 Justine Perrin1†
Justine Perrin1† Marisa Capitao1†
Marisa Capitao1† Marie Mougin-Degraef1,2François Guérard1
Marie Mougin-Degraef1,2François Guérard1 Alain Faivre-Chauvet1,2
Alain Faivre-Chauvet1,2 Latifa Rbah-Vidal1Joëlle Gaschet1
Latifa Rbah-Vidal1Joëlle Gaschet1 Yannick Guilloux1
Yannick Guilloux1 Françoise Kraeber-Bodéré1,2,3
Françoise Kraeber-Bodéré1,2,3 Michel Chérel1,3
Michel Chérel1,3 Jacques Barbet4*
Jacques Barbet4*- 1CRCINA, INSERM, CNRS, Université d'Angers, Université de Nantes, Nantes, France
- 2Nuclear Medicine, University Hospital, Nantes, France
- 3Nuclear Medicine, ICO Cancer Center, Saint-Herblain, France
- 4GIP Arronax, Saint-Herblain, France
The impressive development of cancer immunotherapy in the last few years originates from a more precise understanding of control mechanisms in the immune system leading to the discovery of new targets and new therapeutic tools. Since different stages of disease progression elicit different local and systemic inflammatory responses, the ability to longitudinally interrogate the migration and expansion of immune cells throughout the whole body will greatly facilitate disease characterization and guide selection of appropriate treatment regiments. While using radiolabeled white blood cells to detect inflammatory lesions has been a classical nuclear medicine technique for years, new non-invasive methods for monitoring the distribution and migration of biologically active cells in living organisms have emerged. They are designed to improve detection sensitivity and allow for a better preservation of cell activity and integrity. These methods include the monitoring of therapeutic cells but also of all cells related to a specific disease or therapeutic approach. Labeling of therapeutic cells for imaging may be performed in vitro, with some limitations on sensitivity and duration of observation. Alternatively, in vivo cell tracking may be performed by genetically engineering cells or mice so that may be revealed through imaging. In addition, SPECT or PET imaging based on monoclonal antibodies has been used to detect tumors in the human body for years. They may be used to detect and quantify the presence of specific cells within cancer lesions. These methods have been the object of several recent reviews that have concentrated on technical aspects, stressing the differences between direct and indirect labeling. They are briefly described here by distinguishing ex vivo (labeling cells with paramagnetic, radioactive, or fluorescent tracers) and in vivo (in vivo capture of injected radioactive, fluorescent or luminescent tracers, or by using labeled antibodies, ligands, or pre-targeted clickable substrates) imaging methods. This review focuses on cell tracking in specific therapeutic applications, namely cell therapy, and particularly CAR (Chimeric Antigen Receptor) T-cell therapy, which is a fast-growing research field with various therapeutic indications. The potential impact of imaging on the progress of these new therapeutic modalities is discussed.
Introduction
The origins of immunotherapy go back to early centuries of history as illustrated by the fight against smallpox. Realization that survivors were immune to the disease eventually led to the practice of inoculation or variolation, that spread throughout Europe in the early eighteenth century. The discovery of cowpox vaccination by Edward Jenner in 1796 ultimately resulted, after a global vaccination campaign, in the eradication of the disease announced by the World Health Organization in 1977. Fighting infectious diseases with vaccines proved successful, but eradication of other diseases remains elusive. While Jonas Salk developed the first poliomyelitis vaccine in the 1950, the disease is not yet considered as eradicated and remains endemic in several African countries (1). In the meantime, the role of immunity in other pathologies has been explored and the immune system is now identified as a general defense system that distinguishes self from non-self or altered self. Its ability to recognize normal cells from infected or tumor cells has implications in cancer immune surveillance, graft rejection, and many other pathologies but can also result in autoimmune, and inflammatory diseases. It was also realized that the immune system uses an incredibly complex network of connected cellular and molecular agents, not yet fully known and understood.
The focus of this review is on anti-cancer immunotherapy as it is making impressive progress. However, the concepts can also be paralleled in other immune-mediated disorders and for conditions requiring immunotherapeutic intervention. Therapeutic antibodies and cell-based therapies, such as adoptive immunotherapy and stem-cell therapy, have been developed years ago, but, in the last few years, a more precise understanding of control mechanisms of the immune system triggered an impressive development of immunotherapy (2). Novel therapeutic approaches have recently emerged that reached clinical practice with remarkable success in a variety of cancers (3, 4). The different types of tissue injuries and the different stages of disease progression are more precisely identified, as well as the different local and systemic inflammatory responses. Monitoring the depletion, migration, and expansion of immune cells throughout the whole body should help characterizing the diseases and guiding selection of appropriate treatment regiments (5). Such methods have an important role in basic cancer research, where they serve to elucidate novel biological mechanisms. The development of effective therapeutic strategies, targeting tumor cells as well as their micro-environment, also requires the ability to determine in vivo the location, distribution, and long-term viability of the cell populations as well as their biological fate with respect to cell activation and differentiation.
This process is referred to as cell tracking and is not limited to therapeutic cells but includes all cells related to a specific disease or therapeutic approach, like tumor cells, immune cells or microenvironment. It involves non-invasive methods for monitoring the distribution and migration of biologically active cells in living organisms. In conjunction with various non-invasive imaging modalities, cell-labeling methods, such as exogenous labeling or transfection with a reporter gene, allow visualization of labeled cells in vivo in real time, as well as monitoring and quantifying cell accumulation and function by a variety of imaging approaches. In this Review, we briefly describe the basic principles of cell-tracking methods and explain various approaches to cell tracking. Then we highlight recent examples of application of new technologies in animals, focusing on immune checkpoint inhibitor antibodies and cell-based therapies that use natural or genetically engineered T cells, dendritic cells, macrophages or stem cells, and when documented, the clinical potential of these methods.
Cell Tracking Methods: Looking For Cells in Animal or Human Bodies
Most earlier reviews on this topic have classified imaging techniques as direct or indirect labeling methods. The distinction between direct and indirect labeling is not entirely clear and here we will discuss ex vivo vs. in vivo labeling: ex vivo labeling include labeling cells with paramagnetic, radioactive or fluorescent tracers before injection, while in vivo labeling relates to in situ imaging cells by injecting radioactive, fluorescent, or luminescent tracers, or antibodies.
SPECT and PET imaging with labeled monoclonal antibodies has been used for years to detect cancer cells. With the development of immuno-PET, they are now used to detect, quantify and longitudinally monitor in vivo a variety of cells in the context of immunotherapy of cancer and other diseases (6). Using radiolabeled tracers for in vivo imaging will thus be discussed in this review as one of the possible methods of cell tracking.
The various labeling techniques discussed in this review are presented schematically in Figure 1.
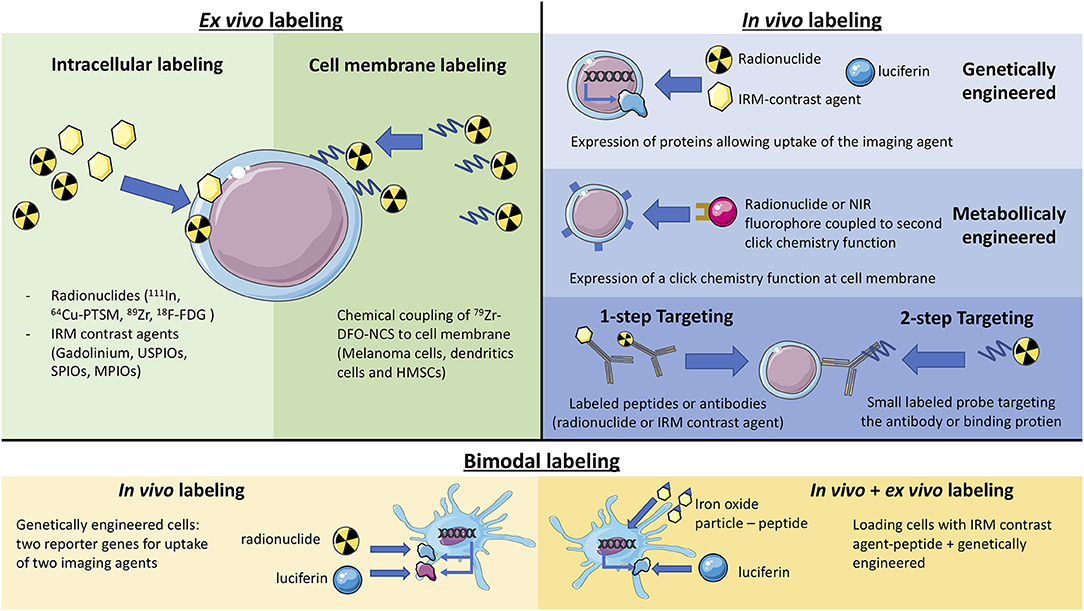
Figure 1. Schematic representation of the different labeling methods (ex vivo labeling, in vivo labeling, and bimodal).
Ex vivo Cell Labeling
While the administration of radiolabeled white blood cells has been a classical nuclear medicine technique for years to detect inflammatory lesions (7), new non-invasive methods for monitoring the distribution and migration of biologically active cells in living organisms have emerged. They aim at improving the detection sensitivity and allowing for a better preservation of cell activity and integrity. These methods have been the subject of many reviews (8). Labeling therapeutic cells for imaging may now be performed in vitro with little impact on cell function nor migration ability, with some limitations on sensitivity and duration of observation (7, 9, 10). Methods based on radioactive imaging or MRI have the highest potential for clinical imaging. They are briefly presented here in this order, highlighting recent progress.
Radioactive (SPECT, PET)
Labeling cells with long-lived radionuclides before re-injection has been used for years in nuclear medicine routine, as mentioned above, but concerns about cell viability and maintenance of cell functions arose. Typically, 111In-oxine is used to label leukocytes (11). Cell labeling yield is good, but a significant efflux rate was reported, and image quality is considered suboptimal with this high energy single photon emitter.
Most recent developments relate to cell labeling using positron emitters because, in human, PET imaging offers better resolution and more precise quantification compared to SPECT. Copper-64 is an interesting candidate, with good imaging properties and a relatively long half-life of 12.7 h. 64Cu-pyruvaldehyde-bis(N4-methylthiosemicarbazone (64Cu-PTSM) was thus used to label C6 glioma cells, as the lipophilic complex is readily taken up in cells. A good cell labeling yield, but a significant efflux rate from cells was observed (12). Zirconium-89 has a half-life of 78.4 h, which is quite convenient to monitor cell trafficking over a few days after administration. Myeloma cells were labeled with 89Zr-oxine using a technique similar to that used for In-111 cell labeling (9). Cell labeling yield was reasonable but contrasting results for efflux rate and cell viability were reported. Sato et al. (10) reported that 89Zr-oxine complex readily labeled dendritic cells (DC) with an efficiency range of 13.0–43.9 and 83.5% ± 1.8 retention 5 days after labeling. In this study, it was considered that labeling did not affect the viability of mouse DCs and Cytotoxic T Lymphocytes (CTLs), nor did it affect functionality. More recently 89Zr-labeled CAR (Chimeric Antigen Receptor) T cells were shown to retain more than 60% of the 89Zr over 6 days while their capacity of in vitro cytokine production, migration, and tumor cytotoxicity, as well as their in vivo antitumor activity (13) were preserved. To further reduce efflux rate and improve viability and cell functions, labeling mixed lymphocyte cell populations with Zr-89 radiolabeled nanoparticles was explored (14, 15).
An alternative approach to loading the radionuclide inside the cells has been proposed. It uses Zr-89-desferrioxamine-NCS, which chemically couples to the membrane of cells. Mouse melanoma cells, dendritic cells and human mesenchymal stem cells were labeled by this method, which was shown to afford stable labeling for 7 days, with little effect of on cell viability and proliferation and to allow for serial PET scans in mouse models (16).
With its fast and efficient uptake and good retention, 18F-labeled fluoro-2-deoxy-2-D-glucose (18F-FDG) may be used to label cells in vitro to monitor cell traffic in vivo. For instance, cardiac stem cells were labeled and their biodistribution and retention was quantified in a pig model of chronic myocardial infarction (17). A potential drawback of 18F-FDG for assessing cell therapies following implantation is the local retention of radiotracer released from the cells. Thus, 3′-deoxy-3′-L-[18F]-fluorothymidine (18F-FLT) has been proposed to label cells instead of 18F-FDG. Human Umbilical Endothelial Vein Cells (HUVECs) incubated with 18F-FLT and injected in mice with hind-limb ischemia were shown to provide a better estimation of HUVECs retention than cells labeled with 18F-FDG (18).
