 Luke A. Law
Luke A. Law Douglas K. Graham2
Douglas K. Graham2 Jorge Di Paola
Jorge Di Paola Brian R. Branchford
Brian R. Branchford- 1Department of Anesthesiology, Mayo Clinic, Rochester, MN, United States
- 2Section of Hematology/Oncology, Department of Pediatrics, Emory University, Atlanta, GA, United States
- 3Section of Hematology/Oncology, Department of Pediatrics, University of Colorado School of Medicine, Aurora, CO, United States
- 4University of Colorado Hemophilia and Thrombosis Center, Aurora, CO, United States
The GAS6/TYRO3-AXL-MERTK (TAM) signaling pathway is essential for full and sustained platelet activation, as well as thrombus stabilization. Inhibition of this pathway decreases platelet aggregation, shape change, clot retraction, aggregate formation under flow conditions, and surface expression of activation markers. Transgenic mice deficient in GAS6, or any of the TAM family of receptors that engage this ligand, exhibit in vivo protection against arterial and venous thrombosis but do not demonstrate either spontaneous or prolonged bleeding compared to their wild-type counterparts. Comparable results are observed in wild-type mice treated with pharmacological inhibitors of the GAS6-TAM pathway. Thus, GAS6/TAM inhibition offers an attractive novel therapeutic option that may allow for a moderate reduction in platelet activation and decreased thrombosis while still permitting the primary hemostatic function of platelet plug formation.
Introduction
The burden of disease from thrombosis is high, with 1 out of every 6 deaths in the United States caused by coronary heart disease and approximately 690,000 ischemic strokes occurring per year (1). Current anti-platelet therapies are complicated by hemorrhagic side effects and interpatient response variability (2), demonstrating a need to explore new pathways that may yield safer and/or more effective therapies.
One such alternative pathway for the modulation of thrombosis and hemostasis is the TAM family of receptors tyrosine kinases (TYRO3, AXL, and MERTK) and their ligands, including growth arrest-specific gene 6 (GAS6) and Protein S. It has been shown that inhibition of the GAS6/TAM pathway decreases platelet activation responses and protects mice against arterial and venous thrombosis, without increasing bleeding (3, 4) thereby representing a promising therapeutic target for novel anti-platelet agents. This pathway is also active in a variety of benign and malignant cell types with functions including regulation of inflammation (5, 6), phagocytosis of apoptotic cells and cellular debris (5), platelet stabilization (3, 7, 8), maintenance of vascular smooth-muscle homeostasis (6, 9–11), spermatogenesis (12), maintenance of the retina (13), and cancer progression (14).
The abrogation of TAM signaling, as evidenced by studying transgenic mice deficient in one or more receptors, is associated with additional biological effects including significantly increased inflammatory response, impaired clearance of cellular debris and apoptotic cells, and systemic autoimmunity (6, 15, 16). Additionally, deleterious Mertk mutations are associated with the development of retinitis pigmentosa in humans, (17–19) a finding also noted in canine (20) and murine models (13). Rodent studies have evaluated the effect of expressing the paralog receptor Tyro3 (21) or gene therapy with human MERTK (22) to abrogate the disease, and recent human studies have involved translational read-through inducing drugs (23). Further information regarding the effects of GAS6/TAM signaling absence or inhibition is shown in Table 1.
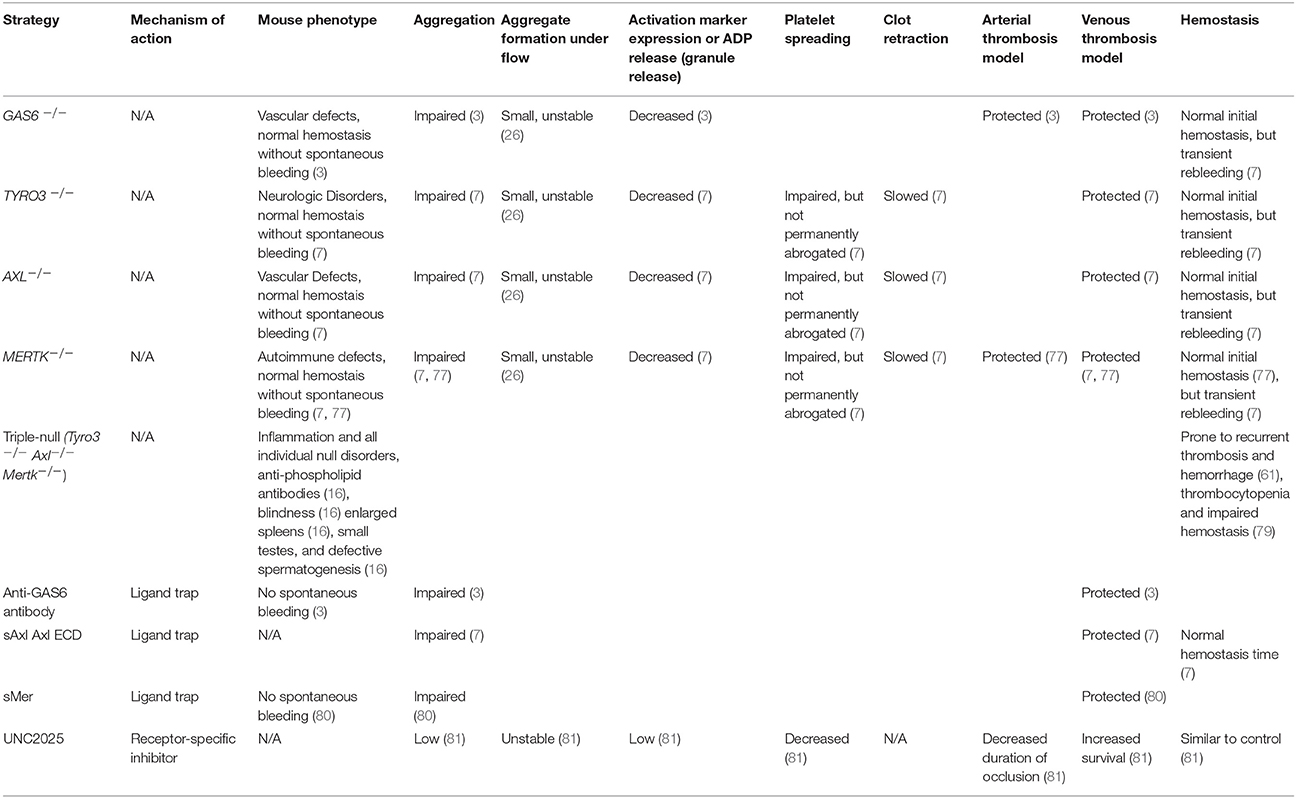
Table 1. Effects of various Gas6/TAM inhibition strategies.
