 Aphrodite Kapurniotu
Aphrodite Kapurniotu Ozgun Gokce
Ozgun Gokce Jürgen Bernhagen
Jürgen Bernhagen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 23 January 2019
Sec. Translational Medicine
Volume 6 - 2019 | https://doi.org/10.3389/fmed.2019.00003
This article is part of the Research Topic Multitasking Biomolecules in Human Pathologies: Known Players on Their Unexpected Journeys View all 6 articles
When the human genome was sequenced, it came as a surprise that it contains “only” 21,306 protein-coding genes. However, complexity and diversity are multiplied by alternative splicing, non-protein-coding transcripts, or post-translational modifications (PTMs) on proteome level. Here, we discuss how the multi-tasking potential of proteins can substantially enhance the complexity of the proteome further, while at the same time offering mechanisms for the fine-regulation of cell responses. Discoveries over the past two decades have led to the identification of “surprising” and previously unrecognized functionalities of long known cytokines, inflammatory mediators, and intracellular proteins that have established novel molecular networks in physiology, inflammation, and cardiovascular disease. In this mini-review, we focus on alarmins and atypical chemokines such as high-mobility group box protein-1 (HMGB-1) and macrophage migration-inhibitory factor (MIF)-type proteins that are prototypical examples of these classes, featuring a remarkable multitasking potential that allows for an elaborate fine-tuning of molecular networks in the extra- and intracellular space that may eventually give rise to novel “task”-based precision medicine intervention strategies.
The human genome project was concluded in the beginning of this millennium (1). It came as a surprise that the genome only contains 21,306 protein-coding genes; this appeared to contradict the remarkable complexity of higher organisms. However, it turned out that complexity and diversity are substantially enhanced by other mechanisms such as alternative splicing, non-protein-coding transcripts, or post-translational modifications (PTMs) on proteome level. For example, protein translation and mRNA stability are regulated and fine-tuned by thousands of non-coding RNAs including microRNAs, small interfering RNAs (siRNAs), and long non-coding RNAs (lncRNAs). The complexity in the available battery of gene products in humans is further multiplied by alternative promoter sites and transcript splicing. Moreover, the size of the proteome is much larger than the predicted 21,306 proteins. It is amplified by a factor of >50-fold by a multitude of PTMs that determine the activity, interaction specificity, and sub-cellular localization of many cellular proteins (Table 1). Not all of these diversity-enhancing mechanisms operate at the same time and on the same protein class, but they certainly lead to a significant expansion of the proteome. While it is well-known that some PTMs alter the interaction profile of a protein and switch-on or switch-off certain protein-protein or protein-substrate binding events, interactions that are subject to PTM-modulated changes are typically confined to binding partners of the same or similar functional class. A prototypical example would be the regulation of the activity of the enzyme phosphofructokinase-2 (PFK-2), which, following post-translational phosphorylation, is converted from a kinase to a phosphatase, consistent with the inversion of substrate specificity from a mono-phosphorylated to a bis-phosphorylated sugar (i.e., the binding partner).
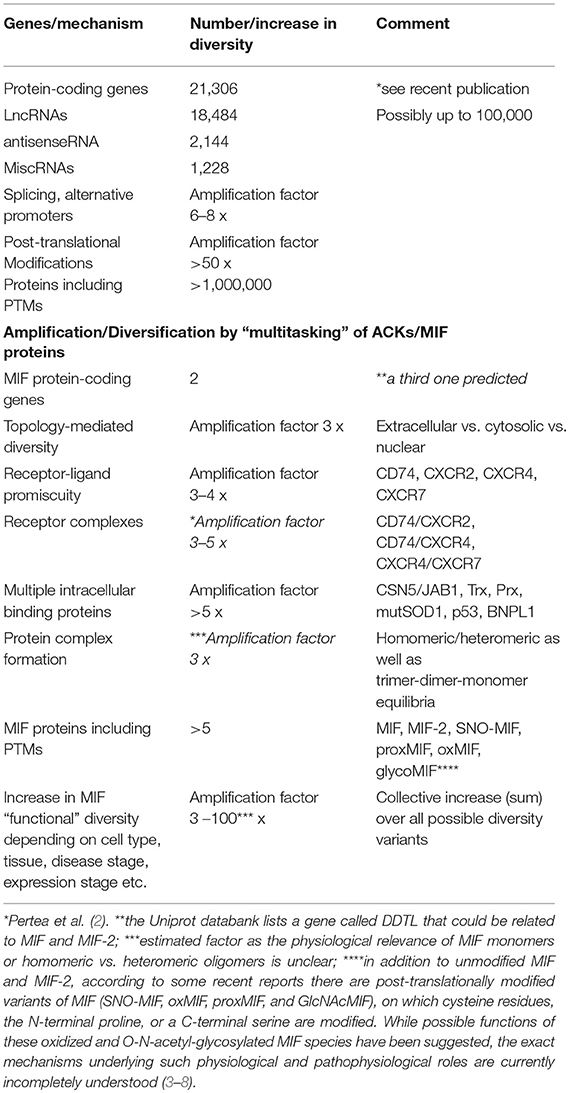
Table 1. The diversity in the human proteome is enlarged by multitasking proteins: atypical chemokines and MIF family proteins as role models.
Research in the last two decades has led to the discovery of numerous molecules that engage in unanticipated—second—activities, thus suggesting a novel concept of “molecular multitasking” or “molecular moonlighting.” The concept holds that certain biomolecules may execute functions in previously unexpected -different- locations, that they may utilize previously unrecognized -different- substrates, or that they may engage in previously unexpected -different- binding interactions, thus previously unexpected functions that often are seemingly unrelated to their established “main” function. The “molecular multitasking” or “molecular moonlighting” concept thus goes beyond the “one molecule, one function” paradigm. With respect to proteins it goes beyond the “one molecule, one-fold, one function” paradigm. The concept is appropriately described by the “moonlighting” definition which refers to the practice of holding a “second regular job” in addition to: one's main job” (https://www.collinsdictionary.com/de/worterbuch/englisch/moonlighting).
Alarmins and certain cytokines and chemokines are protagonistic examples of biomolecules executing “molecular multitasking/moonlighting.” In fact, work in the past two decades has led to the identification of surprising and previously unrecognized functionalities of long known cytokines, inflammatory mediators, and intracellular proteins, unraveling novel molecular networks in physiology, inflammation, and cardiovascular disease. To this end, alarmin research has established the idea that homeostatic intracellular proteins can have additional, non-homeostatic, extracellular functions that signal danger, and promote inflammation. While the alarmin category of molecules not only encompasses proteins, but also small molecule metabolites such as ATP or nucleic acids such as RNA, in this article, we will discuss only protein-type alarmins and atypical chemokines/cytokines with an emphasis on high-mobility group box-1 (HMGB-1) and macrophage migration-inhibitory factor (MIF)-type proteins. We will discuss their remarkable multitasking potential that enhances the diversity of the proteome and allows for a fine-tuning of molecular networks in the extra- and intracellular space in health and disease.
Alarmins are molecules released from a damaged or diseased cell that upon release can stimulate a sterile immune or inflammatory response (9–12). Examples are heat-shock proteins such as HSP-70 or−90, intracellular cytokines such as interleukin-1 α (IL-1α) or IL-33, amino-acyl tRNA synthetase fragments or the p43 auxiliary component of the tRNA multisynthetase complex, chromatin proteins such HMGB1 or histones, whole nucleosomes, phagocyte inflammatory proteins such as the S100 proteins S100A8, S100A9, or S100A12, but also small molecule metabolites such as ATP or uric acid crystals. Alarmins are sometimes referred to as “endokines” to indicate that “endo”genous intracellular molecules acquire cyto'kine'-like functions once release into the extracellular space. There is a significant overlap between alarmins and the molecular class of danger-associated molecular patterns (DAMPs) or danger signals. The DAMP terminology was coined by symmetry to the concept of pathogen-associated molecular patterns (PAMPs), conserved pathogen-derived molecular motifs that are recognized by host pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs) (the “stranger theory”). Accordingly, endogenous “molecular patterns” that signal cell or tissue damage and the need for the development of a sterile host inflammatory response were coined as DAMPs (the “danger theory”) (13). In fact, it has been suggested that alarmins and DAMPs are two terms for the same class of molecules and concept. We will preferentially use the term alarmin in this review article.
There are numerous excellent recent reviews on alarmins/DAMPs (9, 10, 13–18). Briefly, alarmins have a physiological, often homeostatic, role inside the cell, but take on additional, novel, functions when they are exposed to the extracellular environment. They signal “danger” to the host and trigger a local inflammatory response, which in physiologic healing leads over to tissue regeneration, but when dysregulated can cause pathologic inflammation and related disorders. Following tissue injury or cell stress, most alarmins are passively released from dead cells, but the release of some alarmins can also follow a controlled secretion program to signal early, sub-lethal cell stress. For protein-type alarmins, this regulated secretory process follows a so-called “unconventional” endoplasmic reticulum (ER)/Golgi-independent pathway (19).
