 Serena Bugatti
Serena Bugatti Antonio Manzo
Antonio Manzo Carlomaurizio Montecucco
Carlomaurizio Montecucco
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Med. , 03 December 2018
Sec. Rheumatology
Volume 5 - 2018 | https://doi.org/10.3389/fmed.2018.00339
This article is part of the Research Topic Diagnostics and Treatment Outcomes View all 4 articles
Rheumatoid arthritis (RA) is a highly heterogeneous syndrome in terms of clinical presentation, progression, and response to therapy. In such a complicated context, the identification of disease-related biomarkers would be undoubtedly helpful in assisting tailored approaches for every patient. Despite remarkable efforts, however, progress in new biomarker development and validation is dramatically slow. At present, none of the candidate genetic, cellular, or molecular biomarker has yet surpassed the clinical value of RA-specific autoantibodies, including rheumatoid factor (RF) and anti-citrullinated protein autoantibodies (ACPA). Rather, recent years have witnessed significant advancements in our understanding of the multiple roles that RF and ACPA play in RA pathophysiology. This has helped clarifying the mechanistic basis of the clinical associations of autoantibodies in RA. In this short review, we will briefly summarize the effector functions of RF and ACPA, and analyse how autoantibodies may help subclassifying RA patients in terms of clinical presentation and response to therapy.
Our knowledge on rheumatoid arthritis (RA) has dramatically increased over the past 2 decades. Recognition that RA starts early and key processes of damage even develop before clinically apparent arthritis have fuelled the processes of early diagnosis and prompt treatment institution within the window of opportunity (1, 2). In parallel, better understanding of disease pathogenesis has helped developing new targeted therapeutics that allow good control of signs and symptoms and improve the overall outcomes of the disease (3). Despite these remarkable advances, however, the management of patients with RA remains imprecise. It is widely accepted that RA encompasses great heterogeneity in clinical presentation and outcomes. Yet, any attempt of personalizing treatment based on patients' genotypic and phenotypic characteristics has been largely unsuccessful (4, 5). Despite considerable efforts in producing new taxonomies, the most robust distinction of RA subtypes remains the classification based on the autoantibody status. Patients with rheumatoid factor (RF) and anti-citrullinated protein antibodies (ACPA) have specific genetic and environmental risk factors, and differ from autoantibody-negatives in their clinical course and prognosis (6). The recent discovery of the direct pathogenetic roles of autoantibodies in RA (7) has refueled the possibility of using specific characteristics of the RF and ACPA response as clinically useful biomarkers. In this short review, we will try to summarize the clinical associations of autoantibodies in RA also in light of their mechanistic contribution to disease processes.
The past 10 years of research have shed increasing and exciting light on the multiple roles of autoantibodies (both RF and ACPA) in RA pathology, briefly summarized below.
All immunoglobulins (Ig), and in particular those of the IgG class, have effector functions mediated by the interaction of their Fc tail with specific activating or inhibitory receptors (FcR) present on the surface of various cell types. ACPA detected by second-generation assays are mostly of the IgG class, with predominance of the IgG1 subclass (8), which mediates Ig immunological activities (9). Through this mechanism, immune complexes containing ACPA are able to stimulate tumor necrosis factor (TNF) secretion by macrophages (10–12) (Figure 1A). This stimulus appears to be increased by the simultaneous presence of RF of the IgM and IgA classes, suggesting a synergism between ACPA and RF (12–14). In addition to FcR-mediated cell activation, ACPA can directly activate monocytes by binding through their Fab variable portion to a citrullinated GRP78 cell-surface receptor, driving NF-κB activation, and cytokine production (15) (Figure 1A). Another important effector function of Ig is represented by their ability to activate complement via both the classical and the alternative pathway. Complement activation is also demonstrated for ACPA (16), but is particularly effective in the case of the classic pentameric IgM RF (17). Accordingly, early studies have shown that aggregates of IgG-RF within the joints are associated with low complement levels and low C1q and C3 in synovial fluid (18). More recently, ACPA have been shown to influence inflammation and its chronicity also by increasing the generation of neutrophil extracellular traps (NETs) (19). NETs then stimulate joint inflammation by promoting the production of pro-inflammatory cytokines, chemokines, and adhesion molecules by synovial fibroblasts (19, 20).
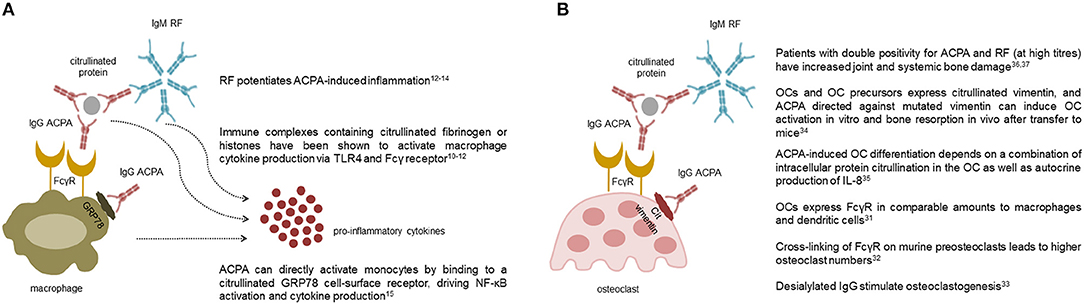
Figure 1. Pro-inflammatory and osteoclastogenic roles of autoantibodies in rheumatoid arthritis. The multiple effects of anti-citrullinated protein antibodies (ACPA) and rheumatoid factor (RF) on macrophages (A) and osteoclasts (B) are shown. Collectively, ACPA are able to stimulate cytokine production by macrophages both through the interaction of their Fc tail with stimulating Fcγ receptors, and through Fab-mediated recognition of membrane citrullinated proteins such as GRP78. Pentameric class M RF potentiates ACPA-induced inflammation likely via activation of the complement cascade (A). ACPA are also able to promote osteoclast differentiation and activation through similar mechanisms mediated by their Fc and Fab portions. Again, clinical studies indicate that RF, especially at high titres, amplifies ACPA-mediate bone damage (B).
Most of the pathological functions of autoantibodies in course of disease have been traditionally attributed to IgGs (macrophage activation through FcγR engagement) and IgMs (complement activation). However, recent evidence has also highlighted important effector functions of antibodies of the IgA class. Indeed, IgA potently activate neutrophils (21), and incubation of RA neutrophils with IgA immune complexes results in the release of NETs (22). These in vitro effects fit with clinical evidences linking the presence of IgA RF to worse disease prognosis (23, 24), and provide intriguing support to the possible mucosal origin of autoantibody responses in RA. Indeed, IgA represents the dominant Ig class in mucosal immune responses, and IgA plasmablasts dominate in ACPA-positive individuals at risk of developing RA (25).
Importantly, the unique effectors functions of autoantibodies are not only mediated by their diversification in classes and subclasses, but also by some specific biochemical characteristics including the sugar moiety attached to the Fc tail (26). In particular, antibodies carrying Fc glycans that lack galactose residues display an increased ability to activate the immune system (27, 28). Interestingly, a decrease in the level of galactosylation is one of the most prominent changes in total serum and ACPA IgG in course of RA (29, 30), whilst levels of anti-inflammatory glycans rise during drug-induced disease remission and pregnancy (31–33).
Altogether, the afore described mechanisms underline the complexity of the inflammatory function of autoantibodies in RA, which depends on intricate and not completely disentangled characteristics including classes, subclasses and biochemical properties. Better understanding of ACPA and RF diversity across the different phases of the disease will hopefully allow important advancements in the biological and clinical understanding of how inflammation develops, evolves and eventually resolves in RA.
