 Shalina Taylor1,2†
Shalina Taylor1,2† A. A. Roger Thompson
A. A. Roger Thompson
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Med. , 03 August 2018
Sec. Pulmonary Medicine
Volume 5 - 2018 | https://doi.org/10.3389/fmed.2018.00217
This article is part of the Research Topic Pulmonary Hypertension: Mechanisms and Management, History and Future View all 11 articles
Pulmonary arterial hypertension (PAH) is a severe vasculopathy characterized by the presence of fibrotic lesions in the arterial wall and the loss of small distal pulmonary arteries. The vasculopathy is accompanied by perivascular inflammation and increased protease levels, with neutrophil elastase notably implicated in aberrant vascular remodeling. However, the source of elevated elastase levels in PAH remains unclear. A major source of neutrophil elastase is the neutrophil, an understudied cell population in PAH. The principal function of neutrophils is to destroy invading pathogens by means of phagocytosis and NET formation, but proteases, chemokines, and cytokines implicated in PAH can be released by and/or prime and activate neutrophils. This review focuses on the contribution of inflammation to the development and progression of the disease, highlighting studies implicating neutrophils, neutrophil elastase, and other neutrophil proteases in PAH. The roles of cytokines, chemokines, and neutrophil elastase in the disease are discussed and we describe new insight into the role neutrophils potentially play in the pathogenesis of PAH.
Pulmonary arterial hypertension (PAH) is a progressive disease characterized by thickening, and progressive occlusion of distal arteries in the lung related to vascular cell dysfunction, and perivascular inflammation. As a consequence of elevation in pulmonary arterial pressure, the right ventricle hypertrophies and later becomes dysfunctional leading to right heart failure (1, 2). Perivascular inflammation has been observed in all subsets of PAH and correlates with clinical markers of disease progression such as an increase in pulmonary vascular resistance and a decrease in the 6-min walk test (3, 4). The consequences of perivascular inflammation include cytokine production by vascular and inflammatory cells, and degradation of the extracellular matrix (ECM) by proteases (5, 6). Both the increased cytokine production and the peptides that are released as a result of ECM degradation cause activation and recruitment of circulating immune cells (6, 7). Neutrophils are among the cells that are recruited, and these cells release proteolytic enzymes, including neutrophil elastase (NE), that cause vascular injury (8). Although many aspects of perivascular inflammation can cause progressive PAH, this review will focus on neutrophils and NE. We will review evidence of neutrophil accumulation in PAH, discuss the role of NE and other proteases in driving vascular remodeling and highlight potential interactions between neutrophils, NE and the key genetic driver of PAH, bone morphogenetic protein receptor type 2 (BMPR2).
Neutrophils are the predominant circulating leukocyte population and are important in modulating innate and adaptive immunity. They are early responders and are recruited to sites of sterile inflammation and infection by environmental cues.
Relatively little attention has been given to the role of neutrophils in the pathogenesis of PAH. Neutrophil and macrophage perivascular accumulation has been observed in murine lungs in association with hypoxic pulmonary hypertension (PH) and in monocrotaline-induced PH in rats (9, 10). It has also been established that the neutrophil to lymphocyte ratio is increased in PAH patients when compared to healthy controls (11) and that increased neutrophil to lymphocyte ratio positively correlates with New York Heart Association (NYHA) functional class (FC) and predicts event free survival (12, 13). It is not clear why this relative increase in circulating neutrophils is associated with PAH progression. However, neutrophils produce a wide range of substances that could contribute to vascular remodeling and promote inflammation in PAH. For example, myeloperoxidase (MPO), a catalyst for reactive oxygen species (ROS) formation, has recently been implicated in the pathophysiology of PAH (14). Two independent cohorts of PAH patients had increased plasma MPO relative to healthy controls and Mpo−/− mice displayed reduced right ventricular pressures following hypoxic exposure (14). Interestingly, although MPO can reduce nitric oxide (NO) bioavailability in other vascular beds (15), there was no evidence supporting a difference in NO availability between wild-type and MPO-deficient mice (14). Instead, activation of a Rho kinase pathway by MPO was found to drive pulmonary vasoconstriction and SMC proliferation (14).
In addition to MPO and ROS, neutrophils produce proteolytic enzymes. Activity of these enzymes is tightly controlled by endogenous inhibitors, but excessive protease activity has the potential to destroy tissue and cause extensive fibrotic remodeling, leading to organ failure (16). Indeed neutrophil proteases are implicated in airway and parenchymal lung diseases [reviewed by Taggart et al. (17)] and are known to have roles in systemic vascular pathology (18, 19).
Neutrophil granule proteases include members of the serine protease family (NE, cathepsin G, proteinase 3) and matrix metalloproteinases [neutrophil collagenase (MMP-8) and gelatinase (MMP-9)] (20). Of these proteases, NE has been most heavily implicated in the pathogenesis of PAH, as described below.
Neutrophils are the dominant cellular source of NE but it is also produced by macrophages and smooth muscle cells (SMC) (21–24). There is evidence of both augmented NE release from neutrophils isolated from PAH patients (25) and upregulation of endogenous NE in PAH patient SMCs and in experimental animal models of PH (23, 26, 27). Macrophages are observed in the plexiform lesions of PAH patient lungs and activated macrophages release leukotriene B4, which induces endothelial cell (EC) injury and results in EC apoptosis, but could also promote neutrophil recruitment (28). Moreover, in vitro work suggests that human alveolar macrophages internalize NE through ingestion of apoptotic neutrophils, and act as a vehicle for the enzyme, transporting it to the tissues and subsequently releasing the active form (29). However, further work is required to show conservation of this mechanism in perivascular or interstitial lung macrophages.
