 Mirjam P. Roffel
Mirjam P. Roffel Ken R. Bracke
Ken R. Bracke Irene H. Heijink
Irene H. Heijink Tania Maes
Tania Maes
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 19 May 2020
Sec. Pulmonary Medicine
Volume 7 - 2020 | https://doi.org/10.3389/fmed.2020.00196
This article is part of the Research Topic Cross-talk In The Lung Microenvironment In Chronic Respiratory Diseases View all 6 articles
Asthma and Chronic Obstructive Pulmonary Disease (COPD) are chronic obstructive respiratory diseases characterized by airway obstruction, inflammation, and remodeling. Recent findings indicate the importance of microRNAs (miRNAs) in the regulation of pathological processes involved in both diseases. MiRNAs have been implicated in a wide array of biological processes, such as inflammation, cell proliferation, differentiation, and death. MiR-223 is one of the miRNAs that is thought to play a role in obstructive lung disease as altered expression levels have been observed in both asthma and COPD. MiR-223 is a hematopoietic cell–derived miRNA that plays a role in regulation of monocyte-macrophage differentiation, neutrophil recruitment, and pro-inflammatory responses and that can be transferred to non-myeloid cells via extracellular vesicles or lipoproteins. In this translational review, we highlight the role of miR-223 in obstructive respiratory diseases, focusing on expression data in clinical samples of asthma and COPD, in vivo experiments in mouse models and in vitro functional studies. Furthermore, we provide an overview of the mechanisms by which miR-223 regulates gene expression. We specifically focus on immune cell development and activation and involvement in immune responses, which are important in asthma and COPD. Collectively, this review demonstrates the importance of miR-223 in obstructive respiratory diseases and explores its therapeutic potential in the pathogenesis of asthma and COPD.
Worldwide, the prevalence of people with chronic respiratory disease is increasing. The most common chronic inflammatory airway diseases are asthma and chronic obstructive pulmonary disease (COPD), with a prevalence of 358 million asthma patients and 174 million COPD patients, respectively (1). Over the last years, it has become clear that both diseases are complex and heterogeneous with different underlying processes (2, 3).
Asthma affects the large and small airways leading to symptoms such as shortness of breath, wheezing, coughing, and chest tightness. The cause of asthma is unknown, however there are several risk factors influencing the development and severity of asthma. The most common risk factors are host factors (including genetics) and environmental factors (allergens, viral/microbial infections, air pollutants, smoke) (4).
Asthma can be divided in several phenotypes depending on presence of allergy, inflammatory profiles, and age of onset (2). Allergic asthma is the most common form of asthma, where allergens such as house dust mite, pollen, and pet dander trigger a type 2-driven inflammatory response in the lungs. This leads to airway remodeling, with increased smooth muscle mass, airway hyperresponsiveness, and mucus hypersecretion (4). Allergic asthma, which generally develops early in life, is characterized by the presence of allergen specific immunoglobulin E, elevated airway infiltration of eosinophils and T helper 2 (Th2) lymphocytes and increased levels of type 2 cytokines e.g., IL-5 and IL-13 (2). Type 2 inflammation can also occur in non-allergic eosinophilic asthma, which develops later in life, is often more severe and is associated with increased numbers of type 2 innate lymphoid cells (ILC2s) that produce IL-5 and IL-13 in response to epithelial damage (5). Other, non-type 2 asthma phenotypes are characterized by elevated sputum neutrophils numbers (called neutrophilic asthma) or by airway remodeling that is not accompanied by higher sputum levels of eosinophils and neutrophils (called paucigranulocytic asthma) (2). The clinical manifestations of asthma can be controlled by inhaled corticosteroids (ICS), combined with bronchodilators in the majority of asthma patients, while a subset of patients with severe asthma is insensitive to ICS (6, 7).
COPD affects around 10% of the world's population and is the third leading cause of death worldwide (8). In COPD, chronic inflammation mainly affects the small airways leading to symptoms such as shortness of breath, chronic cough and/or wheezing (6). It is a heterogeneous disease characterized by irreversible airflow limitation and accelerated lung function decline caused by pulmonary inflammation, chronic bronchitis, (small) airway remodeling and/or destruction of alveolar tissue (emphysema) (9). The major risk factor for developing COPD is chronic exposure to noxious particles and gases such as cigarette smoke, air pollution and occupational exposures. In COPD, the exposure to those noxious particles and gases leads to pulmonary infiltration of predominantly neutrophils and CD8+ T lymphocytes. Furthermore, macrophages play a major role in the inflammatory response in COPD, upon activation macrophages release several cytokines and matrix metalloproteases (10). In the lung of severe COPD patients increased B cell counts and increased number and size of B-cell rich lymphoid follicles have been found (11). The major treatment in COPD patients is long-acting bronchodilators and ICS. However, COPD patients are less sensitive to ICS compared to allergic asthma patients (10). In asthma and COPD, current treatments relieve symptoms and prevent exacerbations, however, they cannot counteract the underlying disease process.
MicroRNAs (miRNAs) are small, non-coding RNA molecules with a length of 21–25 nucleotides. MiRNAs can control gene expression by targeting specific mRNAs for degradation or translational repression. MiRNAs bind to a specific sequence at the 3′UTR of the target messenger RNA (mRNA). The complementary sequences between miRNA and mRNA are usually not fully overlapping, implicating that each miRNA can regulate hundreds of target genes and that several miRNAs can target the same mRNAs (12).
MiRNAs are involved in multiple biological processes including inflammation, cell proliferation, differentiation, and death and may thus regulate various pathological processes (13). Indeed, miRNAs have been implicated in many diseases. Furthermore, miRNAs are present in bodily fluids due to active secretion from living cells in microvesicles, secretion from cells with RNA-binding proteins or leakage of microvesicles from death cells (14). Due to stability and high expression of miRNAs in bodily fluids, they can be potential biomarkers in asthma and COPD. As mentioned above, the development of asthma and COPD is the consequence of an interaction between genetic and environmental factors. By regulating gene expression, miRNAs may constitute an important link between the factors involved in the development of obstructive lung disease.
In this review, we will focus on the potential role of miR-223 in the pathogenesis of asthma and COPD, since miR-223 is differentially expressed in various tissues from asthma patients compared to healthy controls and is among the highest differentially expressed miRNA in COPD patients. Furthermore, miR-223 has a crucial role in innate immunity, myeloid cell differentiation, and cell homeostasis and has several targets that are involved in pathways implicated in the pathogenesis of both asthma and COPD. Finally, miR-223 is easily detectable in multiple human samples (e.g., sputum, bronchial biopsies, and blood) and could therefore serve as a potential biomarker.
Although the role of miR-223 in hematopoietic cell development and innate and adaptive immune responses has recently been extensively reviewed by others (15), we shortly summarize the most important findings with regard to the role of miR-223 in hematopoiesis. MiR-223 is transcribed from an independent promoter located on the X chromosome and is mainly expressed by hematopoietic cells (16). Whereas, levels of miR-223 are low in pluripotent hematopoietic stem cells and common myeloid progenitors, miR-223 expression is induced upon myeloid differentiation (17). In humans, the expression of miR-223 is controlled by two transcriptional factors that compete for binding to the miR-223 promoter, namely nuclear factor I A-type (NFI-A) and CCAAT enhancer protein α (C/EBPα). Under resting conditions, NFI-A is bound to the miR-223 promoter, maintaining low expression of miR-223. During granulocytic differentiation, NFI-A is released from the miR-223 promoter and replaced by C/EBPα, resulting in upregulation of miR-223 expression (16). Interestingly, one of the target genes of miR-223 is NFI-A, implicating that upregulation of miR-223 dampens the expression of NFI-A, resulting in a positive feedback loop. In line with a role of miR-223 in myeloid differentiation, overexpression of miR-223 induces the monocytic and granulocytic differentiation marker CD11b, while inhibition of miR-223 was shown to reduce the expression of CD11b in promyelocytic leukemia cells (16). In addition to the importance in myeloid differentiation, miR-223 is involved in erythropoiesis by dampening the gene expression and protein translation of LIM-only protein 2, a positive regulator of erythropoiesis (18, 19).
MiRNA profiling in human blood demonstrated that miR-223 is expressed in hematopoietic stem cells, granulocytes, dendritic cells and monocytes, while lower levels of miR-223 were also found in naïve and memory T cells (16, 20, 21). In induced sputum high expression of miR-223 was measured in monocytes, macrophages and neutrophils (22). Furthermore, in situ hybridisation in human bronchial biopsies showed that miR-223 expression was mainly expressed in neutrophils localized in the lamina propria (22).
In this review, we will use findings of in vivo murine models to provide mechanistic insight into how miR-223 can contribute to the pathogenesis of asthma and COPD. In mice, the regulation of miR-223 expression is slightly different compared to humans. The myeloid differentiation factors PU.1 together with C/EBPβ enhance the activity of the miR-223 promoter, whereas erythroid transcription factor GATA binding protein 1 represses the miR-223 promoter (23). Depletion of miR-223 in a mouse model does not result in apparent developmental abnormalities, however granulocyte hyperplasia has been observed in the bone marrow (17). The increased numbers of neutrophils in miR-223 deficient mice display morphological changes, including nuclear hypersegmentation and blebbing and are hyperactivated as demonstrated by their increased superoxide production (17). Similar to humans, granulocyte-monocyte progenitors express low levels of miR-223 in blood and bone marrow obtained from mice, but during myeloid differentiation the expression levels of miR-223 are increased. Expression of miR-223 is highest in mature neutrophils, whereas B- and T-cells barely express miR-223 (17, 24, 25).
In summary, the expression of miR-223 is controlled by both enhancers and repressors, is low in haematopoietic progenitor cells and increased during granulopoiesis, indicating its importance in the control of cell homeostasis.
As described above, miR-223 is mainly expressed in myeloid cells and may play a role in innate immunity. Therefore, several studies investigated the expression of miR-223 in asthma and COPD patients (Table 1). The first report on miRNA profiling in asthma patients, where miR-223 was mentioned, was performed in bronchial biopsies of eight mild atopic asthmatic patients and eight healthy controls using a miRNA array. No difference in miRNA expression was observed between asthma patients and healthy controls (26). However, miR-223 was one of highest expressed miRNAs in biopsies and was shown to be expressed in macrophages (26). No differences in miR-223 expression were found in an additional study on bronchial biopsies from 12 mild and 12 severe asthma patients compared to healthy controls using quantitative real time polymerase chain reaction (27), which may be due to the low sample size. In larger cohorts and more specific compartments differences in miR-223 expression between asthma patients and healthy controls were identified. MiRNA profiling in bronchial brushings obtained from 16 steroid naive asthmatics compared to 12 healthy controls demonstrated higher expression of miR-223 in asthma patients compared to healthy controls (28). Two independent asthma cohorts showed that miR-223 is higher expressed in induced sputum supernatant of severe asthma patients compared to healthy controls (22). Moreover, after subdividing the asthma groups into neutrophilic and eosinophilic asthma based on the percentage sputum neutrophils and eosinophils, increased miR-223 levels were found in neutrophilic asthma patients compared to healthy controls and eosinophilic asthmatics. Furthermore, miR-223 was negatively associated with FEV1, FEV1/FVC ratio, and positively associated with the percentage neutrophils in sputum (22). A recent study by Gomez et al. found almost similar results, miR-223 expression was increased in sputum of asthma patients and was associated with a neutrophilic asthma phenotype. Furthermore, miR-223 expression levels were also correlated with multiple features of severe asthma, bronchodilator response, and FeNO levels (29). In contrast, in a study using blood samples, miR-223 expression was lower in blood-derived T cells from mild to moderate asthma patients compared to healthy controls (21). Another study found that in utero exposure to cigarette smoke increases the risk of developing allergic sensitization, which was associated with lower levels of regulatory T cells and with high miR-223 expression in cord blood (34).
