 Ahmed Abdel-Razik1*†
Ahmed Abdel-Razik1*† Nasser Mousa1Rania Elhelaly2Rasha Elzehery2Ahmad S. Hasan2
Nasser Mousa1Rania Elhelaly2Rasha Elzehery2Ahmad S. Hasan2 Mostafa Abdelsalam3Ahmed Salah Seif4Ahmed M. Tawfik5Niveen El-Wakeel6Waleed Eldars6,7
Mostafa Abdelsalam3Ahmed Salah Seif4Ahmed M. Tawfik5Niveen El-Wakeel6Waleed Eldars6,7- 1Tropical Medicine Department, Faculty of Medicine, Mansoura University, Mansoura, Egypt
- 2Clinical Pathology Department, Faculty of Medicine, Mansoura University, Mansoura, Egypt
- 3Nephrology and Dialysis Unit, Internal Medicine Department, Faculty of Medicine, Mansoura University, Mansoura, Egypt
- 4Scientific Fellow of Tropical Medicine, Hepatology and Gastroenterology Department, Shebin Elkom Teaching Hospital, Menoufia, Egypt
- 5Diagnostic & Interventional Radiology Department, Faculty of Medicine, Mansoura University, Mansoura, Egypt
- 6Medical Microbiology and Immunology Department, Faculty of Medicine, Mansoura University, Mansoura, Egypt
- 7Medical Microbiology and Immunology Department, Faculty of Medicine, Delta University for Science and Technology, Talkha, Egypt
Background and Aim: The relationship between liver cirrhosis and Helicobacter pylori (H. pylori) is a debatable matter. The aim of this study is to evaluate the possible association between H. pylori infection and liver cirrhosis.
Methods: A single-center prospective cohort pilot study of 558 patients with cirrhosis was followed up for 1 year. Serum C-reactive protein (CRP), tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), nitric oxide (NO), vascular endothelial growth factor (VEGF) levels and Fecal H. pylori antigen were evaluated by enzyme-linked immunosorbent assay (ELISA). All patients with positive H. pylori were treated and then followed up for 3 months. Participants with eradicated H. pylori were followed up for one further year.
Results: H. pylori-positive patients (48.4%) were associated with increased levels of serum CRP, TNF-α, IL-6, NO, and VEGF, as well as increased incidence of varices, portal hypertensive gastropathy, gastric antral vascular ectasia, hepatocellular carcinoma (HCC), spontaneous bacterial peritonitis, hepatic encephalopathy, portal vein thrombosis (PVT), and hepatorenal syndrome (all P < 0.05). Multivariate analysis models revealed that the presence of H. pylori was an independent risk variable for the development of portal vein thrombosis and hepatocellular carcinoma (P = 0.043, P = 0.037) respectively. After treatment of H. pylori infection, there was a significant reduction in all measured biochemical parameters and reported cirrhotic complications (all P < 0.05).
Conclusion: Incidence of PVT and HCC development increased with H. pylori infection through increased inflammatory markers and vascular mediators. Moreover, its eradication may reduce the incidence of these complications.
Introduction
Helicobacter pylori (H. pylori) is a pathogenic bacteria that affects the human gastric tissue, and it is the most common etiology of peptic ulcer and gastritis (1). However, H. pylori is implicated in extra-gastric disorders, as idiopathic thrombocytopenic purpura, vitamin B12 deficiency, iron deficiency, and can also be related to many other diseases, for example, neurodegenerative syndromes, ischemic heart disease, and diabetes (2). Furthermore, it has been hypothesized that H. pylori could be a risk factor for many liver disorders, for example, nonalcoholic fatty liver disease, isolated hypertransaminasemia, and portosystemic encephalopathy (3–6).
As mentioned in the literature review, the association between liver diseases and H. pylori has been discussed and still remains a matter of debate (7). While the presence of Helicobacter species or H. pylori has been reported in hepatic tissue samples from patients with different hepatic disorders (8–14), nevertheless, a direct association of H. pylori in the development of cirrhotic complications in patients with liver disease has been postulated with a less strong evidence (3). To date, the problem has received scant attention in the research literature. The purpose of this study, therefore, is to evaluate the possible association between H. pylori infection and liver cirrhosis.
Patients and Methods
This prospective single-center cohort pilot study was carried out at the Tropical Medicine Department (Mansoura University-Egypt), between April 2015 and May 2019. We prospectively enrolled 803 consecutive patients with liver cirrhosis who were referred to our center. Only 558 patients who met the inclusion criteria were included in this study. Patients' clinical, radiological, demographic, hematological, and biochemical findings were evaluated at baseline and throughout the follow-up periods.
The inclusion criteria were (1) patients with liver cirrhosis, (2) aged ≥18 years, and (3) had undergone all investigations to confirm H. pylori infection.
The exclusion criteria were (1) patients with history of gastrectomy, (2) had cancer, (3) had alcoholic liver disease, (4) had missing data, (5) pregnancy and lactation, (6) patients with kidney diseases and hematologic disorders; (7) pancreatitis, (8) peritoneal carcinomatosis, (9) abdominal tuberculosis, (10) uncontrolled thyroid disorders, (11) cerebrovascular accident causes, (12) bone marrow suppression, (13) had inherited coagulation abnormalities, (14) collagen vascular diseases, (15) patients with metabolic or cholestatic hepatobiliary disorders, (16) heart failure, (17) patients with portal vein thrombosis (PVT), spontaneous bacterial peritonitis (SBP), hepatocellular carcinoma (HCC), hepatorenal syndrome (HRS), and hepatic encephalopathy (HE) at baseline of the study, (18) usage of hepatotoxic, antiplatelet or anticoagulant treatment, oral contraceptive drugs, and NSAIDs, (19) who had a history of recent upper gastrointestinal (GI) bleeding (in last 6 weeks) were excluded from the study. (20) use of proton-pump inhibitors (PPIs) for at least 4 weeks before enrollment, and (21) recent usage of antibiotics and/or prophylaxis with norfloxacin or rifaximin for SBP within the preceding 3 months prior to the onset of the study were also excluded from this study.
The First Part of the Study: Patients Fulfilling the Inclusion Criteria Follow-Up
All patients (n = 558) who met the criteria were followed up for 1-year. All biochemical parameters and complications of liver cirrhosis were also recorded.
The Second Part of the Study: H. pylori-Positive Patients Treatment and Follow-Up
After a 1-year follow-up, all patients with H. pylori infection (n = 270) were treated with two regimens according to the clinical status, patient's general condition, and possible adverse effects related to drugs. The first one; pantoprazole 40 mg, clarithromycin 500 mg, and amoxicillin 1000 mg, twice daily, were used for 14 days. The second one; pantoprazole 40 mg and amoxicillin 1 g twice daily, and levofloxacin 500 mg once daily, were used also for 2 weeks. Treatment with pantoprazole (dose as mentioned above) was given to all patients for 1 more month, to complete the eradication therapy (15). All patients have been notified of the side effects of the drugs used. Therapeutic compliance was checked by tablet counting and all the patients were given an emergency telephone number, though no complications were reported leading to treatment discontinuation during the study period. To evaluate the effect of H. pylori eradication, all parameters were assessed again in patients with successful H. pylori eradication (n = 212) after a 3-months follow-up.
Patients who had not responded to any of the two above mentioned H. pylori therapeutic protocols (n = 58, 21.5% of patients) have received another H. pylori regimen used in our outpatient clinics (15).