NE is dispensed by neutrophils during degranulation and upon release of neutrophil extracellular traps (NETs). NETs consist of decondensed chromatin decorated with NE and other antimicrobial proteases. NE plays an important role in NET release, evidenced by reduced NET formation in the presence of NE inhibitors and in NE knockout mice (30). Upon stimulation, NE is discharged from azurophilic granules and translocates to the nucleus where it contributes to histone degradation, facilitating chromatin decondensation (30). Depending on the stimulus, chromatin is then expelled via vesicles (31) or suicidal NETosis (32), reviewed by Jorch et al. (33).
NETs assist in microbial trapping and killing (32), but non-infectious roles for NETs are emerging and NETs have been identified in vascular pathologies such as atherosclerosis and PAH (34, 35). Detrimental consequences attributed to NETs include nuclear factor kappa-light-chain-enhancer of activated B cells-dependent PA EC pathological angiogenesis, release of the vasoactive agent endothelin-1, and promotion of SMC proliferation (34). NETs can also directly induce EC cell death (36) and promote thrombus formation (37). In PAH lung tissue, markers of NET formation (DNA, myeloperoxidase, and citrullinated histone H3) have been identified in proximity to plexiform lesions and circulating DNA levels were elevated in IPAH patient plasma (34), although this study did not investigate whether IPAH patient neutrophils are more prone to releasing NETs.
Studies prior to the identification of NETs had suggested that binding of NE to DNA inhibited the activity of the protease (38). However, Kolaczkowska et al. used an in vivo zymography assay to demonstrate that NE associated with NETs remains proteolytically active (39). This implies that NE attached to NETs is shielded from endogenous anti-protease activity and thus high NET levels in PAH may represent an important source of active NE.
Elastolytic activity has been implicated in the pathogenesis of PAH for over three decades, with evidence of fragmented internal elastic laminae in the pulmonary arteries of children with congenital heart defects (40). There is a clear temporal relationship between increased NE activity and vascular changes in PAH experimental rat models (41). Furthermore, inhibition of NE enzymatic activity not only attenuates the progression of PAH but can also reverse the disease process in experimental models. This was first shown in the monocrotaline model where a progressive and fatal form of PH was reversed by elastase inhibitors in association with normalization of hemodynamic changes of PH and structural abnormalities that included extensive occlusive muscularization (26). Nickel et al. also demonstrated reversal of hemodynamic and histological measures of PH in the Sugen/Hypoxia rat model following treatment with recombinant human elafin (42). Increased NE activity was demonstrated in the lungs of rats with Sugen/Hypoxia-induced PH and this was attenuated by elafin (42). Importantly, relevance to human disease was confirmed through use of a human pulmonary artery explant model. Tissue sections containing pulmonary arteries were isolated from the explanted lungs of PAH patients and treatment with elafin induced regression of neointimal changes and improved measures of vessel lumen size (42).
Release of NE can stimulate adverse remodeling by degrading virtually all the components of the ECM in addition to elastin, and including collagen, fibronectin, and laminin (43). As a consequence, degraded ECM releases bioactive peptides and growth factors such as epidermal growth factor (EGF) and fibroblast growth factor (FGF), which have both mitogenic and motogenic effects on SMCs and fibroblasts (44–47). Additionally, heightened NE activity leads to the activation of matrix metalloproteinases (MMPs), which could potentiate ECM degradation, and induction of tenascin C, a glycoprotein associated with the upregulation of growth factor receptors and proliferation of SMCs (48). The sub-endothelial deposition of tenascin C and fibronectin appears to be a chemotactic factor for PA SMCs, facilitating their migration and the formation of neointimal lesions (48, 49). Conversely, inhibition of NE leads to reduced tenascin C, induction of SMC apoptosis, and regression of pulmonary artery (PA) medial hypertrophy (26).
The role of elastase in vascular remodeling was further explored using transgenic mice that over-express the S100A4 protein (23, 27). Following infection with the murine herpes virus, MHV-68, S100A4 mice develop histological features of human PAH including neointima formation (27). These vascular lesions display fragmented elastic laminae associated with heightened lung elastase activity (27). Infusion of recombinant human elafin, an endogenous elastase inhibitor, reduced the number and severity of neointimal lesions in this model (23). Furthermore, using FLAG-tagged elafin, NE was identified as the serine elastase responsible for the elevation in elastase activity in the S100A4 lungs (23). While this finding implies that NE is the dominant target in this model, the relative importance of NE above other proteases in human disease has not been confirmed and the potential roles of other proteases will be discussed below.
The source of NE in the S100A4 lungs was localized not only to neutrophils but also to PA SMCs and, as indicated above, this finding was conserved in human disease, evidenced by cultured PA SMCs from IPAH patients expressing higher levels of NE than cells from control donor lungs (23). Interestingly, ex vivo perfusion of S100A4 lungs with porcine pancreatic elastase suggested that the elastin fibers in these mice were more prone to degradation (23). This implies that certain viral infections, and potentially other inflammatory stimuli, may predispose the vasculature to pathological remodeling upon exposure to elastase.
