether OCA worsened liver injury under cholestatic conditions was carried out (108). BDL and ANIT treatment were studied in rats (108). In both models, OCA treatment exacerbated liver injury in a dose-dependent manner and downregulated the expression of basolateral transporters (108). The non-steroidal FXR agonist GW4064 was also tested in the ANIT cholestasis model. In contrast, GW4064 administration decreased the severity of cholestatic injury compared to OCA and reduced AST, ALT, GGT, and bilirubin (108). This is again consistent with the results published by Liu et al. (75) and suggests that the safety of FXR agonists is impacted by their pharmacokinetic properties (108). OCA, as a semi-synthetic derivative of CDCA, has a high rate of intestinal absorption, which allows it to recirculate like endogenous BAs (108). While synthetic GW4064 undergoes taurine conjugation in the liver which is then not recognized by intestinal transporters thus reducing its bioavailability (108). Under cholestatic conditions, OCA accumulates in the liver where it may reach toxic concentrations (108, 109). In mice, genetic KO of FXR or inhibition of FXR both resulted in protection from injury induced by OCA in an ANIT model of cholestasis (108). After RNAseq analysis, FXR antagonism was shown to reverse the transcription of over 2,000 genes, including V-Maf avian musculoaponeurotic fibrosarcoma oncogene homolog G (Mafg) and its partner nuclear factor erythroid 2-related factor 2 (Nrf2) (108). Mafg expression has been shown to be induced in cholestatic diseases and represses genes involved in the synthesis of antioxidant glutathione (110, 111). The modulation of these transcription factors was then investigated. Pharmacologic or genetic inhibition of Mafg prevented damage caused by ANIT and OCA, while Nrf2 induction was protective. These results support that the negative side effects of OCA treatment are FXR-mediated (108).
There is currently no approved treatment for PSC, and the efficacy of UDCA for PSC remains uncertain (112). Thus, the efficacy and safety of OCA in PSC patients were assessed in a phase II randomized, double-blind, placebo-controlled, dose-finding study (113). Patients (n = 76) were assigned to receive 1.5–3.0 mg OCA, 5–10 mg OCA, or placebo once daily for 24 weeks (113). At 24 weeks, treatment with 5–10 mg OCA significantly reduced serum ALP compared to placebo (113). Dose-related pruritus was reported as the most common side effect, consistent with the earlier clinical studies (113).
The safety and efficacy of the non-steroidal FXR agonist cilofexor (GS-9674) were evaluated in a phase II double-blinded, placebo-controlled study in PSC patients (114). Randomized patients received 100 mg cilofexor, 30 mg cilofexor, or placebo once daily for 12 weeks (114). Treatment with cilofexor was generally well-tolerated, safe, and improved the biochemical markers of cholestasis and inflammation (114). Significant dose-dependent reductions in serum ALP, GGT, ALT, and AST with cilofexor compared to placebo were reported (114). The effect of cilofexor on ALP was independent of UDCA use, and adverse events were similar between treatment groups (114).
Cilofexor was also evaluated in a double-blind, placebo-controlled, phase II trial in patients with NASH (115). Non-cirrhotic patients (n = 140) were randomized to receive 100 mg cilofexor, 50 mg cilofexor, or placebo once daily for 24 weeks (115). Cilofexor was safe and significantly improved hepatic steatosis, liver biochemistry (e.g., GGT), and bile acids (115). Compared to OCA treatment that resulted in increases in serum LDL-C and total cholesterol, cilofexor treatment had no significant effects on serum lipids (115). Moderate to severe pruritus was reported in 14% of the 100-mg cilofexor group and 4% of the 30-mg group (115). In contrast, 23% of the OCA-treated patients reported pruritus (116). However, cilofexor treatment only had modest beneficial effects on liver biochemistry compared to OCA treatment, indication of a potential limitation for efficacy (115).
To evaluate the effect of FXR activation by OCA on insulin resistance and liver lipid metabolism, Zucker (fa/fa) rats that contain a loss-of-function mutation in the hunger hormone leptin receptor were utilized (117). This mutation leads to hyperphagia and hyperleptinemia, resulting in diabetes, insulin resistance, obesity, and liver steatosis; therefore, Zucker (fa/fa) rats are considered a non-alcoholic fatty liver disease (NAFLD) model (117). Daily OCA treatment (10 mg/kg) over 7 weeks reversed insulin resistance and prevented body weight gain and liver fat deposition (117). Moreover, OCA treatment reduced blood triglyceride and plasma aminotransferases and improved liver histopathology (117). Reversal of insulin resistance after the administration of OCA is further supported by in vitro data showing that OCA significantly increases insulin secretion in mouse β-TC6 cells and human pancreatic islets (118). Additionally, OCA activation of FXR in mouse β-TC6 cells leads to AKT (protein kinase B)-dependent translocation of glucose transporter 2 (GLUT2), thus increasing the glucose uptake by these cells (118). Taken together, OCA activation of FXR improves hyperglycemia through enhanced insulin secretion and glucose uptake by the liver (118).
OCA has also been shown to exhibit anti-inflammatory and anti-fibrotic properties. While investigating the NF-κB signaling pathway, a key inflammation pathway, pretreatment of HepG2 cells with OCA (3 μM) inhibited the expression of the cytokine-inducible enzymes COX-2 and iNOS after stimulation with LPS or tumor necrosis factor alpha (TNFα) (67). Inhibition of iNOS by OCA was also confirmed in LPS-treated primary mouse hepatocytes (67).