The epidemiological association between ACPA and the severity of radiographic damage in RA has recently found causal explanation with the identification of the direct osteoclastogenic role of ACPA (34, 35). In general, immune complexes containing IgG, particularly those which have undergone modifications of their glycan residues, are able to bind to activating FcγR present on the surface of immature and mature osteoclasts and promote their activation (36–38) (Figure 1B). In addition, through their Fab portion, ACPA are able to bind to citrullinated vimentin present on the surface of osteoclasts and their monocyte-macrophage precursors (39), stimulating their differentiation and activation through interleukin (IL)-8 mediated circuits (40) (Figure 1B). Again, the process of ACPA-induced osteoclast activation seems to be amplified by the concomitant presence of RF, as indirectly suggested by radiographic studies which have shown increased amounts of joint and systemic bone loss in patients with double autoantibody-positivity, especially at high titres (41, 42). Altogether, such novel insights into the osteoclastogenic properties of ACPA have thus reinforced earlier concepts on the pathophysiological role of autoantibodies in RA by demonstrating their direct involvement in specific process of the disease such as bone damage.
Besides triggering inflammation and enhancing bone damage, a direct role of ACPA in promoting pain has also been suggested. Indeed, at least in experimental mouse models, the inoculation of ACPA triggers a pain reaction. Such mechanism seems to be mediated by the binding of ACPA to citrullinated epitopes on the surface of marrow osteoclast precursors, inducing the expression of CXCL1 (which is the murine analog of human IL-8), which in turn promotes nociceptive mechanisms (43). Although the nociceptive effect of ACPA in vivo remains to be fully proven, clinical evidences showing that ACPA-positive subjects may suffer from joint pain in the absence of overt synovitis (44) offer intriguing confirmation of this concept.
It is now well-established that autoantibodies precede the onset of clinical symptoms by several years in a proportion of patients with RA (45, 46). The autoantibody response significantly evolves in the pre-clinical phase of the disease, with level increase, epitope spreading, isotype switching, affinity maturation, and change in glycosylation over time (47). Although the precise dynamics of such changes are not properly defined, and are likely to vary among different subjects, there is evidence that the extent of the autoantibody response conditions some clinical characteristics also in the very early phases of RA. Indeed, among autoantibody-positive subjects suffering from arthralgia, those at higher risk of quickly developing RA generally have higher autoantibody levels (48), more epitope spreading (49), and more frequently are ACPA and RF double positive (50). Thus, in a broader sense, autoantibody diversification may have an amplifying effect on inflammation and autoimmunity, leading to the transition from asymptomatic autoimmunity to complex responses triggering clinical inflammation. If this is the case, changes in autoantibody characteristics may serve as useful biomarkers to predict the development of RA and may represent valuable targets for preventive therapeutic strategies.
If early, pre-clinical autoimmunity remains an area of investigation, the clinical associations of autoantibodies in course of established RA have been abundantly analyzed. Literature dating back to the'80 was overall concordant in associating RF positivity, especially at high titers, to more inflammation (51, 52). As such, fluctuations in RF levels were proposed as a biomarker of disease activity and response to therapy (53). Also, RF of the IgA class was regarded as a specific diagnostic marker in early arthritis (54) and, in general, as a marker of more aggressive and refractory disease (23, 24). With ACPA capturing the scene from the 2000s, studies on early RA populations have massively focussed on this new autoantibody system, and interest on RF as a possible serological marker of inflammation has temporarily declined. In spite of their well-established association with more erosive disease, however, ACPA have soon appeared scarcely informative for patients' clinical phenotyping at presentation. In early RA patients from the Leiden early arthritis cohort, van der Helm-van Mil and co-authors (55) indeed reported similar symptoms and joint distribution between ACPA-positive and -negative patients. Number of swollen and tender joints, pain, functional status and acute phase reactants also appeared unaffected by the ACPA status in early RA patients from a Swedish cohort (56). Similarly, in 92 RA patients with symptoms of < 3 months' duration, the number of affected joints and inflammatory markers were comparable, and the only observed difference was the prevalence of knee involvement, slightly more common in ACPA-positive patients (57). More recent analyses have however relooked at clinical associations taking into account the interactions between RF and ACPA, rather than the two autoantibodies separately. In this setting, double autoantibody-positive patients seem to be characterized by a more inflammatory phenotype. In the French ESPOIR cohort, ACPA and RF double-positive patients presented with more marked elevation of acute phase reactants (58). Similarly, a large US study of 1,488 RA patients showed that double positivity for RF and ACPA was associated with significantly higher levels of C-reactive protein and pro-inflammatory cytokines (12). This was consistent with the amplifying effect of RF on TNF production triggered by ACPA in vitro (12). Analyzing the clinical data of patients enrolled in several clinical trials, Aletaha and collaborators (59) confirmed the boosting activity of RF on the background of ACPA, as ACPA single-positive patients had lower levels of disease activity. More recently, Derksen et al. (60) have nicely demonstrated in two large independent early RA cohorts that the amount of inflammation is proportional to the number of autoantibody-specificities, as patients with RF, ACPA and anti-carbamylated protein antibodies all positive had the highest levels of acute phase reactants. If these findings are confirmed, the simple distinction between autoantibody-positive and -negative patients would therefore appear insufficient at capturing different aspects of the disease. Rather, within autoantibody-positive patients, differences might exist depending on the diversity of the autoimmune response, as broader responses (indicating a break of tolerance to more autoantigens) would harbor more severe clinical phenotypes. It is however important to emphasize that the clinical association between the extent of the autoimmune response and disease activity described here does not allow to draw any conclusion on the cause-effect relationship between autoantibodies, in particular RF, and inflammation. On the one hand, the classic IgM RF could indeed represent a pro-inflammatory trigger, in light of its ability to activate complement (17, 18). On the other hand, RF production could be boosted by innate inflammatory stimuli outside a T cell-dependent germinal center reaction (61), therefore representing consequence rather than cause of inflammation.
Another aspect of the disease which appears to be strongly (and possibly mechanistically) linked to the presence of autoantibodies is extra-articular involvement, including cardiovascular (CV) comorbidity. Several studies have reported higher rates of CV mortality and events in autoantibody-positive patients (62, 63). This could be attributed to the higher inflammatory burden associated to autoantibodies, contributing to accelerated atherosclerosis, endothelial dysfunction, and plaque vulnerability (64). Supporting their direct pathogenic role, autoantibodies have been associated with CV events even in the absence of clinically apparent RA (65, 66). Indeed, irrespective of inflammation, autoantibodies might affect cardiac function, as suggested by studies indicating left ventricular strain, impaired left ventricular relaxation, and lower left ventricular mass in association with ACPA (67, 68). The direct effects of ACPA are in line with large epidemiological studies showing an increased rate of CV deaths specifically in ACPA-positive RA patients, whilst RF would be more associated to non-CV mortality (63).
It is however important to note here that the aforementioned associations between autoantibody diversity and local and systemic inflammation refer to patients fulfilling the 1987 classification criteria for RA. There is ample evidence now indicating that the introduction of the 2010 classification criteria has profoundly changed the clinical picture of RA (69). In this new setting, the high weigh attributed to autoantibodies in the scoring system has produced the paradoxical effect of making the clinical presentation of autoantibody-positive patients significantly milder compared to autoantibody-negatives (70, 71). Accordingly, ACPA-positive patients diagnosed with the 2010 criteria do not show more severe disease burden in terms of fatigue, pain, well-being, and independence compared to ACPA-negative patients (72). Also, with decreasing rates of incident CVD, the association between autoantibodies and comorbidities tends to disappear (73).
Disease remission is increasingly achievable in patients with RA provided that diagnosis is established early and treatment instituted effectively (74). However, the definition of remission is challenging, as it also incorporates the concepts of sustainability on treatment as well as persistence after drug suspension (75, 76). In this context, the predictive value of autoantibodies is difficult to establish. Furthermore, RF and ACPA could be related to slightly different remission outcomes.
In historical RA cohorts, RF has been shown to be inversely associated with the achievement of point remission in several studies. In the Utrecht RA cohort established in 1990, 36% of the patients achieved at least one period of remission; among other predictors, RF negativity was associated with 1.63 hazards of achieving remission (77). IgM RF positive patients were less likely to achieve remission also in other cohorts (78, 79), and a recent study on early RA patients commencing methotrexate (MTX) as their first disease modifying anti-rheumatic drug (DMARD) within 3 months of their baseline visit confirmed that RF was associated with earlier MTX failure due to inefficacy (80). In contrast, the predictive value of ACPA for achieving point remission is not that obvious. Surprisingly, studies in early RA cohorts diagnosed according to the 2010 classification criteria have shown that ACPA might rather be regarded as a marker of better outcomes, as ACPA-positive patients more frequently achieve remission upon induction therapy with high doses of MTX (81). Furthermore, broader autoantibody profiles in terms of specificities and classes associate with earlier response to treatment (82). Whether such findings also depend on the lower levels of disease activity in ACPA-positive patients diagnosed according to the 2010 criteria (70) remains to be demonstrated.
