 Ekamol Tantisattamo1,2,3*
Ekamol Tantisattamo1,2,3* Miklos Z. Molnar4,5,6
Miklos Z. Molnar4,5,6 Bing T. Ho7Uttam G. Reddy1,2Donald C. Dafoe8Hirohito Ichii8Antoney J. Ferrey1,2Ramy M. Hanna1
Bing T. Ho7Uttam G. Reddy1,2Donald C. Dafoe8Hirohito Ichii8Antoney J. Ferrey1,2Ramy M. Hanna1 Kamyar Kalantar-Zadeh1,2Alpesh Amin9
Kamyar Kalantar-Zadeh1,2Alpesh Amin9- 1Division of Nephrology, Hypertension and Kidney Transplantation, Department of Medicine, Harold Simmons Center for Kidney Disease Research and Epidemiology, University of California Irvine School of Medicine, Orange, CA, United States
- 2Nephrology Section, Department of Medicine, Tibor Rubin Veterans Affairs Medical Center, VA Long Beach Healthcare System, Long Beach, CA, United States
- 3Section of Nephrology, Department of Internal Medicine, Multi-Organ Transplant Center, William Beaumont Hospital, Oakland University William Beaumont School of Medicine, Royal Oak, MI, United States
- 4Division of Nephrology, University of Tennessee Health Science Center, Memphis, TN, United States
- 5Methodist University Hospital Transplant Institute, Memphis, TN, United States
- 6Division of Transplant Surgery, Department of Surgery, University of Tennessee Health Science Center, Memphis, TN, United States
- 7Division of Nephrology and Hypertension, Department of Medicine, Comprehensive Transplant Center, Northwestern University Feinberg School of Medicine, Chicago, IL, United States
- 8Division of Transplantation, Department of Surgery, University of California Irvine School of Medicine, Orange, CA, United States
- 9Department of Medicine, University of California Irvine School of Medicine, Orange, CA, United States
Hypertension is one of the most common cardiovascular co-morbidities after successful kidney transplantation. It commonly occurs in patients with other metabolic diseases, such as diabetes mellitus, hyperlipidemia, and obesity. The pathogenesis of post-transplant hypertension is complex and is a result of the interplay between immunological and non-immunological factors. Post-transplant hypertension can be divided into immediate, early, and late post-transplant periods. This classification can help clinicians determine the etiology and provide the appropriate management for these complex patients. Volume overload from intravenous fluid administration is common during the immediate post-transplant period and commonly contributes to hypertension seen early after transplantation. Immunosuppressive medications and donor kidneys are associated with post-transplant hypertension occurring at any time point after transplantation. Transplant renal artery stenosis (TRAS) and obstructive sleep apnea (OSA) are recognized but common and treatable causes of resistant hypertension post-transplantation. During late post-transplant period, chronic renal allograft dysfunction becomes an additional cause of hypertension. As these patients develop more substantial chronic kidney disease affecting their allografts, fibroblast growth factor 23 (FGF23) increases and is associated with increased cardiovascular and all-cause mortality in kidney transplant recipients. The exact relationship between increased FGF23 and post-transplant hypertension remains poorly understood. Blood pressure (BP) targets and management involve both non-pharmacologic and pharmacologic treatment and should be individualized. Until strong evidence in the kidney transplant population exists, a BP of <130/80 mmHg is a reasonable target. Similar to complete renal denervation in non-transplant patients, bilateral native nephrectomy is another treatment option for resistant post-transplant hypertension. Native renal denervation offers promising outcomes for controlling resistant hypertension with no significant procedure-related complications. This review addresses the epidemiology, pathogenesis, and specific etiologies of post-transplant hypertension including TRAS, calcineurin inhibitor effects, OSA, and failed native kidney. The cardiovascular and survival outcomes related to post-transplant hypertension and the utility of 24-h blood pressure monitoring will be briefly discussed. Antihypertensive medications and their mechanism of actions relevant to kidney transplantation will be highlighted. A summary of guidelines from different professional societies for BP targets and antihypertensive medications as well as non-pharmacological interventions, including bilateral native nephrectomy and native renal denervation, will be reviewed.
Introduction
Barring contraindications, kidney transplantation is the treatment of choice for advanced chronic kidney disease (CKD) and end-stage renal disease (ESRD) (1). Survival benefit and quality of life are significantly improved after a successful kidney transplantation with renal allograft function. Since the introduction of calcineurin inhibitors (CNI) in the 1980's, short-term renal allograft survival has greatly improved, but there has been no significant effect on long-term renal allograft survival (2, 3).
Several immunological and non-immunological causes contribute to long-term renal and patient survival outcomes. Similar to non-transplant patients, cardiovascular diseases (CVD) remain the leading cause of morbidity and mortality in kidney transplant recipients (4). Hypertension (HTN) is a usual finding in this population and one of the most common risk factors for CVD (5).
This article will review the pathogenesis of post-transplant HTN, including transplant renal artery stenosis (TRAS) and differing management options based on the etiology of hypertension in different clinical transplant recipient scenarios. Determining when non-pharmacological interventions, including transplant renal artery angioplasty and/or stenting, bilateral native nephrectomy, and native renal denervation (RDN), are appropriate will also be discussed.
Epidemiology of Post-Transplant Hypertension
Depending on the definition and methods of blood pressure (BP) measurement utilized, the prevalence of post-transplant HTN has been widely reported, and it has generally increased over time. This greater incidence of post-transplant hypertension maybe related to the introduction of cyclosporine (CsA) (6–8). A study in Spain looked at patients transplanted in three different years (1990 vs. 1994 vs. 1998) and noted a progressive increase in the incidence of post-transplant HTN with subsequent years for all those three periods. The number of antihypertensive medications required in more recent transplants also increased compared to patients who were transplanted earlier (9). Overall prevalence of post-transplant HTN has ranged from 24 to 90% (5, 8–19).
Definition of Post-Transplant Hypertension
Post-kidney transplant HTN can be defined as a persistently elevated BP or normotension with use of antihypertensive medications after successful kidney transplantation. However, the main question that remains is what is a normal BP level? Different studies have defined post-transplant HTN with different cutoff levels for systolic and diastolic blood pressure (SBP and DBP) and different requirements for the use of antihypertensive medications. Table 1 summarizes the details of these studies (8, 9, 16–18).
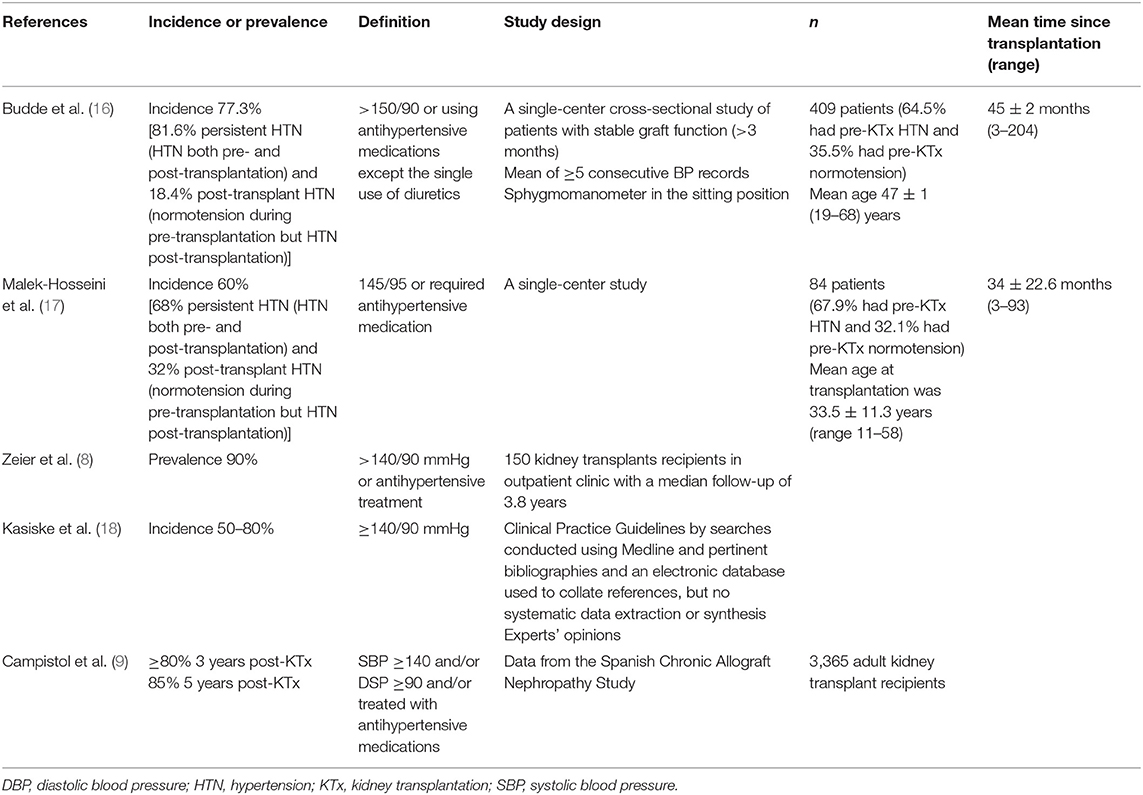
Table 1. Summarized definitions of post-transplant hypertension from studies specifically examining the prevalence of post-transplant hypertension.
In addition to the defining normal BP levels, the presence or absence of HTN during the pre-kidney transplant period may further categorize kidney transplant recipients into four groups: persistent HTN, recovered HTN, persistent normotension, and post-transplant HTN. Persistent HTN occurs in patients with HTN both in the pre- and post-transplant periods, whereas patients with recovered HTN have HTN only during the pre- but not the post-transplant period. Persistent normotensive patients have no history of HTN preceding transplant and remain normotensive post-transplant. Post-transplant HTN requires developing de novo HTN after kidney transplant (Figure 1). Malek-Hosseini et al. (17) reported the incidences of persistent HTN, recovered HTN, persistent normotension, and post-transplant HTN as 40, 28, 13, and 19%, respectively. In this review, post-kidney transplant HTN refers to persistent and post-transplant (de novo) HTN unless otherwise specified.
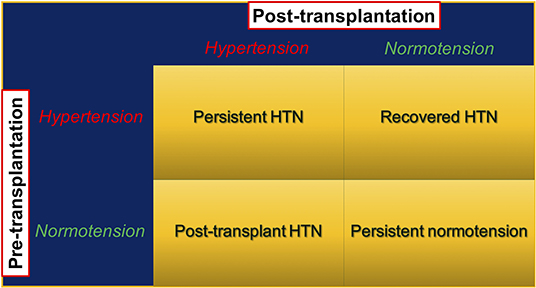
Figure 1. Post-kidney transplant hypertension stratified by presence and absence of pre-transplant hypertension. HTN, hypertension; KTx, kidney transplant.
Isolated forms of HTN both systolic and diastolic still occur after kidney transplantation. The European Society of Hypertension and the European Society of Cardiology guidelines defined isolated systolic HTN in the general population as SBP ≥140 and DBP <90 mmHg (20). This is the most common phenotype of HTN in elderly patients (21). Linear increase in systolic and diastolic BP occurs with age until the fifth or the sixth decades of life when SBP continues increasing, but DBP tends to decrease (22). Pathogenesis of isolated systolic HTN involves in both intrinsic alterations resulting from normal aging process accompanied by development of modifiable risk factors leading to increased arterial stiffness (23).
Alternatively, diastolic HTN is defined as DBP of ≥90 mmHg, with a SBP <140 mmHg, and is more common in younger, sedentary individuals with a higher body mass index (BMI) (24).
A recent large randomized controlled clinical trial of blood pressure management in non-diabetic patients (SPRINT) demonstrated cardiovascular (CV) benefits of tighter BP control (25) leading to new BP guidelines and re-defined HTN for the general population as systolic blood pressure (SBP) >130 or DBP >80 mmHg (26, 27). Although there has been a change in the definition of HTN in the non-transplant population, the definition of HTN in kidney transplant recipients remains controversial, and hard outcomes related to BP levels are still limited. A recent 2017 American College of Cardiology/American Heart Association (ACC/AHA) guidelines recommend a target BP of <130/80 mmHg (26, 27). Until there is stronger evidence of an association between BP level and outcomes in kidney transplant recipients, a BP ≥130/80 mmHg may be a reasonable definition for HTN in this population.
Pathogenesis of Post-Transplant Hypertension
The change in prevalence of post-transplant HTN across different post-transplant periods may reflect the differences in pathogenesis of post-transplant HTN over time (Figure 2). Identifying when post-transplant HTN first occurred can narrow the differential diagnosis for the etiology of post-transplant HTN and lead to tailored therapy.
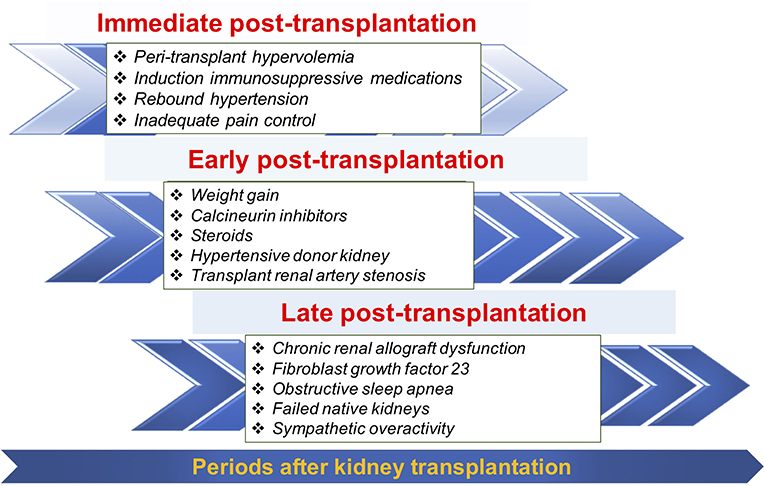
Figure 2. Selected common factors contributing to post-transplant hypertension during three different periods.
Immediate Post-transplant Period
During this time, post-transplant HTN is generally a result of external factors like transplant surgery, IV fluids, and high doses of steroids.
Peri-transplant Hypervolemia
IV fluid given during surgery and in the immediate post-operative period can lead to hypervolemia, particularly in patients with delayed graft function (DGF). A single-center cross-sectional study showed that the prevalence of hypervolemia measured by multifrequency bioimpedance analysis for extracellular fluid in stable kidney transplant recipients was 30%, and up to 5% had severe hypervolemia. This study showed that hypervolemia was significantly associated with elevated systolic, diastolic, and mean arterial pressures (28). Although this study enrolled 123 kidney transplant recipients with a median duration of enrollment at 5 years post-transplant, hypervolemia most commonly occurred during the immediate post-transplant period, and weight gain above the pre-transplant estimated dry weight was associated with HTN.
Several indicators can be used to estimate volume status. These include conventional measurements such as BP, heart rate, urine output, central venous pressure, and pulmonary artery pressure as well as non-conventional measurements such as intraoperative transesophageal echocardiography and non-invasive dynamic cardiac output technology, e.g., pulse contour analysis, pulse wave transit time, thoracic electrical bioimpedance/bioreactance, and carbon dioxide rebreathing technologies (29, 30). Although many of the non-conventional measurements are useful for volume assessment, they are not readily available during peri-transplant period and conventional measurements remain the standard in clinical practice.
High-Dose Steroids
High-dose steroids are commonly used immunosuppressive medications during the peri-transplant period. The incidence of steroid-induced HTN during the immediate post-transplant period is unknown. The mechanism of steroid-induced HTN is unclear, but it may result from alterations in intrinsic pressor response leading to arterial vascular resistance (31). Since a steroid dose of more than 20 mg of prednisone per day is the threshold for having HTN (32), high-dose IV steroids can contribute to HTN during immediate post-transplant period.
Rebound Hypertension
ESRD patients commonly have uncontrolled HTN and require several medications to control their BP. During the immediate post-transplant period, it is common practice to hold some if not all of the pre-transplant BP medications in an effort to avoid early hypotension. However, this abrupt discontinuation of antihypertensive therapy can lead to rebound hypertension, an elevated BP above pretreatment levels, as a result of sympathetic overactivity. Beta-adrenergic agonists, clonidine (both oral and transdermal forms) (33), and beta-blockers are commonly associated with this phenomenon, particularly when abruptly stopped. Geyskes et al. demonstrated that almost all patients who took clonidine 900 mcg per day for more than 1 month developed hypertension after the medication was discontinued. Sympathetic overactivity without renin angiotensin system mediation plays a major role in the mechanism of rebound HTN from clonidine (34).
Similar to clonidine, rebound HTN from beta-blockers causes elevated BP and heart rate. It also leads to cardiac events including angina, myocardial infarction, or sudden cardiac death in patients with underlying coronary artery disease (CAD) (35–39). Increased sympathetic activity associated with upregulated adrenergic receptors in individuals taking beta-blockers is thought to be the mechanism of beta-blockers rebound HTN (36, 38, 40). Given the cardiac risk with peri-operative beta-blocker withdrawal (41, 42), the 2014 ACC/AHA guidelines give a class I recommendation with B level evidence for the continuation of beta-blockers during the peri-operative period in patients who chronically use them (43).
Inappropriate Pain Management
Acute pain is associated with elevated BP via sympathetic nervous system activation, leading to increases in peripheral vascular resistance, heart rate, and stroke volume. In addition stimulation of the neuroendocrine system via hypothalamic-pituitary-adrenal axis leads to pain-induced HTN (44). Inadequate peri-operative pain control is one of the most common causes of HTN immediately post-transplant. Opioid analgesic is commonly used for pain management during immediate post-transplant period. Non-steroidal anti-inflammatory drugs (NSAID) can provide effective pain relief, but should be avoided in the post-transplant period because of their negative renal effects, including reduced renal plasma flow particularly in patient without establishing renal allograft function or those with DGF.
Early Post-transplant Period
There has not been a period after kidney transplantation that has been classically defined as the early post-transplant period. However, we found that systolic HTN (≥140 mmHg) occurred at the mean duration of 26–50 weeks after kidney transplantation (45), while the baseline renal allograft function and stable dose of maintenance immunosuppressive medications are generally reached around 3–6 months post-transplant. Therefore, we define the early post-transplant period for this review as the time between 24 and 52 weeks post-transplantation. During which time several factors contribute to HTN.