After animal studies showed that OCA decreased insulin resistance and hepatic steatosis, the efficacy and safety of OCA were first evaluated in a phase IIa study in patients with type II diabetes and non-alcoholic fatty liver disease (119). The participants were randomly assigned to placebo (n = 23), 25 mg OCA (n = 20), or 50 mg OCA (n = 21) groups for the 6-week treatment period (119). Both OCA groups exhibited reduced GGT and ALT levels along with decreased bodyweight (119). Furthermore, treatment of OCA led to improved insulin sensitivity and elevated FGF19 serum levels. This, in conjunction with the decreased BA precursor C4 and endogenous BAs, again confirmed OCA's FXR agonist activity (119).
Based on previous favorable results, OCA was further investigated in the phase IIb Farnesoid X Receptor Ligand Obeticholic Acid in NASH Treatment (FLINT) trial (116). In this multicenter, double-blind, randomized clinical trial, patients with non-cirrhotic NASH were assigned to receive 25 mg OCA (n = 141) daily or placebo (n = 142) for 72 weeks (116). OCA treatment was shown to improve the biochemical and histological features of NASH when compared with placebo; specifically, 45% of OCA patients improved their NAFLD activity score by two points or greater without worsening of fibrosis compared to the 21% improvement in placebo patients (116). However, there was no significant difference in the histological resolution of NASH between the OCA-treated and placebo groups (120). Adverse outcomes of pruritus and unfavorable dyslipidemia manifested in the OCA treatment group (116). Additionally, the favorable effects on ALP, lipids, and blood glucose seen in the placebo group associated with weight loss were absent or reversed in the OCA-treated patients (120).
Currently, OCA is being evaluated by Intercept in a phase III trial REGENERATE (121). To assess OCA's effect on liver histology and clinical outcomes, 2,065 biopsy-confirmed NASH patients were randomized into a 10-mg OCA, 25-mg OCA, or placebo group (121). Total study duration is estimated to be 6 years, with interim biopsies performed after the first 18 months to evaluate improvement of fibrosis stage and resolution of NASH with no worsening fibrosis (121). Although OCA was recently approved by the FDA for treating PBC, the current American Association of the Study of Liver Diseases guidelines do not recommend the off-label treatment of OCA in NASH patients until further safety and efficacy data are available (122). In February 2019, Intercept announced that OCA achieved the primary endpoint of improving liver fibrosis without worsening of NASH after 18 months (p = 0.0002). This marks the first and largest successful phase 3 study in fibrosis patients due to NASH. Intercept filed a New Drug Application (NDA) with the FDA in September 2019. As of June 2020, the FDA issued a complete response letter stating that the predicted benefit of OCA did not outweigh the potential risks in patients with fibrosis due to NASH and that long-term outcome needs to be evaluated (123). Thus, accelerated approval was not granted at this time.
Tropifexor is a representative of non-steroidal FXR agonists. In mouse models of NASH, tropifexor significantly reduced oxidative stress, steatosis, inflammation, and fibrosis (124). It will be very interesting to see whether, as a non-steroidal FXR agonist, tropifexor will present similar adverse effect to the steroidal FXR agonists.
Conclusion
As a key regulator of BA homeostasis, FXR activation suppresses BA synthesis mainly through the induction of FGF15/19 in the gut and promotes enterohepatic BA circulation. FXR agonism also regulates lipid metabolism, reduces hepatic gluconeogenesis and glycolysis, and increases glycogen synthesis while playing an anti-inflammatory role during liver injury. FGF15/19 favorably increases energy expenditure and glycogen synthesis while decreasing gluconeogenesis and fatty acid synthesis. While FXR and FGF19 have been considered promising targets for the treatment of cholestasis and NASH, the molecular mechanism by which these two factors regulate liver BA transport, steatosis, and inflammation needs to be further determined, and most importantly, an individualized treatment plan is paramount to develop drugs and treatment strategy with better efficacy and less toxic effects.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
MS and GG have contributed to the writing and editing of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH-R01GM104037; NIH-R21ES029258; NIH-T32ES007148; VA-BX002741; Rutgers Busch grant.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We appreciate Dr. Justin D Schumacher for his suggestions and careful review of the manuscript.
Abbreviations
ALP, Alkaline phosphatase; ALT, Alanine aminotransferase; ANIT, α-Naphthylisothiocyanate; ASBT, Apical sodium-dependent bile salt transporter; AST, Aspartate transaminase; BA, Bile acid; BDL, Bile duct ligation; BSEP, Bile salt export pump; C4, 7α-Hydroxy-4-cholesten-3-one; CA, Cholic acid; CDCA, Chenodeoxycholic acid; CY27A1, Cytochrome P450 27A1; CYP7A1, Cytochrome P450 7A1; CYP8B1, Cytochrome P450 8B1; DCA, Deoxycholic acid; FGF, Fibroblast growth factor; FGF15, Fibroblast growth factor 15; FGF19, Fibroblast growth factor 19; FGFR4, Fibroblast growth factor receptor 4; FXR, Farnesoid X receptor; GGT, Gamma-glutamyltransferase; HSC, Hepatic stellate cell; IBABP, Intestinal bile acid binding protein; iNOS, Inducible nitric oxide synthase; KO, Knockout; LCA, Lithocholic acid; LPS, Lipopolysaccharide; LT, Liver transplantation; MCA, Muricholic acid; MRP2, Multidrug resistance-associated protein 2; NASH, Non-alcoholic steatohepatitis; NR, Nuclear receptor; NTCP, Sodium taurocholate co-transporting polypeptide; OCA, Obeticholic acid; OSTα, Organic solute transporter alpha; OSTβ, Organic solute transporter beta; PBC, Primary biliary cholangitis; PFIC2, Progressive familial intrahepatic cholestasis type 2; PSC, Primary sclerosing cholangitis; PXR, Pregnane X receptor; SHP-1, Small heterodimer partner 1; UDCA, Ursodeoxycholic acid; WT, Wild type.