The Third Part of the Study: Follow-Up of all Patients With Successfully Eradicated H. pylori
All patients (n = 205; as seven patients were lost during the follow-up period) who achieved eradication were followed up for one further year in comparison with H. pylori-negative patients (n = 276; as 12 patients were lost during the follow-up period). All biochemical parameters and complications of liver cirrhosis were also recorded again.
The baseline data was captured within 1 week from the time of enrollment and the after-treatment data was captured 3 months after finishing therapy. The end-of-study data was captured in the last week of the 1 year after therapy follow up period. At the end of the study, clinical examination, radiological and biochemical findings of the patients did not show any problems affecting the follow-up parameters in the subjects' health that were not recorded at the start of the study.
Diagnosis of Liver Cirrhosis and Its Complications
Liver cirrhosis was assessed by clinical evaluation, unequivocal biochemical results, abdominal ultrasonography (US), elastography, histopathological evaluation of liver biopsy, or endoscopic results suggesting portal hypertension. The severity of liver cirrhosis was scored according to the Child-Pugh classification and MELD scoring system (16).
A color Doppler ultrasonography using a 3.5–5 MHz convex probe (SonoAce X6 Ultrasound System; Medison Electronics, Seoul, Korea) was done to all patients. Once, PVT was suspected by routine Doppler US examination, the final diagnosis was approved by portal angiography that also enabled a more precise differentiation between complete and partial obstructive thrombosis and their extension (17). Treatment and follow-up of all patients with PVT were carried out according to EASL clinical practice guidelines (18).
Diagnosis of SBP was based on positive ascitic fluid bacterial culture and/or ascitic fluid absolute polymorphonuclear leukocyte (PMN) count of at least 250 cells/mm3 without an intra-abdominal source of infection (19). According to EASL guidelines, patients with suspected SBP received an appropriate empirical antibiotic therapy, and then antibiotic was shifted depending on the results of culture and sensitivity as well as secondary antibiotic prophylaxis in patients with cirrhosis with a prior history of SBP was recommended (20).
In all cirrhotic individuals, US imaging and alpha-fetoprotein (AFP) were utilized every 6 months for surveillance purposes. Two imaging systems, both showing a focal lesion >2 cm in diameter with highlights of arterial hypervascularization, or a solitary radiologic study with these highlights joined with a serum AFP level of >400 ng/ml may be considered as noninvasive diagnostic tools especially for earlier stages of HCC (21). Surveillance and management of patients with HCC were carried out according to EASL guidelines (22).
HRS was diagnosed according to the International Club of Ascites-Acute Kidney Injury (ICA-AKI) criteria (23).
Overt HE is diagnosed depending on excluding other etiologies of altered mental status and by clinical findings (24). The West Haven criteria are considered the cornerstone for classifying the severity of overt HE for most patients (25). The minimal hepatic encephalopathy (MHE) is diagnosed based on specific psychometric testing (26). The management of patients with HE was recommended according to the recent EASL guidelines (24). In this study, HE may be considered as overt HE and/or MHE.
It is generally recommended that all patients with cirrhosis at the time of diagnosis undergo elective esophagogastroduodenoscopy (EGD) screening for varices and periodically thereafter if small or no varices are identified. The New Italian Endoscopic Club has categorized the severity of portal hypertensive gastropathy (PHG) depending on the presence of 4 elementary lesions: mosaic-like pattern, red point lesions, cherry red spots, and black-brown spots, that are typically localized in the gastric mucosa related to the corpus or fundus of the stomach (27). Gastric antral vascular ectasia (GAVE) is characterized by red spots or patches in either a linear or diffuse array in the gastric antrum (28). For EGD, an Olympus GIF-Q200 (Olympus Optical Co. Ltd., Tokyo, Japan) was used.
Diagnosis and Follow-Up of H. pylori
In all patients, the fecal antigen test was used to establish the diagnosis of H. pylori infection as well as to prove its eradication. This test was done one month after antibiotic treatment completion and 2 weeks after stopping therapy with pantoprazole (29). This test was also repeated at the end of therapy to identify any reinfection. Many types of research affirmed that the monoclonal antibodies utilization may have the highest sensitivity and specificity before and after eradication treatment (30, 31), and may also increase the diagnostic value in post-treatment testing and decrease the inter-test variability problem (32).
Data Collection
All patients completed a standardized, self-validated questionnaire from which we collected data about their employment condition, marital status, crowding index, income, alcohol consumption, medical history; especially diabetes, malignancy and hypertension, and smoking habits, as well as previous therapeutic history. BMI was calculated as weight (kg) divided by height squared (m2). Crowding index is the number of individuals/rooms excluding both the kitchen and bathroom, which correlated inversely with socioeconomic status (33).
A professional nutritional team started counseling to the patients with cirrhosis and malnutrition for proper protein and calorie intake. They instructed them to ingest the recommended daily protein intake of 1.5 g/kg of actual body weight. Vitamins and micronutrients were administered to treat clinically suspected or confirmed deficiencies. Patients with cirrhotic ascites following sodium restriction had 5 g of salt added daily to their diet to improve its palatability based on EASL guidelines of recommended daily sodium intake of 80 mmol/ day in these cases (34).
Sampling
Following overnight fasting, fresh venous blood samples (7 mL) were collected from all patients [5 mL with no anticoagulants for serum testing and 1 mL on EDTA for complete blood count (CBC)]. The serum samples were then divided into aliquots and frozen at −20°C until subsequent evaluations. Three ml of venous blood were collected into plain tubes. Samples were then transported immediately to the laboratory, separated within 15 min of collection and analyzed immediately for serum ammonia.
Methodology
Complete liver function tests and serum creatinine were done on a Dimension Xpand Plus Chemistry Analyzer using respective kits (by Siemens Technology, Princeton, New Jersey). Serum C-reactive protein (CRP) was assessed on COBAS C111 using its standard kits (Roche Diagnostics, Basel, Switzerland). Interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) were measured by ELISA utilizing kits supplied by Dia Source (Rue du Bosquet, Louvain-la-Neuve, Belgium. Serum nitric oxide (NO) was evaluated using ELISA kits provided by MyBiosource (San Diego, CA 92195–3308, USA), with its detection range 1.56 nmol/ml−100 nmol/ml (nmol/ml = μmol/L). Serum VEGF was assessed by ELISA kits provided by Invitrogen Corporation (Flynn Road, Camarillo, CA, USA) with its detection range (0–1.5 ng/ml). Estimated glomerular filtration rate (eGFR) was assessed according to the following equation = 186 x (Creatinine/88.4)−1.154 × (Age)−0.203 × (0.742 if female) x (1.210 if black) (35). Serum ammonia was measured using a Colorimetric kit supplied by Elabscience (14780 Memorial Drive, Suite 216, Houston, Texas 77079, USA). Assessment of H. pylori antigens in stool was done by a sandwich ELISA technique using specific monoclonal antibodies for H. pylori from Immundiagnostik AG (Stubenwald-Allee 8a, 64625 Bensheim, Germany). Based on their absorbances (OD), the findings were categorized as follows: positive (OD > 0.170), borderline (OD = 0.130–0.170), and negative (OD < 0.130).
Ethics
The study protocols and all procedures were reviewed and approved by the Mansoura Institutional Review Board. Written consents were obtained from all patients. The work was done in accordance with the Helsinki Declaration's guidelines.