In addition to the impact of NE on SMC migration and proliferation, this enzyme contributes to PAH pathogenesis by proteolytic modification of cytokines. For example, NE promotes IL-1β activity by cleaving the pro-isoform of IL-1β in human coronary ECs, thereby increasing the secretion of the bioactive form in extracellular vesicles (50). Notably, secretion of active IL-1β could promote neutrophil survival, an example of positive regulation of neutrophil activity by cytokines (51).
NE also cleaves CXCL12 (SDF-1 alpha), a chemokine involved in the regulated release of neutrophils from the bone marrow (52–54). CXCL12 inactivation would promote mobilization of neutrophils into the circulation, particularly in the context of elevated plasma IL-8 levels, as observed in PAH patients (4, 54). On the other hand, NE can inactivate tumor-necrosis factor (55) and IL-6 (56), suggesting that there is a balance that must be maintained between elastase and anti-elastase activity. The chronic perivascular inflammatory phenotype of PAH patients characterized by elevated levels of circulating and tissue cytokines, is consistent with persistent neutrophil activation. Indeed, many of the cytokines elevated in PAH are known to enhance neutrophil function and survival and may thus perpetuate neutrophil-mediated inflammation (Table 1).

Table 1. Cytokines elevated in PAH and known to alter neutrophil function.
As mentioned above, other neutrophil proteases may contribute to the vascular remodeling observed in PAH. For example, MMP-9 expression is upregulated in human plexiform pulmonary arterial lesions (61) and in lungs isolated from rats with monocrotaline-induced pulmonary hypertension (62). Furthermore, transgenic overexpression of human MMP-9 exacerbated monocrotaline-induced pulmonary hypertension in mice (63). The role of other MMPs and MMP inhibitors in PAH has been reviewed by Chelladuri et al. (64).
This review has focused mainly on neutrophil proteases, but it should be noted that many other cells release proteases. For example, upregulation of MMP-9 has been detected in natural killer cells from patients with PAH (65) and it is also released by monocytes and macrophages (66). Further examples of important non-neutrophil proteases are chymase and tryptase, the main proteases released by mast cells. Heath and Yacoub (67) noted increased perivascular mast cell numbers in both primary and secondary forms of pulmonary hypertension compared to controls, an observation confirmed more recently by others (68, 69). Circulating tryptase levels were elevated in PAH patients compared to controls and correlated with disease severity as assessed by brain natriuretic peptide level (68). In this study a small number of patients were treated with mast cell stabilizers, but there were no changes in clinical endpoints such as 6-min walk distance or BNP level.
A key question about protease activity in any disease process is how the protease evades suppression by endogenous inhibitors. NE is inhibited by several anti-proteases including α1-antitrypsin, secretory leucocyte peptidase inhibitor (SLPI) and elafin. On the one hand it is possible that localized release of the protease simply overcomes anti-protease activity in the immediate microenvironment. Indeed, markers of pre-inhibited elastase activity have been reported including the cleaved fibrinogen product, Aα-Val360 (70). On the other hand, proteases may be shielded from their inhibitors by attachment to NETS (discussed above), or via transport in exosomes. Neutrophil-derived exosomes degrade NE substrates and induce emphysematous changes in murine lungs following intra-tracheal administration (71). It is also possible that proteases have enzyme-independent functions but the beneficial consequences of elafin treatment in animal models of PH and in lung explant models (23, 42) would favor the hypothesis that there is an excess of protease activity. Interestingly, however, elafin may exert protective effects independently of protease inhibition as described below.
The bone morphogenetic protein receptor 2 (BMPR2) signaling pathway has become a key focus of investigation since BMPR2 gene mutations were identified as the main predisposing risk factor in the heritable forms of PAH (HPAH) (72), with dysfunction in the signaling pathway present in all subtypes of PAH (73). However, although present in a high proportion of HPAH cases—identified in 70% of familial PAH cases (74)—the autosomal dominant BMPR2 mutation exhibits low penetrance with 70–80% of those carrying the mutation never developing PAH (75). This, and the lack of spontaneous PAH in most heterozygous BMPR2 animal models, implies that a second insult or background genetic variants are needed for predisposed individuals to develop PAH. Inflammation has been proposed as a second hit which promotes adverse remodeling when there is a loss of BMPR2.
BMPR2 is expressed by all cells in the arterial wall but by far the highest level of expression is present in endothelial cells (73). Reduced BMPR2 signaling is related both to excessive vascular smooth muscle proliferation (76) and exaggerated PA EC apoptosis (77). A study by Burton et al. (78) investigated the role of BMPR2 in maintaining the barrier function of PA ECs and in suppressing inflammation within the pulmonary vasculature. Using static and flow-based in vitro systems, they were able to demonstrate that a reduction in BMPR2 expression facilitated neutrophil transmigration across the PA EC monolayer and reported that a lack of BMPR2 led to overexpression of IL-8, which in turn led to the recruitment of neutrophils. Similarly, loss of BMPR2 can lead to heightened expression of IL-6, an inducer of SMC proliferation (79). Taken together, BMPR2 plays a role in dampening inflammatory signals in the pulmonary vasculature that could influence neutrophil recruitment and elastase activity.
Conversely, neutrophils may also impact BMPR2 function by releasing NE and degrading BMP9, an anti-angiogenic ligand for the BMPR2/ALK1 heterodimer present on PA ECs (80). BMP9 has been shown to circulate in humans at biologically active levels (81), maintaining vascular quiescence. Li et al. showed that BMP9 is readily cleaved by NE, released by activated neutrophils (80). Further work by this group demonstrated that the administration of recombinant BMP9 reversed established PH in BMPR2-deficient mice, overcoming reduced BMPR2 levels, and preventing lung vascular leakage (82). Paradoxically, Appleby et al. demonstrated that BMP9 could have a pro-inflammatory role and, in fact, facilitate neutrophil recruitment to the pulmonary vasculature by activated endothelial cells (83). However, the mouse model used in that study was acute endotoxemia, so this may not relate directly to PAH pathogenesis.