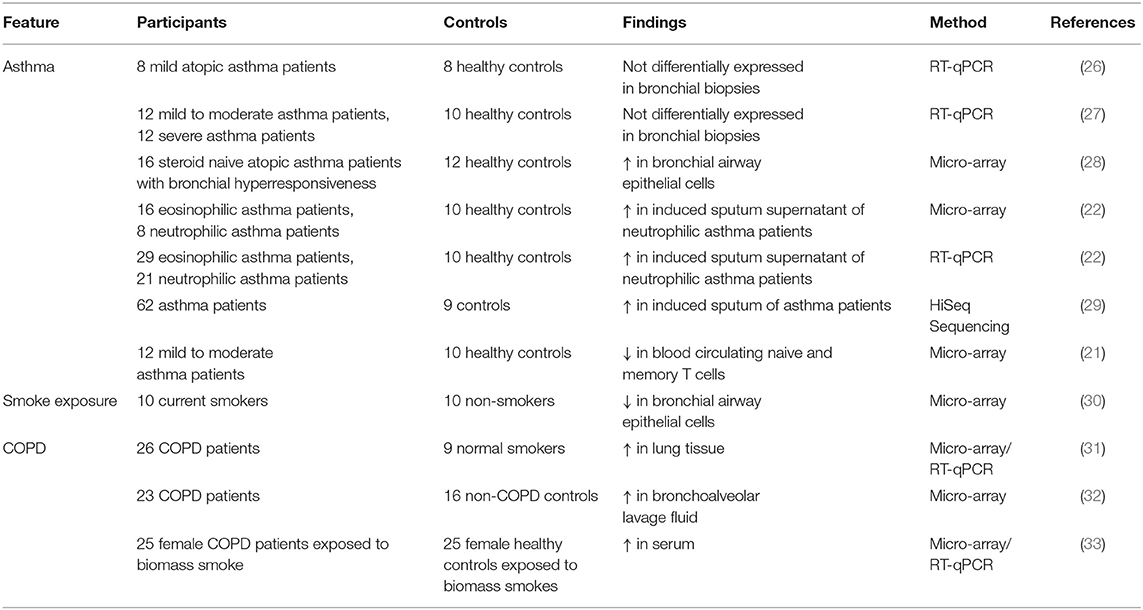
Table 1. Overview of miR-223 expression in asthma and COPD patients compared to controls.
With regard to smoking and COPD, lower miR-223 expression levels have been observed in bronchial brushings from current smokers compared to never smokers (30). Ezzie et al. focusing on differentially expressed miRNAs in lung tissue of smokers with or without COPD, demonstrated higher miR-223 expression in COPD patients compared to smokers without COPD (31). Furthermore, higher levels of miR-223 were also measured in bronchoalveolar lavage fluid (BALF) obtained from COPD patients compared to non-COPD controls (32). In serum of women with COPD due to biomass smoke, miR-223 levels are higher compared to healthy controls exposed to biomass smoke (33).
Taken together, it is clear that miR-223 expression is differently expressed in obstructive lung disease and seems to be associated with neutrophilia (Table 1). However, thus far miR-223 expression was associated with an inflammatory phenotype in only two asthma studies (22, 29). In mild atopic asthma patients the increase in miR-223 could also originate from an eosinophilic component, since both neutrophils and eosinophils were present in the bronchial brushings (28). With regard to COPD, no data are available that link miR-223 expression to disease stage, inflammatory phenotype or presence of emphysema. Furthermore, (biomass) smoke can alter the expression of miR-223, which together with differences in examined samples and patient groups further adds to the complexity of the observed findings.
Both asthma and COPD are characterized by inflammatory responses and airway remodeling upon environmental exposures. In the paragraphs below we will describe how miR-223 can influence these processes. The role of miR-223 in regulating inflammation and cell proliferation, differentiation and death was reviewed before (15), but here we focus on the potential implications for asthma and COPD. We will use findings from in vivo murine models of infection and acute lung injury to speculate on the importance of miR-223 in asthma and COPD and highlight several of the miR-223 targets, which have been validated by luciferase assays (Table 2). Furthermore, we review findings obtained from in vitro studies, which are often within the context of cancer, but can also be relevant within the context of asthma and COPD. Table 3 gives a summary of the experiments done for miR-223 in each cell type and the expression of this target gene in asthma and/or COPD. Furthermore, Figure 1 shows an overview of the potential roles of miR-223 in the pathogenesis of asthma and COPD.

Table 2. Overview of the validated target genes of miR-223.
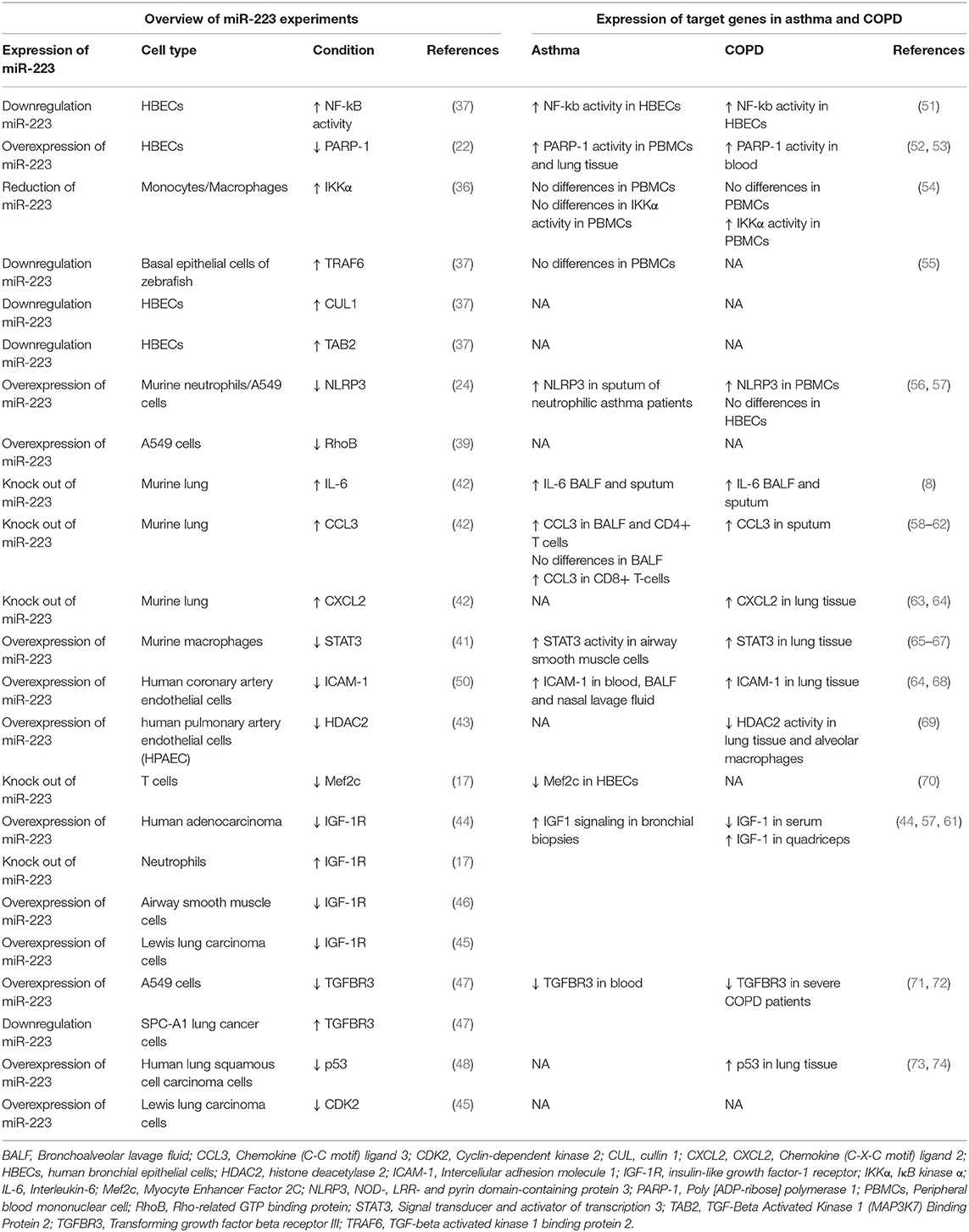
Table 3. Overview of miR-223 experiments and their expression in asthma and COPD.
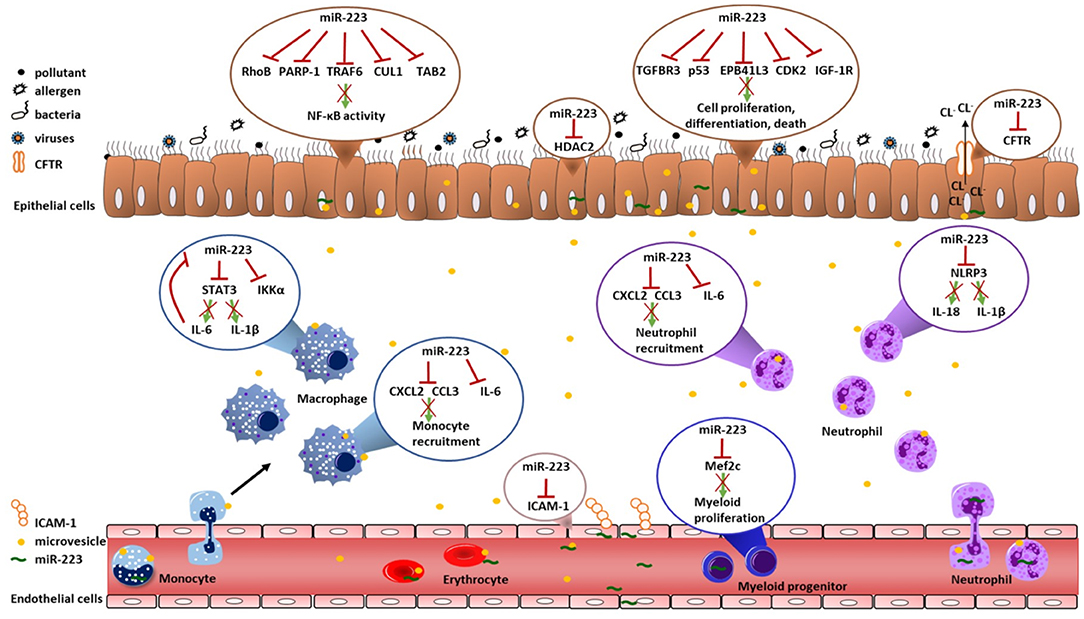
Figure 1. Overview of validated targets of miR-223. This overview illustrates all validated targets genes of miR-223 that could contribute to the pathogenesis of asthma and COPD. There were no studies that investigated validated targets of miR-223 in T-cells and eosinophils. The red cross indicates that miR-223 targets this gene and/or protein. CCL3, Chemokine (C-C motif) ligand 3; CUL1, cullin 1; CDK2, Cyclin-dependent kinase 2; CFTR, cystic fibrosis transmembrane conductance regulator; CXCL2, Chemokine (C-X-C motif) ligand 2; EPB41L3, erythrocyte membrane protein band 4.1 like 3; HDAC2, histone deacetylase 2; ICAM-1, Intercellular adhesion molecule 1; IGF-1R, insulin-like growth factor-1 receptor; IKKα, IκB kinase α; IL-6, Interleukin-6; Mef2c, Myocyte Enhancer Factor 2C; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3; PARP-1, Poly [ADP-ribose] polymerase 1; RhoB, Rho-related GTP binding protein; STAT3, Signal transducer and activator of transcription 3; TAB2, TGF-Beta Activated Kinase 1 (MAP3K7) Binding Protein 2; TGFBR3, Transforming growth factor beta receptor III.