RF positivity not only appears associated with lower rates of point remission, but also with reduced chances of maintaining the state of disease remission over time. In a population of 650 RA patients followed for a mean of 10.3 years, Raheel et al. (83) were able to demonstrate that patients with positive RF had higher rates of flare compared to RF-negatives. In the IMPROVED study, positivity for RF was confirmed to be predictive of losing remission in course of drug tapering (84). Interestingly enough, in the same study, ACPA did not show a similar association, as ACPA-positive patients achieving early remission could taper medications similarly to ACPA-negatives (84). Very recently, data on the ARCTIC trial, in which treatment adjustments were tightly targeted on disease remission, have confirmed that RF, but not ACPA positivity was associated with not reaching sustained remission in univariate analysis (85).
If ACPA thus appear rather ininfluent in the achievement and maintenance of remission on treatment, their role in predicting disease relapse after drug suspension is undoubtedly established. In the Leiden Early Arthritis Clinic and the British Early Rheumatoid Arthritis Study, sustained DMARD-free remission was achieved far less commonly by ACPA-positive patients (86). Similar results were confirmed in the context of clinical trials (87), including again the IMPROVED study in which ACPA-positive patients showed more relapses in disease activity soon after drug suspension (84). This findings have solid evidence also in the recent RETRO trial (88–90). Here, ACPA-positivity and broader ACPA reactivities emerged as the strongest predictors of disease recurrence independent of the level of residual disease activity at drug tapering/discontinuation. Importantly, ACPA appear associated to increased hazards of losing remission irrespective of the type of drug being suspended, as studies on TNF and non-TNF drug tapering have shown similar results compared with conventional synthetic (cs) DMARD suspension (91).
Collectively, data from the literature thus indicate that both RF and ACPA are important factors related to remission outcomes. RF would mostly predict lower chances of achieving and maintaining remission on treatment, whilst ACPA would represent the major obstacle for safe drug suspension.
Despite being unanimously considered as important prognostic markers associated with more rapid evolution of joint damage, RA-specific autoantibodies, and in particular ACPA, have never been recommended as a guidance to assist the choice of first-line treatment in patients with early RA. Indeed, both the European and the American recommendations push for MTX as the anchor drug in all patients irrespective of autoantibody positivity (74, 92). This recommendation funds on evidences indicating that the worse radiographic progression observed in ACPA-positive RA is not a consequence of continuing disease activity, as response to therapy seems overall comparable irrespective of the serological subgroup. Among the several studies published so far, the more robust evidence of this concept probably comes from the long-term follow-up of the BEST strategy trial (93). Here, despite more radiological damage progression, ACPA-positive patients achieved reduction in disease activity similar to that of ACPA-negative patients in all treatment groups. Similarly, in the METEOR database including more than 1,800 patients with MTX as part of their initial treatment strategy, short-term remission in newly diagnosed RA was not affected by neither ACPA nor RF positivity (94). The analysis of ACPA reactivities does not seem to add much to prediction of treatment response, as recently shown in the context of the ARCTIC trial, were no difference in median baseline levels or number of ACPA reactivities was found between patients with different EULAR responses to MTX monotherapy (95).
If MTX efficacy appears overall similar between autoantibody-positive and –negative patients, still some differences in the magnitude of the response according to the serological status might exist. In patients with undifferentiated arthritis according to the 1987 criteria, MTX was effective at delaying progression to RA only in ACPA-positive patients, many of whom could be reclassified as having had RA according to the 2010 criteria at baseline (96). Similarly, in the IMPROVED study, ACPA-positivity was associated with higher chances of achieving remission upon induction treatment with MTX at the dose of 25 mg/week (81). In line with these data, results from our early arthritis inception cohort indicate that higher starting doses of MTX (15 mg/week) are more effective than lower doses (10 mg/week) in autoantibody-positive but not autoantibody-negative patients (97). The differential response to more aggressive strategies with csDMARDs also emerges from the CARDERA trial (98). Here, triple therapy with MTX, cyclosporine and prednisolone was more effective at reducing disease activity compared to MTX monotherapy only in ACPA-positive early RA patients.
Based on the above described evidences, it would be tempting to speculate that, by interfering with both the innate and the adaptive branches of the immune system (99), MTX at high doses is particularly effective in autoantibody-positive RA. However, one cannot exclude that the observed differences are actually related to misdiagnoses in patients lacking autoantibodies, rather than to intrinsic pathogenetic pathways differentially susceptible to different treatment strategies.
If the differential response to csDMARDs according to the autoantibody profile remains questionable, the impact of the serological status on response to biological (b) DMARDs targeting different immune pathways is, at least for certain mechanisms of actions, more defined.
It can be easily appreciated that bDMARDs selectively affecting the process of autoantibody production, including B cell depletion or inhibition of T cell costimulation, are particularly effective in autoantibody-positive patients. A meta-analysis of four randomized placebo-controlled trials from the rituximab clinical programme confirmed the additional treatment benefit in autoantibody-positive RA, particularly in patients for whom at least one TNF inhibitor had failed, and without significant differences between RF and ACPA (100). Similarly, a meta-analysis of 23 clinical trials and observational studies showed that positivity for RF at baseline predicted better response to rituximab according to both the ACR and the EULAR criteria (101). In recent times, ACPA positivity has emerged as particularly relevant in affecting the response to inhibition of T cell co-stimulation. A post-hoc analysis of the AMPLE trial in 2016 initially showed that very high levels of ACPA at baseline were associated with better clinical response to abatacept but not to adalimumamb (102). Such association has found confirmation in several independent real-life studies. In the French ORA registry, the proportion of ACPA-positivity was significantly higher among EULAR responders at 6 months (103). Data from other registries and real-world studies, such as the PanEuropean registry and the ACTION study, have shown that baseline autoantibodies impact on the retention rate of abatacept, as a surrogate marker of efficacy and safety (104, 105). Again, RF and ACPA appear both associated with better efficacy and lower risk of abatacept discontinuation for any reason (105, 106). It is also interesting to note that both B cell depleting agents and co-stimulation blockade selectively and equally affect not only RF, but also ACPA levels, thus confirming direct interference with adaptive immune responses (107). Although the clinical meaning of reduced ACPA titres upon treatment remains to be determined, initial evidence is starting to suggest that conversion to seronegative status may be associated with better clinical and radiographic outcomes (108).
Studies on the prognostic value of autoantibodies for response to therapy to bDMARDs targeting different mechanisms of action are less conclusive. The assessment of response to TNF inhibitors according to RF and/or ACPA status has provided conflicting results. A systematic literature review of 14 studies involving more than 5,000 patients treated with infliximab, etanercept, adalimumab, and golimumab demonstrated that neither RF nor ACPA were associated with differential response (109). However, the heterogeneity of the analysis was high, leaving results largely uncertain. Some studies have indeed highlighted lower rates of response in patients with high levels of RF (24, 110), particularly of the IgA class (24). These results have very recently been confirmed by post-hoc analyses on the RISING study, in which the dose of infliximab was progressively escalated (111, 112). In this study, patients with levels of RF and ACPA both high were characterized by higher levels of TNF, lower levels of infliximab and lower clinical response. Tociluzumab has even more discordant data. Indeed, response to IL-6 targeting does not seem to be affected by the ACPA status (113, 114). In contrast, high levels of RF would appear to favor tociluzimab treatment according to one report (114) and one meta-analysis (101). However, the mechanism of action of IL-6 antagonism in RA is complex and includes interference with both the innate and adaptive arms of the immune system. Accordingly, blockade of IL-6 receptor suppresses T- and B-cell activation signatures in RA synovium similarly to rituximab, but differently from TNF-antagonists (115). It is therefore very likely that those patients with the highest levels of activation of the B cell compartment also show preferential response to IL-6 targeting. In line with this concept, RA patients showing dense infiltration of lymphoid cells in their synovial membrane, as well as elevated serum levels of factors involved in B cell recirculation and positioning (such as the chemokine CXCL13), respond to tociluzumab better than to adalimumamb (116).