This mini-review, however, focuses solely on the role of the GAS6/TAM signaling in hemostasis and thrombosis. Prior to any suggestion of GAS6/TAM-modulation as a reasonable clinical trial candidate for anti-platelet effect, much work remains to be done to establish dose-dependent organ-specific effects to better define any “off-target” considerations.
Platelet exposure to agonists including von Willebrand factor, collagen, thrombin, thromboxane A2, and adenosine diphosphate (ADP) leads to platelet activation and recruitment of subsequent platelets in a dynamic process known as “inside-out” signaling (24, 25). This process involves the rapid engagement of cell surface receptors that activate signal transduction pathways involving Src family kinases, Akt, phosphoinositide 3- kinases (PI3Ks), and the ITAM signaling pathway, ultimately leading to intracellular calcium mobilization and conversion of the fibrinogen receptor, integrin αIIbβ3 from an inactive (low-affinity) to an active (high-affinity) conformation. Upon fibrinogen engagement to the activated αIIbβ3 “outside-in” (24) signaling occurs promoting platelet spreading, clot retraction, granule secretion, and stable adhesion. The combination of these processes leads to a stable clot formation. It has previously been shown that “outside-in” signaling can be inhibited by interruption of the GAS6/TAM receptor pathway (7).
GAS6 Structure and Receptor Interactions
GAS6 (Ligand)
GAS6 (Figure 1), a 75 kDa product of the growth arrest-specific gene 6 (Gas6), is a vitamin K-dependent growth factor expressed in many cell types including vascular smooth muscle cells, mesangial cells, endothelial cells, bone marrow stromal cells, and macrophages (26–28), as well as in human platelets and plasma (3, 26, 29). Cellular secretion of GAS6 in the vasculature is primarily autocrine and is stimulated by several platelet agonists such as thrombin or collagen (3), as well as erythropoietin (30), and the pro-inflammatory cytokine TGFβ (31).
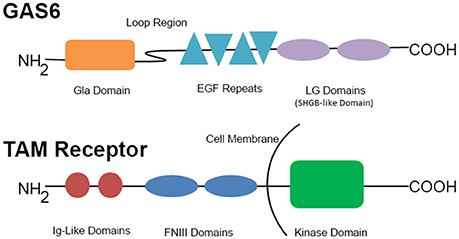
Figure 1. GAS6 and representative TAM family receptor. This schematic represents the components of GAS6 and the TAM receptors. EGF, epidermal growth factor; FNIII, fibronectin III; Ig, immunoglobulin; LG, laminin G-like.
Other components of this signaling pathway are also active and may contribute to the pathologic state known as thromboinflammation (32, 33). GAS6 levels are elevated in inflammatory conditions such as sepsis/systemic inflammatory response syndrome (34), systemic lupus erythematosus (35–37), and atherosclerotic plaques in vascular smooth muscle cells (31). GAS6 universally exhibits anti-inflammatory effects through downregulation of TNFα, interleukin-6, interferon γ, and intercellular adhesion molecule-1 expression, playing a significant role in preserving immune homeostasis (15, 27, 31, 38, 39). In addition to blocking platelet activation and protecting mice from thrombosis (3), inhibition or deficiency of Gas6 has also been shown to prevent liver inflammation, steatohepatitis, and hepatic fibrosis (40) but enhanced colitis-related tumorigenesis (41) in murine models.
Once secreted, GAS6 primarily binds to the TAM family receptor tyrosine on the platelet surface (42) by the C-terminal sex hormone binding globulin (SHBG)-like domain composed of two laminin G domains (Figure 1). This binding triggers dimerization and autophosphorylation (43–45), of these receptors and subsequent activation of the downstream signaling molecules PI3K (46, 47), Rap1 (47–49), and Akt (50–54). As seen in Figure 2, the activation of PI3K/Akt leads to phosphorylation of the cytoplasmic tail of the β3 integrin, promoting propagation and amplification of “outside-in” signaling (7, 55, 56), resulting in shape change, clot retraction, and subsequent platelet plug stabilization.
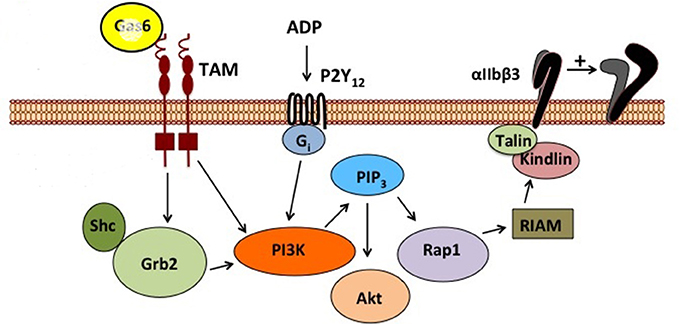
Figure 2. Schematic representation of GAS6/TAM signaling pathway. This figure depicts the signal transduction cascade initiated by GAS6 binding to TYRO3, AXL, or MERTK and the complementary contribution of the ADP/P2Y signaling pathway.
The N-terminal Gla domain of GAS6 (Figure 1) can also undergo calcium-dependent structural transformations allowing for high-affinity binding to phosphatidylserine (PtdS) residues (54, 57–60) exposed on the surface of nearby cells in response to cell activation, stress, and apoptosis (27). This allows GAS6 to target a wide variety of injured or activated cells in clinical settings such as endothelial cell remodeling (11), regulation of innate immunity (12, 61), vascular smooth-muscle homeostasis (9, 62), erythropoiesis (30), and survival regulation of tumor cells from mesenchymal, epithelial and hematopoietic origins (63, 64). Additionally, GAS6 bridges membrane-bound PtdS and TAM receptors (27, 54, 58, 59). The Gla domain of GAS6 [also involved in the regulation of osteoclast function (65, 66) and oligodendrocyte survival (67–69)] is connected to a disulfide-bridged loop, which, in turn, connects to four epidermal growth factor domains and a SHBG-like domain (Figure 1). Protein S, a negative regulator of the clotting cascade, is a close structural analog of GAS6, but has a disulfide-bridged loop that interacts with activated protein C following serine protease cleavage—to which GAS6 is insensitive due to structural constraints.