Several studies have demonstrated increased levels of miR-223 in blood samples and/or inflamed lung samples from mice that were exposed to allergens, bacteria, viruses, or fungi compared to samples from untreated mice, all relevant exposures for the development of obstructive lung disease (35, 42, 43, 75, 76). In this section we will overview the role of miR-223 in inflammatory responses.
The nuclear factor kappa B (NF-κB) pathway plays a major role in inflammatory responses by regulating cell survival, activation, and differentiation of immune cells. The activation of NF-κB induces expression of various pro-inflammatory genes and inflammasome activation (77). In bronchial biopsies obtained from asthma and COPD patients, NF-κB is activated upon exposure to various environmental triggers, leading to pro-inflammatory responses (51) and therefore the activation of NF-κB may be a critical component of the pathogenesis of both diseases.
Several studies suggest that miR-223 is a negative regulator of NF-κB signaling. While overexpression of miR-223 in human bronchial epithelial cells (HBECs) decreased NF-κB activity, downregulation of miR-223 in HBECs resulted in increased NF-κB activity after Pseudomonas aeruginosa stimulation (37). One of the targets of miR-223 involved in the activation of NF-κB pathway is poly (ADP-ribose) polymerase 1 (PARP-1). PARP-1 influences expression of various pro-inflammatory factors and chromatin remodeling, which promotes inflammation (78).
Overexpressing miR-223 in HBECs downregulates PARP-1 expression (22). With regard to the pathogenesis of asthma, it was demonstrated that PARP-1 deficiency in OVA-induced allergic mouse models leads to a reduction of eosinophilia and Th2 cytokine production (79, 80). Furthermore, PARP-1 downregulates STAT-6, leading to downregulation of GATA-3, which is crucial in promoting type 2 differentiation and expression of IL-4, IL-5, and IL-13 (79). Of note, multiple in vivo and in vitro studies found contradicting results on PARP-1 inhibition and IL-17 levels, which is important for the recruitment of neutrophils (78). An in vivo study found elevated IL-17 levels in serum but decreased levels of GM-CSF, IL-22, and KC in BAL and increased levels of regulatory T cells in the spleen of PARP-1 deficient mice exposed to HDM. This suggest that PARP-1 depletion resulted in an anti-inflammatory effect (52). Similar to NF-κB, PARP-1 activity is increased in asthma patients, which was measured in peripheral blood mononuclear cells and lung tissue compared to controls (52). Based on the evidence that miR-223 is especially increased in sputum supernatant of neutrophilic asthma patients compared to healthy controls and eosinophilic asthma patients (22), we suggest that miR-223 acts as a modulator to limit PARP-1 expression, controlling eosinophilic, and neutrophilic responses in asthma. Therefore, it would be of interest to investigate the levels of PARP-1 in eosinophilic and neutrophilic inflammation in asthma and associate this with miR-223 expression. The activity of PARP-1 in COPD patients is systematically increased (53). In vivo experiments demonstrated that PARP-1 inhibition protects against emphysema and elastase induced-inflammation (81) and in vitro experiments showed that PARP-1 is activated upon cigarette smoke and oxidative stress (82). Furthermore, in an acute lung injury model with miR-223 deficiency, repression of PARP-1 reversed acute lung injury (35). However, the exact role of PARP-1 in COPD has not been well-addressed. Therefore, further studies are needed to identify the role and relationship of miR-223 and PARP-1 in COPD and its involvement in specific disease phenotypes.
Another target of miR-223 involved in the activation of NF-κB is IκB kinase α (IKKα), which together with IKKβ forms the IKK kinase complex (36). Amongst others, the exposure to bacteria and viruses can induce phosphorylation of the IKK kinase complex, resulting in NF-κB activation (83). Moreover, IKKα is a transcriptional regulator of Th17 differentiation by interacting with the RORγt transcription factor (84), and inhibition of IKKα results in lower levels of Th17 cells (85). During in vitro differentiation of human monocytes to macrophages, the reduction of miR-223 expression is associated with elevated IKKα levels (36). Upon TLR9 activation, higher levels of IKKα and reduced levels of several pro-inflammatory mediators have been found in bone marrow-derived miR-223 KO neutrophils compared to WT neutrophils (86). In asthma and COPD patients, IKKα levels were not different in peripheral blood mononuclear cells compared to healthy controls. However, higher p-IKKα levels were found in peripheral blood mononuclear cells of COPD patients and control smokers compared to asthma patients and non-smoking controls (54).
Recent studies showed that other miR-223 targets involved in the NF-κB activation are TGF-beta activated kinase 1 binding protein 2 (TRAF6), cullin 1(CUL1), and TGF-beta activated kinase 1 (MAP3K7) binding protein 2 (TAB2) (37, 87). Inhibition of miR-223 in basal epithelial cells from zebrafish induced TRAF6, CUL1, and TAB2 (37). TRAF6 is an intracellular signaling molecule mediating NF-κB activation by toll-like receptor activation of dendritic cells. Depletion of TRAF6 in dendritic cells resulted in higher infiltration of eosinophils in lung tissue and higher levels of IL-5, IL-13, and IGF-1 in an OVA-induced mouse model (88). This suggests that downregulation of TRAF6 by miR-223 contributes to Th2 responses. Moreover, in HBECs overexpression of miR-223 reduced NF-κB activity by targeting CUL1 and TAB2 (37). Furthermore, miR-223 knockdown in zebrafish leads to upregulation of Cul1 expression, and subsequent higher neutrophil recruitment after wounding compared to controls (37). This indicates that in in vivo models miR-223 contributes to homeostasis of neutrophils and eosinophils.
In two different asthma cohorts, there were no significant differences in TRAF6 gene expression in PBMCs-derived from steroid naïve asthmatic children compared to healthy controls (55). For CUL1 and TAB2, it is unknown if there are any differences in expression between asthma, COPD patients, and healthy controls. Therefore, despite evidence from experimental studies, the effect of miR-223 on the target genes IKKα, TRAF6, and CUL1 in asthma and COPD patients, and whether their regulation contributes to the pathogenesis of these diseases, remains unclear.
NF-κB activation results in expression of several genes including the inflammasome NLR family pyrin domain containing 3 (NLRP3). Activation of the NLRP3 inflammasome leads to cleavage of pro-IL-1β and pro-IL-18 by caspase-1 into mature IL-1β and IL-18. IL-1β is a cytokine involved in the initiation and persistence of inflammation. NLRP3 can be activated by cigarette smoke, lipopolysaccharide (LPS), bacterial and viral exposures (89) and is identified in airway epithelial cells and peripheral blood neutrophils (90). Yuan et al. already highlighted that miR-223 represses NLRP3 function (15). Here we will focus on the role of miR-223 and NLRP3 in the context of asthma and COPD. In sputum of neutrophilic asthma patients excessive inflammasome activation and production of IL-1β have been found, which correlate with neutrophil counts in lung tissue and sputum, asthma severity, and steroid resistance in asthma (56, 90, 91). As well-increased gene expression of NLRP3, IL-1β, and IL-18 has been found in PBMCs from COPD patients compared to smokers (92), while no differences of NLRP3, IL-1β, and IL-18 in bronchial epithelial were observed in severe stable COPD patients compared to current smokers (93), see also Table 3. This indicates that IL-1β and NLRP3 play an important role in neutrophilic asthma inflammation and in COPD.
Multiple in vitro and in vivo studies demonstrated that miR-223 targets NLRP3, resulting in reduced inflammation (24, 38, 39, 75). Overexpression of miR-223 in murine neutrophils reduces the activity of NLRP3, leading to less secretion of IL-1β (24), while miR-223 deficient mice have increased levels of NLRP3 in bone-marrow-derived neutrophils (75). However, NLRP3 or caspase-1 deficient mice still have acute pulmonary inflammation upon cigarette exposure (94). A recent study showed that overexpression of miR-223 in human adenocarcinoma alveolar basal epithelial cells reduced caspase-1, IL-1β, and IL-18 levels by targeting NLRP3 and rho-related GTP-binding protein (RhoB) after LPS stimulation (39). In addition, mice infected with Klebsiella pneumoniae and treated with microvesicles loaded with miR-223/miR-142 display reduced levels of caspase-1 activity in lung and reduced levels IL-1β and IL-18 in BALF, which the authors suggest is probably caused by targeting NLRP3 (38). Similar results were recently found in an acute and chronic liver injury mouse model, where a miR-223 mimic reduced NLRP3 and IL-1β expression in mice with endotoxin-induced acute hepatitis and diet-induced chronic fibrosis. Furthermore, this miR-223 mimic also led to less infiltration of macrophages and neutrophils in the liver, possibly due to the lower NLRP3 and IL-1β levels (95). An interesting finding is that in vitro stimulation of a bronchial epithelial cell line carrying a mutation in cystic fibrosis transmembrane conductance regulator (CFTR) gene with IL-1β induces miR-223 expression. Furthermore, they showed that CFTR is a validated target of miR-223 (40), which could be important since recently a link between CFTR and the chronic bronchitis phenotype in COPD was proposed (96).
In contrary to in vitro studies, the mechanisms mediating NLRP3 and IL-1β expression in obstructive lung diseases are more complex and the higher levels of miR-223 in neutrophilic asthma patients and COPD patients may not be sufficient to reduce pro-inflammatory gene expression as is observed in experimental studies.
Infection can contribute to the development of asthma (97) and cause asthma and COPD exacerbations (97, 98). The impact of miR-223 deficiency on combatting infections, however differs depending on the model system and stimulus. In vitro, miR-223 deficient neutrophils have enhanced capacity to kill Candidia albicans compared to WT (wild type) neutrophils (17). However, in vivo infections with mycobacterium tuberculosis in miR-223 deficient mice leads to much higher mortality in contrast to WT littermates, while depletion of polymorphonuclear neutrophils upon infection prolonged the survival of miR-223 deficient littermates (42). In latter study, high bacterial burden, more neutrophils, and increased levels of the validated targets IL-6, C-C Motif Chemokine Ligand 3 (CCL3), and C-X-C Motif Chemokine Ligand 2 (CXCL2) were observed in the lung of miR-223 deficient mice compared to WT animals (42). Notably, in all models of infection and acute lung injury increased inflammation was found in miR-223 deficient mice compared to WT mice (17, 42, 75). This confirms the notion that miR-223 can have a protective role in lung inflammation upon microbial challenge, which could also be relevant in asthma and COPD.
As mentioned, miR-223 expression affected IL-6, Cxcl2, and Ccl3 levels in the above described mycobacterium tuberculosis model (42). This is of interest, since IL-6 is also increased in BALF and induced sputum of asthmatics and COPD patients (8). IL-6 plays a role in the expansion of Th2 and Th17 cells, which play a role in airway inflammation in (severe) asthma and/or COPD. Furthermore, CCL3 (MIP-1α) and CXCL2 (MIP-2) are important mediators in asthma and COPD because they are chemoattractant for monocytes/macrophages and neutrophils. In asthma, the reports on CCL3 expression are not univocal, as increased as well as equal and reduced CCL3 levels have been found depending on the patient cohort, investigated sample or cell type (Table 3) (58–61). In COPD, higher levels of CCL3 were detected in sputum of COPD patients compared to healthy non-smokers and smokers, which correlate with IL-6 and sIL-6R levels in sputum of COPD patients (62, 99). This indicates that elevated miR-223 expression levels in asthma and COPD could thus influence monocytic inflammatory responses. With regard to CXCL2, little is known in asthma but it has been associated with a neutrophilic phenotype in mouse models (100). In COPD patients CXCL2 gene expression is upregulated in lung tissue compared to controls (63, 64).