Given the complex pathophysiology and the clinical heterogeneity of RA, biomarkers capable of guiding personalized treatment approaches are urgently needed. RA would indeed benefit from personalized approaches across all the phases of disease development and evolution (Figure 2). Prompt recognition of at risk subject would indeed pave the way for preventive strategies. Soon after disease diagnosis, the precise identification of those patients in whom conventional, less expensive, and safe drugs such as MTX are more likely to be effective would allow significant savings for individuals and the society as a whole. Also, a more detailed and informed understanding of the phase of clinical remission would improve our ability to tailor treatment and follow-up to the individual patient. Several lines of evidence indeed indicate that, once remission has been achieved, the decision of tapering therapies is largely empirical, with high rates of insuccess. While awaiting new prognostic markers, rheumatologist may still obtain valuable information from traditional factors including autoantibodies. At present, neither RF nor ACPA can be reliably used as absolute markers of prognosis and response to treatment. However, the field of immunology is moving fast, and has opened new perspectives that, in the near future, could have important clinical translation. We believe that further refinement of the analytic procedures capable of capturing the broad spectrum of autoantibody characteristics, including classes, subclasses, specificities and biochemical properties, will rapidly allow better subclassification of patients with RA.
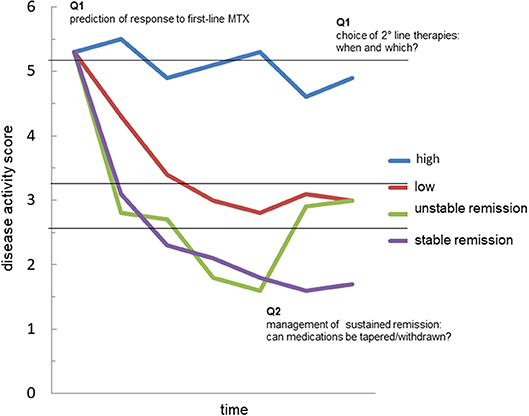
Figure 2. The possible value of autoantibodies as biomarkers in rheumatoid arthritis. The graph represents the possible trajectories of disease activity over time in patients with rheumatoid arthritis (RA): persistently high disease activity despite methotrexate (MTX) treatment (blue); satisfactory control of disease activity with a disease activity score indicative of low disease (red); achievement of remission, which is however unstable at follow-up (green); achievement of sustained remission (purple). Accurate prediction of such trajectories in every patient is currently an unmet need. The analysis of the fine characteristics of the autoantibody response in course of RA, including autoantibody levels, avidity, classes, subclasses and biochemical properties might generate useful biomarkers capable of answering critical questions such as: (Q1) the identification of those patients more likely to respond to first-line treatment with methotrexate (MTX); (Q2) the choice of second-line drug, among several mechanisms of actions available, in MTX-failures; (Q3) the possibility of tapering/stopping medications in patients achieving remission.
✓ The dichotomic classification of patients with RA into autoantibody-positive and -negative solely based on RF and/or ACPA appears too simplistic.
✓ The autoantibody system in RA is likely to expand rapidly with the identification of new antigenic specificities.
✓ The pathogenic properties of autoantibodies depend on several characteristics including levels, avidity, classes, subclasses, and biochemical properties.
✓ All of these properties are likely to be modulated in different patients during different phases of the disease.
✓ Further studies are needed to test whether the different characteristics of autoantibodies, alone or in combination, may serve as biomarkers for:
• prediction of disease development in at risk individuals
• prediction of response to MTX
• choice of the second-line drug in MTX-failures
• identification of patients in whom remission is more likely to persist even after drug tapering/discontinuation
SB and AM prepared a first draft of the manuscript. RC and CM critically reviewed the manuscript. All authors conceived the idea of this review article and approved the final version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Smolen JS, Aletaha D, Barton A, Burmester GR, Emery P, Firestein GS, et al. Rheumatoid arthritis. Nat Rev Dis Primers (2018) 4:18001. doi: 10.1038/nrdp.2018.1
2. Monti S, Montecucco C, Bugatti S, Caporali R. Rheumatoid arthritis treatment: the earlier the better to prevent joint damage. RMD Open (2015) 1(Suppl. 1):e000057. doi: 10.1136/rmdopen-2015-000057
3. Nam JL, Takase-Minegishi K, Ramiro S, Chatzidionysiou K, Smolen JS, van der Heijde D, et al. Efficacy of biological disease-modifying antirheumatic drugs: a systematic literature review informing the 2016 update of the EULAR recommendations for the management of rheumatoid arthritis. Ann Rheum Dis. (2017) 76:1113–36. doi: 10.1136/annrheumdis-2016-210713
4. Bluett J, Barton A. Precision medicine in rheumatoid arthritis. Rheum Dis Clin North Am. (2017) 43:377–87. doi: 10.1016/j.rdc.2017.04.008
5. Romão VC, Vital EM, Fonseca JE, Buch MH. Right drug, right patient, right time: aspiration or future promise for biologics in rheumatoid arthritis? Arthritis Res Ther. (2017) 19:239. doi: 10.1186/s13075-017-1445-3
6. Malmström V, Catrina AI, Klareskog L. The immunopathogenesis of seropositive rheumatoid arthritis: from triggering to targeting. Nat Rev Immunol. (2017) 17:60–75. doi: 10.1038/nri.2016.124
7. Derksen VFAM, Huizinga TWJ, van der Woude D. The role of autoantibodies in the pathophysiology of rheumatoid arthritis. Semin Immunopathol. (2017) 39:437–46. doi: 10.1007/s00281-017-0627-z
8. Engelmann R, Brandt J, Eggert M, Karberg K, Krause A, Neeck G, et al. IgG1 and IgG4 are the predominant subclasses among auto-antibodies against two citrullinated antigens in RA. Rheumatology (2008) 47:1489–92. doi: 10.1093/rheumatology/ken336
9. Nimmerjahn F, Ravetch JV. Antibody-mediated modulation of immune responses. Immunol Rev. (2010) 236:265–75. doi: 10.1111/j.1600-065X.2010.00910.x
10. Clavel C, Nogueira L, Laurent L, Iobagiu C, Vincent C, Sebbag M, et al. Induction of macrophage secretion of tumor necrosis factor alpha through Fcgamma receptor IIa engagement by rheumatoid arthritis-specific autoantibodies to citrullinated proteins complexed with fibrinogen. Arthritis Rheum. (2008) 58:678–88. doi: 10.1002/art.23284
11. Laurent L, Clavel C, Lemaire O, Anquetil F, Cornillet M, Zabraniecki L, et al. Fcγ receptor profile of monocytes and macrophages from rheumatoid arthritis patients and their response to immune complexes formed with autoantibodies to citrullinated proteins. Ann Rheum Dis. (2011) 70:1052–9. doi: 10.1136/ard.2010.142091
12. Sokolove J, Johnson DS, Lahey LJ, Wagner CA, Cheng D, Thiele GM, et al. Rheumatoid factor as a potentiator of anti-citrullinated protein antibody-mediated inflammation in rheumatoid arthritis. Arthritis Rheumatol. (2014) 66:813–21. doi: 10.1002/art.38307
13. Laurent L, Anquetil F, Clavel C, Ndongo-Thiam N, Offer G, Miossec P, et al. IgM rheumatoid factor amplifies the inflammatory response of macrophages induced by the rheumatoid arthritis-specific immune complexes containing anticitrullinated protein antibodies. Ann Rheum Dis. (2015) 74:1425–31. doi: 10.1136/annrheumdis-2013-204543
14. Anquetil F, Clavel C, Offer G, Serre G, Sebbag M. IgM and IgA rheumatoid factors purified from rheumatoid arthritis sera boost the Fc receptor- and complement-dependent effector functions of the disease-specific anti-citrullinated protein autoantibodies. J Immunol. (2015) 194:3664–74. doi: 10.4049/jimmunol.1402334
15. Lu MC, Lai NS, Yu HC, Huang HB, Hsieh SC, Yu CL. Anti-citrullinated protein antibodies bind surface-expressed citrullinated Grp78 on monocyte/macrophages and stimulate tumor necrosis factor alpha production. Arthritis Rheum. (2010) 62:1213–23. doi: 10.1002/art.27386
16. Trouw LA, Haisma EM, Levarht EW, van der Woude D, Ioan-Facsinay A, Daha MR, et al. Anti-cyclic citrullinated peptide antibodies from rheumatoid arthritis patients activate complement via both the classical and alternative pathways. Arthritis Rheum. (2009) 60:1923–31. doi: 10.1002/art.24622
17. Okroj M, Heinegård D, Holmdahl R, Blom AM. Rheumatoid arthritis and the complement system. Ann Med. (2007) 39:517–30. doi: 10.1080/07853890701477546
18. Winchester RJ, Agnello V, Kunkel HG. Gamma globulin complexes in synovial fluids of patients with rheumatoid arthritis. Partial characterization and relationship to lowered complement levels. Clin Exp Immunol. (1970) 6:689–706.
19. Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. (2013) 5:178ra40. doi: 10.1126/scitranslmed.3005580
20. Carmona-Rivera C, Carlucci PM, Moore E, Lingampalli N, Uchtenhagen H, James E, et al. Synovial fibroblast-neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis. Sci Immunol. (2017) 2:eaag3358. doi: 10.1126/sciimmunol.aag3358
21. van der Steen LP, Bakema JE, Sesarman A, Florea F, Tuk CW, Kirtschig G, et al. Blocking Fcα receptor I on granulocytes prevents tissue damage induced by IgA autoantibodies. J Immunol. (2012) 189:1594–601. doi: 10.4049/jimmunol.1101763
22. Aleyd E, Al M, Tuk CW, van der Laken CJ, van Egmond M. IgA complexes in plasma and synovial fluid of patients with rheumatoid arthritis induce neutrophil extracellular traps via FcαRI. J Immunol. (2016) 197:4552–9. doi: 10.4049/jimmunol.1502353
23. Teitsson I, Withrington RH, Seifert MH, Valdimarsson H. Prospective study of early rheumatoid arthritis. I. Prognostic value of IgA rheumatoid factor. Ann Rheum Dis. (1984) 43:673–8. doi: 10.1136/ard.43.5.673
24. Bobbio-Pallavicini F, Caporali R, Alpini C, Avalle S, Epis OM, Klersy C, et al. High IgA rheumatoid factor levels are associated with poor clinical response to tumour necrosis factor alpha inhibitors in rheumatoid arthritis. Ann Rheum Dis. (2007) 66:302–7. doi: 10.1136/ard.2006.060608
25. Kinslow JD, Blum LK, Deane KD, Demoruelle MK, Okamoto Y, Parish MC, et al. Elevated IgA plasmablast levels in subjects at risk of developing rheumatoid arthritis. Arthritis Rheumatol. (2016) 68:2372–83. doi: 10.1002/art.39771
26. Seeling M, Brückner C, Nimmerjahn F. Differential antibody glycosylation in autoimmunity: sweet biomarker or modulator of disease activity? Nat Rev Rheumatol. (2017) 13:621–30. doi: 10.1038/nrrheum.2017.146
27. Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science (2006) 313:670–3. doi: 10.1126/science.1129594
28. Rademacher TW, Williams P, Dwek RA. Agalactosyl glycoforms of IgG autoantibodies are pathogenic. Proc Natl Acad Sci USA. (1994) 91:6123–7. doi: 10.1073/pnas.91.13.6123
29. Tomana M, Schrohenloher RE, Koopman WJ, Alarcón GS, Paul WA. Abnormal glycosylation of serum IgG from patients with chronic inflammatory diseases. Arthritis Rheum. (1988) 31:333–8. doi: 10.1002/art.1780310304
30. Scherer HU, van der Woude D, Ioan-Facsinay A, el Bannoudi H, Trouw LA, Wang J, et al. Glycan profiling of anti-citrullinated protein antibodies isolated from human serum and synovial fluid. Arthritis Rheum. (2010) 62:1620–9. doi: 10.1002/art.27414
31. Ercan A, Cui J, Hazen MM, Batliwalla F, Royle L, Rudd PM, et al. Hypogalactosylation of serum N-glycans fails to predict clinical response to methotrexate and TNF inhibition in rheumatoid arthritis. Arthritis Res Ther. (2012) 14:r43. doi: 10.1186/ar3756
32. Gindzienska-Sieśkiewicz E, Radziejewska I, Domysławska I, Klimiuk PA, Sulik A, Rojewska J, et al. Changes of glycosylation of IgG in rheumatoid arthritis patients treated with methotrexate. Adv Med Sci. (2016) 61:193–7. doi: 10.1016/j.advms.2015.12.009
33. Rook GA, Steele J, Brealey R, Whyte A, Isenberg D, Sumar N, et al. Changes in IgG glycoform levels are associated with remission of arthritis during pregnancy. J Autoimmun. (1991) 4:779–94. doi: 10.1016/0896-8411(91)90173-A
34. Catrina AI, Svensson CI, Malmström V, Schett G, Klareskog L. Mechanisms leading from systemic autoimmunity to joint-specific disease in rheumatoid arthritis. Nat Rev Rheumatol. (2017) 13:79–86. doi: 10.1038/nrrheum.2016.200
35. Bugatti S, Bogliolo L, Montecucco C, Manzo A. B cell autoimmunity and bone damage in rheumatoid arthritis. Reumatismo (2016) 68:117–25. doi: 10.4081/reumatismo.2016.914
36. Grevers LC, de Vries TJ, Everts V, Verbeek JS, van den Berg WB, van Lent PL. Immune complex-induced inhibition of osteoclastogenesis is mediated via activating but not inhibitory Fcγ receptors on myeloid precursor cells. Ann Rheum Dis. (2013) 72:278–85. doi: 10.1136/annrheumdis-2012-201568
37. Seeling M, Hillenhoff U, David JP, Schett G, Tuckermann J, Lux A, et al. Inflammatory monocytes and Fcγ receptor IV on osteoclasts are critical for bone destruction during inflammatory arthritis in mice. Proc Natl Acad Sci USA. (2013) 110:10729–34. doi: 10.1073/pnas.1301001110
38. Harre U, Lang SC, Pfeifle R, Rombouts Y, Frühbeißer S, Amara K, et al. Glycosylation of immunoglobulin G determines osteoclast differentiation and bone loss. Nat Commun. (2015) 6:6651. doi: 10.1038/ncomms7651
39. Harre U, Georgess D, Bang H, Bozec A, Axmann R, Ossipova E, et al. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J Clin Invest. (2012) 122:1791–802. doi: 10.1172/JCI60975
40. Krishnamurthy A, Joshua V, Haj Hensvold A, Jin T, Sun M, Vivar N, et al. Identification of a novel chemokine-dependent molecular mechanism underlying rheumatoid arthritis-associated autoantibody-mediated bone loss. Ann Rheum Dis. (2016) 75:721–9. doi: 10.1136/annrheumdis-2015-208093
41. Hecht C, Englbrecht M, Rech J, Schmidt S, Araujo E, Engelke K, et al. Additive effect of anti-citrullinated protein antibodies and rheumatoid factor on bone erosions in patients with RA. Ann Rheum Dis. (2015) 74:2151–6. doi: 10.1136/annrheumdis-2014-205428
42. Bugatti S, Bogliolo L, Vitolo B, Manzo A, Montecucco C, Caporali R. Anti-citrullinated protein antibodies and high levels of rheumatoid factor are associated with systemic bone loss in patients with early untreated rheumatoid arthritis. Arthritis Res Ther. (2016) 18:226. doi: 10.1186/s13075-016-1116-9
43. Wigerblad G, Bas DB, Fernades-Cerqueira C, Krishnamurthy A, Nandakumar KS, Rogoz K, et al. Autoantibodies to citrullinated proteins induce joint pain independent of inflammation via a chemokine-dependent mechanism. Ann Rheum Dis. (2016) 75:730–8. doi: 10.1136/annrheumdis-2015-208094
44. van Steenbergen HW, Aletaha D, Beaart-van de Voorde LJ, Brouwer E, Codreanu C, Combe B, et al. EULAR definition of arthralgia suspicious for progression to rheumatoid arthritis. Ann Rheum Dis. (2017) 76:491–6. doi: 10.1136/annrheumdis-2016-209846
45. Nielen MM, van Schaardenburg D, Reesink HW, van de Stadt RJ, van der Horst-Bruinsma IE, de Koning MH, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. (2004) 50:380–6. doi: 10.1002/art.20018
46. Rantapää-Dahlqvist S, de Jong BA, Berglin E, Hallmans G, Wadell G, Stenlund H, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. (2003) 48:2741–9. doi: 10.1002/art.11223
47. Falkenburg WJJ, van Schaardenburg D. Evolution of autoantibody responses in individuals at risk of rheumatoid arthritis. Best Pract Res Clin Rheumatol. (2017) 31:42–52. doi: 10.1016/j.berh.2017.07.005