GAS6 does not appear to be a primary effector of platelet activation (70), but enhances and extends the platelet activation response triggered by ADP and other agonists through modulation of “outside-in” signaling via the αIIbβ3 integrin (3, 26) and regulation of granule secretion. It has been proposed that autocrine signaling in platelets is possible through release of GAS6 from α-granules (3, 7, 71–73). The precise source of GAS6 in human blood is not well established. Most studies indicate the presence of GAS6 in human plasma with levels varying from 15 to 65 μg/L (26, 74–76). This variation in levels of GAS6 in plasma had no correlation with extent of platelet activation in humans (70). While at least one study did not reveal physiologically relevant amounts of GAS6 in human platelets (75), others have demonstrated the presence of GAS6 mRNA (3, 77) as well as the protein itself at low concentrations (20 μg/L, equivalent to 5ng per 109 platelets) by various techniques, including immunoelectron microscopy and Western blots (26). While GAS6 levels in murine platelets are 6-fold higher than in human platelets, the plasma levels are comparable (78).
Other Ligands
In addition to GAS6, other ligands are known to stimulate the TAM receptors, including Protein S (82), Tubby, Tubby-like protein (TULP1), and Galectin-3. While GAS6 and Protein S are both vitamin K dependent proteins and share approximately 43% amino acid sequence identity and have the same domain structure (83). Protein S has been shown to be capable of binding TYRO3 (65) and MERTK (84), but has not yet been found to exhibit affinity for AXL (16). Tubby and TULP1 facilitate retinal pigment epithelium and macrophage phagocytosis through MERTK binding (85), while Galectin-3 is a MERTK-specific “eat-me” signal that stimulates phagocytosis of apoptotic cells and cellular debris by macrophages and retinal pigment epithelial cells (86) but is also upregulated after tissue injury such as myocardial infarction or in both acute and chronic liver injury, as described in a recent review (87).
Gas6 Receptors (TAM Subfamily of Tyrosine Kinases)
GAS6 binds to, and promotes tyrosine phosphorylation of, the single transmembrane tyrosine kinases of the TAM receptor subfamily (7, 71). Members of this receptor family exhibit a common structure determined by two extracellular N-terminal immunoglobulin-like domains and two fibronectin-III-like domains followed by a tyrosine kinase domain residing at the C-terminal (cytoplasmic) portion, as seen in Figure 1 (88, 89).
Although, of the TAM receptors, AXL has the highest in vitro demonstrated affinity for GAS6 with Kd = 1.0 nMol/L (followed by TYRO3 with roughly equal affinity) (90) it has been shown in some studies that MERTK (Kd at least 10-fold lower) (91, 92), is the only GAS6 receptor detected on platelets (whether human or murine) (77). Other studies, however, have not only reported the presence of AXL in platelets using immunoelectron microscopy, but also noted that labeling patterns suggest changes in location after stimulation of the platelets (26). A comprehensive review of this signaling pathway describes platelet expression of all three TAM receptors (56). Taken together, these varied results demonstrate a distinct lack of current consensus regarding expression in platelets and further research is warranted.
Genetic Deficiency of GAS6 or TAM Receptor(s)
Gas6−/− mice exhibit normal development, produce adequate-sized litters, and appear phenotypically normal (3). They have coagulation factor levels and clotting times within the normal range and exhibit normal platelet and megakaryocyte counts as well as normal platelet ultra-structural morphology (3). Gas6−/− mice do not demonstrate spontaneous bleeding or thrombosis. In fact, these mice are protected from thrombosis, and their platelets create smaller aggregates under flow conditions in comparison to their wild type (WT) counterparts (an effect reversible with the addition of recombinant human GAS6) (26).
Additionally, Gas6−/− mice exhibit decreased aggregation in response to low agonist concentrations [ADP 2.5–10 μM, collagen 2 μg/mL, and U46619 (thromboxane A2 analog) 10 μM] (3). At the same low agonist concentration, platelets from Gas6−/− mice showed evidence of normal shape change but did not aggregate to the same degree as platelets from WT mice. At high agonist concentrations (ADP 50 μM, collagen 5–15 μg/mL, and U46619 100 μM), irreversible aggregation was comparable between Gas6−/− and WT mice. Interestingly, with stimulation by a strong agonist such as thrombin, aggregation was comparable between Gas6−/− and WT platelets, but electron microscopy showed that Gas6−/− platelets were less densely packed, incompletely degranulated and made fewer connections with neighboring platelets (3). Testing the hypothesis that GAS6 is involved in cancer-induced coagulopathy, Aghourian et al. induced inferior vena cava thrombi with FeCl3 in both WT and GAS6−/− mice with lung carcinoma (93). The Gas6−/− mice formed much smaller thrombi than their WT counterparts, however larger thrombi were seen in Gas6−/− mice by injecting recombinant GAS6, indicating GAS6 inhibition may have anti-thrombotic effects in the setting of lung cancer (93).
Gas6−/− mice also exhibit decreased thrombosis in vivo. In a stasis-induced venous thrombosis model Gas6−/− mice developed significantly smaller thrombi than did WT mice (3). In a Rose Bengal model of arterial thrombosis, the Gas6−/− mice formed thrombi 60% smaller than did their WT counterparts (3). In an induced pulmonary embolism (PE) model histological examination failed to demonstrated the extensive PEs in the Gas6−/− mice that were consistently present in WT mice (3).
Genetic deficiencies of individual receptors for GAS6 also result in decreased platelet activation responses and protection from thrombosis in vivo. Loss of any one of the TAM receptors was sufficient to protect mice from collagen/epinephrine- and stasis-induced thromboembolism (7) without evidence of major hemorrhagic side effects, though GAS6−/− mice did have a tendency to re-bleed after tail clipping (7, 8) likely due to inability to form stable aggregates. After an initial platelet activation response to thrombin, mice deficient in one of the TAM receptors show significant reduction in clot retraction. For example, Mertk−/− mice demonstrate significantly lower platelet aggregation in response to low-moderate concentrations of agonist as compared to wild type mice (7). Additionally, no second wave of platelet aggregation is observed, likely due to impaired granule release. Mertk−/− mice were also protected from carotid artery occlusion in a ferric chloride induced injury model showing increased time to complete occlusion of the vessel as well as increased artery patency after 30 min. Further, bleeding time via tail clipping does not differ significantly between Mertk −/− and WT mice.