Another target of miR-223, which can also be activated by IL-6 and CXCL2 signaling, is STAT3. Overexpression of miR-223 in murine macrophages (Raw264.7) resulted in reduced levels of IL-6 and IL-1β upon LPS and poly(I:C) stimulation by targeting STAT3 (41). STAT3 signaling contributes to the induction and/or response of Th2 and Th17 cells (101–104). Asthma patients have increased STAT3 activity in airway smooth muscle cells (65) and COPD patients have increased STAT3 levels in lung tissue compared to controls (66, 67). Furthermore, a single nucleotide polymorphism (SNP) of STAT3 has been associated with atopic -but not with non-atopic asthma- in children (105). Whether miR-223 modulates STAT-3 in asthma and COPD and whether it is involved in neutrophilic or eosinophilic responses remains to be determined. Finally, miR-223 can also influence release of neutrophils into tissues by reducing the expression of cell adhesion molecule-1 (ICAM-1) in endothelial cells (50). ICAM-1 expression is increased in blood, BAL, and nasal lavage fluid of asthma patients and in lung tissue of COPD patients compared to controls (64, 68).
Taken together, miR-223 targets multiple genes that are involved in the recruitment and activation of neutrophils and monocytes/macrophages, and expansion of Th2 and Th17 cells.
Most studies demonstrated that miR-223 is involved in the NF-κB pathway and has an anti-inflammatory function. However, Leuenberger et al. showed that miR-223 can cause epigenetic changes by targeting histone deacetylase 2 (HDAC2), resulting in activation of the NF-κB pathway. HDAC2 is necessary to deacetylate histones resulting in suppression of several activated inflammatory genes. Increased expression of miR-223 and decreased expression of HDAC2 were found in endothelial cells after stimulation with tumor necrosis factor-α and IL-1β. Furthermore, decreased levels of HDAC and HDAC2 activity were demonstrated in endothelial cells overexpressing miR-223, leading to unwinding of chromatin and activation of several proinflammatory cytokines and chemokines (43, 69). An in vivo study showed that heterozygous HDAC2 mutant mice have increased inflammatory responses, cellular senescence, airspace enlargement, and lung function decline after chronic cigarette exposure, compared to WT mice (106). Moreover, an earlier study found that COPD patients have lower HDAC2 activity in lung tissue and alveolar macropahges compared to healthy controls and that the severity of the disease is negatively correlated with HDAC2 activity (69). HDAC2 activity is an important regulator of corticosteroid sensitivity (107). COPD and asthma patients with lower HDAC2 activity compared to healthy controls are less sensitive to corticosteroids than patients with higher HDAC2 activity levels (108, 109). Overexpression of HDAC2 in alveolar macrophages can restore the corticosteroid sensitivity (69). This indicates that elevated miR-223 expression by e.g., neutrophils contributes to the insensitivity of corticosteroids in asthma and COPD patients by modulating HDAC2, which can worsen the severity of the disease. Notably, early-onset allergic eosinophilic asthmatics are generally corticosteroid-sensitive. It will be interesting to investigate how miR-223 expression correlates with HDAC2 expression in these patients.
Overall, previous paragraphs indicate that miR-223 mainly acts as an anti-inflammatory miRNA. Several studies showed that depletion of miR-223 resulted in enhanced inflammatory responses, while overexpression of miR-223 reduced pro-inflammatory responses. Moreover, miR-223 targets multiple genes that are involved in the NF-κB pathway, e.g., PARP1, IKKα, TRAF6, CUL1, and TAB2. Based on the evidence that miR-223 is increased in multiple samples from asthma and COPD patients, we propose a model where miR-223 acts as a protective miRNA to reduce/prevent pro-inflammatory responses. The increased levels of miR-223 could inhibit PARP-1 levels and modulate the balance of neutrophilic/eosinophilic responses. However, the higher levels of miR-223 in neutrophilic asthma patients may not be sufficient to reduce the levels of IL-6, CCL3, and CXCL2, which contribute to Th17 responses. Since most of the target genes of miR-223 are elevated in asthma and COPD, it might be that the protective effect of miR-223 is impaired or not sufficient to reduce the high inflammatory responses in asthma and COPD, possibly by changes in the local tissue compartments. Another explanation for increased expression of miR-223 target genes in asthma and COPD may be that miR-223 also targets HDAC2, which is important for corticosteroid sensitivity. In asthma and COPD patients with high miR-223 levels, the reduction in HDAC2 could lead to lower corticosteroid sensitivity and less reduction of pro-inflammatory cytokines and chemokines in response to corticosteroids. However, until now it is unknown if there is a direct link between miR-223 levels in the different disease phenotypes and corticosteroid sensitivity due to HDAC2.
Further studies should direct how miR-223 is involved in the pathogenesis of asthma and COPD, since the effects of altered miR-223 expression levels on pro-inflammatory responses remain enigmatic.
In addition to the function of miR-223 in inflammatory processes, several studies have demonstrated that miR-223 contributes to cell proliferation, differentiation, and death. Cell dysfunction and death are also important features in asthma and COPD. In asthma dysregulated apoptosis has been found in T cells, eosinophils, and neutrophils (110). However, the exact contribution of apoptosis in airway epithelial cells to the pathogenesis of asthma is unclear. Oxidative stress induced by smoking can cause damage to the lung matrix and death of structural cells, leading to emphysema. In COPD patients increased apoptosis is present in the lungs, as well as decreased clearance of apoptotic cells (111).
The deficiency of miR-223 in a mouse model leads to hyperproliferation of neutrophils and enhanced differentiation of granulocyte-monocyte progenitor cells (112). MiR-223 targets myocyte enhancer factor 2c (Mef2c), a transcription factor that promotes proliferation of myeloid progenitors, indicating that miR-223 inhibits proliferation of myeloid progenitor cells. Depletion of Mef2c restores the hyperproliferation of neutrophils and expansion of granulocyte-monocyte progenitor cells (17). Interestingly, Mef2c is decreased in bronchial epithelial cells from mild and severe asthma patients compared to healthy controls (70), which could result from the elevated miR-223 levels in bronchial epithelial cells of asthma patients. Cell proliferation can also be controlled by insulin-like growth factor-1 receptor (IGF-1R). In vivo and in vitro studies demonstrated that IGF-1R is a validated target of miR-223 (17, 44–46). IGF-1 upon binding with IGF-1R results in activation of signaling pathway PI3K/Akt and mTOR that controls cell proliferation, growth, survival, and apoptosis. In eosinophil progenitors from miR-223 KO mice upregulated expression in IGF-1R has been found. This results in a delay in differentiation and growth of eosinophil progenitors (113), suggesting that changes in miR-223 can also modulate eosinophil function in asthma. Both in asthma and COPD abnormal IGF-1 signaling has been demonstrated (57). Asthma patients treated with ICS have reduced IGF-1 levels, which is associated with reduced airway wall thickness and inflammation (114). Furthermore, in vivo inhibition of Igf1r reduced airway inflammation, airway resistance, airway wall thickening, and eosinophil levels in blood and BALF in an OVA-induced mouse model (115) and reduced neutrophil and macrophage numbers in a bleomycin-induced mouse model (116). This indicates that miR-223 contributes to airway wall remodeling in asthma. COPD patients with acute exacerbation have decreased IGF-1 levels in serum compared to stable COPD patients and healthy controls (117), while in lung tissue mRNA levels of IGF-1 are increased in COPD patients compared to controls (63). It would be of interest to investigate if miR-223 levels are elevated upon ICS treatment in asthma and COPD.
Besides cell proliferation and differentiation, several in vitro studies in lung cancer cells have investigated the role of miR-223 in cell viability and invasion and apoptosis. However, contradicting results have been found about the function of miR-223 in lung cancer cells, which is probably due to the use of different cell types. Two studies demonstrated that miR-223 promotes cell viability, invasion and reduces apoptosis in adenocarcinoma A549 cells (118) and in SPC-A1 lung cancer cells by targeting transforming growth factor β (TGF-β) receptor 3 (TGFBR3) (47). TGFBR3 is involved in TGF-β signaling, which regulates cell growth, differentiation, and development (47). Reduced expression of miR-223 was demonstrated in airway smooth muscle cells stimulated with TGF-β1 (46), while recently overexpression of miR-223 in DCs increased the levels of TGF-β upon LPS stimulation, which were associated with higher levels of IL-10 and more T regulatory cells (119). TGF-β and the receptors play a key role in airway remodeling in asthma and COPD. TGF-β signaling induces amongst others the expression of collagen and can cause the disruption of airway epithelial integrity, which contribute to epithelial-mesenchymal transition (120). In asthma patients, the expression of TGF-β1 is increased in bronchial biopsies and is associated with the severity of the disease (121). Increased TGF-β1 levels were also found in serum of COPD patients (122), however, in bronchial epithelial cells and alveolar macrophages of COPD patients TGF-β1 levels were decreased compared to control smokers (123).
Furthermore, in asthma and COPD patients several genetic variations (SNPs) were found for TGFBR3 (124, 125). Decreased levels of TGFBR3 were measured in lungs from severe COPD patients with emphysema and in blood of asthma patients compared to controls (71, 72), which might be caused by increased levels of miR-223 in COPD and asthma patients.
Two other studies investigated the role of miR-223 in cell death. Overexpression of miR-223 in human lung squamous cell carcinoma cells inhibit migration and proliferation by targeting p53 (48). Furthermore, the growth rate of Lewis lung carcinoma cells is reduced upon miR-223 overexpression by targeting cyclin-dependent kinase 2 (CDK2). Similar to p53, CDK2 is involved in cell cycle arrest (45). Furthermore, a SNP of CDK2 has been associated with asthma (126). During homeostasis the expression of p53 is low. However, oxidative stress, which can be caused by smoking in airway epithelial cells (127), induces p53 expression and activity, resulting in cell apoptosis. Despite the elevated levels of miR-223 in COPD patients, the validated target p53 is still higher expressed in lung tissue of COPD patients compared to non-COPD controls, especially in smoking COPD patients (73, 74). This suggests that miR-223 could be less sufficient in reducing apoptosis by targeting p53 in COPD patients compared to non-COPD controls.
Overall, miR-223 can target several genes that are involved in cell proliferation, differentiation, viability, invasion, and death by targeting Mef2c, IGF-1R, TGFBR3, CDK2, and p53, genes that have been implicated in the pathogenesis of asthma and/or COPD. However, the function of miR-223 is cell type and tissue dependent. Since structural cells barely express miR-223, the functional differences of miR-223 might be caused by the transfer of miR-223 from myeloid cell to structural cells.