References
1. Bloch K, Berg BN, Rittenberg D. The biological conversion of cholesterol to cholic acid. J Biol Chem. (1943) 149:511–7.
2. Danielsson H, Sjovall J. Bile acid metabolism. Annu Rev Biochem. (1975) 44:233–53. doi: 10.1146/annurev.bi.44.070175.001313
4. Ishibashi S, Schwarz M, Frykman PK, Herz J, Russell DW. Disruption of cholesterol 7alpha-hydroxylase gene in mice. I. Postnatal lethality reversed by bile acid and vitamin supplementation. J Biol Chem. (1996) 271:18017–23. doi: 10.1074/jbc.271.30.18017
5. Russell DW, Setchell KD. Bile acid biosynthesis. Biochemistry. (1992) 31:4737–49. doi: 10.1021/bi00135a001
6. Chiang JY. Regulation of bile acid synthesis: pathways, nuclear receptors, and mechanisms. J Hepatol. (2004) 40:539–51. doi: 10.1016/j.jhep.2003.11.006
7. Li-Hawkins J, Gafvels M, Olin M, Lund EG, Andersson U, Schuster G, et al. Cholic acid mediates negative feedback regulation of bile acid synthesis in mice. J Clin Invest. (2002) 110:1191–200. doi: 10.1172/JCI0216309
8. Axelson M, Aly A, Sjovall J. Levels of 7 alpha-hydroxy-4-cholesten-3-one in plasma reflect rates of bile acid synthesis in man. FEBS Lett. (1988) 239:324–8. doi: 10.1016/0014-5793(88)80944-X
9. Li T, Chiang JY. Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev. (2014) 66:948–83. doi: 10.1124/pr.113.008201
10. Takahashi S, Fukami T, Masuo Y, Brocker CN, Xie C, Krausz KW, et al. Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J Lipid Res. (2016) 57:2130–7. doi: 10.1194/jlr.M071183
11. de Boer JF, Verkade E, Mulder NL, de Vries HD, Huijkman N, Koehorst M, et al. A human-like bile acid pool induced by deletion of hepatic Cyp2c70 modulates effects of FXR activation in mice. J Lipid Res. (2020) 61:291–305. doi: 10.1194/jlr.RA119000243
12. Honda A, Miyazaki T, Iwamoto J, Hirayama T, Morishita Y, Monma T, et al. Regulation of bile acid metabolism in mouse models with hydrophobic bile acid composition. J Lipid Res. (2020) 61:54–69. doi: 10.1194/jlr.RA119000395
13. Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. (2003) 72:137–74. doi: 10.1146/annurev.biochem.72.121801.161712
14. Johnson MR, Barnes S, Kwakye JB, Diasio RB. Purification and characterization of bile acid-CoA:amino acid N-acyltransferase from human liver. J Biol Chem. (1991) 266:10227–33.
15. Killenberg PG, Jordan JT. Purification and characterization of bile acid-CoA:amino acid N-acyltransferase from rat liver. J Biol Chem. (1978) 253:1005–10.
16. Trauner M, Boyer JL. Bile salt transporters: molecular characterization, function, and regulation. Physiol Rev. (2003) 83:633–71. doi: 10.1152/physrev.00027.2002
17. Konig J, Nies AT, Cui Y, Leier I, Keppler D. Conjugate export pumps of the multidrug resistance protein (MRP) family: localization, substrate specificity, and MRP2-mediated drug resistance. Biochim Biophys Acta. (1999) 1461:377–94. doi: 10.1016/S0005-2736(99)00169-8
18. Lam P, Wang R, Ling V. Bile acid transport in sister of P-glycoprotein (ABCB11) knockout mice. Biochemistry. (2005) 44:12598–605. doi: 10.1021/bi050943e
19. Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, Arnell H, et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet. (1998) 20:233–8. doi: 10.1038/3034
20. Imagawa K, Takayama K, Isoyama S, Tanikawa K, Shinkai M, Harada K, et al. Generation of a bile salt export pump deficiency model using patient-specific induced pluripotent stem cell-derived hepatocyte-like cells. Sci Rep. (2017) 7:41806. doi: 10.1038/srep41806
21. Li J, Dawson PA. Animal models to study bile acid metabolism. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:895–911. doi: 10.1016/j.bbadis.2018.05.011
22. Wong MH, Oelkers P, Craddock AL, Dawson PA. Expression cloning and characterization of the hamster ileal sodium-dependent bile acid transporter. J Biol Chem. (1994) 269:1340–7.