Statistical Analyses
Statistical analysis was performed using SPSS software (version 20; SPSS Inc., Chicago, IL, USA). Categorical and continuous parameter differences were expressed using Pearson's chi-square test and Mann–Whitney U-test respectively. Our results are expressed as numbers (%) and mean ± SD. Univariate and multivariate logistic regression analyses were performed to identify independent indicators for the presence of various cirrhotic complications. The Bonferroni correction for multiple comparisons was used where appropriate. P-values < 0.05 were considered statistically significant.
Results
Of the 803 patients with cirrhosis who underwent routine health screening for H. pylori presence, 558 patients had liver ultrasound imaging, EGD report, liver function test, and results of H. pylori infection and were enrolled in this study. Two-hundred and forty five patients met the exclusion criteria. Of the remaining 558 patients, 288 patients (51.6%) were H. pylori-negative and 270 patients (48.4%) were H. pylori-positive (Figure 1).
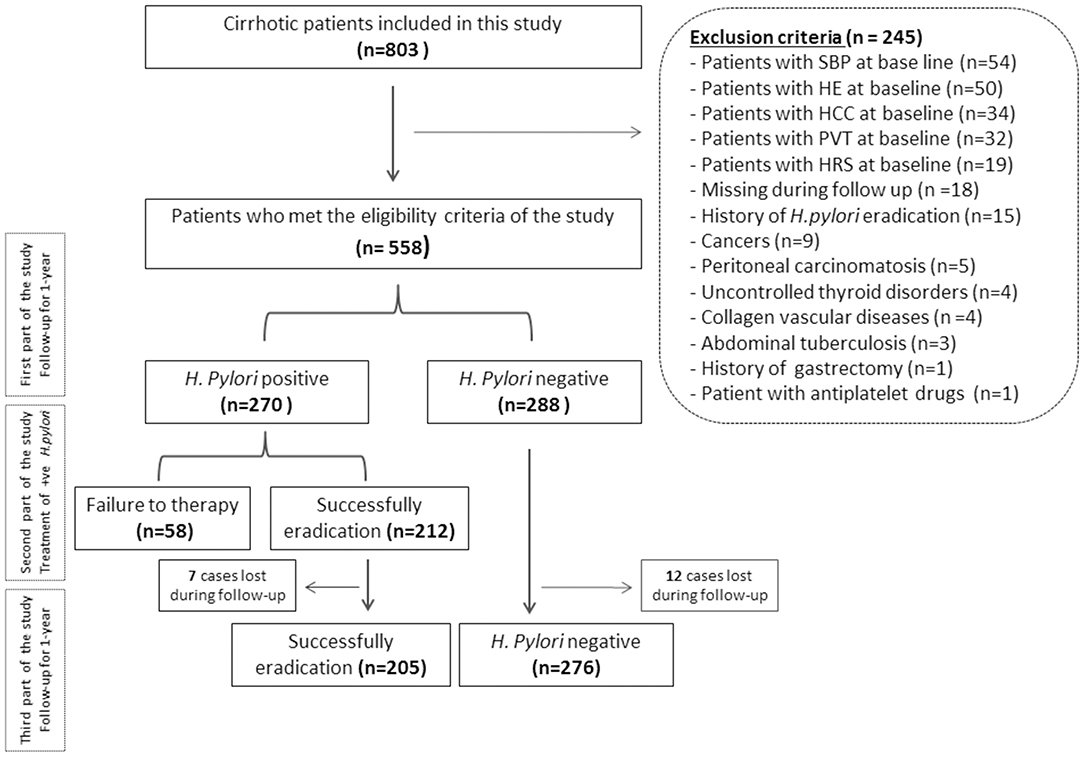
Figure 1. Flowchart of the patients included in this study. SBP, spontaneous bacterial peritonitis; HE, hepatic encephalopathy; HCC, hepatocellular carcinoma; PVT, portal vein thrombosis; HRS, hepatorenal syndrome; H. pylori, Helicobacter pylori.
These two groups were compared and were found to have a significant difference in crowding index, serum ammonia, WBCs, CRP, IL-6, TNF-α, NO, and VEGF (all P < 0.05), as observed in Table 1.
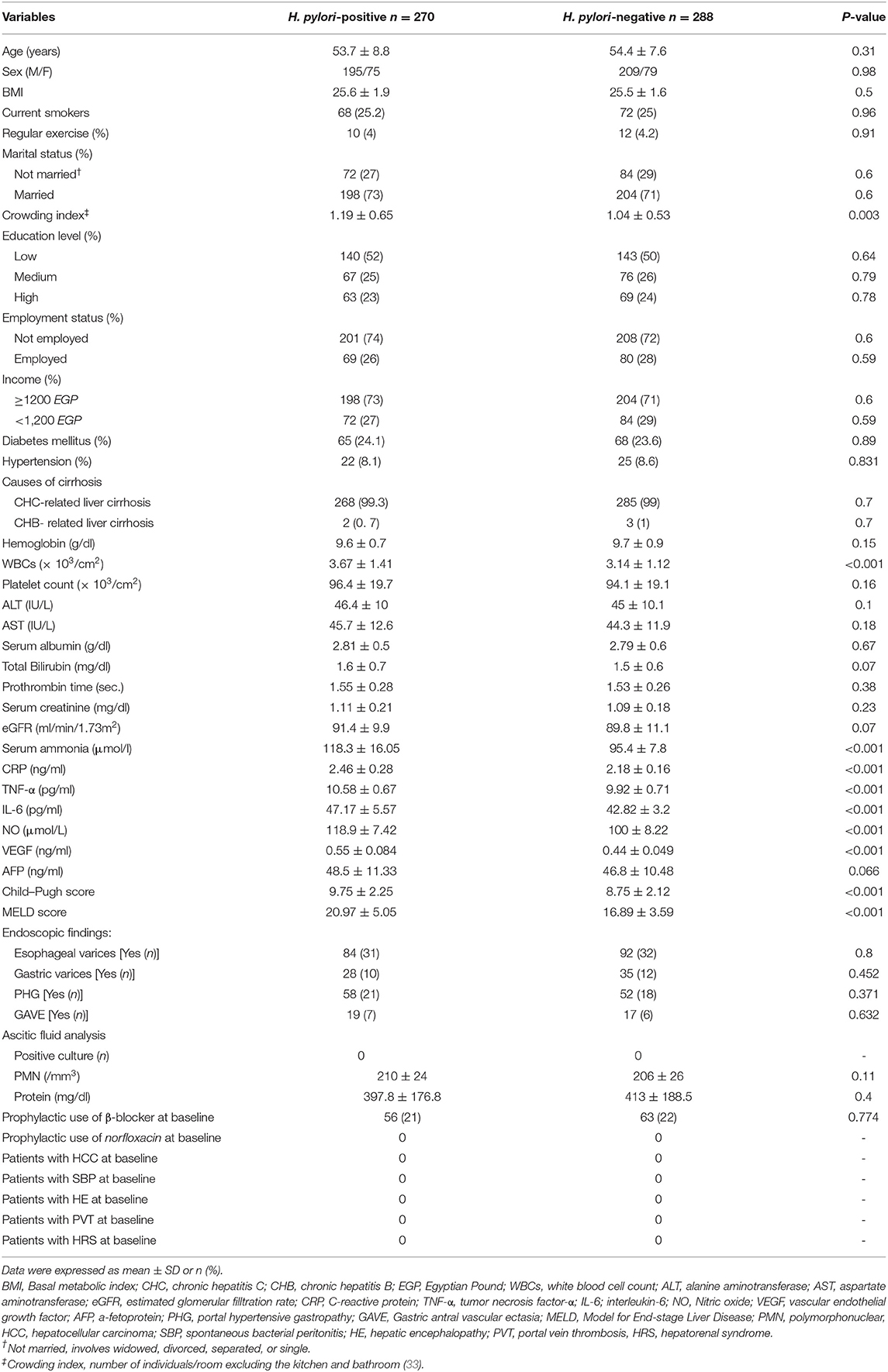
Table 1. Baseline demographic, biochemical, and clinical characteristics of the studied groups.