Here, we focus on the “multitasking” concept as it applies to proteinaceous alarmins and their functional and molecular connection to atypical chemokines (ACKs) (for latter, see next chapter). This principle is best characterized by HMGB1, a typical “multitasking” alarmin and the first identified member of the HMGB family. HMGB1 is a ubiquitous nuclear DNA-binding protein that is highly conserved, present in most cell types, and that serves as nuclear/transcriptional cofactor (20). In addition to its intracellular nuclear function, HMGB1 can be released into the extracellular milieu following cell damage or via regulated secretion via an autophagolysosomal pathway (21). Extracellular HMGB1 fulfills a second independent function as an intercellular-acting cytokine and inflammatory mediator (10, 14, 17, 22–29). In analogy to the “multitasking”/“moonlighting” principle, transcriptional regulation by nuclear HMGB1 represents the physiological “day-time job” (job 1) inside the cell, while its “cytokine” activity outside the cell would be the additional “night-time” or “moonlighting” job (job 2) that HMGB1 takes on to signal cell stress or damage and initiate a rapid local inflammatory response (10, 14, 17) (Figure 1). Weighing the terms “multitasking” vs. “moonlighting,” the latter appears preferable as the definition of “multitasking” asks for the handling of multiple jobs at the same time. However, the “day-time” and “night-time” jobs of HMGB1 (and other “multitaskers”) are clearly separated in time and localization. Transcriptional coregulation by HMGB1 is restricted to the intracellular–nuclear-compartment and healthy homeostatic cell conditions, whereas the inflammation-promoting cytokine function of HMGB1 only occurs outside the cell. Inversely, nuclear HMGB1 is invisible to the host immune and tissue inflammatory system. The two HMGB1 “jobs” are thus distinctly separated in space and phase (Figure 1).
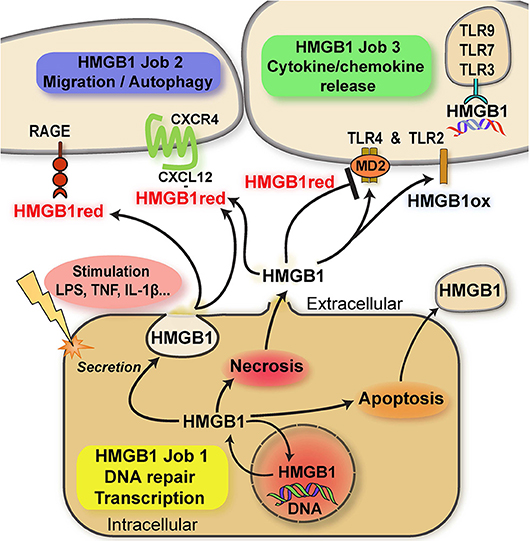
Figure 1. Scheme summarizing the multitasking/moonlighting functions of HMGB1. For details see manuscript text. HMGB1, high mobility group box-1; HMGB1red, fully reduced HMGB1; HMGB1ox, partially or fully oxidized HMGB1; TLR, Toll-like receptor; RAGE, receptor of advanced glycation endproducts (AGEs); MD2, myeloid differentiation factor 2/lymphocyte antigen 96.
HMGB1 contains three conserved cysteines. These residues are redox-sensitive and their differential modification modulates the bioactivity of HMGB1 in the extracellular space via determining the binding specificity to three different receptors due to the induction of specific three-dimensional conformations. Disulfide formation between the C23 and C45 cysteines of HMGB1 and preservation of C106 in its reduced thiol form (“disulfide HMGB1”) enables for binding and signaling through the TLR4/MD-2 receptor axis. This activity leads to the induction of inflammatory cytokines in macrophages and may be termed the “cytokine task” of extracellular HMGB1 (22, 23, 30–32) (Figure 1). In contrast, the all-thiol conformer of HMGB1 may behave as a co-chemokine through heterocomplex formation with the classical CXC chemokine CXCL12. HMGB1/CXCL12 complexes bind to CXCR4 to promote chemotactic cell migration of monocytes/macrophages and T cells (33) (the “chemokine task”) (Figure 1). Alternatively, all-thiol HMGB1 binds to receptor of advanced glycation endproducts (RAGE) to trigger cell migration or autophagy (the “autophagy task”; Figure 1). Interestingly, RAGE and TLR4 function as promiscuous multitaskers themselves, interacting with other DAMPs such as the S100 proteins S100A12 (34) and S100A8/A9, respectively (35), ligands such the Aβ amyloid polypeptide and heat shock proteins (HSPs) or PAMPs.
The post-translational redox modifications of HMGB1 with the exception of the sulfonyl-modification are reversible, enabling HMGB1 to switch from its “cytokine task” to its “chemokine task” or vice versa. Terminally oxidized sulfonyl-HMGB1 fully lacks inflammatory activity. Distribution of HMGB1 between its “day-time job” (nucleus) and its “night-time job” (extracellular milieu) is further regulated by acetylation of lysine residues within its nuclear localization sequence (Figure 1). Acetylation favors the cytosolic fraction over nuclear HMGB1 and thus is a prerequisite to redirect HMGB1 into the extracellular space (14, 17, 24, 36). A similar role for alarmin hyper-acetylation has also been shown for the nuclear Y-Box protein-1 (YB-1), which is secreted by non-conventional pathways following acetylation (37).
The extracellular cytokine/chemokine-like activities of HMGB1 thus represent true multitasking in the “second” job of this alarmin/DAMP protein (Figure 1).
At first glance, alarmins and chemokines, while both playing important roles in host defense and inflammation, are fundamentally different classes of mediators. As outlined above, alarmins are bona fide intracellular effectors that upon abundant rapid release alert the environment about cell stress and danger. In apparent contrast, chemokines (and cytokines in general) are bona fide extracellular mediators that typically have no role within the cell. As discussed above, the IL-1-type cytokines IL-33 and IL-37 that have intrinsic nuclear activities are exceptions to this rule. While classical chemokines of the homeostatic sub-class such as CXCL11 or CXCL12 are stored intracellularly under resting conditions, they do not appear to fulfill intracellular functions, except for “awaiting” their secretion. Moreover, classical chemokines of the inflammatory sub-class, with some exceptions, are not intracellularly measurable at rest; their production is tightly regulated. Transcription and translation are induced by inflammatory or stress stimulation only, which is when levels rise from essentially “zero” by several hundred- or thousand-fold. Induction also is typically directly coupled with the secretion of inflammatory chemokines into the extracellular space, where they drive leukocyte migration and/or promote inflammation through binding to their cognate chemokine receptors. When secreted from inflammatory endothelium, chemokines such as CXCL1 are deposited on the endothelial surface to form an haptotactic gradient and function as arrest chemokines (38, 39). However, beyond regulation at induction level, some inflammatory chemokines are pre-stored following translation. For example, CCL2 is stored under the endothelial surface as intraendothelial chemokine to guide lymphocytes across an inflamed endothelial barrier, circumventing the need for surface-deposited chemokines or extraendothelial chemokine gradients (40). Moreover, some chemokines are stored as proforms. This applies to the platelet chemokines connective tissue-activating protein III (CTAP-III/NAP-2/CXCL7) and platelet factor 4 (PF4/CXCL4), which are pre-stored in platelet granules (41). Furthermore, CX3CL1 and CXCL16 are translocated to the plasma membrane, where they are “stored” as transmembrane proforms that are “activated” by proteolytic processing. In this case, proteolysis represents an important regulated “induction” step (42). Moreover, fine-tuning of several other chemokines has been described at the post-translational level, e.g., by N-terminal processing (43–45).
Alarmin receptors are as diverse structurally as alarmins themselves, spanning classes such as scavenger receptors and PRRs, ligand-gated channels, single-spanning helix-type transmembrane proteins, or chemokine receptors as a sub-group of G protein-coupled receptors (GPCRs) [for detailed overview see 2, 7, 11, 39, 40]. In contrast, classical chemokines (CKs) are 8–10 kD small proteins that are uniformly defined by an N-terminal cysteine motif and a characteristic β-strand-rich structural core, featuring the so-called chemokine-fold. Forty-nine classical chemokines interact with 18 GPCR-type classical chemokine receptors (CKRs) as well as five atypical chemokine receptors (ACKRs). The chemokine network is characterized by a high degree of promiscuity with numerous chemokines binding to several receptors and certain receptors engaging more than one chemokine ligand. Classical chemokines are divided into CC-, CXC-, CX3C-, and C-type sub-classes owing to the positioning of one or two vicinal cysteines at the N-terminal. The receptors are termed correspondingly (46–48). Chemokines form monomers or dimers, but higher-order oligomers also are observed. The receptors also exist as monomers and dimers, but the precise stoichiometry of ligand and receptor oligomeric combinations is not yet fully understood for most chemokine/receptor pairs (49–52). Thus, proteinaceous alarmins and classical chemokines exhibit fundamental structural and functional differences.