During the last decade, it has become clear that miRNAs can be transferred to recipient cells by extracellular vesicles and high-density lipoproteins, influencing biologic processes in recipient cells (128). Extracellular vesicles contain proteins, lipids, and DNA and RNA molecules, including miRNAs, which can be important non-invasive biomarkers for asthma and COPD (129). Recent studies have shown that inflammatory cells can transfer miR-223 to lung epithelial cells via extracellular vesicles. During homeostasis, the expression of miR-223 is low in alveolar and bronchial epithelial cells, however during lung inflammation miRNAs can transfer in extracellular vesicles derived from polymorphonuclear neutrophils to alveolar epithelial cells, increasing miR-223 levels in epithelial cells (35). A functional study on miR-223 transfer in extracellular vesicles found that miR-223 is still functional active after uptake in recipient cells (130). Furthermore, dramatically increased extracellular vesicle numbers were measured in BALF and serum of mice treated with LPS or Klebsiella pneumoniae. These extracellular vesicles are derived from lung macrophages (38). Liang et al. found that activated platelets also release large amounts of extracellular vesicles that contain high levels of miR-223 and can transfer to human adenocarcinoma A549 cells (49). High-density lipoproteins can also transfer miR-223 into endothelial cells (50). A recent study showed that severe asthma patients have less mature miRNA in extracellular vesicles-derived from BALF and that the miRNA profiles are altered compared to healthy controls, with 90 downregulated miRNA (including miR-223) in asthma. Furthermore, increased expression levels of predicted target genes of these 90 miRNAs were demonstrated, including genes involved in inflammation and remodeling, such as the TGF-β receptors (131). Extracellular vesicles derived from mild asthma patients appear to have a different miRNA profile than those from severe asthma patients, since comparison of miR-223 expression between extracellular vesicles derived from mild asthma patients compared to healthy controls showed no differences (132). Furthermore, BALF from allergen-exposed mice contains higher miR-223 levels compared to BALF from control mice (133). Moreover, cigarette smoke exposure can change the composition of extracellular vesicles derived from HBECs (134). Moreover, COPD patients with acute exacerbation release more extracellular vesicles compared to non-smoking healthy controls (135). This indicates that upon stress extracellular vesicles are altered and might contribute to pro-inflammatory responses. Although miR-223 can be transferred in extracellular vesicles or high-density lipoproteins to recipient cells to control or alter biological functions, further study is required to identify the therapeutic potential of extracellular vesicles in asthma and COPD.
Despite the fact that miR-223 is one of the best studied miRNAs in literature, its functional role in pathogenesis of asthma and COPD remains incompletely understood. One important topic that needs to be addressed further is the identification of precise targets of miR-223 in health and disease and to discriminate in which cellular compartments these targets are altered in disease. In this review we only focused on validated targets, implicating that probably many other miR-223-controlled genes that are relevant for asthma and/or COPD were not considered. A second important area that requires future research is the discrimination between different diseases phenotypes; e.g., eosinophilic, neutrophilic, paucigranulocytic asthma and the involvement of different target such as PARP-1 and NLRP3 in the determination of these phenotypes.
In addition to investigating the expression of miR-223 and its targets in samples from well-characterized patient groups, also the use of in vivo murine models can provide more insight into the role of miR-223 in asthma and COPD. MiR-223 deficient mice can be tested in different asthma and COPD mouse models to better understand how miR-223 contributes to asthma and COPD. Here, the classical models of sole allergen expose or combined exposures with air pollution or microbial triggers such as LPS could be applied, which could mimic a predominant eosinophilic or a mixed neutrophilic/eosinophilic phenotype in asthma (136, 137). For COPD, mouse models with exposure to cigarette smoke, LPS or elastase can be used (138, 139). These studies can help to investigate the role of miR-223 in different asthma and COPD phenotypes when using knockouts for miR-223. However, an important point of consideration when using miR-223-deficient mice is the involvement of miR-223 in hematopoietic cell development, which can complicate evaluation of inflammatory processes. A complementary approach is therefore the use of miR-223 mimics or antagomirs to up- or downregulate miR-223 expression in a specific tissues, such as the lung via inhalation. The use of mimics and antagomirs can unravel the role of miR-223 in the pathogenesis of asthma and COPD, but also in in other diseases (140).
Based on the findings of the studies described above, we suggest that it requires further investigation to determine if miR-223 can be a new diagnostic and/or therapeutic opportunity. The high and stable secretion of miR-223 in bodily fluids makes it a potential biomarker in asthma and COPD. The findings for miR-223 in sputum of neutrophilic asthma patients and the correlation between miR-223 expression and lung function (22) and bronchodilator response (29) suggest that miR-223 could be a diagnostic tool to determine asthma severity. However, miR-223 cannot be linked to a particular phenotype as higher levels of miR-223 were found in bronchial airway epithelial cells from atopic asthma patients with bronchial hyperresponsiveness as well (28). Ideally such a biomarker should predict the disease severity or phenotype in e.g., blood plasma or serum, since sputum inductions are not part of standard clinical practice. It therefore requires further investigation whether the correlations between miR-223 expression, inflammatory phenotype and disease severity are similar in other tissue compartments.
Overall, most of the in vitro studies have indicated an anti-inflammatory role of miR-223. Therefore, despite of the increased levels of miR-223 in asthma and COPD patients, we suggest that overexpressing miR-223 (e.g., by a miRNA mimic), rather than inhibiting miR-223, could be a therapeutic approach in asthma and COPD. Recent studies in cancer showed that there are multiple options to deliver the miRNA of interest, e.g., DNA plasmids, lipid vesicles, nanoparticles, or viral vectors (141). In the context of asthma and COPD, overexpression miR-223 in bronchial epithelial cells can be an option. However, main challenges of miRNA therapy are accuracy and efficiency of delivery. Since the miRNAs need to reach the right target cells, their uptake must be efficient and the correct genes must be targeted, without eliciting unwanted innate immune responses (142). Therefore, many hurdles have to be taken to consider miR-223 as a therapeutic option for asthma and COPD.
In this review, we summarized current knowledge on miR-223 and how this miRNA could be involved in the development and pathogenesis of chronic obstructive airway diseases. In asthma patients, higher miR-223 expression levels in bronchial airway epithelial cells and in induced sputum have been observed, while COPD patients have higher miR-223 expression levels in lung tissue compared to healthy controls. In vivo and in vitro experiments showed that miR-223 is involved in pathways associated with cell proliferation, differentiation, and death and remodeling, however, the exact role is still unclear. Furthermore, miR-223 affects neutrophil function. Higher levels of miR-223 protect mice from lung inflammation, probably by reducing the activity of the NF-κB pathway. Therefore, most data point to an anti-inflammatory role of miR-223, and thus suggest that increased miR-223 levels in the airways try to counteract the ongoing inflammation. It might be that in vivo miR-223 is not sufficient to reduce the strong inflammatory responses in asthma and COPD or that epithelial cells from asthma and COPD patients are less able to take up miR-223. In conclusion, miR-223 is a fascinating miRNA and additional studies are required to unravel its exact role in the pathogenesis of asthma and COPD.
MR and TM contributed conception and design of the review. MR drafted and finalized the manuscript. KB, IH, and TM revised the manuscript and provided critical advice on the content of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
This Research at the Department of Respiratory Medicine (Ghent University) was funded by a Ghent University Grant (BOF19-GOA-008), and Fund for Scientific Research in Flanders (FWO Vlaanderen project 3G041819 and FWO-EOS project G0G2318N). MR was funded by U4 project (Ghent University Grant BOF 05U40516).
TM has participated in an advisory board from GlaxoSmithKline. TM is shareholder of Oryzon Genomics and of Mendelion Lifesciences SL.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
BALF, Bronchoalveolar lavage fluid; C/EBPα, CCAAT enhancer protein α; CCL3, C-C Motif Chemokine Ligand 3; CDK2, Cyclin-dependent kinase 2; CFTR, Cystic fibrosis transmembrane conductance regulator; COPD, Chronic obstructive pulmonary disease; CUL1, Cullin 1; CXCL2, C-X-C Motif Chemokine Ligand 2; EPB41L3, Erythrocyte membrane protein band 4.1 like 3; HBECs, Human bronchial epithelial cells; HDAC2, Histone deacetylase 2; ICAM-1, Intercellular adhesion molecule 1; ICS, Inhaled corticosteroids; IGF-1R, Insulin-like growth factor-1 receptor; IKKα, IκB kinase α; IL-6, Interleukin-6; LPS, Lipopolysaccharide; Mef2c, Myocyte enhancer factor 2c; MiRNAs, MicroRNAs; mRNA, Messenger RNA; NF-κB, Nuclear factor kappa B; NFI-A, Nuclear factor I A-type; NLRP3, NLR family pyrin domain containing 3; OVA, Ovalbumin; PARP-1, Poly (ADP-ribose) polymerase 1; PBMCs, Peripheral blood mononuclear cell; RhoB, Rho-related GTP binding protein; RT-qPCR, Quantitative real time polymerase chain reaction; SNP, Single nucleotide polymorphism; STAT3, Signal transducer and activator of transcription 3; TAB2, TGF-beta activated kinase 1 (MAP3K7) binding protein 2; TGF-β, Transforming growth factor β; TGFBR3, TGF-β receptor 3; Th2, T helper 2; TRAF6, Tumor necrosis factor receptor associated factor 6.
1. Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the global burden of disease study 2010. Lancet. (2012) 380:2095–128. doi: 10.1016/S0140-6736(12)61728-0
2. Kuruvilla ME, Lee FEH, Lee GB. Understanding asthma phenotypes, endotypes, and mechanisms of disease. Clin Rev Allergy Immunol. (2019) 56:219–33. doi: 10.1007/s12016-018-8712-1
3. Segal LN, Martinez FJ. Chronic obstructive pulmonary disease subpopulations and phenotyping. J Allergy Clin Immunol. (2018) 141:1961–71. doi: 10.1016/j.jaci.2018.02.035
4. Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol. (2015) 16:45–56. doi: 10.1038/ni.3049
5. Papi A, Brightling C, Pedersen SE, Reddel HK. Asthma. Lancet. (2018) 391:783–800. doi: 10.1016/S0140-6736(17)33311-1
6. Postma DS, Rabe KF. The asthma-COPD overlap syndrome. N Engl J Med. (2015) 373:1241–9. doi: 10.1056/NEJMra1411863
7. Peters SP. Asthma phenotypes: nonallergic (Intrinsic) asthma. J Allergy Clin Immunol Pract. (2014) 2:650–2. doi: 10.1016/j.jaip.2014.09.006
8. Barnes PJ. Targeting cytokines to treat asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. (2018) 18:454–66. doi: 10.1038/s41577-018-0006-6
9. Osei ET, Florez-Sampedro L, Timens W, Postma DS, Heijink IH, Brandsma CA. Unravelling the complexity of COPD by microRNAs: it's a small world after all. Eur Respir J. (2015) 46:807–18. doi: 10.1183/13993003.02139-2014
10. Brusselle GG, Joos GF, Bracke KR. Novel insights into the immunology of chronic obstructive pulmonary disease (COPD). Lancet. (2011) 378:1015–26. doi: 10.1016/S0140-6736(11)60988-4
11. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. (2004) 350:2645–53. doi: 10.1056/NEJMoa032158
12. Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. (2008) 455:58–63. doi: 10.1038/nature07228
13. Baltimore D, Boldin MP, O'Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nat Immunol. (2008) 9:839–45. doi: 10.1038/ni.f.209
14. Turchinovich A, Weiz L, Langheinz A, Burwinkel B. Characterization of extracellular circulating microRNA. Nucleic Acids Res. (2011) 39:7223–33. doi: 10.1093/nar/gkr254
15. Yuan X, Berg N, Lee JW, Le TT, Neudecker V, Jing N, et al. MicroRNA miR-223 as regulator of innate immunity. J Leukoc Biol. (2018) 104:515–24. doi: 10.1002/JLB.3MR0218-079R
16. Fazi F, Rosa A, Fatica A, Gelmetti V, de Marchis ML, Nervi C, et al. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPα regulates human granulopoiesis. Cell. (2005) 123:819–31. doi: 10.1016/j.cell.2005.09.023
17. Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. (2008) 451:1125–9. doi: 10.1038/nature06607
18. Felli N, Pedini F, Romania P, Biffoni M, Morsilli O, Castelli G, et al. MicroRNA 223-dependent expression of LMO2 regulates normal erythropoiesis. Haematologica. (2009) 94:479–86. doi: 10.3324/haematol.2008.002345
19. Yuan JY, Wang F, Yu J, Yang GH, Liu XL, Zhang JW. MicroRNA-223 reversibly regulates erythroid and megakaryocytic differentiation of K562 cells. J Cell Mol Med. (2009) 13:4551–9. doi: 10.1111/j.1582-4934.2008.00585.x
20. Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, et al. Library sequencing. (2009) 129:1401–14. doi: 10.1016/j.cell.2007.04.040
21. Seumois G, Vijayanand P, Eisley CJ, Omran N, Kalinke L, North M, et al. An integrated nano-scale approach to profile miRNAs in limited clinical samples. Am J Clin Exp Immunol. (2012) 1:70–89.