Results of H. pylori-Positive Patients 1-Year Follow-Up
There was a significant increase in serum ammonia, CRP, IL-6, TNF-α, NO, VEGF, AFP, ascitic PMNs, and Child–Pugh score as well as there was a significant decrease in WBCs, platelet count, AST, serum albumin, eGFR, and ascitic fluid protein (all P < 0.05) as highlighted in Table 2.

Table 2. Demographic, biochemical, and clinical characteristics of the studied groups after a 1-year follow-up.
Results of H. pylori-Negative Patients 1-Year Follow-Up
There was a significant increase in total bilirubin, serum ammonia, CRP, VEGF, NO, AFP, and ascitic PMNs as well as there was a significant decrease in WBCs, platelet count, ALT, AST, serum albumin, prothrombin time, serum creatinine, and e GFR (all P < 0.05) as listed in Table 2.
Comparison of 1-Year Follow-Up Results in Both Groups
There was a significant increase WBCs, platelet count, ALT, AST, serum albumin, prothrombin time, serum creatinine, eGFR, serum ammonia, CRP, IL-6, TNF-α, NO, AFP, VEGF levels, Child–Pugh score, and ascitic PMNs as well as there was a significant decrease in ascitic fluid protein in H. pylori-positive vs. negative patients (all P < 0.05), as observed in Table 2.
There was a significant increase in the incidence of esophageal varices (EV), gastric varices (GV), PHG, GAVE, HCC, SBP and its recurrence, HE, PVT, and HRS in H. pylori-positive vs. negative patients (all P < 0.05), as highlighted in Table 2.
According to ascitic fluid analysis, among the 47 patients who developed SBP in both groups, 18 (38%) had positive ascitic fluid cultures; the isolated organism was only Escherichia coli (E. coli). The rest of the SBP patients had culture-negative SBP (n = 29). Ascitic fluid with positive cultures was significantly increased in H. pylori-positive vs. negative patients (P = 0.027) as demonstrated in Table 2.
3-Months Follow-Up Results After H. pylori-Positive Patients Treatment
There was a significant decrease in WBCs count, serum albumin, creatinine, ammonia, CRP, IL-6, TNF-α, NO, AFP, and VEGF levels as well as there was a significant increase in hemoglobin level (all P < 0.001), as reported in Table 3.
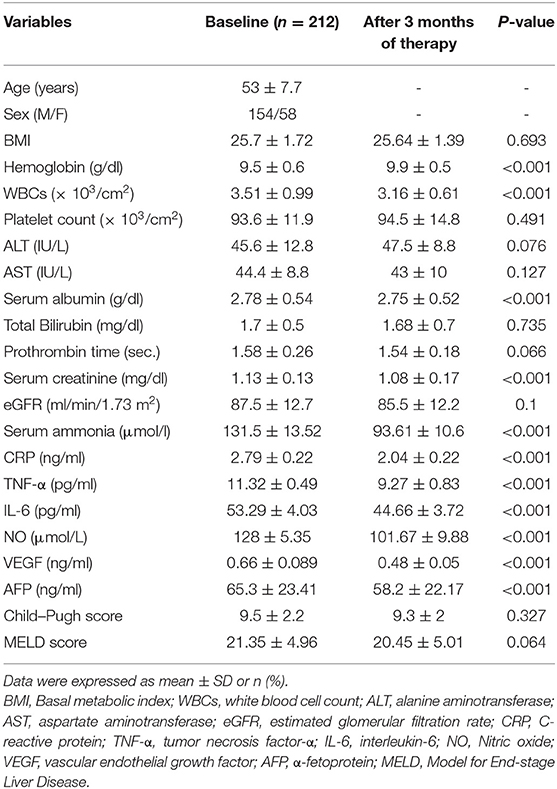
Table 3. Changes in biochemical, clinical, and demographic characteristics of the H. pylori positive group after 3 months of therapy.
Data From an Extra 1-Year Follow-Up Assessment After H. pylori Eradication
There was no significant changes regarding all variables as well as there was no statistically significant difference in the incidence of EV, GV, PHG, GAVE, HCC, SBP and their recurrence, HE, PVT, and HRS in H. pylori-positive patients with eradication vs. H. pylori-negative patients (all P > 0.05), as observed in Table 4.
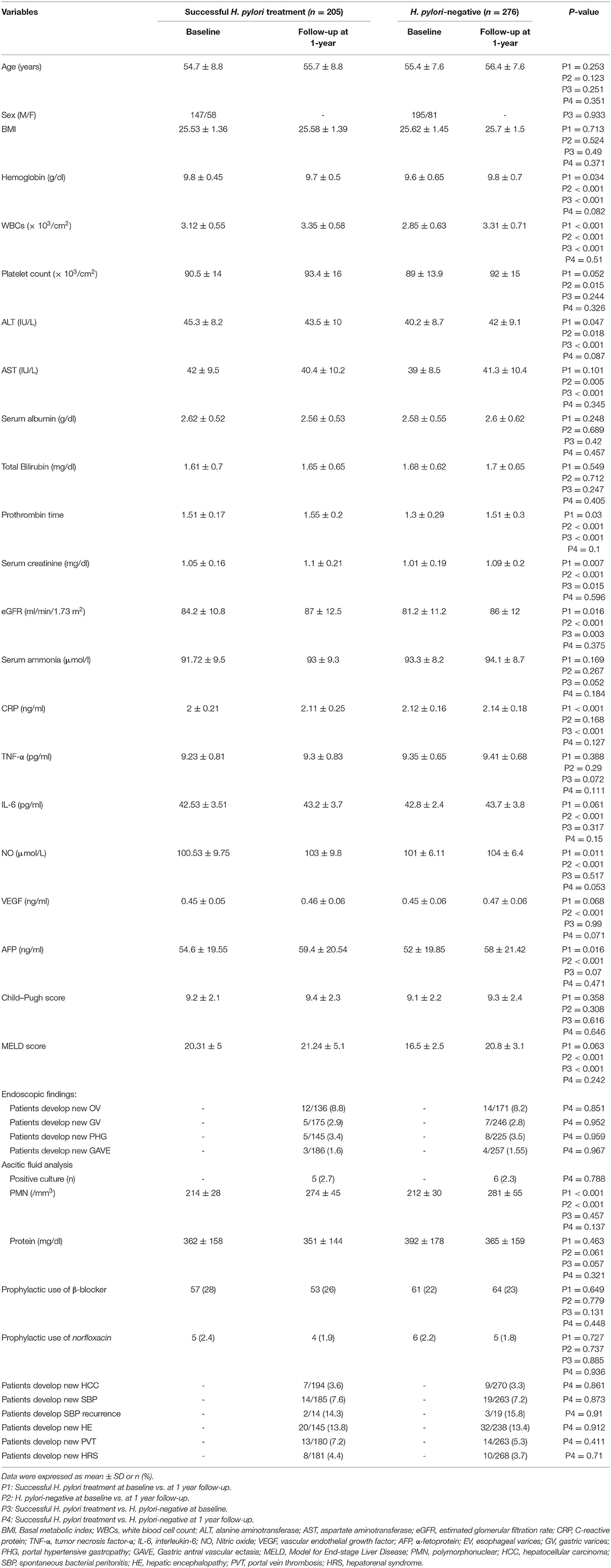
Table 4. Baseline demographic, biochemical, and clinical characteristics of the studied groups for 1 year after treatment of H. pylori-positive patients (Successful H. pylori treatment).