23. Gong YZ, Everett ET, Schwartz DA, Norris JS, Wilson FA. Molecular cloning, tissue distribution, and expression of a 14-kDa bile acid-binding protein from rat ileal cytosol. Proc Natl Acad Sci USA. (1994) 91:4741–5. doi: 10.1073/pnas.91.11.4741
24. Dawson PA, Hubbert M, Haywood J, Craddock AL, Zerangue N, Christian WV, et al. The heteromeric organic solute transporter alpha-beta, ost alpha-ost beta, is an ileal basolateral bile acid transporter. J Biol Chem. (2005) 280:6960–8. doi: 10.1074/jbc.M412752200
25. Hagenbuch B, Meier PJ. Molecular cloning, chromosomal localization, and functional characterization of a human liver Na+/bile acid cotransporter. J Clin Invest. (1994) 93:1326–31. doi: 10.1172/JCI117091
26. Smit JJ, Schinkel AH, Oude Elferink RP, Groen AK, Wagenaar E, van Deemter L, et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell. (1993) 75:451–62. doi: 10.1016/0092-8674(93)90380-9
27. Oude Elferink RP, Ottenhoff R, van Wijland M, Smit JJ, Schinkel AH, Groen AK. Regulation of biliary lipid secretion by mdr2 P-glycoprotein in the mouse. J Clin Invest. (1995) 95:31–8. doi: 10.1172/JCI117658
28. Vree JML, de Jacquemin E, Sturm E, Cresteil D, Bosma PJ, Aten J, et al. Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad Sci USA. (1998) 95:282–7. doi: 10.1073/pnas.95.1.282
29. Trauner M, Fickert P, Wagner M. MDR3 (ABCB4) defects: a paradigm for the genetics of adult cholestatic syndromes. Semin Liver Dis. (2007) 27:077–98. doi: 10.1055/s-2006-960172
30. Popov Y, Patsenker E, Fickert P, Trauner M, Schuppan D. Mdr2 (Abcb4)-/- mice spontaneously develop severe biliary fibrosis via massive dysregulation of pro- and antifibrogenic genes. J Hepatol. (2005) 43:1045–54. doi: 10.1016/j.jhep.2005.06.025
31. Ikenaga N, Liu SB, Sverdlov DY, Yoshida S, Nasser I, Ke Q, et al. A new Mdr2–/– mouse model of sclerosing cholangitis with rapid fibrosis progression, early-onset portal hypertension, and liver cancer. Am J Pathol. (2015) 185:325–34. doi: 10.1016/j.ajpath.2014.10.013
32. Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. (2006) 47:241–59. doi: 10.1194/jlr.R500013-JLR200
33. Dowling RH. The enterohepatic circulation of bile acids as they relate to lipid disorders. J Clin Pathol Suppl Assoc Clin Pathol. (1973) 5:59–67. doi: 10.1136/jcp.s1-5.1.59
34. Guo GL, Lambert G, Negishi M, Ward JM, Brewer HB Jr, Kliewer SA, et al. Complementary roles of farnesoid X receptor, pregnane X receptor, and constitutive androstane receptor in protection against bile acid toxicity. J Biol Chem. (2003) 278:45062–71. doi: 10.1074/jbc.M307145200
35. Maruyama T, Miyamoto Y, Nakamura T, Tamai Y, Okada H, Sugiyama E, et al. Identification of membrane-type receptor for bile acids (M-BAR). Biochem Biophys Res Commun. (2002) 298:714–9. doi: 10.1016/S0006-291X(02)02550-0
36. Liu R, Zhao R, Zhou X, Liang X, Campbell DJ, Zhang X, et al. Conjugated bile acids promote cholangiocarcinoma cell invasive growth through activation of sphingosine 1-phosphate receptor 2. Hepatology. (2014) 60:908–18. doi: 10.1002/hep.27085
37. Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science. (2001) 294:1866–70. doi: 10.1126/science.294.5548.1866
38. Edwards PA, Kast HR, Anisfeld AM. BAREing it all: the adoption of LXR and FXR and their roles in lipid homeostasis. J Lipid Res. (2002) 43: 2–12.
39. Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, et al. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell. (2000) 6:507–15. doi: 10.1016/S1097-2765(00)00050-2
40. Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science. (1999) 284:1365–8. doi: 10.1126/science.284.5418.1365
41. Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell. (1999) 3:543–53. doi: 10.1016/S1097-2765(00)80348-2
42. Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, et al. Identification of a nuclear receptor for bile acids. Science. (1999) 284:1362–5. doi: 10.1126/science.284.5418.1362
43. Thomas AM, Hart SN, Kong B, Fang J, Zhong XB, Guo GL. Genome-wide tissue-specific farnesoid X receptor binding in mouse liver and intestine. Hepatology. (2010) 51:1410–9. doi: 10.1002/hep.23450
44. Kim I, Ahn SH, Inagaki T, Choi M, Ito S, Guo GL, et al. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J Lipid Res. (2007) 48:2664–72. doi: 10.1194/jlr.M700330-JLR200
45. Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. (2005) 2:217–25. doi: 10.1016/j.cmet.2005.09.001
46. Kong B, Wang L, Chiang JY, Zhang Y, Klaassen CD, Guo GL. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology. (2012) 56:1034–43. doi: 10.1002/hep.25740
47. Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. (2000) 6:517–26. doi: 10.1016/S1097-2765(00)00051-4
48. Plass JR, Mol O, Heegsma J, Geuken M, Faber KN, Jansen PL, et al. Farnesoid X receptor and bile salts are involved in transcriptional regulation of the gene encoding the human bile salt export pump. Hepatology. (2002) 35:589–96. doi: 10.1053/jhep.2002.31724
49. Landrier JF, Eloranta JJ, Vavricka SR, Kullak-Ublick GA. The nuclear receptor for bile acids, FXR, transactivates human organic solute transporter-alpha and -beta genes. Am J Physiol Gastrointest Liver Physiol. (2006) 290:G476–85. doi: 10.1152/ajpgi.00430.2005
50. Zollner G, Wagner M, Moustafa T, Fickert P, Silbert D, Gumhold J, et al. Coordinated induction of bile acid detoxification and alternative elimination in mice: role of FXR-regulated organic solute transporter-alpha/beta in the adaptive response to bile acids. Am J Physiol Gastrointest Liver Physiol. (2006) 290:G923–32. doi: 10.1152/ajpgi.00490.2005
51. Denson LA, Sturm E, Echevarria W, Zimmerman TL, Makishima M, Mangelsdorf DJ, et al. The orphan nuclear receptor, shp, mediates bile acid-induced inhibition of the rat bile acid transporter, ntcp. Gastroenterology. (2001) 121:140–7. doi: 10.1053/gast.2001.25503
52. Chen F, Ma L, Dawson PA, Sinal CJ, Sehayek E, Gonzalez FJ, et al. Liver receptor homologue-1 mediates species- and cell line-specific bile acid-dependent negative feedback regulation of the apical sodium-dependent bile acid transporter. J Biol Chem. (2003) 278:19909–16. doi: 10.1074/jbc.M207903200
53. Wang L, Lee Y-K, Bundman D, Han Y, Thevananther S, Kim CS, et al. Redundant pathways for negative feedback regulation of bile acid production. Dev Cell. (2002) 2:721–31. doi: 10.1016/S1534-5807(02)00187-9
54. Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. (2004) 113:1408–18. doi: 10.1172/JCI21025
55. Schoenfield LJ, Lachin JM. Chenodiol (chenodeoxycholic acid) for dissolution of gallstones: the National Cooperative Gallstone Study. A controlled trial of efficacy and safety. Ann Intern Med. (1981) 95:257–82. doi: 10.7326/0003-4819-95-3-257
56. Prieur X, Coste H, Rodriguez JC. The human apolipoprotein AV gene is regulated by peroxisome proliferator-activated receptor-alpha and contains a novel farnesoid X-activated receptor response element. J Biol Chem. (2003) 278:25468–80. doi: 10.1074/jbc.M301302200
57. Kast HR, Nguyen CM, Sinal CJ, Jones SA, Laffitte BA, Reue K, et al. Farnesoid X-activated receptor induces apolipoprotein C-II transcription: a molecular mechanism linking plasma triglyceride levels to bile acids. Mol Endocrinol. (2001) 15:1720–8. doi: 10.1210/mend.15.10.0712
58. Claudel T, Inoue Y, Barbier O, Duran-Sandoval D, Kosykh V, Fruchart J, et al. Farnesoid X receptor agonists suppress hepatic apolipoprotein CIII expression. Gastroenterology. (2003) 125:544–55. doi: 10.1016/S0016-5085(03)00896-5
59. Sirvent A, Claudel T, Martin G, Brozek J, Kosykh V, Darteil R, et al. The farnesoid X receptor induces very low density lipoprotein receptor gene expression. FEBS Lett. (2004) 566:173–7. doi: 10.1016/j.febslet.2004.04.026
60. Pineda Torra I, Claudel T, Duval C, Kosykh V, Fruchart JC, Staels B. Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Mol Endocrinol. (2003) 17:259–72. doi: 10.1210/me.2002-0120
61. Jiao Y, Lu Y, Li XY. Farnesoid X receptor: a master regulator of hepatic triglyceride and glucose homeostasis. Acta Pharmacol Sin. (2015) 36:44–50. doi: 10.1038/aps.2014.116