Weight Gain
Between 6- and 12-months post-transplantation, weight gain commonly occurs (46, 47). The mean weight gain and increase in BMI at 1 year post-transplant is 6.2 ± 10.7 kg and 2.1 ± 3.8 kg/m2, respectively (47). Obesity (BMI ≥ 30 kg/m2) after kidney transplantation is significantly associated with post-transplant HTN (48). Positive fluid intake from intra-and post-operative IV fluid is a common cause of weight gain during the early post-transplant period (49). The amount of sodium in the IV fluid can contribute to HTN. However, after regaining renal allograft function, urinary excretion of the fluid gain can mobilize sodium and water. This can partly help to control BP.
Calcineurin Inhibitors
The prevalence of HTN in kidney transplant recipients is between 70 and 90% (50, 51), which is greater than the prevalence during pre-CNI era of 40–50% (52, 53). There are two main mechanism of CNI-induced post-transplant HTN resulting from interfering vascular tone and renal sodium transport handling.
Altered vascular tone
Both increased vasoconstriction and impaired vasodilation contribute to CNI-induced post-transplant HTN; although, the latter is thought to be the main mechanism (54).
Renal vasoconstriction. Renal vasoconstriction is mediated by endothelin, a vasoconstrictor, rather than angiotensin II, as captopril does not prevent CsA-induced renal vasoconstriction (55). However, the vasoconstrictive effect on renal or systemic vasculature remains unclear (56). There are inconsistent reports on the effect of angiotensin II on blood vessels. CsA causes local effect on smooth muscle cells by increasing the number of angiotensin II type 1 receptors resulting in vasoconstriction (57).
Renal vasodilation. Impaired vasodilation is a result of CNI-induced reduction of nitric oxide, a vasodilator. CNIs inhibit inducible nitric oxide synthase in vascular smooth muscle cells (58).
Increased renal sodium transport handling
Sympathetic nervous system activation. CsA causes sympathetic excitation and subsequent sodium retention (59). There is a link between CNI-induced HTN and phospho-protein synapsin found on microvesicles in renal sensory nerve endings (60).
With-No-K(Lys)—STE20/SPS1-related proline/alanine-rich kinase—Sodium Chloride Cotransporter (WNK-SPAK-NCC) pathway. CNI induces salt-sensitive HTN via activation of the WNK- SPAK-NCC pathway similarly to a rare genetic form of HTN, called familial hyperkalemic hypertension (FHHt, also called Gordon syndrome or pseudohypoaldosteronism type 2) (61). FHHt results from a loss-of-function mutation of WNK kinases that activate NCC (62) and manifests as hyperkalemic hypertension with a non-anion gap metabolic acidosis and hypercalciuria (61). In normal circumstances, calcineurin, a phosphatase, inhibits some kinases, including the kinases WNK3, WNK4, and SPAK in the distal convoluted tubule (DCT), which interact to phosphorylate and activate NCC (54, 63). CNI inhibits calcineurin and leads to phosphorylation and activation of WNK and SPAK kinases and NCC. Therefore, sodium and chloride reabsorption in the DCT is increased and salt-sensitive HTN occurs. Low fractional excretion of chloride supports increased NCC activity (64), and a decreased plasma aldosterone level is consistent with volume expansion (54). From a mechanistic standpoint, thiazide diuretics should be effective for CNI-induced HTN (54, 65).
Steroids
Since steroids can cause HTN, steroid avoidance or withdrawal (SAW) maintenance immunosuppressive medication regimens can be considered. However, the effect of SAW on post-transplant HTN has yielded conflicting data. Curtis et al. (10) showed that the prevalence of HTN decreased in patients taking alternate-day steroid therapy. A systematic review and meta-analysis revealed that steroid avoidance or withdrawal significantly decreases CV outcomes including HTN but increases risk of acute rejection (66). It is common for steroids to be reintroduced after a diagnosis of acute rejection in recipients who were initially managed with a SAW regimen. A randomized control trial, however, demonstrated no difference in blood pressure change between alternative day and daily prednisone (67). SAW protocols should be considered in select patients, specifically those who would be at greater risk for CV outcomes but be immunologically lower risk of rejection.
Hypertensive Donor Kidney
Salt intake can lead to water retention and HTN, especially in salt-sensitive individuals (68). In addition, several animal studies have demonstrated a renal role in the development of HTN (69–73). Kidney transplantation from idiopathic hypertensive rat donors to genetically normotensive recipients led to post-transplant HTN resulting from decreased renal salt excretion. On the other hand, transplantation from genetically normotensive rat donors to hypertensive rats with pre-transplant bilateral native nephrectomy led to normotension after transplantation (74).
A study in kidney transplant recipients who had undergone native nephrectomy before kidney transplantation from normotensive donors found that all recipients were normotensive post-transplant without need for antihypertensive therapy (75). Another study in kidney transplant recipients from normotensive families found a net increase in antihypertensive requirement antihypertensive requirement after kidney transplantation when kidneys came from donors with hypertensive families compared to those receiving kidneys from donors with normotensive families. However, in recipients with familial HTN, kidney transplantation from any type of kidney did not influence the prevalence of post-transplant HTN (76). In addition, among kidney transplant recipients with no family history of HTN, patients receiving kidneys from donors with family history of HTN required 10 times greater antihypertensive medication requirement as compared to those who receive kidneys from donors without a family history of HTN (77). This all suggests a role for kidney and genetic kidney disease in developing HTN.
Transplant Renal Artery Stenosis (TRAS)
Vascular complications are one of the major causes of poorer transplant outcomes. TRAS is a well-recognized and common vascular complication, which leads to worse renal allograft function and CV complications including post-transplant HTN. It is crucial to recognize this early since treatment cause reverse those negative outcomes. Approximately 1–5% of post-transplant HTN is secondary to TRAS (78, 79). However, because of differing definitions applied in studies, the incidence of TRAS was reported to increase from 1 to 23% (80). Wong et al. (81) reported that the prevalence of TRAS increased from 2.4 to 12.4% after the introduction of color Doppler ultrasonography (CDU) in 1985, likely related to improved detection with non-invasive testing. TRAS may occur at any time after kidney transplantation but is generally diagnosed between 3 and 24 months post transplantation (82–84). Unlike renal artery stenosis (RAS) in non-transplant patients, pathogenesis of TRAS is complex and involves non-immunological and immunological factors (81, 85). The non-immunological factors include vascular damage at the time of surgical anastomosis between the donor renal artery and recipient artery as well as the presence of native vascular diseases in both donor and recipient arteries (86). Because the recipient iliac artery, not the abdominal aorta, is the most common vascular target for donor renal artery anastomosis, the connection between these smaller arteries may be predisposed to narrowing and subsequent development of TRAS physiology (87). Fibromuscular dysplasia is not a common cause of RAS in transplant patients. Immunological factors leading to vascular endothelial dysfunction can cause TRAS. Other transplant-related risk factors have been reported including cytomegalovirus (CMV) infection (88). Similar to non-transplant patients, atherosclerotic disease can cause TRAS, but pathogenesis of atherosclerotic TRAS may be different. Atherosclerotic TRAS is unlikely to occur in the early post-transplant period unless there is pre-existing donor and/or recipient atherosclerotic diseases (89). In addition to traditional risk factors for atherosclerotic disease, some immunologic factors may be at play in atherosclerotic TRAS. For example, diffuse stenosis may suggest immune-mediated vascular endothelial injury (90). Moreover, similar histological findings between vascular rejection and stenotic transplant renal arteries (82, 91) as well as an association between post anastomotic TRAS and de novo class II donor-specific antibodies (92) raise the possibility of immunologic contribution to atherosclerotic TRAS.
Symptoms and signs of TRAS are non-specific; however, common clinical clues that should lead to a work-up for TRAS are unexplained worsening renal allograft function or uncontrolled HTN (79). Since renal hypoperfusion causes increased renin, angiotensin, and aldosterone, salt retention can lead to peripheral edema, congestive heart failure, and flash pulmonary edema. Notably, paradoxical normotension or hypotension can be seen with use of high-dose diuretics and/or angiotensin-converting enzyme inhibitors (ACEI) or angiotensin II receptor blockers (ARB) (93). Bruits over transplant renal allografts site are common but non-specific. Bruits may be related to other causes like arteriovenous fistula (AVF) in a kidney after biopsy (94).
Several imaging studies can be used to diagnose TRAS. CDU is often the initial imaging study used because it is non-invasive, widely available, and relatively inexpensive. However, the image quality and interpretation are dependent on ultrasonographer technique and experience. Peak systolic velocity (PSV) of the main renal artery and poststenotic intrarenal arterial resistive index (RI) are used to determine and grade the severity of TRAS (79). However, since the diagnostic value of CDU is operator dependent, other imaging modalities may be utilized to verify the diagnosis. Renal artery computed tomography (CT) or magnetic resonance angiography (MRA) should be utilized to further delineate or confirm the diagnosis. However, the risk of contrast-induced nephropathy (CIN) and nephrogenic systemic fibrosis would need to be considered and may limit the use of these imaging studies. Renal artery angiography remains the gold standard diagnostic test for TRAS, but it is invasive and can lead to CIN. Carbon dioxide (CO2) angiography can mitigate some of the risk of CIN, but, in most cases, small amounts of IV contrast are still required to attain sufficiently detailed images.
Three therapeutic options for TRAS are pharmacological therapy alone or pharmacologic therapy in addition to renal artery angioplasty with stenting or surgical revascularization (90).
For pharmacological therapy, the pathophysiology of TRAS is similar to that of bilateral RAS in the non-transplant population. A decrease in renal blood flow to a transplant renal allograft causes increased RAAS activation resulting in salt and water retention and subsequent HTN. ACEI or ARB plus diuretics are a likely effective regimen for BP control. However, this pharmacologic therapy is limited by decreasing renal function resulting from decreases in systemic BP leading to reduced renal perfusion and intraglomerular pressure below the limitation of autoregulation. This increases efferent arteriolar resistance mediated by angiotensin II. By blocking the action of angiotensin II, autoregulation is blunted, and GFR is reduced (95). Volume depletion from diuretics may also contribute to rising serum creatinine in these instances. For this reason, it is not common to utilize RAAS and/or diuretics during early post-transplant period. Once baseline renal allograft function is established, however, and there are no contraindications for RAAS and/or diuretics, such as rising serum creatinine, hyperkalemia, or volume depletion, medical therapy may be utilized to control blood pressure (79). Statins and acetylsalicylic acid may also be part of pharmacological therapy although there is no clear evidence for these use specifically in TRAS (79).
Patients with worsening serum creatinine and/or uncontrolled HTN attributable to TRAS should undergo renal artery angioplasty with stenting. There is no randomized controlled clinical trial (RCT) comparing the efficacy of angioplasty ± stenting vs. surgical revascularization vs. pharmacological therapy alone in the kidney transplant population. Data from non-transplant patients from 4 RCT (96–99) did not show the benefit of angioplasty on BP control, and 4 RCT (97, 99–101) did not demonstrate better renal outcomes. However, several observational studies demonstrate highly successful both technical and clinical outcomes (88–100 and 65–94%) with varied procedure-related complications (0–25.5%) in kidney transplant recipients (102–105). Long-term renal allograft and patient survival with up to 21 years of follow-up is not different between patient with TRAS undergoing percutaneous angioplasty or stent placement and patients that without TRAS (105). A single study showed that both immediate and long-term success rate were lower in angioplasty compared to surgical revascularization; however, the former was still a preferred procedure when TRAS is recent, linear, and distal. Whereas, surgical revascularization is performed primarily in individuals with kinking and proximal TRAS (106). There are several surgical techniques, including resection and revision of the anastomosis, saphenous vein bypass graft of the stenotic segment, localized endarterectomy, and excision/reimplantation of the renal artery (90, 107). Generally, surgical revascularization is reserved for cases of unsuccessful angioplasty. Long-term renal allograft function and survival with a mean follow-up of 9.8 ± 2.1 years has demonstrated the efficacy and safety of surgical revascularization (108).
Late Post-transplant Period
Apart from the above-discussed factors contributing to HTN in the early post-transplant period, some factors may contribute to HTN in the late post-transplant period.
Chronic Renal Allograft Dysfunction
A causal relationship between chronic renal allograft dysfunction and HTN has not been proven but follows on from a logical examination of pathophysiology. Renal allograft injury—both acute and chronic—is associated with HTN. Common causes of renal allograft injury include acute allograft rejection—both acute antibody-mediated—and acute cellular rejection. Chronic renal allograft injury results from persistent antibody-mediated rejection, interstitial fibrosis/tubular atrophy, thrombotic microangiopathy, and recurrent glomerular disease in the renal allograft (19). An animal study in rats showed that HTN alone led to chronic allograft nephropathy (CAN). This study utilized rats with deoxycorticosterone acetate and salt-induced hypertension and compared them to normotensive rats. The hypertensive rats had higher proteinuria, smooth muscle cell-growth factors, platelet-derived growth factor (PDGF), tubular cell expression of proliferating cell nuclear antigen, extracellular matrix deposition, and presence of class I and II major histocompatibility complex (MHC). This suggests that HTN and immunologic factors affect the expression of growth factors in renal allografts and may be the cause of chronic renal allograft dysfunction (109).
Fibroblast Growth Factor (FGF) 23
After kidney transplantation, FGF23 levels decrease. The rate at which FGF23 levels normalize after transplantation is dependent on shorter dialysis vintage before kidney transplantation and the time to normalization of renal allograft function including mineral metabolism (110, 111). FGF 23 is a known independent risk factor of renal allograft loss, cardiovascular and all-cause mortality in kidney transplant recipients (112, 113). The relationship of FGF 23 levels in the pre- and post-kidney transplant period to BP level or development of HTN after kidney transplantation is unknown. However, given that a higher level of FGF23 is associated with increased SBP and DBP as well as an incident HTN in 1,758 non-hypertensive young adults without CKD or CVD (114), this association may hold true for the transplant population. Despite this, further studies are necessary.
Obstructive Sleep Apnea
Prevalence of obstructive sleep apnea (OSA) in kidney transplant recipients varies with the severity of OSA. The prevalence of mild, moderate, and severe OSA in kidney transplant recipients participating in Sleep disorders Evaluation in Patients after kidney Transplantation (SLEPT) Study who had a mean eGFR of 52 ± 19 ml/min/1.73 m2 was 18, 11, and 14%, respectively. In this study, kidney transplant patients with OSA required a significantly higher number of antihypertensive medications and tended to have higher SBP as compared to non-OSA patients (115).
Similar to the non-transplant population, risk factors for OSA in kidney transplant recipients are male gender, obesity, use of hypnotic drugs, presence of severe comorbidity (e.g., heart disease, cerebrovascular disease, peripheral vascular disease, diabetes mellitus), and impaired kidney function (116). There is a relationship between HTN, CKD, and OSA, and they have common risk factors and pathophysiology, including increased sympathetic activity, endothelial dysfunction, increased inflammatory markers, hyperaldosteronism, and chronic volume overload. In addition, these three conditions are associated with CV risks, morbidity, and mortality (117). If any of these three diseases is uncontrolled or progress the other two conditions tend to worsen or become more difficult to manage. Therefore, appropriate management of OSA is an essential component of the antihypertensive therapy in kidney transplant recipients who later develop renal allograft dysfunction especially with resistant HTN. Moreover, kidney transplant recipients who have risk factors for OSA should undergo early screening (115, 118) and be appropriately treated.
In summary, the pathogenesis of post-transplant HTN may be categorized by the time period during which hypertension develops after kidney transplantation. Another approach to differentiate the etiologies of post-transplant HTN is by dividing the causes into immunological and non-immunological factors. Immunological factors, as discussed, mainly include renal allograft dysfunction and immunosuppressive medications; while non-immunological factors involve the donor, recipient, and surgical related factors (Figure 3).
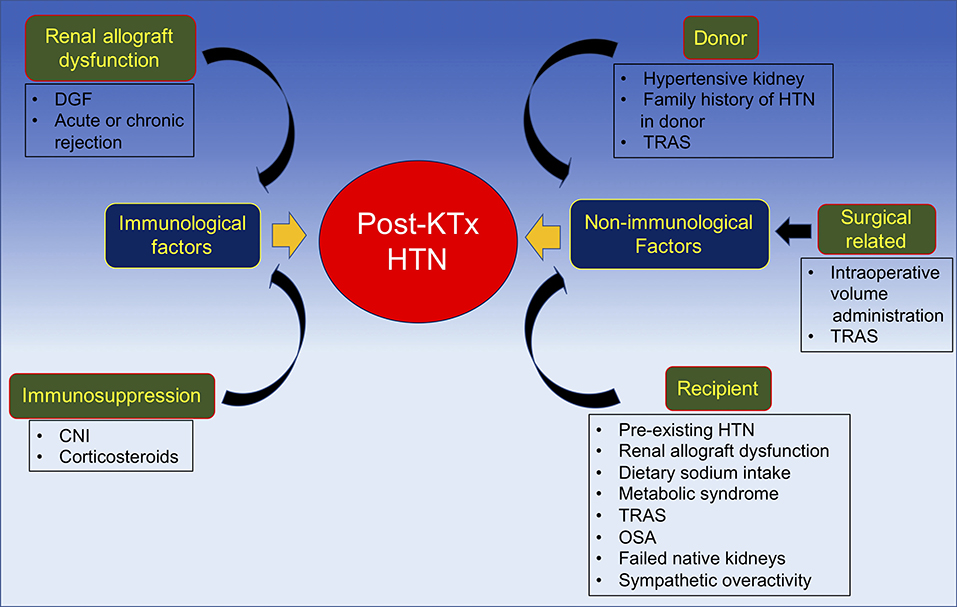
Figure 3. Pathogenesis of post-kidney transplant hypertension divided into immunological and non-immunological factors. CNI, calcineurin inhibitor; DGF, delayed graft function; HTN, hypertension; KTx, kidney transplantation; OSA, obstructive sleep; TRAS, transplant renal artery stenosis.
Outcomes of Hypertension After Kidney Transplantation
Pre-transplant BP is associated with renal allograft and patient survival outcomes after kidney transplantation. Very low pre-transplant SBP (<110 mmHg) and DBP (<50 mmHg) are associated with a decrease in renal allograft loss. Specifically during dialysis lower pre- and post-dialysis DBP are associated with better patient survival post-transplantation (119).