Overall, it appears that inflammation-driven release of NE can suppress BMPR2 signaling. Nickel and colleagues investigated the impact of the NE inhibitor elafin on BMPR2 signaling in PAH (42). In the Sugen/Hypoxia rat model of PH, elafin improved pulmonary endothelial expression of apelin, a BMPR2 target gene. Loss of apelin expression in the pulmonary endothelium during PAH is associated with the failure to repress the release of fibroblast growth factor-2 (FGF2) (84). Nickel et al. also found that in human cells, elafin augmented BMPR2 interactions with caveolin-1, another downstream target of BMPR2 signaling (42). Unfortunately, changes in BMP9 levels following elafin administration were not measured. Nonetheless, the findings suggest that improving BMPR2 signaling in addition to the beneficial sequelae of inhibiting NE, described earlier in this review, could reverse the vascular remodeling observed in PAH. Interestingly, targeting NE activity may be of particular importance in patients with BMPR2 mutations, as histological assessment of pulmonary vessels in such patients demonstrates a reduction in elastin and fibrillin-1, the two major constituents of elastic fibers (85). Furthermore, pulmonary artery elastic fibers from mice with compound Bmpr2/1a mutations were more susceptible to elastase-mediated degradation of elastic fibers compared to wild-type mice (85).
Collectively the studies discussed above implicate the neutrophil and neutrophil products such as MPO, proteases and NETs in the pathogenesis of PAH. Neutrophils are attractive candidates in contributing to vascular changes seen in PAH because they are among the first cells to arrive at sites of inflammation. The evidence of increased circulating elastase in PAH patients and fragmented elastic lamina in PAH vessels, highlights a key pathogenic role for proteolytic enzymes and in particular NE. NE has a wide range of targets, but we describe evidence for how it could contribute to PAH pathogenesis by modulating the activity of cytokines and degrading ECM releasing growth factors that promote remodeling. Moreover, the release of NE and NETs is likely to alter the local inflammatory environment, augmenting leukocyte responses and further driving inflammation in PAH. It is also evident that reduced BMPR2 receptor signaling cooperatively interacts with the sequelae of inflammation and neutrophil activation in contributing to adverse vascular remodeling. Figure 1 provides a summary of the role of neutrophils, NE and NETs in PAH.
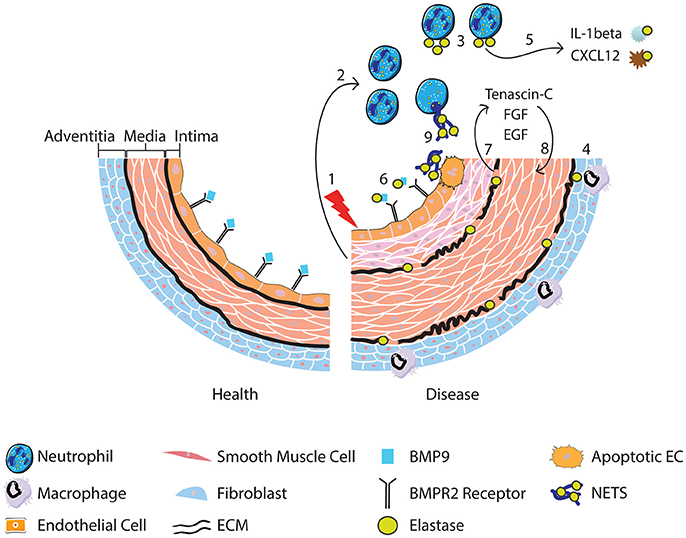
Figure 1. Following vascular injury (1), the release of chemokines and chemotactic peptides from surrounding tissue recruits and activates neutrophils (2). Neutrophils release elastase (3) although it can also be released from activated macrophages that engulf neutrophil elastase (4) and from smooth muscle cells. Release of neutrophil elastase leads to cleavage of cytokines resulting in the conversion of the pro-to active form of IL-1b, promoting neutrophil survival, and degradation of CXCL12, favoring release of neutrophils from the bone marrow (5). BMP9 is also cleaved, impacting upon BMPR2 receptor signaling (6). Degradation of the ECM by elastase (7) releases SMC mitogenic growth factors promoting SMC proliferation (8). Neutrophil elastase is also involved in the release of NETs, which may induce EC apoptosis (9) and therefore contribute to endothelial dysfunction.
While increases in NE activity coincide with changes in vascular remodeling in animal models, it remains unclear whether intrinsic neutrophil abnormalities are necessary to initiate aberrant remodeling in human lungs or whether alterations in NET and NE release by neutrophils are a consequence of other features of the disease. To better understand the role of neutrophils at different stages of PAH pathogenesis, a longitudinal study to evaluate how they become increased and activated would be important. However, directly targeting neutrophils as a therapeutic strategy would be challenging given their vital role in host defense and as mentioned above, cells other than neutrophils can produce damaging proteases. Of these proteases, NE provides an attractive target as evidence from animal models suggests that NE inhibition has the potential to inhibit aberrant remodeling of the pulmonary vessels and indirectly dampen persistent inflammation in PAH. These findings have encouraged initiation of clinical trials of elastase inhibitors such as elafin for PAH (NCT03522935). Further, the potential for NE inhibitors such as elafin to enhance BMPR2 signaling and target pathology driven by BMPR2 deficiency, identifies these drugs as promising novel therapies for PAH.