22. Maes T, Cobos FA, Schleich F, Sorbello V, Henket M, de Preter K, et al. Asthma inflammatory phenotypes show differential microRNA expression in sputum. J Allergy Clin Immunol. (2016) 137:1433–46. doi: 10.1016/j.jaci.2016.02.018
23. Fukao T, Fukuda Y, Kiga K, Sharif J, Hino K, Enomoto Y, et al. An evolutionarily conserved mechanism for microRNA-223 expression revealed by microRNA gene profiling. Cell. (2007) 129:617–31. doi: 10.1016/j.cell.2007.02.048
24. Bauernfeind F, Rieger A, Schildberg FA, Knolle PA, Schmid-Burgk JL, Hornung V. NLRP3 inflammasome activity is negatively controlled by miR-223. J Immunol. (2012) 189:4175–81. doi: 10.4049/jimmunol.1201516
25. Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. (2004) 303:83–6. doi: 10.1126/science.1091903
26. Williams AE, Larner-Svensson H, Perry MM, Campbell GA, Herrick SE, Adcock IM, et al. MicroRNA expression profiling in mild asthmatic human airways and effect of corticosteroid therapy. PLoS ONE. (2009) 4:e5889. doi: 10.1371/journal.pone.0005889
27. Rijavec M, Korošec P, Žavbi M, Kern I, Malovrh MM. Let-7a is differentially expressed in bronchial biopsies of patients with severe asthma. Sci Rep. (2014) 4:6103. doi: 10.1038/srep06103
28. Solberg OD, Ostrin EJ, Love MI, Peng JC, Bhakta NR, Hou L, et al. Airway epithelial miRNA expression is altered in asthma. Am J Respir Crit Care Med. (2012) 186:965–74. doi: 10.1164/rccm.201201-0027OC
29. Gomez JL, Chen A, Diaz MP, Zirn N, Gupta A, Britto C, et al. A network of sputum microRNAs is associated with neutrophilic airway inflammation in Asthma. Am J Respir Crit Care Med. (2020). doi: 10.1164/rccm.201912-2360OC
30. Schembri F, Sridhar S, Perdomo C, Gustafson AM, Zhang X, Ergun A, et al. MicroRNAs as modulators of smoking-induced gene expression changes in human airway epithelium. Proc Natl Acad Sci USA. (2009) 106:2319–24. doi: 10.1073/pnas.0806383106
31. Ezzie ME, Crawford M, Cho JH, Orellana R, Zhang S, Gelinas R, et al. Gene expression networks in COPD: microRNA and mRNA regulation. Thorax. (2012) 67:122–31. doi: 10.1136/thoraxjnl-2011-200089
32. Molina-Pinelo S, Pastor MD, Suarez R, Romero-Romero B, Gonzalez De La Peña M, Salinas A, et al. MicroRNA clusters: dysregulation in lung adenocarcinoma and COPD. Eur Respir J. (2014) 43:1740–9. doi: 10.1183/09031936.00091513
33. Velasco-Torres Y, López VR, Pérez-Bautista O, Buendía-Roldan I, Ramírez-Venegas A, Pérez-Ramos J, et al. MiR-34a in serum is involved in mild-to-moderate COPD in women exposed to biomass smoke. BMC Pulm Med. (2019) 19:227. doi: 10.1186/s12890-019-0977-5
34. Herberth G, Bauer M, Gasch M, Hinz D, Röder S, Olek S, et al. Maternal and cord blood miR-223 expression associates with prenatal tobacco smoke exposure and low regulatory T-cell numbers. J Allergy Clin Immunol. (2014) 133:543–50. doi: 10.1016/j.jaci.2013.06.036
35. Neudecker V, Brodsky KS, Clambey ET, Schmidt EP, Packard TA, Davenport B, et al. Neutrophil transfer of miR-223 to lung epithelial cells dampens acute lung injury in mice. Sci Transl Med. (2017) 9:eaah5360. doi: 10.1126/scitranslmed.aah5360
36. Li T, Morgan MJ, Choksi S, Zhang Y, Kim Y-S, Liu Z-G. MicroRNAs modulate the noncanonical NF-κB pathway by regulating IKKα expression during macrophage differentiation. Nat Immunol. (2010) 11:799–805. doi: 10.1038/ni.1918
37. Zhou W, Pal AS, Hsu AYH, Gurol T, Zhu X, Wirbisky-Hershberger SE, et al. MicroRNA-223 suppresses the canonical NF-κB pathway in basal keratinocytes to dampen neutrophilic inflammation. Cell Rep. (2018) 22:1810–23. doi: 10.1016/j.celrep.2018.01.058
38. Zhang D, Lee H, Wang X, Groot M, Sharma L, Dela Cruz CS, et al. A potential role of microvesicle-containing miR-223/142 in lung inflammation. Thorax. (2019) 74:865–74. doi: 10.1136/thoraxjnl-2018-212994
39. Yan Y, Lu K, Ye T, Zhang Z. MicroRNA-223 attenuates LPS-induced inflammation in an acute lung injury model via the NLRP3 inflammasome and TLR4/NF-κB signaling pathway via RHOB. Int J Mol Med. (2019) 43:1467–77. doi: 10.3892/ijmm.2019.4075
40. Oglesby IK, Chotirmall SH, McElvaney NG, Greene CM. Regulation of cystic fibrosis transmembrane conductance regulator by microRNA-145,−223, and−494 is altered in ΔF508 cystic fibrosis airway epithelium. J Immunol. (2013) 190:3354–62. doi: 10.4049/jimmunol.1202960
41. Chen Q, Wang H, Liu Y, Song Y, Lai L, Han Q, et al. Inducible microRNA-223 down-regulation promotes TLR-triggered IL-6 and IL-1β production in macrophages by targeting STAT3. PLoS ONE. (2012) 7:e42971. doi: 10.1371/journal.pone.0042971
42. Dorhoi A, Iannaccone M, Farinacci M, Faé KC, Schreiber J, Moura-Alves P, et al. MicroRNA-223 controls susceptibility to tuberculosis by regulating lung neutrophil recruitment. J Clin Invest. (2013) 123:4836–48. doi: 10.1172/JCI67604
43. Leuenberger C, Schuoler C, Bye H, Mignan C, Rechsteiner T, Hillinger S, et al. MicroRNA-223 controls the expression of histone deacetylase 2: a novel axis in COPD. J Mol Med. (2016) 94:725–734. doi: 10.1007/s00109-016-1388-1
44. Han J, Zhao F, Zhang J, Zhu H, Ma H, Li X, et al. MiR-223 reverses the resistance of EGFR-TKIs through IGF1R/PI3K/Akt signaling pathway. Int J Oncol. (2016) 48:1855–67. doi: 10.3892/ijo.2016.3401
45. Nian W, Ao X, Wu Y, Huang Y, Shao J, Wang Y, et al. miR-223 functions as a potent tumor suppressor of the Lewis lung carcinoma cell line by targeting insulin-like growth factor-1 receptor and cyclin-dependent kinase 2. Oncol Lett. (2013) 6:359–66. doi: 10.3892/ol.2013.1375
46. Liu D, Pan J, Zhao DY, Liu F. MicroRNA-223 inhibits deposition of the extracellular matrix by airway smooth muscle cells through targeting IGF-1R in the Pi3k/Akt pathway. Am J Transl Res. (2018) 10:744–52.
47. Liu C, Yang Z, Deng Z, Zhou Y, Gong Q, Zhao R, et al. Upregulated lncRNA ADAMTS9-AS2 suppresses progression of lung cancer through inhibition of miR-223-3p and promotion of TGFBR3. IUBMB Life. (2018) 70:536–46. doi: 10.1002/iub.1752
48. Luo P, Wang Q, Ye Y, Zhang J, Lu D, Cheng L, et al. MiR-223-3p functions as a tumor suppressor in lung squamous cell carcinoma by miR-223-3p-mutant p53 regulatory feedback loop. J Exp Clin Cancer Res. (2019) 38:74. doi: 10.1186/s13046-019-1079-1
49. Liang H, Yan X, Pan Y, Wang Y, Wang N, Li L, et al. MicroRNA-223 delivered by platelet-derived microvesicles promotes lung cancer cell invasion via targeting tumor suppressor EPB41L3. Mol Cancer. (2015) 14:58. doi: 10.1186/s12943-015-0327-z
50. Tabet F, Vickers KC, Cuesta Torres LF, Wiese CB, Shoucri BM, Lambert G, et al. HDL-transferred microRNA-223 regulates ICAM-1 expression in endothelial cells. Nat Commun. (2014) 5:3292. doi: 10.1038/ncomms4292
51. Edwards MR, Bartlett NW, Clarke D, Birrell M, Belvisi M, Johnston SL. Targeting the NF-κB pathway in asthma and chronic obstructive pulmonary disease. Pharmacol Ther. (2009) 121:1–13. doi: 10.1016/j.pharmthera.2008.09.003
52. Ghonim MA, Pyakurel K, Ibba SV, Wang J, Rodriguez P, Al-Khami AA, et al. PARP is activated in human asthma and its inhibition by olaparib blocks house dust mite-induced disease in mice. Clin Sci. (2015) 129:951–62. doi: 10.1042/CS20150122
53. Hageman GJ, Larik I, Pennings HJ, Haenen GRMM, Wouters EFM, Bast A. Systemic poly(ADP-ribose) polymerase-1 activation, chronic inflammation, and oxidative stress in COPD patients. Free Radic Biol Med. (2003) 35:140–8. doi: 10.1016/S0891-5849(03)00237-5
54. Gagliardo R, Chanez P, Profita M, Bonanno A, Albano GD, Montalbano AM, et al. IκB kinase-driven nuclear factor-κB activation in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2011) 128:635–45.e2. doi: 10.1016/j.jaci.2011.03.045
55. Krusche J, Twardziok M, Rehbach K, Böck A, Tsang MS, Schröder PC, et al. TNF-α-induced protein 3 is a key player in childhood asthma development and environment-mediated protection. J Allergy Clin Immunol. (2019) 144:1684–96.e12. doi: 10.1016/j.jaci.2019.07.029
56. Baines KJ, Simpson JL, Wood LG, Scott RJ, Fibbens NL, Powell H, et al. Sputum gene expression signature of 6 biomarkers discriminates asthma inflammatory phenotypes. J Allergy Clin Immunol. (2014) 133:997–1007. doi: 10.1016/j.jaci.2013.12.1091
57. Wang Z, Li W, Guo Q, Wang Y, Ma L, Zhang X. Insulin-like growth factor-1 signaling in lung development and inflammatory lung diseases. Biomed Res Int. (2018) 2018:17–9. doi: 10.1155/2018/6057589
58. Alam R, York J, Boyars M, Stafford S, Grant JA, Lee J, et al. Increased MCP-1, RANTES, and MIP-1α in bronchoalveolar lavage fluid of allergic asthmatic patients. Am J Respir Crit Care Med. (1996) 153:1398–404. doi: 10.1164/ajrccm.153.4.8616572
59. Rojas-Dotor S, Segura-Méndez NH, Miyagui-Namikawa K, Mondragón-González R. Expression of resistin, CXCR3, IP-10, CCR5 and MIP-1 α in obese patients with different severity of asthma. Biol Res. (2013) 46:13–20. doi: 10.4067/S0716-97602013000100002
60. Grob M, Schmid-Grendelmeier P, Joller-Jemelka HI, Ludwig E, Dubs RW, Grob PJ, et al. Altered intracellular expression of the chemokines MIP-1α, MIP-1β and IL-8 by peripheral blood CD4+ and CD8+ T cells in mild allergic asthma. Allergy Eur J Allergy Clin Immunol. (2003) 58:239–45. doi: 10.1034/j.1398-9995.2003.00035.x