During the post-transplant period, elevated BP is associated with poorer renal allograft and patient outcomes. However, as discussed above, various forms of renal allograft injury are also associated with post-transplant HTN. Several studies have demonstrated an association between post-transplant HTN and renal allograft failure (120–122). Opelz et al. (120) conducted a retrospective study of 29,751 kidney transplant recipients followed for over 7 years. Increased post-transplant SBP and DBP were associated with progressively decreased renal allograft function and death-censored chronic graft failure. Another study from the same cohort database examined the association between changes in BP levels at 1- and 3-years post-transplantation and long-term graft outcomes up to 10 years following transplantation. They found that patients with a SBP >140 mmHg at 1 year who were controlled to a SBP ≤ 140 mmHg at 3 years post-transplantation had improved renal allograft outcomes and reduced CV death compared to those with persistent SBP of >140 mmHg both at 1 and 3 years post-transplantation (123).
There is insufficient information about CV and mortality outcomes related to isolated diastolic HTN in kidney transplant recipients, and more studies are required to determine appropriate management targets for isolated diastolic HTN in this population.
Cardiovascular Outcomes-Related to Post-transplant Hypertension
HTN is one of the most important risk factors for heart failure, particularly heart failure with preserved ejection fraction (HFpEF) (124). Kidney transplant recipients with left ventricular hypertrophy (LVH) or HFpEF have increased morbidity and mortality and are at high risk for cardiac-related events. Both pressure and volume overload contributed to LVH, which is further exacerbated by age, genetic factors, vascular hemodialysis access, dialysis vintage, diabetes, and blood pressure (125).
Renal insufficiency is involved in the pathogenesis of HFpEF (126) and causes salt-sensitive HTN (126, 127). Importantly, a vicious cycle of cardio-renal dysfunction can result from salt and volume overload (126). Therefore, BP control, renal allograft function, and heart function are closely related. Uncontrolled HTN after kidney transplantation leads to structural damage to both the renal allograft and heart eventually resulting in decreased renal and cardiac functions.
Blood Pressure Measurement
BP is one of the most common vital signs obtained in all clinical settings; however, it may frequently be unreliable due to variation of physiologic response to internal and external stimuli as well as inappropriate BP measurement techniques. Reliable BP measurement should be mandatory in clinical practice and can be standardized with the following definitions: the mean of three non-invasive BP measurements is referred to as office blood pressure (OBP); recording at least twice the daily average of two home blood pressure readings over a minimum of 4 days is referred to as home blood pressure monitoring (HBPM), and 24-h ambulatory blood pressure monitoring (24-h ABPM), which requires wearing an electronic blood pressure measurement device to record and averages multiple readings over a 24 h period (128). Purpose, clinical context, and practicality should be taken into consideration when selecting the appropriate BP measurement methods. The different BP measurement methods provide different information, which can be useful for sorting out the pathophysiology of post-transplant HTN. It is important to be knowledgeable about the different definitions for HTN with each method in order to appropriately manage BP after kidney transplantation.
A 24-h ABPM provides the average of both day and night BP readings. Physiological decreases in nocturnal BP further classifies patients into dippers, non-dippers, and reverse dippers. Lee et al. (129) demonstrated a significant decrease in the nocturnal reduction in SBP (ΔSBP) after kidney transplantation. In addition, decrease in ΔSBP was associated with a lower renal allograft function. In this study, the mean OBP and 24-h ABPM did not change at 1-year post-transplantation when compared to the BP measurements before kidney transplantation. However, they found that the proportion of patients who took antihypertensive medications and the numbers of antihypertensive medications required were significantly decreased after kidney transplantation.
A 24-h ABPM can address and assist with the common misclassification of HTN diagnosed traditionally by OBP or HBPM. Compared with OBP, a 24-h ABPM led to 61% disagreement in diagnosis (58% and 3% due to masked and white-coat HTN, respectively) (130).
Although a 24-h ABPM can provide useful information to diagnose patterns of HTN like white coat and masked HTN, OBP and HBPM are more commonly used in clinical practice. Since transplant renal allografts are very sensitive to BP hemodynamic changes, HBPM appears to be a common utilized method of following BP after kidney transplantation. One study in patients who were 1–10 years after kidney transplantation revealed a higher correlation between a 24-h ABPM and HBPM than 24-h ABPM and OBP (131).
Though elevations in both day and night SBP obtained from 24-h ABPM were associated with risk of declining renal allograft function, and nighttime elevation in SBP exhibited a stronger association (132). An elevated 24-h average SBP was significantly associated with a composite endpoint of graft loss, cardiovascular events and death over a 5-year follow-up period in kidney transplant recipients with diabetes, lower eGFR, proteinuria, young age, and who were female (133). An average (day and night) 24-h DBP between 65 and 80 mmHg was associated with greater long term survival during a 9-year follow-up period after kidney transplantation when compared to those who had average DBP <65 or >80 mmHg (134).
Although HBPM is widely available, better correlated to 24-h ABPM, and superior to OBP when predicting hard outcomes, we used HBPM in conjunction with OBP for our kidney transplant patients to minimize misclassification of HTN diagnosis, monitor possible white coat or masked HTN, and adjust antihypertensive medications.
Blood Pressure Management
Non-pharmacologic interventions, such as diet, exercise, and stress reduction, should always be part of treatment of HTN. Since the majority of kidney transplant recipients have pre-transplant HTN requiring antihypertensive medications (persistent HTN) and only small number of patients become normotensive without blood pressure medications (recovered HTN), pharmacological intervention remains the cornerstone of BP control in this population. Additionally, other interventions specific to certain etiologies of resistant HTN, such as transplant renal artery angioplasty ± stenting and treatment for OSA, should be implemented. Renal sympathetic denervation of the native kidneys either by bilateral native nephrectomy or catheter ablation is also treatment option for resistant HTN in this population (Table 2). In this review, we focus on antihypertensive medications and briefly review bilateral native nephrectomy and RDN of native kidneys.
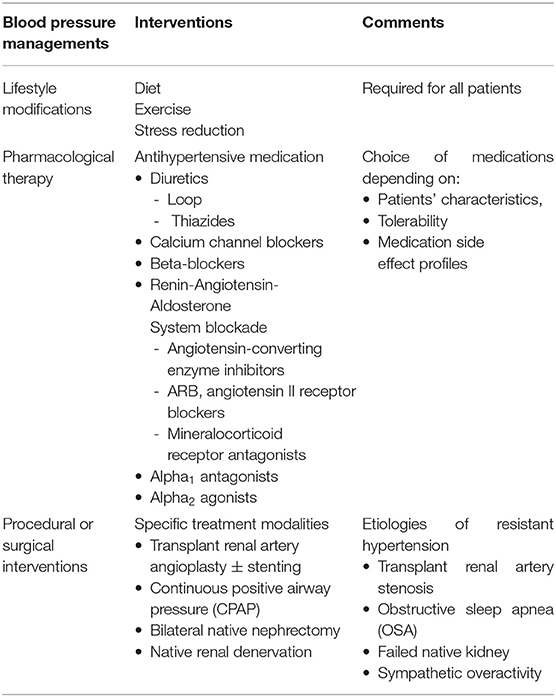
Table 2. Common interventions for post-kidney transplant hypertension.
Pharmacological Managements
Use of Antihypertensive Medications
A recent retrospective cross-sectional analysis from a single transplant center in Poland reported the trend of commonly used antihypertensive medications in their kidney transplant recipients over 14 years. They examined antihypertensive therapy data from transplant recipients first outpatient visits from 2001, 2006, 2011, and 2014 (135). Beta-blockers were the most common antihypertensive medication used in this cohort followed by calcium channel blockers. Uses of ACEI, diuretics, and alpha-blockers were about the same. ARB therapy was utilized least. The average number of antihypertensive medications required was reported to be 2.24 ± 1.03–2.55 ± 1.25. This is slightly lower than what has been reported for CKD patients who on average require 3.5 medications (136). The lower number of antihypertensive medications is likely from regaining kidney function after transplantation and improved solute clearance, including salt and volume excretion by renal allograft.
Blood Pressure Control During Peri-Transplant Period
Antihypertensive Medications
The majority of advanced CKD and ESRD patients have HTN. Volume overload is the most common cause of uncontrolled BP in ESRD patients (137). In the immediate pre-transplant period, particularly in deceased donor renal transplantation when surgeries are often performed emergently (138), BP can be uncontrolled particularly if the patient was due for dialysis at the time of surgery. Volume-dependent HTN is managed with fluid removal during dialysis. However, it is common practice to remove less fluid from patients who are about to undergo transplant, leaving them with a post-dialysis weight slightly above their established dry weight in an effort to avoid intra- and post-operative hypotension. Additionally, ACEI and ARB are generally held following a transplant. An alpha2 agonist like clonidine, however, may need to be continued during peri-transplant period to avoid rebound HTN.
Providers taking care of kidney transplant recipients have different opinions when selecting antihypertensive medications during peri- and post-transplant period. Their decision may mainly be driven by protocols or common practices followed at their kidney transplant centers. Specific information relevant to kidney transplantation should guide the selection of antihypertensive medications, but, as in other scenarios, it is rational to individualize BP management for certain kidney transplant recipients.
Diuretics
Diuretics are not commonly used as the first line antihypertensive medication in kidney transplant recipients. They may cause volume depletion, electrolyte disturbances, and worsening renal allograft function. However, they are indicated and should not be avoided in certain patients in the peri-transplant period.
Loop Diuretics
Volume control rather than BP control is the indication for loop diuretics in kidney transplant recipients, especially during immediate and early post-transplant periods. Kidney transplant recipients generally receive peri-transplant IV fluid to keep up with an increased urine output from a new functioning renal allograft. Volume overload presenting with peripheral edema, pulmonary congestion, or HTN may occur when the establishment allograft function lags behind the volume resuscitation provided. Diuretics can be used to control both volume and BP in this common scenario.
There is also evidences that loop diuretics have a vasodilator effect and can decrease edema, congestion, and oxygen requirements. Theoretically they can decrease ischemic renal injury and risk of DGF in kidney transplant recipients. However, despite their common use in clinical practice, there is no consistent data to support using loop diuretic to increase urine output and prevent DGF. In addition, there is no strong evidence showing an association between loop diuretic and improvement in initial or long-term graft function (139). On the other hand, loop diuretic use has been associated with increased risk of UTI during the first 5 years after kidney transplantation (140). This is because there use depletes medullary NaCl gradient (140), which is known to modulate the adaptive and innate immune response (141). Renal medullary myeloid mononuclear cell phagocytes are classified as pro-inflammatory (M1) and reparative/profibrotic (M2) cells. Decreasing M1/M2 ratio from altering renal medullary NaCl gradient with loop diuretics impairs the natural antibacterial host response (142–144).
Furosemide is the most commonly used loop diuretic. It has been used to predicting progression of AKI with the so-called furosemide stress test (FST) in non-transplant patients (145, 146). Loop diuretics can also provide prognostic value in regard to renal allograft function in the immediate post-transplant period. An inadequate response, urine output <350 ml within 4 h, to a single IV furosemide dose of 1.5 mg/kg given 3 h after renal allograft anastomosis predicts increased risk of DGF (146).
Given the wide range of risks and benefits with loop diuretics, clinicians should individualize their use in transplant recipients. Additional RCTs are required to determine the efficacy and safety of loop diuretic in kidney transplant recipients.
Thiazides
Thiazide diuretics are commonly used antihypertensives in general population. They are however uncommon in the management of kidney transplant recipients because of their metabolic side effects which include hyperglycemia, hyperuricemia, hypercalcemia, and hyponatremia (147, 148).
Thiazides however, may theoretically control CNI-induced HTN. Since CNI-induced HTN is salt sensitive (59, 149) via activation of the WNK-SPAK-NCC pathway (63, 150), thiazides, which inhibit Na-Cl co-transporter, should control CNI-induced HTN. A randomized non-inferiority crossover trial comparing the effects of chlorthalidone and amlodipine in kidney transplant recipients taking tacrolimus showed that chlorthalidone was not inferior to amlodipine in controlling BP (147). Given the vasodilator effect of calcium channel blockers, they are commonly used antihypertensives in kidney transplant recipients intended to counteract the systemic and renal vasoconstrictive effect of CNI via endothelin I (151–153). However, their side effects, including peripheral edema and proteinuria, may make thiazides a suitable alternative for BP control particularly in individuals with peripheral edema (147).
Although loop diuretics are generally favored over thiazide diuretic for volume control, they can lead to additional renal magnesium wasting. Thiazides can be considered in transplant patients who already have other reasons for and often struggle with hypomagnesemia. However, because CNI and loop diuretics act on the common mechanism to lower luminal electro-positivity decreasing paracellular magnesium reabsorption, using a loop diuretic in concert with CNI may not cause further urinary magnesium loss (154). A retrospective study in heart transplant recipients taking CNI showed that the group receiving loop diuretic did not have lower serum magnesium levels or require higher magnesium supplementation compared to the group not receiving loop diuretics (154). Thiazide diuretics, on the other hand, increase serum magnesium when used with CNI. In non-transplant patients without CNI, acute thiazide diuretic use results in increased magnesium reabsorption. However, long-term thiazide diuretic use can result in urinary magnesium wasting when there is concomitant hypokalemia (154). Transplant recipients on CNIs commonly have hyperkalemia, and, therefore, thiazide diuretics may not worsen hypomagnesemia from CNI but instead increase magnesium levels. The previously mentioned study demonstrated that heart transplant recipients receiving thiazide diuretics had higher serum magnesium level and required less magnesium replacement compared to those not on thiazides (154). Therefore, in the setting of volume overload and concomitant hypomagnesemia, thiazide diuretic may be used to mitigate the hypomagnesemic effect of CNI therapy (154).
Thiazides may be considered for kidney transplant recipients with CNI-induced salt-sensitive HTN and hypomagnesemia.
Mineralocorticoid Receptor Antagonists
Mineralocorticoid receptor antagonists (MCRA) have CV benefits (155, 156) and antiproteinuric effect but are not commonly used antihypertensives in kidney transplant recipients, especially those with impaired renal allograft function. Hyperkalemia is a common side effects of MCRA and it may be worse in kidney transplant recipients who have CNI-induced hyperkalemia.
One pilot study evaluating the antiproteinuric effect of spironolactone 25 mg/day given to 11 kidney transplant recipients whose mean proteinuria was 4.4 ± 1.4 g/day on both ACEI and ARB. Proteinuria was decreased more than 50% with a mean reduction of 85% in nine patients after 6 months on spironolactone. Renal allograft function slightly decreased with GFR, changing from 52 ± 12.7 to 48 ± 14.2 mg/dL, and serum potassium did not significantly increase (4.6 ± 0.4–5 ± 0.62 mEq/L) (157). Another study looked at the safety of eplerenone in kidney transplant recipients with eGFR between 30 and 50 mL/min/ 1.73 m2. After 8 weeks of eplerenone 25 mg/d, nine out of 31 patients had serum potassium of >5 mmol/l, and one patient had serum potassium of >5.5 mmol/l. Mean baseline eGFR was 41 (26–59) ml/min/1.73 m2 after 8 week of eplerenone therapy and was not significantly different between patients who did and did not develop hyperkalemia (36.0 [95% CI 26.0–53.0] vs. 44.5 [95% CI 26.0–59.0], p = 0.17). Having a baseline serum potassium >4.35 mmol/l was associated with increased serum potassium of >5 mmol/l once starting eplerenone (158).
CNIs can cause vascular vasoconstriction via activation of mineralocorticoid receptor in smooth muscle leading to impaired renal allograft function. MCRAs bind mineralocorticoid receptors in principle cells of collecting ducts and are potentially reno-protective by blunting the renal vascular resistance induced by CNI therapy (159).
Spironolactone decreases cardiac left ventricular volume and mass, but studies are required to determine its effect in kidney transplant recipients (160). In addition, there is a lack of evidence in terms of CV and mortality benefits in kidney transplant recipients.
With some evidence for antiproteinuric effect and their established safety in transplant recipients even when in combination with ACEI or ARB, MCRA may be a new option for BP control in individual with CNI-induced HTN and proteinuria.
Beta-Blockers
The cardioprotective effects and survival benefit of beta-blockers make them a favored medication in the general (161) and ESRD populations (162–165). In kidney transplant recipients, a recent retrospective study from 2001 to 2014 showed that beta-blockers are the most common used antihypertensive medication (135). In addition, Aftab et al. (166) conducted a single center retrospective study in 321 kidney transplant recipients over a 10 ± 4 year follow-up period and found that those who took beta-blockers had a significant survival advantage compared to those who did not. Moreover, the authors showed that beta-blocker has additive effect on ACEI or ARB with greater survival in kidney transplant patients on this combination compared to those who received either medication alone or neither. The possible protective mechanism of beta-blocker is via mitigation of the sympathetic nervous system, which is stimulated in failed native kidneys (167–169). In addition, beta-blockers decrease proinflammatory cytokines, which are known to increase the risk for atherosclerosis (170).
Although beta-blockers provide survival benefit in kidney transplant recipients, they can cause metabolic side effects, including proteinuria, hyperkalemia, and masking of hypoglycemic symptoms. Therefore, caution should be used when using beta blockers in kidney transplant recipients who are at risk to develop these side effects.
Calcium Channel Blockers
Calcium channel blockers inhibit entry of calcium into vascular smooth muscle cells, resulting in vascular vasodilation (171). Since the vasoconstrictive effect of CNIs leads to post-transplant HTN (172), calcium channel blockers are thought to be an appropriate agent for post-transplant HTN. Theoretically their vasodilatory effect can counteract the vasoconstrictive effect of CNIs and improve BP control (171, 173, 174).
In addition to blood pressure control, calcium channel blockers also prevent post-transplant acute tubular injury (ATI) or DGF. One prospective trial looked at diltiazem use in patients who received deceased donor renal transplantation (DDRT). Participants were randomized to kidneys that had been given diltiazem (Euro-collin's solution (20 mg/l) at time donor nephrectomy; meanwhile, the recipients received a bolus injection of diltiazem (0.28 mg/kg) pre-operatively followed by an infusion 0.0022 mg/min/kg for 2 days before transitioning to oral diltiazem vs. a control group, which did not receive diltiazem but otherwise received the same induction immunosuppression regimen with CsA. The diltiazem group had lower incidence of ATI, higher GFR with primary function, and lower incidences of rejection at 1 month post-transplant. They however had higher CsA level compared to control group. The total CsA required was lower in the diltiazem group, as might be expected, to achieve comparable CsA levels between the two groups. The lower post-transplant ATI in the diltiazem group was postulated to be secondary to lower ischemic damage in the renal allograft and reduced CNI nephrotoxicity (175). A Cochran review of 13 trials with a total of 724 participants also concluded that peri-operative calcium channel blocker significantly decreased incidence of post-transplant ATI and DGF. There was no difference in graft loss, mortality, or requirement for hemodialysis, but there was insufficient evidence to draw conclusions regarding adverse drug reactions (176). There was another randomized placebo control study comparing renal allograft outcomes between patients who received pre-transplant isradipine vs. those who received a placebo. The former group had greater renal allograft function as compared to the latter. However, the rate of DGF and biopsy-proven acute rejection was not different in this study (177).
