 Todd S. Ing1Kavitha Ganta2Gautam Bhave3
Todd S. Ing1Kavitha Ganta2Gautam Bhave3 Susie Q. Lew4Emmanuel I. Agaba5
Susie Q. Lew4Emmanuel I. Agaba5 Christos Argyropoulos6*
Christos Argyropoulos6* Antonios H. Tzamaloukas7
Antonios H. Tzamaloukas7- 1Department of Medicine, Stritch School of Medicine, Loyola University Chicago, Chicago, IL, United States
- 2Medicine Service, Department of Medicine, Raymond G. Murphy Veterans Affairs Medical Center, University of New Mexico School of Medicine, Albuquerque, NM, United States
- 3Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, United States
- 4Department of Medicine, George Washington University School of Medicine, Washington, DC, United States
- 5Department of Medicine, University of Jos, Jos, Nigeria
- 6Department of Medicine, University of New Mexico School of Medicine, Albuquerque, NM, United States
- 7Research Service, Department of Medicine, Raymond G. Murphy Veterans Affairs Medical Center, University of New Mexico School of Medicine, Albuquerque, NM, United States
In hyperglycemia, hypertonicity results from solute (glucose) gain and loss of water in excess of sodium plus potassium through osmotic diuresis. Patients with stage 5 chronic kidney disease (CKD) and hyperglycemia have minimal or no osmotic diuresis; patients with preserved renal function and diabetic ketoacidosis (DKA) or hyperosmolar hyperglycemic state (HHS) have often large osmotic diuresis. Hypertonicity from glucose gain is reversed with normalization of serum glucose ([Glu]); hypertonicity due to osmotic diuresis requires infusion of hypotonic solutions. Prediction of the serum sodium after [Glu] normalization (the corrected [Na]) estimates the part of hypertonicity caused by osmotic diuresis. Theoretical methods calculating the corrected [Na] and clinical reports allowing its calculation were reviewed. Corrected [Na] was computed separately in reports of DKA, HHS and hyperglycemia in CKD stage 5. The theoretical prediction of [Na] increase by 1.6 mmol/L per 5.6 mmol/L decrease in [Glu] in most clinical settings, except in extreme hyperglycemia or profound hypervolemia, was supported by studies of hyperglycemia in CKD stage 5 treated only with insulin. Mean corrected [Na] was 139.0 mmol/L in 772 hyperglycemic episodes in CKD stage 5 patients. In patients with preserved renal function, mean corrected [Na] was within the eunatremic range (141.1 mmol/L) in 7,812 DKA cases, and in the range of severe hypernatremia (160.8 mmol/L) in 755 cases of HHS. However, in DKA corrected [Na] was in the hypernatremic range in several reports and rose during treatment with adverse neurological consequences in other reports. The corrected [Na], computed as [Na] increase by 1.6 mmol/L per 5.6 mmol/L decrease in [Glu], provides a reasonable estimate of the degree of hypertonicity due to losses of hypotonic fluids through osmotic diuresis at presentation of DKH or HHS and should guide the tonicity of replacement solutions. However, the corrected [Na] may change during treatment because of ongoing fluid losses and should be monitored during treatment.
Introduction
Imbalances that develop in patients with severe hyperglycemia and preserved renal function include extracellular gain of solute (glucose) and deficits of water, sodium, potassium, and other ions resulting from glycosuria. These imbalances, which cause extracellular and intracellular volume deficits, changes in the concentrations of key serum ions, and hypertonicity, constitute major treatment targets (1).
This report addresses the management of hyperglycemic hypertonicity. Hypertonicity is seen routinely in severe hyperglycemia (1–5), causes potentially life-threatening neurological manifestations (6, 7), and represents an important component of the treatment (1, 2).
Corrected Sodium Level, Glucose Concentration, and Tonicity
In experimental studies, tonicity of a fluid can be measured directly by rapid photographic recordings of changes in the volume of cells, usually red blood cells, suspended in the fluid of interest (8). In clinical practice, tonicity is evaluated by surrogate biochemical measurements, including serum osmolality and sodium concentration ([Na]) (8). In the absence of solutes with extracellular distribution other than sodium salts (e.g., glucose), [Na] represents the index expressing tonicity (8). In hyperglycemia, glucose accumulation in the extracellular compartment contributes to tonicity (Ton), which is expressed by the formula (9):
where the serum glucose concentration ([Glu]) is in mmol/L (1 mmol/L = 18 mg/dL).
Formula 1 provides accurate information on tonicity in hyperglycemia, except when high levels of plasma solids lower plasma water content (e.g., in hyperlipidemia) causing falsely low measurement of [Na] by indirect potentiometry (10). However, this formula should not be used to guide the composition of the replacement solution. Hypertonicity in hyperglycemia results from gain of extracellular solute (glucose) and osmotic diuresis (2, 5). Correction of hyperglycemia results in extracellular solute loss (11) and decrease in tonicity (12). The tonicity of the replacement solutions should correct the component of hypertonicity resulting from osmotic diuresis (2, 5). [Na] rises during correction of hyperglycemia as water is transferred osmotically from the extracellular into the intracellular compartment (12). The tonicity of replacement solutions should be based on the projected value of [Na] after normalization of [Glu] (2, 6, 8). The corrected [Na] is calculated using a predicted value of the change in [Na] (Δ[Na]) that results directly from the required change in [Glu] (Δ[Glu]) and is applied in the evaluation of the component of hyperglycemic hypertonicity that results from osmotic diuresis. The value of the coefficient used to calculate the corrected [Na] is a point of dispute (13).
This perspective article reviews the sources of various estimates of the coefficient for the corrected [Na] and clinical studies providing evidence for the appropriate coefficient. Then, it computes the corrected [Na] in reports of various categories of hyperglycemic crises, and based on these last reports, provides a frame for the clinical application of the corrected [Na].
Derivations of the Corrected [Na] in Hyperglycemia
Closed System of Hyperglycemia: Theoretical Calculations
This section addresses the modeling of the effect on [Na] from change in [Glu] not accompanied by changes in external balance of body water or monovalent cations, i.e., in a closed system. The theoretical calculations explore the principle asserting that in hyperglycemia [Glu] represents the ratio (baseline amount of extracellular glucose plus amount of glucose gained)/ECFV, where ECFV is hyperglycemic extracellular volume, while Ton (formula 1) represents the ratio (baseline effective body solute plus amount of glucose gained)/TBW, where TBW is total body water (7). Applying this principle and considering that the amounts of sodium in the extracellular compartment and effective solute in the intracellular compartment remain constant during development of hyperglycemia, Katz calculated that [Na] changes by 1.6 mmol/L in the opposite direction of every 5.6 mmol/L (100 mg/dL) change in [Glu] (Δ[Na]/Δ[Glu]) = −1.6 mmol/L per 5.6 mmol/L) (14).
Goldberg proposed using the Katz coefficient to predict the value of [Na] after correction of hyperglycemia (15). Subsequently, Al-Kudsi et al. provided the following formula to calculate this corrected [Na] (16):
The Al-Kudsi formula predicts the value of [Na] after correction of [Glu] to 5.6 mmol/L (100 mg/dL) without any change in body water, sodium or potassium (17). The corrected [Na] at any desired final value of [Glu] can be calculated by substituting this desired [Glu] for 5.6 mmol/L in the numerator of the Al-Kudsi formula.
Δ[Na]/Δ[Glu] values numerically lower than 2.8 mmol/L per 5.6 mmol/L indicate rise in tonicity, while values numerically higher than 2.8 indicate decrease in tonicity during development of hyperglycemia. The Katz report created new insights into the change in tonicity produced by glucose gain. Prior to this report, the value of Δ[Na]/Δ[Glu] used by clinicians, at −2.8 mmol/L per 5.6 mmol/L (18), indicated no change in tonicity as [Glu] rises because a rise in [Glu] by 5.6 mmol/L would be completely offset by a 2.8 mmol/L decrease in [Na] (formula 1). This Δ[Na]/Δ[Glu] value stemmed from the faulty premise that osmotic transfer of intracellular water into the extracellular space as a result of extracellular glucose gain was not associated with an increase in intracellular tonicity. As a matter of fact, the increase in total body effective solute (baseline solute plus glucose gain) causes equal rises in both intracellular and extracellular fluid tonicities. The glucose-induced gain in extracellular solute causes water exit from the cells (19) to bring about hypertonic hyponatremia (20). Katz's coefficient computes tonicity increase (ΔTon) of 2.4 (5.6 – 2 ×1.6) mOsm/L for every 5.6 mmol/L increase in [Glu] (2).
Several guidelines for managing hyperglycemia (21–24) and other reports (25–29) have adopted the Katz coefficient for calculating the corrected [Na]. Alternate guidelines for treating hyperglycemia (30, 31) and hyponatremia (32), and various other reports (33–35) advocate other coefficients. The proposed correction coefficients for [Na] range between −1.35 and −4.0 mmol/L per 5.6 mmol/L Δ[Glu] (36). The variation of these coefficients resulted from both theoretical calculations and clinical studies. This section addresses the theoretical calculations. All theoretical calculations of Δ[Na]/Δ[Glu] are based on the principle stating that in hyperglycemia the volume determining Δ[Glu] is ECFV2 while the volume determining the rise in tonicity is TBW (7).
Calculations based on the same principles as those applied by Katz revealed that Δ[Na]/Δ[Glu] can obtain several values in a closed system (37–40). In these calculations, the important influences on Δ[Na], Δ[Glu], Δ[Na]/Δ[Glu] derive from the degree of hyperglycemia (38) and the state of euglycemic extracellular volume (ECFV1) (39), or more specifically the glucose gained per liter of ECFV1 ([Glu]A) (17, 40) and the baseline, at euglycemia, ratio of intracellular volume (ICFV1) to ECFV1 (the volume ratio α1) (17, 39, 40). Note: In the text and Table 1, the subscripts 1 and 2 denote respectively baseline euglycemia and hyperglycemia.

Table 1. Tonicity-related and body fluid variables in a closed system of hyperglycemia.
The ICVF/ECVF volume ratio α increases during development of hypovolemia or correction of hyperglycemia and decreases during development of hypervolemia or of hyperglycemia. Total glucose gain during development of hyperglycemia is the product of ECFV1 and [Glu]A. [Glu]A, which is not a glucose concentration encountered clinically because ECFV increases during development of hyperglycemia, was entered in the calculations because the same [Glu]A leads to comparable [Glu]2 values in hypothetical subjects with widely different euglycemic volume ratio α1 (17).
Table 1 shows general formulas used by Katz for computation of tonicity-related and volume-related parameters. Total intracellular and extracellular solutes in this Table are the total solutes determining tonicity (14). Solutes with body water distribution, e.g., urea, do not contribute to tonicity (8, 9) and are not included in the calculations of the tonicity-related variables (9, 14). The formula for calculating the hyperglycemic ICFV2/ECFV2 ratio α2 in Table 1 as the corresponding ratio of effective intracellular-to-extracellular solute is based on the principle asserting that body water is apportioned between the intracellular and extracellular compartments in proportion to the amount of solute in each compartment (41). The calculations for the various Δ[Na]/Δ[Glu] coefficients in hyperglycemia (37–40) and for the following examples were based on the formulas of Table 1. Theoretical examples will show the effects of the ICFV1/ECFV1 volume ratio α1 and of the degree of hyperglycemia on tonicity-related parameters in hypothetical patients. For all examples the baseline values were 5.6 mmol/L for [Glu]1, 140 mmol/L per [Na]1, and 285.6 mOsm/L for Ton1.
For the same volume ratio α1, the same degree of hyperglycemia results in the same hypertonicity values regardless of the size of extracellular volume. Example: Two hypothetical patients, with TBW 48 L and 92 L and both with α1 = 2.0, as in Katz's report (14), and [Glu]A = 100 mmol/L. ECFV1 values are 16 and 32 L and glucose loads are 1,600 (16 ×100) and 3,200 (32 ×100) mmol, respectively. In both patients, [Glu]2 = 87.3 mmol/L (1,571 mg/dL), [Na]2 = 115.7 mmol/L, Δ[Na]/Δ[Glu] = −1.66 mmol/L per 5.6 mmol/L, and Ton2 = 318.7 mOsm/L.
For comparable degrees of hyperglycemia, hypertonicity is higher in hypervolemia and lower in hypovolemia compared to euvolemia. [Glu]A was 100 mmol/L in the euvolemic, hypervolemic, and hypovolemic examples. Euvolemic values are shown in the previous example. A hypothetical patient with TBW = 64 L, α1 = 1.0, ECFV1 = 32 L, and glucose load 3,200 (32 ×100) mmol represents severe hypervolemia. In this patient, [Glu]2 = 91.8 mmol/L (1,653 mg/dL), [Na]2 = 127.9 mmol/L, Δ[Na]/Δ[Glu] = −1.18 mmol/L per 5.6 mmol/L, and Ton2 = 335.6 mOsm/L. Another hypothetical patient with TBW = 40 L, α1 = 4.0, ECFV1 = 8 L, and glucose load 800 (8 ×100) mmol represents severe hypovolemia. In this patient, [Glu]2 = 83.8 mmol/L (1,509 mg/dL), [Na]2 = 111.1 mmol/L, Δ[Na]/Δ[Glu] = −2.10 mmol/L per 5.6 mmol/L, and Ton2 = 305.6 mOsm/L.
Although the rises in tonicity are progressively equal at progressive equal glucose loads, the increases in the [Glu] and the decreases in both [Na] and Δ[Na]/Δ[Glu] decrease progressively (38). The change in Δ[Na]/Δ[Glu] at progressive hyperglycemia is very gradual. For example, in the hypothetical patient with α1 = 2.0, the rate of rise in tonicity for each 100 mmol/L of [Glu]A is 33.3 mOsm/L; for the first [Glu]A of 100 mmol/L, [Glu] rises by 81.7 mmol/L from 5.6 to 87.3 mmol/L and [Na] decreases by 24.2 mmol/L from 140 to 115.8 mmol/L for a Δ[Na]/Δ[Glu] of −1.66 mmol/L per 5.6 mmol/L; for a subsequent [Glu]A of another 100 mmol/L, [Glu] rises by 60.2 mmol/L from 87.3 to 149.1 mmol/L (2,684 mg/dL), and [Na] decreases by 14.2 mmol/L from 115.8 to 101.6 mmol/L for a Δ[Na]/Δ[Glu] of −1.29 mmol/L per 5.6 mmol/L in this step and −1.50 mmol/L per 5.6 mmol/L from the euglycemic state.