48. Rakieh C, Nam JL, Hunt L, Hensor EM, Das S, Bissell LA, et al. Predicting the development of clinical arthritis in anti-CCP positive individuals with non-specific musculoskeletal symptoms: a prospective observational cohort study. Ann Rheum Dis. (2015) 74:1659–66. doi: 10.1136/annrheumdis-2014-205227
49. van de Stadt LA, van der Horst AR, de Koning MH, Bos WH, Wolbink GJ, van de Stadt RJ, et al. The extent of the anti-citrullinated protein antibody repertoire is associated with arthritis development in patients with seropositive arthralgia. Ann Rheum Dis. (2011) 70:128–33. doi: 10.1136/ard.2010.132662
50. Bos WH, Wolbink GJ, Boers M, Tijhuis GJ, de Vries N, van der Horst-Bruinsma IE, et al. Arthritis development in patients with arthralgia is strongly associated with anti-citrullinated protein antibody status: a prospective cohort study. Ann Rheum Dis. (2010) 69:490–4. doi: 10.1136/ard.2008.105759
51. Withrington RH, Teitsson I, Valdimarsson H, Seifert MH. Prospective study of early rheumatoid arthritis. II. Association of rheumatoid factor isotypes with fluctuations in disease activity. Ann Rheum Dis. (1984) 43:679–85. doi: 10.1136/ard.43.5.679
52. Robbins DL, Feigal DW Jr, Leek JC. Relationship of serum IgG rheumatoid factor to IgM rheumatoid factor and disease activity in rheumatoid arthritis. J Rheumatol. (1986) 13:259–62.
53. Mikuls TR, O'Dell JR, Stoner JA, Parrish LA, Arend WP, Norris JM, et al. Association of rheumatoid arthritis treatment response and disease duration with declines in serum levels of IgM rheumatoid factor and anti-cyclic citrullinated peptide antibody. Arthritis Rheum. (2004) 50:3776–82. doi: 10.1002/art.20659
54. Bas S, Genevay S, Meyer O, Gabay C. Anti-cyclic citrullinated peptide antibodies, IgM and IgA rheumatoid factors in the diagnosis and prognosis of rheumatoid arthritis. Rheumatology (2003) 42:677–80. doi: 10.1093/rheumatology/keg184
55. van der Helm-van Mil AH, Verpoort KN, Breedveld FC, Toes RE, Huizinga TW. Antibodies to citrullinated proteins and differences in clinical progression of rheumatoid arthritis. Arthritis Res Ther. (2005) 7:R949–58. doi: 10.1186/ar1767
56. Rönnelid J, Wick MC, Lampa J, Lindblad S, Nordmark B, Klareskog L, et al. Longitudinal analysis of citrullinated protein/peptide antibodies (anti-CP) during 5 year follow up in early rheumatoid arthritis: anti-CP status predicts worse disease activity and greater radiological progression. Ann Rheum Dis. (2005) 64:1744–9. doi: 10.1136/ard.2004.033571
57. Cader MZ, Filer AD, Buckley CD, Raza K. The relationship between the presence of anti-cyclic citrullinated peptide antibodies and clinical phenotype in very early rheumatoid arthritis. BMC Musculoskelet Disord. (2010) 11:187. doi: 10.1186/1471-2474-11-187
58. Mouterde G, Lukas C, Goupille P, Flipo RM, Rincheval N, Daurès JP, et al. Association of anticyclic citrullinated peptide antibodies and/or rheumatoid factor status and clinical presentation in early arthritis: results from the ESPOIR cohort. J Rheumatol. (2014) 41:1614–22. doi: 10.3899/jrheum.130884
59. Aletaha D, Alasti F, Smolen JS. Rheumatoid factor, not antibodies against citrullinated proteins, is associated with baseline disease activity in rheumatoid arthritis clinical trials. Arthritis Res Ther. (2015) 17:229. doi: 10.1186/s13075-015-0736-9
60. Derksen VF, Ajeganova S, Trouw LA, van der Helm-van Mil AH, Hafström I, Huizinga TW, et al. Rheumatoid arthritis phenotype at presentation differs depending on the number of autoantibodies present. Ann Rheum Dis. (2017) 76:716–20. doi: 10.1136/annrheumdis-2016-209794
61. Elliott SE, Kongpachith S, Lingampalli N, Adamska JZ, Cannon BJ, Mao R, et al. Affinity maturation drives epitope spreading and generation of pro-inflammatory anti-citrullinated protein antibodies in rheumatoid arthritis. Arthritis Rheumatol. (2018). doi: 10.1002/art.40587. [Epub ahead of print].
62. López-Longo FJ, Oliver-Mi-arro D, de la Torre I, González-Díaz de Rábago E, Sánchez-Ramón S, Rodríguez-Mahou M, et al. Association between anti-cyclic citrullinated peptide antibodies and ischemic heart disease in patients with rheumatoid arthritis. Arthritis Rheum. (2009) 61:419–24. doi: 10.1002/art.24390
63. Ajeganova S, Humphreys JH, Verheul MK, van Steenbergen HW, van Nies JA, Hafström I, et al. Anticitrullinated protein antibodies and rheumatoid factor are associated with increased mortality but with different causes of death in patients with rheumatoid arthritis: a longitudinal study in three European cohorts. Ann Rheum Dis. (2016) 75:1924–32. doi: 10.1136/annrheumdis-2015-208579
64. Ridker PM. From C-reactive protein to Interleukin-6 to Interleukin-1: moving upstream to identify novel targets for atheroprotection. Circ Res. (2016) 118:145–56. doi: 10.1161/CIRCRESAHA.115.306656
65. Majka DS, Vu TT, Pope RM, Teodorescu M, Karlson EW, Liu K, et al. Association of rheumatoid factors with subclinical and clinical atherosclerosis in African American women: the multiethnic study of atherosclerosis. Arthritis Care Res. (2017) 69:166–174. doi: 10.1002/acr.22930
66. Cambridge G, Acharya J, Cooper JA, Edwards JC, Humphries SE. Antibodies to citrullinated peptides and risk of coronary heart disease. Atherosclerosis (2013) 228:243–6. doi: 10.1016/j.atherosclerosis.2013.02.009
67. Marasovic-Krstulovic D, Martinovic-Kaliterna D, Fabijanic D, Morovic-Vergles J. Are the anti-cyclic citrullinated peptide antibodies independent predictors of myocardial involvement in patients with active rheumatoid arthritis? Rheumatology (2011) 50:1505–12. doi: 10.1093/rheumatology/ker121
68. Giles JT, Malayeri AA, Fernandes V, Post W, Blumenthal RS, Bluemke D, et al. Left ventricular structure and function in patients with rheumatoid arthritis, as assessed by cardiac magnetic resonance imaging. Arthritis Rheum. (2010) 62:940–51. doi: 10.1002/art.27349
69. Mjaavatten MD, Bykerk VP. Early rheumatoid arthritis: the performance of the 2010 ACR/EULAR criteria for diagnosing RA. Best Pract Res Clin Rheumatol. (2013) 27:451–66. doi: 10.1016/j.berh.2013.09.001
70. Nordberg LB, Lillegraven S, Lie E, Aga AB, Olsen IC, Hammer HB, et al. Patients with seronegative RA have more inflammatory activity compared with patients with seropositive RA in an inception cohort of DMARD-naïve patients classified according to the 2010 ACR/EULAR criteria. Ann Rheum Dis. (2017) 76:341–5. doi: 10.1136/annrheumdis-2015-208873