However, intriguing overlaps between these categories of mediators have been identified. First and as outlined above, alarmins, once released into the extracellular milieu, and classical chemokines can directly interact to form heterodimers; all-thiol HMGB1 binds to CXCL12 and HMGB1/CXCL12 dimers elicit CXCR4 signaling responses that are different from those triggered by CXCL12 alone (33); CCL5/HNP1 heterocomplexes represent another example of a dimer between a classical chemokine (in this case the CC chemokine CCL5/RANTES) and a prototypical alarmin (in this case the human neutrophilic peptide HNP1) (53). Secondly, the four classical chemokine categories (i.e., the CC-, CXC-, CX3C-, and C-type classes) have more recently been amended by a fifth “functional” class of chemokines, called the chemokine-like function (CLF) chemokines or innate chemokines, or atypical chemokines (ACKs), which share significant functional similarities with classical chemokines, i.e., exhibiting chemotactic activity. Here, we use the term ACK in analogy to ACKRs.
ACKs are a structurally diverse class of small 8–25 kD proteins that lack the classifying N-terminal cysteine residues. Some ACKs share gross architectural similarity with classical chemokines such as extended β-pleated sheet structures with juxtaposing α-helices, but do not contain a cognate chemokine-fold (9, 54–56). Despite these structural differences to classical chemokines, ACKs/CLF chemokines bind to CKRs with high affinity and elicit chemotactic cell migration.
A typical ACK would be a constitutively expressed cellular protein with a bona fide intracellular function that can be released or secreted upon cell injury or during inflammatory stimulation to act as a chemokine-like mediator in the extracellular milieu, pinpointing significant similarities with proteinaceous alarmins.
The concept of ACK/CLF chemokines dates back to the work of Wakasugi and Schimmel who discovered that proteolytically cleaved fragments of tyrosine amino-acyl-tRNA synthetase (TyrRS) are secreted into the extracellular milieu, where they act as chemokines by binding to the classical chemokine receptor CXCR1 on neutrophils and monocytes (57). Subsequent work demonstrated similar activities for TrpRS, HisRs, and AsnRS, as well as acting through CXCR1, CXCR3, CCR5, and CCR3, respectively (11, 58–60). Moreover, the p43 auxiliary component of the tRNA multisynthetase complex is identical to endothelial monocyte-activating polypeptide-II (EMAP-II), an inflammatory cytokine with anti-angiogenic properties (61). It was further demonstrated that EMAP-II is identical or similar to the C-domain fragment of TyrRS but also certain non-mammalian proteins (62). Together, this indicated that intracellular housekeeping proteins such as amino-acyl-tRNA synthetases (AaaRS) needed for the ribosomal protein biosynthesis machinery have a “second job” in the extracellular environment as cytokines/chemokines. In the following chapter, we will also discuss the ribosomal protein RPS19, which after release serves as a cytokine antagonist in the extracellular space.
Defensins, in particular, β-defensins, are small cysteine-rich cationic proteins abundantly produced by epithelial and endothelial cells that act as pore-forming amphipathic antimicrobial peptides, also have important “second job” functions as chemokine-like factors. Human β-defensin-1 (HBD1), HBD3, and mouse β-defensin-14 (MBD14) bind to and signal through CCR6 on T cells and dendritic cells, while HBD2 and HBD3 as well as MBD4 and MBD14 additionally interact with CCR2. Moreover, HBD3 acts as an alternative ligand for CXCR4 (63–67). These studies also identified an important structural determinant for the promiscuous usage of classical CKRs by non-classical chemotactic mediators. HBD1 and 2 express a positive charge cluster on their surface that is very similar to that of the cognate CCR6 ligand CCL20/MIP-3α, representing a remarkable example of structural mimicry (68). Additional examples of ACK/CLF-type chemokines are thioredoxin (for which the CKR has not yet been identified) (69), the cold shock protein YB-1, which upon secretion not only interacts with Notch-3 but also with CXCR2 (70), as well as cathelicidin-related antimicrobial peptides such as LL-37/Cramp-1, which bind to CKR-like formyl-peptide receptors, although it may be argued that cathelicidins share more similarity with inflammatory neutrophilic cytokines secreted by conventional pathways than with alarmins or ACKs (18, 71–73). Similarly, serum amyloid A (SAA), a prominent liver-derived pro-inflammatory acute phase protein exhibits ACK-like properties owing to its signaling activity through the chemoattractant receptor formyl-peptide receptor-2 (FPR2), although part of its initially described chemotactic activities were more recently redefined as “indirect” effects via TLR2 (74).
In any case, all these ACK-type proteins have typical “moonlighting” characteristics, featuring “night-time (second) jobs” as chemokine-like mediators in inflammation, autoimmunity, and host defense. Also, although they are structurally diverse, some of them share mixed β-sheet/α-helix structural elements that are reminiscent of the β-sheet/α-helix architecture of classical CKs. Table 2 summarizes known ACK, indicating their moonlighting functions and receptors vs. intracellular job 1-interactors.
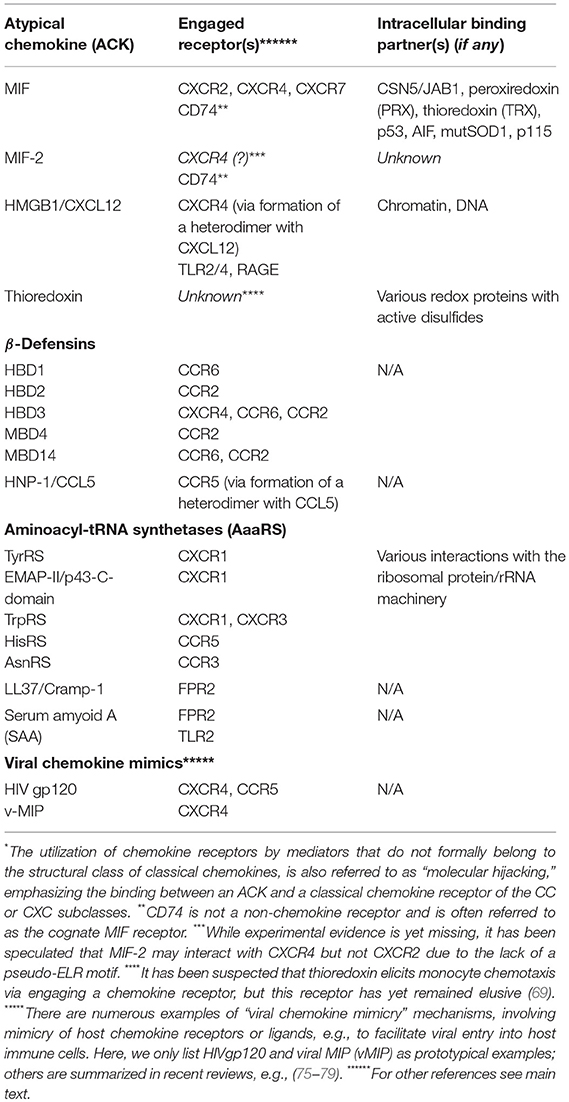
Table 2. List of known atypical chemokines (ACKs), their “hijacked”*chemokine receptors, other receptors, and intracellular interaction partners.
Work in the last decade has demonstrated that MIF family proteins are prototypical ACKs that feature all characteristics and multitasking properties of these proteins.
Macrophage migration-inhibitory factor (MIF) was identified in 1966 as one of the first cytokines to be discovered (80). Initially described as a T-cell-derived inhibitor of random macrophage migration, MIF has been redefined as a pleiotropic chemokine-like inflammatory cytokine that acts as an upstream mediator of innate and adaptive immunity. This also suggested that the eponymous migration-inhibitory activity measured 50 years ago, probably is some kind of “desensitization effect” of a chemokine-type mediator. Owing to its potent inflammatory activity profile, MIF has been found to be a pivotal mediator of acute and chronic inflammatory diseases such as septic shock, acute respiratory distress syndrome, neuroinflammation, or rheumatoid arthritis. Dysregulation of MIF also drives atherogenesis and other cardiovascular conditions as well as tumorigenesis. Several excellent review articles have summarized the role of MIF and its homolog MIF-2/D-dopachrome tautomerase (D-DT) as inflammatory cytokines and their significance in numerous diseases (39, 81–92).