Although calcium channel blockers provide better renal allograft function, several studies showed no difference in terms of BP control when using verapamil compared to enalapril or doxazosin (178). No difference in GFR, serum creatinine level, protein excretion, or BP when nitrendipine or nifedipine was compared to the placebo (179). Moreover, one retrospective study showed 2.26 time greater risk of ischemic heart disease in kidney transplant recipients who received dihydropyridine calcium channel blockers (180).
The evidence for the benefits of calcium channel blockers appears inconsistent; however, a recent systematic review and meta-analysis of 60 trials including 3,802 patients showed that, compared to placebo or no treatment, calcium channel blockers decreased graft loss [risk ratio (RR) of 0.75, 95% confidence intervals (CI) 0.57–0.99] and increased GFR [mean difference (MD) 4.5 mL/min, 95% CI 2.2–6.7]. Compared to ACEIs, patients treated with calcium channel blockers tend to have a higher GFR; although there is inconclusive data regarding graft loss between these two groups (181). Given the available information, calcium channel blockers remain a preferred antihypertensive agent for kidney transplant recipients barring there are no specific indications for other antihypertensive agents or contraindications to calcium channel blocker therapy.
Adverse drug reactions and drug–drug interaction between calcium channel blockers and other commonly used medications should be taken into consideration. Calcium channel blockers can result in peripheral edema and muscle weakness especially when used in combination with steroids. Gum hyperplasia is also more common when calcium channel blockers are used with CsA (171). Although dihydropyridine calcium channel blockers do not inhibit the cytochrome P450 (CYP) 3A4 isoenzyme, they are metabolized by a CYP3A4 isoenzyme and can compete with CNI. This leads to increase in both calcium channel blocker and CNI exposure levels. Non-dihydropyridine calcium channel blockers inhibit CYP3A4 isoenzyme and significantly increase CNI level.
Angiotensin-Converting Enzyme Inhibitors (ACEI) and Angiotensin II Receptor Blockers (ARB)
Since proteinuria is a surrogate marker for renal disease, lowering proteinuria is one strategy to slow the progression of CKD. ACEIs and ARBs are antihypertensive agents with known antiproteinuric effect. ACEIs inhibit angiotensin-converting enzyme, which converts renin to angiotensin. ARBs act on angiotensin II receptors and subsequently inhibit RAAS pathway. A systematic review and meta-analysis of 21 clinical trials, including 1,549 patients, revealed no difference in the MAP change between ACEI or ARB group and the control group. Serum potassium was also not different between the two groups. However, the ACEI or ARB group had a decrease in proteinuria, eGFR, and hematocrit when compared to the control group. There was insufficient data regarding the benefits of ACEI or ARB therapy in terms of patient or renal allograft survival (182). In the aforementioned systematic review and meta-analysis, eGFR in the ACEI group was not different from that in the placebo group but was lower than the calcium channel blocker group. A comparison of data regarding graft loss between ACEI and calcium channel blocker therapy was inconclusive (181).
RAAS activation is associated with interstitial fibrosis and tubular atrophy (IF/TA), one of the most common causes of renal allograft loss (183, 184). The ACEI perindopril was shown to prevent cortical interstitial expansion, a marker of fibrosis in CKD patients with biopsy-proven diabetic nephropathy (185). One RCT in kidney transplant recipients who received losartan 100 mg daily within 3 months post-transplant and continued it for 5 years compared to matched placebo controls did not show a benefit of ARB therapy in terms of reduction in a composite interstitial expansion or ESRD from IF/TA (186).
ACEIs and ARBs can lead to regression of LVH in kidney transplant recipients (187, 188), but these same studies did not demonstrate an improvement in all-cause mortality (189). A recent systematic review and meta-analysis demonstrated a survival benefit with ACEI or ARB therapy in kidney transplant recipients but only from pooled cohort studies and not RCTs (190).
Although ACEIs and ARBs provide antiproteinuric effect, they are generally not the drugs of choice during the immediate and early post-transplant periods. This is mainly because they are known to decrease GFR. This side effect is reversible, but it leads to a confusion early on differentiating their role from other causes of renal allograft dysfunction and may lead to unnecessary workups, including invasive investigation, e.g., transplant renal biopsy.
While there is no robust evidence for CV or survival advantages with the use of ACEIs, ARBs, and MCRAs, these antihypertensive medications may be considered. They may be considered in transplant recipients, particularly those with LVH, congestive heart failure, and proteinuria.
Alpha1-Antagonists
Alpha1-antagonists are rarely used as the initial or as a single antihypertensive agent in kidney transplant recipients. Although they reduce BP by decreasing peripheral vasoconstriction and theoretically should counteract with vasoconstrictive effect of CNI, there is no evidence that alpha1-antagonists provide superior antihypertensive effect or slows progression of renal allograft dysfunction when compared to other antihypertensive medications. A study with long-term follow up in 88 hypertensive kidney transplant recipients who were divided into three groups, verapamil, enalapril, and doxazosin, demonstrated that doxazosin provided the same efficacy in BP control compared with the others while maintaining an excellent safety profile. However, up to 38% of patients in the doxazosin group needed additional antihypertensive medications compared to only 8 and 13% in verapamil and enalapril groups, respectively (178). Alpha1-antagonist may have a role as an adjunctive therapy rather than first line antihypertensive agent in transplant recipients.
Alpha2 Agonists
Centrally acting alpha2 agonists work on presynaptic alpha2 adrenoceptors in the central nervous system and suppress central sympathetic activity (191). Specifically, activation of alpha2A receptors causes a sympatho-inhibitory effect and lowers BP. However, stimulating alpha2A receptors in blood vessels causes a peripheral vasoconstriction (192). On the other hand, alpha2B receptor activation leads to a sympatho-excitatory effect (192).
Two of the oldest alpha2 agonists, methyldopa and clonidine, have long been used for BP control (193). Clonidine is currently the most commonly used alpha2 agonist. When used as monotherapy, methyldopa is associated with antihypertensive tolerance, edema, and weight gain. Clonidine is also associated with weight gain and progressive resistance with continued use. Chronic use is not associated with sodium and water retention or body fluid volume expansion (193). It instead causes a decrease in exchangeable body sodium and plasma volume, which may be another antihypertensive mechanism of clonidine (194).
Similar to other groups of antihypertensive medications, clonidine has an effect on renal hemodynamics and lowers renal vascular resistance. A study in 13 essential hypertensive patients who received long-term clonidine showed that it lowered plasma renin activity, modulated renal vascular resistance, and subsequently lowered MAP (195). A study of the effect of clonidine on renal hemodynamics was conducted in six kidney transplant recipients who initially received furosemide for 2 weeks before the addition of clonidine titrated to BP control found that GFR and effective renal plasma flow measured by inulin and amino-hippurate sodium clearances was unchanged (196).
Clonidine is an effective antihypertensive medication. Many ESRD patients who are on kidney transplant waiting lists have uncontrolled HTN and are on clonidine in addition to their other antihypertensive agents. However, as previously discussed, rebound HTN is common after discontinuation of clonidine, and patients who are on clonidine prior to kidney transplant tend to have uncontrolled HTN resulting from this rebound phenomenon. In these patients, clonidine is often re-started and be tapered off during early post-transplant period. Therefore, clonidine is rarely used as a single antihypertensive agent following kidney transplantation.
In summary, there is no drug of choice for BP control after kidney transplantation. Several factors are involved in selecting the appropriate antihypertensive medications include immunologic and non-immunologic factors as well as the time after kidney transplantation.
Different from the non-transplant CKD population, beta-blockers and calcium channel blockers are the most frequently used combination in kidney transplant recipients. Beta-blockers provide a cardioprotective effect for kidney transplant patients, who likely have underlying CAD (166). Calcium channel blockers have vasodilatory effect that counteracts the vasoconstrictive effect of CNIs (197). ACEIs and ARBs, on the other hand, are not routinely used antihypertensive medications in kidney transplant recipients. This is especially true during the early post-transplant period when baseline renal allograft function is not well-established. They can, however, be considered when there is a specific indication for their use, such as proteinuria and posttransplant erythrocytosis. Rising serum creatinine from ACEI or ARB therapy, although reversible, is the main reason they are avoid. This change is serum creatinine can be difficult to differentiate from other causes of worsening renal allograft function, particularly acute renal allograft rejection, leading to unnecessary workups and possibly unnecessary renal allograft biopsies. Similarly to ACEIs and ARBs, diuretics are not generally used as the first line for BP control in kidney transplant recipients. It may be used for volume control at immediate or early post-transplant.
From the characteristics, clinical outcomes, and side effect profiles of antihypertensive medications, we routinely use dihydropyridine calcium channel blockers and/or beta-blockers as first-line antihypertensive medications. Since the majority of patients do not achieve BP control during the immediate or early post-transplant period, the combination of dihydropyridine channel blockers with beta-blockers is commonly used in our transplant center. Once renal allograft function is established and stable, we consider adding or replacing ACEI or ARB for dihydropyridine channel blockers and beta-blockers if there are appropriate indications, such as proteinuria or post-transplant polycythemia.
Table 3 summarizes characteristics of the most commonly used antihypertensive medications and rationales for selecting each BP medication in kidney transplant recipients (154, 175, 176, 198–204).
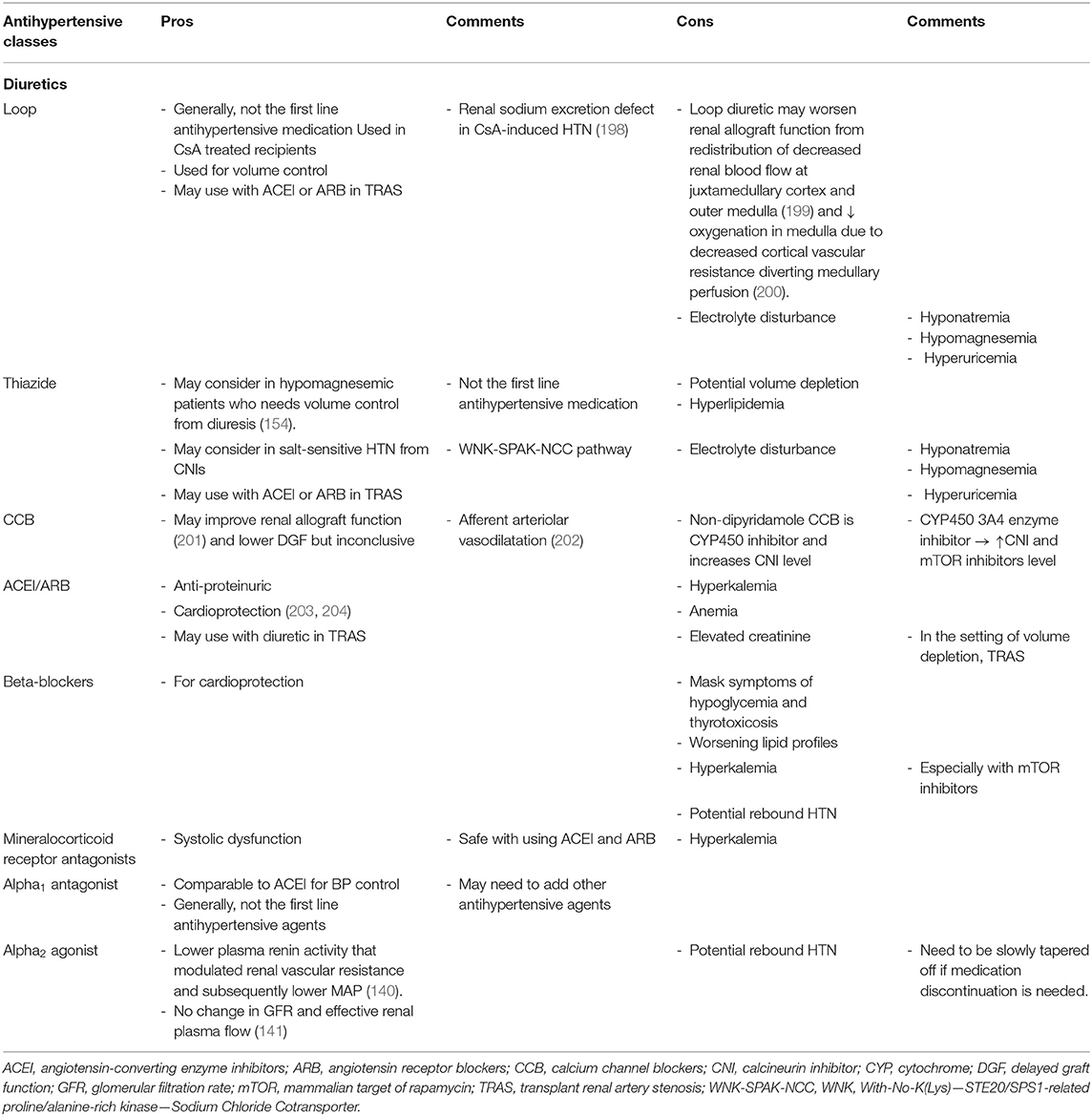
Table 3. Summarized common antihypertensive medications used in kidney transplant patients.
Blood Pressure Guideline for Kidney Transplant Recipients
BP targets have been a controversial topic not only in the non-transplant population but also in kidney transplant recipients. Several professional societies from different countries have created clinical practice guidelines with some similarities and differences (Table 4) (26, 27, 205–212).
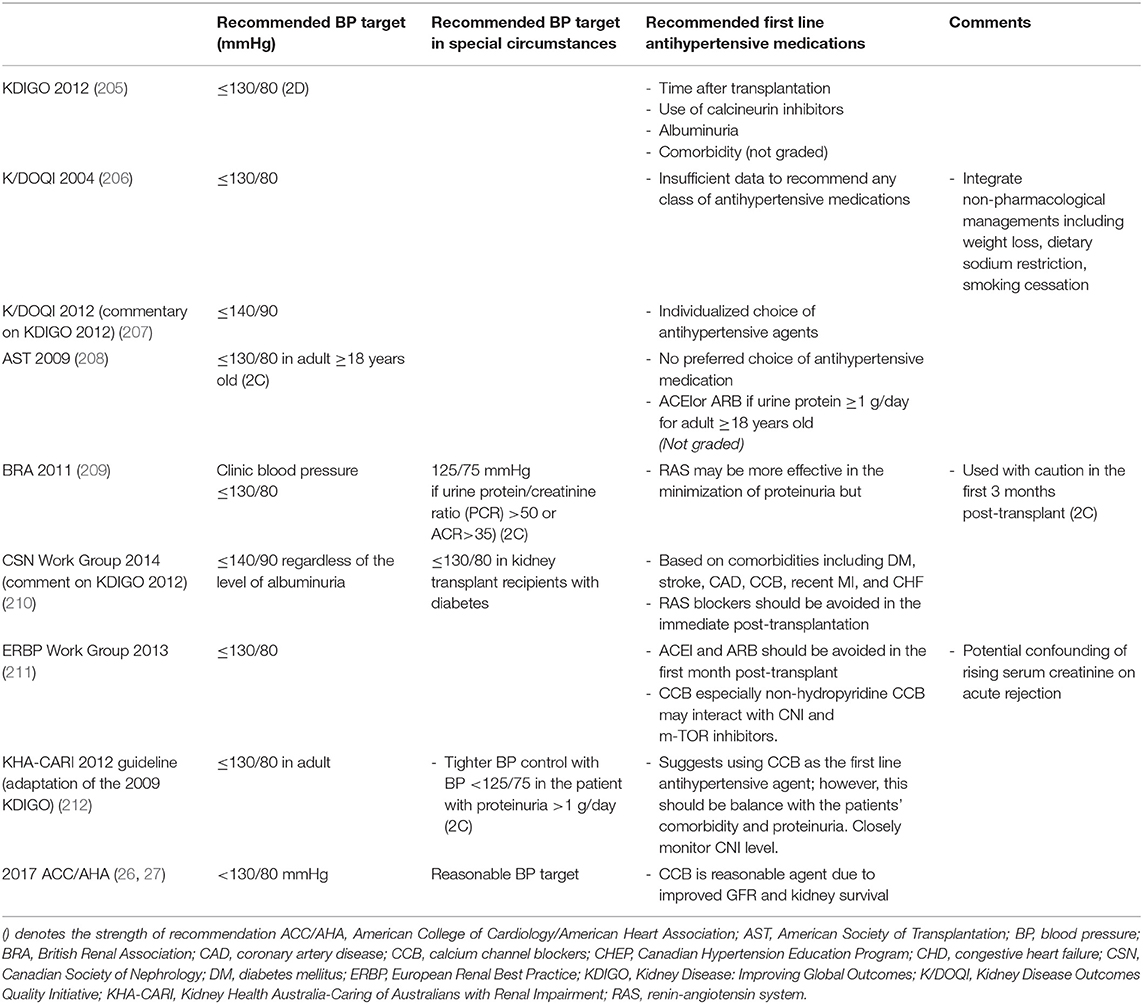
Table 4. Summarized blood pressure guideline for kidney transplant recipients from different scientific medical societies.
Until there is strong clinical outcome evidence in terms of CV, patients, or renal allograft survival, BP targets should be individualized, taking into account immunologic and non-immunologic factors that are contributing to HTN in each kidney transplant recipient.
Other Management for Blood Pressure
Native Nephrectomy
In ESRD patients with resistant HTN, defined as uncontrolled BP with at least three antihypertensive medications of which one is a diuretic, secondary HTN should be considered. Apart from renovascular and hormonal causes of secondary HTN, failed kidneys can contribute to HTN or be the cause of uncontrolled HTN in ESRD patients. In ESRD secondary to ADPKD, the mechanism of HTN from failed kidneys is related to intra-renal renin instead of systemic renin (213, 214). In kidney transplantation, the presence of failed native kidneys is associated with post-transplant resistant HTN. This is presumed in part related to the effect of angiotensin II (215). In these kidney transplant recipients with resistant HTN, ACEIs and native nephrectomy may need to be considered.