71. van der Helm-van Mil AH, Zink A. What is rheumatoid arthritis? Considering consequences of changed classification criteria. Ann Rheum Dis. (2017) 76:315–7. doi: 10.1136/annrheumdis-2016-209629
72. Boer AC, Boonen A, van der Helm van Mil AHM. Is anti-citrullinated protein antibody-positive rheumatoid arthritis still a more severe disease than anti-citrullinated protein antibody-negative rheumatoid arthritis? A longitudinal cohort study in rheumatoid arthritis patients diagnosed from 2000 onward. Arthritis Care Res. (2018) 70:987–96. doi: 10.1002/acr.23497
73. Barra LJ, Pope JE, Hitchon C, Boire G, Schieir O, Lin D, et al. The effect of rheumatoid arthritis-associated autoantibodies on the incidence of cardiovascular events in a large inception cohort of early inflammatory arthritis. Rheumatology (2017) 56:768–76. doi: 10.1093/rheumatology/kew474
74. Smolen JS, Landewé R, Bijlsma J, Burmester G, Chatzidionysiou K, Dougados M, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. (2017) 76:960–77. doi: 10.1136/annrheumdis-2016-210715
75. Ajeganova S, Huizinga T. Sustained remission in rheumatoid arthritis: latest evidence and clinical considerations. Ther Adv Muscuoloskelet Dis. (2017) 9:249–62. doi: 10.1177/1759720X17720366
76. Bugatti S, Sakellariou G, Luvaro T, Greco MI, Manzo A. Clinical, imaging, and pathological suppression of synovitis in rheumatoid arthritis: is the disease curable? Front Med. (2018) 5:140. doi: 10.3389/fmed.2018.00140
77. Verstappen SM, van Albada-Kuipers GA, Bijlsma JW, Blaauw AA, Schenk Y, Haanen HC, et al. A good response to early DMARD treatment of patients with rheumatoid arthritis in the first year predicts remission during follow up. Ann Rheum Dis. (2005) 64:38–43. doi: 10.1136/ard.2003.014928
78. Pease CT, Bhakta BB, Devlin J, Emery P. Does the age at onset of rheumatoid arthritis influence phenotype?: a prospective study of outcome and prognostic factors. Rheumatology (1999) 38:228–34. doi: 10.1093/rheumatology/38.3.228
79. Forslind K, Hafstrom I, Ahlmen M, Svensson B, and the BARFOT Study Group. Sex: a major predictor of remission in early rheumatoid arthritis? Ann Rheum Dis. (2007) 66:46–52. doi: 10.1136/ard.2006.056937
80. Bluett J, Sergeant JC, MacGregor AJ, Chipping JR, Marshall T, Symmons DPM, et al. Risk factors for oral methotrexate failure in patients with inflammatory polyarthritis: results from a UK prospective cohort study. Arthritis Res Ther. (2018) 20:50. doi: 10.1186/s13075-018-1544-9
81. Wevers-de Boer K, Visser K, Heimans L, Ronday HK, Molenaar E, Groenendael JH, et al. Remission induction therapy with methotrexate and prednisone in patients with early rheumatoid and undifferentiated arthritis (the IMPROVED study). Ann Rheum Dis. (2012) 71:1472–7. doi: 10.1136/annrheumdis-2011-200736
82. De Moel EC, Derksen VFAM, Stoeken G, Trouw LA, Bang H, Goekoop RJ, et al. Baseline autoantibody profile in rheumatoid arthritis is associated with early treatment response but not long-term outcomes. Arthritis Res Ther. (2018) 20:33. doi: 10.1186/s13075-018-1520-4
83. Raheek S, matteson EL, Crowson CS, Myasoedova E. Improved flare and remission pattern in rheumatoid arthritis over recent decades: a population-based study. Rheumatology (2017) 56:2154–61. doi: 10.1093/rheumatology/kex352
84. Wevers-de Boer KVC, Heimans L, Visser K, Schouffoer AA, Molenaar ETH, van Groenendael JHLM, et al. Determinants of reaching drug-free remission in patients with early rheumatoid or undifferentiated arthritis after one year of remission-steered treatment. Rheumatology (2015) 54:1380–4. doi: 10.1093/rheumatology/keu477
85. Paulshus Sundlisæter N, Olsen IC, Aga AB, Hammer HB, Uhlig T, van der Heijde D, et al. Predictors of sustained remission in patients with early rheumatoid arthritis treated according to an aggressive treat-to-target protocol. Rheumatology (2018) 57:2022–31. doi: 10.1093/rheumatology/key202
86. Van der Woude D, Young A, Jayakumar K, Mertens BJ, Toes RE, van der Heijde D, et al. Prevalence and predictive factors for sustained disease-modifying antirheumatic drug-free remission in rheumatoid arthritis: results from two large early arthritis cohorts. Arthritis Rheum. (2009) 60:2262–71. doi: 10.1002/art.24661
87. Klarenbeek NB, van der Kooij SM, Güler-Yüksel M, van Groenendael JH, Han KH, Kerstens PJ, et al. Discontinuing treatment in patients with rheumatoid arthritis in sustained clinical remission: exploratory analyses from the BeSt study. Ann Rheum Dis. (2011) 70:315–9. doi: 10.1136/ard.2010.136556
88. Haschka J, Englbrecht M, Hueber AJ, Manger B, Kleyer A, Reiser M, et al. Relapse rates in patients with rheumatoid arthritis in stable remission tapering or stopping antirheumatic therapy: interim results from the prospective randomized controlled RETRO study. Ann Rheum Dis. (2016) 75:45–51. doi: 10.1136/annrheumdis-2014-206439
89. Rech J, Hueber AJ, Finzel S, Englbrecht M, Haschka J, Manger B, et al. Prediction of disease relapses by multibiomarker disease activity and autoantibody status in patients with rheumatoid arthritis on tapering DMARD treatment. Ann Rheum Dis. (2016) 75:1637–44. doi: 10.1136/annrheumdis-2015-207900
90. Figueiredo CP, Bang H, Cobra JF, Englbrecht M, Hueber AJ, Haschka J, et al. Antimodified protein antibody response pattern influences the risk of disease relapse in patients with rheumatoid arthritis tapering disease modifying antirheumatic drugs. Ann Rheum Dis. (2017) 76:399–407. doi: 10.1136/annrheumdis-2016-209297
91. Schett G, Emery P, Tanaka Y, Burmester G, Pisetsky DS, Naredo E, et al. Tapering biologic and conventional DMARD therapy in rheumatoid arthritis: current evidence and future directions. Ann Rheum Dis. (2016) 75:1428–37. doi: 10.1136/annrheumdis-2016-209201
92. Singh JA, Saag KG, Bridges SL Jr, Akl EA, Bannuru RR, Sullivan MC, et al. 2015 American college of rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Rheumatol. (2016) 68:1–26. doi: 10.1002/art.39480
93. van den Broek M, Dirven L, Klarenbeek NB, Molenaar TH, Han KH, Kerstens PJ, et al. The association of treatment response and joint damage with ACPA-status in recent-onset RA: a subanalysis of the 8-year follow-up of the BeSt study. Ann Rheum Dis. (2012) 71:245–8. doi: 10.1136/annrheumdis-2011-200379
94. Dekkers JS, Bergstra SA, Chopra A, Tikly M, Fonseca JE, Salomon-Escoto K, et al. Autoantibody status is not associated with early treatment response to first-line methotrexate in patients with early rheumatoid arthritis. Rheumatology (2018). doi: 10.1093/rheumatology/key263. [Epub ahead of print].