Interestingly, a more significant difference between WT and Mertk−/− was observed at lower agonist concentrations (2.5 μg/mL collagen, 10 μM U46619, and 0.25 mMoL/L PAR4 thrombin receptor agonist peptide AYPGKF) (7) indicating that strong stimulation from higher agonist concentrations may overcome the functional deficit, similar to that described Gas6−/− mice. Finally, Mertk−/− mice are viable, fertile and do not exhibit significant differences in litter size, mouse weight, cell counts in any lineage or coagulation tests such as the prothrombin time and partial thromboplastin time when compared to WT mice. In addition, the binding of Gas6−/− platelets to fibrinogen on initial activation with various agonists was not impaired, hence “inside-out” platelet signaling and subsequent activation of αIIbβ3, does not appear to be affected. However, αIIbβ3 tyrosine phosphorylation as well as Akt phosphorylation are significantly decreased and platelet spreading was impaired in mice deficient in one of the TAM receptors. Taken together, these data indicate that absence of any of the TAM receptors in platelets appears to cause inhibition of the “outside-in” signaling pathway. The GAS6/TAM pathway is not a primary activator of platelets, as this signal transduction cascade does not stimulate platelets by itself, but rather augments and potentiates activation processes initiated by other agonists (26, 81). Its inhibition (with or without additional antagonism of the ADP/P2Y12 pathway), therefore, has a mild anti-platelet and anti-thrombotic effect, with the benefit of minimal observable hemorrhagic effect.
Tyro3−/− mice likewise demonstrate reduced thrombus formation and decreased platelet aggregation stability (but not initial aggregation), due to decreased outside-in signaling and subsequent limitation of platelet granule secretion (7). Effects on inflammation in this strain are not well described, but neurologic effects of Tyro3 knockout include neural degeneration with seizures and paralysis (12).
Axl−/− mice exhibit hematologic effects such as reduced thrombus formation and decreased platelet aggregate stability, similar to the Tyro3−/− and Mertk−/− mice (7). Effects on inflammation include an increase in apoptosis and enhanced inflammation in the CNS because of delayed removal of myelin debris during experimental autoimmune encephalomyelitis (94). Additionally, these mice exhibit elevated vascular permeability and impaired vascular remodeling (including Increase in CD45+ cells and decrease in VSMC, macrophages, and neutrophils) (44).
Indeed, genetic disruption of any one of the three receptors is associated with a significant reduction in expression of the other two, and a total reduction in the platelet's ability to bind to and activate from GAS6 stimulation (7). Finally, decreased alpha granule secretion has been reported in Tyro3−/−, Axl−/−, and Mertk−/− mice (8).
Transgenic murine models with genetic deletion of all three TAM receptors have been described with platelet function inhibition similar to those mice with single receptor deletions. Additionally, they are prone to recurrent thrombosis and hemorrhage (61), thrombocytopenia and impaired hemostasis (79). They also exhibit elevated inflammation/auto-immunity, anti-phospholipid antibodies, blindness, enlarged spleens, small testes, and defective spermatogenesis (16).
Pharmacologic Inhibition of the GAS6/TAM Pathway
Anti-GAS6 Antibodies and GAS6 Traps
Anti-GAS6 antibodies have been shown to be effective inhibitors of platelet activation. Mice pre-treated with anti-GAS6 antibodies displayed protection from PE in a similar manner as that seen with Gas6−/− mice, and neither group showed evidence of increased bleeding (3). Additionally, it was demonstrated that GAS6 neutralizing antibodies inhibited aggregation in human washed platelets (3, 71). Not surprisingly, inhibition of aggregation with anti-GAS6 antibodies was observed at low agonist concentrations which was reversed with increasing concentration of agonists (71).
Trapping of GAS6 with a recombinant extracellular domain of AXL (hAxl-EC-Fc) resulted in a dose-dependent blockade of platelet aggregation in response to ADP, but had no effect in the absence of ADP (7). These mice were protected from collagen/epinephrine induced PE in a manner similar to the GAS6−/− mice, and showed no spontaneous bleeding or increase in bleeding times (7). Purified preparations of cell-surface cleavage products comprising plasma-derived soluble extracellular domains of MERTK (sMer) (80) and AXL(sAxl) (29) also have been shown to bind and sequester GAS6, resulting in decreased platelet aggregation and protection of mice from collagen-epinephrine induced PE.
Receptor-Specific Small Molecule Inhibitors
Recent studies have shown that MERTK-specific small molecule inhibitors UNC2025 and UNC2881 inactivate the MERTK tyrosine kinase domain, disrupting subsequent signaling (4, 81, 95). Of note, UNC2025 is equipotent against MERTK and FLT3, with 50-fold greater selectivity in cell-based assays than for AXL (95). Since FLT3 expression has not been reported in human or murine megakaryocytes or platelets, the antiplatelet effect of this compound is unlikely to be mediated through FLT3 inhibition.
Mice treated with these molecules demonstrated protection from PE in the collagen/epinephrine model, and the average patency time of the carotid after injury with FeCl3 was significantly increased (81). Additionally, treated mice did not show increased bleeding by tail clipping or spontaneous bleeding. Platelets from mice treated with these inhibitors also demonstrated decreased activation responses, decreased surface activation markers, and reduced stability of aggregates formed under physiologic flow conditions. UNC2025 caused a significant decrease in p-selectin positive platelets and total platelet counts (CD41 positive) at vascular injury locations via a laser intravital vascular microscopy model (81). These findings suggest that MERTK-specific small molecule inhibitors cause decreased accumulation without affecting platelet adhesion at sites of vascular injury.
The inhibition of platelet activation and protection from thrombosis afforded by the GAS6/TAM antagonists described above is encouraging but should be considered with caution. The limited predictive value of murine tail bleed hemostasis models are well-known (96) and may underestimate the bleeding risk imparted by certain compounds, such as the anti-factor VIII therapeutic antibody LE2E9 that prevented thrombosis without increased bleeding risk in animal models but increased hemorrhagic side effects compared to controls in a human therapeutic trial (97).
GAS6/TAM Antagonists Combined With ADP/P2Y Inhibitors
Washed human platelets pre-treated with a combination of UNC2025 and ADP/P2Y inhibitors (ADPis) demonstrated a statistically significant synergistic decrease in platelet aggregation and mice treated with the same combination exhibited a longer time until artery occlusion and shorter duration of occlusion in a FeCl3-induced carotid artery injury model than would be expected if combining the effects of the two agents independently, (81) suggesting a synergistic interaction. Similarly, the mice given a combination of low dose ADPi and UNC2025 were better protected against mortality in the collagen/epinephrine model than mice given either agent alone. As opposed to increased bleeding noted after tail clip in the high dose ADPi group, mice treated with a combination of ADPis and UNC2025 demonstrated no increased spontaneous bleeding or tail clip-induced bleeding.
The synergistic effect noted in these studies likely relates to the combination of effects on different areas of a thrombus that may disrupt its stable formation. When considered in the context of a hierarchical description of thrombus development proposed by Stalker et al. GAS6/TAM inhibitors appear to affect the thrombin-stimulated platelet core while ADP/P2Y inhibitors act on the shell (98).