There is evidence that kidney transplant recipients with pre- or post-transplant native nephrectomy have decreased BP when compared to those without native nephrectomy (Table 5) (216–221). The majority of these studies were notably conducted in patients with ADPKD as the cause of ESRD.
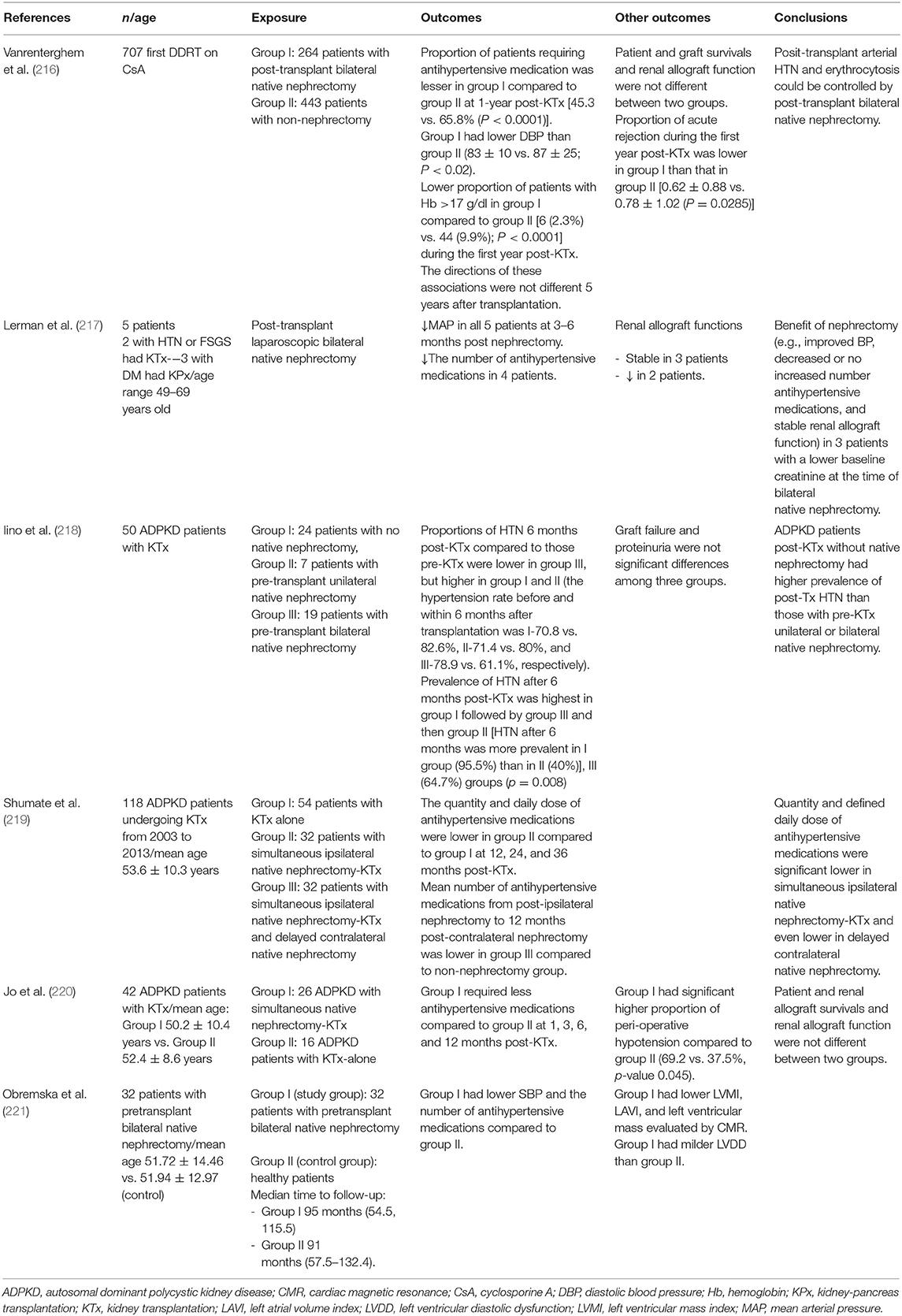
Table 5. Studies examining effectiveness of native nephrectomy and blood pressure control in kidney transplant recipients.
Native Renal Sympathetic Denervation
Apart from the angiotensin II pathway, sympathetic overactivity from failed native kidneys is another mechanism that leads to resistant HTN. Renal and systemic sympathetic hyperactivity contributes to the pathophysiology of resistant HTN. The effect of RDN on BP control was established (222). In kidney transplant recipients with native non-functioning kidneys, sympathetic nervous system activation from afferent signal of native kidneys was preserved (223).
Surgical Renal Denervation by Bilateral Native Nephrectomy
Complete RDN can be performed by bilateral native nephrectomy. A retrospective study in 32 kidney transplant recipients undergoing pre-transplant bilateral native nephrectomy revealed lower SBP, a number of antihypertensive medications, left ventricular mass index, and left atrial volume index but higher left ventricular diastolic dysfunction compared to a matched (age, gender, creatinine levels, eGFR, immunosuppressive treatment, and the time of renal replacement therapy) control group (221). This data supports the effect of RDN as treatment options for HTN in this population.
Although native nephrectomy can improve post-transplant HTN, both pre- and post-transplant native nephrectomy can lead to surgical complications, which can cause impaired renal allograft function. Therefore, native nephrectomy for the purpose of BP control in kidney transplant recipients should be performed in select patients with severely uncontrolled post-transplant HTN whom, without treatment, would have deterioration of renal allograft function or be at increased risk of CV complications. Patients who undergo bilateral native nephrectomy for alternative indications such as recurrent infection, discomfort from large polycystic kidneys, or suspicious native renal tumors may have the added benefit of improved BP control.
Catheter Ablative Renal Denervation
Partial reinnervation of the human transplanted kidney has been shown histologically. Axonal regeneration in a human transplant kidney started at 28 days to 5 months post-kidney transplantation and was complete within 8–12 months to 2 years (224, 225). The function of efferent renal nerves that reinnervate transplanted renal arteries, however, may not be the same as what occurs in native renal nerves (226, 227). One study of renal hemodynamics, sodium excretion, and tubular function after noradrenaline infusion (2 μg h−1 kg−1) and lower body negative pressure (−27 mmHg) in 25 kidney transplant recipients and 10 normal subjects concluded that the reinnervated efferent renal nerves in the human transplanted kidney are still essentially functionally denervated (228). Since complete RDN by bilateral native nephrectomy in kidney transplant recipients is invasive and carries a risk of additional operation, native RDN is another option, and it should be reserved for selected patients.
Two reported kidney transplant recipients with resistant HTN had improved BP control after RDN of native kidneys (229, 230). An investigator-initiated, prospective, single-center RCT with 6-month follow-up of 18 kidney transplant recipients with resistant HTN compared the feasibility and efficacy of renal sympathetic denervation as compared to medical therapy alone. The former group had a significantly decreased office SBP of 23.3 ± 14.5 mmHg and a higher proportion of patients who converted from non-dippers to dippers. Nocturnal BP recorded with 24-h ABPM, but not daytime BP, was also lower by 10.38 ± 12.8 mmHg in the RDN group, though this was not statistically significant. Safety endpoints including changes in renal allograft function and renovascular complications were not different between the 2 groups (231). Although native RDN is effective in controlling BP, larger RCTs with a sham-control are required.
Conclusions
HTN is a very common disease in CKD and ESRD and remains so after kidney transplantation. The pathogenesis of post-transplant HTN is complex. BP measurement is still the main barrier to accurately diagnose and follow-up in HTN management. A 24-h ABPM, though the gold standard, is inconvenient and not wildly utilized. The choice of antihypertensive medication requires the clinician to take transplant and immunological factors into the consideration. Management for OSA and interventions for resistant HTN, such as transplant renal artery angioplasty ± stenting, bilateral native nephrectomy, and native RDN, remain options for resistant HTN in selected kidney transplant recipients. There is no conclusive BP target for this population and therapy targets need to be individualized. Further research to develop a stronger body of evidence for the pathogenesis of post-transplant HTN and guide clinicians on the appropriate BP measurement method, use of antihypertensive medications, surgical or procedural interventions, and establish BP targets for kidney transplant recipients.
Author Contributions
ET participated in designing the topics and detail of the manuscript, writing the manuscript, and preparing figures and tables. MM participated in the design of the topics, detail of the manuscript, and the editing of the manuscript. BH, UR, DD, HI, and AA participated in preparing and reviewing the manuscript. AF and RH participated in preparing, editing, and reviewing the manuscript. KK-Z participated in designing the topics and detail of the manuscript as well as preparing and reviewing the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a research grant from the Division of Nephrology, Hypertension and Kidney Transplantation, Department of Medicine, University of California, Irvine, School of Medicine.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the Division of Nephrology, Hypertension and Kidney Transplantation, Department of Medicine, University of California, Irvine, School of Medicine for research grant support and Multi-Organ Transplant Center, Section of Nephrology, Department of Internal Medicine, William Beaumont Hospital, Oakland University William Beaumont School of Medicine for supporting the authors in writing this review.
Abbreviations
24-h ABPM, 24-hour ambulatory blood pressure monitoring; AVF, arteriovenous; ACC/AHA, American College of Cardiology/American Heart Association; ACEI, angiotensin-converting enzyme inhibitors; ADPKD, autosomal dominant polycystic kidney disease; AHI, apnea-hypopnia index; ARB, angiotensin II receptor blockers; ATI, acute tubular injury; AVF, arteriovenous fistula; BMI, body mass index; BP, blood pressure; CAD, coronary artery disease; CAN, chronic allograft nephropathy; CCB, calcium channel blockers; CDU, color Doppler ultrasonography; CI, confidence interval; CIN, contrast-induced nephropathy; CKD, chronic kidney disease; CMR, cardiac magnetic resonance; CMV, cytomegalovirus; CNI, calcineurin inhibitor; CNS, central nervous system; CO2, carbon dioxide; Cr, creatinine; CrCl, creatinine clearance; CsA, cyclosporine A; CT, computed tomography; CV, cardiovascular; CVD, cardiovascular diseases; CYP, cytochrome P (CYP); DBP, diastolic blood pressure; DCT, distal convoluted tubule; DDRT, deceased donor renal transplantation; ESRD, end-stage renal disease; FGF23, fibroblast growth factor 23; FHHt, familial hyperkalemic hypertension; FST, furosemide stress test; HBPM, home blood pressure monitoring; Hb, hemoglobin; HFpEF, heart failure with preserved ejection fraction; HTN, hypertension; IV, intravenous; KPx, kidney-pancreas transplantation; KTx, kidney transplantation; LAVI, left atrial volume index; LVDD, left ventricular diastolic dysfunction; LVH, left ventricular hypertrophy; LVMI, left ventricular mass index; MAP, mean arterial pressure; MCRA, mineralocorticoid receptor antagonist; MD, mean difference; MHC, major histocompatibility complex; MI, myocardial infarction; MRA, magnetic resonance angiography; mTOR, mammalian target of rapamycin; NaCl, sodium chloride; Na-K-ATPase, sodium-potassium adenosine triphosphase; Na-K-2Cl, sodium-potassium-2 chloride; NCC, sodium chloride cotransporter; OBP, office blood pressure; OSA, obstructive sleep apnea; PAMORA, Peripherally acting mu opioid receptor antagonists; PDGF, platelet-derived growth factor; PSV, peak systolic velocity; RAAS, Renin-Angiotensin-Aldosterone System; RAS, renal artery stenosis; RCT, randomized controlled clinical trial; RDN, renal denervation; RI, resistive index; ROMK, renal outer medullary potassium; RRT, renal replacement therapy; SAW, steroid avoidance or withdrawal; SBP, systolic blood pressure; SLEPT, SLeep disorders Evaluation in Patients after kidney Transplantation; SPAK, STE20/SPS1-related proline/alanine-rich kinase; TRAS, transplant renal artery stenosis; WNK, With-No-K(Lys).
References
1. Suthanthiran M, Strom TB. Renal transplantation. N Engl J Med. (1994) 331:365–76. doi: 10.1056/NEJM199408113310606
2. Lamb KE, Lodhi S, Meier-Kriesche HU. Long-term renal allograft survival in the United States: a critical reappraisal. Am J Transplant. (2011) 11:450–62. doi: 10.1111/j.1600-6143.2010.03283.x
3. Molnar-Varga M, Molnar MZ, Szeifert L, Kovacs AZ, Kelemen A, Becze A, et al. Health-related quality of life and clinical outcomes in kidney transplant recipients. Am J Kidney Dis. (2011) 58:444–52. doi: 10.1053/j.ajkd.2011.03.028
4. Briggs JD. Causes of death after renal transplantation. Nephrol Dial Transplant. (2001) 16:1545–9. doi: 10.1093/ndt/16.8.1545
5. Kasiske BL, Anjum S, Shah R, Skogen J, Kandaswamy C, Danielson B, et al. Hypertension after kidney transplantation. Am J Kidney Dis. (2004) 43:1071–81. doi: 10.1053/j.ajkd.2004.03.013
6. Tantisattamo E, Molnar MZ, Ho BT, Reddy UG, Dafoe DC, Ichii H. Cyclosporin in cadaveric renal transplantation: one-year follow-up of a multicentre trial. Lancet. (1983) 2:986–9. doi: 10.1016/S0140-6736(83)90978-9
7. The Canadian Multicentre Transplant Study Group. A randomized clinical trial of cyclosporine in cadaveric renal transplantation. Analysis at three years. N Engl J Med. (1986) 314:1219–25. doi: 10.1056/NEJM198605083141904
8. Zeier M, Mandelbaum A, Ritz E. Hypertension in the transplanted patient. Nephron. (1998) 80:257–68. doi: 10.1159/000045184
9. Campistol JM, Romero R, Paul J, Gutierrez-Dalmau A. Epidemiology of arterial hypertension in renal transplant patients: changes over the last decade. Nephrol Dial Transplant. (2004) 19(Suppl. 3):iii62–6. doi: 10.1093/ndt/gfh1018
10. Curtis JJ, Galla JH, Kotchen TA, Lucas B, McRoberts JW, Luke RG. Prevalence of hypertension in a renal transplant population on alternate-day steroid therapy. Clin Nephrol. (1976) 5:123–7.
11. Luke RG. Hypertension in renal transplant recipients. Kidney Int. (1987) 31:1024–37. doi: 10.1038/ki.1987.102
12. Ponticelli C, Montagnino G, Aroldi A, Angelini C, Braga M, Tarantino A. Hypertension after renal transplantation. Am J Kidney Dis. (1993) 21:73–8. doi: 10.1016/0272-6386(93)70098-J
13. Vianello A, Mastrosimone S, Calconi G, Gatti PL, Calzavara P, Maresca MC. The role of hypertension as a damaging factor for kidney grafts under cyclosporine therapy. Am J Kidney Dis. (1993) 21:79–83. doi: 10.1016/0272-6386(93)70099-K
14. Raine AE. Does antihypertensive therapy modify chronic allograft failure? Kidney Int Suppl. (1995) 52:S107–11.
15. Sanders CE Jr, Curtis JJ. Role of hypertension in chronic renal allograft dysfunction. Kidney Int Suppl. (1995) 52:S43–7.