Magnetic Resonance Imaging (MRI)
Gadolinium(III) chelates, such as gadopentetate dimeglumine, are effective paramagnetic contrast agents owing to their unpaired electrons. These electrons confer a magnetic moment that increases the relaxivity of water protons, shortens the longitudinal relaxation rate (T1) and, therefore, increases the signal by creating a positive contrast in T1-weighted MRI images (19). The amount of gadolinium that may be loaded into cells obviously limits the sensitivity. As an example, rat mesenchymal stem cells (MSC) were loaded in vitro with Gd-DTPA using the lipidic transfection agent Effectene. Electron microscopy detected the presence of Gd-DTPA particles in the MSCs and no difference was observed in cell viability or proliferation between the labeled and unlabeled MSCs. T1-weighted MRI was then used to detect the labeled cells in vitro and in the rat brain (20).
Superparamagnetic iron-oxide particles have an inherently larger effect on MRI relaxivity than soluble paramagnetic agents. Their core may contain several thousand iron atoms, which increases the local iron concentration and sensitivity. These particles may be coated with dextran, siloxan, citrate, or polymers to improve biodistribution. The superparamagnetic agent results in negative contrast in T2-weighted sequences by causing inhomogeneities in the local magnetic field and spin–spin dephasing, which shortens transverse relaxation times (21). Ultra-small superparamagnetic iron oxide (USPIO) of 10–50 nm, superparamagnetic iron oxide (SPIO) of 50–100 nm and micrometer-sized iron oxide (MPIOs) up to >1 μm particles have been used (8). Again, cell viability limits the intracellular particle concentrations and thus cell detection sensitivity. Phagocytic cells, such as dendritic cells or pancreatic islet cells, can accumulate large amounts of nanoparticles to allow for their detection in animals and patients (22). Macrophages were easily and efficiently labeled with micrometer-sized particles of iron-oxide (MPIO) in situ and analyzed via ex vivo magnetic resonance microscopy (MRM) and in vivo monitoring by magnetic resonance imaging (MRI). The results were confirmed by fluorescence with an anti-macrophage phenotype marker F4/80 antibody (23). Technological improvements in the sensitivity of MRI equipment afforded promising results in detecting smaller numbers of cells that are difficult to label, including T lymphocytes (24).
Chemical exchange saturation transfer has been proposed as a new mechanism for contrast enhancement in MRI (25) in diamagnetic CEST or paramagnetic CEST (PARACEST), exchangeable protons resonate at a chemical shift different from that of water. Radiofrequency applied at their frequency saturates exchangeable protons, which transfer into water and reduce MRI signal in their vicinity. Although the sensitivity is rather low, the possibility of switching the signal “on” and “off” has attracted much interest (26).
Magnetic resonance also allows for high sensitivity detection of non-radioactive fluorine (19F). Human NK cells were cultured for 24 or 48 h with a commercially available emulsified PFPE perfluorocarbon (CS-ATM-1000) under conditions where labeling had no measurable effect on cell viability and cytotoxicity against K562 leukemia cells. 19F-labeled NK cells could then be detected at the site of injection and shown to migrate (27).
In vivo Labeling
Even if in vitro cell labeling looks rather easy and if progress has been made, direct labeling of cells prior to injection does not allow for long term in vivo imaging. Sensitivity is limited, especially for MRI, when cell viability and functionality is preserved. One drawback has been repeatedly mentioned: macrophages can take up cells or cell debris at the site of injection and migrate. The dilution of the imaging probe during cell division and its release from the cell eventually lead to the disappearance of the signal. Thus, finding alternative routes for tracking cells of interest in vivo has been the subject of many technical developments. One such alternative is the in vitro cell transfection with genes coding for transporters or enzymes as well as metabolic engineering that allow in vivo cell detection using various molecular imaging techniques after injection of a specific tracer.
Genetically Engineered Cells for Radioactive, MRI, or Bioluminescence Imaging
To achieve long term labeling, cells can be genetically engineered to express reporter genes. This reporter gene will allow the targeting of the cells by administering an imaging probe. A stable expression of this reporter allows for a virtually unlimited number of imaging sessions, without any impact of cell division.
Radioactive imaging
Iodine is taken up by the thyroid and by a few other tissues through the sodium-iodine symporter (NIS). Thus, cells were transfected with the NIS gene, most often the human gene (hNIS), injected and imaged by SPECT using a variety of radioactive tracers including iodine-123 (sodium iodide) and technetium-99m (sodium pertechnetate) in a variety of animal models (28). NIS may also be used for PET with iodine-124 or 18F-tetrafluoroborate (29, 30). This approach was used recently to label tumor cells in vivo (31) and to monitor dendritic cell traffic from the skin to lymph nodes (32). This approach has some limitations, though. First, as mentioned above, NIS is expressed by a variety of normal cells, particularly in thyroid, salivary glands and stomach. Thus, imaging cells in these organs is excluded due to background signal. Second, sensitivity for the detection and quantification of transfected cells expressing NIS in vivo is limited because, in the transfected cells, the radioactive tracer does not become linked to tyrosine as iodine is in the thyroid.
Another reporter gene that has attracted much interest is the herpes simplex virus type 1 thymidine kinase (HSV1-tk). With this kind of genes that code for intracellular proteins, the risk of immune reactions is reduced. HSV1-tk allows for PET and SPECT using a variety of anti-viral agents specific for the virus kinase and not recognized by the human enzyme. They enter cells and become phosphorylated and trapped intracellularly only in HSV1-tk-transfected cells. Compounds such as FIAU (5-iodo-2-fluoro-2-deoxy-1-D-arabino-furanosyl-uracil), FEAU (2-fluoro-2-deoxyarabinofuranosyl-5-ethyluracil) or acycloguanosine derivatives (e.g., FPCV: fluoropenciclovir, FHBG: 9-[4-fluoro-3-(hydroxymethyl) butyl] guanine) may be labeled with 18F and used for in vivo PET imaging. Sensitivity may be improved by using a mutated gene, HSV1-sr39tk, that codes for a more potent enzyme. HSV1-sr39tk may be used with [18F]-FHBG as a tracer (33).
In a similar approach to the transfection of cells with viral thymidine kinase, animals may be engineered to express thymidine kinases in specific cells. As an example, Rosa26-mT/sr39tk mice were generated and HSV1-sr39tk expression in platelets, T lymphocytes or cardiomyocytes was induced. Longitudinal PET imaging and quantification of T-cell homing during inflammation and cardiomyocyte viability after myocardial infarction could then be monitored using [18F]-FHBG, a cell-permeable tracer that is phosphorylated by HSV1-tk and retained inside the cells (34).
Alternatively, cells may be transfected to express cell-surface receptors for peptides as, for instance, the human glucagon-like peptide 1 receptor gene and imaged with the peptide labeled with fluorine-18 (35). A similar approach was used to detect transplanted pancreatic islet cells that express glucagon-like peptide 1 receptor (GLP-1R) by PET imaging after the injection of 64Cu-DO3A-VS-Cys40-Exendin-4, showing persistent and specific uptake in the mouse pancreas (36). The mutated version of the dopamine receptor, D2R80A, that internalize 18F-Fallypride, has also been proposed for imaging mesenchymal stem cells (37, 38).
Magnetic resonance imaging
Reporter-gene transfection has been proposed for MRI. The transferrin receptor has been used to capture transferrin-conjugated SPIO particles (39). Dendritic cells transfected with the ferritin gene show increased iron uptake that may be detected by MRI (40, 41). A very similar approach to the NIS system may be used for MRI, by transfecting cells with the Divalent Metal Transporter 1 (DMT1) that can import manganese (42). In the same setting, radioactive manganese (52Mn), may be used for PET imaging (43).
Optical imaging
Bioluminescence imaging (BLI) consists in the use of a luciferase enzyme, which reacts with its substrate, luciferin, and emits light between 480 and 600 nm, depending on the type of enzyme (firefly, Renilla, or bacterial) and substrate (44). This method implies the insertion of the luciferase gene inside the genome of the tracked cells by cell transfection during in vitro culture or by engineering mice to express the luciferase in target cells. In this later case, the mouse itself allow for visualizing intrinsic cells during the development of a pathology. In the case of adoptive cellular therapy, the cells can be isolated from the mouse before the adoptive transfer without need for in vitro transfection. Although the insertion and expression of luciferase is stable, so far adoptively transferred cells have only been followed up to a week, due to the decay of the signal. This may be linked to the death of transferred cells (45). However, after the disappearance of the BLI signal, mice were sacrificed, and histology or flow cytometry was performed. It has been reported that, although the cells are still present and express luciferase, the BLI signal is no more detectable (46). Metabolic changes may be suspected as luciferases need energy and cofactors. Due to this lack of sensitivity, BLI is very often associated with another reporter gene, like Green Fluorescent Protein (GFP), which allow the ex vivo detection by flow cytometry or immunostaining of the organs.
Indeed, these reporter genes are most of the times not used alone but in association, either to enhance the signal (39) or to confirm its specificity by a different imaging approach (47, 48). Most of these proteins are endogenous and not toxic (dopamine receptor, NIS, ferritin). They can be expressed naturally in some organs of the human body, limiting their use. On the other side, inducing their expression in cells implies a possible impact on the functions of the cells.
Animals may also be made to express fluorescent proteins or luciferase in specific cells. This approach has been extensively developed for many different studies, including oncogenesis and cancer therapy (49). For instance, the photoconvertible fluorescent protein Kikume green-red protein was used to track dendritic cells in vivo. The KikGR protein changes its color from green to red upon UV illumination. Then, migration of dendritic cells, specifically CD103+ dendritic cells, from the skin to lymph nodes could be monitored after UV illumination of the skin of knock-in mice expressing the protein (50).
Metabolically Engineered Cells and Click Chemistry
Metabolic engineering and click chemistry (also known as bio-orthogonal chemistry) takes advantage of fast and high yield chemical reactions that may take place in aqueous media and even in vivo. A variety of chemical reagents have been developed that allow for highly specific reactions that are not hindered by biological conditions. Cells of interest were labeled by glycoengineering and bioorthogonal click chemistry by incubation in vitro with tetra-acetylated N-azidoacetyl-D-mannosamine to generate unnatural sialic acids with azide groups on their surface. The cells may then be injected in vivo and detected by the second click chemistry reagent, coupled to a fluorochrome such as dibenzyl cyclooctyne-conjugated Cy5 (DBCO-Cy5) for near-infrared fluorescence imaging or to iron-loaded nanoparticles for MRI (51). This approach was shown to improve labeling efficacy and to reduce false signals generated by macrophage phagocytosis of in vitro labeled cell debris. It does not require genetic modifications. So far, this approach has only been used for near-Infrared fluorescence (NIR) with stem cells and tumor cells (52, 53). Although NIR imaging is non-toxic and cheap, its limited spatial resolution and poor penetration through tissue complicate its use in clinical imaging.
Indirect Methods: Labeled Antibodies and Tracers
Labeled antibodies may be used to detect cells in vivo by SPECT, PET, or NIR fluorescence. They have mainly been used for tumor diagnosis, staging or tumor response monitoring (54). It has been reported that labeled antibodies allow the tracking of T cells in vivo (55).
The first step is to choose the target antigen. Ideally, this antigen should be exclusively expressed on target cells, but most of the time other tissue also express it. For T lymphocytes, many targets have been tested, e.g., CD3, CD8, CD2, and CD7 (56–58).
Once the target is chosen, the antibody must be radiolabeled. Ideally, the radionuclide has a half-life compatible with the biological half-life of the antibody. In human, 89Zr and 64Cu, with half-lives of 78.4 and 12.7 h, respectively, have been used for PET imaging. The radiolabeling method also has an important impact on the quality of the images, since free radionuclide can lead to enhanced background noise, or worse, false positive signal in normal organs, where the target antigen is not expressed. For instance, 89Zr shows a natural tropism to the bone (59) that can impede the tracking of bone marrow cells.
Multistep labeling techniques using antibodies have been developed to improve target to normal tissues ratio. Among these pretargeting approaches, the affinity enhancement system (AES) has been shown to be an excellent method for in vivo tumor imaging by SPECT and PET (60). Recently, new pretargeting approaches have been developed. One is based on the in vivo formation of an oligonucleotide duplex. A first oligonucleotide analog (e.g., peptide nucleic acid or PNA) is coupled to an antibody or a small binding protein (e.g., an anti-HER2 Affibody) for pretargeting of a radiolabeled complementary oligonucleotide analog (61). Another approach is based on bio-orthogonal chemistry (62). The CC49 antibody recognizing the tag72 antigen derivatized with trans-cyclooctene (TCO) was used for pretargeting 111In-labeled DOTA-dipyridyltetrazine, demonstrating fast and high tumor activity uptake and high tumor to muscle ratio in a mouse model. Using small binding proteins such as diabodies or affibodies instead of intact IgG antibodies improves the pretargeting performances for PET (62, 63). Pretargeting may also be applied to NIR fluorescence imaging (63).