Conclusion
Inhibition of the Gas6/TAM pathway leads to decreased platelet activation responses in vitro and protects mice from arterial or venous thrombosis in vivo. As mice with knockout of either the ligand or any of the TAM receptors are known to have normal hemostasis labs and do not exhibit spontaneous bleeding or prolonged time to cessation in bleeding models, this pathway represents an attractive potential target for novel anti-platelet agents with reduced risk of hemorrhagic complications compared to current agents. Further research is needed to address the potential effects of deletion or inhibition of this pathway outside of the hemostatic system.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported in part by the National Hemophilia Foundation/Baxter Clinical Research Fellowship, American Society of Hematology Scholar Award, Hemostasis, and Thrombosis Research Society Mentored Research Award (HTRS-MRA), CSL Behring/Prof. Heimburger Award in Hemostasis, and NIH K12HD068372-03 Child Health Research Career Development grant (BB), NIH R01HL084086, R01HL120728 and the Postle Family Chair of Pediatric Cancer and Blood Disorders (JD), and 5H30MC00008-20-00 HRSA/MCHB (BB and JD).
DG has filed a patent application for use of MERTK inhibition as cancer therapy.
References
1. Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ. Heart disease and stroke statistics−2014 update: a report from the American Heart Association. Circulation (2014) 129:e28–292. doi: 10.1161/01.cir.0000441139.02102.80
2. De Miguel A, Ibanez B, Badimon JJ. Clinical implications of clopidogrel resistance. Thromb Haemost. (2008) 100:196–203. doi: 10.1160/TH08-01-0049
3. Angelillo-Scherrer A, de Frutos P, Aparicio C, Melis E, Savi P, Lupu F, et al. Deficiency or inhibition of Gas6 causes platelet dysfunction and protects mice against thrombosis. Nat Med. (2001) 7:215–21. doi: 10.1038/84667
4. Zhang W, McIver AL, Stashko MA, DeRyckere D, Branchford BR, Hunter D, et al. Discovery of Mer specific tyrosine kinase inhibitors for the treatment and prevention of thrombosis. J Med Chem. (2013) 56:9693–700. doi: 10.1021/jm4013888
5. Paolino M, Penninger JM. The role of TAM family receptors in immune cell function: implications for cancer therapy. Cancers (2016) 8: doi: 10.3390/cancers8100097
6. Rothlin CV, Carrera-Silva EA, Bosurgi L, Ghosh S. TAM receptor signaling in immune homeostasis. Annu Rev Immunol. (2015) 33:355–91. doi: 10.1146/annurev-immunol-032414-112103
7. Angelillo-Scherrer A, Burnier L, Flores N, Savi P, DeMol M, Schaeffer P, et al. Role of Gas6 receptors in platelet signaling during thrombus stabilization and implications for antithrombotic therapy. J Clin Invest (2005) 115:237–46. doi: 10.1172/JCI22079
8. Saller F, Burnier L, Schapira M, Angelillo-Scherrer A. Role of the growth arrest-specific gene 6 (gas6) product in thrombus stabilization. Blood Cells Mol Dis. (2006) 36:373–8. doi: 10.1016/j.bcmd.2005.12.038
9. Melaragno MG, Cavet ME, Yan C, Tai LK, Jin ZG, Haendeler J, et al. Gas6 inhibits apoptosis in vascular smooth muscle: role of Axl kinase and Akt. J Mol Cell Cardiol. (2004) 37:881–7. doi: 10.1016/j.yjmcc.2004.06.018
10. Melaragno MG, Fridell YW, Berk BC. The Gas6/Axl system: a novel regulator of vascular cell function. Trends Cardiovasc Med. (1999) 9:250–3. doi: 10.1016/S1050-1738(00)00027-X
11. Melaragno MG, Wuthrich DA, Poppa V, Gill D, Lindner V, Berk BC, et al. Increased expression of Axl tyrosine kinase after vascular injury and regulation by G protein-coupled receptor agonists in rats. Circ Res. (1998) 83:697–704. doi: 10.1161/01.RES.83.7.697
12. Lu Q, Gore M, Zhang Q, Camenisch T, Boast S, Casagranda F, Lai C, et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature (1999) 398:723–8. doi: 10.1038/19554
13. Feng W, Yasumura D, Matthes MT, LaVail MM, Vollrath D. Mertk triggers uptake of photoreceptor outer segments during phagocytosis by cultured retinal pigment epithelial cells. J Biol Chem. (2002) 277:17016–22. doi: 10.1074/jbc.M107876200
14. Cook RS, Jacobsen KM, Wofford AM, DeRyckere D, Stanford J, Prieto AL, et al. MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J Clin Invest. (2013) 123:3231–42. doi: 10.1172/JCI67655
15. Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell (2007) 131:1124–36. doi: 10.1016/j.cell.2007.10.034
16. van der Meer JH, van der Poll T, van 't Veer C. TAM receptors, Gas6, and protein S: roles in inflammation and hemostasis. Blood (2014) 123:2460–9. doi: 10.1182/blood-2013-09-528752
17. Ksantini M, Lafont E, Bocquet B, Meunier I, Hamel CP. Homozygous mutation in MERTK causes severe autosomal recessive retinitis pigmentosa. Eur J Ophthalmol. (2012) 22:647–53. doi: 10.5301/ejo.5000096
18. Gal A, Li Y, Thompson DA, Weir J, Orth U, Jacobson SG, et al. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat Genet. (2000) 26:270–1. doi: 10.1038/81555
19. McHenry CL, Liu Y, Feng W, Nair AR, Feathers KL, Ding X, et al. MERTK arginine-844-cysteine in a patient with severe rod-cone dystrophy: loss of mutant protein function in transfected cells. Invest Ophthalmol Vis Sci. (2004) 45:1456–63. doi: 10.1167/iovs.03-0909
20. Everson R, Pettitt L, Forman OP, Dower-Tylee O, McLaughlin B, Ahonen S, et al. An intronic LINE-1 insertion in MERTK is strongly associated with retinopathy in Swedish Vallhund dogs. PLoS ONE (2017) 12:e0183021. doi: 10.1371/journal.pone.0183021