61. Holgate ST, Bodey KS, Janezic A, Frew AJ, Kaplan AP, Teran LM. Release of RANTES, MIP-1 α, and MCP-1 into asthmatic airways following endobronchial allergen challenge. Am J Respir Crit Care Med. (1997) 156:1377–83. doi: 10.1164/ajrccm.156.5.9610064
62. Ravi AK, Khurana S, Lemon J, Plumb J, Booth G, Healy L, et al. Increased levels of soluble interleukin-6 receptor and CCL3 in COPD sputum. Respir Res. (2014) 15:103. doi: 10.1186/s12931-014-0103-4
63. Brandsma CA, Guryev V, Timens W, Ciconelle A, Postma DiS, Bischoff R, et al. Integrated proteogenomic approach identifying a protein signature of COPD and a new splice variant of SORBS1. Thorax. (2020) 75:180–3. doi: 10.1136/thoraxjnl-2019-213200
64. Tiwari N, Marudamuthu AS, Tsukasaki Y, Ikebe M, Fu J, Shetty S. P53-and PAI-1-mediated induction of C-X-C chemokines and CXCR2: Importance in pulmonary inflammation due to cigarette smoke exposure. Am J Physiol Lung Cell Mol Physiol. (2016) 310:L496–506. doi: 10.1152/ajplung.00290.2015
65. Lv J, Sun B, Mai Z, Jiang M, Du J. STAT3 potentiates the ability of airway smooth muscle cells to promote angiogenesis by regulating VEGF signalling. Exp Physiol. (2017) 102:598–606. doi: 10.1113/EP086136
66. Qu P, Roberts J, Li Y, Albrecht M, Cummings OW, John N. Stat3 downstream genes serve as biomarkers in human lung Carcinomas and chronic obstructive pulmonary disease. Lung Cancer. (2009) 63:341–7. doi: 10.1016/j.lungcan.2008.05.025
67. Yew-Booth L, Birrell MA, Lau MS, Baker K, Jones V, Kilty I, et al. JAK-STAT pathway activation in COPD. Eur Respir J. (2015) 46:843–5. doi: 10.1183/09031936.00228414
68. Mukhopadhyay S, Malik P, Arora SK, Mukherjee TK. Intercellular adhesion molecule-1 as a drug target in asthma and rhinitis. Respirology. (2014) 19:508–13. doi: 10.1111/resp.12285
69. Ito K, Ito M, Elliott WM, Cosio B, Caramori G, Onn MK, et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med. (2005) 352:1967–76. doi: 10.1056/NEJMoa041892
70. Sheu CC, Tsai MJ, Chen FW, Chang KF, Chang WA, Chong IW, et al. Identification of novel genetic regulations associated with airway epithelial homeostasis using next-generation sequencing data and bioinformatics approaches. Oncotarget. (2017) 8:82674–88. doi: 10.18632/oncotarget.19752
71. Golpon HA, Coldren CD, Zamora MR, Cosgrove GP, Moore MD, Tuder RM, et al. Emphysema lung tissue gene expression profiling. Am J Respir Cell Mol Biol. (2004) 31:595–600. doi: 10.1165/rcmb.2004-0008OC
72. Al Nadawi WA, Alabassi HM. TGF-BRIII gene expression and TGF-B1 serum level in Iraqi children with asthma. J Pharm Sci Res. (2019) 11:2292–4.
73. Gouda MM, Shaikh SB, Chengappa D, Kandhal I, Shetty A, Bhandary Y. Changes in the expression level of IL-17A and p53-fibrinolytic system in smokers with or without COPD. Mol Biol Rep. (2018) 45:2835–41. doi: 10.1007/s11033-018-4398-y
74. Mizuno S, Bogaard HJ, Gomez-Arroyo J, Alhussaini A, Kraskauskas D, Cool CD, et al. MicroRNA-199a-5p is associated with hypoxia-inducible factor-1α expression in lungs from patients with COPD. Chest. (2012) 142:663–72. doi: 10.1378/chest.11-2746
75. Feng Z, Qi S, Zhang Y, Qi Z, Yan L, Zhou J, et al. Ly6G+ neutrophil-derived miR-223 inhibits the NLRP3 inflammasome in mitochondrial DAMP-induced acute lung injury. Cell Death Dis. (2017) 8:e3170. doi: 10.1038/cddis.2017.549
76. Garbacki N, di Valentin E, Huynh-Thu VA, Geurts P, Irrthum A, Crahay C, et al. MicroRNAs profiling in murine models of acute and chronic asthma: a relationship with mRNAs targets. PLoS ONE. (2011) 6:e16509. doi: 10.1371/journal.pone.0016509
77. Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. (2017) 2:17023. doi: 10.1038/sigtrans.2017.23
78. Sethi GS, Dharwal V, Naura AS. Poly(ADP-ribose)polymerase-1 in lung inflammatory disorders: a review. Front Immunol. (2017) 8:1172. doi: 10.3389/fimmu.2017.01172
79. Datta R, Naura AS, Zerfaoui M, Errami Y, Oumouna M, Kim H, et al. PARP-1 deficiency blocks IL-5 expression through calpain-dependent degradation of STAT-6 in a murine asthma model. Allergy. (2011) 66:853–61. doi: 10.1111/j.1398-9995.2011.02549.x
80. Mustapha O, Datta R, Oumouna-Benachour K, Suzuki Y, Hans C, Matthews K, et al. Poly(ADP-ribose) polymerase-1 inhibition prevents eosinophil recruitment by modulating Th2 cytokines in a murine model of allergic airway inflammation: a potential specific effect on IL-5. J Immunol. (2006) 177:6489–96. doi: 10.4049/jimmunol.177.9.6489
81. Dharwal V, Naura AS. PARP-1 inhibition ameliorates elastase induced lung inflammation and emphysema in mice. Biochem Pharmacol. (2018) 150:24–34. doi: 10.1016/j.bcp.2018.01.027
82. Hwang J, Chung S, Sundar IK, Yao H, Arunachalam G, McBurney MW, et al. Cigarette smoke-induced autophagy is regulated by SIRT1–PARP-1-dependent mechanism: implication in pathogenesis of COPD. Arch Biochem Biophys. (2010) 500:203–9. doi: 10.1016/j.abb.2010.05.013
83. Adli M, Merkhofer E, Cogswell P, Baldwin AS. IKKα and IKKβ each function to regulate NF-κB activation in the TNF-induced/canonical pathway. PLoS ONE. (2010) 5:e9428. doi: 10.1371/journal.pone.0009428
84. Li L, Ruan Q, Hilliard B, DeVirgiliis J, Karin M, Chen YH. Transcriptional regulation of the Th17 immune response by IKKα. J Exp Med. (2011) 208:787–96. doi: 10.1084/jem.20091346
85. He Z, Wang F, Zhang J, Sen S, Pang Q, Luo S, et al. Regulation of Th17 differentiation by IKKα-dependent and -independent phosphorylation of RORγt. J Immunol. (2017) 199:955–64. doi: 10.4049/jimmunol.1700457
86. He Y, Feng D, Li M, Gao Y, Ramirez T, Cao H, et al. Hepatic mitochondrial DNA/Toll-like receptor 9/MicroRNA-223 forms a negative feedback loop to limit neutrophil overactivation and acetaminophen hepatotoxicity in mice. Hepatology. (2017) 66:220–34. doi: 10.1002/hep.29153
87. Zhou K, Feng X, Wang Y, Liu Y, Tian L, Zuo W, et al. miR-223 is repressed and correlates with inferior clinical features in mantle cell lymphoma through targeting SOX11. Exp Hematol. (2018) 58:27–34.e1. doi: 10.1016/j.exphem.2017.10.005
88. Han D, Walsh MC, Kim KS, Hong SW, Lee J, Yi J, et al. Dendritic cell expression of the signaling molecule TRAF6 is required for immune tolerance in the lung. Int Immunol. (2017) 29:71–8. doi: 10.1093/intimm/dxx011
89. Pinkerton JW, Kim RY, Robertson AAB, Hirota JA, Wood LG, Knight DA, et al. Inflammasomes in the lung. Mol Immunol. (2017) 86:44–55. doi: 10.1016/j.molimm.2017.01.014
90. Simpson JL, Phipps S, Baines KJ, Oreo KM, Gunawardhana L, Gibson PG. Elevated expression of the NLRP3 inflammasome in neutrophilic asthma. Eur Respir J. (2014) 43:1067–76. doi: 10.1183/09031936.00105013
91. Kim RY, Pinkerton JW, Essilfie AT, Robertson AAB, Baines KJ, Brown AC, et al. Role for NLRP3 inflammasome-mediated, IL-1β-dependent responses in severe, steroid-resistant asthma. Am J Respir Crit Care Med. (2017) 196:283–97. doi: 10.1164/rccm.201609-1830OC
92. Wang H, Lv C, Wang S, Ying H, Weng Y, Yu W. NLRP3 inflammasome involves in the acute exacerbation of patients with chronic obstructive pulmonary disease. Inflammation. (2018) 41:1321–33. doi: 10.1007/s10753-018-0780-0
93. Di Stefano A, Caramori G, Barczyk A, Vicari C, Brun P, Zanini A, et al. Innate immunity but not NLRP3 inflammasome activation correlates with severity of stable COPD. Thorax. (2014) 69:516–24. doi: 10.1136/thoraxjnl-2012-203062
94. Pauwels NS, Bracke KR, Dupont LL, Van Pottelberge GR, Provoost S, Vanden Berghe T, et al. Role of IL-1 and the Nlrp3/caspase-1/IL-1 axis in cigarette smoke-induced pulmonary inflammation and COPD. Eur Respir J. (2011) 38:1019–28. doi: 10.1183/09031936.00158110
95. Jimenez Calvente C, Del Pilar H, Tameda M, Johnson CD, Feldstein AE. MicroRNA 223 3p negatively regulates the NLRP3 inflammasome in acute and chronic liver injury. Mol Ther. (2020) 28:653–63. doi: 10.1016/j.ymthe.2019.09.013
96. Solomon GM, Fu L, Rowe SM, Collawn JF. The therapeutic potential of CFTR modulators for COPD and other airway diseases. Curr Opin Pharmacol. (2017) 34:132–9. doi: 10.1016/j.coph.2017.09.013
97. Matsumoto K, Inoue H. Viral infections in asthma and COPD. Respir Investig. (2014) 52:92–100. doi: 10.1016/j.resinv.2013.08.005
98. Toews GB. Impact of bacterial infections on airway diseases. Eur Respir Rev. (2005) 14:62–8. doi: 10.1183/09059180.05.00009504
99. Wang F, He B. CCR1 and CCR5 expression on inflammatory cells is related to cigarette smoking and chronic obstructive pulmonary disease severity. Chin Med J. (2012) 125:4277–82. doi: 10.3760/cma.j.issn.0366-6999.2012.23.021
100. Mizutani N, Nabe T, Yoshino S. IL-17A promotes the exacerbation of IL-33–induced airway hyperresponsiveness by enhancing neutrophilic inflammation via CXCR2 signaling in mice. J Immunol. (2014) 192:1372–84. doi: 10.4049/jimmunol.1301538
101. Alcorn JF, Crowe CR, Kolls JK. T H 17 cells in Asthma and COPD . Annu Rev Physiol. (2010) 72:495–516. doi: 10.1146/annurev-physiol-021909-135926
102. Geraghty P, Wyman AE, Garcia-Arcos I, Dabo AJ, Gadhvi S, Foronjy R. STAT3 modulates cigarette smoke-induced inflammation and protease expression. Front Physiol. (2013) 4:267. doi: 10.3389/fphys.2013.00267
103. Gavino AC, Nahmod K, Bharadwaj U, Makedonas G, Tweardy DJ. STAT3 inhibition prevents lung inflammation, remodeling, and accumulation of Th2 and Th17 cells in a murine asthma model. Allergy. (2016) 3:1684–92. doi: 10.1111/all.12937