In addition to the degree of hyperglycemia and the volume ratio α1, the following potential sources of variations in Δ[Na]/Δ[Glu] and therefore in corrected [Na] have been discussed: (a) differences in the apparent volumes of distribution of glucose and sodium due to intracellular entry of sodium ions, which is larger in some chronic illnesses than in the normal state (17), or to absence of insulin requirement for glucose uptake by the cells of certain organs, which was the source of the proposed value of −1.35 mmol/L per 5.6 mmol/L for Δ[Na]/Δ[Glu] (37). The difference in corrected [Na]2 values calculated with Δ[Na]/Δ[Glu] values of −1.6 and −1.35 mmol/L per 5.6 mmol/L has little clinical significance even in extreme hyperglycemia. For example, if [Na]1 is 140 mmol/L at a [Glu]1 of 5.6 mmol/L (100 mg/dL), [Na]2 values calculated, respectively by Δ[Na]/Δ[Glu] of −1.6 and −1.35 mmol/L per 5.6 mmol/L are 109.6 and 114.4 mmol/L if [Glu] rises to 111.1 mmol/L (2,000 mg/dL). (b) Differences in sodium concentration between plasma and interstitial compartment due to Gibbs-Donnan equilibrium between these two sub-compartments of the ECFV (17); (c) Exit of potassium from cells and changes in intracellular solute during development of hyperglycemia (39). Potassium transfer may alter the value of Δ[Na]/Δ[Glu], but this effect will be small in hyperglycemia developing in a closed system (17). (d) Sodium exchanges between sodium stores in proteoglycans (mainly glycosaminoglycan) in skin, bones and cartilage (42) and sodium in the extracellular compartment, which could influence Δ[Na]/Δ[Glu], but have not been studied in hyperglycemia (17).
Finally, both definition and methods of measurement of ECFV encounter difficulties (43, 44). Nevertheless, the average normal ECFV is in the order of 40–45% of TBW, not 33% as in Katz's calculations (43, 44), and therefore at euvolemia the ICFV1/ECFV1 volume ratio α1 should be between 1.50 and 1.22, not 2.00. For a [Glu]A of 100 mmol/L, Δ[Na]/Δ[Glu] is −1.44 mmol/L per 5.6 mmol/L if α1 is 1.22. In a patient presenting with [Glu]2 = 105.6 mmol/L (1,100 mg/dL) and [Na]2 = 111.4 mmol/L, the corrected [Na], for Δ[Glu] = 100 mmol/L, is 140.0 mmol/L using the Katz coefficient for Δ[Na]/Δ[Glu] of −1.6 and 137.1 mmol/L using a Δ[Na]/Δ[Glu] of −1.22 mmol/L per 5.6 mmol/L. The difference between these corrected [Na] values has minimal clinical significance.
Closed System of Hyperglycemia: Clinical Observations
Hyperglycemia in patients with advanced renal failure allows study of the theoretical predictions in a closed system because it can be treated with insulin infusion and with no or minimal changes in the external balance of sodium, potassium and water (45, 46). One report analyzed 43 episodes of severe hyperglycemia ([Glu] > 33.3 mmol/L or 600 mg/dL), treated with insulin and no other interventions, in patients on chronic dialysis with no or minimal fluid intake and urine loss and no change in body weight during treatment (47). Mean ± standard deviation values at presentation and end of observation, respectively, were as follows: [Glu] 50.7 ± 10.9 mmol/L (913 ± 197 mg/dL) and 9.4 ± 4.3 mmol/L (170 ± 78 mg/dL); [Na] 125 ± 5 and 136 ± 5 mmol/L; and Ton 300 ± 13 and 282 ± 11 mOsm/L. Δ[Na]/Δ[Glu] was −1.50 ± 0.22 mmol/L per 5.6 mmol/L. In a review, which analyzed 148 hyperglycemic episodes in patients on dialysis, Δ[Na]/Δ[Glu] was −1.61 ± 0.36 mmol/L per 5.6 mmol/L (48). In both reports, a small number of patients with pronounced edema had Δ[Na]/Δ[Glu] values numerically much lower than 1.6 per 5.6 mmol/L in agreement with theoretical predictions (47, 48); no episode of extreme hyperglycemia with Δ[Na]/Δ[Glu] substantially different from 1.6 mmol/L per 5.6 mmol/L was observed.
Another study analyzed the relationship between [Glu] and [Na] by linear regression in 208 patients on dialysis who had at least three measurements of [Glu] and [Na] and a difference between the lowest and highest value of [Glu] exceeding 16.7 mmol/L (300 mg/dL). In this study, the 5th−95th percentile range of [Glu] was 4.5–30.5 mmol/L (81–549 mg/dL) and Δ[Na]/Δ[Glu] was −1.47±0.82 mmol/L per 5.6 mmol/L (49).
The reports of hyperglycemic episodes in dialysis patients provided support for Katz's Δ[Na]/Δ[Glu] coefficient of −1.6 mmol/L per 5.6 mmol/L for computation of the corrected [Na] in oligoanuric patients. Hyperglycemic episodes in patients with preserved renal function represent a different entity. The next section addresses these patients.
Open System of Hyperglycemia: Theoretical Calculations
Severe hyperglycemia in patients with preserved renal function causes deficits in body sodium, potassium, and water, which are the key determinants of [Na] at euglycemia (50). Balance abnormalities specific to hyperglycemia develop from water gain in the gastrointestinal tract and losses of water, sodium and potassium from the urinary tract. Thirst is caused by hyperglycemic hypertonicity and hypovolemia from urinary losses. Hyperglycemic hypertonicity caused thirst in animal experiments (51). Polydipsia is a prominent clinical manifestation of hyperglycemic crises (7, 52, 53). Water intake from hyperglycemia led to hyponatremia after correction with insulin of approximately one-third of the hyperglycemic episodes in dialysis patients (47).
A major rise in tonicity in hyperglycemia results from osmotic diuresis, in which water loss is relatively greater than loss of sodium plus potassium (5, 17, 54). Thus, in hyperglycemic crises occurring in patients with preserved renal function, who represent an open system, [Na] receives influences from three pathophysiologic processes: rise in [Glu] and water gain cause [Na] decreases, while osmotic diuresis causes [Na] increase. In these patients, quantitating the isolated effect of glucose gain is imperative because this effect is predictable with a reasonable degree of certainty, as shown in the previous section, and more importantly, it will disappear with correction of hyperglycemia without requiring additional measures.
Prediction of the quantitative effects of water intake and particularly of osmotic diuresis, which is the dominant effect on tonicity in severe hyperglycemic episodes (2, 3), is difficult because the magnitude of these processes varies greatly (2, 17). The effects of osmotic diuresis on [Na] require correction by fluid infusion. One report calculated the effects of osmotic diuresis on tonicity-related values in a hypothetical subject with extreme hyperglycemia ([Glu] of 137.9 mmol/L or 2,483 mg/dL) by using the average reported values of urinary water loss (25% of baseline body water) and urinary sodium plus potassium concentration (60 mmol/L) in severe hyperglycemia (17). In this hypothetical subject, [Na] after correction of hyperglycemia calculated from the body contents of water, glucose, sodium, and potassium at baseline and after their changes from osmotic diuresis, was 167 mmol/L, while corrected [Na] calculated by the Al-Kudsi formula was 169 mmol/L. This finding suggests that the corrected [Na] by the Al-Kudsi formula provides a reasonable prediction of the part of hypertonicity that is due to osmotic diuresis.
Open System of Hyperglycemia: Clinical Observations
Accounting for changes in external balances of water, sodium, and potassium during development and treatment of hyperglycemia is necessary for any evaluation of the corrected [Na] in patients with renal function. There is a paucity of studies in this area. In a prospective study, hyperglycemia was produced in normal volunteers by infusion of somatostatin and 20% dextrose in 0.45% saline and the relation between [Glu] and [Na] was evaluated by linear regression (55); the overall slope of Δ[Na]/Δ[Glu was −2.4 mmol/L per 5.6 mmol/L. However, in a piecewise linear regression, this slope was −1.6 mmol/L per 5.6 mmol/L up to a [Glu] of 24.4 mmol/L (440 mg/dL) and −4.0 mmol/L per 5.6 mmol/L between [Glu] levels of 24.4 and 44.4 mmol/L (440–800 mg/dL). These findings were used in the development of several guidelines (30–32).
Assuming baseline values of 5.6 mmol/L for [Glu], 140 mmol/L for [Na], and 285.6 mOsm/L for tonicity, we calculated Ton2 values in hyperglycemia using the Δ[Na]/Δ[Glu] slopes −1.6 and −4.0 per 5.6 mmol/L (53). A slope of −1.6 mmol/L per 5.6 mmol/L for [Glu] between 5.6 and 24.4 mmol/L, calculates at a [Glu]2 of 24.4 mmol/L a [Na]2 of 134.6 mmol/L and a Ton2 293.5 mOsm/L (formula 1). A slope of −4.0 mmol/L per 5.6 mmol/L between [Glu] 24.4 and 44.4 mmol/L, calculates at a [Glu]2 of 44.4 mmol/L a [Na]2 of 120.2 mmol/L and a Ton2 of 284.8 mOsm/L. According to these calculations, tonicity, after rising appropriately with [Glu] rising from 5.6 to 24.4 mmol/L, decreased to its baseline euglycemic level after further [Glu] rise to 44.4 mmol/L. As noted earlier, Δ[Na]/Δ[Glu] values numerically larger than 2.8 mmol/L per 5.6 mmol/L are inconsistent with the principle of rise in tonicity as [Glu] rises. Consequently, the Δ[Na]/Δ[Glu] value of −4.0 mmol/L per 5.6 mmol/L could not be the exclusive result of a rise in [Glu] from 24.4 to 44.4 mmol/L. Water gain added to development of hyperglycemia is the most probable mechanism for this Δ[Na]/Δ[Glu] value. Another prospective study of rising [Glu] in patients with diabetes mellitus computed a Δ[Na]/Δ[Glu of −1.50 mmol/L per 5.6 mmol/L (56).
Clinical Application of the Corrected [Na]: Definition of the Hyperglycemic Syndromes
The guidelines for hyperglycemic crises address diabetic ketoacidosis (DKA) and hyperosmolar hyperglycemic state (HHS) (1, 21–24). The diagnostic features of DKA include low arterial blood pH and serum bicarbonate, presence of ketone bodies in serum and urine, a wide serum anion gap, and variable tonicity (1). [Glu] > 13.9 mmol/L (250 mg/dL) or > 11.1 mmol/L (200 mg/dL) was also included in these criteria (1, 21). However, euglycemic DKA has become more frequent after the introduction of sodium glucose cotransporter 2 (SGLT-2) inhibitors in the treatment of diabetes mellitus (57).
The criteria for diagnosis of HHS include Ton ≥ 320 mOsm/L, [Glu] ≥ 33.3 mmol/L (600 mg/dL) and serum bicarbonate ≥ 15 mmol/L (1, 20, 58), or Ton ≥ 330 mOsm/L (59) and [Glu] ≥ 30 mmol/L (540 mg/dL) (27, 59). Hypertonicity may cause coma in hyperglycemic syndromes (60, 61). Ton ≥ 320 mOsm/L causes coma frequently (52), although neurological manifestations from hypertonicity may occur in hyperglycemic patients with lower tonicity levels (62, 63).
At equal levels of hyperglycemic hypertonicity, elevated [Na] indicates severe water deficit (64, 65). The corrected [Na] illustrates the difference in water deficit between high [Na] and high [Glu] in this case. For example, for a given Ton of 320 mOsm/L, if [Glu] is 30 mmol/L (540 mg/dL) [Na] will be 145 mmol/L and the corrected [Na], at 152 mmol/L, indicates a water deficit, relative to the effective solute state, of 8.5% (= {152/140} × 100), while if [Glu] is 90 mmol/L (1,620 mg/dL), [Na] will be 115 mmol/L and the corrected [Na], at 141 mmol/L, indicates essentially similar status of body water and body effective solute.
Clinical Application of the Corrected [Na]: Corrected [Na] in Hyperglycemic Syndromes
Table 2 shows presenting values for [Glu], [Na], tonicity, and corrected [Na] in reports of DKA (66–171), HHS (3, 9, 13, 75–78, 110, 129, 169, 172–251), and hyperglycemia in chronic kidney disease (CKD) stage V (12, 16, 47–49, 156, 171, 252–277), which was included in Table 2 as the control group because it causes limited or no water and electrolyte losses through osmotic diuresis. All but three of the cases in this last group were on maintenance dialysis. To show the range of the tonicity-related values, Table 2 includes studies as well as case reports. Reports of combined DKA and HHS were included in the DKA part of the table. Studies reporting median, instead of mean, tonicity-related values were not included in this table. The DKA category does not include reports on euglycemic DKA, while the HHS category contains six cases with Ton ≥ 320 mOsm/L and [Glu] between 17.2 mmol/L (310 mg/dL) and 30 mmol/L (540 mg/dL). The reason for including these cases was explained above. The corrected [Na] was calculated de novo in reports that did not calculate the corrected [Na] and was recalculated using the Al-Kudsi formula in reports that had used Δ[Na]/Δ[Glu] coefficients different from −1.6 mmol/L per 100 mg/dL to calculate it.