According to ascitic fluid analysis, among the 33 patients who develop SBP in both groups, 11 (33%) had positive ascitic fluid cultures; the isolated organism was only Escherichia coli (E. coli). The rest of the SBP patients had culture-negative SBP (n = 22). No significant differences were found between the two groups (P = 0.873) as shown in Table 4.
All patients with PVT in both groups had been subjected to measurements of activated partial thromboplastin time, fibrinogen, protein C, protein S, antithrombin III, serum homocysteine, D-dimer, antinuclear antibody, anticardiolipin IgG antibodies, anti-double-strand DNA after 1 year of the follow-up and after 1 year of H. pylori eradication. No significant difference between the two groups was evident in both conditions. The details of diagnostic biochemical criteria were described in Tables S1, S2.
Univariate and Multivariate Regression Analysis Models Predicting Different Cirrhotic Complications Within the Follow-Up Period
Univariate analysis revealed that the presence of H. pylori infection was associated independently with the development of SBP, PVT, and HCC (all P < 0.05) while multivariate regression analysis model reported that the presence of H. pylori infection was associated independently with the development of only PVT and HCC, after adjustment for age, BMI, inflammatory markers (CRP, IL-6, and TNF-α), vascular mediators (VEGF and NO), Child-Pugh score and MELD score as shown in Table 5.
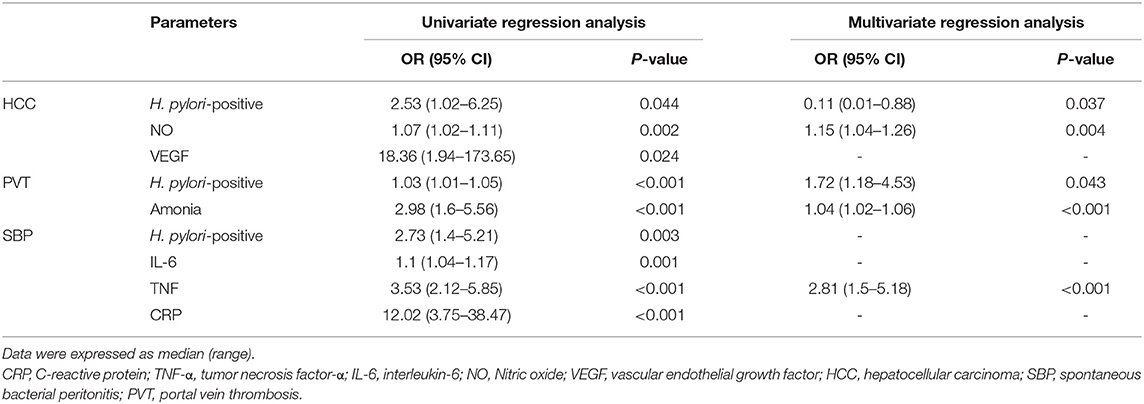
Table 5. Univariate and multivariate regression analysis models predict cirrhotic complications within the follow-up period after adjusting for multiple confounding variables.
Incidence Rate (IR), Incidence Rate Difference (IRD), and Incidence Rate Ratio (IRR) of Cirrhotic Complications Resulting From Multivariate Regression Analysis Models Before and After Therapy Were Demonstrated as Follows
As regards to PVT, the IR was 0.115 (95% CI, 0.078–0.163) before therapy, and changed to 0.072 (95% CI, 0.0385–0.124) after therapy. The IRD was 0.043 (95% CI, −0.016–0.102) with P = 0.042. The IRR was 1.59 (95% CI, 0.81–3.31).
As regards to HCC, the IR was 0.059 (95% CI, 0.034–0.096) before therapy, and changed to 0.036 (95% CI, 0.015–0.074) after therapy. The IRD was 0.023 (95% CI, −0.018–0.064) with P = 0.037. The IRR was 1.64 (95% CI, 0.64–4.72).
Discussion
H. pylori infection leads to an increase of proinflammatory cytokines interleukin (IL)-1, IL-2, IL-4, IL-6, IL-8, IL-10, IL-17, TNF-α, and interferon-β (36). It has been hypothesized that infection with H. pylori has a systemic effect via the activity of these cytokines and exacerbation of different inflammatory reactions (7).
Portal hypertension (PH) plays a major role in the development of cirrhotic complications. In this regard, recent papers have suggested that H. pylori could lead to a condition of chronic vasculitis, thus causing endothelial dysfunction (37). The involvement of PV may prompt the increased stiffness of the vessel, with a decreased capacity to adapt to blood flow changes, leading to increased portal pressure, regardless of inadequate direct experimental evidence (38, 39). The systemic inflammatory conditions, actuated by H. pylori, enhance NO production. Recent research has reported that the concentration of NO was increased in cirrhotic H. pylori-positive patients (40). This finding was concurrent with our outcomes. NO is a common vasodilator and is implicated in the regulation of systemic and splanchnic hemodynamics in PH (41, 42). In addition, lipopolysaccharide of H. pylori, in a cultured cell model of liver tissue, could initiate a microvascular inflammatory response by stimulating the inducible nitric oxide synthase (iNOS), a mechanism that may share in the pathogenesis of PH (43).
H. pylori is believed to magnify the expression of VEGF, via a signaling pathway involving the MEK-ERK and NF-kB cascade (44, 45). Elevated VEGF concentration was related to a significant increment in neo-angiogenesis as estimated by of CD34-positive micro-vessels determination. Increased levels of the VEGF in this study corroborate these earlier findings (44). A strong correlation between serum VEGF levels and PH was demonstrated, and the proof that H. pylori may lead to a local increment in VEGF in gastric tissue might bolster a unifying hypothesis in which experimental and human models help a direct mediation in VEGF-induced PH (46, 47).
The impact of H. pylori on PH may be multifactorial. Alterations in the vasodilatation dynamics, endothelial dysfunction, and vascular overgrowth are the most engaging speculations about these issues.
The results of this study indicate that the incidence of PVT increased in H. pylori-positive patients. The possible explanation may be due to the fact that H. Pylori secretes a neutrophilic activating protein that leads to neutrophilic infiltration of vascular walls resulting in venous thrombosis (48). The bacterium was reported to enhance the secretion of adhesion molecules, for example, E-selectin, intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1), as well as growth-related oncogene alpha, IL-6, and IL-8 (49). All these agents can enhance the induction of neutrophils through the endothelium, an occasion which is an initiating etiology of endothelial dysfunction. Moreover, increased stiffness of the PV induced by H. pylori (39), the “angiogenetic drive” of H. pylori (50) and the reduction in mean portal flow velocity in cirrhotics (51) support our hypothesis in the development of PVT.
The most important clinically relevant finding was the increased incidence of HCC in H. pylori-positive patients. These relationships may partly be explained by the role of H. pylori infection in activating the transforming growth factor β1-dependent oncogenic pathway, affecting the equilibrium between hepatocyte proliferation and apoptosis in experimental models (52). These results were reported by Zhang et al. that H. pylori infection leads to a direct cytopathic effect on HepG2 hepatoma cells by upregulating the expression of various proteins associated with signal transduction and gene transcription (53). Excess generation of NO is well-recognized as an essential step initiating neoplastic transformation (54). In addition, NO plays a pivotal role in the development of HCC and its progression (55). In this study, the multivariate analysis revealed that NO can be considered as an independent risk factor for the development of HCC in H. pylori-positive patients. The role of H. pylori in production of NO was previously discussed (40, 43).