16. Budde K, Waiser J, Fritsche L, Zitzmann J, Schreiber M, Kunz R, et al. Hypertension in patients after renal transplantation. Transplant Proc. (1997) 29:209–11. doi: 10.1016/S0041-1345(96)00066-8
17. Malek-Hosseini SA, Ghahramani N, Behzadi S, Rais-Jalali GA, Ahmad E, Salahi H, et al. Incidence of posttransplant hypertension among renal allograft recipients. Transplant Proc. (1998) 30:775–6. doi: 10.1016/S0041-1345(98)00043-8
18. Kasiske BL, Vazquez MA, Harmon WE, Brown RS, Danovitch GM, Gaston RS, et al. Recommendations for the outpatient surveillance of renal transplant recipients. American Society of Transplantation. J Am Soc Nephrol. (2000) 11(Suppl. 15):S1–86. Retrieved from: https://jasn.asnjournals.org
19. Weir MR, Burgess ED, Cooper JE, Fenves AZ, Goldsmith D, McKay D, et al. Assessment and management of hypertension in transplant patients. J Am Soc Nephrol. (2015) 26:1248–60. doi: 10.1681/ASN.2014080834
20. Mancia G, Fagard R, Narkiewicz K, Redon J, Zanchetti A, Bohm M, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J. (2013) 34:2159–219. doi: 10.1093/eurheartj/eht151
21. Bavishi C, Goel S, Messerli FH. Isolated systolic hypertension: an update after SPRINT. Am J Med. (2016) 129:1251–8. doi: 10.1016/j.amjmed.2016.08.032
22. Duprez DA. Systolic hypertension in the elderly: addressing an unmet need. Am J Med. (2008) 121:179–84 e3. doi: 10.1016/j.amjmed.2007.10.027
23. Franklin SS. Elderly hypertensives: how are they different? J Clin Hypertens. (2012) 14:779–86. doi: 10.1111/j.1751-7176.2012.00703.x
24. Midha T, Lalchandani A, Nath B, Kumari R, Pandey U. Prevalence of isolated diastolic hypertension and associated risk factors among adults in Kanpur, India. Indian Heart J. (2012) 64:374–9. doi: 10.1016/j.ihj.2012.06.007
25. Group SR, Wright JT Jr, Williamson JD, Whelton PK, Snyder JK, Sink KM, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. (2015) 373:2103–16. doi: 10.1056/NEJMoa1511939
26. Whelton PK, Carey RM, Aronow WS, Casey DE Jr, Collins KJ, Dennison Himmelfarb C, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. (2018) 71:e13–115. doi: 10.1161/HYP.0000000000000065
27. Whelton PK, Carey RM, Aronow WS, Casey DE Jr, Collins KJ, Dennison Himmelfarb C, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. (2018) 71:e127–e248. doi: 10.1016/j.jacc.2017.11.006
28. Chan W, Bosch JA, Jones D, McTernan PG, Inston N, Moore S, et al. Hypervolemia and blood pressure in prevalent kidney transplant recipients. Transplantation. (2014) 98:320–7. doi: 10.1097/TP.0000000000000066
29. Clement RP, Vos JJ, Scheeren TWL. Minimally invasive cardiac output technologies in the ICU: putting it all together. Curr Opin Crit Care. (2017) 23:302–9. doi: 10.1097/MCC.0000000000000417
30. Calixto Fernandes MH, Schricker T, Magder S, Hatzakorzian R. Perioperative fluid management in kidney transplantation: a black box. Crit Care. (2018) 22:14. doi: 10.1186/s13054-017-1928-2
31. Pirpiris M, Sudhir K, Yeung S, Jennings G, Whitworth JA. Pressor responsiveness in corticosteroid-induced hypertension in humans. Hypertension. (1992) 19:567–74. doi: 10.1161/01.HYP.19.6.567
32. Taler SJ, Textor SC, Canzanello VJ, Schwartz L, Porayko M, Wiesner RH, et al. Role of steroid dose in hypertension early after liver transplantation with tacrolimus (FK506) and cyclosporine. Transplantation. (1996) 62:1588–92. doi: 10.1097/00007890-199612150-00011
33. Metz S, Klein C, Morton N. Rebound hypertension after discontinuation of transdermal clonidine therapy. Am J Med. (1987) 82:17–9. doi: 10.1016/0002-9343(87)90371-8
34. Geyskes GG, Boer P, Dorhout Mees EJ. Clonidine withdrawal. Mechanism and frequency of rebound hypertension. Br J Clin Pharmacol. (1979) 7:55–62. doi: 10.1111/j.1365-2125.1979.tb00897.x
35. Miller RR, Olson HG, Amsterdam EA, Mason DT. Propranolol-withdrawal rebound phenomenon. Exacerbation of coronary events after abrupt cessation of antianginal therapy. N Engl J Med. (1975) 293:416–8. doi: 10.1056/NEJM197508282930902
36. Houston MC. Abrupt cessation of treatment in hypertension: consideration of clinical features, mechanisms, prevention and management of the discontinuation syndrome. Am Heart J. (1981) 102:415–30. doi: 10.1016/0002-8703(81)90317-3
37. Rangno RE, Nattel S, Lutterodt A. Prevention of propranolol withdrawal mechanism by prolonged small dose propranolol schedule. Am J Cardiol. (1982) 49:828–33. doi: 10.1016/0002-9149(82)91965-8
38. Lefkowitz RJ, Caron MG, Stiles GL. Mechanisms of membrane-receptor regulation. Biochemical, physiological, and clinical insights derived from studies of the adrenergic receptors. N Engl J Med. (1984) 310:1570–9. doi: 10.1056/NEJM198406143102406
39. Psaty BM, Koepsell TD, Wagner EH, LoGerfo JP, Inui TS. The relative risk of incident coronary heart disease associated with recently stopping the use of beta-blockers. JAMA. (1990) 263:1653–7. doi: 10.1001/jama.263.12.1653
40. Frishman WH, Klein N, Strom J, Cohen MN, Shamoon H, Willens H, et al. Comparative effects of abrupt withdrawal of propranolol and verapamil in angina pectoris. Am J Cardiol. (1982) 50:1191–5. doi: 10.1016/0002-9149(82)90442-8
41. Shammash JB, Trost JC, Gold JM, Berlin JA, Golden MA, Kimmel SE. Perioperative beta-blocker withdrawal and mortality in vascular surgical patients. Am Heart J. (2001) 141:148–53. doi: 10.1067/mhj.2001.111547
42. Hoeks SE, Scholte Op Reimer WJ, van Urk H, Jorning PJ, Boersma E, Simoons ML, et al. Increase of 1-year mortality after perioperative beta-blocker withdrawal in endovascular and vascular surgery patients. Eur J Vasc Endovasc Surg. (2007) 33:13–9. doi: 10.1016/j.ejvs.2006.06.019
43. Fleisher LA, Fleischmann KE, Auerbach AD, Barnason SA, Beckman JA, Bozkurt B, et al. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines. J Am Coll Cardiol. (2014) 64:e77–137. doi: 10.1016/j.jacc.2014.07.944
44. Sacco M, Meschi M, Regolisti G, Detrenis S, Bianchi L, Bertorelli M, et al. The relationship between blood pressure and pain. J Clin Hypertens. (2013) 15:600–5. doi: 10.1111/jch.12145
45. Tantisattamo E, Vutthikraivit P, Mopuru HR. Post-transplant weight control and hypertension after kidney transplantation. Hypertension. (2017) 70:AP200. doi: 10.1161/hyp.70.suppl_1.p200
46. Costa B, Moratelli L, Silva LB, Paiva AC, Silva AN, Carminatti M, et al. Body mass index in the first year after kidney transplantation. Transplant Proc. (2014) 46:1750–2. doi: 10.1016/j.transproceed.2014.05.021
47. Cashion AK, Sanchez ZV, Cowan PA, Hathaway DK, Lo Costello A, Gaber AO. Changes in weight during the first year after kidney transplantation. Prog Transplant. (2007) 17:40–7. doi: 10.7182/prtr.17.1.2433464056125h72
48. el-Agroudy AE, Wafa EW, Gheith OE, Shehab el-Dein AB, Ghoneim MA. Weight gain after renal transplantation is a risk factor for patient and graft outcome. Transplantation. (2004) 77:1381–5. doi: 10.1097/01.TP.0000120949.86038.62
49. Tantisattamo E. Post-transplant weight gain and obesity: an opportunity for renal dietary management. Adv Obes Weight Manag Control. (2017) 7:276–9. doi: 10.15406/aowmc.2017.07.00195
50. First MR, Neylan JF, Rocher LL, Tejani A. Hypertension after renal transplantation. J Am Soc Nephrol. (1994) 4:S30–6.
51. Midtvedt K, Hartmann A. Hypertension after kidney transplantation: are treatment guidelines emerging? Nephrol Dial Transplant. (2002) 17:1166–9. doi: 10.1093/ndt/17.7.1166
52. Bachy C, Alexandre GP, van Ypersele de Strihou C. Hypertension after renal transplantation. Br Med J. (1976) 2:1287–9. doi: 10.1136/bmj.2.6047.1287
53. Wauthier M, Vereerstraeten P, Pirson Y, Toussaint C, Alexandre GP, Kinnaert P, et al. Prevalence and causes of hypertension late after renal transplantation. Proc Eur Dial Transplant Assoc. (1983) 19:566–71.
54. Hoorn EJ, Walsh SB, McCormick JA, Zietse R, Unwin RJ, Ellison DH. Pathogenesis of calcineurin inhibitor-induced hypertension. J Nephrol. (2012) 25:269–75. doi: 10.5301/jn.5000174
55. Murray BM, Paller MS, Ferris TF. Effect of cyclosporine administration on renal hemodynamics in conscious rats. Kidney Int. (1985) 28:767–74. doi: 10.1038/ki.1985.196
56. Forslund T, Hannonen P, Reitamo S, Fyhrquist F. Hypertension in cyclosporin A-treated patients is independent of circulating endothelin levels. J Intern Med. (1995) 238:71–5. doi: 10.1111/j.1365-2796.1995.tb00901.x
57. Avdonin PV, Cottet-Maire F, Afanasjeva GV, Loktionova SA, Lhote P, Ruegg UT. Cyclosporine A up-regulates angiotensin II receptors and calcium responses in human vascular smooth muscle cells. Kidney Int. (1999) 55:2407–14. doi: 10.1046/j.1523-1755.1999.00481.x
58. Marumo T, Nakaki T, Hishikawa K, Suzuki H, Kato R, Saruta T. Cyclosporin A inhibits nitric oxide synthase induction in vascular smooth muscle cells. Hypertension. (1995) 25:764–8. doi: 10.1161/01.HYP.25.4.764
59. Moss NG, Powell SL, Falk RJ. Intravenous cyclosporine activates afferent and efferent renal nerves and causes sodium retention in innervated kidneys in rats. Proc Natl Acad Sci USA. (1985) 82:8222–6. doi: 10.1073/pnas.82.23.8222
60. Zhang W, Li JL, Hosaka M, Janz R, Shelton JM, Albright GM, et al. Cyclosporine A-induced hypertension involves synapsin in renal sensory nerve endings. Proc Natl Acad Sci USA. (2000) 97:9765–70. doi: 10.1073/pnas.170160397
61. Hadchouel J, Delaloy C, Faure S, Achard JM, Jeunemaitre X. Familial hyperkalemic hypertension. J Am Soc Nephrol. (2006) 17:208–17. doi: 10.1681/ASN.2005030314
62. Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, et al. Human hypertension caused by mutations in WNK kinases. Science. (2001) 293:1107–12. doi: 10.1126/science.1062844
63. Hoorn EJ, Walsh SB, McCormick JA, Furstenberg A, Yang CL, Roeschel T, et al. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med. (2011) 17:1304–9. doi: 10.1038/nm.2497
64. Colussi G, Bettinelli A, Tedeschi S, De Ferrari ME, Syren ML, Borsa N, et al. A thiazide test for the diagnosis of renal tubular hypokalemic disorders. Clin J Am Soc Nephrol. (2007) 2:454–60. doi: 10.2215/CJN.02950906
65. Luft FC. How calcineurin inhibitors cause hypertension. Nephrol Dial Transplant. (2012) 27:473–5. doi: 10.1093/ndt/gfr679
66. Knight SR, Morris PJ. Steroid avoidance or withdrawal after renal transplantation increases the risk of acute rejection but decreases cardiovascular risk. A meta-analysis. Transplantation. (2010) 89:1–14. doi: 10.1097/TP.0b013e3181c518cc
67. McDonald FD, Horensten ML, Mayor GB, Turcotte JG, Selezinka W, Schork MA. Effect of alternate-day steroids on renal transplant function. A controlled study. Nephron. (1976) 17:415–29. doi: 10.1159/000180750
68. Meneton P, Jeunemaitre X, de Wardener HE, MacGregor GA. Links between dietary salt intake, renal salt handling, blood pressure, and cardiovascular diseases. Physiol Rev. (2005) 85:679–715. doi: 10.1152/physrev.00056.2003
69. Ball CO, Meneely GR. Observations on dietary sodium chloride. J Am Diet Assoc. (1957) 33:366–70.
70. Cherchovich GM, Capek K, Jefremova Z, Pohlova I, Jelinek J. High salt intake and blood pressure in lower primates (Papio hamadryas). J Appl Physiol. (1976) 40:601–4. doi: 10.1152/jappl.1976.40.4.601
71. Corbett WT, Kuller LH, Blaine EH, Damico FJ. Utilization of swine to study the risk factor of an elevated salt diet on blood pressure. Am J Clin Nutr. (1979) 32:2068–75. doi: 10.1093/ajcn/32.10.2068
72. Srinivasan SR, Dalferes ER Jr, Wolf RH, Radhakrishnamurthy B, Foster TA, Berenson GS. Variability in blood pressure response to dietary sodium intake among African green monkeys (>Cercopithecus aethiops). Am J Clin Nutr. (1984) 39:792–6. doi: 10.1093/ajcn/39.5.792
73. Denton D, Weisinger R, Mundy NI, Wickings EJ, Dixson A, Moisson P, et al. The effect of increased salt intake on blood pressure of chimpanzees. Nat Med. (1995) 1:1009–16. doi: 10.1038/nm1095-1009
74. Rettig R, Bandelow N, Patschan O, Kuttler B, Frey B, Uber A. The importance of the kidney in primary hypertension: insights from cross-transplantation. J Hum Hypertens. (1996) 10:641–4.
75. Curtis JJ, Luke RG, Dustan HP, Kashgarian M, Whelchel JD, Jones P, et al. Remission of essential hypertension after renal transplantation. N Engl J Med. (1983) 309:1009–15. doi: 10.1056/NEJM198310273091702
76. Guidi E, Bianchi G, Rivolta E, Ponticelli C, Quarto di Palo F, Minetti L, et al. Hypertension in man with a kidney transplant: role of familial versus other factors. Nephron. (1985) 41:14–21. doi: 10.1159/000183539
77. Guidi E, Menghetti D, Milani S, Montagnino G, Palazzi P, Bianchi G. Hypertension may be transplanted with the kidney in humans: a long-term historical prospective follow-up of recipients grafted with kidneys coming from donors with or without hypertension in their families. J Am Soc Nephrol. (1996) 7:1131–8.
78. Aguera Fernandez LG, Zudaire JJ, Isa WA, Sanchez de la Muela PL, Rosell D, de Castro F, et al. Vascular complications in 237 recipients of renal transplant from cadaver. Actas Urol Esp. (1992) 16:292–5.
79. Bruno S, Remuzzi G, Ruggenenti P. Transplant renal artery stenosis. J Am Soc Nephrol. (2004) 15:134–41. doi: 10.1097/01.ASN.0000099379.61001.F8
80. Fervenza FC, Lafayette RA, Alfrey EJ, Petersen J. Renal artery stenosis in kidney transplants. Am J Kidney Dis. (1998) 31:142–8. doi: 10.1053/ajkd.1998.v31.pm9428466
81. Wong W, Fynn SP, Higgins RM, Walters H, Evans S, Deane C, et al. Transplant renal artery stenosis in 77 patients–does it have an immunological cause? Transplantation. (1996) 61:215–9. doi: 10.1097/00007890-199601270-00009
82. Lacombe M. Arterial stenosis complicating renal allotransplantation in man: a study of 38 cases. Ann Surg. (1975) 181:283–8. doi: 10.1097/00000658-197503000-00007
83. Roberts JP, Ascher NL, Fryd DS, Hunter DW, Dunn DL, Payne WD, et al. Transplant renal artery stenosis. Transplantation. (1989) 48:580–3.
84. Hurst FP, Abbott KC, Neff RT, Elster EA, Falta EM, Lentine KL, et al. Incidence, predictors and outcomes of transplant renal artery stenosis after kidney transplantation: analysis of USRDS. Am J Nephrol. (2009) 30:459–67. doi: 10.1159/000242431
85. O'Connell TX, Mowbray JF. Arterial intimal thickening produced by alloantibody and xenoantibody. Transplantation. (1973) 15:262–3. doi: 10.1097/00007890-197302000-00017
86. Tantisattamo E, Ratanasrimetha P, Spanuchart I, Shrivastava P, Samarapungavan D, Gallon L. Transplant renal artery stenosis: an overlooked cause of resistant hypertension in a new kidney. Ann Clin Exp Hypertension. (2015) 3:1030. Retrieved from: http://www.jscimedcentral.com/ExperimentalHypertension/
87. Aslam S, Salifu MO, Ghali H, Markell MS, Friedman EA. Common iliac artery stenosis presenting as renal allograft dysfunction in two diabetic recipients. Transplantation. (2001) 71:814–7. doi: 10.1097/00007890-200103270-00022
88. Pouria S, State OI, Wong W, Hendry BM. CMV infection is associated with transplant renal artery stenosis. QJM. (1998) 91:185–9. doi: 10.1093/qjmed/91.3.185
89. Becker BN, Odorico JS, Becker YT, Leverson G, McDermott JC, Grist T, et al. Peripheral vascular disease and renal transplant artery stenosis: a reappraisal of transplant renovascular disease. Clin Transplant. (1999) 13:349–55. doi: 10.1034/j.1399-0012.1999.130412.x
90. Chen W, Kayler LK, Zand MS, Muttana R, Chernyak V, DeBoccardo GO. Transplant renal artery stenosis: clinical manifestations, diagnosis and therapy. Clin Kidney J. (2015) 8:71–8. doi: 10.1093/ckj/sfu132
91. Porter KA, Thomson WB, Owen K, Kenyon JR, Mowbray JF, Peart WS. Obliterative vascular changes in four human kidney homotransplants. Br Med J. (1963) 2:639–45. doi: 10.1136/bmj.2.5358.639
92. Willicombe M, Sandhu B, Brookes P, Gedroyc W, Hakim N, Hamady M, et al. Postanastomotic transplant renal artery stenosis: association with de novo class II donor-specific antibodies. Am J Transplant. (2014) 14:133–43. doi: 10.1111/ajt.12531
93. Curtis JJ, Luke RG, Whelchel JD, Diethelm AG, Jones P, Dustan HP. Inhibition of angiotensin-converting enzyme in renal-transplant recipients with hypertension. N Engl J Med. (1983) 308:377–81. doi: 10.1056/NEJM198302173080707
94. Alfrey E, Smythe R, Friedman A. Two masqueraders of transplant renal artery stenosis (TRAS). Clin Transpl. (1993) 7:183.
95. Hall JE, Guyton AC, Jackson TE, Coleman TG, Lohmeier TE, Trippodo NC. Control of glomerular filtration rate by renin-angiotensin system. Am J Physiol. (1977) 233:F366–72. doi: 10.1152/ajprenal.1977.233.5.F366
96. Plouin PF, Chatellier G, Darne B, Raynaud A. Blood pressure outcome of angioplasty in atherosclerotic renal artery stenosis: a randomized trial. Essai Multicentrique Medicaments vs. Angioplastie (EMMA) Study Group. Hypertension. (1998) 31:823–9. doi: 10.1161/01.HYP.31.3.823
97. Webster J, Marshall F, Abdalla M, Dominiczak A, Edwards R, Isles CG, et al. Randomised comparison of percutaneous angioplasty vs continued medical therapy for hypertensive patients with atheromatous renal artery stenosis. Scottish and Newcastle Renal Artery Stenosis Collaborative Group. J Hum Hypertens. (1998) 12:329–35. doi: 10.1038/sj.jhh.1000599
98. van Jaarsveld BC, Krijnen P, Pieterman H, Derkx FH, Deinum J, Postma CT, et al. The effect of balloon angioplasty on hypertension in atherosclerotic renal-artery stenosis. Dutch Renal Artery Stenosis Intervention Cooperative Study Group. N Engl J Med. (2000) 342:1007–14. doi: 10.1056/NEJM200004063421403
99. Investigators A, Wheatley K, Ives N, Gray R, Kalra PA, Moss JG, et al. Revascularization versus medical therapy for renal-artery stenosis. N Engl J Med. (2009) 361:1953–62. doi: 10.1056/NEJMoa0905368
100. Bax L, Woittiez AJ, Kouwenberg HJ, Mali WP, Buskens E, Beek FJ, et al. Stent placement in patients with atherosclerotic renal artery stenosis and impaired renal function: a randomized trial. Ann Intern Med. (2009) 150:840–8; W150–1. doi: 10.7326/0003-4819-150-12-200906160-00119
101. Cooper CJ, Murphy TP, Cutlip DE, Jamerson K, Henrich W, Reid DM, et al. Stenting and medical therapy for atherosclerotic renal-artery stenosis. N Engl J Med. (2014) 370:13–22. doi: 10.1056/NEJMoa1310753
102. Leonardou P, Gioldasi S, Pappas P. Transluminal angioplasty of transplanted renal artery stenosis: a review of the literature for its safety and efficacy. J Transplant. (2011) 2011:693820. doi: 10.1155/2011/693820
103. Chew LL, Tan BS, Kumar K, Htoo MM, Wong KS, Cheng CW, et al. Percutaneous transluminal angioplasty of transplant renal artery stenosis. Ann Acad Med Singapore. (2014) 43:39–43. Retrieved from: http://annals.edu.sg.