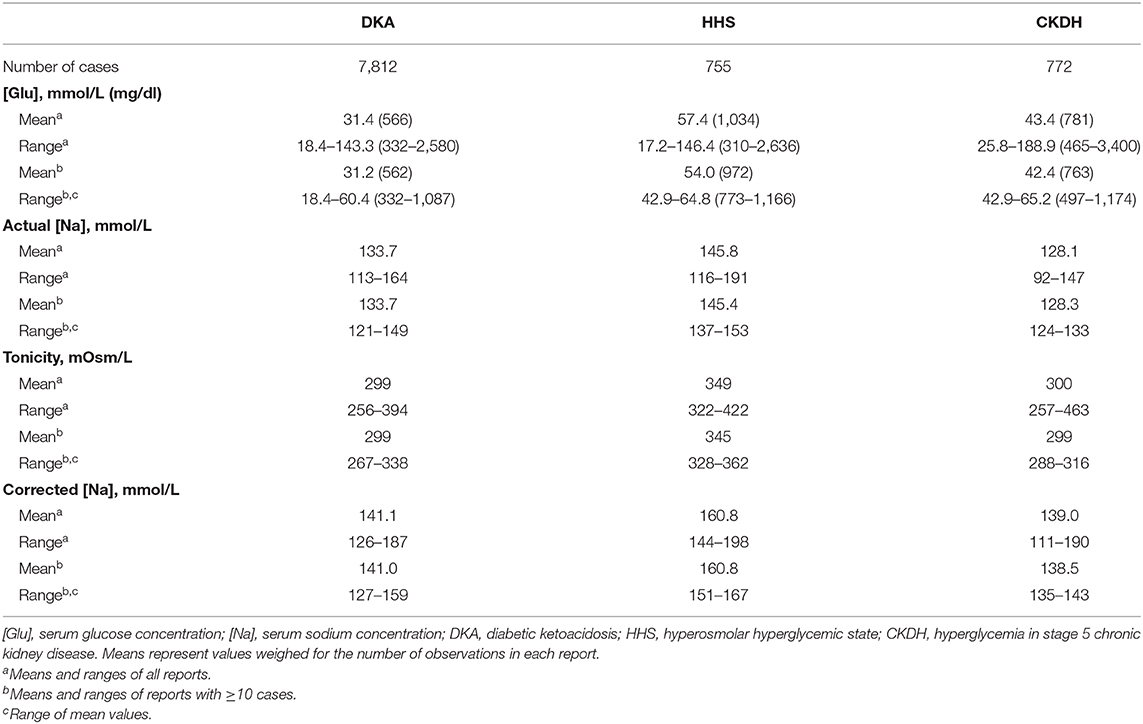
Table 2. Presenting serum glucose, sodium, tonicity, and corrected sodium levels in reported hyperglycemic crises.
In Table 2, there exists considerable overlap of [Glu], [Na] tonicity, and corrected [Na] ranges in the three categories of hyperglycemia. DKA combined with HHS occurred in many instances. The term Diabetic Hyperosmolar Ketoacidosis (DHKA) was proposed for DKA combined with HHS (52). Patients on dialysis who presented with hyperglycemia and elevated corrected [Na] have usually lost hypotonic fluids through hemodialysis (252), or peritoneal dialysis (253–256, 274), with high glucose concentration in the dialysate.
The second important finding in Table 2 is in the mean corrected [Na] values. In DKA the overall mean corrected [Na] was within the normal range of [Na] (137–143 mmol/L). Among DKA series with ≥10 cases, mean corrected [Na] was in the eunatremic range in 45 series (66–69, 71, 74, 75, 80, 83, 84, 90, 91, 93, 104–106, 108, 109, 112, 113, 118, 120, 122, 125–127, 129, 135, 137, 138, 140, 143–146, 151, 155, 156, 158, 160, 163–165, 168–171) reporting 6,355 episodes, including the pivotal PECARN study in which mean corrected [Na] was 140.8 mmol/L in one study group and 140.7 mmol/L in each of the other three study groups (163); and in the hypernatremic range in the remaining 18 series (74, 76, 78, 80, 85, 91, 93, 98, 100–102, 104, 116, 120, 129, 133, 139, 169) reporting 1,301 episodes. Thus, although many patients have water deficits in excess of sodium and potassium deficits, an equal or even larger number of patients do not have excessive water deficits at presentation with DKA. This finding has important consequences in the choice of the tonicity of replacement solutions. In the HHS group, the mean corrected [Na], at > 160 mmol/L, is in the range of [Na] that constitutes a medical emergency (7). Mean corrected [Na] was in the eunatremic range in hyperglycemia of patients with CKD stage 5.
Discussion
Importance of the Corrected [Na] in Hyperglycemic Syndromes
Preventing cerebral edema is a key concern during treatment of hyperglycemic crises. Tonicity-related parameters have received attention in the studies of the pathogenesis of this complication. In 100 cases of cerebral edema developing during treatment of DKA, weighed means at presentation were as follows: [Glu] 34 mmol/L (612 mg/dL), [Na] 132.4 mmol/L, tonicity 299 mOsm/L, and corrected [Na] 140.6 mmol/L (72, 73, 94–96, 98, 102, 106, 109, 112, 117, 123, 124, 126, 131, 138). These values do not differ substantially from the mean values of all DKA cases in Table 2. However, factors related to tonicity statistically associated with brain edema during treatment of DKA include decrease in tonicity, large early infusion volumes, very high [Glu] at presentation, rapid decline in [Glu], very low [Na], and administration of large doses of insulin (102, 106, 118). The change in corrected [Na] during treatment of DKA was the best discriminator for the development of severe coma in one study (126). Deterioration of neurological manifestations associated with substantial rises of the corrected [Na] has been reported during treatment of both DKA (2, 126) and HHS (205, 223, 231).
Other reported factors associated with cerebral edema in DKA include the degree of acidosis (96, 106, 118, 278), high levels of blood urea at presentation (118, 277), and vasogenic factors (112). One study found no effect of the rate of replacement fluid infusion (138). The PECARN study found no significant differences in neurological manifestations during and following treatment of DKA between using 0.9% saline and 0.45% saline as replacement solutions and between fast and slow initial infusion rates (163). Vascular endothelial changes caused by elevated blood levels of cytokines and chemokines secondary to inflammatory status associated with DKA were proposed by the authors of the PECARN study as the main mechanism for the development of cerebral edema.
High value of corrected [Na] at presentation with DKA is associated with increased incidence and severity of acute kidney injury (AKI) (168, 279). Weighed mean values at presentation with DKA and AKI were 36.9 mmol/L (665 mg/dL) for [Glu], 135.5 mmol/L for actual [Na], 312 mOsm/L for tonicity, and 146.7 mmol/L for corrected [Na] in 93 patients (2, 92, 97, 121, 134, 142, 143, 158, 168). AKI occurs frequently in HHS (3, 213, 217, 218, 223, 227, 244).
Management of Hyperglycemic Hypertonicity
Tonicity Targets During Treatment of Hyperglycemic Crises
Attention to tonicity plays a role in prevention of severe neurological manifestations during treatment of hyperglycemic emergencies. Decrease in tonicity from extracellular solute loss leads to osmotic entry of fluid into cells and could contribute to the development of cerebral edema (278). For this reason, one report proposed a very slow decrease in tonicity during the early stages of treatment (280). The optimal rate of decline in tonicity, however, has not been clarified. The proposed guideline for the maximal rate of decline in osmolality (tonicity should be targeted instead of total osmolality) during treatment of hyperglycemia is 3 mOsm/kg hourly (22, 23). One set of guidelines proposed a 3–8 mOsm/L range in hourly rate of decrease in tonicity (31).
The change in tonicity due exclusively to correction of hyperglycemia has two components, a fall in [Glu] and a rise in [Na]. Guidelines propose hourly rates of 2.8–4.2 mmol/L (50–75 mg/dL) for the decrease in [Glu] (1) and of 0.5 mmol/L for the commensurate rise in [Na] (30). Note that Katz's Δ[Na]/Δ[Glu] coefficient predicts a 0.8–1.2 mmol/L increase in [Na] for each 2.8–4.2 mmol/L decrease in [Glu].
Use of the Corrected [Na] During Treatment of Hyperglycemic Crises
The corrected [Na] predicts the relation between effective body solute and total body water after decrease of [Glu] to its desired level (2, 17) and should be used as a guide for the composition of replacement solutions in the same fashion as actual [Na] values are used to guide fluid management of dysnatremias (7, 281–285). Evidence presented earlier supports the use of the Al-Kudsi formula for calculation of the corrected [Na]. Two limitations of the corrected [Na] should be addressed during treatment: First, the corrected [Na] using the Al-Kudsi formula is not accurate in some conditions, mainly in advanced extracellular volume disturbances. Second, and more importantly, the corrected [Na] reflects the relation between effective body solute and body water at the moment of blood sampling (2, 17, 36). Correction of the extracellular volume deficit improves renal function and in the face of persistent hyperglycemia leads to large volume osmotic diuresis, which causes further water deficit and rises in the corrected [Na] (2).
We propose the following scheme for use of the corrected [Na] during treatment of hyperglycemic crises: The initial measurement of serum values should include osmolality in addition to basic metabolic panel. In the absence of an exogenous solute (e.g., ethanol) an osmol gap, that is the difference between measured osmolality and osmolarity calculated as 2 × [Na] + [Glu] + serum urea, where [Glu] and urea are in mmol/L, exceeding 10–12 mOsm/L indicates either presence of another non-dissociated compound in the serum (e.g., acetone) or a condition causing decrease in the water fraction of the serum (e.g., hyperlipidemia, hyperproteinemia). In the second case, falsely low [Na] values are reported when this measurement is performed in an autoanalyzer that requires dilution of the samples measured (286). If there is a large osmol gap, [Na] should be measured again in an apparatus that does not require dilution of the measured specimen, e.g., a blood gas machine, to obtain an accurate estimate of the presenting tonicity.
The tonicity of replacement solutions should be based on repeated calculations of the corrected [Na]. If the corrected [Na] at presentation is in the eunatremic range, infusion of isotonic saline should be started at a rate dictated by clinical manifestations of hypovolemia. Prevention of either decline or rise in the corrected [Na] is critical. Patients with corrected [Na] values within the normal range of [Na], like the average patient with DKA (Table 2), do not have relatively larger deficit of water compared to monovalent cations. In these patients, use of isotonic solutions as initial treatment of DKA and slow decline of [Glu], as proposed in the guidelines (1), leads to rapid correction of severe extracellular volume deficits and prevents sharp changes in the corrected [Na].
In subjects with initial corrected [Na] in the eunatremic range, tonicity should decline at a low rate. Maintenance of the corrected [Na] at the same level and decrease in [Glu] at the rate proposed in the guidelines (2.8–4.2 mmol/L hourly), will lead to a 0.8–1.2 mmol/L per hour rate of increase in [Na] and, from Equation (1), to a 1.2–1.8 mOsm/L per hour rate of decrease in tonicity. This conservative rate of decline in tonicity, which is slower than the hourly rate of 3 mOsm/L proposed in guidelines, may assist in preventing cerebral edema. In the rare instance of low presenting corrected [Na], or for treatment of cerebral edema, hypertonic saline infusion may be used (111). During treatment, urine volume should be monitored and [Glu], [Na], serum potassium concentration, and other relevant parameters should be measured frequently, initially every 1–2 h. The corrected [Na] should be calculated after each measurement of [Glu] and [Na] and should guide changes in the tonicity of the infusate. Development of large osmotic diuresis may lead to increases in the corrected [Na] and the need for hypotonic infusions later in the course of treatment.
A corrected [Na] in the hypernatremic range at presentation with hyperglycemia indicates excessive water deficit that must be corrected. Initially, infusion of isotonic fluids will correct rapidly volume deficits and will also decrease the level of hypertonicity. However, the subsequent development of large volume osmotic diuresis may lead to rise in the corrected [Na]. Monitoring urine volume, frequent measurement of the relevant serum biochemical values, and repeated calculation of the corrected [Na] after each measurement of [Glu] and [Na] is imperative. The corrected [Na] should not rise further; however, deciding whether it should remain at the same level at least early during the decrease in [Glu] or it should decrease at a slow rate (e.g., by 0.5 mmol/L every 1 or 2 h) from the start of the treatment requires prospective studies. Infusion of hypotonic solutions will eventually be needed regardless of whether the early phase of treatment aims at maintaining or decreasing the corrected [Na]. Addition of potassium salts to the infused saline should be guided by repeated measurements of the serum potassium concentration. In deciding the concentration of sodium in the replacement solutions, it is important to take into account the concentration of potassium salts in the infusate (2).
Conclusions
The corrected [Na] calculated by the Al-Kudsi formula should guide the tonicity of replacement solutions. This use should be tempered by the knowledge that rarely encountered extreme volume disturbances can cause [Na] changes substantially different from those predicted by the corrected [Na] and, more importantly, that the corrected [Na] can vary greatly during treatment depending on changes in the external balances of water, sodium and potassium. For these reasons, frequent measurements of [Glu] and [Na], repeated calculation of the corrected [Na] after each measurement, and changes in the tonicity of replacement solutions based on the corrected [Na] are critical steps in the management of tonicity issues in hyperglycemia.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data came from tables of publications cited in the text.
Author Contributions
TI: conceptualization. TI, KG, GB, CA, and AT: literature review. TI, GB, SL, and AT: methodology. SL, CA, and AT: visualization. TI and AT: writing-original draft preparation. KG, GB, SL, EA, and CA: writing-review and editing. All authors contributed to the article and approved the submitted version.
Funding
GB was supported by a Burrows Wellcome Fund Career Award for Medical Scientists (13030995) and NIH grant RO1 DK116964.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer DM declared a past co-authorship with several of the authors TI, AT, and CA to the handling editor.
Acknowledgments
The authors acknowledge Dialysis Clinic Inc. for supporting this work by covering publication expenses [DCI #C-3765].