In addition, VEGF is a master regulator of angiogenesis in malignant and normal tissues. It plays an essential function in enhancing the proliferation of endothelial cells, thus favoring neovascularization within and around malignant cells. It participates in many other conditions such as activation of receptors related to the proliferation of tumor cells and recruitment of endothelial cells (56, 57). H. pylori is associated with the production of VEGF (44), which may lead to HCC development. No doubt these findings will be much scrutinized, but there are some immediate dependable conclusions for the role of H. pylori infection and the development of HCC in this study.
Our findings reported a significant decrease in serum ammonia, pro-inflammatory mediators, NO, and VEGF levels as well as decreased incidence of all studied cirrhotic complications after H. pylori infection treatment. Overall, this study strengthens the idea that H. pylori infection induces hepatic decompensation. The precise mechanism of H. pylori in hepatic decompensation remains to be elucidated. This new understanding should help in improving the predictions of H. pylori infection impact on liver cirrhosis.
If we assume the treatment with different antibiotics/PPIs is the cause of the decrease in the inflammatory parameters, they would have increased again after the stoppage of the treatment. Even after the 1 year follow up these mediators/markers didn't show any increase again in comparison with the H. pylori negative patients. This means that H. pylori was the only factor behind this elevation.
This study, to the best of our knowledge, is the first to contribute to this growing area of research by exploring the impact of H. pylori on liver cirrhosis. These findings have significant implications in the understanding of how H. pylori was implicated in the pathogenesis of various cirrhotic complications.
The generalizability of these results are subject to certain limitations. First; single-center study. Second; GI endoscopy was used to diagnose PHG and GAVE with no histopathological assessment. Third; after therapy and successful H. pylori eradication, recurrence of SBP may have been affected by the use of PPI (58) and the prophylactic use of β-blockers (58, 59). Fourth; some patients were non-adherent to the standard recommendations (at least to receive norfloxacin for secondary prophylaxis of SBP and prophylactic use of β-blockers for varices). Fifth, several studies have obtained controversial results, on the relationship between H. pylori infection and cirrhosis, on the basis of etiology (60). Because most of the cases have the same etiology which is hepatitis C virus, we cannot incorporate the different etiologies in our multivariate analysis, or correlate them with our results. Finally, the probability of false negatives becoming positives during reassessment/follow-up may occur, but repeated and strict measures were followed to avoid this bias.
In spite of its limitations, the study certainly adds to our understanding of the role of H. pylori in different complications of liver cirrhosis. Even if there were non- H. pylori factors contributing to the outcome seen, we think it is a great achievement to draw the attention of the clinicians to the important role played by H. pylori in the pathophysiology of cirrhotic complications and consequently the importance of H. pylori treatment and eradication in these patients.
In conclusion, H. pylori infection was evidently related to increased incidence of various cirrhotic complications, especially hepatocellular carcinoma and portal vein thrombosis development through increased secretion of a lot of inflammatory markers and vascular mediators. Moreover, its eradication may reduce the incidence of these complications.
Data Availability Statement
The data that support the findings of this study have restrictions and so are not publicly available. Data are however available from the authors upon reasonable request.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of the Mansoura University, Egypt (Approval no. R.19.08.573). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
Guarantor of the article: AA-R. AA-R and NM acquired, analyzed, and interpreted data, performed statistical analysis, and wrote, edited, and reviewed the manuscript. MA and AS recruited and followed up with patients, acquired, analyzed, and interpreted data, performed statistical analysis, and critically revised the manuscript. AH and AT recruited and followed up with patients, acquired, analyzed, and interpreted data, critically revised the manuscript. RElh and RElz acquired, analyzed and interpreted data, underwent laboratory investigations, and revised the manuscript. WE and NE-W acquired, analyzed, and interpreted data, performed statistical analysis, underwent laboratory investigations, and critically revised the manuscript. All authors approved the final version of the article, including the authorship list.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the staff and patients of the Tropical Medicine Department and the lab specialists for their significant aid.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2020.00096/full#supplementary-material
Abbreviations
BMI, Basal metabolic index; CHC, chronic hepatitis C; CHB, chronic hepatitis B; WBCs, white blood cell count; ALT, alanine aminotransferase; AST, aspartate aminotransferase; eGFR, estimated glomerular filtration rate; CRP, C-reactive protein; TNF-α, tumor necrosis factor-α; IL-6, interleukin-6; NO, Nitric oxide; VEGF, vascular endothelial growth factor; AFP, α-fetoprotein; PPIs, proton-pump inhibitors; PHG, portal hypertensive gastropathy; GAVE, Gastric antral vascular ectasia; MELD, Model for End-stage Liver Disease; PMN, polymorphonuclear; HCC, hepatocellular carcinoma; SBP, spontaneous bacterial peritonitis; HE, hepatic encephalopathy; PVT, portal vein thrombosis; HRS, hepatorenal syndrome.
References
1. Ierardi E, Goni E, Losurdo G, di Mario F. Helicobacter pylori and nonmalignant diseases. Helicobacter. (2014) 19:27–31. doi: 10.1111/j.1523–5378.2011.00878.x
2. Franceschi F, Zuccalà G, Roccarina D, Gasbarrini A. Clinical effects of Helicobacter pylori outside the stomach. Nat Rev Gastroenterol Hepatol. (2014) 11:234–42. doi: 10.1038/nrgastro.2013.243
3. Abdel-Razik A, Mousa N, Shabana W, Refaey M, Elhelaly R, Elzehery R, et al. Helicobacter pylori and non-alcoholic fatty liver disease: a new enigma? Helicobacter. (2018) 23:e12537. doi: 10.1111/hel.12537
4. Zullo A, Hassan C, Morini S. Helicobacter pylori infection in patients with liver cirrhosis: facts and fictions. Dig Liver Dis. (2003) 35:197–205. doi: 10.1016/s1590–8658(03)00029-x
5. Francavilla A, Ierardi E. Helicobacter species in liver and biliary diseases. Dig Liver Dis. (2000) 32:217–19. doi: 10.1016/s1590–8658(00)80283–2
6. Caviglia GP, Olivero A, Rosso C, Bosco C, Ribaldone DG, Fagoonee S. Laboratory evidence of helicobacter species infection in hepatocellular carcinoma. Minerva Biotecnol. (2018) 30:88–93. doi: 10.23736/S1120–4826.18.02428-X
7. Waluga M, Kukla M, Zorniak M, Bacik A, Kotulski R. From the stomach to other organs: Helicobacter pylori and the liver. World J Hepatol. (2015) 7:2136–46. doi: 10.4254/wjh.v7.i18.2136
8. Nilsson HO, Mulchandani R, Tranberg KG, Stenram U, Wadström T. Helicobacter species identified in liver from patients with cholangiocarcinoma and hepatocellular carcinoma. Gastroenterology. (2001) 120:323–4. doi: 10.1053/gast.2001.21382