95. Jonsson MK, Hensvold AH, Hansson M, Aga AB, Sexton J, Mathsson-Alm L, et al. The role of anti-citrullinated protein antibody reactivities in an inception cohort of patients with rheumatoid arthritis receiving treat-to-target therapy. Arthritis Res Ther. (2018) 20:146. doi: 10.1186/s13075-018-1635-7
96. van Dongen H, van Aken J, Lard LR, Visser K, Ronday HK, Hulsmans HM, et al. Efficacy of methotrexate treatment in patients with probable rheumatoid arthritis: a double-blind, randomized, placebo-controlled trial. Arthritis Rheum. (2007) 56:1424–32. doi: 10.1002/art.22525
97. Crepaldi G, Bugatti S, Benaglio F, Sakellariou G, Manzo A, Montecucco C, et al. The 2010 classification criteria and a more aggressive treatment strategy improve clinical outcomes in seropositive but not seronegative rheumatoid arthritis. Ann Rheum Dis. (2017) 76(Suppl. 2):237. doi: 10.1136/annrheumdis-2017-eular.5101
98. Seegobin SD, Ma MH, Dahanayake C, Cope AP, Scott DL, Lewis CM, et al. ACPA-positive and ACPA-negative rheumatoid arthritis differ in their requirements for combination DMARDs and corticosteroids: secondary analysis of a randomized controlled trial. Arthritis Res Ther. (2014) 16:r13. doi: 10.1186/ar4439
99. Brown PM, Pratt AG, Isaacs JD. Mechanism of action of methotrexate in rheumatoid arthritis, and the search for biomarkers. Nat Rev Rheumatol. (2016) 12:731–42. doi: 10.1038/nrrheum.2016.175
100. Isaacs JD, Cohen SB, Emery P, Tak PP, Wang J, Lei G, et al. Effect of baseline rheumatoid factor and anticitrullinated peptide antibody serotype on rituximab clinical response: a meta-analysis. Ann Rheum Dis. (2013) 72:329–36. doi: 10.1136/annrheumdis-2011-201117
101. Maneiro RJ, Salgado E, Carmona L, Gomez-Reino JJ. Rheumatoid factor as predictor of response to abatacept, rituximab and tocilizumab in rheumatoid arthritis: Systematic review and meta-analysis. Semin Arthritis Rheum. (2013) 43:9–17. doi: 10.1016/j.semarthrit.2012.11.007
102. Sokolove J, Schiff M, Fleischmann R, Weinblatt ME, Connolly SE, Johnsen A, et al. Impact of baseline anti-cyclic citrullinated peptide-2 antibody concentration on efficacy outcomes following treatment with subcutaneous abatacept or adalimumab: 2-year results from the AMPLE trial. Ann Rheum Dis. (2016) 75:709–14. doi: 10.1136/annrheumdis-2015-207942
103. Gottenberg JE, Ravaud P, Cantagrel A, Combe B, Flipo RM, Schaeverbeke T, et al. Positivity for anti-cyclic citrullinated peptide is associated with a better response to abatacept: data from the 'Orencia and Rheumatoid Arthritis' registry. Ann Rheum Dis. (2012) 71:1815–9. doi: 10.1136/annrheumdis-2011-201109
104. Gottenberg JE, Courvoisier DS, Hernandez MV, Iannone F, Lie E, Canhão H, et al. Brief report: association of rheumatoid factor and anti-citrullinated protein antibody positivity with better effectiveness of abatacept: results from the Pan-European registry analysis. Arthritis Rheumatol. (2016) 68:1346–52. doi: 10.1002/art.39595
105. Nüßlein HG, Alten R, Galeazzi M, Lorenz HM, Nurmohamed MT, Bensen WG, et al. Efficacy and prognostic factors of treatment retention with intravenous abatacept for rheumatoid arthritis: 24-month results from an international, prospective, real-world study. Clin Exp Rheumatol. (2016) 34:489–99.
106. Alten R, Nüßlein HG, Mariette X, Galeazzi M, Lorenz HM, Cantagrel A, et al. Baseline autoantibodies preferentially impact abatacept efficacy in patients with rheumatoid arthritis who are biologic naïve: 6-month results from a real-world, international, prospective study. RMD Open (2017) 3:e000345. doi: 10.1136/rmdopen-2016-000345
107. Wunderlich C, Oliviera I, Figueiredo CP, Rech J, Schett G. Effects of DMARDs on citrullinated peptide autoantibody levels in RA patients-A longitudinal analysis. Semin Arthritis Rheum. (2017) 46:709–14. doi: 10.1016/j.semarthrit.2016.09.011
108. Jansen DTSL, Emery P, Smolen JS, Westhovens R, Le Bars M, Connolly SE, et al. Conversion to seronegative status after abatacept treatment in patients with early and poor prognostic rheumatoid arthritis is associated with better radiographic outcomes and sustained remission: post hoc analysis of the AGREE study. RMD Open (2018) 4:e000564. doi: 10.1136/rmdopen-2017-000564
109. Lv Q, Yin Y, Li X, Shan G, Wu X, Liang D, et al. The status of rheumatoid factor and anti-citrullinated peptide antibody are not associated with the effect of anti-TNFα agent treatment in patients with rheumatoid arthritis: a meta-analysis. PLoS ONE (2014) 9:e89442. doi: 10.1371/journal.pone.0089442
110. De Rycke L, Verhelst X, Kruithof E, Van den Bosch F, Hoffman IE, Veys EM, et al. Rheumatoid factor, but not anti-cyclic citrullinated peptide antibodies, is modulated by infliximab treatment in rheumatoid arthritis. Ann Rheum Dis. (2005) 64:299–302. doi: 10.1136/ard.2004.023523
111. Takeuchi T, Miyasaka N, Tatsuki Y, Yano T, Yoshinari T, Abe T, et al. Baseline tumour necrosis factor alpha levels predict the necessity for dose escalation of infliximab therapy in patients with rheumatoid arthritis. Ann Rheum Dis. (2011) 70:1208–15. doi: 10.1136/ard.2011.153023
112. Takeuchi T, Miyasaka N, Inui T, Yano T, Yoshinari T, Abe T, et al. High titers of both rheumatoid factor and anti-CCP antibodies at baseline in patients with rheumatoid arthritis are associated with increased circulating baseline TNF level, low drug levels, and reduced clinical responses: a post hoc analysis of the RISING study. Arthritis Res Ther. (2017) 19:194. doi: 10.1186/s13075-017-1401-2
113. Kawashiri SY, Kawakami A, Iwamoto N, Fujikawa K, Aramaki T, Tamai M, et al. Disease activity score 28 may overestimate the remission induction of rheumatoid arthritis patients treated with tocilizumab: comparison with the remission by the clinical disease activity index. Mod Rheumatol. (2011) 21:365–69. doi: 10.3109/s10165-010-0402-7
114. Pers YM, Fortunet C, Constant E, Lambert J, Godfrin-Valnet M, De Jong A, et al. Predictors of response and remission in a large cohort of rheumatoid arthritis patients treated with tocilizumab in clinical practice. Rheumatology (2014) 53:76–84. doi: 10.1093/rheumatology/ket301
115. Ducreux J, Durez P, Galant C, Nzeusseu Toukap A, Van den Eynde B, Houssiau FA, et al. Global molecular effects of tocilizumab therapy in rheumatoid arthritis synovium. Arthritis Rheumatol. (2014) 66:15–23. doi: 10.1002/art.38202
Keywords: rheumatoid arthritis, autoantibodies, rheumatoid factor, anti-citrullinated protein antibodies, remission
Citation: Bugatti S, Manzo A, Montecucco C and Caporali R (2018) The Clinical Value of Autoantibodies in Rheumatoid Arthritis. Front. Med. 5:339. doi: 10.3389/fmed.2018.00339
Received: 30 June 2018; Accepted: 19 November 2018;
Published: 03 December 2018.
Edited by:
Francesco Ciccia, University of Palermo, ItalyReviewed by:
Javier Rodríguez-Carrio, Universidad de Oviedo Mieres, SpainCopyright © 2018 Bugatti, Manzo, Montecucco and Caporali. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Serena Bugatti, c2VyZW5hLmJ1Z2F0dGlAdW5pcHYuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.