References
1. Kitabchi AE, Umpierrez GE, Miles JM, Fisher JN. Hyperglycemic crises in adult patients with diabetes. Diabetes Care. (2009) 32:1335–43. doi: 10.2337/dc09-9032
2. Tzamaloukas AH, Sun Y, Konstantinov NK, Ing TS, Dorin RI, Malhotra D, et al. Principles of quantitative fluid and cation replacement in extreme hyperglycemia. Cureus. (2013) 5:e110. doi: 10.7759/cureus.110
3. Arieff AI, Carroll HJ. Nonketotic hyperosmolar coma with hyperglycemia: clinical features, pathophysiology, renal function, acid-base balance, plasma-cerebrospinal fluid equilibria and the effects of therapy in 37 cases. Medicine (Baltimore). (1972) 51:73–94. doi: 10.1097/00005792-197203000-00001
4. Popli S, Tzamaloukas AH, Ing TS. Osmotic diuresis-induced hypernatremia: better explained by solute-free water clearance or electrolyte-free water clearance. Int Urol Nephrol. (2013) 45:1687–92. doi: 10.1007/s11255-012-0353-3
5. Roumelioti M-E, Ing TS, Rondon-Berrios H, Glew RH, Khitan ZJ, Sun Y, et al. Principles of quantitative water and electrolyte replacement of losses from osmotic diuresis. Int Urol Nephrol. (2018) 50:1263–70. doi: 10.1007/s11255-018-1822-0
6. Feig PU, McCurdy DK. The hypertonic state. N Engl J Med. (1977) 297:1444–54. doi: 10.1056/NEJM197712292972608
7. Rondon-Berrios H, Argyropoulos C, Ing TS, Raj DS, Malhotra D, Agaba EI, et al. Hypertonicity: Clinical entities, manifestations and treatment. World J Nephrol. (2017) 6:1–13. doi: 10.5527/wjn.v6.i1.1
8. Rohrscheib M, Rondon-Berrios H, Argyropoulos C, Glew RH, Murata GH, Tzamaloukas AH. Indices of serum tonicity in clinical practice. Am J Med Sci. (2015) 349:537–44. doi: 10.1097/MAJ.0000000000000470
9. McCurdy DK. Hyperosmolar hyperglycemic nonketotic diabetic coma. Med Clin North Am. (1970) 54:683–99. doi: 10.1016/S0025-7125(16)32636-0
10. Goldman MH, Kashani M. Spurious hyponatremia in diabetic ketoacidosis with massive lipid elevations. J Med Soc N J. (1982) 9:591–2.
11. Tomkins AM, Dormandy TL. Osmolal pattern during recovery from diabetic coma. Lancet. (1971) 2:952–5. doi: 10.1016/S0140-6736(71)90271-6
12. Tzamaloukas AH, Levinstone AR, Gardner KD Jr. Hyperglycemia in advanced renal failure: sodium and water metabolism. Nephron. (1982) 31:40–4. doi: 10.1159/000182612
13. Baldrighi M, Sainaghi PP, Bellan M, Bartoli E, Castello LM. Hyperglycemic hyperosmolar state: a pragmatic approach to properly manage sodium derangements. Curr Diabetes Rev. (2018) 14:534–41. doi: 10.2174/1573399814666180320091451
14. Katz MA. Hyperglycemia-induced hyponatremia: calculation of the expected serum sodium depression. N Engl J Med. (1973) 289:843–4. doi: 10.1056/NEJM197310182891607
15. Goldberg M. Hyponatremia. Med Clin North Am. (1981) 65:251–69. doi: 10.1016/S0025-7125(16)31523-1
16. Al-Kudsi RR, Daugirdas JT, Ing TS, Kheirbek AO, Popli JE, Hano JE, et al. Extreme hyperglycemia in dialysis patients. Clin Nephrol. (1982) 17:228–31.
17. Tzamaloukas AH, Khitan Z, Glew RH, Roumelioti M-A, Rondon-Berrios H, Elisaf MS, et al. Serum sodium concentration and tonicity in hyperglycemic crises: major influences and treatment implications. J Am Heart Assoc. (2019) 8:e011786. doi: 10.1161/JAHA.118.011786
18. Welt LG. Clinical Disorders of Hydration and Acid-Base Balance. Boston, MA: Little, Brown and Company (1959).
19. Seldin DW, Tarail R. Effect of hypertonic solutions on metabolism and excretion of electrolytes. Am J Physiol. (1949) 59:160–74. doi: 10.1152/ajplegacy.1949.159.1.160
20. Rondon-Berrios H, Agaba EI, Tzamaloukas AH. Hyponatremia: pathophysiology, classification, manifestations and management. Int Urol Nephrol. (2014) 46:2153–65. doi: 10.1007/s11255-014-0839-2
21. Kitabchi AE, Umpierrez GE, Murphy MB, Barrett EJ, Kreisberg RA, Malone JI, et al. Management of hyperglycemic crises in patients with diabetes. Diabetes Care. (2001) 24:131–53. doi: 10.2337/diacare.24.1.131
22. Chiasson JL, Aris-Jilwan N, Bélanger R, Bertrand S, Beauregard H, Ekoé JM, et al. Diagnosis and treatment of diabetic ketoacidosis and the hyperglycemic hyperosmolar state. CMAJ. (2003) 168:859–66.
23. Diabetes Canada Clinical Practice Guidelines Expert Committee, Goguen J, Gilbert J. Hyperglycemic emergencies in adults. Can J Diabetes. (2018) 42:S109–14. doi: 10.1016/j.jcjd.2017.10.013
24. Wolfsdorf JI, Glaser N, Agus M, Fritsch M, Hanas R, Rewers A, et al. ISPAD Clinical Practice Consensus Guidelines 2018: Diabetic ketoacidosis and the hyperglycemic hyperosmolar state. Pediatr Diabetes. (2018) 19:155–77. doi: 10.1111/pedi.12701
25. Harris GD, Fiordalisi I, Harris WL, Mosovich LL, Fibeg L. Minimizing the risk of brain herniation during treatment of diabetic ketoacidemia: a retrospective and prospective study. J Pediatr. (1990) 117:22–31. doi: 10.1016/S0022-3476(05)82439-4
26. Harris GD, Fiordalisi I. Physiologic management of diabetic ketoacidemia. A 5-year prospective pediatric experience in 231 episodes. Arch Pediatr Adolesc Med. (1994) 48:1046–52. doi: 10.1001/archpedi.1994.02170100044009
27. Oster JR, Singer I. Hyponatremia, hyposmolality, and hypotonicity: tables and fables. Arch Intern Med. (1999) 159:333–6. doi: 10.1001/archinte.159.4.333
28. Milionis HJ, Liamis G, Elisaf MS. Appropriate treatment of hypernatraemia in diabetic hyperglycaemic hyperosmolar syndrome. J Intern Med. (2001) 249:273–6. doi: 10.1046/j.1365-2796.2001.0799a.x
29. Al-Mawed S, Pankratz VS, Chong K, Sandoval M, Roumelioti ME, Unruh M. Low serum sodium levels at hospital admission: outcomes among 2.3 million hospitalized patients. PLoS ONE. (2018) 13:e0194379. doi: 10.1371/journal.pone.0194379
30. Wolfsdorf J, Glaser N, Sperling MA; American Diabetes Association. Diabetic ketoacidosis in infants, children, and adolescents: a consensus statement from the American Diabetes Association. Diabetes Care. (2006) 29:1150–9. doi: 10.2337/dc06-9909
31. Scott A on behalf of the Joint British Diabetes Societies (JBDS) for Inpatient Care. Management of hyperosmolar hyperglycaemic state in adults with diabetes. Diabet Med. (2015) 32:714–24. doi: 10.1111/dme.12757
32. Spasovski G, Vanholder R, Allolio B, Annane D, Ball S, Bichet D, et al. Hyponatremia Guideline Development Group. Clinical practice guidelines on diagnosis and treatment of hyponatraemia. Eur J Clin Endocrinol. (2014) 170:G1–47. doi: 10.1530/EJE-13-1020
33. Liamis G, Liberopoulos E, Barkas F, Elisaf M. Diabetes mellitus and electrolyte disorders. World J Clin Cases. (2014) 16:488–96. doi: 10.12998/wjcc.v2.i10.488
34. Mohan S, Gu S, Parikh A, Radhakrishnan J. Prevalence of hyponatremia and association with mortality: results from NHANES. Am J Med. (2013) 126:1127–37.e1. doi: 10.1016/j.amjmed.2013.07.021
35. Pipeleers L, Wissing KM, Hilbrands R. Acid-base and electrolyte disturbances in patients with diabetes mellitus. Acta Clin Belg. (2019) 74:28–33. doi: 10.1080/17843286.2018.1546983
36. Palmer BF, Clegg DJ. Electrolyte and acid-base disturbances in patients with diabetes mellitus. N Engl J Med. (2015) 373:548–59. doi: 10.1056/NEJMra1503102
37. Roscoe JM, Halperin ML, Rolleston FS, Goldstein MB. Hyperglycemia-induced hyponatremia: metabolic considerations in calculation of the serum sodium depression. Can Med Assoc J. (1975) 112:452–3.
38. Robin AP, Ing TS, Lancaster GA, Soung LS, Sparagana M, Geis WP, et al. Hyperglycemia-induced hyponatremia: a fresh look. Clin Chem. (1979) 25:496–7. doi: 10.1093/clinchem/25.3.496a
39. Moran SM, Jamison RL. The variable hyponatremic response to hyperglycemia. West J Med. (1985) 142:49–53. doi: 10.1177/002581728505300407
40. Tzamaloukas AH, Kyner WT, Galley WR Jr. Determinants of osmotic phenomena created by an isolated change in extracellular solute in anuria. Min Electrolyte Metab. (1987) 13:117–25.
41. Darrow DC, Yannett H. The change in the distribution of body water accompanying increase and decrease in extracellular electrolyte. J Clin Invest. (1935) 14:266–75. doi: 10.1172/JCI100674
42. Sterns RH. Disorders of plasma sodium- causes, consequences and correction. N Engl J Med. (2015) 372:55–65. doi: 10.1056/NEJMra1404489
43. Bhave G, Neilson EG. Body fluid dynamics: back to the future. J Am Soc Nephrol. (2011) 22:2166–81. doi: 10.1681/ASN.2011080865
44. Roumelioti ME, Glew RH, Khitan ZJ, Rondon-Berrios H, Argyropoulos CP, Malhotra D, et al. Fluid balance concepts in medicine: principles and practice. World J Nephrol. (2018) 7:1–28. doi: 10.5527/wjn.v7.i1.1
45. Tzamaloukas AH, Ing TS, Siamopoulos KC, Raj DS, Elisaf MS, Rohrscheib M, et al. Pathophysiology and management of fluid and electrolyte disturbances in patients on chronic dialysis with severe hyperglycemia. Semin Dial. (2008) 21:431–9. doi: 10.1111/j.1525-139X.2008.00464.x
46. Sun Y, Roumelioti ME, Ganta K, Glew RH, Gibb J, Vigil D, et al. Dialysis-associated hyperglycemia: manifestations and treatment. Int Urol Nephrol. (2020) 52:505–17. doi: 10.1007/s11255-019-02373-1
47. Tzamaloukas AH, Rohrscheib M, Ing TS, Siamopoulos KC, Elisaf MF, Spalding CT. Serum tonicity, extracellular volume and clinical manifestations in symptomatic dialysis-associated hyperglycemia treated only with insulin. Int J Artif Organs. (2004) 27:751–8. doi: 10.1177/039139880402700904
48. Tzamaloukas AH, Ing TS, Siamopoulos KC, Rohrscheib M, Elisaf MS, Raj DSC, et al. Body fluid abnormalities in severe hyperglycemia in patients on chronic dialysis: review of published reports. J Diabetes Complications. (2008) 22:29–37. doi: 10.1016/j.jdiacomp.2007.06.012
49. Penne EL, Thijssen S, Raimann JG, Levin NW, Kotanko P. Correction of serum sodium for glucose concentration in hemodialysis patients with poor glucose control. Diabetes Care. (2010) 33:e91. doi: 10.2337/dc10-0557
50. Edelman IS, Leibman J, O'Meara MP, Birkenfeld LW. Interrelations between serum sodium concentration, serum osmolarity and total exchangeable sodium, total exchangeable potassium and total body water. J Clin Invest. (1958) 37:1236–56. doi: 10.1172/JCI103712
51. Fitzsimons JT. The physiological basis of thirst. Kidney Int. (1976) 10:3–11. doi: 10.1038/ki.1976.74
52. Nyenwe EA, Kitabchi AE. The evolution of diabetic ketoacidosis: an update of its etiology, pathogenesis and management. Metabolism. (2016) 65:507–21. doi: 10.1016/j.metabol.2015.12.007
53. Fayfman M, Pasquel FJ, Umpierrez GE. Management of hyperglycemic crises: diabetic ketoacidosis and hyperglycemic hyperosmolar state. Med Clin North Am. (2017) 101:587–606. doi: 10.1016/j.mcna.2016.12.011
54. Gennari FJ, Kassirer JP. Osmotic diuresis. N Engl J Med. (1974) 291:714–20. doi: 10.1056/NEJM197410032911408
55. Hillier TA, Abbott RD, Barrett EJ. Hyponatremia: evaluating the correction factor for hyperglycemia. Am J Med. (1999) 106:399–403. doi: 10.1016/S0002-9343(99)00055-8
56. Shalwitz RA, Gingerich RL, McGill JB, McDonald JM. Effect of hyperglycemia in plasma sodium and potassium concentration revisited. Clin Chem. (1991) 37:293–4. doi: 10.1093/clinchem/37.2.293a
57. Yu X, Zhang S, Zhang L. Newer perspectives of mechanisms for euglycemic diabetic ketoacidosis. Int J Endocrinol. (2018) 2018:7074868. doi: 10.1155/2018/7074868
58. Rosenbloom AL. Hyperglycemic hyperosmolar state: an emerging pediatric problem. J Pediatr. (2010) 156:180–4. doi: 10.1016/j.jpeds.2009.11.057
59. Zeitler P, Haqq A, Rosenbloom A, Glaser N, Drugs and Therapeutics Committee of the Lawson Wilkins Pediatric Endocrine Society. Hyperglycemic hyperosmolar syndrome in children: pathophysiological considerations and suggested guidelines for treatment. J Pediatr. (2011) 158:9–14, 14:e1–2. doi: 10.1016/j.jpeds.2010.09.048
60. Cruz-Caudillo JC, Sabatini S. Diabetic hyperosmolar syndrome. Nephron. (1995) 69:201–10. doi: 10.1159/000188458
61. Fulop M, Tannenbaum H, Dreyer N. Ketotic hyperosmolar coma. Lancet. (1973) 2:635–9. doi: 10.1016/S0140-6736(73)92478-1
62. Maccario M, Messis CP, Vastola EF. Focal seizures as a manifestation of hyperglycemia without ketoacidosis. A report of seven cases with review of the literature. Neurology. (1965) 15:195–206. doi: 10.1212/WNL.15.3.195
63. Miloy P. Hyperosmolar non-ketotic coma in acromegaly. JAMA. (1972) 222:814–6. doi: 10.1001/jama.1972.03210070044013
64. Daugirdas JT, Kronfol NO, Tzamaloukas AH, Ing TS. Hyperosmolar coma: cellular dehydration and the serum sodium concentration. Ann Intern Med. (1989) 110:855–7. doi: 10.7326/0003-4819-110-11-855
65. Elisaf M, Papagalanis ND, Siamopoulos KC. The importance of serum sodium in the symptomatology of hyperglycemic-induced hypertonicity. J Nephrol. (1993) 6:202–5.