104. Wang L, Liu B, Yan J, Wang Y, Wang W, Wang W, et al. Interventional therapy for transplant renal artery stenosis is safe and effective in preserving allograft function and improving hypertension. Vasc Endovascular Surg. (2017) 51:4–11. doi: 10.1177/1538574416682157
105. Patel U, Kumar S, Johnson OW, Jeon JH, Das R. Long-term graft and patient survival after percutaneous angioplasty or arterial stent placement for transplant renal artery stenosis: a 21-year matched cohort study. Radiology. (2019) 290:555–63. doi: 10.1148/radiol.2018181320
106. Benoit G, Moukarzel M, Hiesse C, Verdelli G, Charpentier B, Fries D. Transplant renal artery stenosis: experience and comparative results between surgery and angioplasty. Transpl Int. (1990) 3:137–40. doi: 10.1007/BF00355459
107. Shames BD, Odorico JS, D'Alessandro AM, Pirsch JD, Sollinger HW. Surgical repair of transplant renal artery stenosis with preserved cadaveric iliac artery grafts. Ann Surg. (2003) 237:116–22. doi: 10.1097/00000658-200301000-00016
108. Rouer M, Godier S, Monnot A, Etienne I, Bertrand D, Guerrot D, et al. Long-term outcomes after transplant renal artery stenosis surgery. Ann Vasc Surg. (2019) 54:261–8. doi: 10.1016/j.avsg.2018.05.066
109. Schindler R, Tanriver Y, Frei U. Hypertension and allograft nephropathy–cause, consequence, or both? Nephrol Dial Transplant. (2000) 15:8–10. doi: 10.1093/ndt/15.1.8
110. Evenepoel P, Naesens M, Claes K, Kuypers D, Vanrenterghem Y. Tertiary 'hyperphosphatoninism' accentuates hypophosphatemia and suppresses calcitriol levels in renal transplant recipients. Am J Transplant. (2007) 7:1193–200. doi: 10.1111/j.1600-6143.2007.01753.x
111. Prasad N, Jaiswal A, Agarwal V, Kumar S, Chaturvedi S, Yadav S, et al. FGF23 is associated with early post-transplant hypophosphataemia and normalizes faster than iPTH in living donor renal transplant recipients: a longitudinal follow-up study. Clin Kidney J. (2016) 9:669–76. doi: 10.1093/ckj/sfw065
112. Wolf M, Molnar MZ, Amaral AP, Czira ME, Rudas A, Ujszaszi A, et al. Elevated fibroblast growth factor 23 is a risk factor for kidney transplant loss and mortality. J Am Soc Nephrol. (2011) 22:956–66. doi: 10.1681/ASN.2010080894
113. Baia LC, Humalda JK, Vervloet MG, Navis G, Bakker SJ, de Borst MH, et al. Fibroblast growth factor 23 and cardiovascular mortality after kidney transplantation. Clin J Am Soc Nephrol. (2013) 8:1968–78. doi: 10.2215/CJN.01880213
114. Akhabue E, Montag S, Reis JP, Pool LR, Mehta R, Yancy CW, et al. FGF23 (fibroblast growth factor-23) and incident hypertension in young and middle-aged adults: the CARDIA study. Hypertension. (2018) 72:70–6. doi: 10.1161/HYPERTENSIONAHA.118.11060
115. Molnar MZ, Lazar AS, Lindner A, Fornadi K, Czira ME, Dunai A, et al. Sleep apnea is associated with cardiovascular risk factors among kidney transplant patients. Clin J Am Soc Nephrol. (2010) 5:125–32. doi: 10.2215/CJN.04030609
116. Molnar MZ, Szentkiralyi A, Lindner A, Czira ME, Szabo A, Mucsi I, et al. High prevalence of patients with a high risk for obstructive sleep apnoea syndrome after kidney transplantation–association with declining renal function. Nephrol Dial Transplant. (2007) 22:2686–92. doi: 10.1093/ndt/gfm246
117. Aziz F, Chaudhary K. The triad of sleep apnea, hypertension, and chronic kidney disease: a spectrum of common pathology. Cardiorenal Med. (2016) 7:74–82. doi: 10.1159/000450796
118. Huang HC, Walters G, Talaulikar G, Figurski D, Carroll A, Hurwitz M, et al. Sleep apnea prevalence in chronic kidney disease - association with total body water and symptoms. BMC Nephrol. (2017) 18:125. doi: 10.1186/s12882-017-0544-3
119. Molnar MZ, Foster CE III, Sim JJ, Remport A, Krishnan M, Kovesdy CP, et al. Association of pre-transplant blood pressure with post-transplant outcomes. Clin Transplant. (2014) 28:166–76. doi: 10.1111/ctr.12292
120. Opelz G, Wujciak T, Ritz E. Association of chronic kidney graft failure with recipient blood pressure. Collaborative Transplant Study. Kidney Int. (1998) 53:217–22. doi: 10.1046/j.1523-1755.1998.00744.x
121. Mange KC, Cizman B, Joffe M, Feldman HI. Arterial hypertension and renal allograft survival. JAMA. (2000) 283:633–8. doi: 10.1001/jama.283.5.633
122. Mange KC, Feldman HI, Joffe MM, Fa K, Bloom RD. Blood pressure and the survival of renal allografts from living donors. J Am Soc Nephrol. (2004) 15:187–93. doi: 10.1097/01.ASN.0000104574.04006.08
123. Opelz G, Dohler B, Collaborative Transplant S. Improved long-term outcomes after renal transplantation associated with blood pressure control. Am J Transplant. (2005) 5:2725–31. doi: 10.1111/j.1600-6143.2005.01093.x
124. Yancy CW, Lopatin M, Stevenson LW, De Marco T, Fonarow GC, Committee ASA, et al. Clinical presentation, management, and in-hospital outcomes of patients admitted with acute decompensated heart failure with preserved systolic function: a report from the Acute Decompensated Heart Failure National Registry (ADHERE) Database. J Am Coll Cardiol. (2006) 47:76–84. doi: 10.1016/j.jacc.2005.09.022
125. Hernandez D. Left ventricular hypertrophy after renal transplantation: new approach to a deadly disorder. Nephrol Dial Transplant. (2004) 19:1682–6. doi: 10.1093/ndt/gfh283
126. Fang JC. Heart failure with preserved ejection fraction: a kidney disorder? Circulation. (2016) 134:435–7. doi: 10.1161/CIRCULATIONAHA.116.022249
127. Hall JE. Renal dysfunction, rather than nonrenal vascular dysfunction, mediates salt-induced hypertension. Circulation. (2016) 133:894–906. doi: 10.1161/CIRCULATIONAHA.115.018526
128. Williams B, Mancia G, Spiering W, Agabiti Rosei E, Azizi M, Burnier M, et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur Heart J. (2018) 39:3021–104. doi: 10.1093/eurheartj/ehy339
129. Lee MH, Ko KM, Ahn SW, Bae MN, Choi BS, Park CW, et al. The impact of kidney transplantation on 24-hour ambulatory blood pressure in end-stage renal disease patients. J Am Soc Hypertens. (2015) 9:427–34. doi: 10.1016/j.jash.2015.04.001
130. Ahmed J, Ozorio V, Farrant M, Van Der Merwe W. Ambulatory vs office blood pressure monitoring in renal transplant recipients. J Clin Hypertens. (2015) 17:46–50. doi: 10.1111/jch.12448
131. Agena F, Prado Edos S, Souza PS, da Silva GV, Lemos FB, Mion D Jr, et al. Home blood pressure (BP) monitoring in kidney transplant recipients is more adequate to monitor BP than office BP. Nephrol Dial Transplant. (2011) 26:3745–9. doi: 10.1093/ndt/gfr143
132. Mallamaci F, D'Arrigo G, Tripepi R, Leonardis D, Porto G, Testa A, et al. Office, standardized and 24-h ambulatory blood pressure and renal function loss in renal transplant patients. J Hypertens. (2018) 36:119–25. doi: 10.1097/HJH.0000000000001530
133. Balasubramanian S, Cherukuri A, Parker A, Baker R. 24hr ambulatory blood pressure monitoring (ABPM) risk stratifies hypertensive kidney transplant recipients (KTRs). : Abstract# 1443. Transplantation. (2014) 98:101. doi: 10.1097/00007890-201407151-00314
134. Basdeo l, Wadei H, Prendergast M, Mai M, Oshel C, Jarmi T. Diastolic blood pressure derived from twenty four hour ambulatory blood pressure monitoring predicts survival after kidney transplantation [abstract]. Am J Transplant. (2019) 19(Suppl. 3):963–4. doi: 10.1111/ajt.15406
135. Kuzmiuk-Glembin I, Adrych D, Tylicki L, Heleniak Z, Garnier H, Wisniewski J, et al. Treatment of hypertension in renal transplant recipients in four independent cross-sectional analyses. Kidney Blood Press Res. (2018) 43:45–54. doi: 10.1159/000486905
136. Jafar TH, Stark PC, Schmid CH, Landa M, Maschio G, de Jong PE, et al. Progression of chronic kidney disease: the role of blood pressure control, proteinuria, and angiotensin-converting enzyme inhibition: a patient-level meta-analysis. Ann Intern Med. (2003) 139:244–52. doi: 10.7326/0003-4819-139-4-200308190-00006
137. Ekinci C, Karabork M, Siriopol D, Dincer N, Covic A, Kanbay M. Effects of volume overload and current techniques for the assessment of fluid status in patients with renal disease. Blood Purif . (2018) 46:34–47. doi: 10.1159/000487702
138. Tantisattamo E, Bailey JL, Gallon L. Living donation mitigates poorer outcomes in marginal recipients. Curr Transpl Rep. (2016) 3:121–38. doi: 10.1007/s40472-016-0097-6
139. Sandal S, Bansal P, Cantarovich M. The evidence and rationale for the perioperative use of loop diuretics during kidney transplantation: a comprehensive review. Transplant Rev. (2018) 32:92–101. doi: 10.1016/j.trre.2017.11.002
140. Casper J, Schmitz J, Brasen JH, Khalifa A, Schmidt BMW, Einecke G, et al. Renal transplant recipients receiving loop diuretic therapy have increased urinary tract infection rate and altered medullary macrophage polarization marker expression. Kidney Int. (2018) 94:993–1001. doi: 10.1016/j.kint.2018.06.029
141. Sands JM, Layton HE. Advances in understanding the urine-concentrating mechanism. Annu Rev Physiol. (2014) 76:387–409. doi: 10.1146/annurev-physiol-021113-170350
142. Jantsch J, Schatz V, Friedrich D, Schroder A, Kopp C, Siegert I, et al. Cutaneous Na+ storage strengthens the antimicrobial barrier function of the skin and boosts macrophage-driven host defense. Cell Metab. (2015) 21:493–501. doi: 10.1016/j.cmet.2015.02.003
143. Binger KJ, Gebhardt M, Heinig M, Rintisch C, Schroeder A, Neuhofer W, et al. High salt reduces the activation of IL-4- and IL-13-stimulated macrophages. J Clin Invest. (2015) 125:4223–38. doi: 10.1172/JCI80919
144. Berry MR, Mathews RJ, Ferdinand JR, Jing C, Loudon KW, Wlodek E, et al. Renal sodium gradient orchestrates a dynamic antibacterial defense zone. Cell. (2017) 170:860–74. e19. doi: 10.1016/j.cell.2017.07.022
145. McMahon BA, Koyner JL, Novick T, Menez S, Moran RA, Lonze BE, et al. The prognostic value of the furosemide stress test in predicting delayed graft function following deceased donor kidney transplantation. Biomarkers. (2018) 23:61–9. doi: 10.1080/1354750X.2017.1387934
146. Udomkarnjananun S, Townamchai N, Iampenkhae K, Petchlorlian A, Srisawat N, Katavetin P, et al. Furosemide stress test as a predicting biomarker for delayed graft function in kidney transplantation. Nephron. (2019) 141:236–48. doi: 10.1159/000495765
147. Moes AD, Hesselink DA, van den Meiracker AH, Zietse R, Hoorn EJ. Chlorthalidone versus amlodipine for hypertension in kidney transplant recipients treated with tacrolimus: a randomized crossover trial. Am J Kidney Dis. (2017) 69:796–804. doi: 10.1053/j.ajkd.2016.12.017
148. Rodenburg EM, Hoorn EJ, Ruiter R, Lous JJ, Hofman A, Uitterlinden AG, et al. Thiazide-associated hyponatremia: a population-based study. Am J Kidney Dis. (2013) 62:67–72. doi: 10.1053/j.ajkd.2013.02.365
149. van den Berg E, Geleijnse JM, Brink EJ, van Baak MA, Homan van der Heide JJ, Gans RO, et al. Sodium intake and blood pressure in renal transplant recipients. Nephrol Dial Transplant. (2012) 27:3352–9. doi: 10.1093/ndt/gfs069
150. Melnikov S, Mayan H, Uchida S, Holtzman EJ, Farfel Z. Cyclosporine metabolic side effects: association with the WNK4 system. Eur J Clin Invest. (2011) 41:1113–20. doi: 10.1111/j.1365-2362.2011.02517.x
151. Kaye D, Thompson J, Jennings G, Esler M. Cyclosporine therapy after cardiac transplantation causes hypertension and renal vasoconstriction without sympathetic activation. Circulation. (1993) 88:1101–9. doi: 10.1161/01.CIR.88.3.1101
152. Perico N, Dadan J, Remuzzi G. Endothelin mediates the renal vasoconstriction induced by cyclosporine in the rat. J Am Soc Nephrol. (1990) 1:76–83.
153. Lanese DM, Conger JD. Effects of endothelin receptor antagonist on cyclosporine-induced vasoconstriction in isolated rat renal arterioles. J Clin Invest. (1993) 91:2144–9. doi: 10.1172/JCI116440
154. Arthur JM, Shamim S. Interaction of cyclosporine and FK506 with diuretics in transplant patients. Kidney Int. (2000) 58:325–30. doi: 10.1046/j.1523-1755.2000.00169.x
155. Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. (2003) 348:1309–21. doi: 10.1056/NEJMoa030207
156. Zannad F, McMurray JJ, Krum H, van Veldhuisen DJ, Swedberg K, Shi H, et al. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med. (2011) 364:11–21. doi: 10.1056/NEJMoa1009492
157. Gonzalez Monte E, Andres A, Polanco N, Toribio MJ, Santana R, Gutierrez Martinez E, et al. Addition of spironolactone to dual blockade of renin angiotensin system dramatically reduces severe proteinuria in renal transplant patients: an uncontrolled pilot study at 6 months. Transplant Proc. (2010) 42:2899–901. doi: 10.1016/j.transproceed.2010.08.024
158. Bertocchio JP, Barbe C, Lavaud S, Toupance O, Nazeyrollas P, Jaisser F, et al. Safety of eplerenone for kidney-transplant recipients with impaired renal function and receiving cyclosporine A. PLoS ONE. (2016) 11:e0153635. doi: 10.1371/journal.pone.0153635
159. Amador CA, Bertocchio JP, Andre-Gregoire G, Placier S, Duong Van Huyen JP, El Moghrabi S, et al. Deletion of mineralocorticoid receptors in smooth muscle cells blunts renal vascular resistance following acute cyclosporine administration. Kidney Int. (2016) 89:354–62. doi: 10.1038/ki.2015.312
160. Tsutamoto T, Wada A, Maeda K, Mabuchi N, Hayashi M, Tsutsui T, et al. Effect of spironolactone on plasma brain natriuretic peptide and left ventricular remodeling in patients with congestive heart failure. J Am Coll Cardiol. (2001) 37:1228–33. doi: 10.1016/S0735-1097(01)01116-0
161. Haas SJ, Vos T, Gilbert RE, Krum H. Are beta-blockers as efficacious in patients with diabetes mellitus as in patients without diabetes mellitus who have chronic heart failure? A meta-analysis of large-scale clinical trials. Am Heart J. (2003) 146:848–53. doi: 10.1016/S0002-8703(03)00403-4
162. Foley RN, Herzog CA, Collins AJ, United States Renal Data S. Blood pressure and long-term mortality in United States hemodialysis patients: USRDS Waves 3 and 4 Study. Kidney Int. (2002) 62:1784–90. doi: 10.1046/j.1523-1755.2002.00636.x
163. Cice G, Ferrara L, D'Andrea A, D'Isa S, Di Benedetto A, Cittadini A, et al. Carvedilol increases two-year survivalin dialysis patients with dilated cardiomyopathy: a prospective, placebo-controlled trial. J Am Coll Cardiol. (2003) 41:1438–44. doi: 10.1016/S0735-1097(03)00241-9
164. Abbott KC, Trespalacios FC, Agodoa LY, Taylor AJ, Bakris GL. beta-Blocker use in long-term dialysis patients: association with hospitalized heart failure and mortality. Arch Intern Med. (2004) 164:2465–71. doi: 10.1001/archinte.164.22.2465
165. Wali RK, Iyengar M, Beck GJ, Chartyan DM, Chonchol M, Lukas MA, et al. Efficacy and safety of carvedilol in treatment of heart failure with chronic kidney disease: a meta-analysis of randomized trials. Circ Heart Fail. (2011) 4:18–26. doi: 10.1161/CIRCHEARTFAILURE.109.932558
166. Aftab W, Varadarajan P, Rasool S, Kore A, Pai RG. Beta and angiotensin blockades are associated with improved 10-year survival in renal transplant recipients. J Am Heart Assoc. (2013) 2:e000091. doi: 10.1161/JAHA.112.000091
167. Zoccali C, Mallamaci F, Parlongo S, Cutrupi S, Benedetto FA, Tripepi G, et al. Plasma norepinephrine predicts survival and incident cardiovascular events in patients with end-stage renal disease. Circulation. (2002) 105:1354–9. doi: 10.1161/hc1102.105261
168. Campese VM, Krol E. Neurogenic factors in renal hypertension. Curr Hypertens Rep. (2002) 4:256–60. doi: 10.1007/s11906-002-0016-3
169. Xu J, Li G, Wang P, Velazquez H, Yao X, Li Y, et al. Renalase is a novel, soluble monoamine oxidase that regulates cardiac function and blood pressure. J Clin Invest. (2005) 115:1275–80. doi: 10.1172/JCI24066
170. Ohtsuka T, Hamada M, Hiasa G, Sasaki O, Suzuki M, Hara Y, et al. Effect of beta-blockers on circulating levels of inflammatory and anti-inflammatory cytokines in patients with dilated cardiomyopathy. J Am Coll Cardiol. (2001) 37:412–7. doi: 10.1016/S0735-1097(00)01121-9
171. Baroletti SA, Gabardi S, Magee CC, Milford EL. Calcium channel blockers as the treatment of choice for hypertension in renal transplant recipients: fact or fiction. Pharmacotherapy. (2003) 23:788–801. doi: 10.1592/phco.23.6.788.32180
172. de Mattos AM, Olyaei AJ, Bennett WM. Pharmacology of immunosuppressive medications used in renal diseases and transplantation. Am J Kidney Dis. (1996) 28:631–67. doi: 10.1016/S0272-6386(96)90246-2