ST and OD wrote sections of the manuscript; AT initiated the work and drafted sections of the manuscript; AT, MR and RZ revised the manuscript critically for important intellectual content.
AT was supported by a JG Graves Fellowship (University of Sheffield) and a British Heart Foundation-Fulbright Award. ST was supported by the T32 NIH/NHLBI Stanford Training Program in Lung Biology (HL129970-02).
AT has received funds to attend educational events from Actelion. RZ has a patent for use of FK506 to treat pulmonary hypertension. RZ has performed consultancy work for Actelion and Vivus and has stock options with Selten.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer JS and handling Editor declared their shared affiliation.
1. Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. (2008) 118:2372–9. doi: 10.1172/JCI33452
2. Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. (2012) 122:4306–13. doi: 10.1172/JCI60658
3. Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med. (2012) 186:261–72. doi: 10.1164/rccm.201201-0164OC
4. Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation (2010) 122:920–7. doi: 10.1161/CIRCULATIONAHA.109.933762
5. Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. (2004) 43(12 Suppl. S):13S−24. doi: 10.1016/j.jacc.2004.02.029
6. Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res. (2014) 115:165–75. doi: 10.1161/CIRCRESAHA.113.301141
7. Senior RM, Griffin GL, Mecham RP. Chemotactic activity of elastin-derived peptides. J Clin Invest. (1980) 66:859–62. doi: 10.1172/JCI109926
8. Korkmaz B, Horwitz MS, Jenne DE, Gauthier F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev. (2010) 62:726–59. doi: 10.1124/pr.110.002733
9. Frid MG, Brunetti JA, Burke DL, Carpenter TC, Davie NJ, Reeves JT, et al. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol. (2006) 168:659–69. doi: 10.2353/ajpath.2006.050599
10. Schultze AE, Wagner JG, White SM, Roth RA. Early indications of monocrotaline pyrrole-induced lung injury in rats. Toxicol Appl Pharmacol. (1991) 109:41–50.
11. Yildiz A, Kaya H, Ertas F, Oylumlu M, Bilik MZ, Yuksel M, et al. Association between neutrophil to lymphocyte ratio and pulmonary arterial hypertension. Turk Kardiyol Dern Ars. (2013) 41:604–9. doi: 10.5543/tkda.2013.93385
12. Ozpelit E, Akdeniz B, Ozpelit ME, Tas S, Bozkurt S, Tertemiz KC, et al. Prognostic value of neutrophil-to-lymphocyte ratio in pulmonary arterial hypertension. J Int Med Res. (2015) 43:661–71. doi: 10.1177/0300060515589394
13. Harbaum L, Baaske KM, Simon M, Oqueka T, Sinning C, Glatzel A, et al. Exploratory analysis of the neutrophil to lymphocyte ratio in patients with pulmonary arterial hypertension. BMC Pulm Med. (2017) 17:72. doi: 10.1186/s12890-017-040
14. Klinke A, Berghausen E, Fr7-5iedrichs K, Molz S, Lau D, Remane L, et al. Myeloperoxidase aggravates pulmonary arterial hypertension by activation of vascular Rho-kinase. JCI Insight (2018) 3:530. doi: 10.1172/jci.insight.97530
15. Eiserich JP, Baldus S, Brennan ML, Ma W, Zhang C, Tousson A, et al. Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science (2002) 296:2391–4. doi: 10.1126/science.1106830
16. Segal AW. How neutrophils kill microbes. Annu Rev Immunol. (2005) 23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653
17. Taggart C, Mall MA, Lalmanach G, Cataldo D, Ludwig A, Janciauskiene S, et al. Protean proteases: at the cutting edge of lung diseases. Eur Respir J. (2017) 49:2015. doi: 10.1183/13993003.01200-2015
18. Yan H, Zhou HF, Akk A, Hu Y, Springer LE, Ennis TL, et al. Neutrophil proteases promote experimental abdominal aortic aneurysm via extracellular trap release and plasmacytoid dendritic cell activation. Arterioscler Thromb Vasc Biol. (2016) 36:1660–9. doi: 10.1161/ATVBAHA.116.307786
19. Soehnlein O. Multiple roles for neutrophils in atherosclerosis. Circ Res. (2012) 110:875–88. doi: 10.1161/CIRCRESAHA.111.257535
20. Cowland JB, Borregaard N. Granulopoiesis and granules of human neutrophils. Immunol Rev. (2016) 273:11–28. doi: 10.1111/imr.12440
21. Belaaouaj A, Kim KS, Shapiro SD. Degradation of outer membrane protein A in Escherichia coli killing by neutrophil elastase. Science (2000) 289:1185–8. doi: 10.1126/science.289.5482.1185
22. Dollery CM, Owen CA, Sukhova GK, Krettek A, Shapiro SD, Libby P. Neutrophil elastase in human atherosclerotic plaques: production by macrophages. Circulation (2003) 107:2829–36. doi: 10.1161/01.CIR.0000072792.65250.4A
23. Kim YM, Haghighat L, Spiekerkoetter E, Sawada H, Alvira CM, Wang L, et al. Neutrophil elastase is produced by pulmonary artery smooth muscle cells and is linked to neointimal lesions. Am J Pathol. (2011) 179:1560–72. doi: 10.1016/j.ajpath.2011.05.051
24. Kobayashi J, Wigle D, Childs T, Zhu L, Keeley FW, Rabinovitch M. Serum-induced vascular smooth muscle cell elastolytic activity through tyrosine kinase intracellular signalling. J Cell Physiol. (1994) 160:121–31. doi: 10.1002/jcp.1041600115