The feasibility of detecting cells in vivo using MRI and contrast agents targeted using antibodies or antibody fragments has been tested. Magnetic iron oxide nanoparticles were coated with ethylene oxide polymers and coupled to a ScFv targeting the epidermal growth factor receptor. The product showed a long blood circulation time and low accumulation in liver and spleen. Although in vitro binding and internalization was specific, 24 h after administration to mice bearing EGFR-positive breast cancer 4T1 mouse mammary tumors, MRI signal reduction resulting from uptake of the reagents in the tumor was observed but this signal reduction was equivalent for the targeted and the control products (64). More recently, the same approach was improved by site-selective scFv conjugation to SPION PEG nanoparticles. In vivo, the decrease of MR signals in HER2+ xenograft tumor was about 30% at 24 h after the injection, while non-targeted SPION PEG nanoparticles showed no effect (65).
Bi(multi)Modal Imaging
Multimodality approaches deserve specific attention, even if they are generally limited to preclinical studies. Not only can they combine various imaging modalities, such as radioactive, MRI or optical imaging, but also ex vivo and in vivo labeling as well as post-mortem studies. Thus, bimodal systems have emerged that combine magnetic resonance imaging (MRI) or PET with fluorescence or bioluminescence.
Genetically engineered dendritic cells (DC) have been developed for MRI. Proteins which have an affinity for iron compounds may be used as MRI reporters. In a recent study, DC were engineered to express human ferritin heavy chain (FTH), which chelates iron and acts as an endogenous MRI contrast agent, and GFP genes to allow both fluorescence and MRI cell tracking (40). Reporter genes can also be an enzyme like the Drosophila melanogaster 2′-deoxynucleoside kinase (Dm–dNK) that phosphorylates native deoxynucleosides and a wide range of synthetic nucleoside analogs, including fluorescent nucleosides (66). In this study, the fluorescent nucleoside analog, 2′-deoxycytidine (pyrrolo-dC), generated highly specific CEST MRI signal and fluorescence for bimodal imaging (67).
DC can be loaded by phagocytosis of an antigen labeled with an MRI contrast agent (68). It is possible to effectively load DC with multifunctional polymeric nanoparticles. Nanoparticles composed of iron oxide bearing the OVA antigen coupled to a NIR fluorophore (MNP-OVA) allowed the monitoring of the migration of DCs to lymph nodes in DC adoptive transfer immunotherapy using NIR fluorescence imaging and MRI (69).
PET tracking of genetically engineered DC in combination with bioluminescence has also been developed. In a study, DC were made to express both human NIS and effluc genes. DC migration is then made possible by using 18F-tetrafluoroborate (TFB), a substrate for the NIS reporter gene. Bioluminescence imaging is performed to confirm PET results (32). A combination of PET and Cerenkov luminescence has also been described (70).
Non-phagocytic regulatory T cells (Tregs) have been imaged in vivo after transduction by human NIS and the fluorescent protein mCherry. NIS expressing Tregs were labeled in vitro with technetium-99m pertechnetate (99mTcO4−) and imaged in vivo in C57BL/6 mice by SPECT/CT. After 24 h, Tregs were detected in the spleen and the bimodal labeling confirmed their localization by organ biodistribution studies and flow cytometry (71). In a similar way, bone marrow stem cells were labeled with gadodiamide (Omniscan), a non-ionic complex of gadolinium, using the fluorescent Arrest-In transfection reagent (72).
Nanoparticle systems can integrate therapeutic and imaging agents in a single formulation. They may be particularly useful as multimodal imaging agents. They have been used to deliver these agents through passive or active targeting to cells in vitro and in vivo. The different kinds of such nanoparticles, which include polymeric nanoparticles, micelles, liposomes and dendrimers and their potential applications in cancer immunotherapy, and immune cell tracking have been reviewed in detail (73).
Cell Tracking Achievements: What Happened in Cell Tracking Over The Last Ten Years?
New methods have been developed, but has in vivo cell tracking advanced (cancer) immunotherapy? In vivo imaging has the potential to contribute as a drug development tool to improve the understanding of complex mechanisms of action, as a tool to improve efficacy, for example, by stratifying patients as possible responders or non-responders, and as a non-invasive treatment response biomarker to guide immunotherapy and recognize early signs of loss of efficacy. In cell therapy, a series of questions are asked about the delivery of the cells, their viability, differentiation of proliferation, as well as about the immune responses they may trigger. At this point, preclinical studies have been numerous, but transfer to the clinic remains quite limited (74). This part of the review aims at providing a non-exhaustive survey of achievements in cell tracking using the current tracking methods summarized in Table 1.
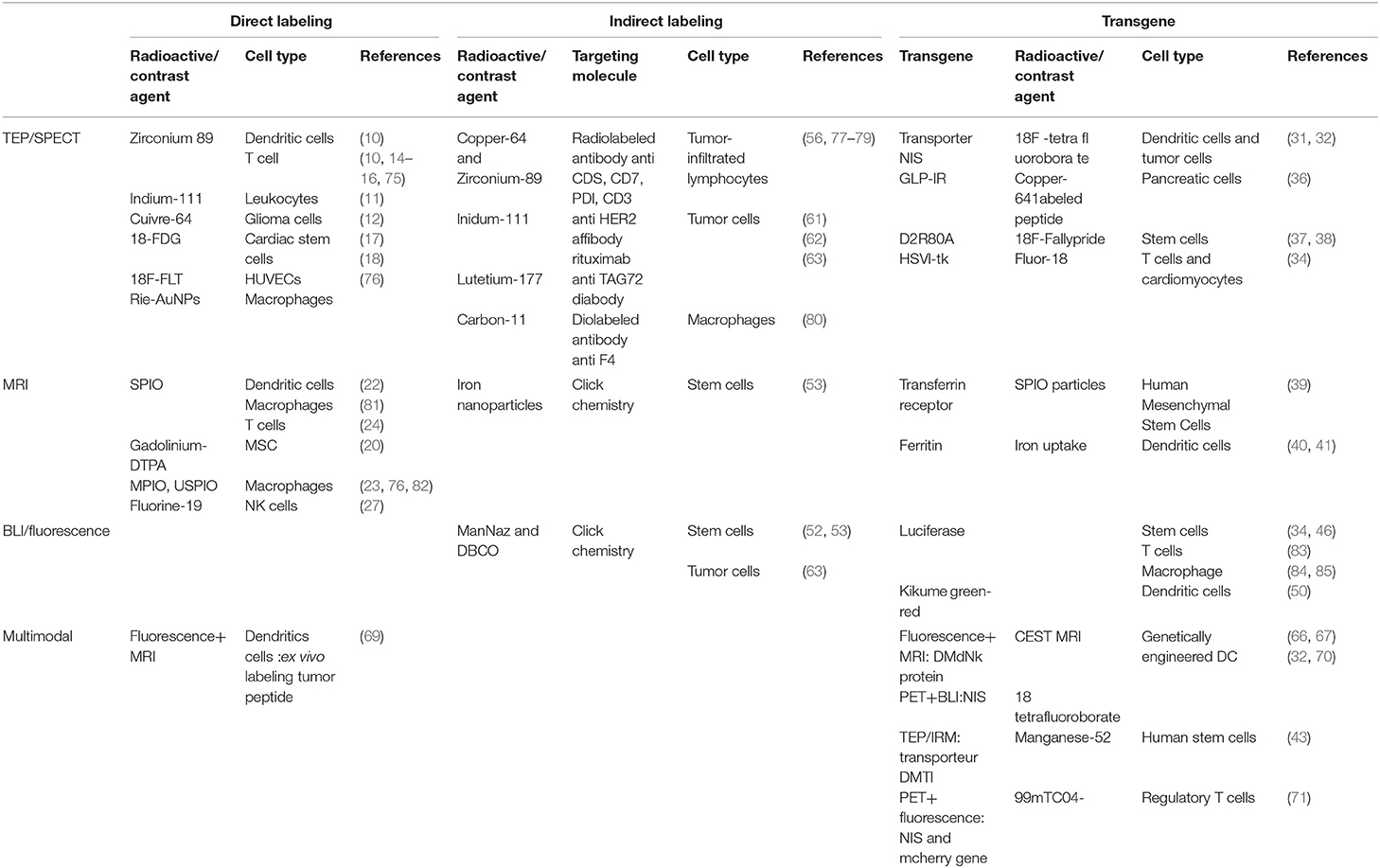
Table 1. Current tracking cell methods in pre-clinic, depending on the type of labeling (direct, indirect, and transgene) and the modality of imaging (TEP/SPECT, MRI, BLI/fluorescence, and multimodal imaging).
Investigating the Tumor and Its Microenvironment
The evaluation of tumor volume and demonstration of tumor shrinkage remains the basis for tumor response assessment with the so-called RECIST criteria. It can be easily performed by CT-scans or MRI when the lesions are measurable, which is by far not always the case. In addition, tumor shrinkage may be delayed and some effective treatments (e.g., some kinase inhibitors) do not result in prominent tumor volume changes. Alternative response criteria, PERCIST, have been proposed (86). In addition, new imaging technologies offer possibilities to look at tumor lesions not as a non-descript mass of tumor cells, but as a complex body of interacting cells of different origins.
Imaging Tumor Cellular Composition
Measuring the relative number of tumor cells in the tumor lesion before and after treatment, may be useful in response assessment. Highly specific markers are needed. For instance, compounds that target melanin biosynthesis (benzamides) (87) and metallopeptides (88) binding to melanocortin type 1 receptor (labeled MSH analogs) have been used in melanoma, but many other labeled molecules, including antibodies, labeled for SPECT and PET, have shown high imaging performances in terms of sensitivity and specificity (89, 90).
Imaging TILs
Monitoring the phenotype and function of tumor infiltrating lymphocytes has long been recognized to be important in adoptive tumor cell therapy (91). This was achieved, in animals as well as in human, by the administration of radiolabeled tracers, usually antibodies or analogs, and SPECT or PET. For example, 64Cu-labeled diabody specific for CD8 was used to assess CD8 T cell density in tumors in mice and treatment related changes (92). Whole antibodies, labeled with zirconium-89 afford similar results (56). Many target antigens have been tested in animal models (56, 58) and CD7 seems so far to be the best candidate to target T lymphocytes, with the lowest toxicity (56).
Surprisingly, in patients, immuno-PET has not been used to detect lymphocytes in tumors, other than through their expression of immune checkpoints, as discussed below. However, labeled IL-2 has been used to visualize lymphocyte infiltrating tumors (77, 93). In a pilot study, patients with metastatic melanoma receiving ipilimumab (IPI) or pembrolizumab (PEMBRO) were subjected to SPECT/CT imaging with 99mTc-labeled interleukin-2 in an attempt to detect TILs. In 5 patients (2 treated with IPI and 3 with PEMBRO), metastatic lesions could be visualized with a positive correlation between size and 99mTc-HYNIC-IL2 uptake, both before and after 12 weeks of therapy (93).
Texture analysis and radiomics may also, without administration of tracer, provide molecular information about infiltration of lymphocytes in tumors. In cancer patients, evidence of for the presence of CD3 T cells in tumors have been obtained by MRI texture analysis (94) and for the presence of CD8 T cells by CT radiomics and Artificial Intelligence analysis (95).
Macrophages
Macrophages are tissue-resident cells of the innate immune systems that perform a variety of functions in host tissue repair and maintenance of homeostasis. Macrophages are associated with auto-immune and inflammatory diseases and, in oncology, one of the tumor escape factors is the presence of pro-tumor macrophage, tumor-associated macrophages (TAM) that support tumor growth (96). In vivo studies have analyzed the biological role and migration of macrophages using different imaging methods such as fluorescent imaging (97), PET, MRI, and multimodal imaging. Macrophages migration to the inflammatory site after an induction of inflammation was analyzed by in vitro labeling with radioactive iodide-embedded gold nanoparticles (RIe-AuNPs) and PET imaging (76). During inflammatory disease such as arthritis, atherosclerotic plaques, in vivo staining of the macrophage with 111In- or 64Cu-labeled antibodies allowed imaging follow-up, evaluation of therapeutic efficacy and therapy adaption (98, 99). For acute or chronic obstructive pulmonary disease, the recruitment of macrophages was monitored by labeling with amine-modified PEGylated dextran-coated SPIO and MRI (81).
In oncology, macrophages are an important part of the tumor microenvironment and thus a therapeutic target. Indeed, the presence of TAM favors tumor escape. In order to assess their presence in tumors and to analyze the efficacy of therapy, these cells were tracked by immuno-SPECT using 111In-labeled anti F4/80 (100) antibody, by MRI using the contrast agents MPIO (82) and ultra-small iron oxide nanoparticles (USPIO) (101), by BLI in transgenic luciferase mice (84) or by multimodal imaging combining MRI and BLI (85). PET imaging using labeled ligands targeting receptors overexpressed in macrophages, such as the Translocator protein (peripheral benzodiazepine receptor), has also been proposed (80).