66. Greenman L, Mateer FM, Gow RC, Peters JH, Danowski TS. Some observations on the development of hypokaliemia during therapy of diabetic acidosis in juvenile and young adult subjects. J Clin Invest. (1949) 28:409–14. doi: 10.1172/JCI102084
67. Seldin DW, Tarail W. The metabolism of glucose and electrolytes in diabetic acidosis. J Clin Invest. (1950) 29:552–65. doi: 10.1172/JCI102293
68. Cohen AS, Vance VK, Runyan JW, Hurwitz D. Diabetic acidosis: an evaluation of the cause, course and therapy of 73 cases. Ann Intern Med. (1960) 52:55–86. doi: 10.7326/0003-4819-52-1-55
69. Shaw CE, Hurwitz GE, Schmukler M, Brager SH, Bessman SP. A clinical and laboratory study of insulin dosage in diabetic acidosis: comparison with small and large doses. Diabetes. (1962) 11:23–30.
70. Abramson E, Arky R. Diabetic acidosis with initial hypokalemia: therapeutic implications. JAMA. (1966) 196:401–3. doi: 10.1001/jama.1966.03100180069021
72. Young E, Bradley R. Cerebral edema with irreversible coma in severe diabetic ketoacidosis. N Engl J Med. (1967) 276:665–9. doi: 10.1056/NEJM196703232761204
73. Hayes TM, Woods CJ. Unexpected death during treatment of uncomplicated diabetic ketoacidosis. Br Med J. (1968) 4:32–33. doi: 10.1136/bmj.4.5622.32
74. Beigelman PM. Severe diabetic ketoacidosis (diabetic “coma”). 482 episodes in 257 patients; experience of three years. Diabetes. (1971) 20:490–500. doi: 10.2337/diab.20.7.490
75. Gerich JE, Martin MM, Recant L. Clinical and metabolic characteristics of the hyperosmolar nonketotic coma. Diabetes. (1971) 20:228–38. doi: 10.2337/diab.20.4.228
76. Alberti KG, Hockaday TD, Turner RC. Small doses of intramuscular insulin in the treatment of diabetic “coma”. Lancet. (1973) 2:515–22. doi: 10.1016/S0140-6736(73)92346-5
77. Ginsberg-Fellner F, Primack WA. Recurrent hyperosmolar nonketotic episodes in a young diabetic. Am J Dis Child. (1975) 129:240–3. doi: 10.1001/archpedi.1975.02120390066014
78. Keller U, Berger W, Ritz E, Truog P. Course and prognosis of 86 episodes of diabetic coma. A five year experience with a uniform schedule of treatment. Diabetologia. (1975) 11:93–100. doi: 10.1007/BF00429830
79. Powner D, Snyder JV, Grenvik A. Altered pulmonary capillary permeability complicating recovery from diabetic ketoacidosis. Chest. (1975) 68:253–6. doi: 10.1378/chest.68.2.253
80. Kitabchi AE, Young R, Sacks H, Morris L. Diabetic ketoacidosis: reappraisal of therapeutic approach. Annu Rev Med. (1979) 30:339–57. doi: 10.1146/annurev.me.30.020179.002011
81. Sprung CL, Rackow EC, Fein IH. Pulmonary edema; a complication of diabetic ketoacidosis. Chest. (1980) 77:687–8. doi: 10.1378/chest.77.5.687
82. Russell Follansbee S, Matthay M. Adult respiratory distress syndrome complicating diabetic ketoacidosis. West J Med. (1981) 135:148–50.
83. Adrogué HJ, Wilson H, Boyd AE III, Suki WN, Eknoyan G. Plasma acid-base patterns in diabetic ketoacidosis. N Engl J Med. (1982) 307:1603–10. doi: 10.1056/NEJM198212233072603
84. Foster DW, McGarry JD. The metabolic derangements and treatment of diabetic ketoacidosis. N Engl J Med. (1983) 309:159–69. doi: 10.1056/NEJM198307213090307
85. Japan and Pittsburgh Childhood Diabetes Research Groups. Coma at the onset of young insulin-dependent diabetes in Japan. Diabetes. (1985) 34:1241–6. doi: 10.2337/diab.34.12.1241
86. Krane EJ, Rockoff MA, Wallman JK, Wolfsdorf JI. Subclinical brain swelling in children during treatment of diabetic ketoacidosis. N Engl J Med. (1985) 312:1147–51. doi: 10.1056/NEJM198505023121803
87. Breidbart S, Singer L, St. Louis Y, Saenger P. Adult respiratory distress syndrome in an adolescent with diabetic ketoacidosis. J Pediatr. (1987) 111:736–8. doi: 10.1016/S0022-3476(87)80256-1
88. Dorin EI, Crapo LM. Hypokalemic respiratory arrest in diabetic ketoacidosis. JAMA. (1987) 257:1517–8. doi: 10.1001/jama.257.11.1517
89. Leventhal RI, Goldman JM. Immediate plasma potassium levels in treating diabetic ketoacidosis. Arch Intern Med. (1987) 147:1501–2. doi: 10.1001/archinte.147.8.1501
90. Duck S, Wyatt D. Factors associated with brain herniation in the treatment of diabetic ketoacidosis. J Pediatr. (1988) 113:10–4. doi: 10.1016/S0022-3476(88)80521-3
91. Adrogué HJ, Barrero J, Eknoyan G. Salutary effects of modest fluid replacement in the treatment of adults with diabetic ketoacidosis. Use in patients without extreme volume deficit. JAMA. (1989) 262:2108–13. doi: 10.1001/jama.1989.03430150076029
92. Kinoshita O, Masuda I, Suzuki M, Tsushima M, Nishioeda Y, Matsuyama T, et al. A case of diabetic non-ketotic hyperosmolar coma with an increase with plasma 3-hydroxybutyrate. Endocrinol Jpn. (1991) 38:465–70. doi: 10.1507/endocrj1954.38.465
93. Wachtel TJ, Tetu-Mouradjian LM, Goldman DL, Ellis SE, O'Sullivan PS. Hyperosmolarity and acidosis in diabetes mellitus: a three-year experience in Rhode Island. J Gen Intern Med. (1991) 6:492–502. doi: 10.1007/BF02598216
94. Couch R, Acott W, Wong G. Early onset of fatal cerebral edema in diabetic ketoacidosis. Diabetes Care. (1991) 14:78–9. doi: 10.2337/diacare.14.1.78b
95. Glasgow A. Devastating cerebral edema in diabetic ketoacidosis before therapy. Diabetes Care. (1991) 14:77–8. doi: 10.2337/diacare.14.1.77
96. Durr JA, Hoffman WH, Sklar AH, El Gammal T, Steinhart CM. Correlates of brain edema in uncontrolled IDDM. Diabetes. (1992) 41:627–32. doi: 10.2337/diab.41.5.627
97. Murdoch IA, Pryor D, Haycock GB, Cameron SJ. Acute renal failure complicating diabetic ketoacidosis. Acta Paediatr. (1993) 82:498–500. doi: 10.1111/j.1651-2227.1993.tb12734.x
98. Mel J, Werther G. Incidence and outcome of diabetic cerebral oedema in childhood: are there predictors? J Paediatr Child Health. (1995) 31:17–20. doi: 10.1111/j.1440-1754.1995.tb02905.x
99. Young MC. Simultaneous acute cerebral and pulmonary edema complicating diabetic ketoacidosis. Diabetes Care. (1995) 18:1288–90. doi: 10.2337/diacare.18.9.1288
100. Elisaf MS, Tsatsoulis AA, Katopodis KP, Siamopoulos KC. Acid-base and electrolyte disturbances in patients with diabetic ketoacidosis. Diabetes Res Clin Pract. (1996) 34:23–7. doi: 10.1016/S0168-8227(96)01332-0
101. Caputo DG, Villarejo F, Valle CB, Diaz Aquiar P, Apeztequia CJ. Hidratation en la cetoacidosis diabetic. Como influye ls velocidad de infusion? (Hydration in diabetic ketoacidosis. What is the effect of infusion rate?). Medicina. (1997) 57:15–20.
102. Hale PM, Rezvani I, Braunstein AW, Lipman TH, Martinez N, Garibaldi L. Factors predicting cerebral edema in young children with diabetic ketoacidosis and new onset type I diabetes. Acta Paediatr. (1997) 86:626–31. doi: 10.1111/j.1651-2227.1997.tb08946.x
103. Hosclaw DS Jr, Torcato B. Acute pulmonary edema in juvenile diabetic ketoacidosis. Pediatr Pulmonol. (1997) 24:438–43.
104. Umpierrez GE, Kelly JP, Navarrette JE, Kaslas MM, Kitabchi AE. Hyperglycemic crises in urban blacks. Arch Intern Med. (1997) 157:669–75. doi: 10.1001/archinte.1997.00440270117011
105. Balasubramanyam A, Zern JW, Hyman DJ, Pavlik V. New profiles of diabetic ketoacidosis: type 1 vs type 2 diabetes and the effects of ethnicity. Arch Intern Med. (1999) 159:2317–22. doi: 10.1001/archinte.159.19.2317
106. Mahoney CP, Vlcek BW, DelAquila M. Risk factors for developing brain herniation during diabetic ketoacidosis. Pediatr Neurol. (1999) 21:721–7. doi: 10.1016/S0887-8994(99)00079-X
107. Vanelli M, Chiari G, Ghizzoni L, Capuano C, Bonetti L, Costi G, et al. Ketoacidosis and hyperosmolarity as first symptoms of type 1 diabetes mellitus following ingestion of high-carbohydrate-containing fluids. J Pediatr Endocrinol Metab. (1999) 12:691–4. doi: 10.1515/JPEM.1999.12.5.691
108. Wagner A, Risse A, Brill HL, Wienhausen-Wilke V, Rottmann M, Sondern K, et al. Therapy of severe diabetic ketoacidosis. Zero-mortality under very-low-dose insulin application. Diabetes Care. (1999) 22:674–7. doi: 10.2337/diacare.22.5.674
109. Azzopardi J, Gatt A, Zammit A, Alberti G. Lack of evidence of cerebral oedema in adults treated for diabetic ketoacidosis with fluids of different tonicity. Diabetes Res Clin Pract. (2002) 57:87–92. doi: 10.1016/S0168-8227(02)00019-0
110. Hollander AS, Olney RC, Blackett PR, Marshall BA. Fatal malignant hyperthermia-like syndrome with rhabdomyolysis complicating the presentation of diabetes mellitus in adolescent males. Pediatrics. (2003) 111:1447–52. doi: 10.1542/peds.111.6.1447
111. Kamat P, Vats A, Gross M, Checchia PA. Use of hypertonic saline for the treatment of altered mental status associated with diabetic ketoacidosis. Pediatr Crit Care Med. (2003) 4:239–42. doi: 10.1097/01.PCC.0000059340.19010.CE
112. Glaser NS, Wooton-Gorges SL, Marcin JP, Buonocore MH, Dicarlo J, Neely EK, et al. Mechanism of cerebral edema in children with diabetic ketoacidosis. J Pediatr. (2004) 145:164–71. doi: 10.1016/j.jpeds.2004.03.045
113. Newton CA, Raskin P. Diabetic ketoacidosis in type 1 and type 2 diabetes mellitus: clinical and biochemical differences. Arch Intern Med. (2004) 164:1925–31. doi: 10.1001/archinte.164.17.1925
114. McDonnell CM, Pedreira CC, Vadamalayan B, Cameron FJ, Werther GA. Diabetic ketoacidosis, hyperosmolarity and hypernatremia: are high-carbohydrate drinks worsening initial presentation? Pediatr Diabetes. (2005) 6:90–4. doi: 10.1111/j.1399-543X.2005.00107.x
115. Murthy K, Harrington JT, Siegel RD. Profound hypokalemia in diabetic ketoacidosis: a therapeutic challenge. Endocr Pract. (2005) 11:331–4. doi: 10.4158/EP.11.5.331
116. Yordam N, Gönç EN, Kandemir N, Alikaşifoglu A, Ozön A. Ten-year experience in management of diabetic ketoacidosis and ketosis: 140 episodes at pediatric age. Turk J Pediatr. (2005) 47:334–8.
117. Dixon AN, Jude EB, Banerjee AK, Bain SC. Simultaneous pulmonary and cerebral oedema, and multiple CNS infarctions as complications of diabetic ketoacidosis: a case report. Diabet Med. (2006) 23:571–3. doi: 10.1111/j.1464-5491.2006.01822.x
118. Edge JA, Jakes RW, Roy Y, Hawkins M, Winters D, Ford-Adams ME, et al. The UK case-control study of cerebral oedema complicating diabetic ketoacidosis in children. Diabetologia. (2006) 49:2002–9. doi: 10.1007/s00125-006-0363-8
119. Bhowmick SK, Hundley OT, Rettiq KR. Severe hypernatremia and hyperosmolality exacerbated by an herbal preparation in a patient with diabetic ketoacidosis. Clin Pediatr. (Phila). (2007) 46:831–4. doi: 10.1177/0009922807303042
120. Hoorn EJ, Carlotti AP, Costa LA, MacMahon B, Bohn G, Zietse R, et al. Preventing a drop in effective plasma osmolality to minimize the likelihood of cerebral edema during treatment of children with diabetic ketoacidosis. J Pediatr. (2007) 150:467–73. doi: 10.1016/j.jpeds.2006.11.062
121. Al-Matrafi J, Vethamuthu J, Feber J. Severe acute renal failure in a patient with diabetic ketoacidosis. Saudi J Kidney Dis Transpl. (2009) 20:831–4.