9. Pellicano R, Mazzaferro V, Grigioni WF, Cutufia MA, Fagoonee S, Silengo L, et al. Helicobacter species sequences in liver samples from patients with and without hepatocellular carcinoma. World J Gastroenterol. (2004) 10:598–601. doi: 10.3748/wjg.v10.i4.598
10. Huang Y, Fan XG, Wang ZM, Zhou JH, Tian XF, Li N. Identification of helicobacter species in human liver samples from patients with primary hepatocellular carcinoma. J Clin Pathol. (2004) 57:1273–7. doi: 10.1136/jcp.2004.018556
11. Xuan SY, Li N, Qiang X, Zhou RR, Shi YX, Jiang WJ. Helicobacter infection in hepatocellular carcinoma tissue. World J Gastroenterol. (2006) 12:2335–40. doi: 10.3748/wjg.v12.i15.2335
12. Cindoruk M, Cirak MY, Unal S, Karakan T, Erkan G, Engin D, et al. Identification of helicobacter species by 16S rDNA PCR and sequence analysis in human liver samples from patients with various etiologies of benign liver diseases. Eur J Gastroenterol Hepatol. (2008) 20:33–6. doi: 10.1097/MEG.0b013e3282efa4f2
13. Pirouz T, Zounubi L, Keivani H, Rakhshani N, Hormazdi M. Detection of Helicobacter pylori in paraffin-embedded specimens from patients with chronic liver diseases, using the amplification method. Dig Dis Sci. (2009) 54:1456–9. doi: 10.1007/s10620–008-0522–5
14. Esmat G, El-Bendary M, Zakarya S, Ela MA, Zalata K. Role of Helicobacter pylori in patients with HCV-related chronic hepatitis and cirrhosis with or without hepatocellular carcinoma: possible association with disease progression. J Viral Hepat. (2012) 19:473–9. doi: 10.1111/j.1365–2893.2011.01567.x
15. Chey WD, Leontiadis GI, Howden CW, Moss SF. ACG clinical guideline: treatment of Helicobacter pylori infection. Am J Gastroenterol. (2017) 112:212–39. doi: 10.1038/ajg.2016.563
16. Kamath PS, Wiesner RH, Malinchoc M, Kremers W, Therneau TM, Kosberg CL, et al. A model to predict survival in patients with end-stage liver disease. Hepatology. (2001) 33:464–70. doi: 10.1053/jhep.2001.22172
17. Zocco MA, di Stasio E, de Cristofaro R, Novi M, Ainora ME, Ponziani F, et al. Thrombotic risk factors in patients with liver cirrhosis: correlation with MELD scoring system and portal vein thrombosis development. J Hepatol. (2009) 51:682–9. doi: 10.1016/j.jhep.2009.03.013
18. European Association for the Study of the Liver. EASL clinical practice guidelines: vascular diseases of the liver. J Hepatol. (2016) 64:179–202. doi: 10.1016/j.jhep.2015.07.040
19. Dever JB, Sheikh MY. Review article: spontaneous bacterial peritonitis–bacteriology, diagnosis, treatment, risk factors and prevention. Aliment Pharmacol Ther. (2015) 41:1116–31. doi: 10.1111/apt.13172
20. European Association for the Study of the Liver. EASL clinical practice guidelines on the management of ascites, spontaneous bacterial peritonitis, and hepatorenal syndrome in cirrhosis. J Hepatol. (2010) 53:397–417. doi: 10.1016/j.jhep.2010.05.004
21. Bruix J, Sherman M, Lloret JM, Beaugrand M, Lencioni R, Burroughs AK, et al. Clinical management of hepatocellular carcinoma. Conclusions of the barcelona-2000 EASL conference. European Association for the Study of the Liver. J Hepatol. (2001) 35:421–30. doi: 10.1016/s0168–8278(01)00130–1
22. European Association for the Study of the Liver. EASL clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. (2018) 69:182–236. doi: 10.1016/j.jhep.2018.03.019
23. Angeli P, Ginès P, Wong F, Bernardi M, Boyer TD, Gerbes A, et al. Diagnosis and management of acute kidney injury in patients with cirrhosis: revised consensus recommendations of the International Club of Ascites. J Hepatol. (2015) 62:968–974. doi: 10.1136/gutjnl-2014–308874
24. Vilstrup H, Amodio P, Bajaj J, Cordoba J, Ferenci P, Mullen KD, et al. Hepatic encephalopathy in chronic liver disease: 2014 practice guideline by the American association for the study of liver diseases and the European association for the study of the liver. Hepatology. (2014) 60:715–35. doi: 10.1002/hep.27210
25. Ferenci P, Lockwood A, Mullen K, Tarter R, Weissenborn K, Blei AT. Hepatic encephalopathy–definition, nomenclature, diagnosis, and quantification: final report of the working party at the 11th World Congresses of Gastroenterology, Vienna, 1998. Hepatology. (2002) 35:716–21. doi: 10.1053/jhep.2002.31250
26. Weissenborn K. Psychometric tests for diagnosing minimal hepatic encephalopathy. Metab Brain Dis. (2013) 28:227–9. doi: 10.1007/s11011–012-9336–4
27. Carpinelli L, Primignani M, Preatoni P, Angeli P, Battaglia G, Beretta L, et al. Portal hypertensive gastropathy: reproducibility of a classification, prevalence of elementary lesions, sensitivity and specificity in the diagnosis of cirrhosis of the liver. A NIEC multicentre study. New Italian Endoscopic Club. Ital J Gastroenterol Hepatol. (1997) 29:533–40.
28. Payen JL, Cales P. Gastric modifications in cirrhosis. Gastroenterol Clin Biol. (1991) 15:285–95.
29. Kato M, Ota H, Okuda M, Kikuchi S, Satoh K, Shimoyama T, et al. Guidelines for the management of Helicobacter pylori infection in Japan: 2016 revised edition. Helicobacter. (2019) 24:e12597. doi: 10.1111/hel.12597
30. Makristathis A, Barousch W, Pasching E, Binder C, Kuderna C, Apfalter P, et al. Two enzyme immunoassays and PCR for detection of Helicobacter pylori in stool specimens from pediatric patients before and after eradication therapy. J Clin Microbiol. (2000) 38:3710–14.
31. Agha-Amiri K, Peitz U, Mainz D, Kahl S, Leodolter A, Malfertheiner P. A novel immunoassay based on monoclonal antibodies for the detection of Helicobacter pylori antigens in human stool. Z Gastroenterol. (2001) 39:555–60. doi: 10.1055/s-2001–16629
32. Lehmann FS, Beglinger C. Current role of Helicobacter pylori stool tests. Digestion. (2003) 68:119–23. doi: 10.1159/000074682
33. Uday J. Introduction. In: Uday J, editor. The Psychological Consequences of Crowding. New Delhi: Sage (1987). p. 15–46.
34. European Association for the Study of the Liver. EASL clinical practice guidelines on nutrition in chronic liver disease. J Hepatol. (2019) 70:172–93. doi: 10.1016/j.jhep.2018.06.024
35. Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of diet in renal disease study groupm. Ann Intern Med. (1999) 130:461–70. doi: 10.7326/0003–4819-130–6-199903160–00002
36. Sun X, Xu Y, Wang L, Zhang F, Zhang J, Fu X, et al. Association between TNFA gene polymorphisms and Helicobacter pylori infection: a meta-analysis. PLoS ONE. (2016) 1:e0147410. doi: 10.1371/journal.pone.0147410
37. Blum A, Tamir S, Mualem K, Ben-Shushan RS, Keinan-Boker L, Paritsky M. Endothelial dysfunction is reversible in Helicobacter pylori-positive subjects. Am J Med. (2011) 124:1171–4. doi: 10.1016/j.amjmed.2011.08.015
38. Akimoto M, Hashimoto H, Shigemoto M, Yokoyama I. Relationship between recurrence of gastric ulcer and the microcirculation. J Cardiovasc Pharmacol. (1998) 31:S507–8. doi: 10.1097/00005344–199800001-00145
39. Mehta G, Gustot T, Mookerjee RP, Garcia-Pagan JC, Fallon MB, Shah VH, et al. Inflammation and portal hypertension – the undiscovered country. J Hepatol. (2014) 61:155–63. doi: 10.1016/j.jhep.2014.03.014
40. Serin E, Pourbagher MA, Gümürdülü Y, Tamer L, Ozer B, Yilmaz U. The effect of decrease in serum nitric oxide concentration on portal hemodynamics after Helicobacter pylori treatment: an open-label pilot study. Turk J Gastroenterol. (2008) 19:158–62.