104. Simeone-Penney MC, Severgnini M, Tu P, Homer RJ, Mariani TJ, Cohn L, et al. Airway epithelial STAT3 is required for allergic inflammation in a murine model of Asthma. J Immunol. (2007) 178:6191–9. doi: 10.4049/jimmunol.178.10.6191
105. Hussein YM, Shalaby SM, Mohamed RH, Hassan TH. Association between genes encoding components of the IL-10 / IL-0 receptor pathway and asthma in children. Ann Allergy Asthma Immunol. (2011) 106:474–80. doi: 10.1016/j.anai.2011.02.021
106. Sundar IK, Rashid K, Gerloff J, Rangel-Moreno J, Li D, Rahman I. Genetic ablation of histone deacetylase 2 leads to lung cellular senescence and lymphoid follicle formation in COPD/emphysema. FASEB J. (2018) 32:4955–71. doi: 10.1096/fj.201701518R
107. Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2013) 131:636–45. doi: 10.1016/j.jaci.2012.12.1564
108. Ito J, Moriyama H, Shimada K. Morphological evaluation of the human facial muscles. Okajimas Folia Anat Jpn. (2006) 83:7–14. doi: 10.2535/ofaj.83.7
109. Hew M, Bhavsar P, Torrego A, Meah S, Khorasani N, Barnes PJ, et al. Relative corticosteroid insensitivity of peripheral blood mononuclear cells in severe asthma. Am J Respir Crit Care Med. (2006) 174:134–41. doi: 10.1164/rccm.200512-1930OC
110. Sauler M, Bazan IS, Lee PJ. Cell death in the lung: the apoptosis–necroptosis Axis. Annu Rev Physiol. (2019) 81:375–402. doi: 10.1146/annurev-physiol-020518-114320
111. Henson PM, Vandivier RW, Douglas IS. Cell death, remodeling, and repair in chronic obstructive pulmonary disease? Proc Am Thorac Soc. (2006) 3:713–7. doi: 10.1513/pats.200605-104SF
112. Haneklaus M, Gerlic M, O'Neill LAJ, Masters SL. MiR-223: infection, inflammation and cancer. J Intern Med. (2013) 274:215–26. doi: 10.1111/joim.12099
113. Lu TX, Lim E-J, Besse JA, Itskovich S, Plassard AJ, Fulkerson PC, et al. miR-223 deficiency increases eosinophil progenitor proliferation. J Immunol. (2013) 190:1576–82. doi: 10.4049/jimmunol.1202897
114. Hoshino M, Nakamura Y, Sim JJ, Yamashiro Y, Uchida K, Hosaka K, et al. Inhaled corticosteroid reduced lamina reticularis of the basement membrane by modulation of insulin-like growth factor (IGF)-I expression in bronchial asthma. Clin Exp Allergy. (1998) 28:568–77. doi: 10.1046/j.1365-2222.1998.00277.x
115. Piñeiro-Hermida S, Gregory JA, López IP, Torrens R, Ruíz-Martínez C, Adner M, et al. Attenuated airway hyperresponsiveness and mucus secretion in HDM-exposed Igf1r-deficient mice. Allergy Eur J Allergy Clin Immunol. (2017) 72:1317–26. doi: 10.1111/all.13142
116. Piñeiro-Hermida S, López IP, Alfaro-Arnedo E, Torrens R, Iñiguez M, Alvarez-Erviti L, et al. IGF1R deficiency attenuates acute inflammatory response in a bleomycin-induced lung injury mouse model. Sci Rep. (2017) 7:4290. doi: 10.1038/s41598-017-04561-4
117. Ye M, Yu H, Yu W, Zhang G, Xiao L, Zheng X, et al. Evaluation of the significance of circulating insulin-like growth factor-1 and C-reactive protein in patients with chronic obstructive pulmonary disease. J Int Med Res. (2012) 40:1025–35. doi: 10.1177/147323001204000321
118. Huang L, Li F, Deng P, Hu C. MicroRNA-223 promotes tumor progression in lung cancer A549 cells via activation of the NF-κB signaling pathway. Oncol Res. (2016) 24:405–13. doi: 10.3727/096504016X14685034103437
119. Chen L, Hou X, Zhang M, Zheng Y, Zheng X, Yang Q, et al. MicroRNA-223-3p modulates dendritic cell function and ameliorates experimental autoimmune myocarditis by targeting the NLRP3 inflammasome. Mol Immunol. (2020) 117:73–83. doi: 10.1016/j.molimm.2019.10.027
120. Bartis D, Mise N, Mahida RY, Eickelberg O, Thickett DR. Epithelial-mesenchymal transition in lung development and disease: does it exist and is it important? Thorax. (2014) 69:760–5. doi: 10.1136/thoraxjnl-2013-204608
121. Minshall EM, Leung DYM, Martin RJ, Song YL, Cameron L, Ernst P, et al. Eosinophil-associated TGF- β 1 mRNA expression and airways fibrosis in bronchial Asthma. Am J Respir Cell Mol Biol. (1997) 17:326–33. doi: 10.1165/ajrcmb.17.3.2733
122. Mak JCW, Chan-Yeung MMW, Ho SP, Chan KS, Choo K, Yee KS, et al. Elevated plasma TGF-β1 levels in patients with chronic obstructive pulmonary disease. Respir Med. (2009) 103:1083–9. doi: 10.1016/j.rmed.2009.01.005
123. Di Stefano A, Sangiorgi C, Gnemmi I, Casolari P, Brun P, Ricciardolo FLM, et al. TGF-β signaling pathways in different compartments of the lower airways of patients with stable COPD. Chest. (2018) 153:851–62. doi: 10.1016/j.chest.2017.12.017
124. Kim HK, Jang TW, Jung MH, Park HW, Lee JE, Shin ES, et al. Association between genetic variations of the transforming growth factor β receptor type III and asthma in a Korean population. Exp Mol Med. (2010) 42:420–7. doi: 10.3858/emm.2010.42.6.043
125. Hersh CP, Hansel NN, Barnes KC, Lomas DA, Pillai SG, Coxson HO, et al. Transforming growth factor-β receptor-3 is associated with pulmonary emphysema. Am J Respir Cell Mol Biol. (2009) 41:324–31. doi: 10.1165/rcmb.2008-0427OC
126. Hirota T, Takahashi A, Kubo M, Tsunoda T, Tomita K, Doi S, et al. Genome-wide association study identifies three new susceptibility loci for adult asthma in the Japanese population. Nat Genet. (2011) 43:893–6. doi: 10.1038/ng.887
127. Van Der Toorn M, Rezayat D, Kauffman HF, Bakker SJL, Gans ROB, Koëter GH, et al. Lipid-soluble components in cigarette smoke induce mitochondrial production of reactive oxygen species in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. (2009) 297:109–14. doi: 10.1152/ajplung.90461.2008
128. Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol. (2011) 13:423–35. doi: 10.1038/ncb2210
129. Gon Y, Shimizu T, Mizumura K, Maruoka S, Hikichi M. Molecular techniques for respiratory diseases: microRNA and extracellular vesicles. Respirology. (2020) 25:149–60. doi: 10.1111/resp.13756
130. Ismail N, Wang Y, Dakhlallah D, Moldovan L, Agarwal K, Batte K, et al. Macrophage microvesicles induce macrophage differentiation and miR-223 transfer. Blood. (2013) 121:984–95. doi: 10.1182/blood-2011-08-374793
131. Francisco-Garcia AS, Garrido-Martín EM, Rupani H, Lau LCK, Martinez-Nunez RT, Howarth PH, et al. Small RNA species and micro RNA profiles are altered in severe asthma nanovesicles from broncho alveolar lavage and associate with impaired lung function and inflammation. Non-coding RNA. (2019) 5:51. doi: 10.3390/ncrna5040051
132. Levänen B, Bhakta NR, Torregrosa Paredes P, Barbeau R, Hiltbrunner S, Pollack JL, et al. Altered microRNA profiles in bronchoalveolar lavage fluid exosomes in asthmatic patients. J Allergy Clin Immunol. (2013) 131:894–903. doi: 10.1016/j.jaci.2012.11.039
133. Pua HH, Happ HC, Gray CJ, Mar DJ, Chiou NT, Hesse LE, et al. Increased hematopoietic extracellular RNAs and vesicles in the lung during allergic airway responses. Cell Rep. (2019) 26:933–44.e4. doi: 10.1016/j.celrep.2019.01.002
134. Fujita Y, Araya J, Ito S, Kobayashi K, Kosaka N, Yoshioka Y, et al. Suppression of autophagy by extracellular vesicles promotes myofibroblast differentiation in COPD pathogenesis. J Extracell Vesicles. (2015) 4:28388. doi: 10.3402/jev.v4.28388
135. Tan DBA, Armitage J, Teo TH, Ong NE, Shin H, Moodley YP. Elevated levels of circulating exosome in COPD patients are associated with systemic inflammation. Respir Med. (2017) 132:261–4. doi: 10.1016/j.rmed.2017.04.014
136. Haspeslagh E, Debeuf N, Hammad H, Lambrecht BN. Murine models of allergic asthma. Methods Mol Biol. 1559:121–36. doi: 10.1007/978-1-4939-6786-5_10
137. de Grove KC, Provoost S, Hendriks RW, McKenzie ANJ, Seys LJM, Kumar S, et al. Dysregulation of type 2 innate lymphoid cells and TH2 cells impairs pollutant-induced allergic airway responses. J Allergy Clin Immunol. (2017) 139:246–57.e4. doi: 10.1016/j.jaci.2016.03.044
138. Ghorani V, Boskabady MH, Khazdair MR, Kianmeher M. Experimental animal models for COPD: a methodological review. Tob Induc Dis. (2017) 15:25. doi: 10.1186/s12971-017-0130-2
139. de Smet EG, Van Eeckhoutte HP, Avila Cobos F, Blomme E, Verhamme FM, Provoost S, et al. The role of miR-155 in cigarette smoke-induced pulmonary inflammation and COPD. Mucosal Immunol. (2019) 13:426–36. doi: 10.1038/s41385-019-0241-6
140. Boscaino V, Fiannaca A, La Paglia L, La Rosa M, Rizzo R, Urso A. MiRNA therapeutics based on logic circuits of biological pathways. BMC Bioinform. (2019) 20:1–22. doi: 10.1186/s12859-019-2881-7
141. Hosseinahli N, Aghapour M, Duijf PHG, Baradaran B. Treating cancer with microRNA replacement therapy: a literature review. J Cell Physiol. (2018) 233:5574–88. doi: 10.1002/jcp.26514
Keywords: asthma, COPD, miRNAs, miR-223, inflammation
Citation: Roffel MP, Bracke KR, Heijink IH and Maes T (2020) miR-223: A Key Regulator in the Innate Immune Response in Asthma and COPD. Front. Med. 7:196. doi: 10.3389/fmed.2020.00196
Received: 17 January 2020; Accepted: 23 April 2020;
Published: 19 May 2020.
Edited by:
Kian Fan Chung, Imperial College London, United KingdomReviewed by:
Bernahrd Ryffel, Centre National de la Recherche Scientifique (CNRS), FranceCopyright © 2020 Roffel, Bracke, Heijink and Maes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tania Maes, dGFuaWEubWFlc0BVR2VudC5iZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.