122. Toledo JD, Modesto V, Peinador M, Alvarez P, López-Prats JL, Sanchis R, et al. Sodium concentration in rehydration fluids for children with ketoacidotic diabetes: effect on serum sodium concentration. J Pediatr. (2009) 154:895–900. doi: 10.1016/j.jpeds.2008.12.042
123. Eid YM, Abdel Salam MM. Diabetic ketoacidosis presenting with emphysematous pyelonephritis. J Diabetes Complications. (2010) 24:214–6. doi: 10.1016/j.jdiacomp.2008.12.010
124. Seewi O, Vierzig A, Roth B, Schönau E. Symptomatic cerebral oedema during treatment of diabetic ketoacidosis: effect of adjuvant infusion. Diabetol Metab Syndr. (2010) 2:56. doi: 10.1186/1758-5996-2-56
125. Deeter KH, Roberts JS, Bradford H, Richards T, Shaw K, Marro K, et al. Hypertension despite dehydration during severe pediatric diabetic ketoacidosis. Pediatr Diabetes. (2011) 12:295–301. doi: 10.1111/j.1399-5448.2010.00695.x
126. Durward A, Ferguson LP, Taylor IA, Tibby SM. The temporal relationship between glucose-corrected serum sodium and neurological status in severe diabetic ketoacidosis. Arch Dis Child. (2011) 96:50–7. doi: 10.1136/adc.2009.170530
127. Savaş-Erdeve S, Berberoglu M, Oygar P, Siklar Z, Kendirli T, Hacihamdioglu B, et al. Efficiency of fluid treatments with different sodium concentration in children with type 1 diabetic ketoacidosis. J Clin Res Pediatr Endocrinol. (2011) 3:149–53. doi: 10.4274/jcrpe.v3i3.29
128. Abdulaziz S, Dabbagh O, Al Daker MO, Hassan I. Hypokalaemia and refractory asystole complicating diabetic ketoacidosis, lessons for prevention. BMJ Case Rep. (2012) 2012:bcr-2012–007312. doi: 10.1136/bcr-2012-007312
129. Anthanont P, Khawcharoenporn T, Tharavanij T. Incidences and outcomes of hyperglycemic crises in Thailand. J Med Assoc Thai. (2012) 95:995–1002.
130. Cebeci AN, Güven A. Delirium in diabetic ketoacidosis: a case report. J Clin Res Pediatr Endocrinol. (2012) 4:39–41. doi: 10.4274/Jcrpe.478
131. Christodoulidou M, Selmi E. Severe diabetic ketoacidosis leading to cardiac failure, pulmonary oedema and spinal cord oedema resulting in tetraplegia. BMJ Case Rep. (2012) 2012:bcr2012006769. doi: 10.1136/bcr-2012-006769
132. Hashimoto K, Ito Y, Tanahashi H, Hayashi M, Yamakita N, Yasuda K. Hyperglycemic chorea-ballism or acute exacerbation chorea? Huntington's disease unmasked by diabetic ketoacidosis in type 1 diabetes mellitus. J Clin Endocrinol Metab. (2012) 97:3016–20. doi: 10.1210/jc.2012-1190
133. Kanwal SK, Bando A, Kumar V. Clinical profile of diabetic ketoacidosis in Indian children. Indian J Pediatr. (2012) 79:901–4. doi: 10.1007/s12098-011-0634-3
134. Nwosu BU, Adhami S, Rogol AD. Stroke in a child with Adams-Oliver syndrome and mixed diabetic ketoacidosis and hyperglycemic hyperosmolar syndrome. J Pediatr Endocrinol Metab. (2012) 25:357–61. doi: 10.1515/jpem-2011-0465
135. Sottosanti M, Morrison GC, Singh RN, Sharma AP, Fraser DD, Alawi K, et al. Dehydration in children with diabetic ketoacidosis: a prospective study. Arch Dis Child. (2012) 97:96–100. doi: 10.1136/archdischild-2011-300173
136. Bin Salleh H, Mujawar QM. Hypertension in severe pediatric diabetic ketoacidosis: case report and review of literature. Pediatr Emerg Care. (2013) 29:82–3. doi: 10.1097/PEC.0b013e31827b564e
137. Elmehdawi RR, Ehmida M, Elmagrehi H, Alaysh A. Incidence and mortality of diabetic ketoacidosis in Benghazi-Libya in 2007. Oman Med J. (2013) 28:178–83. doi: 10.5001/omj.2013.50
138. Glaser NS, Wootton-Gorges SL, Buonocore MH, Tancredi DJ, Marcin JP, Caltagirone R, et al. Subclinical cerebral edema in children with diabetic ketoacidosis randomized to 2 different rehydration protocols. Pediatrics. (2013) 131:e73–80. doi: 10.1542/peds.2012-1049
139. Azevedo LC, Choi H, Simmonds K, Davidow J, Bagshaw SM. Incidence and long-term outcomes of critically ill adult patients with moderate-to-severe diabetic ketoacidosis: retrospective matched cohort study. J Crit Care. (2014) 29:971–7. doi: 10.1016/j.jcrc.2014.07.034
140. Basnet S, Venepali PK, Andoh J, Velhulst S, Koirala J. Effect of normal saline and half normal saline on serum electrolytes during recovery phase of diabetic ketoacidosis. J Intensive Care Med. (2014) 29:38–42. doi: 10.1177/0885066612467149
141. Kim HJ, Kim DH, Jun YH, Lee JE. A rare diabetes ketoacidosis in combined severe hypernatremic hyperosmolarity in a new onset Asian adolescent with type 1 diabetes. BMJ Case Rep. (2014) 2014:bcr2014208016. doi: 10.1136/bcr-2014-208016
142. Li W, Gong C, Wu D, Liu M. Two case reports of severe pediatric hyperosmolar hyperglycemia and diabetic ketoacidosis accompanied with rhabdomyolysis and acute renal failure. J Pediatr Endocrinol Metab. (2014) 27:1227–31. doi: 10.1515/jpem-2014-0131
143. Orban JC, Maizière EM, Ghaddad A, Van Obberghen E, Ichai C. Incidence and characteristics of acute kidney injury in severe diabetic ketoacidosis. PLoS ONE. (2014) 9:e110925. doi: 10.1371/journal.pone.0110925
144. Rodriguez-Gutiérrez R, Cámara-Lemarroy CR, Quiantanilla-Flores DL, González-Moreno EI, González-Chávez JM, Lavalle-González FJ, et al. Severe ketoacidosis (pH ≤ 6.9) in type 2 diabetes: more frequent and less ominous than previously thought. Biomed Res Int. (2015) 2015:134780. doi: 10.1155/2015/134780
145. Bakes K, Haukoos JS, Deakyne SJ, Hopkins E, Easter J, McFann K, et al. Effect of volume of fluid resuscitation on metabolic normalization in children presenting in diabetic ketoacidosis: a randomized controlled trial. J Emerg Med. (2016) 50:551–9. doi: 10.1016/j.jemermed.2015.12.003
146. Cheng YC, Huang CH, Lin WR, Lu PL, Chang K, Tsai JJ, et al. Clinical outcomes of septic patients with diabetic ketoacidosis between 2004 and 2013 in a tertiary hospital in Taiwan. J Microbiol Immunol Infect. (2016) 49:663–71. doi: 10.1016/j.jmii.2014.08.018
147. Davis SM, Maddux AB, Alonso GT, Okada CR, Mourani PM, Maahs DM. Profound hypokalemia associated with severe diabetic ketoacidosis. Pediatr Diabetes. (2016) 17:61–5. doi: 10.1111/pedi.12246
148. Sedhai YR, Reddy K, Patel D, Lozada JA. Unusual case of pheochromocytoma presenting with diabetic ketoacidosis. BMJ Case Rep. (2016) 2016:bcr2016216961. doi: 10.1136/bcr-2016-216961
149. Singh D, Cantu M, Marx MH, Akingbola O. Diabetic ketoacidosis and fluid refractory hypotension. Clin Pediatr. (Phila). (2016) 55:182–4. doi: 10.1177/0009922815584549
150. Arya MK, Ahmed SM, Shah KN, Ali US. Diabetic ketoacidosis with extreme hyperglycemia in a 4-year-old child. Indian J Crit Care Med. (2017) 21:610–2. doi: 10.4103/ijccm.IJCCM_226_17
151. Kruljac I, Caćić P, Ostojić V, Štefanović M, Šikić A, Vrkljan M. Diabetic ketosis during hyperglycemic crisis is associated with decreased all-cause mortality in patients with type 2 diabetes mellitus. Endocrine. (2017) 55:139–43. doi: 10.1007/s12020-016-1082-7
152. Majid A, Wheeler BJ. Severe diabetic ketoacidosis complicated by hypocapnic seizure. Endocrinol Diabetes Metab Case Rep. (2017) 2017:17-0048. doi: 10.1530/EDM-17-0048
153. Nambam B, Menefee E, Gungor N, Movie R. Severe complications after initial management of hyperglycemic hyperosmolar syndrome and diabetic ketoacidosis with a standard diabetic ketoacidosis protocol. J Pediatr Endocrinol Metab. (2017) 30:1141–5. doi: 10.1515/jpem-2017-0183
154. Omar HR, El-Khabiry E, Dalvi P, Mangar D, Camporesi EM. Brugada ECG pattern during hyperkalemic diabetic ketoacidosis. Ther Adv Endocrinol Metab. (2017) 8:20–1. doi: 10.1177/2042018816680589
155. Rashid MO, Sheikh A, Salam A, Farooq S, Kiran Z, Islam N. Diabetic ketoacidosis characteristics and differences in type 1 diabetes versus type 2 diabetes patients. J Ayub Med Coll Abbottabad. (2017) 29:398–402.
156. Schaapveld-Davis CM, Negrete AL, Hudson JQ, Saikumar J, Finch CK, Kocak M, et al. End-stage renal disease increases rates of adverse glucose events when treating diabetic ketoacidosis or hyperosmolar hyperglycemic state. Clin Diabetes. (2017) 35:202–8. doi: 10.2337/cd16-0060
157. Van de Vyver C, Damen J, Haentjens C, Bailaux D, Bouts B. An exceptional case of diabetic ketoacidosis. Case Rep Emerg Med. (2017) 2017:4351620. doi: 10.1155/2017/4351620
158. Baalaaji M, Jayashree M, Nallasamy K, Singhi S, Bansai A. Predictors and outcome of acute kidney injury in children with diabetic ketoacidosis. Indian Pediatr. (2018) 55:311–4. doi: 10.1007/s13312-018-1274-8
159. Choi HS, Kwon A, Chae HW, Suh J, Kim DH, Kim HS. Respiratory failure in a diabetic ketoacidosis patient with severe hypophosphatemia. Ann Pediatr Endocrinol Metab. (2018) 23:103–6. doi: 10.6065/apem.2018.23.2.103
160. George JT, Mishra AK, Iyadurai R. Correlation between the outcomes and severity of diabetic ketoacidosis: a retrospective pilot study. J Family Med Prim Care. (2018) 7:787–90. doi: 10.4103/jfmpc.jfmpc_116_18
161. Hörber S, Hudak S, Kächele M, Overkamp D, Fritsche A, Häring HU, et al. Unusual high blood glucose in ketoacidosis as first presentation of type 1 diabetes mellitus. Endocrinol Diabetes Metab Case Rep. (2018) 2018:18-0094. doi: 10.1530/EDM-18-0094
162. Kumar V, Nanavati SM, Melki G, Upadhyaya M, Dhillon R, Gibiezaite S, et al. A case of diabetic ketoacidosis presenting with hypernatremia, hyperosmolarity, and altered sensorium. Case Rep Endocrinol. (2018) 2018:4806598. doi: 10.1155/2018/4806598
163. Kupperman N, Ghetti S, Schunk JE, Stoner MJ, Rewers A, McManemy JK, et al. Clinical trial of fluid infusion rates for pediatric diabetic ketoacidosis. N Engl J Med. (2018) 378:2275–87. doi: 10.1056/NEJMoa1716816
164. Ndebele NFM, Naidoo M. The management of diabetic ketoacidosis at a rural regional hospital in KwlaZulu-Natal. Afr J Prim Health Care Fam Med. (2018) 10:e1–6. doi: 10.4102/phcfm.v10i1.1612
165. Shafi O, Kumar V. Initial fluid therapy in pediatric diabetic ketoacidosis: a comparison of hypertonic saline solution and normal saline solution. Pediatr Endocrinol Diabetes Metab. (2018) 24:56–64. doi: 10.18544/PEDM-24.02.0103
166. Yesilbas O, Cem E, Cimbek EA. Successful treatment of life-threatening severe metabolic acidosis by continuous veno-venous hemodialysis in a child with diabetic ketoacidosis. J Pediatr Endocrinol Metab. (2018) 31:1043–5. doi: 10.1515/jpem-2018-0232
167. Halloum A, Al Neyadi S. Myocardial dysfunction associated with diabetic ketoacidosis in a 5-year-old girl. SAGE Open Med Case Rep. (2019) 7:2050313X19847797. doi: 10.1177/2050313X19847797
168. Weissbach A, Zur N, Kaplan E, Kadmon G, Gendler Y, Nahum E. Acute kidney injury in critically ill children admitted to the PICU for diabetic ketoacidosis. A retrospective study. Pediatr Crit Care Med. (2019) 20:e10–4. doi: 10.1097/PCC.0000000000001758
169. Pasquel FJ, Tsegka K, Wang H, Cardona S, Galindo RJ, Fayfman M, et al. Clinical outcomes in patients with isolated or combined diabetic ketoacidosis and hyperosmolar hyperglycemic state: a retrospective, hospital-based cohort study. Diabetes Care. (2020) 43:349–57. doi: 10.2337/dc19-1168
170. Basnet S, Musaitif R, Khanal A, Powell K, Caprirolo G, Majcina R, et al. Effect of potassium infusions on plasma levels during treatment of diabetic ketoacidosis. J Pediatr Intensive Care. (2020) 9:113–118. doi: 10.1055/s-0039-1700981
171. Galindo RJ, Pasquel FJ, Fayfman M, Tsegka K, Dhruv N, Cardona S, et al. Clinical characteristics and outcomes of patients with end-stage renal disease hospitalized with diabetic ketoacidosis. BMJ Open Diabetes Res Care. (2000) 8:e000763. doi: 10.1136/bmjdrc-2019-000763
172. Evans EI, Butterfield WJH. Stress response in severely burned: interim report. Ann Surg. (1951) 134:588–613. doi: 10.1097/00000658-195113440-00007
174. Arney GK, Pearson E, Sutherland AB. Burn stress pseudodiabetes. Ann Surg. (1960) 152:77–90. doi: 10.1097/00000658-196007000-00012
175. Davidson AJG. Diabetic coma without ketoacidosis in a patient with acute pancreatitis. Br Med J. (1964) 1:356. doi: 10.1136/bmj.1.5379.356
176. Danowski TS, Nabarro JDN. Hyperosmolar and other types of ketoacidotic coma in diabetes. Diabetes. (1965) 14:162–5. doi: 10.2337/diab.14.3.162
177. DiBenedetto RJ, Crocco JA, Soscia JL. Hyperglycemia nonketotic coma. Arch Intern Med. (1965) 116:74–82. doi: 10.1001/archinte.1965.03870010076010
178. Rosenberg SA, Brief DK, Kinney JM, Herrera MG, Wilson RE, Moore FD. The syndrome of dehydration, coma and severe hyperglycemia without ketosis in patients convalescing from burns. N Engl J Med. (1965) 272:931–8. doi: 10.1056/NEJM196505062721802
179. Halmos PB, Nelson JK, Lowry RC. Hyperosmolar non-ketoacidotic coma in diabetes. Lancet. (1966) 1:675–9. doi: 10.1016/S0140-6736(66)91626-6
180. Boyer MH. Hyperosmolar anacidotic coma in association with glucocorticoid therapy. JAMA. (1967) 202:1007–9. doi: 10.1001/jama.202.11.1007
181. Ehrlich RM, Bain HW. Hyperglycemia and hyperosmolarity in an eighteen-month-old child. N Engl J Med. (1967) 276:683–4. doi: 10.1056/NEJM196703232761208
182. Kumar RS. Hyperosmolar non-ketotic coma. Lancet. (1968) 1:48–9. doi: 10.1016/S0140-6736(68)90042-1
183. Lotz M, Geraghty M. Hyperglycemic, hyperosmolar nonketotic coma in a ketosis-prone juvenile diabetic. Ann Intern Med. (1968) 69:1245–6. doi: 10.7326/0003-4819-69-6-1245
184. Johnson RD, Conn JW, Dykman DJ, Pek S, Starr JI. Mechanisms and management of hyperosmolar coma without ketoacidosis in the diabetic. Diabetes. (1969) 18:111–6. doi: 10.2337/diab.18.2.111
185. Lausier J, Grenier B, Dupin M, Desbuquois G. Coma hyperosmolaire chez un enfant diabétique (hyperosmolar coma in a diabetic child). Ann Pediatr. (1969) 16:604–7.