21. Vollrath D, Yasumura D, Benchorin G, Matthes MT, Feng W, Nguyen NM, et al. Tyro3 modulates Mertk-Associated Retinal Degeneration. PLoS Genet. (2015) 11:e1005723. doi: 10.1371/journal.pgen.1005723
22. Ghazi NG, Abboud EB, Nowilaty SR, Alkuraya H, Alhommadi A, Cai HM, et al. Treatment of retinitis pigmentosa due to MERTK mutations by ocular subretinal injection of adeno-associated virus gene vector: results of a phase I trial. Hum Genet. (2016) 135:327–43. doi: 10.1007/s00439-016-1637-y
23. Ramsden CM, Nommiste B, R Lane A, Carr AF, Powner MB, J K Smart M, et al. Rescue of the MERTK phagocytic defect in a human iPSC disease model using translational read-through inducing drugs. Sci Rep. (2017) 7:51. doi: 10.1038/s41598-017-00142-7
24. Li Z, Delaney MK, O'Brien KA, Du X. Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol. (2010) 30:2341–9. doi: 10.1161/ATVBAHA.110.207522
25. Ren Q, Ye S, Whiteheart SW. The platelet release reaction: just when you thought platelet secretion was simple. Curr Opin Hematol. (2008) 15:537–41. doi: 10.1097/MOH.0b013e328309ec74
26. Cosemans JM, Van Kruchten R, Olieslagers S, Schurgers LJ, Verheyen FK, Munnix IC, et al. Potentiating role of Gas6 and Tyro3, Axl and Mer (TAM) receptors in human and murine platelet activation and thrombus stabilization. J Thromb Haemost. (2010) 8:1797–808. doi: 10.1111/j.1538-7836.2010.03935.x
27. Laurance S, Lemarie CA, Blostein MD. Growth arrest-specific gene 6 (gas6) and vascular hemostasis. Adv Nutr. (2012) 3:196–203. doi: 10.3945/an.111.001826
28. Nagai K, Miyoshi M, Kake T, Fukushima N, Matsuura M, Shibata E, et al. Dual involvement of growth arrest-specific gene 6 in the early phase of human IgA nephropathy. PLoS ONE (2013) 8:e66759. doi: 10.1371/journal.pone.0066759
29. Ekman C, Stenhoff J, Dahlback B. Gas6 is complexed to the soluble tyrosine kinase receptor Axl in human blood. J Thromb Haemost. (2010) 8:838–44. doi: 10.1111/j.1538-7836.2010.03752.x
30. Angelillo-Scherrer A, Burnier L, Lambrechts D, Fish RJ, Tjwa M, Plaisance S, et al. Role of Gas6 in erythropoiesis and anemia in mice. J Clin Invest. (2008) 118:583–96. doi: 10.1172/JCI30375
31. Clauser S, Meilhac O, Bieche I, Raynal P, Bruneval P, Michel JB, et al. Increased secretion of Gas6 by smooth muscle cells in human atherosclerotic carotid plaques. Thromb Haemost. (2012) 107:140–9. doi: 10.1160/TH11-05-0368
32. Blostein MD, Rajotte I, Rao DP, Holcroft CA, Kahn SR. Elevated plasma gas6 levels are associated with venous thromboembolic disease. J Thromb Thrombolysis (2011) 32:272–8. doi: 10.1007/s11239-011-0597-2
33. Laurance S, Bertin FR, Ebrahimian T, Kassim Y, Rys RN, Lehoux S, et al. Gas6 promotes inflammatory (CCR2(hi)CX3CR1(lo)) monocyte recruitment in venous thrombosis. Arterioscler Thromb Vasc Biol. (2017) 37:1315–22. doi: 10.1161/ATVBAHA.116.308925
34. Ekman C, Linder A, Akesson P, Dahlback B. Plasma concentrations of Gas6 (growth arrest specific protein 6) and its soluble tyrosine kinase receptor sAxl in sepsis and systemic inflammatory response syndromes. Crit Care (2010) 14:R158. doi: 10.1186/cc9233
35. Ekman C, Jonsen A, Sturfelt G, Bengtsson AA, Dahlback B. Plasma concentrations of Gas6 and sAxl correlate with disease activity in systemic lupus erythematosus. Rheumatology (2011) 50:1064–9. doi: 10.1093/rheumatology/keq459
36. Recarte-Pelz P, Tassies D, Espinosa G, Hurtado B, Sala N, Cervera R, et al. Vitamin K-dependent proteins GAS6 and protein S and TAM receptors in patients of systemic lupus erythematosus: correlation with common genetic variants and disease activity. Arthritis Res Ther. (2013) 15:R41. doi: 10.1186/ar4199
37. Wu CS, Hu CY, Tsai HF, Chyuan IT, Chan CJ, Chang SK, et al. Elevated serum level of growth arrest-specific protein 6 (Gas6) in systemic lupus erythematosus patients is associated with nephritis and cutaneous vasculitis. Rheumatol Int. (2014) 34:625–9. doi: 10.1007/s00296-013-2882-1
38. Giangola MD, Yang WL, Rajayer SR, Nicastro J, Coppa GF, Wang P. Growth arrest-specific protein 6 attenuates neutrophil migration and acute lung injury in sepsis. Shock (2013) 40:485–91. doi: 10.1097/SHK.0b013e3182a588c1
39. Alciato F, Sainaghi PP, Sola D, Castello L, Avanzi GC. TNF-alpha, IL-6, and IL-1 expression is inhibited by GAS6 in monocytes/macrophages. J Leukocyte Biol. (2010) 87:869–75. doi: 10.1189/jlb.0909610
40. Fourcot A, Couchie D, Chobert MN, Zafrani ES, Mavier P, Laperche Y, et al. Gas6 deficiency prevents liver inflammation, steatohepatitis, and fibrosis in mice. Am J Physiol Gastrointest Liver Physiol. (2011) 300:G1043–53. doi: 10.1152/ajpgi.00311.2010
41. Akitake-Kawano R, Seno H, Nakatsuji M, Kimura Y, Nakanishi Y, Yoshioka T, et al. Inhibitory role of Gas6 in intestinal tumorigenesis. Carcinogenesis (2013) 34:1567–74. doi: 10.1093/carcin/bgt069
42. Migdall-Wilson J, Bates C, Schlegel J, Brandao L, Linger RM, DeRyckere D, et al. Prolonged exposure to a Mer ligand in leukemia: Gas6 favors expression of a partial Mer glycoform and reveals a novel role for Mer in the nucleus. PLoS ONE (2012) 7:e31635. doi: 10.1371/journal.pone.0031635
43. Foley JH, Conway EM. Gas6 gains entry into the coagulation cascade. Blood (2013) 121:570–1. doi: 10.1182/blood-2012-11-468678
44. Korshunov VA. Axl-dependent signalling: a clinical update. Clin Sci. (2012) 122:361–8. doi: 10.1042/CS20110411
45. Brown JE, Krodel M, Pazos M, Lai C, Prieto AL. Cross-phosphorylation, signaling and proliferative functions of the Tyro3 and Axl receptors in Rat2 cells. PLoS ONE (2012) 7:e36800. doi: 10.1371/journal.pone.0036800