Imaging Tumor Metabolic Activity
18F-FDG is the most commonly used radiopharmaceutical for imaging tumor metabolism in clinical practice. Its use is based on the increased glycolytic rate in tumors compared to physiologic cells, known as the Warburg effect. However, inflammatory and other metabolically active effector immune cells may contribute to activity uptake in tumor lesions (102). By contrast, lesions with high numbers of proliferative tumor cells are 18F-FDG avid, whereas low 18F-FDG avid lesions have been shown by immunohistology to be infiltrated by activated immune cells. As a result, 18F-FDG is not considered as a marker of immune response and new markers such as amino acids, nucleotides, choline, and receptor ligands have been studied. In hematolymphoid tissues, however, increased levels of deoxycytidine kinase (DCK) expression is found; DCK is the rate-limiting step in the deoxycytidine salvage pathway. The tissue-specific expression of this enzyme allows more specific targeting by, for example, 18F−2-fluoro-D-(arabinofuranosyl)cytosine (18F-FAC), which has been shown to accumulate preferentially in CD8+ T cells and in innate immune cells in mice (103).
18F-labeled 3-fluoro-3-deoxythymidine (18F-FLT) is trapped intracellularly after phosphorylation by thymidine kinase 1 (TK-1) but is not incorporated into DNA since 18F-FLT-monophosphate is a very poor substrate of thymidylate kinase (TMPK). Imaging with 18F-FLT has been evaluated to show proliferation more specifically (18), but effector immune cells that infiltrate tumors are mostly of a differentiated phenotype and do not proliferate. Thus, 18F-FLT uptake in the lymph nodes of vaccinated patients only increased in the presence of antigen-loaded DC, providing the first clinical demonstration that immune responses induced by antigen-specific therapy can be imaged in vivo (102).
In bladder cancer, and presumably in other cancers, correlations have been observed between tumor 18F-FDG uptake and expression of PD-1/PD-L1 (104). Such a correlation may be useful for the selection of appropriate therapeutic strategies.
Immune Checkpoint Inhibitors: Assessment of Immune Status in Tumor Lesions
Immunotherapy agents do not directly attack tumors but re-activate the immune system by targeting adaptive or innate immunity. Immuno-oncology has been revolutionized by the introduction of immune checkpoint inhibitors (ICI) and the approval of ipilimumab in 2011. ICI are monoclonal antibodies targeting immuno-regulatory molecules on the surface of T cells, antigen-presenting cells, and neoplastic cell populations. Clinical success of reagents blocking the CTLA-4 (cytotoxic T lymphocyte-associated protein 4, CD152) and PD-1/PD-L1 checkpoints (programmed cell death protein 1, CD279; programmed death-ligand 1, CD274) has driven rapid regulatory approval for treatment of patients with both solid and hematologic malignancies (105). Patients treated with immune checkpoint inhibitors (ICI) have objective response rates of 20–40% for solid tumors, lymphomas, and malignant melanomas. Thus, 60% of patients do not respond to treatment. It may of course be expected that patients with tumors presenting a higher load of tumor infiltrating lymphocytes (TIL) are more likely to respond to anti-PD-1/PD-L1 check point inhibitors (106).
A detailed understanding of the tumor microenvironment, including the identification and quantification of different immune cell subsets, their spatial context, and the expression of these immune checkpoint markers is obviously required to go further with these new therapies (107). Changes in immune cell infiltration and biomarker expression before and after therapeutic intervention are critical parameters for clinical development (108). Thus, assessment of PD-L1 expression by IHC has emerged as an important predictive biomarker for patients with various cancers including non-small cell lung cancer (NSCLC) and renal cell cancer (78).
Immuno-detection using antibodies labeled with zirconium-89 or copper-64 for PET, as well as indium-111 for SPECT, has been used to assess the CTLA-4 and PD-1 status of TIL in vivo and the expression of PD-L1 by tumor cells in order to predict the therapeutic efficacy of the administration of immune checkpoint inhibitors in mice and in human (79, 109–111). This approach was also proposed in the context of anti CTLA-4 therapy (107). Based on tumor biopsies, it appears that some patients with PD-L1-negative tumors show clinical benefit of anti-PD-L1 treatment. Thus, a zirconium-89-labeled anti-PD-L1 antibody (atezolizumab), was used to image 22 cancer patients before atezolizumab therapy. High PET signal was observed in lymphoid tissues and inflammation sites. In tumors, high but heterogeneous and variable across tumors uptake was observed and clinical responses could be better correlated with PET than with immunohistochemistry or other biomarkers (112).
In mice, the presence of CD8+ T-cells was monitored using 89Zr-labeled an anti-CD8 single domain antibody after treatment of B16 melanoma with an anti-CTLA-4, showing that response correlated with the homogeneity of the distribution of CD8+ T-cells through the tumors (58). In mice with B6-F10 syngeneic melanoma, an anti-mouse PD-1 antibody labeled with copper-64 showed tumor uptake (79).
Monitoring the Activation and Expansion of Immune Effector Cells
Activation and expansion of the immune system may be monitored by imaging changes in the expression of various receptors to cytokines and growth factors as well as changes in the amounts of interstitial water resulting from inflammation. Immune cell trafficking is another aspect of immune system activation.
Imaging Immune Cell Activation
Several examples may be found in preclinical and clinical studies. In mice, an antibody against the cytokine IFNγ, which becomes sequestered at the surface of tumor cells after its production by T lymphocytes, was shown to reflect the activation status of cytotoxic T cells (113).
Reactive lymph nodes also express and secrete chemokines that induce immune cells relocation. Among others, the CCR7 chemokine and chemotactic agents, which play a key role in directing cell trafficking, are suitable imaging targets. For example, CXCL12 is a key chemotaxis factor for lymphocytes with expression of the CXCR4 receptor on their cell membrane (114). PET tracers targeting CXCR4 were thus used in cardiovascular disease and infections. Interestingly, Radiolabeled CXCR4 ligands are also very effective for cancer cell imaging (e.g., 68Ga-labeled pentixafor) and CXCR4-trageting therapeutics labeled with 177Lu are currently under clinical development (114).
Activation of the immune system also results in VEGF release and, subsequently, in significant lymph node volume increase. Lymph node volume can be measured using various techniques including MRI, CT, and ultrasound. Ultrasound imaging using targeted microbubbles improves the evaluation of the microvasculature (115). Dynamic contrast-enhanced (DCE)-MRI using gadolinium (Gd) or USPIO-based contrast agents may also be used to monitor angiogenesis: expansion of lymph node size, total blood flow and blood volume, permeability of perfused capillaries, and total surface of perfused capillaries. MRI measures of vascularity using iron-based contrast agents have been validated against histology, the gold standard in angiogenesis assessment. Diffusion-weighted (DW)-MRI detects metastatic lymph nodes (116) and may be able to image reactive LNs in immune responses.
Imaging the expression of VEGF receptor may also be a way to monitor the activation of endothelial cells in LN resulting from immunotherapy. This was achieved in preclinical models by using anti-VEGFR (bevacizumab) labeled with indium-111 for SPECT (117) or by using RGD peptides labeled with various radionuclides for SPECT and PET imaging (118, 119). These approaches have shown potential in mice, for instance, to image inflammation-induced expansion and regression of lymphatic networks by PET, they have not yet been translated into human.
Changes that occur in the tumor due to an increased immune response can also be imaged using MRI, for example through changes in relaxation times, contrast, or apparent diffusion coefficient (120). These changes have been shown to correlate with conventional histological measures in mice after treatment by transferred cytotoxic T cells that expressed a modified TCR specific for a tumor antigen.
Imaging Trafficking of Immune Effector Cells
Antigen presenting cells (APC) are cells of the immune system that present pathogen peptides linked to class I or class II major histocompatibility complex (MHC) molecules to T lymphocytes (TL) to initiate adaptive immune responses. They are dendritic cells (DC), macrophages, and B lymphocytes. Analyzing antigen capture, migration to the lymph nodes and antigen presentation by APC started with fluorescently labeled cells using in vivo intravital optical imaging (121, 122). Regardless of the microscope type used, this system remains an invasive process, limited in depth penetration and restricted to a specific area of the body. Thus, APC trafficking has been monitored mostly by MRI and, more recently, tracking methods using PET have been reported. Either the cells or mice are genetically engineered, or labeled antigens are loaded in vivo or ex vivo into APC thanks to their phagocytic capacity. DC may be loaded ex vivo with pathogen peptides or irradiated tumor cells and reinjected to the patient. As an alternative, vaccination using labeled irradiated tumor cells or inactivated pathogens have been used to quantify antigen capture and delivery to lymph nodes by MRI (123).
Imaging lymphocyte trafficking is most easily achieved with ex vivo labeled cells. Transfused cells often traffic initially to the lungs, bone marrow, liver, and spleen. In mice, labeling Th1 cells with 64Cu-PTSM was shown to permit their detection in single LNs and to monitor T-cell homing in vivo over 48 h (117). Changes in cell trafficking resulting from treatments with cyclophosphamide or IL-12 may be monitored by in vivo imaging. A similar method, using zirconium-89, was used to monitor γδ T-cells homing into tumor lesions in mice (75). IL13Rα2-CAR T cells delivered intraventricularly were detectable by PET for at least 6 days throughout the central nervous system and within intracranial tumors. When intravenously administered, PSCA-CAR T cells also showed tumor tropism, with a nine-fold greater tumor-to-muscle ratio than for CAR-negative T cells. Bone marrow uptake of 89Zr-labeled hematopoietic stem cells could also be monitored in mice (124) and bone marrow cell uptake in acute fractures in mice could be inhibited, rather than accelerated, by a CXCR4 antagonist, plerixafor (125).
The use of reporter gene expression is another way to study cell trafficking, because imaging is independent of factors lifetime and distribution of the tracer and an enzymatic reporter allows for amplification of a weak signal. Antigen-specific T-cells were made to express a viral Tk gene could be tracked in mice, over a period of 3 weeks, using an 18F-tagged probe specific to this variant of Tk. Detection of 104 T cells was claimed (67).
Lymphocytes may also be imaged by targeting cell surface markers. 99mTc-labeled IL-2 was used to detect tumor-infiltrating lymphocytes in melanoma patients (77, 93). Non-depleting 111In-labeled anti-CD4 antibodies have been used to track CD4+ T cells by SPECT in mice with good correlation with pathologic measures (126). In vivo 19F MRI was also used to track homing to draining lymph nodes of T cells that were intracellularly labeled ex vivo with a perfluoropolyether (PFPE) nanoemulsion (127). Time-lapse 19F MRI was used to calculate the number of T-cells in lymph nodes over 21 days and correlated with in vitro fluorescence measurements to compensate for in vivo T-cell division. MRI also allowed visualization of CD8+ cytotoxic T cells, regulatory T cells, and myeloid-derived suppressor cells loaded with to monitor the effect of vaccination. Increased recruitment of cytotoxic T cells and decreased recruitment of myeloid-derived suppressor cells and regulatory T cells to the tumor was observed (128).
Cell-Based Therapies
Earlier Results in Cell-Based Therapy
So far, most clinical studies have used 99mTc or 111In or superparamagnetic iron oxide to label therapeutic cells for in vivo cell tracking using SPECT or MRI, as reviewed by Srinivas et al. (129). Adoptive T-cell therapy (ACT) using expanded autologous tumor-infiltrating lymphocytes (TIL) and tumor antigen-specific T cell expanded from peripheral blood are complex but powerful immunotherapies. Clinical trials that included cell tracking have compared various routes of administration, the effect of the number of injected cells or host pretreatment with cyclofosfamide and compared various therapeutic cell preparation and encapsulation methods.
Tracking Antigen-Presenting Cells in vivo
DC are the most effective professional antigen presenting cells for the priming of naïve T cells in vitro and in vivo. These properties are the consequence of constitutive expression of MHC molecules class I and II and co-stimulatory molecules (CD80, CD86, CD40) and of their ability to secrete regulatory cytokines such as interleukin 12 upon recognition by the T cell receptor. In the immature stage, DC have the ability to capture the antigen by phagocytosis or endocytosis, migrate to the lymph nodes where they become mature and prime T lymphocytes inducing the adaptive immunity. This is the reason why, in recent years, immunotherapy targeting dendritic cells has developed.
Imaging demonstrated the ability of intradermally or subcutaneously administered therapeutic DC to migrate from the sites of injection into lymph nodes with about 4% of DC reaching draining lymph nodes (130). Actual contact of the DC with T-cells cannot be demonstrated by in vivo imaging, but ex vivo only after lymph node resection. By contrast, intravenously injected mature DC are trapped in the lungs and redistribute to the liver, spleen, and bone marrow. No lymph node localization has been detected so far, which does not mean that DC completely fail to reach the lymph nodes. The techniques may not be sufficiently sensitive to detect the small numbers of cells that do reach the lymph nodes. Direct intranodal administration of therapeutic DC is also common in clinical studies. Then in vivo imaging has been used to study the migration of DC from the primary injected node to secondary nodes. The large variability in the fraction of injected cells (from 0 to 84 %) that was shown to migrate cast doubts on the accuracy of intranodal injections. Labeling the antigen to monitor its fate after DC delivery has been proposed in preclinical in vivo models. When DTPA was conjugated to the epsilon NH2 group of the Lys154 residue, MHC binding of the peptide was preserved and could still be recognized by cytotoxic T cells. These studies allowed the non-invasive determination of the behavior of MHC–peptide complexes expressed by DC in cell vaccination (131) but has not yet been reproduced in the clinic.