186. Manzano F, Kozak GP. Acute quadriplegia in diabetic hyperosmolar coma with hypokalemia. JAMA. (1969) 207:2278–81. doi: 10.1001/jama.1969.03150250108018
187. Monteleone JA, Keefe DM. Transient hyperglycemia and aketotic hyperosmolar acidosis with heat stroke. Pediatrics. (1969) 44:737–41.
188. Oakes DD, Schreibman PH, Hoffman RS, Arky RA. Hyperglycemic, non-ketotic coma in the patient with burns: factors in pathogenesis. Metabolism. (1969) 18:103–9. doi: 10.1016/0026-0495(69)90102-4
189. Rubin HM, Kramer R, Drash A. Hyperosmolality complicating diabetes mellitus in childhood. J Pediatr. (1969) 74:177–86. doi: 10.1016/S0022-3476(69)80065-X
190. Belmonte MM, Colle E, Murphy DA, Wiglesworth FW. Nonketotic hyperosmolar diabetic come in Down's syndrome. J Pediatr. (1970) 77:879–81. doi: 10.1016/S0022-3476(70)80254-2
191. Jacobson R, Horenstein M, Kassel L. Hyperglycemia and hyperosmolarity in a brittle diabetic with thyrotoxicosis. Diabetes. (1970) 19:70–1. doi: 10.2337/diab.19.1.70
192. Vinik A, Seftel H, Joffe BI. Metabolic findings in hyperosmolar non-ketotic diabetic stupor. Lancet. (1970) 2:797–9. doi: 10.1016/S0140-6736(70)91461-3
193. Balsam MJ, Baker L, Kaye R. Hyperosmolar nonketotic come associated with diazoxide therapy for hypoglycemia. J Pediatr. (1971) 78:523–5. doi: 10.1016/S0022-3476(71)80241-X
194. Dagnone LE, Reid JD, Green RN. Non-ketotic hyperosmolar coma with insulin resistance in a 12-year-old girl. Can Med Assoc J. (1971) 105:940:943–5.
195. Evans CC, Littler WA, Meade JB. Hyperosmolar hyperglycemic non-ketotic diabetic coma complicating open heart surgery. Br Heart J. (1972) 34:1075–7. doi: 10.1136/hrt.34.10.1075
196. Doromal NM, Canter JW. Hyperosmolar hyperglycemic nonketotic coma complicating intravenous hyperalimentation. Surg Gynecol Obstet. (1973) 136:729–32.
197. Heggarty H, Trindade P, Bryan EM. Hyperglycaemia in hyperosmolar dehydration. Arch Dis Child. (1973) 48:740–1. doi: 10.1136/adc.48.9.740
198. Hobin FP, Maraist FB. Diabetic hyperosmolar nonketoacidotic coma complicated by mesenteric thrombosis. Case report. J Florida Med Assoc. (1973) 60:25–6.
199. Fernandez F, Hughes ER. Letter: non-ketotic hyperosmolar diabetic coma in an infant. J Pediatr. (1974) 84:606–7. doi: 10.1016/S0022-3476(74)80690-6
200. Lampe EW II, Ruiz JO, Simmons RL, Najarian JS. Hyperglycemic non-ketotic coma after renal transplantation. Am J Surg. (1974) 127:342–4. doi: 10.1016/0002-9610(74)90046-4
201. Tchertkoff V, Nayak SV, Kamath C, Salomon MI. Hyperosmolar nonketotic diabetic coma: vascular complications. J Am Geriatr Soc. (1974) 22:462–6. doi: 10.1111/j.1532-5415.1974.tb04815.x
202. Dorchy H, Pardou A, Weemaes I, Loeb H. Coma hyperosmolaire acido-cétosique sans cétonurie initiale: evolution favorable (Acido-ketotic hyperosmolar coma without initial ketonuria: favorable course). Pediatrie. (1975) 30:19–27.
203. Khaleeli AA, Wyman AL. Hyperosmolar non-ketotic diabetic coma induced by furosemide in modest dosage. Postgrad Med J. (1978) 54:43–4. doi: 10.1136/pgmj.54.627.43
204. Goldman SL. Hyperglycemic hyperosmolar coma in a 9-month-old child. Am J Dis Child. (1979) 133:181–3. doi: 10.1001/archpedi.1979.02130020073015
205. Joosten R, Frank M, Hörnchen H, Bertrams J. Hyperosmolar nonketotic diabetic coma. Eur J Pediatr. (1981) 137:233–6.
206. Yamashiro Y, Yamamoto T, Mayama H. Nonketotic hyperosmolar coma in two diabetic children. Acta Paediatr Scand. (1981) 70:337–40.
207. Asplund K, Eriksson S, Hägg E, Lithner F, Strand T, Wester PO. Hyperosmolar non-ketotic coma in diabetic stroke patients. Acta Med Scand. (1982) 212:407–11. doi: 10.1111/j.0954-6820.1982.tb03238.x
208. Altomonte L, Ghirlanda G, Palumbo P, Mann A, Greco V. Coma diabetico iperosmolare. Contributo casistico e revione della litteratura (Hyperosmolar diabetic coma. Case reports and review of the literature). Minerva Med. (1983) 74:227–33.
209. Fujikawa LS, Meister DM, Nozik RA. Hyperosmolar hyperglycemic nonketotic coma. A complication of short-term systemic corticosteroid use. Ophthalmology. (1983) 90:1239–42. doi: 10.1016/S0161-6420(83)34403-1
210. Hoffman WH, Fernados SS. Hyperglycemic hyperosmolar non-ketotic coma in a non-diabetic child. Diabetologia. (1983) 25:531. doi: 10.1007/BF00284466
211. Khardori T, Soller NG. Hyperosmolar hyperglycemic nonketotic syndrome. Am J Med. (1984) 77:899–904. doi: 10.1016/0002-9343(84)90539-4
212. Flynn JT, Bachynski BN, Rodrigues MM, Curless RG, Joshi B. Hyperglycemic acidotic comma in Kearns-Sayre syndrome. Trans Am Ophthalmol Soc. (1985) 83:131–61.
213. Schlepphorst E, Levin ME. Rhabdomyolysis associated with hyperosmolar nonketotic coma. Diabetes Care. (1985) 8:198–200. doi: 10.2337/diacare.8.2.198
214. Vernon DD, Postellon DC. Nonketotic hyperosmolal diabetic coma in a child: management with low-dose insulin infusion and intracranial pressure monitoring. Pediatrics. (1986) 77:770–2.
215. Wachtel TJ, Silliman RA, Lamberton P. Prognostic factors in the diabetic hyperosmolar state. J Amer Geriatr Soc. (1987) 35:737–41. doi: 10.1111/j.1532-5415.1987.tb06351.x
216. McComb RD, Pfeiffer RF, Casey JH, Wolcott G, Till DJ. Lateral pontine and extrapontine myelinolysis associated with hypernatremia and hyperglycemia. Clin Neuropathol. (1989) 8:284–8.
217. Lustman CC, Guérin JM, Barbotin-Larrieu FE. Hyperosmolar nonketotic syndrome associated with rhabdomyolysis and acute kidney failure. Diabetes Care. (1991) 14:146–7. doi: 10.2337/diacare.14.2.146
218. Leung CB, Li PKT, Lui SF, Lai KN. Acute renal failure (ARF) caused by rhabdomyolysis due to diabetic hyperosmolar nonketotic coma: a case report and literature review. Renal Failure. (1992) 14:81–5. doi: 10.3109/08860229209039121
219. Eidlitz-Markus T, Nussinovitch M, Varsano I, Kauschansky A. Nonketotic hyperosmolar coma in children. Isr J Med Sci. (1994) 30:585–7.
220. Piniés JA, Cairo G, Gaztambide S, Vazquez JA. Course and prognosis of 132 patients with diabetic nonketotic hyperosmolar state. Diabete Metab. (1994) 20:43–8.
221. Yang JY, Cui XL, He XJ. Non-ketotic hyperosmolar coma complicating steroid treatment in childhood nephrosis. Pediatr Nephrol. (1995) 9:621–2. doi: 10.1007/BF00860958
222. Rother KI, Schwenk F II. An unusual case of nonketotic hyperglycemic syndrome during childhood. Mayo Clin Proc. (1995) 70:62–5. doi: 10.1016/S0025-6196(11)64667-3
223. Gottschalk ME, Ros SP, Zeller WP. The emergency management of hyperglycemic-hyperosmolar nonketotic coma in the pediatric patient. Pediatr Emerg Care. (1996) 12:48–51. doi: 10.1097/00006565-199602000-00014
224. Tanaka S, Kobayashi T, Kawanami D, Hori A, Okubo N, Nakanishi K, et al. Paradoxical glucose infusion for hypernatraemia in diabetic hyperglycemic hyperosmolar syndrome. J Intern Med. (2000) 248:166–8. doi: 10.1046/j.1365-2796.2000.00710-2.x
225. Milionis HJ, Liamis GL, Elisaf MS. Hyperosmolar syndrome in a patient with uncontrolled diabetes mellitus. Am J Kidney Dis. (2001) 37:E41. doi: 10.1016/S0272-6386(05)90005-X
226. Pettigrew DC. Index of suspicion. Case 2. Diagnosis: hyperglycemic nonketotic hypertonicity (HNKH). Pediatr Rev. (2001) 22:169–73. doi: 10.1542/pir.22-5-169
227. Ka T, Takahashi S, Tsutsumi Z, Moriwaki Y, Yamamoto T, Fukuchi M. Hyperosmolar non-ketotic diabetic syndrome associated with rhabdomyolysis and acute renal failure: a case report and review of the literature. Diabetes Nutr Metab. (2003) 16:317–22.