46. Kim S, Mangin P, Dangelmaier C, Lillian R, Jackson SP, Daniel JL, et al. Role of phosphoinositide 3-kinase beta in glycoprotein VI-mediated Akt activation in platelets. J Biol Chem. (2009) 284:33763–72. doi: 10.1074/jbc.M109.048553
47. Moore SF, Hunter RW, Harper MT, Savage JS, Siddiq S, Westbury SK, et al. Dysfunction of the PI3 kinase/Rap1/integrin alpha(IIb)beta(3) pathway underlies ex vivo platelet hypoactivity in essential thrombocythemia. Blood (2013) 121:1209–19. doi: 10.1182/blood-2012-05-431288
48. Zhang G, Xiang B, Ye S, Chrzanowska-Wodnicka M, Morris AJ, Gartner TK, et al. Distinct roles for Rap1b protein in platelet secretion and integrin alphaIIbbeta3 outside-in signaling. J Biol Chem. (2011) 286:39466–77. doi: 10.1074/jbc.M111.239608
49. Joo SJ. Mechanisms of Platelet Activation and Integrin alphaIIbeta3. Korean Circ J. (2012) 42:295–301. doi: 10.4070/kcj.2012.42.5.295
50. Woulfe DS. Akt signaling in platelets and thrombosis. Expert Rev Hematol. (2010) 3:81–91. doi: 10.1586/ehm.09.75
51. O'Brien KA, Stojanovic-Terpo A, Hay N, Du XP. An important role for Akt3 in platelet activation and thrombosis. Blood (2011) 118:4215–23. doi: 10.1182/blood-2010-12-323204
52. Kovacsovics TJ, Bachelot C, Toker A, Vlahos CJ, Duckworth B, Cantley LC, et al. Phosphoinositide 3-kinase inhibition spares actin assembly in activating platelets but reverses platelet aggregation. J Biol Chem. (1995) 270:11358–66. doi: 10.1074/jbc.270.19.11358
53. Trumel C, Payrastre B, Plantavid M, Hechler B, Viala C, Presek P, et al. A key role of adenosine diphosphate in the irreversible platelet aggregation induced by the PAR1-activating peptide through the late activation of phosphoinositide 3-kinase. Blood (1999) 94:4156–65.
54. Lemke G, Rothlin CV. Immunobiology of the TAM receptors. Nat Rev Immunol (2008) 8:327–36. doi: 10.1038/nri2303
55. Shattil SJ, Newman PJ. Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood (2004) 104:1606–15. doi: 10.1182/blood-2004-04-1257
56. Linger RM, Keating AK, Earp HS, Graham DK. Taking aim at Mer and Axl receptor tyrosine kinases as novel therapeutic targets in solid tumors. Expert Opin Ther Targets (2010) 14:1073–90. doi: 10.1517/14728222.2010.515980
57. Huang M, Rigby AC, Morelli X, Grant MA, Huang G, Furie B, et al. Structural basis of membrane binding by Gla domains of vitamin K-dependent proteins. Nat Struct Biol. (2003) 10:751–6. doi: 10.1038/nsb971
58. Nakano T, Ishimoto Y, Kishino J, Umeda M, Inoue K, Nagata K, et al. Cell adhesion to phosphatidylserine mediated by a product of growth arrest-specific gene 6. J Biol Chem. (1997) 272:29411–4. doi: 10.1074/jbc.272.47.29411
59. Rajotte I, Hasanbasic I, Blostein M. Gas6-mediated signaling is dependent on the engagement of its gamma-carboxyglutamic acid domain with phosphatidylserine. Biochem Biophys Res Commun. (2008) 376:70–3. doi: 10.1016/j.bbrc.2008.08.083
60. Morizono K, Xie Y, Olafsen T, Lee B, Dasgupta A, Wu AM, et al. The soluble serum protein Gas6 bridges virion envelope phosphatidylserine to the TAM receptor tyrosine kinase Axl to mediate viral entry. Cell Host Microbe (2011) 9:286–98. doi: 10.1016/j.chom.2011.03.012
61. Lu Q, Lemke G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science (2001) 293:306–11. doi: 10.1126/science.1061663
62. Korshunov VA, Mohan AM, Georger MA, Berk BC. Axl, a receptor tyrosine kinase, mediates flow-induced vascular remodeling. Circ Res. (2006) 98:1446–52. doi: 10.1161/01.RES.0000223322.16149.9a
63. O'Bryan JP, Frye RA, Cogswell PC, Neubauer A, Kitch B, Prokop C, et al. axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol Cell Biol. (1991) 11:5016–31. doi: 10.1128/MCB.11.10.5016
64. Hafizi S, Dahlback B. Gas6 and protein S. Vitamin K-dependent ligands for the Axl receptor tyrosine kinase subfamily. FEBS J. (2006) 273:5231–44. doi: 10.1111/j.1742-4658.2006.05529.x
65. Nakamura YS, Hakeda Y, Takakura N, Kameda T, Hamaguchi I, Miyamoto T, et al. Tyro 3 receptor tyrosine kinase and its ligand, Gas6, stimulate the function of osteoclasts. Stem Cells (1998) 16:229–38. doi: 10.1002/stem.160229
66. Katagiri M, Hakeda Y, Chikazu D, Ogasawara T, Takato T, Kumegawa M, et al. Mechanism of stimulation of osteoclastic bone resorption through Gas6/Tyro 3, a receptor tyrosine kinase signaling, in mouse osteoclasts. J Biol Chem. (2001) 276:7376–82. doi: 10.1074/jbc.M007393200
67. Binder MD, Xiao J, Kemper D, Ma GZ, Murray SS, Kilpatrick TJ. Gas6 increases myelination by oligodendrocytes and its deficiency delays recovery following cuprizone-induced demyelination. PLoS ONE (2011) 6:e17727. doi: 10.1371/journal.pone.0017727
68. Shankar SL, O'Guin K, Cammer M, McMorris FA, Stitt TN, Basch RS, et al The growth arrest-specific gene product Gas6 promotes the survival of human oligodendrocytes via a phosphatidylinositol 3-kinase-dependent pathway. J Neurosci. (2003) 23:4208–18. doi: 10.1523/JNEUROSCI.23-10-04208.2003
69. Shankar SL, O'Guin K, Kim M, Varnum B, Lemke G, Brosnan CF, et al. Gas6/Axl signaling activates the phosphatidylinositol 3-kinase/Akt1 survival pathway to protect oligodendrocytes from tumor necrosis factor alpha-induced apoptosis. J Neurosci. (2006) 26:5638–48. doi: 10.1523/JNEUROSCI.5063-05.2006
70. Clauser S, Bachelot-Lozat C, Fontana P, Gaussem P, Remones V, Aiach M, et al. Physiological plasma Gas6 levels do not influence platelet aggregation. Arterioscler Thromb Vasc Biol. (2006) 26:e22. doi: 10.1161/01.ATV.0000201968.72967.f9