25. Rose F, Hattar K, Gakisch S, Grimminger F, Olschewski H, Seeger W, et al. Increased neutrophil mediator release in patients with pulmonary hypertension–suppression by inhaled iloprost. Thromb Haemost. (2003) 90:1141–9. doi: 10.1160/TH03-03-0173
26. Cowan KN, Heilbut A, Humpl T, Lam C, Ito S, Rabinovitch M. Complete reversal of fatal pulmonary hypertension in rats by a serine elastase inhibitor. Nat Med (2000) 6:698–702. doi: 10.1038/76282
27. Spiekerkoetter E, Alvira CM, Kim YM, Bruneau A, Pricola KL, Wang L, et al. Reactivation of gammaHV68 induces neointimal lesions in pulmonary arteries of S100A4/Mts1-overexpressing mice in association with degradation of elastin. Am J Physiol Lung Cell Mol Physiol. (2008) 294:L276–89. doi: 10.1152/ajplung.00414.2007
28. Tian W, Jiang X, Tamosiuniene R, Sung YK, Qian J, Dhillon G, et al. Blocking macrophage leukotriene b4 prevents endothelial injury and reverses pulmonary hypertension. Sci Transl Med. (2013) 5:200ra117. doi: 10.1126/scitranslmed.3006674
29. Campbell EJ, Wald MS. Hypoxic injury to human alveolar macrophages accelerates release of previously bound neutrophil elastase. Implications for lung connective tissue injury including pulmonary emphysema. Am Rev Respir Dis. (1983) 127:631–5. doi: 10.1164/arrd.1983.127.5.631
30. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. (2010) 191:677–91. doi: 10.1083/jcb.201006052
31. Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol. (2010) 185:7413–25. doi: 10.4049/jimmunol.1000675
32. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303:1532–5. doi: 10.1126/science.1092385
33. Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. (2017) 23:279–87. doi: 10.1038/nm.4294
34. Aldabbous L, Abdul-Salam V, McKinnon T, Duluc L, Pepke-Zaba J, Southwood M, et al. Neutrophil extracellular traps promote angiogenesis: evidence from vascular pathology in pulmonary hypertension. Arterioscler Thromb Vasc Biol. (2016) 36:2078–87. doi: 10.1161/ATVBAHA.116.307634
35. Borissoff JI, Joosen IA, Versteylen MO, Brill A, Fuchs TA, Savchenko AS, et al. Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arterioscler Thromb Vasc Biol. (2013) 33:2032–40. doi: 10.1161/ATVBAHA.113.301627
36. Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS ONE (2012) 7:e32366. doi: 10.1371/journal.pone.0032366
37. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
38. Belorgey D, Bieth JG. DNA binds neutrophil elastase and mucus proteinase inhibitor and impairs their functional activity. FEBS Lett. (1995) 361:265–8.
39. Kolaczkowska E, Jenne CN, Surewaard BG, Thanabalasuriar A, Lee WY, Sanz MJ, et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun. (2015) 6:6673. doi: 10.1038/ncomms7673
40. Rabinovitch M, Bothwell T, Hayakawa BN, Williams WG, Trusler GA, Rowe RD, et al. Pulmonary artery endothelial abnormalities in patients with congenital heart defects and pulmonary hypertension. A correlation of light with scanning electron microscopy and transmission electron microscopy. Lab Invest. (1986) 55:632–53.
41. Zhu L, Wigle D, Hinek A, Kobayashi J, Ye C, Zuker M, et al. The endogenous vascular elastase that governs development and progression of monocrotaline-induced pulmonary hypertension in rats is a novel enzyme related to the serine proteinase adipsin. J Clin Invest. (1994) 94:1163–71. doi: 10.1172/JCI117432
42. Nickel NP, Spiekerkoetter E, Gu M, Li CG, Li H, Kaschwich M, et al. Elafin reverses pulmonary hypertension via caveolin-1-dependent bone morphogenetic protein signaling. Am J Respir Crit Care Med. (2015) 191:1273–86. doi: 10.1164/rccm.201412-2291OC
43. Lee KM, Tsai KY, Wang N, Ingber DE. Extracellular matrix and pulmonary hypertension: control of vascular smooth muscle cell contractility. Am J Physiol. (1998) 274(1 Pt 2):H76–82.
44. Tsukamoto Y, Helsel WE, Wahl SM. Macrophage production of fibronectin, a chemoattractant for fibroblasts. J Immunol. (1981) 127:673–8.
45. Thompson K, Rabinovitch M. Exogenous leukocyte and endogenous elastases can mediate mitogenic activity in pulmonary artery smooth muscle cells by release of extracellular-matrix bound basic fibroblast growth factor. J Cell Physiol. (1996) 166:495–505. doi: 10.1002/(SICI)1097-4652(199603)166:3<495::AID-JCP4>3.0.CO;2-K
46. Tu L, Dewachter L, Gore B, Fadel E, Dartevelle P, Simonneau G, et al. Autocrine fibroblast growth factor-2 signaling contributes to altered endothelial phenotype in pulmonary hypertension. Am J Respir Cell Mol Biol. (2011) 45:311–22. doi: 10.1165/rcmb.2010-0317OC
47. Chen PY, Qin L, Li G, Tellides G, Simons M. Fibroblast growth factor (FGF) signaling regulates transforming growth factor beta (TGFbeta)-dependent smooth muscle cell phenotype modulation. Sci Rep. (2016) 6:33407. doi: 10.1038/srep33407
48. Jones PL, Cowan KN, Rabinovitch M. Tenascin-C, proliferation and subendothelial fibronectin in progressive pulmonary vascular disease. Am J Pathol. (1997) 150:1349–60.