Here, we will mainly address MIF's CLFs and its role as ACK and alarmin-type mediator. MIF is an evolutionarily conserved protein that is expressed in most species and kingdoms. Its structure is unique and cannot be grouped into any of the known cytokine classes, but despite an only 20–30% sequence homology, MIF proteins show a remarkable architectural similarity to a class of bacterial isomerases/tautomerase (83, 93). MIF also shares with these proteins a conserved catalytic tautomerase pocket that contains an unusually acidic proline residue, but the significance of this activity in mammals is unclear (82, 83, 91). There is also a remote similarity between the three-dimensional structure of CXC chemokines such as CXCL8 and MIF (39, 93, 94). The architecture of the MIF monomer resembles that of the CXCL8 dimer. MIF proteins are semi-constitutively expressed in many cell types. Accordingly, upregulation of MIF in inflammation and other stress conditions is mostly regulated at the level of MIF release rather than transcriptional induction, although remarkable exceptions such as in neonatal cells have been noted (82, 95, 96). In fact, following translation, MIF is localized in the cytosol and does not enter the ER/Golgi pathway. In analogy, to the above-discussed storage of some classical inflammatory chemokines, it could be argued that cytosolic deposition of MIF following its translation represents “cytosolic storage” awaiting secretory signals. In fact, MIF secretion occurs by a non-conventional pathway involving ABCA1 transporters, p115 and JAB1/CSN5, but the mechanistic details are still unclear (95, 97, 98). Despite the non-canonical nature of the MIF secretion process, release of MIF into the extracellular space is tightly controlled by inflammatory and immune stimulation and occurs in a regulated fashion. In addition, it has been suggested that some tumor cells as well as certain endothelial and epithelial cells release low concentrations of MIF in a semi-constitutive -autocrine- manner that would be independent of a specific and acute trigger.
Once released into the interstitial space and/or circulation, MIF functions to modulate the activity of numerous immune and inflammatory cells. When secretion occurs from inflamed endothelium, e.g., in atherogenesis, MIF is immobilized on the endothelial surface to form an haptotactic gradient. Deposition is similar to atherogenic arrest chemokines and involves interaction of basic surface charges of MIF with negatively charged proteoglycans (94). Circulating or immobilized MIF then engages one or more of its receptors, typically expressed on myeloid cells or lymphocytes. This is the surface-expressed form of CD74/invariant-chain, which also acts as an MHC class II chaperone facilitating antigen loading to class II complexes in the endoplasmic-reticulum (99), as well as the CKRs CXCR2 and CXCR4, the cognate receptors for the ELR+ CXC chemokines CXCL1, CXCL2, or CXCL8, and the ELR- CXC chemokine CXCL12/SDF-1α, respectively (94). CD74 may be also expressed in class II-negative cells, i.e., upon inflammatory stimulation, and exhibits a major role as cytokine receptor for MIF and MIF-2, driving proliferative responses (100, 101). CXCR2 is mainly expressed on neutrophils and monocytes/macrophages, but can also be upregulated on a number of other cell types under inflammatory stimulation. CXCR4 is ubiquitously expressed, but expression levels can be elevated in inflammation. High-affinity binding of MIF to CXCR2 and CXCR4 represents non-cognate interactions between an ACK and CKRs (Table 2) and gives rise to leukocyte recruitment responses in inflammation and atherogenesis (39, 86, 92, 94). Binding of MIF to CXCR2 drives atherogenic recruitment of monocytes and neutrophils (94, 102, 103). MIF/CXCR4 binding supports the recruitment of atherogenic T and B lymphocytes (94, 104, 105), but also regulates the egress of metastatic tumor cells and progenitor cell recruitment (39). All three receptors have important roles in inflammation and cardiovascular disease. The surprising AMP kinase-mediated cardioprotective effects of MIF and MIF-2 in myocardial ischemia/reperfusion injury are mediated by cardiac-expressed CD74 (106–108). The binding of MIF-2 to CXCR2 and CXCR4 has not been addressed, while MIF has also been suggested to interact with CXCR7-mediated pathways (109).
The structural determinants underlying the promiscuous binding of MIF to CXCR2 have been elucidated: MIF/CXCR2 binding is similar but not identical to that of the cognate ligand CXCL8 and requires an N-like loop and a pseudo-ELR motif (39, 103, 110, 111). This represents another intriguing example of molecular mimicry that is reminiscent of how the ACKs HBD1 and 2 mimic the receptor binding motif that CCL20 utilizes to engage CCR6 (68). Recent data also inform about the molecular determinants that govern the MIF/CXCR4 interaction. They show that the MIF/CXCR4 interface involves an extended N-like loop of MIF, an RLR motif, and the N-terminal Pro-2 of MIF as well as sequences from two extracellular loops of CXCR4 (112, 113). Overall, the binding motifs controlling MIF/CXCR4 binding appear to be less similar to those of the cognate binding pair CXCL12/CXCR4, thus representing an example of “remote” molecular mimicry (75, 114, 115).
In conjunction, these features of MIF proteins are reminiscent of bona fide alarmins such as HMGB1 or IL-33, with independent intra- and extracellular roles. In fact, while only speculated about for a long time (116), the potential intracellular activities of MIF proteins have been defined more recently. Intracellular activities were identified both in the cytosolic and nuclear compartment. Cytosolic MIF interacts with CSN5/JAB1 to regulate COP9 signalosome signaling, while inversely, CSN5 is involved in MIF secretion (97, 117). MIF/CSN5 interactions are further linked to apoptotic processes via p53 (97, 118–120). Depending on the oxidative environment, cytosolic MIF also interacts with redox-regulating proteins thioredoxin (Trx) and peroxiredoxin (Prx), and this is mediated by MIF's redox-active cysteine residues (121). While the activity spectrum of MIF's cytosolic interactions is diverse, they may be collectively designated as “cell-protective,” “homeostasis-promoting” activities (Figure 2). This assignment was recently confirmed by an unexpected novel cytosolic interaction partner of MIF. MIF binds to a mutant form of superoxide dismutase (mutSOD1) and inhibits the accumulation of misfolded SOD1, protecting from motoneuron damage (122–124). It is currently unclear whether MIF exerts this function also in non-neuronal cells. The recent discovery of a nuclear function of MIF came as a big surprise. Neurons under ischemic or excitotoxic stress accumulate cytosolic poly-ADP ribose (PAR) trees following DNA damage. This leads to mitochondrial release of apoptosis-inducing factor (AIF), which binds to MIF and escorts it into the nuclear compartment. Nuclear MIF further promotes DNA damage by direct nuclease regulation, leading to cell death by parthanatos (125). While this study has yet to be independently confirmed and while it is unclear whether MIF-2 exhibits similar activities, it supports the concept that MIF proteins can exert their functions in three independent compartments: (i) in the extracellular space, (ii) in the cytosol, and (iii) in the nucleus, representing a intriguing example of “topological multitasking” (Figure 2). With respect to the moonlighting concept, it also means that the intracellular functions (cytosolic and nuclear) would be the intracellular job of MIF (“job 1”), with cytosolic functions designated as “job 1a” and the nuclear activity being “job 1b”. The cytokine/chemokine functions of MIF in the extracellular environment would be MIF's second task (“job 2”) (Figure 2). The activities of MIF may be further fine-tuned by (PTMs), which have been suggested to modulate both intra- and extracellular MIF in a context-dependent manner (Table 1). Interestingly, the intra- and extracellular MIF pools appear to be further linked through other ACK-type/multitasking proteins. MIF and HMGB1 both engage CXCR4 and also are functionally related in a reciprocal manner (33, 94, 126). Moreover, the ribosomal protein RPS19 may be released into the extracellular space, where it binds to and attenuates MIF-mediated inflammatory activities (127).
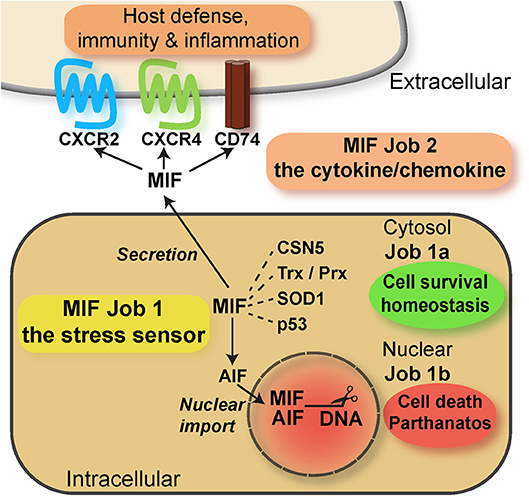
Figure 2. Scheme summarizing the multitasking/moonlighting functions of MIF proteins. For overview purposes, only MIF but not MIF-2/D-DT is depicted. For details see manuscript text. MIF, macrophage migration-inhibitory factor; CSN, COP9 signalosome; Trx, thioredoxin; Prx, peroxiredoxin; SOD, superoxide dismutase; AIF, apoptosis-inducing factor.
In conclusion, although the mechanisms regulating the interplay between the different MIF tasks have not yet been elucidated in detail, it is clear that multitasking by MIF over three different topologies and involving several different binding proteins is an impressive example of molecular multitasking, illustrating the functional versatility of MIF proteins and atypical chemokines in general. The ACK characteristics of MIF proteins are also summarized in Table 2.
The potential roles of HMGB1 and MIF-family proteins in inflammatory, autoimmune and cardiovascular diseases have been extensively studied and various therapeutic approaches to interfere with their pathologic activities, i.e., antibodies and small molecule inhibitors (SMD) have been explored. This has been summarized in several recent review articles (14, 91, 92, 128–140). Here, we will only briefly allude to the translational opportunities that may arise from specifically targeting the moonlighting characteristics of HMGB1 or MIF proteins.