CAR (Chimeric Antigen Receptor) T-Cell Therapy
CAR T-cell therapy is a fast-growing research field with various therapeutic indications in autoimmunity, allotransplantation, infection and cancer. Enhancing the functionality and the safety of the injected cells is an important aspect of the clinical development of this very potent therapy. Therefore, there is a real need to develop in vivo molecular imaging to better visualize, predict and improve the efficiency of this type of immunotherapy (5). However, so far, clinical studies of CAR T-cell tracking have only established proofs of concept of its feasibility (74).
SPECT and PET imaging are two possible modalities for tracking the fate of T-cells injected for therapeutic use. Labeling T-cells has been extensively investigated and radiolabeling is possible with little impact on cell function or migration ability (13). However, the radionuclide half-life is a limitation to track the cells more than for a few days after injection.
A variety of solutions to this limitation have been proposed (132). While multimodal imaging has been shown possible (133), CAR T-cell tracking in animals has demonstrated homing and persistence in the tumors and spleen by ex vivo MRI of tissue samples after CAR T-cell labeling with perfluorocarbon (134) and in whole animals by immuno-PET (135). Although the context of CAR T-cell therapy would be appropriate to develop genetic modification of the T-cells to express reporter genes as discussed above, the outcome of therapy remains monitored mostly by functional imaging and especially by MRI (136).
Other Adoptive T-Cell Transfer Therapies
Although BLI is most of the time used to follow tumor and stem cells, T cells could also be monitored thanks to the luciferase gene; for instance, the migration of CD8 T cells toward tumor site was evaluated in a xenograft mouse model (83), and the migration of tumor associated macrophages has been visualized in a transgenic mouse model of ovarian cancer (84). Also, by optical imaging, but using a fluorophore targeted to NIS-transfected cells, tracking of ex vivo-expanded NK cells has been performed in vitro and in vivo showing fast NK cell accumulation in tumors in triple-negative in breast cancer xenografts (137).
89Zr-oxinate labeling was used to track Vγ9Vδ2 T cells in vivo by PET. In a mouse xenograft model of human breast cancer, the Vγ9Vδ2 T cells could be tracked over 1 week and it was shown that injection of PEGylated liposomal alendronate increased homing of the T cells to the tumors, which was confirmed by histology (75).
Stem Cell Therapies
Mesenchymal stem cells (MSC) have been proposed for cardiac regeneration after myocardial infarction (MI). Mesenchymal stem cells derived from rat fetal heart have the potential to differentiate into cardiomyocytes, endothelial cells and smooth muscle cells in vitro. These cells were labeled with technetium-99m for in vivo tracking that revealed a focal uptake of cells in the anterior mid-ventricular region of the heart in line with subsequent ventricular functional recovery (138). Cardiac stem cells were also loaded with 18F-FDG and imaged by PET to quantify their biodistribution and assess the retention of implanted cells in a model of chronic myocardial infarction in pigs. Acute cell retention was shown not to correlate with cell engraftment, which is improved by IM injection (17).
Stem cells have been tracked in various models with BLI, for instance in acute liver injury or acute kidney injury, to study the migration and persistence of human bone-marrow derived stem cells to the liver and kidney (45, 139). Luciferase-transfected adipose-derived stem cells could be transplanted in liver and brain and monitored in vivo by bioluminescence for several days. Ex vivo, immunofluorescence detected the continued expression of luciferase for 4 months, demonstrating that the transplanted cells do not dye, even if the bioluminescence signal is lost (46).
In a tumor graft model, the migration of mesenchymal stem cells toward the subcutaneous tumor could also be observed (140). This study took advantage of a different type of luciferase that metabolizes different substrate, allowing them to follow the migration of stem cells with the Firefly Luciferase, and the tumor progression with the Renilla Luciferase.
BLI imaging has allowed to investigate the impact of stem cells injection modalities, showing that the intravenous route often leads to sequestration in the lung (141) preventing the migration of stem cells to other organs, while the intracardiac route seems to prevent this phenomenon.
Discussion
Cell tracking has a long history of routine clinical use in Nuclear Medicine and it serves a purpose in infectious and inflammatory diseases despite its limitations (142). Imaging has an increasing role in the context of personalized medicine, which becomes the approach to take, at least in developed countries. CT-scan, MRI and ultrasonography are now inescapable and Nuclear Medicine modalities have gained larger recognition, particularly in cancer. However, the number of tracers of frequent, routine use remains quite limited. In addition to bone and thyroid scans, 18F-FDG is certainly the tracer that has the biggest impact in cancer management, with a few other PET tracers for those cancers in which 18F-FDG does not perform so well, such as prostate cancer. In view of the incredible number of preclinical and early clinical studies about cancer imaging, this seems not much. There are many obvious reasons, the major one being the difficulty of demonstrating that a new imaging technique has its place in medicine as compared to all existing ones. If the imaging technique needs an injectable tracer, such as a radiopharmaceutical, the situation is worse, because of the cost of developing a product that has the regulatory status of a drug and by far not the sales and price of a therapeutic compound.
Will immunotherapy change this situation? Most of the very large number of original publications and reviews that deal with immunotherapy advocate for more imaging, especially more specific imaging of receptors, antigens and other biomarkers that characterize the function of cells in vivo. With the progress of cellular therapies, whether regenerative or cancer-oriented, many papers call for cell tracking in vivo as a way to understand their behavior and mechanisms of action and, by the way, to design improved therapies. Indeed, if not all novel immunotherapies are cell therapies, they all bear upon complex cellular interactions at the tumor sites and in immunologic tissues and better knowledge of the nature of cancer and non-cancer cells residing in tumors, their activation, proliferation and migration in living animals and, of course, in humans, must be a way of progress in therapy.
This realization triggered a lot of developments that made possible better cell labeling, mostly to make them visible by MRI and PET, for longer times after re-injection, as well as improved tracers to target specific biomarkers in vivo, using SPECT and PET, but also MRI, not only on tumor cells but on those cells that make the tumor microenvironment, e.g., endothelial cells, infiltrating antigen-presenting cells, lymphocytes, macrophages and other cells of the immune system. New techniques have been developed and the use of reporter genes to make cells detectable any time after their inoculation using specific tracers is a particularly elegant and powerful one. This review has rapidly depicted these approaches and it is expected that it convinces the reader that they are feasible and effective.
There is no best technique, though. It depends on the objective and, obviously, the most powerful one, for instance the use of reporter genes, are associated with complex manipulations, cost and regulatory hurdles. Interestingly, radioactive (SPECT, PET) and non-radioactive (MRI, optical imaging, ultrasonography) methods have been proposed, which all have advantages and drawbacks. Of course, bimodal and even multimodal agents have been developed. Multimodal imaging is clearly the way to go, with SPECT and PET now always associated with CT and PET-MRI systems developing. It is also clear that multimodal imaging experiments in animals that allowed for in vivo imaging and ex vivo in-depth investigation of the fate of injected cells and confirmation of in vivo imaging results are most convincing. However, such studies do not necessarily need bifunctional tracers, which sometimes look more like “tours de force” than candidates for further development.
One question here is: have these developments and studies been useful for the development of immunotherapy? The answer is not obvious. Such studies have pointed out to some problems and they have been mostly confirmatory, when they have not merely been proofs of concept for feasibility. This review has attempted at presenting clinical results in cell tracking and, while it may have missed some, it is in line with other recent reviews on the subject to conclude that the number of clinical studies is quite small. It may be considered that this is only the beginning of a new story and that groundbreaking discoveries in immunotherapy will be made thanks to imaging and that at least some of the approaches reported here will find their application. Very few cell tracking techniques will become routine. The introduction of reporter genes in therapeutic cells is probably the technique with the highest sensitivity for long term monitoring of cell trafficking, proliferation, and persistence. For cell therapies in which the cells are genetically modified, namely CAR-T cells, the addition of a second gene and cell tracking may be considered in the context of clinical trials. Whether this approach will be used in routine clinical practice is not likely. Conversely, tracers for in vivo imaging, particularly PET imaging, designed to detect and quantify specific cell populations are being developed and some will find a routine use. It is always difficult to make predictions, but it seems logical that expensive therapies or therapies that may be efficacious but associated with serious side-effects will not be given to patients who have no chance to benefit from them. The imaging of tumor microenvironment may give answers to how the patients will respond to such therapies, especially immunotherapy. Expression of immune checkpoints, like anti-PD-1, is already assessed from biopsies prior to immunotherapy, but the use of PET-imaging or MRI could allow a non-invasive assessment of the immune state of the tumor. This could provide new insights into the prediction of the response to treatment in patients. This is the theragnostic approach, which is not a reality today, but most probably be one in the future. It is also quite probable that future research in immunotherapy will take advantage of all these technological advances, certainly for preclinical studies, but also in the clinic. Indeed, it is time to combine the novel therapeutic approaches, which afford impressive remissions but not yet to all patients and this will call for precise, specific understanding of what is really going on in the living organism.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported in part by grants from the French National Agency for Research called Investissements d'Avenir IRON Labex no ANR-11-LABX-0018-01 and ArronaxPlus Equipex no ANR-11-EQPX-0004.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
2. Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: Immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol. (2016) 13:273–90. doi: 10.1038/nrclinonc.2016.25
3. Hoos A. Development of immuno-oncology drugs—from CTLA4 to PD1 to the next generations. Nat Rev Drug Discov. (2016) 15:235–47. doi: 10.1038/nrd.2015.35
4. Maldini CR, Ellis GI, Riley JL. CAR T cells for infection, autoimmunity and allotransplantation. Nat Rev Immunol. (2018) 18:605–16. doi: 10.1038/s41577-018-0042-2
5. Fruhwirth GO, Kneilling M, de Vries IJM, Weigelin B, Srinivas M, Aarntzen EHJG. The potential of in vivo imaging for optimization of molecular and cellular anti-cancer immunotherapies. Mol Imaging Biol. (2018) 20:696–704. doi: 10.1007/s11307-018-1254-3
6. van Dongen GAMS, Visser GWM, Lub-de Hooge MN, de Vries EG, Perk LR. Immuno-PET: a navigator in monoclonal antibody development and applications. Oncologist. (2007) 12:1379–89. doi: 10.1634/theoncologist.12-12-1379
7. Roca M, de Vries EFJ, Jamar F, Israel O, Signore A. Guidelines for the labelling of leucocytes with 111In-oxine. Inflammation/Infection Taskgroup of the European Association of Nuclear Medicine. Eur J Nucl Med Mol Imaging. (2010) 37:835–41. doi: 10.1007/s00259-010-1393-5
8. Kircher MF, Gambhir SS, Grimm J. Noninvasive cell-tracking methods. Nat Rev Clin Oncol. (2011) 8:677–88. doi: 10.1038/nrclinonc.2011.141
9. Charoenphun P, Meszaros LK, Chuamsaamarkkee K, Sharif-Paghaleh E, Ballinger JR, Ferris TJ, et al. [89Zr]Oxinate4 for long-term in vivo cell tracking by positron emission tomography. Eur J Nucl Med Mol Imaging. (2014) 42:278–87. doi: 10.1007/s00259-014-2945-x
10. Sato N, Wu H, Asiedu KO, Szajek LP, Griffiths GL, Choyke PL. 89 Zr-Oxine complex PET cell imaging in monitoring cell-based therapies. Radiology. (2015) 275:490–500. doi: 10.1148/radiol.15142849
11. McAfee JG, Subramanian G, Gagne G. Technique of leukocyte harvesting and labeling: problems and perspectives. Semin Nucl Med. (1984) 14:83–106. doi: 10.1016/S0001-2998(84)80023-9
12. Phelps ME, Walsh J, McCarthy T, Adonai N, Iyer M, Toyokuni T, et al. Ex vivo cell labeling with 64Cu-pyruvaldehyde-bis(N4-methylthiosemicarbazone) for imaging cell trafficking in mice with positron-emission tomography. Proc Natl Acad Sci USA. (2002) 99:3030–5. doi: 10.1073/pnas.052709599
13. Weist MR, Starr R, Aguilar B, Chea J, Miles JK, Poku E, et al. PET of adoptively transferred chimeric antigen receptor T cells with 89Zr-Oxine. J Nucl Med. (2018) 59:1531–7. doi: 10.2967/jnumed.117.206714
14. Brown G, Ellis B, Locatelli P, Jones AKP, Fairclough M, Prenant C, et al. A new technique for the radiolabelling of mixed leukocytes with zirconium-89 for inflammation imaging with positron emission tomography. J Label Compd Radiopharm. (2016) 59:270–6. doi: 10.1002/jlcr.3392
15. Gennari A, Boutin H, Alzabin S, Jones AKP, Prenant C, McMahon A, et al. Development of a method for the preparation of zirconium-89 radiolabelled chitosan nanoparticles as an application for leukocyte trafficking with positron emission tomography. Appl Radiat Isot. (2017) 130:7–12. doi: 10.1016/j.apradiso.2017.09.004
16. Bansal A, Pandey MK, Demirhan YE, Nesbitt JJ, Crespo-Diaz RJ, Terzic A, et al. Novel 89Zr cell labeling approach for PET-based cell trafficking studies. EJNMMI Res. (2015) 5:19. doi: 10.1186/s13550-015-0098-y
17. Collantes M, Pelacho B, García-Velloso MJ, Gavira JJ, Abizanda G, Palacios I, et al. Non-invasive in vivo imaging of cardiac stem/progenitor cell biodistribution and retention after intracoronary and intramyocardial delivery in a swine model of chronic ischemia reperfusion injury. J Transl Med. (2017) 15:1–11. doi: 10.1186/s12967-017-1157-0
18. MacAskill MG, Tavares AS, Wu J, Lucatelli C, Mountford JC, Baker AH, et al. PET cell tracking using 18F-FLT is not limited by local reuptake of free radiotracer. Sci Rep. (2017) 7:44233. doi: 10.1038/srep44233
19. Caravan P, Ellison JJ, McMurry TJ, Lauffer RB. Gadolinium(III) chelates as MRI contrast agents: structure, dynamics, and applications. Chem Rev. (1999) 99:2293–352. doi: 10.1021/cr980440x
20. Yang ZX, Cheng X, Jia Y, Geng K, Wu R, Yi M, et al. Tracking of mesenchymal stem cells labeled with gadolinium diethylenetriamine pentaacetic acid by 7T magnetic resonance imaging in a model of cerebral ischemia. Mol Med Rep. (2014) 11:954–60. doi: 10.3892/mmr.2014.2805
21. Weissleder R, Cheng HC, Bogdanova A, Bogdanov A Jr. Magnetically labeled cells can be detected by MR imaging. J Magn Reson Imaging. 7:258–63. doi: 10.1002/jmri.1880070140
22. Sipe JC, Filippi M, Martino G, Furlan R, Rocca MA, Rovaris M, et al. Method for intracellular magnetic labeling of human mononuclear cells using approved iron contrast agents. Magn Reson Imaging. (1999) 17:1521–3. doi: 10.1016/S0730-725X(99)00085-5
23. Foley LM, Hitchens TK, Ho C, Janesko-Feldman KL, Melick JA, Bayir H, et al. Magnetic resonance imaging assessment of macrophage accumulation in mouse brain after experimental traumatic brain injury. J Neurotrauma. (2009) 26:1509–19. doi: 10.1089/neu.2008.0747
24. Smirnov P. Cellular magnetic resonance imaging using superparamagnetic anionic iron oxide nanoparticles: applications to in vivo trafficking of lymphocytes and cell-based anticancer therapy. Methods Mol Biol. (2009) 512:333–53. doi: 10.1007/978-1-60327-530-9_19
25. Ward KM, Aletras AH, Balaban RS. A new class of contrast agents for MRI based on proton chemical exchange dependent saturation transfer (CEST). J Magn Reson. (2000) 143:79–87. doi: 10.1006/jmre.1999.1956
26. Winter PM. Magnetic resonance chemical exchange saturation transfer imaging and nanotechnology. Wiley Interdiscip Rev Nanomed Nanobiotechnol. (2012) 4:389–98. doi: 10.1002/wnan.1167
27. Bouchlaka MN, Ludwig KD, Gordon JW, Kutz MP, Bednarz BP, Fain SB, et al. 19F-MRI for monitoring human NK cells in vivo. Oncoimmunology. (2016) 5:1–12. doi: 10.1080/2162402X.2016.1143996
28. Penheiter AR, Russell SJ, Carlson SK. The sodium iodide symporter (NIS) as an imaging reporter for gene, viral, and cell-based therapies. Curr Gene Ther. (2012) 12:33–47. doi: 10.2174/156652312799789235
29. Jauregui-Osoro M, Sunassee K, Weeks AJ, Berry DJ, Paul RL, Cleij M, et al. Synthesis and biological evaluation of [18F]tetrafluoroborate: a PET imaging agent for thyroid disease and reporter gene imaging of the sodium/iodide symporter. Eur J Nucl Med Mol Imaging. (2010) 37:2108–16. doi: 10.1007/s00259-010-1523-0
30. Youn H, Jeong JM, Chung J-K. A new PET probe, (18)F-tetrafluoroborate, for the sodium/iodide symporter: possible impacts on nuclear medicine. Eur J Nucl Med Mol Imaging. (2010) 37:2105–7. doi: 10.1007/s00259-010-1601-3
31. Diocou S, Volpe A, Jauregui-Osoro M, Boudjemeline M, Chuamsaamarkkee K, Man F, et al. [18F]tetrafluoroborate-PET/CT enables sensitive tumor and metastasis in vivo imaging in a sodium iodide symporter-expressing tumor model. Sci Rep. (2017) 7:946. doi: 10.1038/s41598-017-01044-4
32. Lee SB, Lee HW, Lee H, Jeon YH, Lee S-W, Ahn B-C, et al. Tracking dendritic cell migration into lymph nodes by using a novel PET probe 18F-tetrafluoroborate for sodium/iodide symporter. EJNMMI Res. (2017) 7:32. doi: 10.1186/s13550-017-0280-5
33. Collins SA, Hiraoka K, Inagaki A, Kasahara N, Tangney M. PET imaging for gene and cell therapy. Curr Gene Ther. (2012) 12:20–32. doi: 10.2174/156652312799789271
34. Thunemann M, Schörg BF, Feil S, Lin Y, Voelkl J, Golla M, et al. Cre/lox-assisted non-invasive in vivo tracking of specific cell populations by positron emission tomography. Nat Commun. (2017) 8:444. doi: 10.1038/s41467-017-00482-y
35. Pan Y, Lv J, Pan D, Xu Y, Yang M, Ju H, et al. Evaluating the utility of human glucagon-like peptide 1 receptor gene as a novel radionuclide reporter gene: a promising molecular imaging tool. Appl Microbiol Biotechnol. (2019) 103:1311–24. doi: 10.1007/s00253-018-9562-8
36. Wu Z, Todorov I, Li L, Bading JR, Li Z, Nair I, et al. In vivo imaging of transplanted islets with 64Cu-DO3A-VS-Cys 40 -exendin-4 by targeting GLP-1 receptor. Bioconjug Chem. (2011) 22:1587–94. doi: 10.1021/bc200132t
37. Haralampieva D, Betzel T, Dinulovic I, Salemi S, Stoelting M, Kramer SD, et al. Noninvasive PET imaging and tracking of engineered human muscle precursor cells for skeletal muscle tissue engineering. J Nucl Med. (2016) 57:1467–73. doi: 10.2967/jnumed.115.170548
38. Schonitzer V, Haasters F, Kasbauer S, Ulrich V, Mille E, Gildehaus FJ, et al. In vivo mesenchymal stem cell tracking with PET using the dopamine type 2 receptor and 18F-fallypride. J Nucl Med. (2014) 55:1342–7. doi: 10.2967/jnumed.113.134775
39. Guo R, Li Q, Yang F, Hu X, Jiao J, Guo Y, et al. In vivo MR imaging of dual MRI reporter genes and deltex-1 gene-modified human mesenchymal stem cells in the treatment of closed penile fracture. Mol Imaging Biol. (2018) 20:417–27. doi: 10.1007/s11307-017-1128-0
40. Kim HS, Woo J, Lee JH, Joo HJ, Choi YS, Kim H, et al. In vivo tracking of dendritic cell using MRI reporter gene, ferritin. PLoS ONE. (2015) 10:1–13. doi: 10.1371/journal.pone.0125291
41. He X, Cai J, Liu B, Zhong Y, Qin Y. Cellular magnetic resonance imaging contrast generated by the ferritin heavy chain genetic reporter under the control of a Tet-On switch. Stem Cell Res Ther. (2015) 6:207. doi: 10.1186/s13287-015-0205-z
42. Bartelle BB, Mana MD, Suero-Abreu GA, Rodriguez JJ, Turnbull DH. Engineering an effective Mn-binding MRI reporter protein by subcellular targeting. Magn Reson Med. (2015) 74:1750–7. doi: 10.1002/mrm.25566
43. Lewis CM, Graves SA, Hernandez R, Valdovinos HF, Barnhart TE, Cai W, et al. 52Mn production for PET/MRI tracking of human stem cells expressing divalent metal transporter 1 (DMT1). Theranostics. (2015) 5:227–39. doi: 10.7150/thno.10185
44. Kim JE, Kalimuthu S, Ahn B-C. In vivo cell tracking with bioluminescence imaging. Nucl Med Mol Imaging. (2015) 49:3–10. doi: 10.1007/s13139-014-0309-x
45. Xie P, Hu X, Li D, Xie S, Zhou Z, Meng X, et al. Bioluminescence imaging of transplanted mesenchymal stem cells by overexpression of hepatocyte nuclear factor4α: tracking biodistribution and survival. Mol Imaging Biol. (2019) 21:44–53. doi: 10.1007/s11307-018-1204-0
46. Muñoz MF, Argüelles S, Guzman-Chozas M, Guillén-Sanz R, Franco JM, Pintor-Toro JA, et al. Cell tracking, survival, and differentiation capacity of adipose-derived stem cells after engraftment in rat tissue. J Cell Physiol. (2018) 233:6317–28. doi: 10.1002/jcp.26439
47. Qin C, Cheng K, Chen K, Hu X, Liu Y, Lan X, et al. Tyrosinase as a multifunctional reporter gene for Photoacoustic/MRI/PET triple modality molecular imaging. Sci Rep. (2013) 3:1490. doi: 10.1038/srep01490
48. Sanches PG, Peters S, Rossin R, Kaijzel EL, Que I, Löwik CWGM, et al. Bone metastasis imaging with SPECT/CT/MRI: a preclinical toolbox for therapy studies. Bone. (2015) 75:62–71. doi: 10.1016/j.bone.2015.02.002
49. Kocher B, Piwnica-Worms D. Illuminating cancer systems with genetically engineered mouse models and coupled luciferase reporters in vivo. Cancer Discov. (2013) 3:616–29. doi: 10.1158/2159-8290.CD-12-0503