62. Yamagata K, Daitoku H, Shimamoto Y, Matsuzaki H, Hirota K, Ishida J, et al. Bile acids regulate gluconeogenic gene expression via small heterodimer partner-mediated repression of hepatocyte nuclear factor 4 and Foxo1. J Biol Chem. (2004) 279:23158–65. doi: 10.1074/jbc.M314322200
63. Stayrook KR, Bramlett KS, Savkur RS, Ficorilli J, Cook T, Christe ME, et al. Regulation of carbohydrate metabolism by the farnesoid X receptor. Endocrinology. (2005) 146:984–91. doi: 10.1210/en.2004-0965
64. Zhan L, Liu H-X, Fang Y, Kong B, He Y, Zhong X, et al. Genome-wide binding and transcriptome analysis of human farnesoid X receptor in primary human hepatocytes. PLoS ONE. (2014) 9:e105930. doi: 10.1371/journal.pone.0105930
65. Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest. (2006) 116:1102–9. doi: 10.1172/JCI25604
66. Zhang Y, Lee FY, Barrera G, Lee H, Vales C, Gonzalez FJ, et al. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci USA. (2006) 103:1006–11. doi: 10.1073/pnas.0506982103
67. Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology. (2008) 48:1632–43. doi: 10.1002/hep.22519
68. Zhang S, Wang J, Liu Q, Harnish DC. Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J Hepatol. (2009) 51:380–8. doi: 10.1016/j.jhep.2009.03.025
69. Daly C, Rollins BJ. Monocyte chemoattractant protein-1 (CCL2) in inflammatory disease and adaptive immunity: therapeutic opportunities and controversies. Microcirculation. (2003) 10:247–57. doi: 10.1080/mic.10.3-4.247.257
70. Liu T, Zhang L, Joo D, Sun SC. NF-kappaB signaling in inflammation. Signal Transduct Target Ther. (2017) 2:17023. doi: 10.1038/sigtrans.2017.23
71. Fiorucci S, Rizzo G, Antonelli E, Renga B, Mencarelli A, Riccardi L, et al. Cross-talk between farnesoid-X-receptor (FXR) and peroxisome proliferator-activated receptor gamma contributes to the antifibrotic activity of FXR ligands in rodent models of liver cirrhosis. J Pharmacol Exp Ther. (2005) 315:58–68. doi: 10.1124/jpet.105.085597
72. Renga B, Mencarelli A, Migliorati M, Cipriani S, D'Amore C, Distrutti E, et al. SHP-dependent and -independent induction of peroxisome proliferator-activated receptor-gamma by the bile acid sensor farnesoid X receptor counter-regulates the pro-inflammatory phenotype of liver myofibroblasts. Inflamm Res. (2011) 60:577–87. doi: 10.1007/s00011-010-0306-1
73. Schumacher JD, Kong B, Pan Y, Zhan L, Sun R, Aa J, et al. The effect of fibroblast growth factor 15 deficiency on the development of high fat diet induced non-alcoholic steatohepatitis. Toxicol Appl Pharmacol. (2017) 330:1–8. doi: 10.1016/j.taap.2017.06.023
74. Schumacher JD, Kong B, Wu J, Rizzolo D, Armstrong LE, Chow MD, et al. Direct and indirect effects of fibroblast growth factor (FGF) 15 and FGF19 on liver fibrosis development. Hepatology. (2019) 71:670–85. doi: 10.1002/hep.30810
75. Liu Y, Binz J, Numerick MJ, Dennis S, Luo G, Desai B, et al. Hepatoprotection by the farnesoid X receptor agonist GW4064 in rat models of intra- and extrahepatic cholestasis. J Clin Invest. (2003) 112:1678–87. doi: 10.1172/JCI18945
76. Stedman C, Liddle C, Coulter S, Sonoda J, Alvarez JG, Evans RM, et al. Benefit of farnesoid X receptor inhibition in obstructive cholestasis. Proc Natl Acad Sci USA. (2006) 103:11323–8. doi: 10.1073/pnas.0604772103
77. Cui YJ, Aleksunes LM, Tanaka Y, Goedken MJ, Klaassen CD. Compensatory induction of liver efflux transporters in response to ANIT-induced liver injury is impaired in FXR-null mice. Toxicol Sci. (2009) 110:47–60. doi: 10.1093/toxsci/kfp094
78. van Golen RF, Olthof PB, Lionarons DA, Reiniers MJ, Alles LK, Uz Z, et al. FXR agonist obeticholic acid induces liver growth but exacerbates biliary injury in rats with obstructive cholestasis. Sci Rep. (2018) 8:16529. doi: 10.1038/s41598-018-33070-1
79. Jones H, Alpini G, Francis H. Bile acid signaling and biliary functions. Acta Pharm Sin B. (2015) 5:123–8. doi: 10.1016/j.apsb.2015.01.009
80. Boyer JL. Bile formation and secretion. Compr Physiol. (2013) 3:1035–78. doi: 10.1002/cphy.c120027
81. Jung D, York JP, Wang L, Yang C, Zhang A, Francis HL, et al. FXR-induced secretion of FGF15/19 inhibits CYP27 expression in cholangiocytes through p38 kinase pathway. Pflugers Arch. (2014) 466:1011–9. doi: 10.1007/s00424-013-1364-3
82. Cheng L, Tian F, Tian F, Tang L, Chen G, Luo Z, et al. Repression of farnesoid X receptor contributes to biliary injuries of liver grafts through disturbing cholangiocyte bile acid transport. Am J Transplant. (2013) 13:3094–102. doi: 10.1111/ajt.12479
83. Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. (2009) 8:235–53. doi: 10.1038/nrd2792