41. Iwakiri Y. Endothelial dysfunction in the regulation of cirrhosis and portal hypertension. Liver Int. (2012) 32:199–213. doi: 10.1111/j.1478–3231.2011.02579.x
42. Hu LS, George J, Wang JH. Current concepts on the role of nitric oxide in portal hypertension. World J Gastroenterol. (2013) 19:1707–17. doi: 10.3748/wjg.v19.i11.1707
43. Boonyanugomol W, Chomvarin C, Baik SC, Song JY, Hahnvajanawong C, Kim KM, et al. Role of cagA positive Helicobacter pylori on cell proliferation, apoptosis, and inflammation in biliary cells. Dig Dis Sci. (2011) 56:1682–92. doi: 10.1007/s10620–010-1512-y
44. Kang MJ, Song EJ, Kim BY, Kim DJ, Park JH. Helicobacter pylori induce vascular endothelial growth factor production in gastric epithelial cells through hypoxia-inducible factor-1a-dependent pathway. Helicobacter. (2014) 19:476–83. doi: 10.1111/hel.12169
45. Strowski MZ, Cramer T, Sch€afer G, Jüttner S, Walduck A, Schipani E, et al. Helicobacter pylori stimulates host vascular endothelial growth factor-A (Vegf-A) gene expression via MEK/ERK-dependent activation of Sp1 and Sp3. FASEB J. (2004) 18:218–20. doi: 10.1096/fj.03–0055fje
46. Muti LA, Pâârvu AE, Crăciun AM, Miron N, Acalovschi M. Nitro-oxidative stress, VEGF and MMP-9 in patients with cirrhotic and non-cirrhotic portal hypertension. Clujul Med. (2015) 88:140–5. doi: 10.15386/cjmed-458
47. Yakut M, Örmeci N, Erdal H, Keskin O, Karayel Z, Tutkak H, et al. The association between precancerous gastric lesions and serum pepsinogens, serum gastrin, vascular endothelial growth factor, serum interleukin-1 beta, serum toll-like receptor-4 levels and Helicobacter pylori Cag A status. Clin Res Hepatol Gastroenterol. (2013) 37:302–11. doi: 10.1016/j.clinre.2012.09.013
48. Mozzini C, Garbin U, Fratta Pasini A, Cominacini L. An exploratory look at NETosis in atherosclerosis. Intern Emerg Med. (2017) 12:13–22. doi: 10.1007/s11739–016-1543–2
49. Innocenti M, Thoreson AC, Ferrero RL, Strömberg E, Bölin I, Eriksson L, et al. Helicobacter pylori induced activation of human endothelial cells. Infect Immun. (2002) 70:4581–90. doi: 10.1128/IAI.70.8.4581–4590.2002
50. Tuccillo C, Cuomo A, Rocco A, Martinelli E, Staibano S, Mascolo M, et al. Vascular endothelial growth factor and neo-angiogenesis in H. pylori gastritis in humans. J Pathol. (2005) 207:277–84. doi: 10.1002/path.1844
51. Abdel-Razik A, Mousa N, Elhelaly R, Tawfik A. De-novo portal vein thrombosis in liver cirrhosis: risk factors and correlation with the model for end-stage liver disease scoring system. Eur J Gastroenterol Hepatol. (2015) 27:585–92. doi: 10.1097/MEG.0000000000000325
52. Ki MR, Goo MJ, Park JK, Hong IH, Ji AR, Han SY, et al. Helicobacter pylori accelerates hepatic fibrosis by sensitizing transforming growth factor-β1-induced inflammatory signaling. Lab Invest. (2010) 90:1507–16. doi: 10.1038/labinvest.2010.109
53. Zhang Y, Fan XG, Chen R, Xiao ZQ, Feng XP, Tian XF, et al. Comparative proteome analysis of untreated and Helicobacter pylori treated HepG2. World J Gastroenterol. (2005) 11:3485–9. doi: 10.3748/wjg.v11.i22.3485
54. Fukumura D, Kashiwagi S, Jain RK. The role of nitric oxide in tumour progression. Nat Rev Cancer. (2006) 6:521–34. doi: 10.1038/nrc1910
55. Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, et al. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. (2004) 116:617–28. doi: 10.1016/s0092–8674(04)00131-x
56. Tseng PL, Tai MH, Huang CC, Wang CC, Lin JW, Hung CH, et al. Overexpression of VEGF is associated with positive p53 immunostaining in hepatocellular carcinoma (HCC) and adverse outcome of HCC patients. J Surg Oncol. (2008) 98:349–57. doi: 10.1002/jso.21109
57. Lamszus K, Ulbricht U, Matschke J, Brockmann MA, Fillbrandt R, Westphal M. Levels of soluble vascular endothelial growth factor (VEGF) receptor 1 in astrocytic tumors and its relation to malignancy, vascularity, and VEGF-A. Clin Cancer Res. (2003) 9:1399–405.
58. Abdel-Razik A, Abdelsalam M, Gad DF, Abdelwahab A, Tawfik M, Elzehery R, et al. Recurrence of spontaneous bacterial peritonitis in cirrhosis: novel predictors. Eur J Gastroenterol Hepatol. (2019). doi: 10.1097/MEG.0000000000001578. [Epub ahead of print].
59. Perez-Paramo M, Munoz J, Albillos A, Freile I, Portero F, Santos M, et al. Effect of propranolol on the factors promoting bacterial translocation in cirrhotic rats with ascites. Hepatology. (2000) 31:43–8. doi: 10.1002/hep.510310109
Keywords: Helicobacter pylori, liver cirrhosis, C-reactive protein, hepatocellular carcinoma, portal vein thrombosis, spontaneous bacterial peritonitis, hepatic encephalopathy
Citation: Abdel-Razik A, Mousa N, Elhelaly R, Elzehery R, Hasan AS, Abdelsalam M, Seif AS, Tawfik AM, El-Wakeel N and Eldars W (2020) Helicobacter pylori as an Initiating Factor of Complications in Patients With Cirrhosis: A Single-Center Observational Study. Front. Med. 7:96. doi: 10.3389/fmed.2020.00096
Received: 12 January 2020; Accepted: 04 March 2020;
Published: 24 March 2020.
Edited by:
Pedro M. Baptista, University of Zaragoza, SpainReviewed by:
Rinaldo Pellicano, Molinette Hospital, ItalyNazri Mustaffa, University of Science Malaysia, Malaysia
Copyright © 2020 Abdel-Razik, Mousa, Elhelaly, Elzehery, Hasan, Abdelsalam, Seif, Tawfik, El-Wakeel and Eldars. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ahmed Abdel-Razik, YWhtZWRhYmRlbHJhemlrNzZAZ21haWwuY29t
†ORCID: Ahmed Abdel-Razik orcid.org/0000-0001-9924-9727