50. Tomura M, Hata A, Matsuoka S, Shand FHW, Nakanishi Y, Ikebuchi R, et al. Tracking and quantification of dendritic cell migration and antigen trafficking between the skin and lymph nodes. Sci Rep. (2015) 4:6030. doi: 10.1038/srep06030
51. Kang S-W, Lee S, Na JH, Yoon HI, Lee D-E, Koo H, et al. Cell labeling and tracking method without distorted signals by phagocytosis of macrophages. Theranostics. (2014) 4:420–31. doi: 10.7150/thno.7265
52. Lee SY, Lee S, Lee J, Yhee JY, Yoon HI, Park S-J, et al. Non-invasive stem cell tracking in hindlimb ischemia animal model using bio-orthogonal copper-free click chemistry. Biochem Biophys Res Commun. (2016) 479:779–86. doi: 10.1016/j.bbrc.2016.09.132
53. Lee S, Yoon HI, Na JH, Jeon S, Lim S, Koo H, et al. In vivo stem cell tracking with imageable nanoparticles that bind bioorthogonal chemical receptors on the stem cell surface. Biomaterials. (2017) 139:12–29. doi: 10.1016/j.biomaterials.2017.05.050
54. Bailly C, Gouard S, Lacombe M, Saëc P, Chalopin B, Bourgeois M, et al. Comparison of Immuno-PET of CD138 and PET imaging with 64CuCl2 and 18F-FDG in a preclinical syngeneic model of multiple myeloma. Oncotarget. (2018) 9:9061–72. doi: 10.18632/oncotarget.23886
55. McCracken MN, Tavaré R, Witte ON, Wu AM. Advances in PET detection of the antitumor T cell response. Adv Immunol. (2016) 131:187–231. doi: 10.1016/bs.ai.2016.02.004
56. Beckford Vera DR, Smith CC, Bixby LM, Glatt DM, Dunn SS, Saito R, et al. Immuno-PET imaging of tumor-infiltrating lymphocytes using zirconium-89 radiolabeled anti-CD3 antibody in immune-competent mice bearing syngeneic tumors. PLoS ONE. (2018) 13:e0193832. doi: 10.1371/journal.pone.0193832
57. Mayer KE, Mall S, Yusufi N, Gosmann D, Steiger K, Russelli L, et al. T-cell functionality testing is highly relevant to developing novel immuno-tracers monitoring T cells in the context of immunotherapies and revealed CD7 as an attractive target. Theranostics. (2018) 8:6070–87. doi: 10.7150/thno.27275
58. Rashidian M, Ingram JR, Dougan M, Dongre A, Whang KA, LeGall C, et al. Predicting the response to CTLA-4 blockade by longitudinal noninvasive monitoring of CD8 T cells. J Exp Med. (2017) 214:2243–55. doi: 10.1084/jem.20161950
59. Abou DS, Ku T, Smith-Jones PM. In vivo biodistribution and accumulation of 89Zr in mice. HHS public access. Nucl Med Biol. (2011) 3812:675–81. doi: 10.1016/j.nucmedbio.2010.12.011
60. Bailly C, Bodet-Milin C, Rousseau C, Faivre-Chauvet A, Kraeber-Bodéré F, Barbet J. Pretargeting for imaging and therapy in oncological nuclear medicine. EJNMMI Radiopharm Chem. (2017) 2:6. doi: 10.1186/s41181-017-0026-8
61. Honarvar H, Westerlund K, Altai M, Sandström M, Orlova A, Tolmachev V, et al. Feasibility of affibody molecule-based PNA-mediated radionuclide pretargeting of malignant tumors. Theranostics. (2016) 6:93–103. doi: 10.7150/thno.12766
62. Rossin R, Renart Verkerk P, van den Bosch SM, Vulders RCM, Verel I, Lub J, et al. In vivo chemistry for pretargeted tumor imaging in live mice. Angew Chemie Int Ed. (2010) 49:3375–8. doi: 10.1002/anie.200906294
63. Zhang X, Wang B, Zhao N, Tian Z, Dai Y, Nie Y, et al. Improved tumor targeting and longer retention time of NIR fluorescent probes using bioorthogonal chemistry. Theranostics. (2017) 7:20912. doi: 10.7150/thno.20912
64. Chen H, Wang L, Yu Q, Qian W, Tiwari D, Yi H, et al. Anti-HER2 antibody and ScFvEGFR-conjugated antifouling magnetic iron oxide nanoparticles for targeting and magnetic resonance imaging of breast cancer. Int J Nanomed. (2013) 8:3781–94. doi: 10.2147/IJN.S49069
65. Alric C, Hervé-Aubert K, Aubrey N, Melouk S, Lajoie L, Même W, et al. Targeting HER2-breast tumors with scFv-decorated bimodal nanoprobes. J Nanobiotechnol. (2018) 16:18. doi: 10.1186/s12951-018-0341-6
66. Li Y, Soni PB, Liu L, Zhang X, Liotta DC, Lutz S. Synthesis of fluorescent nucleoside analogs as probes for 2′-deoxyribonucleoside kinases. Bioorg Med Chem Lett. (2010) 20:841–3. doi: 10.1016/j.bmcl.2009.12.097
67. Bar-Shir A, Alon L, Korrer MJ, Lim HS, Yadav NN, Kato Y, et al. Quantification and tracking of genetically engineered dendritic cells for studying immunotherapy. Magn Reson Med. (2018) 79:1010–9. doi: 10.1002/mrm.26708
68. van Krieken JH, Oyen WJG, de Vries IJM, Verdijk P, Figdor CG, Barentsz JO, et al. Magnetic resonance tracking of dendritic cells in melanoma patients for monitoring of cellular therapy. Nat Biotechnol. (2005) 23:1407–13. doi: 10.1038/nbt1154
69. Noh YW, Jang YS, Ahn KJ, Lim YT, Chung BH. Simultaneous in vivo tracking of dendritic cells and priming of an antigen-specific immune response. Biomaterials. (2011) 32:6254–63. doi: 10.1016/j.biomaterials.2011.05.013
70. Ahn B-C, Jeong SY, Lee J, Jeong H-J, Ahn SB, Lee SB, et al. Radionuclide-embedded gold nanoparticles for enhanced dendritic cell-based cancer immunotherapy, sensitive and quantitative tracking of dendritic cells with PET and Cerenkov luminescence. NPG Asia Mater. (2016) 8:e281. doi: 10.1038/am.2016.80
71. Sharif-Paghaleh E, Ratnasothy K, Sunassee K, Tavaré R, Alhabbab R, Mullen GE, et al. In vivo SPECT reporter gene imaging of regulatory T cells. PLoS ONE. (2011) 6:e25857. doi: 10.1371/journal.pone.0025857
72. Li YQ, Tang Y, Fu R, Meng QH, Zhou X, Ling ZM, et al. Efficient labeling in vitro with non-ionic gadolinium magnetic resonance imaging contrast agent and fluorescent transfection agent in bone marrow stromal cells of neonatal rats. Mol Med Rep. (2015) 12:913–20. doi: 10.3892/mmr.2015.3532
73. Conniot J, Gaspar R, Silva LC, Florindo HF, Silva JM, Fernandes JG, et al. Cancer immunotherapy: nanodelivery approaches for immune cell targeting and tracking. Front Chem. (2014) 2:1–27. doi: 10.3389/fchem.2014.00105
74. Bulte JWM, Daldrup-Link HE. Clinical tracking of cell transfer and cell transplantation: trials and tribulations. Radiology. (2018) 289:604–15. doi: 10.1148/radiol.2018180449
75. Man F, Lim L, Volpe A, Gabizon A, Shmeeda H, Draper B, et al. In vivo PET tracking of 89Zr-labeled Vγ9Vδ2 T cells to mouse xenograft breast tumors activated with liposomal alendronate. Mol Ther. (2019) 27:219–29. doi: 10.1016/j.ymthe.2018.10.006
76. Lee SB, Lee HW, Singh TD, Li Y, Kim SK, Cho SJ, et al. Visualization of macrophage recruitment to inflammation lesions using highly sensitive and stable radionuclide-embedded gold nanoparticles as a nuclear bio-imaging platform. Theranostics. (2017) 7:926–34. doi: 10.7150/thno.17131
77. Signore A, Annovazzi A, Barone R, Bonanno E, D'Alessandria C, Chianelli M, et al. 99mTc-interleukin-2 scintigraphy as a potential tool for evaluating tumor-infiltrating lymphocytes in melanoma lesions: a validation study. J Nucl Med. (2004) 45:1647–52.
78. Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. (2015) 14:847–56. doi: 10.1158/1535-7163.MCT-14-0983
79. Natarajan A, Mayer AT, Xu L, Reeves RE, Gano J, Gambhir SS. Novel radiotracer for immunoPET imaging of PD-1 checkpoint expression on tumor infiltrating lymphocytes. Bioconjug Chem. (2015) 26:2062–9. doi: 10.1021/acs.bioconjchem.5b00318
80. Lanfranca MP, Lazarus J, Shao X, Nathan H, Di Magliano MP, Zou W, et al. Tracking macrophage infiltration in a mouse model of pancreatic cancer with the positron emission tomography tracer [11C]PBR28. J Surg Res. (2018) 232:570–7. doi: 10.1016/j.jss.2018.07.015
81. Al Faraj A, Sultana Shaik A, Pureza MA, Alnafea M, Halwani R. Preferential macrophage recruitment and polarization in LPS-induced animal model for COPD: noninvasive tracking using MRI. PLoS ONE. (2014) 9:e90829. doi: 10.1371/journal.pone.0090829
82. Valable S, Barbier EL, Bernaudin M, Roussel S, Segebarth C, Petit E, et al. In vivo MRI tracking of exogenous monocytes/macrophages targeting brain tumors in a rat model of glioma. Neuroimage. (2008) 40:973–83. doi: 10.1016/j.neuroimage.2008.01.005
83. Perez C, Jukica A, Listopad JJ, Anders K, Kühl AA, Loddenkemper C, et al. Permissive expansion and homing of adoptively transferred T cells in tumor-bearing hosts. Int J Cancer. (2015) 137:359–71. doi: 10.1002/ijc.29401
84. He H, Kanada M, Contag CH, Krishnan V, Schaar BT, Dorigo O, et al. Imaging of tumor-associated macrophages in a transgenic mouse model of orthotopic ovarian cancer. Mol Imaging Biol. (2017) 19:694–702. doi: 10.1007/s11307-017-1061-2
85. Yang R, Sarkar S, Yong VW, Dunn JF. In vivo MR imaging of tumor-associated macrophages: the next frontier in cancer imaging. Magn Reson Insights. (2018) 11:1178623X1877197. doi: 10.1177/1178623X18771974
86. Wahl RL, Jacene H, Kasamon Y, Lodge MA. From RECIST to PERCIST: evolving Considerations for PET response criteria in solid tumors. J Nucl Med. (2009) 50(Suppl. 1):122S−50S. doi: 10.2967/jnumed.108.057307
87. Cammilleri S, Labeille B, Kelly A, Isnardi V, D'Incan M, Tychyj C, et al. 123I-BZA2 as a melanin-targeted radiotracer for the identification of melanoma metastases: results and perspectives of a multicenter phase III clinical trial. J Nucl Med. (2013) 55:15–22. doi: 10.2967/jnumed.113.123554
88. Ren G, Liu Z, Miao Z, Liu H, Subbarayan M, Chin FT, et al. PET of malignant melanoma using 18F-labeled metallopeptides. J Nucl Med. (2009) 50:1865–72. doi: 10.2967/jnumed.109.062877
89. Lee ST, Burvenich I, Scott AM. Novel target selection for nuclear medicine studies. Semin Nucl Med. (2019) 49:357–68. doi: 10.1053/j.semnuclmed.2019.06.004
90. Verhoeff SR, van den Heuvel MM, van Herpen CML, Piet B, Aarntzen EHJG, Heskamp S. Programmed cell death-1/ligand-1 PET imaging. PET Clin. (2019) 15:35–43. doi: 10.1016/j.cpet.2019.08.008
91. Hinrichs CS, Turcotte S, Wunderlich JR, Dudley ME, Hogan K, Gros A, et al. Phenotype and function of T cells infiltrating visceral metastases from gastrointestinal cancers and melanoma: implications for adoptive cell transfer therapy. J Immunol. (2013) 191:2217–25. doi: 10.4049/jimmunol.1300538
92. Seo JW, Tavaré R, Mahakian LM, Silvestrini MT, Tam S, Ingham ES, et al. CD8+ T-cell density imaging with 64Cu-labeled cys-diabody informs immunotherapy protocols. Clin Cancer Res. (2018) 24:4976–87. doi: 10.1158/1078-0432.CCR-18-0261
93. Markovic S, Galli F, Suman VJ, Nevala WK, Paulsen AM, Hung JC, et al. Non-invasive clinical visualization of tumor infiltrating lymphocytes in patients with metastatic melanoma undergoing immune checkpoint inhibitor therapy: a pilot study. J Clin Oncol. (2017) 35:3034. doi: 10.1200/JCO.2017.35.15_suppl.3034
94. Narang S, Kim D, Aithala S, Heimberger AB, Ahmed S, Rao D, et al. Tumor image-derived texture features are associated with CD3 T-cell infiltration status in glioblastoma. Oncotarget. (2017) 8:101244–54. doi: 10.18632/oncotarget.20643
95. Sun R, Limkin EJ, Vakalopoulou M, Dercle L, Champiat S, Han SR, et al. A radiomics approach to assess tumour-infiltrating CD8 cells and response to anti-PD-1 or anti-PD-L1 immunotherapy: an imaging biomarker, retrospective multicohort study. Lancet Oncol. (2018) 19:1180–91. doi: 10.1016/S1470-2045(18)30413-3
96. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. (2017) 14:399–416. doi: 10.1038/nrclinonc.2016.217
97. Eisenblatter M, Ehrchen J, Varga G, Sunderkotter C, Heindel W, Roth J, et al. In vivo optical imaging of cellular inflammatory response in granuloma formation using fluorescence-labeled macrophages. J Nucl Med. (2009) 50:1676–82. doi: 10.2967/jnumed.108.060707