84. Owen BM, Mangelsdorf DJ, Kliewer SA. Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends Endocrinol Metab. (2015) 26:22–9. doi: 10.1016/j.tem.2014.10.002
85. Goetz R, Beenken A, Ibrahimi OA, Kalinina J, Olsen SK, Eliseenkova AV, et al. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol Cell Biol. (2007) 27:3417–28. doi: 10.1128/MCB.02249-06
86. Nishimura T, Utsunomiya Y, Hoshikawa M, Ohuchi H, Itoh N. Structure and expression of a novel human FGF, FGF-19, expressed in the fetal brain. Biochim Biophys Acta. (1999) 1444:148–51. doi: 10.1016/S0167-4781(98)00255-3
87. Potthoff MJ, Kliewer SA, Mangelsdorf DJ. Endocrine fibroblast growth factors 15/19 and 21: from feast to famine. Genes Dev. (2012) 26:312–24. doi: 10.1101/gad.184788.111
88. Lin BC, Wang M, Blackmore C, Desnoyers LR. Liver-specific activities of FGF19 require Klotho beta. J Biol Chem. (2007) 282:27277–84. doi: 10.1074/jbc.M704244200
89. Yang C, Jin C, Li X, Wang F, McKeehan WL, Luo Y. Differential specificity of endocrine FGF19 and FGF21 to FGFR1 and FGFR4 in complex with KLB. PLoS One. (2012) 7:e33870. doi: 10.1371/journal.pone.0033870
90. Schumacher JD, Guo GL. Pharmacologic modulation of bile acid-FXR-FGF15/FGF19 pathway for the treatment of nonalcoholic steatohepatitis. Handb Exp Pharmacol. (2019) 256:325–57. doi: 10.1007/164_2019_228
91. Naugler WE, Tarlow BD, Fedorov LM, Taylor M, Pelz C, Li B, et al. Fibroblast growth factor signaling controls liver size in mice with humanized livers. Gastroenterology. (2015) 149:728–40 e15. doi: 10.1053/j.gastro.2015.05.043
92. Tomlinson E, Fu L, John L, Hultgren B, Huang X, Renz M, et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology. (2002) 143:1741–7. doi: 10.1210/endo.143.5.8850
93. Fu L, John LM, Adams SH, Yu XX, Tomlinson E, Renz M, et al. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology. (2004) 145:2594–603. doi: 10.1210/en.2003-1671
94. Marcelin G, Jo Y-H, Li X, Schwartz GJ, Zhang Y, Dun NJ, et al. Central action of FGF19 reduces hypothalamic AGRP/NPY neuron activity and improves glucose metabolism. Mol Metab. (2014) 3:19–28. doi: 10.1016/j.molmet.2013.10.002
95. Kir S, Beddow SA, Samuel VT, Miller P, Previs SF, Suino-Powell K, et al. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science. (2011) 331:1621–4. doi: 10.1126/science.1198363
96. Benoit B, Meugnier E, Castelli M, Chanon S, Vieille-Marchiset A, Durand C, et al. Fibroblast growth factor 19 regulates skeletal muscle mass and ameliorates muscle wasting in mice. Nat Med. (2017) 23:990–6. doi: 10.1038/nm.4363
97. Potthoff MJ, Boney-Montoya J, Choi M, He T, Sunny NE, Satapati S, et al. FGF15/19 regulates hepatic glucose metabolism by inhibiting the CREB-PGC-1alpha pathway. Cell Metab. (2011) 13:729–38. doi: 10.1016/j.cmet.2011.03.019
98. Bhatnagar S, Damron HA, Hillgartner FB. Fibroblast growth factor-19, a novel factor that inhibits hepatic fatty acid synthesis. J Biol Chem. (2009) 284:10023–33. doi: 10.1074/jbc.M808818200
99. Zhou M, Learned RM, Rossi SJ, DePaoli AM, Tian H, Ling L. Engineered fibroblast growth factor 19 reduces liver injury and resolves sclerosing cholangitis in Mdr2-deficient mice. Hepatology. (2016) 63:914–29. doi: 10.1002/hep.28257
100. Zhou M, Learned RM, Rossi SJ, DePaoli AM, Tian H, Ling L. Engineered FGF19 eliminates bile acid toxicity and lipotoxicity leading to resolution of steatohepatitis and fibrosis in mice. Hepatol Commun. (2017) 1:1024–42. doi: 10.1002/hep4.1108
101. Pellicciari R, Costantino G, Camaioni E, Sadeghpour BM, Entrena A, Willson TM, et al. Bile acid derivatives as ligands of the farnesoid X receptor. Synthesis, evaluation, and structure-activity relationship of a series of body and side chain modified analogues of chenodeoxycholic acid. J Med Chem. (2004) 47:4559–69. doi: 10.1021/jm049904b
102. Suraweera D, Rahal H, Jimenez M, Viramontes M, Choi G, Saab S. Treatment of primary biliary cholangitis ursodeoxycholic acid non-responders: a systematic review. Liver Int. (2017) 37:1877–86. doi: 10.1111/liv.13477
103. Hirschfield GM, Mason A, Luketic V, Lindor K, Gordon SC, Mayo M, et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology. (2015) 148:751–761.e8. doi: 10.1053/j.gastro.2014.12.005
104. Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus C, Invernizzi P, et al. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med. (2016) 375:631–43. doi: 10.1056/NEJMoa1509840
105. Manne V, Kowdley KV. Obeticholic acid in primary biliary cholangitis: where we stand. Curr Opin Gastroenterol. (2019) 35:191–6. doi: 10.1097/MOG.0000000000000525
106. Kowdley KV, Luketic V, Chapman R, Hirschfield GM, Poupon R, Schramm C, et al. A randomized trial of obeticholic acid monotherapy in patients with primary biliary cholangitis. Hepatology. (2018) 67:1890–902. doi: 10.1002/hep.29569