49. Cowan KN, Jones PL, Rabinovitch M. Elastase and matrix metalloproteinase inhibitors induce regression, and tenascin-C antisense prevents progression, of vascular disease. J Clin Invest. (2000) 105:21–34. doi: 10.1172/JCI6539
50. Alfaidi M, Wilson H, Daigneault M, Burnett A, Ridger V, Chamberlain J, et al. Neutrophil elastase promotes interleukin-1beta secretion from human coronary endothelium. J Biol Chem. (2015) 290:24067–78. doi: 10.1074/jbc.M115.659029
51. Prince LR, Allen L, Jones EC, Hellewell PG, Dower SK, Whyte MK, et al. The role of interleukin-1beta in direct and toll-like receptor 4-mediated neutrophil activation and survival. Am J Pathol. (2004) 165:1819–26. doi: 10.1016/S0002-9440(10)63437-2
52. Valenzuela-Fernandez A, Planchenault T, Baleux F, Staropoli I, Le-Barillec K, Leduc D, et al. Leukocyte elastase negatively regulates Stromal cell-derived factor-1 (SDF-1)/CXCR4 binding and functions by amino-terminal processing of SDF-1 and CXCR4. J Biol Chem. (2002) 277:15677–89. doi: 10.1074/jbc.M111388200
53. Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest. (2010) 120:2423–31. doi: 10.1172/JCI41649
54. Martin C, Burdon PC, Bridger G, Gutierrez-Ramos JC, Williams TJ, Rankin SM. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity (2003) 19:583–93. doi: 10.1016/S1074-7613(03)00263-2
55. Scuderi P, Nez PA, Duerr ML, Wong BJ, Valdez CM. Cathepsin-G and leukocyte elastase inactivate human tumor necrosis factor and lymphotoxin. Cell Immunol. (1991) 135:299–313.
56. Bank U, Kupper B, Reinhold D, Hoffmann T, Ansorge S. Evidence for a crucial role of neutrophil-derived serine proteases in the inactivation of interleukin-6 at sites of inflammation. FEBS Lett. (1999) 461:235–40.
57. Sanchez O, Marcos E, Perros F, Fadel E, Tu L, Humbert M, et al. Role of endothelium-derived CC chemokine ligand 2 in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. (2007) 176:1041–7. doi: 10.1164/rccm.200610-1559OC
58. Cracowski JL, Chabot F, Labarere J, Faure P, Degano B, Schwebel C, et al. Proinflammatory cytokine levels are linked to death in pulmonary arterial hypertension. Eur Respir J. (2014) 43:915–7. doi: 10.1183/09031936.00151313
59. Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, et al. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med. (1995) 151:1628–31. doi: 10.1164/ajrccm.151.5.7735624
60. Heresi GA, Aytekin M, Hammel JP, Wang S, Chatterjee S, Dweik RA. Plasma interleukin-6 adds prognostic information in pulmonary arterial hypertension. Eur Respir J. (2014) 43:912–4. doi: 10.1183/09031936.00164713
61. Jonigk D, Golpon H, Bockmeyer CL, Maegel L, Hoeper MM, Gottlieb J, et al. Plexiform lesions in pulmonary arterial hypertension composition, architecture, and microenvironment. Am J Pathol. (2011) 179:167–79. doi: 10.1016/j.ajpath.2011.03.040
62. Schermuly RT, Yilmaz H, Ghofrani HA, Woyda K, Pullamsetti S, Schulz A, et al. Inhaled iloprost reverses vascular remodeling in chronic experimental pulmonary hypertension. Am J Respir Crit Care Med. (2005) 172:358–63. doi: 10.1164/rccm.200502-296OC
63. George J, D'Armiento J. Transgenic expression of human matrix metalloproteinase-9 augments monocrotaline-induced pulmonary arterial hypertension in mice. J Hypertens. (2011) 29:299–308. doi: 10.1097/HJH.0b013e328340a0e4
64. Chelladurai P, Seeger W, Pullamsetti SS. Matrix metalloproteinases and their inhibitors in pulmonary hypertension. Eur Respir J. (2012) 40:766–82. doi: 10.1183/09031936.00209911
65. Ormiston ML, Chang C, Long LL, Soon E, Jones D, Machado R, et al. Impaired natural killer cell phenotype and function in idiopathic and heritable pulmonary arterial hypertension. Circulation (2012) 126:1099–109. doi: 10.1161/CIRCULATIONAHA.112.110619