228. Ahlsson F, Gedeborg R, Hesselager G, Tuvemo T, Enblad P. Treatment of extreme hyperglycemia monitored with intracerebral microdialysis. Pediatr Crit Care Med. (2004) 5:89–92. doi: 10.1097/01.PCC.0000102396.02043.22
229. Morales AE, Rosenbloom AL. Death caused by hyperglycemic hyperosmolar state at the onset of type 2 diabetes. J Pediatr. (2004) 144:270–3. doi: 10.1016/j.jpeds.2003.10.061
230. Bhowmick SK, Levens KL, Retig KR. Hyperosmolar hyperglycemic crisis: an acute life-threatening event in children and adolescents with type 2 diabetes mellitus. Endocr Pract. (2005) 11:23–9. doi: 10.4158/EP.11.1.23
231. Carchman RM, Dechert-Zeger M, Calikoglu AS, Harris BD. A new challenge in pediatric obesity: pediatric hyperglycemic hyperosmolar syndrome. Pediatr Crit Care Med. (2005) 6:20–4. doi: 10.1097/00130478-200501000-00047
232. Fourtner SH, Weinzimer SA, Katz LEL. Hyperglycemic hyperosmolar non-ketotic syndrome in children with type 2 diabetes. Pediatr Diabetes. (2005) 6:129–35. doi: 10.1111/j.1399-543X.2005.00113.x
233. Kershaw MJR, Newton T, Barrett TG, Berry K, Kirk J. Childhood diabetes presenting with hyperosmolar dehydration but without ketoacidosis: a report of three cases. Diabet Med. (2005) 22:645–7. doi: 10.1111/j.1464-5491.2005.01517.x
234. Venkatraman R, Jayashree M, Singhi S, Marwaha RK. Hyperglycemic hyperosmolar nonketotic syndrome in a child with acute lymphoblastic leukemia undergoing induction chemotherapy. J Pediatr Hematol Oncol. (2005) 27:234–5. doi: 10.1097/01.mph.0000161530.40065.2f
235. Cochran JB, Walters S, Losek JD. Pediatric hyperglycemic hyperosmolar syndrome: diagnostic difficulties and high mortality rate. Am J Emerg Med. (2006) 24:297–301. doi: 10.1016/j.ajem.2005.10.007
236. Kilbane BJ, Mehta S, Backeljauw PF, Shanley TP, Crimmins NA. Approach to management of malignant hyperthermia-like syndrome in pediatric diabetes mellitus. Pediatr Crit Care Med. (2006) 7:169–73. doi: 10.1097/01.PCC.0000192340.09136.82
237. Pinhas-Hamiel O, Chernausek SD, Zeitler P. Acute necrotizing pancreatitis in an adolescent with type 2 diabetes. Curr Opin Pediatr. (2006) 18:206–8. doi: 10.1097/01.mop.0000193286.79399.43
238. Baluch AR, Oommen SP. Malignant hyperthermia associated with diabetic hyperosmolar hyperglycemic nonketotic state in a young man. J Clin Anesth. (2007) 19:470–2. doi: 10.1016/j.jclinane.2006.12.008
239. Canarie MF, Boque CW, Basaniak KJ, Weinzimer SA, Tamborlane WV. Decompensated hyperglycemic hyperosmolarity without significant ketoacidosis in the adolescent and young adult population. J Pediatr Endocrinol Metab. (2007) 20:1115–24. doi: 10.1515/JPEM.2007.20.10.1115
240. Bartoli E, Guidetti F, Bergamasco L. Estimating excess glucose, sodium and water deficits in non-ketotic hyperglycemia. Nephrol Dial Transplant. (2007) 22:3478–86. doi: 10.1093/ndt/gfm427
241. O'Malley G, Moran C, Draman MS, King T, Smith D, Thompson CJ, et al. Central pontine myelinolysis complicating treatment of the hyperglycaemic hyperosmolar state. Ann Clin Biochem. (2008) 45:440–3. doi: 10.1258/acb.2008.007171
242. Murthy S, Sharara-Chami R. Aggressive fluid resuscitation in severe pediatric hyperglycemic hyperosmolar syndrome: a case report. Int J Pediatr Endocrinol. (2010) 2010:379063. doi: 10.1186/1687-9856-2010-379063
243. Fadini GP, de Kreutzenberg SV, Rigato M, Brocco S, Marchesan M, Tiengo A, et al. Characteristics and outcomes of the hyperglycemic hyperosmolar non-ketotic syndrome in a cohort of 51 consecutive cases in a single center. Diabetes Res Clin Pract. (2011) 94:172–9. doi: 10.1016/j.diabres.2011.06.018
244. Stunkard ME, Pikul VT, Foley K. Hyperosmolar hyperglycemic syndrome with rhabdomyolysis. Clin Lab Sci. (2011) 24:8–13. doi: 10.29074/ascls.24.1.8
245. Bassham B, Estrada C, Abramo T. Hyperglycemic hyperosmolar syndrome in the pediatric patient: a case report and review of the literature. Pediatr Emerg Care. (2012) 28:699–702. doi: 10.1097/PEC.0b013e31825d23c9
246. Alharfi IM, Singh R, Clarson C, Foster JR. Hyperosmolar hyperglycemic state without ketosis in a toddler with type 1 diabetes. Pediatr Emerg Care. (2014) 30:485–7. doi: 10.1097/PEC.0000000000000167
247. Krujiac I, Rinčic G, Pećina HI. Hyperosmolar coma in a patient with hypothalamic Langerhans cell histiocytosis. Endocrine. (2016) 52:176–7. doi: 10.1007/s12020-015-0675-x
248. Price A, Losek J, Jackson B. Hyperglycaemic hyperosmolar syndrome in children: patient characteristics, diagnostic delays and associated complications. J Paediatr Child Health. (2016) 52:80–4. doi: 10.1111/jpc.12980
249. Brzezniak CE, Vietor N, Hogan PE, Oronsky B, Thilagar B, Ray CM, et al. A case of paraneoplastic syndrome presenting as hyperglycemic hyperosmolar nonketotic syndrome. Case Rep Oncol. (2017) 10:321–4. doi: 10.1159/000467390
250. Galili Y, Gonzalez C, Lytle M, Carlan SJ, Madruga M. Full recovery from extreme hypernatremia in an elderly woman with hyperosmolar hyperglycemic syndrome and abnormal electroencephalogram. Sage Open Med Case Rep. (2019) 7:1–4. doi: 10.1177/2050313X19848887
251. Xiao H, Barmanray R, Qian S, De Alwis D, Fennesy G. Survival following extreme hypernatraemia associated with severe dehydration and undiagnosed diabetes mellitus. Case Rep Endocrinol. (2019) 2019:4174259. doi: 10.1155/2019/4174259
252. Potter DJ. Death as a result of hyperglycemia without ketosis. A complication of hemodialysis. Ann Intern Med. (1966) 64:399–401. doi: 10.7326/0003-4819-64-2-399
253. Boyer J, Gill GN, Epstein FH. Hyperglycemia and hyperosmolality complicating peritoneal dialysis. Ann Intern Med. (1967) 67:568–72. doi: 10.7326/0003-4819-67-3-568
254. Whang R. Hyperglycemic nonketotic coma induced by peritoneal dialysis. J Lancet. (1967) 87:453–6.
255. Handa JP, Kushner GB. Hyperosmolar hyperglycemic nonketotic coma during peritoneal dialysis. South Med J. (1968) 61:700–2. doi: 10.1097/00007611-196807000-00008
256. Gault MH, Ferguson EL, Sidhu JS, Corbin RP. Fluid and electrolyte complications of peritoneal dialysis. Choice of dialysis solutions. Ann Intern Med. (1971) 75:253–62. doi: 10.7326/0003-4819-75-2-253
257. Axelrod L. Response of congestive heart failure to correction of hyperglycemia in the presence of diabetic nephropathy. N Engl J Med. (1975) 293:1243–5. doi: 10.1056/NEJM197512112932408
258. Kaldany A, Curt GA, Estes NM, Weinrauch LA, Christlieb AR, D'Elia JA. Reversible acute pulmonary edema due to uncontrolled hyperglycemia in diabetic individuals with renal failure. Diabetes Care. (1982) 5:506–11. doi: 10.2337/diacare.5.5.506
259. Ryder RE, Hayes TM. Normo-osmolar, nonketotic, hyponatremic diabetic syndrome associated with impaired renal function. Diabetes Care. (1983) 6:402–4. doi: 10.2337/diacare.6.4.402
260. Montoliu J, Revert L. Lethal hyperkalemia associated with severe hyperglycemia in diabetic patients with renal failure. Am J Kidney Dis. (1985) 5:47–8. doi: 10.1016/S0272-6386(85)80135-9
261. Krediet RT, Struijk DG, Arisz L. Hyponatremia in continuous ambulatory peritoneal dialysis patients with diabetic nephropathy during hyperglycemic episodes. Transplant Proc. (1986) 18:1702–4.
262. Emder PJ, Howard BJ, Rosenberg AR. Nonketotic hyperosmolar diabetic pre-coma due to pancreatitis in a boy on continuous ambulatory peritoneal dialysis. Nephron. (1986) 44:355–7. doi: 10.1159/000184019
263. Tzamaloukas AH, Avasthi PS. Acid-base disorders in hyperglycemia of insulin-dependent diabetic patients on chronic dialysis. J Diabet Complications. (1988) 2:75–8. doi: 10.1016/0891-6632(88)90007-4
264. Crawford BA, Roy LP, Knight JF. Hyperglycaemia complicating haemolytic uraemic syndrome. J Paediatr Child Health. (1990) 26:225–6. doi: 10.1111/j.1440-1754.1990.tb02435.x
265. Popli S, Leehey DJ, Daugirdas JT, Bansal VK, Ho DS, Hano JE, et al. Asymptomatic, nonketotic, severe hyperglycemia with hyponatremia. Arch Intern Med. (1990) 150:1962–4. doi: 10.1001/archinte.1990.00390200138027
266. Catalano C, Fabbian F, Di Landro D. Acute pulmonary oedema occurring in association with diabetic ketoacidosis in a diabetic patient with chronic renal failure. Nephrol Dial Transplant. (1998) 13:491–2. doi: 10.1093/ndt/13.2.491
267. Blicker J, Herd AM, Talbot J. Diabetic ketoacidosis in the dialysis-dependent patient: two case reports and recommendations for treatment. CJEM. (2004) 6:281–4. doi: 10.1017/S1481803500009271
268. Park JI, Yi JH, Han SW, Kim HJ. Recurrent symptomatic hyperglycemia on maintenance hemodialysis is not necessarily related to hypertonicity: a case report. Electrolyte Blood Press. (2008) 6:56–9. doi: 10.5049/EBP.2008.6.1.56
269. Popli S, Sun Y, Tang HL, Kjellstrand CM, Tzamaloukas AH, Ing TS. Acidosis and coma in adult diabetic maintenance dialysis patients with extreme hyperglycemia. Int Urol Nephrol. (2013) 45:1687–92. doi: 10.1007/s11255-013-0390-6
270. Konstantinov NK, Sun Y, Vigil D, Agaba EI, Servilla KS, Murata GH, et al. Hyperkalemia in two patients with diabetes mellitus and chronic kidney disease and the role of disrupted internal potassium balance. Cureus. (2014) 6:e162. doi: 10.7759/cureus.162
271. Gupta A, Rohrscheib M, Tzamaloukas AH. Extreme hyperglycemia with ketoacidosis and hyperkalemia in a patient on chronic hemodialysis. Hemodial Int. (2008) 12:S43–7. doi: 10.1111/j.1542-4758.2008.00324.x
272. Varma R, Karim M. Lesson of the month 2:diabetic ketoacidosis in established renal failure. Clin Med. (2016) 16:392–3. doi: 10.7861/clinmedicine.16-4-392
273. Yamada H, Funazaki S, Kakei M, Hara K, Ishikawa SE. Diabetic ketoacidosis producing extreme hyperkalemia in a patient with type 1 diabetes on hemodialysis. Endocrinol Diabetes Metab Case Rep. (2017) 2017:17-0068. doi: 10.1530/EDM-17-0068
274. Gibb J, Xu Z, Rohrscheib M, Tzamaloukas AH. Hyperglycemic crisis in an anuric peritoneal dialysis patient with profound and symptomatic hypertonicity. Cureus. (2018) 10:e2566. doi: 10.7759/cureus.2566
275. Thiruppathy P. Management of diabetic ketoacidosis in a patient on hemodialysis. In: Mohan V, Unikrishnan R, editors. Diabetes-Clinical Case Series II. New Delhi: Jayppee Brothers Medical Publishers (2018). p. 75–7.
276. Vigil D, Kien C, Bibb J, Glew RH, Tzamaloukas AH. Symptomatic hyperglycemia in a patient with dialysis ascites. Am J Med Sci. (2019) 357:512–6. doi: 10.1016/j.amjms.2019.02.016
277. Seddik AA, Bashier A, Alhadari AK, AlAlawi F, Alnour HH, Bin Hussain AA, et al. Challenges in management of diabetic ketoacidosis in hemodialysis patients, case presentation and review of literature. Diabetes Metab Syndr. (2019) 13:2481–7. doi: 10.1016/j.dsx.2019.06.022
278. Glaser N, Barnett P, McCaslin I, Nelson D, Trainor J, Louie J, et al. Risk factors for cerebral edema in children with diabetic ketoacidosis. N Engl J Med. (2001) 344:264–9. doi: 10.1056/NEJM200101253440404
279. Hursh BE, Ronsley B, Islam N, Mammen C, Panagiotopoulos C. Acute kidney injury in children with type 1 diabetes hospitalized for diabetic ketoacidosis. JAMA Pediatr. (2017) 171:e170020. doi: 10.1001/jamapediatrics.2017.0020
280. Halperin ML, Maccari C, Kamel KS, Carlotti AP, Bohn D. Strategies to diminish the danger of cerebral edema in a pediatric patient presenting with diabetic ketoacidosis. Pediatr Diabetes. (2006) 7:191–5. doi: 10.1111/j.1399-5448.2006.00190.x
281. Liamis G, Gianoutsos C, Elisaf MS. Hyperosmolar nonketotic syndrome with hypernatremia: how can we monitor treatment? Diabetes Metab. (2000) 26:403–5.
282. Milionis HJ, Liamis GL, Elisaf M. Plasma sodium changes in the hyperglycaemic state: clinical aspects of pathophysiology and management. Int J Diabetes Metab. (2001) 9:60–6. doi: 10.1159/000497542
283. Adrogué HJ, Madias NE. Hypernatremia. N Engl J Med. (2000) 342:1493–9. doi: 10.1056/NEJM200005183422006
284. Chautham K, Pattharannitima P, Patel N, Duffy A, Saha A, Chaudhary K, et al. Rate of correction of hypernatremia and health outcomes in critically ill patients. Clin J Am Soc Nephrol. (2019) 14:656–63. doi: 10.2215/CJN.10640918
285. Seay NW, Lehrich RW, Greenberg A. Diagnosis and management of disorders of body tonicity–hyponatremia and hypernatremia: core curriculum 2020. Am J Kidney Dis. (2020) 75:272–86. doi: 10.1053/j.ajkd.2019.07.014
Keywords: sodium concentration, hyperglycemia, dysnatremia, hypertonicity, diabetic ketoacidosis, hyperosmolar hyperglycemia
Citation: Ing TS, Ganta K, Bhave G, Lew SQ, Agaba EI, Argyropoulos C and Tzamaloukas AH (2020) The Corrected Serum Sodium Concentration in Hyperglycemic Crises: Computation and Clinical Applications. Front. Med. 7:477. doi: 10.3389/fmed.2020.00477
Received: 20 April 2020; Accepted: 15 July 2020;
Published: 25 August 2020.
Edited by:
Ying-Yong Zhao, Northwest University, ChinaReviewed by:
Francis M. Finucane, National University of Ireland Galway, IrelandDeepak Malhotra, University of Toledo, United States
Copyright © 2020 Ing, Ganta, Bhave, Lew, Agaba, Argyropoulos and Tzamaloukas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christos Argyropoulos, Q0FyZ3lyb3BvdWxvc0BzYWx1ZC51bm0uZWR1