It seems overall advisable to try to specifically target the extracellular activities of HMGB1 or MIF proteins, i.e., “job 2” and “job 3” (Figures 1, 2). These are the disease-promoting, inflammatory activities of these alarmins/ACKs. HMGB1 enhances immune cell migration (and thus inflammatory recruitment) via job 2 and triggers the release of inflammatory cytokines and chemokines via job 3. MIF, with the exception of its CD74/AMPK-mediated cardioprotective activity in the early phase of cardiac ischemia, amplifies inflammation via various job 2-mediated pathways. These include e.g., cytokine/chemokine upregulation, leukocyte recruitment, and macrophage survival. In contrast, the job 1 activities of HMGB1 and MIF proteins have been suggested to generally be physiologic, maintaining cell homeostasis. Pharmacologic approaches to tackle HMGB1 or MIF should therefore generally preserve their intracellular activities. Antibody-based approaches appear suitable in this respect. Moreover, targeting the receptors appears prudent at first sight, but specificity issues will have to be considered. This is particularly true for MIF receptor-targeted strategies, as the cardioprotective activities of the CD74 pathway and the homeostatic role of CXCR4 should be preserved (92, 141) (Figure 2). Although HMGB1/CXCR4 heterodimers have been found to drive inflammatory leukocyte recruitment (33), this also holds true for CXCR4 blockade strategies as a means to interfere with job 2 HMGB1 pathways (Figure 1).
Additionally, small molecule inhibitors (SMD), directed at the tautomerase site of MIF proteins, represent an interesting strategy to interfere with the pro-inflammatory job 2 MIF activities. However, considering the above-said, those anti-MIF SMDs that are not membrane-penetrable—thus specifically directed at extracellular -job 2- MIFs—overall appear preferable, although exceptions may apply to applications in cancer and stroke, in which intracellular MIF has been suggested to exert detrimental effects as well (125, 131).
In conclusion, while the moonlighting characteristics of alarmins and ACKs constitute complex mechanistic networks that are subject to differential regulation in health and disease, they principally offer intriguing translational opportunities with “job”- and thus disease phase-specific targeting options. Multiple clinical applications are in sight, but also require efforts to further elucidate the multitasking circuits of these proteins.
JB conceived the concept and wrote the manuscript. AK co-conceived the concept, edited and improved the manuscript, and drafted the table. OG co-conceived the concept, edited and improved the manuscript, and drafted a figure.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by Deutsche Forschungsgemeinschaft (DFG) grant SFB1123-A03 to JB and AK, and by DFG within the framework of Munich Cluster for Systems Neurology (EXC 1010 SyNergy) to JB; OG received funding from a NARSAD Young Investigator Grant.
1. Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The sequence of the human genome. Science (2001) 291:1304–51. doi: 10.1126/science.1058040
2. Pertea M, Shumate A, Pertea G, Varabyou A, Chang Y-C, Madugundu AK, et al. Thousands of large-scale RNA sequencing experiments yield a comprehensive new human gene list and reveal extensive transcriptional noise. bioRxiv [preprint] (2018). doi: 10.1101/332825
3. Luedike P, Hendgen-Cotta UB, Sobierajski J, Totzeck M, Reeh M, Dewor M, et al. Cardioprotection through S-nitros(yl)ation of macrophage migration inhibitory factor. Circulation (2012) 125:1880–9. doi: 10.1161/CIRCULATIONAHA.111.069104
4. Pohl J, Hendgen-Cotta UB, Rammos C, Luedike P, Mull E, Stoppe C, et al. Targeted intracellular accumulation of macrophage migration inhibitory factor in the reperfused heart mediates cardioprotection. Thromb Haemost. (2016) 115:200–12. doi: 10.1160/TH15-05-0436
5. Schindler L, Dickerhof N, Hampton MB, Bernhagen J. Post-translational regulation of macrophage migration inhibitory factor: basis for functional fine-tuning. Redox Biol. (2018) 15:135–42. doi: 10.1016/j.redox.2017.11.028
6. Dickerhof N, Schindler L, Bernhagen J, Kettle AJ, Hampton MB. Macrophage migration inhibitory factor (MIF) is rendered enzymatically inactive by myeloperoxidase-derived oxidants but retains its immunomodulatory function. Free Radic Biol Med. (2015) 89:498–511. doi: 10.1016/j.freeradbiomed.2015.09.009
7. Thiele M, Kerschbaumer RJ, Tam FW, Volkel D, Douillard P, Schinagl A, et al. Selective targeting of a disease-related conformational isoform of macrophage migration inhibitory factor ameliorates inflammatory conditions. J Immunol. (2015) 195:2343–52. doi: 10.4049/jimmunol.1500572
8. Zheng Y, Li X, Qian X, Wang Y, Lee JH, Xia Y, et al. Secreted and O-GlcNAcylated MIF binds to the human EGF receptor and inhibits its activation. Nat Cell Biol. (2015) 17:1348–55. doi: 10.1038/ncb3222
9. Oppenheim JJ, Yang D. Alarmins: chemotactic activators of immune responses. Curr Opin Immunol. (2005) 17:359–65. doi: 10.1016/j.coi.2005.06.002
10. Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. (2007) 81:1–5. doi: 10.1189/jlb.0306164
11. Becker R. Beyond building proteins: tRNA synthetases outside of translation. Nat Med. (2016) 22:452–3. doi: 10.1038/nm0516-452
12. Foell D, Wittkowski H, Vogl T, Roth J. S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol. (2007) 81:28–37. doi: 10.1189/jlb.0306170
13. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. (1994) 12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015
14. Venereau E, Ceriotti C, Bianchi ME. DAMPs from cell death to new life. Front Immunol. (2015) 6:422. doi: 10.3389/fimmu.2015.00422
15. Liston A, Masters SL. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat Rev Immunol. (2017) 17:208–14. doi: 10.1038/nri.2016.151
16. Chan JK, Roth J, Oppenheim JJ, Tracey KJ, Vogl T, Feldmann M, et al. Alarmins: awaiting a clinical response. J Clin Invest. (2012) 122:2711–9. doi: 10.1172/JCI62423
17. Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. (2005) 5:331–42. doi: 10.1038/nri1594
18. Oppenheim JJ, Tewary P, de la Rosa G, Yang D. Alarmins initiate host defense. Adv Exp Med Biol. (2007) 601:185–94. doi: 10.1007/978-0-387-72005-0_19
19. Nickel W, Rabouille C. Mechanisms of regulated unconventional protein secretion. Nat Rev Mol Cell Biol. (2009) 10:148–55. doi: 10.1038/nrm2617
20. Stros M. HMGB proteins: interactions with DNA and chromatin. Biochim Biophys Acta (2010) 1799:101–13. doi: 10.1016/j.bbagrm.2009.09.008
21. Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. Embo J. (2011) 30:4701–11. doi: 10.1038/emboj.2011.398
22. Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science (1999) 285:248–51. doi: 10.1126/science.285.5425.248
23. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature (2002) 418:191–5. doi: 10.1038/nature00858
24. Yang H, Antoine DJ, Andersson U, Tracey KJ. The many faces of HMGB1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J Leukoc Biol. (2013) 93:865–73. doi: 10.1189/jlb.1212662
25. Czura CJ, Wang H, Tracey KJ. Dual roles for HMGB1: DNA binding and cytokine. J Endotoxin Res. (2001) 7:315–21. doi: 10.1177/09680519010070041401
26. Yang H, Wang H, Tracey KJ. HMG-1 rediscovered as a cytokine. Shock (2001) 15:247–53. doi: 10.1097/00024382-200115040-00001
27. Andersson U, Erlandsson-Harris H, Yang H, Tracey KJ. HMGB1 as a DNA-binding cytokine. J Leukoc Biol. (2002) 72:1084–91. doi: 10.1189/jlb.72.6.1084
28. Yang H, Wang H, Czura CJ, Tracey KJ. HMGB1 as a cytokine and therapeutic target. J Endotoxin Res. (2002) 8:469–72. doi: 10.1179/096805102125001091
29. Yang H, Wang H, Czura CJ, Tracey KJ. The cytokine activity of HMGB1. J Leukoc Biol. (2005) 78:1–8. doi: 10.1189/jlb.1104648
30. Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci USA. (2010) 107:11942–7. doi: 10.1073/pnas.1003893107
31. Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med. (2012) 209:1519–28. doi: 10.1084/jem.20120189
32. Yang H, Lundback P, Ottosson L, Erlandsson-Harris H, Venereau E, Bianchi ME, et al. Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol Med. (2012) 18:250–9. doi: 10.2119/molmed.2011.00389
33. Schiraldi M, Raucci A, Munoz LM, Livoti E, Celona B, Venereau E, et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med. (2012) 209:551–63. doi: 10.1084/jem.20111739
34. Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell (1999) 97:889–901. doi: 10.1016/S0092-8674(00)80801-6
35. Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. (2007) 13:1042–9. doi: 10.1038/nm1638
36. Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. Embo J. (2003) 22:5551–60. doi: 10.1093/emboj/cdg516
37. Frye BC, Halfter S, Djudjaj S, Muehlenberg P, Weber S, Raffetseder U, et al. Y-box protein-1 is actively secreted through a non-classical pathway and acts as an extracellular mitogen. EMBO Rep. (2009) 10:783–9. doi: 10.1038/embor.2009.81
39. Tillmann S, Bernhagen J, Noels H. Arrest functions of the MIF ligand/receptor axes in atherogenesis. Front Immunol. (2013) 4:115. doi: 10.3389/fimmu.2013.00115
40. Shulman Z, Cohen SJ, Roediger B, Kalchenko V, Jain R, Grabovsky V, et al. Transendothelial migration of lymphocytes mediated by intraendothelial vesicle stores rather than by extracellular chemokine depots. Nat Immunol. (2011) 13:67–76. doi: 10.1038/ni.2173
41. Gleissner CA, von Hundelshausen P, Ley K. Platelet chemokines in vascular disease. Arterioscler Thromb Vasc Biol. (2008) 28:1920–7. doi: 10.1161/ATVBAHA.108.169417
42. Ludwig A, Weber C. Transmembrane chemokines: versatile 'special agents' in vascular inflammation. Thromb Haemost. (2007) 97:694–703. doi: 10.1160/TH07-01-0035
43. Loos T, Mortier A, Proost P. Chapter 1. Isolation, identification, and production of posttranslationally modified chemokines. Methods Enzymol. (2009) 461:3–29. doi: 10.1016/S0076-6879(09)05401-9
44. Proost P, Struyf S, Van Damme J. Natural post-translational modifications of chemokines. Biochem Soc Trans. (2006) 34:997–1001. doi: 10.1042/BST0340997
45. Proost P, Struyf S, Couvreur M, Lenaerts JP, Conings R, Menten P, et al. Posttranslational modifications affect the activity of the human monocyte chemotactic proteins MCP-1 and MCP-2: identification of MCP-2(6-76) as a natural chemokine inhibitor. J Immunol. (1998) 160:4034–41.
46. Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. (2006) 354:610–21. doi: 10.1056/NEJMra052723
47. Bachelerie F, Ben-Baruch A, Burkhardt AM, Combadiere C, Farber JM, Graham GJ, et al. International Union of Basic and Clinical Pharmacology. LXXXIX Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol Rev. (2014) 66:1–79. doi: 10.1124/pr.113.007724
48. Rajagopalan L, Rajarathnam K. Structural basis of chemokine receptor function–a model for binding affinity and ligand selectivity. Biosci Rep. (2006) 26:325–39. doi: 10.1007/s10540-006-9025-9
50. Thelen M, Munoz LM, Rodriguez-Frade JM, Mellado M. Chemokine receptor oligomerization: functional considerations. Curr Opin Pharmacol. (2010) 10:38–43. doi: 10.1016/j.coph.2009.09.004
51. Thelen M, Stein JV. How chemokines invite leukocytes to dance. Nat Immunol. (2008) 9:953–9. doi: 10.1038/ni.f.207
52. Kraemer S, Alampour-Rajabi S, El Bounkari O, Bernhagen J. Hetero-oligomerization of chemokine receptors: diversity and relevance for function. Curr Med Chem. (2013) 20:2524–36. doi: 10.2174/09298673113209990117
53. Alard JE, Ortega-Gomez A, Wichapong K, Bongiovanni D, Horckmans M, Megens R. T, et al. Recruitment of classical monocytes can be inhibited by disturbing heteromers of neutrophil HNP1 and platelet CCL5. Sci Transl Med. (2015) 7:317ra196. doi: 10.1126/scitranslmed.aad5330
54. Degryse B, de Virgilio M. The nuclear protein HMGB1, a new kind of chemokine? FEBS Lett. (2003) 553:11–7. doi: 10.1016/S0014-5793(03)01027-5
55. Noels H, Bernhagen J, Weber C. Macrophage migration inhibitory factor: a noncanonical chemokine important in atherosclerosis. Trends Cardiovasc Med. (2009) 19:76–86. doi: 10.1016/j.tcm.2009.05.002
56. Schober A, Bernhagen J, Weber C. Chemokine-like functions of MIF in atherosclerosis. J Mol Med. (2008) 86:761–70. doi: 10.1007/s00109-008-0334-2
57. Wakasugi K, Schimmel P. Two distinct cytokines released from a human aminoacyl-tRNA synthetase. Science (1999) 284:147–51. doi: 10.1126/science.284.5411.147
58. Wakasugi K, Slike BM, Hood J, Otani A, Ewalt KL, Friedlander M, et al. A human aminoacyl-tRNA synthetase as a regulator of angiogenesis. Proc Natl Acad Sci USA. (2002) 99:173–7. doi: 10.1073/pnas.012602099
59. Howard OM, Dong HF, Yang D, Raben N, Nagaraju K, Rosen A, et al. Histidyl-tRNA synthetase and asparaginyl-tRNA synthetase, autoantigens in myositis, activate chemokine receptors on T lymphocytes and immature dendritic cells. J Exp Med. (2002) 196:781–91. doi: 10.1084/jem.20020186
60. Howard OM, Dong HF, Su SB, Caspi RR, Chen X, Plotz P, et al. Autoantigens signal through chemokine receptors: uveitis antigens induce CXCR3- and CXCR5-expressing lymphocytes and immature dendritic cells to migrate. Blood (2005) 105:4207–14. doi: 10.1182/blood-2004-07-2697
61. van Horssen R, Eggermont AM, ten Hagen TL. Endothelial monocyte-activating polypeptide-II and its functions in (patho)physiological processes. Cytokine Growth Factor Rev. (2006) 17:339–48. doi: 10.1016/j.cytogfr.2006.08.001
62. Wakasugi K, Schimmel P. Highly differentiated motifs responsible for two cytokine activities of a split human tRNA synthetase. J Biol Chem. (1999) 274:23155–9. doi: 10.1074/jbc.274.33.23155
63. Yang D, Chertov O, Bykovskaia SN, Chen Q, Buffo MJ, Shogan J, et al. Beta-defensins: linking innate and adaptive immunity through dendritic and T cell CCR6. Science (1999) 286:525–8. doi: 10.1126/science.286.5439.525
64. Oppenheim JJ, Biragyn A, Kwak LW, Yang D. Roles of antimicrobial peptides such as defensins in innate and adaptive immunity. Ann Rheum Dis. (2003) 62(Suppl. 2):ii17–21. doi: 10.1136/ard.62.suppl_2.ii17
65. Yang D, Rosenberg HF, Chen Q, Dyer KD, Kurosaka K, Oppenheim JJ. Eosinophil-derived neurotoxin (EDN), an antimicrobial protein with chemotactic activities for dendritic cells. Blood (2003) 102:3396–403. doi: 10.1182/blood-2003-01-0151
66. Rohrl J, Yang D, Oppenheim JJ, Hehlgans T. Identification and Biological Characterization of Mouse beta-defensin 14, the orthologue of human beta-defensin 3. J Biol Chem. (2008) 283:5414–9. doi: 10.1074/jbc.M709103200
67. Rohrl J, Yang D, Oppenheim JJ, Hehlgans T. Human beta-defensin 2 and 3 and their mouse orthologs induce chemotaxis through interaction with CCR2. J Immunol. (2010) 184:6688–94. doi: 10.4049/jimmunol.0903984
68. Hoover DM, Boulegue C, Yang D, Oppenheim JJ, Tucker K, Lu W, et al. The structure of human macrophage inflammatory protein-3alpha /CCL20. Linking antimicrobial and CC chemokine receptor-6-binding activities with human beta-defensins. J. Biol. Chem. (2002) 277:37647–54. doi: 10.1074/jbc.M203907200
69. Bertini R, Howard OMZ, Dong H-F, Oppenheim JJ, Bizzarri C, Caselli G, et al. Thioredoxin, a redox enzyme released in infection and inflammation, is a unique chemoattractant for neutrophils, monocytes, and T cells. J Exp Med. (1999) 189:1783–9. doi: 10.1084/jem.189.11.1783
70. Rauen T, Raffetseder U, Frye BC, Djudjaj S, Muhlenberg PJ, Eitner F, et al. YB-1 acts as a ligand for Notch-3 receptors and modulates receptor activation. J Biol Chem. (2009) 284:26928–40. doi: 10.1074/jbc.M109.046599
71. Yang D, Biragyn A, Hoover DM, Lubkowski J, Oppenheim JJ. Multiple roles of antimicrobial defensins, cathelicidins, and eosinophil-derived neurotoxin in host defense. Annu Rev Immunol. (2004) 22:181–215. doi: 10.1146/annurev.immunol.22.012703.104603