98. Terry SYA, Nayak TK, Oyen WJ, Klein C, Laverman P, Koenders MI, et al. Monitoring therapy response of experimental arthritis with radiolabeled tracers targeting fibroblasts, macrophages, or integrin v 3. J Nucl Med. (2015) 57:467–72. doi: 10.2967/jnumed.115.162628
99. Nahrendorf M, Zhang H, Hembrador S, Panizzi P, Sosnovik DE, Aikawa E, et al. Nanoparticle PET-CT imaging of macrophages in inflammatory atherosclerosis. Circulation. (2008) 117:379–87. doi: 10.1161/CIRCULATIONAHA.107.741181
100. Terry SYA, Boerman OC, Gerrits D, Franssen GM, Metselaar JM, Lehmann S, et al. 111In-anti-F4/80-A3-1 antibody: a novel tracer to image macrophages. Eur J Nucl Med Mol Imaging. (2015) 42:1430–8. doi: 10.1007/s00259-015-3084-8
101. Yang R, Sarkar S, Korchinski DJ, Wu Y, Yong VW, Dunn JF. MRI monitoring of monocytes to detect immune stimulating treatment response in brain tumor. Neuro Oncol. (2016) 19:now180. doi: 10.1093/neuonc/now180
102. Aarntzen EHJG, Srinivas M, Radu CG, Punt CJA, Boerman OC, Figdor CG, et al. In vivo imaging of therapy-induced anti-cancer immune responses in humans. Cell Mol Life Sci. (2013) 70:2237–57. doi: 10.1007/s00018-012-1159-2
103. Radu CG, Shu CJ, Nair-Gill E, Shelly SM, Barrio JR, Satyamurthy N, et al. Molecular imaging of lymphoid organs and immune activation by positron emission tomography with a new [18F]-labeled 2'-deoxycytidine analog. Nat Med. (2008) 14:783–8. doi: 10.1038/nm1724
104. Chen R, Zhou X, Liu J, Huang G. Relationship between the expression of PD-1/PD-L1 and 18F-FDG uptake in bladder cancer. Eur J Nucl Med Mol Imaging. (2019) 11:12270–7. doi: 10.1007/s00259-018-4208-8
105. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. (2012) 12:252–64. doi: 10.1038/nrc3239
106. Katz SC, Pillarisetty V, Bamboat ZM, Shia J, Hedvat C, Gonen M, et al. T cell infiltrate predicts long-term survival following resection of colorectal cancer liver metastases. Ann Surg Oncol. (2009) 16:2524–30. doi: 10.1245/s10434-009-0585-3
107. Ehlerding EB, England CG, Majewski RL, Valdovinos HF, Jiang D, Liu G, et al. ImmunoPET imaging of CTLA-4 expression in mouse models of non-small cell lung cancer. Mol Pharm. (2017) 14:1782–9. doi: 10.1021/acs.molpharmaceut.7b00056
108. Buder-Bakhaya K, Hassel JC. Biomarkers for clinical benefit of immune checkpoint inhibitor treatment-a review from the melanoma perspective and beyond. Front Immunol. (2018) 9:1474. doi: 10.3389/fimmu.2018.01474
109. Kikuchi M, Clump DA, Srivastava RM, Sun L, Zeng D, Diaz-Perez JA, et al. Preclinical immunoPET/CT imaging using Zr-89-labeled anti-PD-L1 monoclonal antibody for assessing radiation-induced PD-L1 upregulation in head and neck cancer and melanoma. Oncoimmunology. (2017) 6:1–13. doi: 10.1080/2162402X.2017.1329071
110. Broos K, Lecocq Q, Raes G, Devoogdt N, Keyaerts M, Breckpot K. Noninvasive imaging of the PD-1: PD-L1 immune checkpoint: embracing nuclear medicine for the benefit of personalized immunotherapy. Theranostics. (2018) 8:3559–70. doi: 10.7150/thno.24762
111. Faure M, Rochigneux P, Olive D, Taix S, Brenot-Rossi I, Gilabert M. Hyperprogressive disease in anorectal melanoma treated by PD-1 inhibitors. Front Immunol. (2018) 9:797. doi: 10.3389/fimmu.2018.00797
112. Bensch F, van der Veen EL, Lub-de Hooge MN, Jorritsma-Smit A, Boellaard R, Kok IC, et al. 89Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat Med. (2018) 24:1852–8. doi: 10.1038/s41591-018-0255-8
113. Gibson HM, McKnight BN, Malysa A, Dyson G, Wiesend WN, McCarthy CE, et al. IFNg PET imaging as a predictive tool for monitoring response to tumor immunotherapy. Cancer Res. (2018) 78:5706–17. doi: 10.1158/0008-5472.CAN-18-0253
114. Kircher M, Herhaus P, Schottelius M, Buck AK, Werner RA, Wester H-J, et al. CXCR4-directed theranostics in oncology and inflammation. Ann Nucl Med. (2018) 32:503–11. doi: 10.1007/s12149-018-1290-8
115. Streeter JE, Gessner RC, Tsuruta J, Feingold S, Dayton PA. Assessment of molecular imaging of angiogenesis with three-dimensional ultrasonography. Mol Imaging. (2011) 10:460–8. doi: 10.2310/7290.2011.00015
116. Sui WF, Chen X, Peng ZK, Ye J, Wu JT. The diagnosis of metastatic axillary lymph nodes of breast cancer by diffusion weighted imaging: a meta-analysis and systematic review. World J Surg Oncol. (2016) 14:155. doi: 10.1186/s12957-016-0906-5
117. Griessinger CM, Kehlbach R, Bukala D, Wiehr S, Bantleon R, Cay F, et al. In vivo tracking of Th1 cells by PET reveals quantitative and temporal distribution and specific homing in lymphatic tissue. J Nucl Med. (2014) 55:301–7. doi: 10.2967/jnumed.113.126318
118. Sancey L, Ardisson V, Riou LM, Ahmadi M, Marti-Batlle D, Boturyn D, et al. In vivo imaging of tumour angiogenesis in mice with the αvβ3 integrin-targeted tracer 99mTc-RAFT-RGD. Eur J Nucl Med Mol Imaging. (2007) 34:2037–47. doi: 10.1007/s00259-007-0497-z
119. Jin Z-H, Furukawa T, Claron M, Boturyn D, Coll J-L, Fukumura T, et al. Positron emission tomography imaging of tumor angiogenesis and monitoring of antiangiogenic efficacy using the novel tetrameric peptide probe 64Cu-cyclam-RAFT-c(-RGDfK-)4. Angiogenesis. (2012) 15:569–80. doi: 10.1007/s10456-012-9281-1
120. Galbán CJ, Hoff BA, Chenevert TL, Ross BD. Diffusion MRI in early cancer therapeutic response assessment. NMR Biomed. (2017) 30:e3458. doi: 10.1002/nbm.3458
121. Pham W, Xie J, Gore JC. Tracking the migration of dendritic cells by in vivo optical imaging. Neoplasia. (2007) 9:1130–7. doi: 10.1593/neo.07586
122. Muccioli M, Pate M, Omosebi O, Benencia F. Generation and labeling of murine bone marrow-derived dendritic cells with Qdot nanocrystals for tracking studies. J Vis Exp. (2011) e2785. doi: 10.3791/2785
123. Long CM, van Laarhoven HWM, Bulte JWM, Levitsky HI. Magnetovaccination as a novel method to assess and quantify dendritic cell tumor antigen capture and delivery to lymph nodes. Cancer Res. (2009) 69:3180–7. doi: 10.1158/0008-5472.CAN-08-3691
124. Asiedu KO, Koyasu S, Szajek LP, Choyke PL, Sato N. Bone marrow cell trafficking analyzed by 89 Zr-oxine positron emission tomography in a murine transplantation model. Clin Cancer Res. (2017) 23:2759–68. doi: 10.1158/1078-0432.CCR-16-1561
125. Asiedu KO, Ferdousi M, Ton PT, Adler SS, Choyke PL, Sato N. Bone marrow cell homing to sites of acute tibial fracture: 89Zr-oxine cell labeling with positron emission tomographic imaging in a mouse model. EJNMMI Res. (2018) 8:109. doi: 10.1186/s13550-018-0463-8
126. Kanwar B, Gao DW, Hwang AB, Grenert JP, Williams SP, Franc B, et al. In vivo imaging of mucosal CD4+ T cells using single photon emission computed tomography in a murine model of colitis. J Immunol Methods. (2008) 329:21–30. doi: 10.1016/j.jim.2007.09.008
127. Srinivas M, Turner MS, Janjic JM, Morel PA, Laidlaw DH, Ahrens ET. In vivo cytometry of antigen-specific T cells using 19F MRI. Magn Reson Med. (2009) 62:747–53. doi: 10.1002/mrm.22063
128. Tremblay M-L, Davis C, Bowen CV, Stanley O, Parsons C, Weir G, et al. Using MRI cell tracking to monitor immune cell recruitment in response to a peptide-based cancer vaccine. Magn Reson Med. (2018) 80:304–16. doi: 10.1002/mrm.27018
129. Srinivas M, Aarntzen EHJG, Bulte JWM, Oyen WJ, Heerschap A, de Vries IJM, et al. Imaging of cellular therapies. Adv Drug Deliv Rev. (2010) 62:1080–93. doi: 10.1016/j.addr.2010.08.009
130. Verdijk P, Aarntzen EHJG, Lesterhuis WJ, Boullart ACI, Kok E, van Rossum MM, et al. Limited amounts of dendritic cells migrate into the T-cell area of lymph nodes but have high immune activating potential in melanoma patients. Clin Cancer Res. (2009) 15:2531–40. doi: 10.1158/1078-0432.CCR-08-2729
131. Laverman P, de Vries IJM, Scharenborg NM, de Boer A, Broekema M, Oyen WJG, et al. Development of 111In-labeled tumor-associated antigen peptides for monitoring dendritic-cell-based vaccination. Nucl Med Biol. (2006) 33:453–8. doi: 10.1016/j.nucmedbio.2006.02.005
132. Emami-Shahri N, Papa S. Dynamic imaging for CAR-T-cell therapy. Biochem Soc Trans. (2016) 44:386–90. doi: 10.1042/BST20150257
133. Bhatnagar P, Alauddin M, Bankson JA, Kirui D, Seifi P, Huls H, et al. Tumor lysing genetically engineered T cells loaded with multi-modal imaging agents. Sci Rep. (2014) 4:4502. doi: 10.1038/srep04502
134. Chapelin F, Gao S, Okada H, Weber TG, Messer K, Ahrens ET. Fluorine-19 nuclear magnetic resonance of chimeric antigen receptor T cell biodistribution in murine cancer model. Sci Rep. (2017) 7:17748. doi: 10.1038/s41598-017-17669-4
135. Mall S, Yusufi N, Wagner R, Klar R, Bianchi H, Steiger K, et al. Immuno-PET imaging of engineered human T cells in tumors. Cancer Res. (2016) 76:4113–23. doi: 10.1158/0008-5472.CAN-15-2784
136. Wang S, O'Rourke DM, Chawla S, Verma G, Nasrallah MP, Morrissette JJD, et al. Multiparametric magnetic resonance imaging in the assessment of anti-EGFRvIII chimeric antigen receptor T cell therapy in patients with recurrent glioblastoma. Br J Cancer. (2019) 120:54–6. doi: 10.1038/s41416-018-0342-0
137. Uong TNT, Lee KH, Ahn SJ, Kim KW, Min JJ, Hyun H, et al. Real-time tracking of ex vivo-expanded natural killer cells toward human triple-negative breast cancers. Front Immunol. (2018) 9:1–14. doi: 10.3389/fimmu.2018.00825
138. Garikipati VNS, Jadhav S, Pal L, Prakash P, Dikshit M, Nityanand S. Mesenchymal stem cells from fetal heart attenuate myocardial injury after infarction: an in vivo serial pinhole gated SPECT-CT study in rats. PLoS ONE. (2014) 9:1–10. doi: 10.1371/journal.pone.0100982
139. Schubert R, Sann J, Frueh JT, Ullrich E, Geiger H, Baer PC. Tracking of adipose-derived mesenchymal stromal/stem cells in a model of cisplatin-induced acute kidney injury: comparison of bioluminescence imaging vs. qRT-PCR. Int J Mol Sci. (2018) 19:1–12. doi: 10.3390/ijms19092564
140. Kalimuthu S, Zhu L, Oh JM, Gangadaran P, Lee HW, Baek SH, et al. Migration of mesenchymal stem cells to tumor xenograft models and in vitro drug delivery by doxorubicin. Int J Med Sci. (2018) 15:1051–61. doi: 10.7150/ijms.25760
141. Scarfe L, Taylor A, Sharkey J, Harwood R, Barrow M, Comenge J, et al. Non-invasive imaging reveals conditions that impact distribution and persistence of cells after in vivo administration. bioRxiv [Preprint]. (2018). doi: 10.1101/202101
Keywords: cell tracking, immunotherapy, PET, SPECT, MRI, adoptive transfer, tumor microenvironment, cancer
Citation: Perrin J, Capitao M, Mougin-Degraef M, Guérard F, Faivre-Chauvet A, Rbah-Vidal L, Gaschet J, Guilloux Y, Kraeber-Bodéré F, Chérel M and Barbet J (2020) Cell Tracking in Cancer Immunotherapy. Front. Med. 7:34. doi: 10.3389/fmed.2020.00034
Received: 04 March 2019; Accepted: 23 January 2020;
Published: 14 February 2020.
Edited by:
Anil Kumar Mishra, Institute of Nuclear Medicine & Allied Sciences (DRDO), IndiaReviewed by:
Baljinder Singh, Post Graduate Institute of Medical Education and Research (PGIMER), IndiaPuja Panwar Hazari, Institute of Nuclear Medicine & Allied Sciences (DRDO), India
Copyright © 2020 Perrin, Capitao, Mougin-Degraef, Guérard, Faivre-Chauvet, Rbah-Vidal, Gaschet, Guilloux, Kraeber-Bodéré, Chérel and Barbet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jacques Barbet, YmFyYmV0QGFycm9uYXgtbmFudGVzLmZy
†These authors have contributed equally to this work