107. Fiorucci S, Di Giorgio C, Distrutti E. Obeticholic acid: an update of its pharmacological activities in liver disorders. Handb Exp Pharmacol. (2019) 256:283–95. doi: 10.1007/164_2019_227
108. Carino A, Biagioli M, Marchianò S, Fiorucci C, Bordoni M, Roselli R, et al. Opposite effects of the FXR agonist obeticholic acid on Mafg and Nrf2 mediate the development of acute liver injury in rodent models of cholestasis. Biochim Biophys Acta. (2020) 1865:158733. doi: 10.1016/j.bbalip.2020.158733
109. Edwards J, LaCerte C, Peyret T, Gosselin N, Marier J, Hofmann A, et al. Modeling and experimental studies of obeticholic acid exposure and the impact of cirrhosis stage. Clin Transl Sci. (2016) 9:328–36. doi: 10.1111/cts.12421
110. Liu T, Yang H, Fan W, Tu J, Li TWH, Wang J, et al. Mechanisms of MAFG dysregulation in cholestatic liver injury and development of liver cancer. Gastroenterology. (2018) 155:557–71.e14. doi: 10.1053/j.gastro.2018.04.032
111. Yang H, Ko K, Xia M, Li TWH, Oh P, Li J, et al. Induction of avian musculoaponeurotic fibrosarcoma proteins by toxic bile acid inhibits expression of glutathione synthetic enzymes and contributes to cholestatic liver injury in mice. Hepatology. (2010) 51:1291–301. doi: 10.1002/hep.23471
112. Santiago P, Scheinberg A, Levy C. Cholestatic liver diseases: new targets, new therapies. Ther Adv Gastroenterol. (2018) 11:175628481878740. doi: 10.1177/1756284818787400
113. Kowdley KV, Vuppalanchi R, Levy C, Floreani A, Andreone P, LaRusso NF, et al. A randomized, placebo-controlled, phase II study of obeticholic acid for primary sclerosing cholangitis. J Hepatol. (2020) 73:94–101. doi: 10.1016/j.jhep.2020.02.033
114. Trauner M, Gulamhusein A, Hameed B, Caldwell S, Shiffman ML, Landis C, et al. The nonsteroidal farnesoid X receptor agonist cilofexor (GS-9674) improves markers of cholestasis and liver injury in patients with primary sclerosing cholangitis. Hepatology. (2019) 70:788–801. doi: 10.1002/hep.30509
115. Patel K, Harrison SA, Elkashab M, Trotter JF, Herring R, Rojter S, et al. Cilofexor, a nonsteroidal FXR agonist, in non-cirrhotic patients with nonalcoholic steatohepatitis: a phase 2 randomized controlled trial. Hepatology. (2020). 72:58–71. doi: 10.1002/hep.31205
116. Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. (2015) 385:956–65. doi: 10.1016/S0140-6736(14)61933-4
117. Cipriani S, Mencarelli A, Palladino G, Fiorucci S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J Lipid Res. (2010) 51:771–84. doi: 10.1194/jlr.M001602
118. Renga B, Mencarelli A, Vavassori P, Brancaleone V, Fiorucci S. The bile acid sensor FXR regulates insulin transcription and secretion. Biochim Biophys Acta. (2010) 1802:363–72. doi: 10.1016/j.bbadis.2010.01.002
119. Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall HU, Kipnes M, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. (2013) 145:574–82 e1. doi: 10.1053/j.gastro.2013.05.042
120. Hameed B, Terrault NA, Gill RM, Loomba R, Chalasani N, Hoofnagle JH, et al. Clinical and metabolic effects associated with weight changes and obeticholic acid in non-alcoholic steatohepatitis. Aliment Pharmacol Ther. (2018) 47:645–56. doi: 10.1111/apt.14492
121. Ratziu V, Sanyal A, Macconell L, Shringarpure R, Marmon T, Shapiro D, et al. Regenerate: a phase 3, double-blind, randomized, placebo-controlled multicenter study of obeticholic acid therapy for nonalcoholic steatohepatitis. J Hepatol. (2016) 64:294–5. doi: 10.1016/S0168-8278(16)00372-X
122. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology. (2018) 67:328–57. doi: 10.1002/hep.29367
123. Intercept Receives Complete Response Letter from FDA for Obeticholic Acid for the Treatment of Fibrosis Due to NASH - Intercept Pharmaceuticals, Inc. [online]. Available online at: https://ir.interceptpharma.com/news-releases/news-release-details/intercept-receives-complete-response-letter-fda-obeticholic-acid (accessed July 6, 2020).
Keywords: bile acids, FGF15/19, FXR, agonist, non-alcoholic fatty liver disease, species difference
Citation: Stofan M and Guo GL (2020) Bile Acids and FXR: Novel Targets for Liver Diseases. Front. Med. 7:544. doi: 10.3389/fmed.2020.00544
Received: 12 May 2020; Accepted: 30 July 2020;
Published: 11 September 2020.
Edited by:
Shannon Glaser, Texas A&M University College of Medicine, United StatesReviewed by:
Roberto Gramignoli, Karolinska Institutet (KI), SwedenLindsey Kennedy, Indiana University Bloomington, United States
Copyright © 2020 Stofan and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Grace L. Guo, Z3VvQGVvaHNpLnJ1dGdlcnMuZWR1