173. Luke RG. Pathophysiology and treatment of posttransplant hypertension. J Am Soc Nephrol. (1991) 2:S37–44.
174. Textor SC, Canzanello VJ, Taler SJ, Wilson DJ, Schwartz LL, Augustine JE, et al. Cyclosporine-induced hypertension after transplantation. Mayo Clin Proc. (1994) 69:1182–93. doi: 10.1016/S0025-6196(12)65772-3
175. Wagner K, Albrecht S, Neumayer HH. Prevention of posttransplant acute tubular necrosis by the calcium antagonist diltiazem: a prospective randomized study. Am J Nephrol. (1987) 7:287–91. doi: 10.1159/000167487
176. Shilliday IR, Sherif M. Calcium channel blockers for preventing acute tubular necrosis in kidney transplant recipients. Cochrane Database Syst Rev. (2007) CD003421. doi: 10.1002/14651858.CD003421.pub4
177. van Riemsdijk IC, Mulder PG, de Fijter JW, Bruijn JA, van Hooff JP, Hoitsma AJ, et al. Addition of isradipine (Lomir) results in a better renal function after kidney transplantation: a double-blind, randomized, placebo-controlled, multi-center study. Transplantation. (2000) 70:122–6. Retrieved from: https://journals.lww.com/transplantjournal/pages/default.aspx
178. Martinez-Castelao A, Hueso M, Sanz V, Rejas J, Alsina J, Grinyo JM. Treatment of hypertension after renal transplantation: long-term efficacy of verapamil, enalapril, and doxazosin. Kidney Int Suppl. (1998) 68:S130–4. doi: 10.1046/j.1523-1755.1998.06826.x
179. Rump LC, Oberhauser V, Schwertfeger E, Speidel L, Zimmerhackl L, Kirste G, et al. Dihydropyridine calcium antagonists and renal function in hypertensive kidney transplant recipients. J Hypertens. (2000) 18:1115–9. doi: 10.1097/00004872-200018080-00017
180. Kasiske BL, Chakkera HA, Roel J. Explained and unexplained ischemic heart disease risk after renal transplantation. J Am Soc Nephrol. (2000) 11:1735–43. Retrieved from: https://jasn.asnjournals.org
181. Cross NB, Webster AC, Masson P, O'Connell PJ, Craig JC. Antihypertensives for kidney transplant recipients: systematic review and meta-analysis of randomized controlled trials. Transplantation. (2009) 88:7–18. doi: 10.1097/TP.0b013e3181a9e960
182. Hiremath S, Fergusson D, Doucette S, Mulay AV, Knoll GA. Renin angiotensin system blockade in kidney transplantation: a systematic review of the evidence. Am J Transplant. (2007) 7:2350–60. doi: 10.1111/j.1600-6143.2007.01928.x
183. Nankivell BJ, Chapman JR. Chronic allograft nephropathy: current concepts and future directions. Transplantation. (2006) 81:643–54. doi: 10.1097/01.tp.0000190423.82154.01
184. El-Zoghby ZM, Stegall MD, Lager DJ, Kremers WK, Amer H, Gloor JM, et al. Identifying specific causes of kidney allograft loss. Am J Transplant. (2009) 9:527–35. doi: 10.1111/j.1600-6143.2008.02519.x
185. Cordonnier DJ, Pinel N, Barro C, Maynard M, Zaoui P, Halimi S, et al. Expansion of cortical interstitium is limited by converting enzyme inhibition in type 2 diabetic patients with glomerulosclerosis. The Diabiopsies Group. J Am Soc Nephrol. (1999) 10:1253–63.
186. Ibrahim HN, Jackson S, Connaire J, Matas A, Ney A, Najafian B, et al. Angiotensin II blockade in kidney transplant recipients. J Am Soc Nephrol. (2013) 24:320–7. doi: 10.1681/ASN.2012080777
187. Hernandez D, Lacalzada J, Salido E, Linares J, Barragan A, Lorenzo V, et al. Regression of left ventricular hypertrophy by lisinopril after renal transplantation: role of ACE gene polymorphism. Kidney Int. (2000) 58:889–97. doi: 10.1046/j.1523-1755.2000.00239.x
188. Klingbeil AU, Muller HJ, Delles C, Fleischmann E, Schmieder RE. Regression of left ventricular hypertrophy by AT1 receptor blockade in renal transplant recipients. Am J Hypertens. (2000) 13:1295–300. doi: 10.1016/S0895-7061(00)01213-9
189. Hiremath S, Fergusson DA, Fergusson N, Bennett A, Knoll GA. Renin-angiotensin system blockade and long-term clinical outcomes in kidney transplant recipients: a meta-analysis of randomized controlled trials. Am J Kidney Dis. (2017) 69:78–86. doi: 10.1053/j.ajkd.2016.08.018
190. Jiang YM, Song TR, Qiu Y, Liu JP, Wang XD, Huang ZL, et al. Effect of renin-angiotensin system inhibitors on survival in kidney transplant recipients: a systematic review and meta-analysis. Kaohsiung J Med Sci. (2018) 34:1–13. doi: 10.1016/j.kjms.2017.07.007
191. Gavras I, Manolis AJ, Gavras H. The alpha2 -adrenergic receptors in hypertension and heart failure: experimental and clinical studies. J Hypertens. (2001) 19:2115–24. doi: 10.1097/00004872-200112000-00001
192. Gavras I, Gavras H. Role of alpha2-adrenergic receptors in hypertension. Am J Hypertens. (2001) 14:171S−7. doi: 10.1016/S0895-7061(01)02085-4
193. Goldberg M, Gehr M. Effects of alpha-2 agonists on renal function in hypertensive humans. J Cardiovasc Pharmacol. (1985) 7(Suppl. 8):S34–7. doi: 10.1097/00005344-198500078-00006
194. Campese VM, Romoff M, Telfer N, Weidmann P, Massry SG. Role of sympathetic nerve inhibition and body sodium-volume state in the antihypertensive action of clonidine in essential hypertension. Kidney Int. (1980) 18:351–7. doi: 10.1038/ki.1980.145
195. Cohen IM, O'Connor DT, Preston RA, Stone RA. Reduced renovascular resistance by clonidine. Clin Pharmacol Ther. (1979) 26:572–7. doi: 10.1002/cpt1979265572
196. Green S, Zawada ET Jr, Muakkassa W, Johnson M, Mackenzie T, McClanahan M, et al. Effect of clonidine therapy on renal hemodynamics in renal transplant hypertension. Arch Intern Med. (1984) 144:1205–8. doi: 10.1001/archinte.144.6.1205
197. Grzesk G, Wicinski M, Malinowski B, Grzesk E, Manysiak S, Odrowaz-Sypniewska G, et al. Calcium blockers inhibit cyclosporine A-induced hyperreactivity of vascular smooth muscle cells. Mol Med Rep. (2012) 5:1469–74. doi: 10.3892/mmr.2012.847
198. Curtis JJ, Luke RG, Jones P, Diethelm AG. Hypertension in cyclosporine-treated renal transplant recipients is sodium dependent. Am J Med. (1988) 85:134–8. doi: 10.1016/S0002-9343(88)80331-0
199. Birtch AG, Zakheim RM, Jones LG, Barger AC. Redistribution of renal blood flow produced by furosemide and ethacrynic acid. Circ Res. (1967) 21:869–78. doi: 10.1161/01.RES.21.6.869
200. Solomon R, Werner C, Mann D, D'Elia J, Silva P. Effects of saline, mannitol, and furosemide on acute decreases in renal function induced by radiocontrast agents. N Engl J Med. (1994) 331:1416–20. doi: 10.1056/NEJM199411243312104
201. Midtvedt K, Hartmann A, Foss A, Fauchald P, Nordal KP, Rootwelt K, et al. Sustained improvement of renal graft function for two years in hypertensive renal transplant recipients treated with nifedipine as compared to lisinopril. Transplantation. (2001) 72:1787–92. doi: 10.1097/00007890-200112150-00013
202. Olyaei AJ, de Mattos AM, Bennett WM. Nephrotoxicity of immunosuppressive drugs: new insight and preventive strategies. Curr Opin Crit Care. (2001) 7:384–9. doi: 10.1097/00075198-200112000-00003
203. Suwelack B, Gerhardt U, Hausberg M, Rahn KH, Hohage H. Comparison of quinapril versus atenolol: effects on blood pressure and cardiac mass after renal transplantation. Am J Cardiol. (2000) 86:583–5; A10. doi: 10.1016/S0002-9149(00)01024-9
204. Midtvedt K, Ihlen H, Hartmann A, Bryde P, Bjerkely BL, Foss A, et al. Reduction of left ventricular mass by lisinopril and nifedipine in hypertensive renal transplant recipients: a prospective randomized double-blind study. Transplantation. (2001) 72:107–11. doi: 10.1097/00007890-200107150-00021
205. Zucchelli P, Zuccala A, Borghi M, Fusaroli M, Sasdelli M, Stallone C, et al. Long-term comparison between captopril and nifedipine in the progression of renal insufficiency. Kidney Int. (1992) 42:452–8. doi: 10.1038/ki.1992.309
206. Kidney Disease Outcomes Quality I. K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis. (2004) 43:S1–290. doi: 10.1053/j.ajkd.2004.03.003
207. Taler SJ, Agarwal R, Bakris GL, Flynn JT, Nilsson PM, Rahman M, et al. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for management of blood pressure in CKD. Am J Kidney Dis. (2013) 62:201–13. doi: 10.1053/j.ajkd.2013.03.018
208. Kidney Disease: Improving Global Outcomes Transplant Work G. KDIGO clinical practice guideline for the care of kidney transplant recipients. Am J Transplant. (2009) 9(Suppl. 3):S1–155. doi: 10.1111/j.1600-6143.2009.02834.x
209. Baker R, Jardine A, Andrews P. Renal Association Clinical Practice Guideline on post-operative care of the kidney transplant recipient. Nephron Clin Pract. (2011) 118(Suppl. 1):c311–47. doi: 10.1159/000328074
210. Ruzicka M, Quinn RR, McFarlane P, Hemmelgarn B, Ramesh Prasad GV, Feber J, et al. Canadian Society of Nephrology commentary on the 2012 KDIGO clinical practice guideline for the management of blood pressure in CKD. Am J Kidney Dis. (2014) 63:869–87. doi: 10.1053/j.ajkd.2014.03.003
211. Verbeke F, Lindley E, Van Bortel L, Vanholder R, London G, Cochat P, et al. A european renal best practice (ERBP) position statement on the kidney disease: improving global outcomes (KDIGO) clinical practice guideline for the management of blood pressure in non-dialysis-dependent chronic kidney disease: an endorsement with some caveats for real-life application. Nephrol Dial Transplant. (2014) 29:490–6. doi: 10.1093/ndt/gft321
212. Chadban SJ, Barraclough KA, Campbell SB, Clark CJ, Coates PT, Cohney SJ, et al. KHA-CARI guideline: KHA-CARI adaptation of the KDIGO Clinical Practice Guideline for the Care of Kidney Transplant Recipients. Nephrology. (2012) 17:204–14. doi: 10.1111/j.1440-1797.2011.01559.x
213. Loghman-Adham M, Soto CE, Inagami T, Sotelo-Avila C. Expression of components of the renin-angiotensin system in autosomal recessive polycystic kidney disease. J Histochem Cytochem. (2005) 53:979–88. doi: 10.1369/jhc.4A6494.2005
214. Goto M, Hoxha N, Osman R, Dell KM. The renin-angiotensin system and hypertension in autosomal recessive polycystic kidney disease. Pediatr Nephrol. (2010) 25:2449–57. doi: 10.1007/s00467-010-1621-z
215. Coffman TM, Himmelstein S, Best C, Klotman PE. Post-transplant hypertension in the rat: effects of captopril and native nephrectomy. Kidney Int. (1989) 36:35–40. doi: 10.1038/ki.1989.157
216. Vanrenterghem Y, Waer M, Christiaens MR, Michielsen P. Bilateral nephrectomy of the native kidneys reduces the incidence of arterial hypertension and erythrocytosis in kidney graft recipients treated with cyclosporin. Leuven Collaborative Group for Transplantation. Transpl Int. (1992) 5(Suppl. 1):S35–7. doi: 10.1111/tri.1992.5.s1.35
217. Lerman MJ, Hinton S, Aronoff R. Bilateral native nephrectomy for refractory hypertension in kidney transplant and kidney pancreas transplant patients. Int J Surg Case Rep. (2015) 15:127–9. doi: 10.1016/j.ijscr.2015.08.001
218. Iino Y, Hayashi M, Kawamura T, Shiigai T, Tomino Y, Yamada K, et al. Renoprotective effect of losartan in comparison to amlodipine in patients with chronic kidney disease and hypertension–a report of the Japanese Losartan Therapy Intended for the Global Renal Protection in Hypertensive Patients (JLIGHT) study. Hypertens Res. (2004) 27:21–30. doi: 10.1291/hypres.27.21
219. Shumate AM, Bahler CD, Goggins WC, Sharfuddin AA, Sundaram CP. Native nephrectomy with renal transplantation is associated with a decrease in hypertension medication requirements for autosomal dominant polycystic kidney disease. J Urol. (2016) 195:141–6. doi: 10.1016/j.juro.2015.07.114
220. Zandbergen AA, Baggen MG, Lamberts SW, Bootsma AH, de Zeeuw D, Ouwendijk RJ. Effect of losartan on microalbuminuria in normotensive patients with type 2 diabetes mellitus. A randomized clinical trial. Ann Intern Med. (2003) 139:90–6. doi: 10.7326/0003-4819-139-2-200307150-00008
221. Obremska M, Boratynska M, Zysko D, Szymczak M, Kurcz J, Gozdzik A, et al. Beneficial effect of bilateral native nephrectomy as complete denervation on left ventricular mass and function in renal transplant recipients. Pol Arch Med Wewn. (2016) 126:58–67. doi: 10.20452/pamw.3269
222. Esler M, Jennings G, Korner P, Willett I, Dudley F, Hasking G, et al. Assessment of human sympathetic nervous system activity from measurements of norepinephrine turnover. Hypertension. (1988) 11:3–20. doi: 10.1161/01.HYP.11.1.3
223. Chatzikyrkou C, Menne J, Gwinner W, Schmidt BM, Lehner F, Blume C, et al. Pathogenesis and management of hypertension after kidney transplantation. J Hypertens. (2011) 29:2283–94. doi: 10.1097/HJH.0b013e32834bd1e7
224. Gazdar AF, Dammin GJ. Neural degeneration and regeneration in human renal transplants. N Engl J Med. (1970) 283:222–4. doi: 10.1056/NEJM197007302830502
225. Mauriello A, Rovella V, Borri F, Anemona L, Giannini E, Giacobbi E, et al. Hypertension in kidney transplantation is associated with an early renal nerve sprouting. Nephrol Dial Transplant. (2017) 32:1053–60. doi: 10.1093/ndt/gfx069
226. DiBona GF. Renal innervation and denervation: lessons from renal transplantation reconsidered. Artif Organs. (1987) 11:457–62. doi: 10.1111/j.1525-1594.1987.tb02710.x
227. Olsen LK, Kamper AL, Svendsen JH, Feldt-Rasmussen B. Renal denervation. Eur J Intern Med. (2015) 26:95–105. doi: 10.1016/j.ejim.2015.01.009
228. Hansen JM, Abildgaard U, Fogh-Andersen N, Kanstrup IL, Bratholm P, Plum I, et al. The transplanted human kidney does not achieve functional reinnervation. Clin Sci. (1994) 87:13–20. doi: 10.1042/cs0870013
229. Protasiewicz M, Poczatek K, Banasik M, Poreba R, Podgorski M, Kurcz J, et al. Successful renal artery denervation in a renal transplant recipient with refractory hypertension. Am J Hypertens. (2014) 27:982–4. doi: 10.1093/ajh/hpt291
230. Dobrowolski LC, Bemelman FJ, Ten Berge IJ, van den Born BJ, Reekers JA, Krediet CT. Renal denervation of the native kidneys for drug-resistant hypertension after kidney transplantation. Clin Kidney J. (2015) 8:79–81. doi: 10.1093/ckj/sfu134
Keywords: antihypertensive medications, bilateral native nephrectomy, blood pressure targets, cardiovascular diseases, kidney transplantation, post-kidney transplant hypertension, native renal sympathetic denervation, 24-h blood pressure monitoring
Citation: Tantisattamo E, Molnar MZ, Ho BT, Reddy UG, Dafoe DC, Ichii H, Ferrey AJ, Hanna RM, Kalantar-Zadeh K and Amin A (2020) Approach and Management of Hypertension After Kidney Transplantation. Front. Med. 7:229. doi: 10.3389/fmed.2020.00229
Received: 26 March 2019; Accepted: 04 May 2020;
Published: 16 June 2020.
Edited by:
Asha Moudgil, Children's National Hospital, United StatesReviewed by:
Gaurav Gupta, Virginia Commonwealth University, United StatesBassam G. Abu Jawdeh, University of Cincinnati, United States
Copyright © 2020 Tantisattamo, Molnar, Ho, Reddy, Dafoe, Ichii, Ferrey, Hanna, Kalantar-Zadeh and Amin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ekamol Tantisattamo, ZXRhbnRpc2FAaHMudWNpLmVkdQ==