72. Soehnlein O, Zernecke A, Eriksson EE, Rothfuchs AG, Pham CT, Herwald H, et al. Neutrophil secretion products pave the way for inflammatory monocytes. Blood (2008) 112:1461–71. doi: 10.1182/blood-2008-02-139634
73. Wantha S, Alard JE, Megens RT, van der Does AM, Doring Y, Drechsler M, et al. Neutrophil-derived cathelicidin promotes adhesion of classical monocytes. Circ Res. (2013) 112:792–801. doi: 10.1161/CIRCRESAHA.112.300666
74. De Buck M, Gouwy M, Wang JM, Van Snick J, Proost P, Struyf S, et al. The cytokine-serum amyloid A-chemokine network. Cytokine Growth Factor Rev. (2016) 30:55–69. doi: 10.1016/j.cytogfr.2015.12.010
75. Qin L, Kufareva I, Holden LG, Wang C, Zheng Y, Zhao C, et al. Structural biology. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science (2015) 347:1117–22. doi: 10.1126/science.1261064
76. Pawig L, Klasen C, Weber C, Bernhagen J, Noels H. Diversity and inter-connections in the CXCR4 chemokine receptor/ligand family: molecular perspectives. Front Immunol. (2015) 6:429. doi: 10.3389/fimmu.2015.00429
77. Lusso P. HIV and the chemokine system: 10 years later. Embo J. (2006) 25:447–56. doi: 10.1038/sj.emboj.7600947
78. Zaitseva M, Peden K, Golding H. HIV coreceptors: role of structure, posttranslational modifications, and internalization in viral-cell fusion and as targets for entry inhibitors. Biochim Biophys Acta (2003) 1614:51–61. doi: 10.1016/S0005-2736(03)00162-7
79. Horuk R. Chemokine receptors and HIV-1: the fusion of two major research fields. Immunol Today (1999) 20:89–94. doi: 10.1016/S0167-5699(98)01396-6
80. David JR. Delayed hypersensitivity in vitro: its mediation by cell-free substances formed by lymphoid cell-antigen interaction. Proc Natl Acad Sci USA. (1966) 56:72–7. doi: 10.1073/pnas.56.1.72
81. Bucala R. MIF re-discovered: pituitary hormone and glucocorticoid-induced regulator of cytokine production. Cytokine Growth Factor Rev. (1996) 7:19–24. doi: 10.1016/1359-6101(96)00008-1
82. Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. (2003) 3:791–800. doi: 10.1038/nri1200
83. Lolis E, Bucala R. Macrophage migration inhibitory factor. Exp Opin Ther Targets (2003) 7:153–64. doi: 10.1517/14728222.7.2.153
84. Morand EF, Leech M, Bernhagen J. MIF: a new cytokine link between rheumatoid arthritis and atherosclerosis. Nat Rev Drug Discov. (2006) 5:399–410. doi: 10.1038/nrd2029
85. Flaster H, Bernhagen J, Calandra T, Bucala R. The MIF-glucocorticoid dyad: regulation of inflammation and immunity. Mol Endocrinol. (2007) 21:1267–80. doi: 10.1210/me.2007-0065
86. Zernecke A, Bernhagen J, Weber C. Macrophage migration inhibitory factor in cardiovascular disease. Circulation (2008) 117:1594–602. doi: 10.1161/CIRCULATIONAHA.107.729125
87. Kleemann R, Bucala R. Macrophage migration inhibitory factor: critical role in obesity, insulin resistance, and associated comorbidities. Mediators Inflamm. (2010) 2010:610479. doi: 10.1155/2010/610479
88. Merk M, Mitchell RA, Endres S, Bucala R. D-dopachrome tautomerase (D-DT or MIF-2): Doubling the MIF cytokine family. Cytokine (2012) 59:10–7. doi: 10.1016/j.cyto.2012.03.014
89. Dayawansa NH, Gao X-M, White DA, Dart AM, Du X-J. Role of MIF in myocardial ischaemia and infarction: insight from recent clinical and experimental findings. Clin Sci. (2014) 127:149–61. doi: 10.1042/CS20130828
90. Rassaf T, Weber C, Bernhagen J. Macrophage migration inhibitory factor in myocardial ischaemia/reperfusion injury. Cardiovasc Res. (2014) 102:321–8. doi: 10.1093/cvr/cvu071
91. Bloom J, Sun S, Al-Abed Y. MIF, a controversial cytokine: a review of structural features, challenges, and opportunities for drug development. Exp Opin Ther Targets (2016) 20:1463–75. doi: 10.1080/14728222.2016.1251582
92. Tilstam PV, Qi D, Leng L, Young L, Bucala R. MIF family cytokines in cardiovascular diseases and prospects for precision-based therapeutics. Exp Opin Ther Targets (2017) 21:671–83. doi: 10.1080/14728222.2017.1336227
93. Sun HW, Bernhagen J, Bucala R, Lolis E. Crystal structure at 2.6-A resolution of human macrophage migration inhibitory factor. Proc Natl Acad Sci USA. (1996) 93:5191–6. doi: 10.1073/pnas.93.11.5191
94. Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. (2007) 13:587–96. doi: 10.1038/nm1567
95. Flieger O, Engling A, Bucala R, Lue H, Nickel W, Bernhagen J. Regulated secretion of macrophage migration inhibitory factor is mediated by a non-classical pathway involving an ABC transporter. FEBS Lett. (2003) 551:78–86. doi: 10.1016/S0014-5793(03)00900-1
96. Roger T, Schneider A, Weier M, Sweep FC, Le Roy D, Bernhagen J, et al. High expression levels of macrophage migration inhibitory factor sustain the innate immune responses of neonates. Proc Natl Acad Sci USA. (2016) 113:E997–1005. doi: 10.1073/pnas.1514018113
97. Lue H, Thiele M, Franz J, Dahl E, Speckgens S, Leng L, et al. Macrophage migration inhibitory factor (MIF) promotes cell survival by activation of the Akt pathway and role for CSN5/JAB1 in the control of autocrine MIF activity. Oncogene (2007) 26:5046–59. doi: 10.1038/sj.onc.1210318
98. Merk M, Baugh J, Zierow S, Leng L, Pal U, Lee SJ, et al. The Golgi-associated protein p115 mediates the secretion of macrophage migration inhibitory factor. J Immunol. (2009) 182:6896–906. doi: 10.4049/jimmunol.0803710
99. Bertolino P, Rabourdin-Combe C. The MHC class II-associated invariant chain: a molecule with multiple roles in MHC class II biosynthesis and antigen presentation to CD4+ T cells. Crit Rev Immunol. (1996) 16:359–79.
100. Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, et al. MIF signal transduction initiated by binding to CD74. J Exp Med. (2003) 197:1467–76. doi: 10.1084/jem.20030286
101. Merk M, Zierow S, Leng L, Das R, Du X, Schulte W, et al. The D-dopachrome tautomerase (DDT) gene product is a cytokine and functional homolog of macrophage migration inhibitory factor (MIF). Proc Natl Acad Sci USA. (2011) 108:E577–585. doi: 10.1073/pnas.1102941108
102. Cho Y, Crichlow GV, Vermeire JJ, Leng L, Du X, Hodsdon ME, et al. Allosteric inhibition of macrophage migration inhibitory factor revealed by ibudilast. Proc Natl Acad Sci USA. (2010) 107:11313–8. doi: 10.1073/pnas.1002716107
103. Xu L, Li Y, Li D, Xu P, Tian S, Sun H, et al. Exploring the binding mechanisms of MIF to CXCR2 using theoretical approaches. Phys Chem Chem Phys. (2015) 17:3370–82. doi: 10.1039/C4CP05095A
104. Klasen C, Ohl K, Sternkopf M, Shachar I, Schmitz C, Heussen N, et al. MIF promotes B cell chemotaxis through the receptors CXCR4 and CD74 and ZAP-70 signaling. J Immunol. (2014) 192:5273–84. doi: 10.4049/jimmunol.1302209
105. Schmitz C, Noels H, El Bounkari O, Straussfeld E, Megens RTA, Sternkopf M, et al. Mif-deficiency favors an atheroprotective autoantibody phenotype in atherosclerosis. FASEB J. (2018) 32:4428–43. doi: 10.1096/fj.201800058R
106. Miller EJ, Li J, Leng L, McDonald C, Atsumi T, Bucala R, et al. Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature (2008) 451:578–82. doi: 10.1038/nature06504
107. Qi D, Atsina K, Qu L, Hu X, Wu X, Xu B, et al. The vestigial enzyme D-dopachrome tautomerase protects the heart against ischemic injury. J Clin Invest. (2014) 124:3540–50. doi: 10.1172/JCI73061
108. Qi D, Hu X, Wu X, Merk M, Leng L, Bucala R, et al. Cardiac macrophage migration inhibitory factor inhibits JNK pathway activation and injury during ischemia/reperfusion. J Clin Invest. (2009) 119:3807–16. doi: 10.1172/JCI39738
109. Alampour-Rajabi S, El Bounkari O, Rot A, Muller-Newen G, Bachelerie F, Gawaz M, et al. MIF interacts with CXCR7 to promote receptor internalization, ERK1/2 and ZAP-70 signaling, and lymphocyte chemotaxis. FASEB J. (2015) 29:4497–511. doi: 10.1096/fj.15-273904
110. Weber C, Kraemer S, Drechsler M, Lue H, Koenen RR, Kapurniotu A, et al. Structural determinants of MIF functions in CXCR2-mediated inflammatory and atherogenic leukocyte recruitment. Proc Natl Acad Sci USA. (2008) 105:16278–83. doi: 10.1073/pnas.0804017105
111. Kraemer S, Lue H, Zernecke A, Kapurniotu A, Andreetto E, Frank R, et al. MIF-chemokine receptor interactions in atherogenesis are dependent on an N-loop-based 2-site binding mechanism. FASEB J. (2011) 25:894–906. doi: 10.1096/fj.10-168559
112. Lacy M, Kontos C, Brandhofer M, Hille K, Groning S, Sinitski D, et al. Identification of an Arg-Leu-Arg tripeptide that contributes to the binding interface between the cytokine MIF and the chemokine receptor CXCR4. Sci Rep. (2018) 8:5171. doi: 10.1038/s41598-018-23554-5
113. Rajasekaran D, Groning S, Schmitz C, Zierow S, Drucker N, Bakou M, et al. Macrophage Migration Inhibitory Factor-CXCR4 Receptor Interactions: EVIDENCE FOR PARTIAL ALLOSTERIC AGONISM IN COMPARISON WITH CXCL12 CHEMOKINE. J Biol Chem. (2016) 291:15881–95. doi: 10.1074/jbc.M116.717751
114. Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science (2010) 330:1066–71. doi: 10.1126/science.1194396
115. Crump MP, Gong JH, Loetscher P, Rajarathnam K, Amara A, Arenzana-Seisdedos F, et al. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. Embo J. (1997) 16:6996–7007. doi: 10.1093/emboj/16.23.6996
116. Panstruga R, Baumgarten K, Bernhagen J. Phylogeny and evolution of plant macrophage migration inhibitory factor/D-dopachrome tautomerase-like proteins. BMC Evol Biol. (2015) 15:64. doi: 10.1186/s12862-015-0337-x
117. Kleemann R, Hausser A, Geiger G, Mischke R, Burger-Kentischer A, Flieger O, et al. Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature (2000) 408:211–6. doi: 10.1038/35041591
118. Hudson JD, Shoaibi MA, Maestro R, Carnero A, Hannon GJ, Beach DH. A proinflammatory cytokine inhibits p53 tumor suppressor activity. J Exp Med. (1999) 190:1375–82. doi: 10.1084/jem.190.10.1375
119. Bech-Otschir D, Kraft R, Huang X, Henklein P, Kapelari B, Pollmann C, et al. COP9 signalosome-specific phosphorylation targets p53 to degradation by the ubiquitin system. Embo J. (2001) 20:1630–9. doi: 10.1093/emboj/20.7.1630
120. Nemajerova A, Mena P, Fingerle-Rowson G, Moll UM, Petrenko O. Impaired DNA damage checkpoint response in MIF-deficient mice. Embo J. (2007) 26:987–97. doi: 10.1038/sj.emboj.7601564
121. Thiele M, Bernhagen J. Link between macrophage migration inhibitory factor and cellular redox regulation. Antiox Redox Signal. (2005) 7:1234–48. doi: 10.1089/ars.2005.7.1234
122. Shvil N, Banerjee V, Zoltsman G, Shani T, Kahn J, Abu-Hamad S, et al. MIF inhibits the formation and toxicity of misfolded SOD1 amyloid aggregates: implications for familial ALS. Cell Death Dis. (2018) 9:107. doi: 10.1038/s41419-017-0130-4
123. Israelson A, Ditsworth D, Sun S, Song S, Liang J, Hruska-Plochan M, et al. Macrophage migration inhibitory factor as a chaperone inhibiting accumulation of misfolded SOD1. Neuron (2015) 86:218–32. doi: 10.1016/j.neuron.2015.02.034
124. Leyton-Jaimes MF, Benaim C, Abu-Hamad S, Kahn J, Guetta A, Bucala R, et al. Endogenous macrophage migration inhibitory factor reduces the accumulation and toxicity of misfolded SOD1 in a mouse model of ALS. Proc Natl Acad Sci USA. (2016) 113:10198–203. doi: 10.1073/pnas.1604600113
125. Wang Y, An R, Umanah GK, Park H, Nambiar K, Eacker SM, et al. A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science (2016) 354:aad6872. doi: 10.1126/science.aad6872
126. Ma F, Kouzoukas DE, Meyer-Siegler KL, Hunt DE, Leng L, Bucala R, et al. Macrophage migration inhibitory factor mediates protease-activated receptor 4-induced bladder pain through urothelial high mobility group box 1. Physiol Rep. (2017) 5:e13549. doi: 10.14814/phy2.13549
127. Filip AM, Klug J, Cayli S, Frohlich S, Henke T, Lacher P, et al. Ribosomal protein S19 interacts with macrophage migration inhibitory factor and attenuates its pro-inflammatory function. J Biol Chem. (2009) 284:7977–85. doi: 10.1074/jbc.M808620200
128. Trivedi-Parmar V, Jorgensen WL. Advances and insights for small molecule inhibition of macrophage migration inhibitory factor. J Med Chem. (2018) 61:8104–19. doi: 10.1021/acs.jmedchem.8b00589
129. Kok T, Wasiel AA, Cool RH, Melgert BN, Poelarends GJ, Dekker FJ. Small-molecule inhibitors of macrophage migration inhibitory factor (MIF) as an emerging class of therapeutics for immune disorders. Drug Discov Today (2018) 23:1910–8. doi: 10.1016/j.drudis.2018.06.017
130. Hertelendy J, Reumuth G, Simons D, Stoppe C, Kim BS, Stromps JP, et al. Macrophage migration inhibitory factor - A favorable marker in inflammatory diseases? Curr Med Chem. (2018) 25:601–5. doi: 10.2174/0929867324666170714114200
131. O'Reilly C, Doroudian M, Mawhinney L, Donnelly SC. Targeting MIF in cancer: therapeutic strategies, current developments, and future opportunities. Med Res Rev. (2016) 36:440–60. doi: 10.1002/med.21385
132. Greven D, Leng L, Bucala R. Autoimmune diseases: MIF as a therapeutic target. Exp Opin Ther Targets (2010) 14:253–64. doi: 10.1517/14728220903551304
133. Bianchi ME, Crippa MP, Manfredi AA, Mezzapelle R, Rovere Querini P, Venereau E. High-mobility group box 1 protein orchestrates responses to tissue damage via inflammation, innate and adaptive immunity, and tissue repair. Immunol Rev. (2017) 280:74–82. doi: 10.1111/imr.12601
134. Venereau E, De Leo F, Mezzapelle R, Careccia G, Musco G, Bianchi ME. HMGB1 as biomarker and drug target. Pharmacol Res. (2016) 111:534–44. doi: 10.1016/j.phrs.2016.06.031
135. Pandolfi F, Altamura S, Frosali S, Conti P. Key role of DAMP in inflammation, cancer, and tissue repair. Clin Ther. (2016) 38:1017–28. doi: 10.1016/j.clinthera.2016.02.028
136. Lu B, Wang C, Wang M, Li W, Chen F, Tracey KJ, et al. Molecular mechanism and therapeutic modulation of high mobility group box 1 release and action: an updated review. Expert Rev Clin Immunol. (2014) 10:713–27. doi: 10.1586/1744666X.2014.909730
137. Yu SL, Wong CK, Tam LS. The alarmin functions of high-mobility group box-1 and IL-33 in the pathogenesis of systemic lupus erythematosus. Expert Rev Clin Immunol. (2013) 9:739–49. doi: 10.1586/1744666X.2013.814428
138. Harris HE, Andersson U, Pisetsky DS. HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat Rev Rheumatol. (2012) 8:195–202. doi: 10.1038/nrrheum.2011.222
139. Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. (2011) 29:139–62. doi: 10.1146/annurev-immunol-030409-101323
140. Yang H, Tracey KJ. Targeting HMGB1 in inflammation. Biochim Biophys Acta (2010) 1799:149–56. doi: 10.1016/j.bbagrm.2009.11.019
Keywords: alarmin, chemokine, cytokine, moonlighting, promiscuity, inflammation, cardiovascular disease, MIF protein family
Citation: Kapurniotu A, Gokce O and Bernhagen J (2019) The Multitasking Potential of Alarmins and Atypical Chemokines. Front. Med. 6:3. doi: 10.3389/fmed.2019.00003
Received: 09 November 2018; Accepted: 04 January 2019;
Published: 23 January 2019.
Edited by:
Klaus T. Preissner, University of Giessen, GermanyReviewed by:
Rory R. Koenen, Maastricht University, NetherlandsCopyright © 2019 Kapurniotu, Gokce and Bernhagen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jürgen Bernhagen, anVlcmdlbi5iZXJuaGFnZW5AbWVkLnVuaS1tdWVuY2hlbi5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.