66. Goetzl EJ, Banda MJ, Leppert D. Matrix metalloproteinases in immunity. J Immunol. (1996) 156:1–4.
67. Heath D, Yacoub M. Lung mast cells in plexogenic pulmonary arteriopathy. J Clin Pathol. (1991) 44:1003–6.
68. Farha S, Sharp J, Asosingh K, Park M, Comhair SA, Tang WH, et al. Mast cell number, phenotype, and function in human pulmonary arterial hypertension. Pulm Circ. (2012) 2:220–8. doi: 10.4103/2045-8932.97609
69. Kosanovic D, Dahal BK, Peters DM, Seimetz M, Wygrecka M, Hoffmann K, et al. Histological characterization of mast cell chymase in patients with pulmonary hypertension and chronic obstructive pulmonary disease. Pulm Circ. (2014) 4:128–36. doi: 10.1086/675642
70. Carter RI, Mumford RA, Treonze KM, Finke PE, Davies P, Si Q, et al. The fibrinogen cleavage product Aalpha-Val360, a specific marker of neutrophil elastase activity in vivo. Thorax (2011) 66:686–91. doi: 10.1136/thx.2010.154690
71. Russell D, Genschmer KR, Szul T, Noerager B, Xu X, Viera L, et al. Neutrophil-derived exosomes purified from COPD patient bronchoalveolar fluid cause a COPD like phenotype in a mouse model via a neutrophil elastase dependent mechanism. Am J Respir Crit Care Med. (2018) 197:A2701–A. doi: 10.1164/ajrccm-conference.2018.197.1_MeetingAbstracts.A2701
72. Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. (2000) 67:737–44. doi: 10.1086/303059
73. Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, et al. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation (2002) 105:1672–8. doi: 10.1161/01.Cir.0000012754.72951.3d
74. Cogan JD, Pauciulo MW, Batchman AP, Prince MA, Robbins IM, Hedges LK, et al. High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterial hypertension. Am J Respir Crit Care Med. (2006) 174:590–8. doi: 10.1164/rccm.200602-165OC
75. Hamid R, Cogan JD, Hedges LK, Austin E, Phillips JA 3rd, Newman JH, et al. Penetrance of pulmonary arterial hypertension is modulated by the expression of normal BMPR2 allele. Hum Mutat. (2009) 30:649–54. doi: 10.1002/humu.20922
76. Morrell NW, Yang X, Upton PD, Jourdan KB, Morgan N, Sheares KK, et al. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-beta(1) and bone morphogenetic proteins. Circulation (2001) 104:790–5. doi: 10.1161/hc3201.094152
77. Teichert-Kuliszewska K, Kutryk MJ, Kuliszewski MA, Karoubi G, Courtman DW, Zucco L, et al. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ Res. (2006) 98:209–17. doi: 10.1161/01.RES.0000200180.01710.e6
78. Burton VJ, Ciuclan LI, Holmes AM, Rodman DM, Walker C, Budd DC. Bone morphogenetic protein receptor II regulates pulmonary artery endothelial cell barrier function. Blood (2011) 117:333–41. doi: 10.1182/blood-2010-05-285973
79. Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res. (2009) 104:236–44. doi: 10.1161/CIRCRESAHA.108.182014
80. Li W, Hoenderdos K, Salmon RM, Upton PD, Condliffe AM, Chilvers ER, et al. Neutrophil and redox dependent proteolysis of bone morphogenetic protein 9: potential role in the pathogenesis of pulmonary arterial hypertension. Thorax (2013) 68:A146–A. doi: 10.1136/thoraxjnl-2013-204457.307
81. David L, Mallet C, Keramidas M, Lamande N, Gasc JM, Dupuis-Girod S, et al. Bone morphogenetic protein-9 is a circulating vascular quiescence factor. Circ Res. (2008) 102:914–22. doi: 10.1161/CIRCRESAHA.107.165530
82. Long L, Ormiston ML, Yang X, Southwood M, Graf S, Machado RD, et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat Med. (2015) 21:777–85. doi: 10.1038/nm.3877
83. Appleby SL, Mitrofan CG, Crosby A, Hoenderdos K, Lodge K, Upton PD, et al. Bone morphogenetic protein 9 enhances lipopolysaccharide-induced leukocyte recruitment to the vascular endothelium. J Immunol. (2016) 197:3302–14. doi: 10.4049/jimmunol.1601219
84. Alastalo TP, Li M, Perez Vde J, Pham D, Sawada H, Wang JK, et al. Disruption of PPARgamma/beta-catenin-mediated regulation of apelin impairs BMP-induced mouse and human pulmonary arterial EC survival. J Clin Invest. (2011) 121:3735–46. doi: 10.1172/JCI43382
85. Tojais NF, Cao A, Lai YJ, Wang L, Chen PI, Alcazar MAA, et al. Codependence of bone morphogenetic protein receptor 2 and transforming growth factor-beta in elastic fiber assembly and its perturbation in pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol. (2017) 37:1559–69. doi: 10.1161/ATVBAHA.117.309696
Keywords: neutrophils, neutrophil elastase, pulmonary hypertension, pulmonary arterial hypertension, vascular remodeling
Citation: Taylor S, Dirir O, Zamanian RT, Rabinovitch M and Thompson AAR (2018) The Role of Neutrophils and Neutrophil Elastase in Pulmonary Arterial Hypertension. Front. Med. 5:217. doi: 10.3389/fmed.2018.00217
Received: 26 April 2018; Accepted: 16 July 2018;
Published: 03 August 2018.
Edited by:
Paul Anthony Corris, Newcastle University, United KingdomReviewed by:
Eleni Papakonstantinou, Aristotle University of Thessaloniki, GreeceCopyright © 2018 Taylor, Dirir, Zamanian, Rabinovitch and Thompson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: A. A. Roger Thompson, ci50aG9tcHNvbkBzaGVmZmllbGQuYWMudWs=
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.