71. Gould WR, Baxi SM, Schroeder R, Peng YW, Leadley RJ, Peterson JT, et al. Gas6 receptors Axl, Sky and Mer enhance platelet activation and regulate thrombotic responses. J Thromb Haemost. (2005) 3:733–41. doi: 10.1111/j.1538-7836.2005.01186.x
72. Coppinger JA, Cagney G, Toomey S, Kislinger T, Belton O, McRedmond JP, et al. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood (2004) 103:2096–104. doi: 10.1182/blood-2003-08-2804
73. Ombrello C, Block RC, Morrell CN. Our expanding view of platelet functions and its clinical implications. J Cardiovas Transl Res (2010) 3:538–46. doi: 10.1007/s12265-010-9213-7
74. Alciato F, Sainaghi PP, Castello L, Bergamasco L, Carnieletto S, Avanzi GC. Development and validation of an ELISA method for detection of growth arrest specific 6(GAS6) protein in human plasma. J Immunoassay Immunochem. (2008) 29:167–80. doi: 10.1080/15321810801888480
75. Balogh I, Hafizi S, Stenhoff J, Hansson K, Dahlback B. Analysis of Gas6 in human platelets and plasma. Arterioscler Thromb Vasc Biol. (2005) 25:1280–6. doi: 10.1161/01.ATV.0000163845.07146.48
76. Clauser S, Peyrard S, Gaussem P, Crespin M, Emmerich J, Aiach M, et al. Development of a novel immunoassay for the assessment of plasma Gas6 concentrations and their variation with hormonal status. Clin Chem. (2007) 53:1808–13. doi: 10.1373/clinchem.2007.089102
77. Chen CL, Li Q, Darrow AL, Wang YP, Derian CK, Yang J, et al. Mer receptor tyrosine kinase signaling participates in platelet function. Arterioscl Throm Vasc Biol. (2004) 24:1118–123. doi: 10.1161/01.ATV.0000130662.30537.08
78. Tjwa M, Bellido-Martin L, Lin Y, Lutgens E, Plaisance S, Bono F, et al. Gas6 promotes inflammation by enhancing interactions between endothelial cells, platelets, and leukocytes. Blood (2008) 111:4096–105. doi: 10.1182/blood-2007-05-089565
79. Wang H, Chen S, Chen Y, Wang H, Wu H, Tang H, et al. The role of Tyro 3 subfamily receptors in the regulation of hemostasis and megakaryocytopoiesis. Haematologica (2007) 92:643–50. doi: 10.3324/haematol.10939
80. Sather S, Kenyon KD, Lefkowitz JB, Liang X, Varnum BC, Henson PM, et al. A soluble form of the Mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood (2007) 109:1026–33. doi: 10.1182/blood-2006-05-021634
81. Branchford BR, Stalker TJ, Law L, Acevedo G, Sather S, Brzezinski C, et al. The small molecule MERTK inhibitor UNC2025 decreases platelet activation and prevents thrombosis. J Thromb Haemost. (2017) 16:352–63. doi: 10.1111/jth.13875.
82. Stitt TN, Conn G, Gore M, Lai C, Bruno J, Radziejewski C, et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell (1995) 80:661–70. doi: 10.1016/0092-8674(95)90520-0
83. Dahlback B, Villoutreix BO. Regulation of blood coagulation by the protein C anticoagulant pathway: novel insights into structure-function relationships and molecular recognition. Arterioscler Thromb Vasc Biol. (2005) 25:1311–20. doi: 10.1161/01.ATV.0000168421.13467.82
84. Burstyn-Cohen T, Lew ED, Traves PG, Burrola PG, Hash JC, Lemke G. Genetic dissection of TAM receptor-ligand interaction in retinal pigment epithelial cell phagocytosis. Neuron (2012) 76:1123–32. doi: 10.1016/j.neuron.2012.10.015
85. Caberoy NB, Zhou Y, Li W. Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J. (2010) 29:3898–910. doi: 10.1038/emboj.2010.265
86. Caberoy NB, Alvarado G, Bigcas JL, Li W. Galectin-3 is a new MerTK-specific eat-me signal. J Cell Physiol. (2012) 227:401–7. doi: 10.1002/jcp.22955
87. Mukherjee SK, Wilhelm A, Antoniades CG. TAM receptor tyrosine kinase function and the immunopathology of liver disease. Am J Physiol Gastrointest Liver Physiol. (2016) 310:G899–905. doi: 10.1152/ajpgi.00382.2015
88. Cunningham BA, Hemperly JJ, Murray BA, Prediger EA, Brackenbury R, Edelman GM. Neural cell adhesion molecule: structure, immunoglobulin-like domains, cell surface modulation, and alternative RNA splicing. Science (1987) 236:799–806. doi: 10.1126/science.3576199
89. Fischer EH, Charbonneau H, Tonks NK. Protein tyrosine phosphatases: a diverse family of intracellular and transmembrane enzymes. Science (1991) 253:401–6. doi: 10.1126/science.1650499
90. Fisher PW, Brigham-Burke M, Wu SJ, Luo J, Carton J, Staquet K, et al. A novel site contributing to growth-arrest-specific gene 6 binding to its receptors as revealed by a human monoclonal antibody. Biochem J. (2005) 387:727–35. doi: 10.1042/BJ20040859
91. Nagata K, Ohashi K, Nakano T, Arita H, Zong C, Hanafusa H, et al. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J Biol Chem. (1996) 271:30022–7. doi: 10.1074/jbc.271.47.30022
92. Chen J, Carey K, Godowski PJ. Identification of Gas6 as a ligand for Mer, a neural cell adhesion molecule related receptor tyrosine kinase implicated in cellular transformation. Oncogene (1997) 14:2033–9. doi: 10.1038/sj.onc.1201039
93. Aghourian MN, Lemarie CA, Bertin FR, Blostein MD. Prostaglandin E synthase is upregulated by Gas6 during cancer-induced venous thrombosis. Blood (2016) 127:769–77. doi: 10.1182/blood-2015-02-628867
94. Weinger JG, Brosnan CF, Loudig O, Goldberg MF, Macian F, Arnett HA, et al. Loss of the receptor tyrosine kinase Axl leads to enhanced inflammation in the CNS and delayed removal of myelin debris during experimental autoimmune encephalomyelitis. J Neuroinflammation (2011) 8:49. doi: 10.1186/1742-2094-8-49
95. Zhang W, DeRyckere D, Hunter D, Liu J, Stashko MA, Minson KA, et al. UNC2025, a Potent and Orally Bioavailable MER/FLT3 Dual Inhibitor. J Med Chem. (2014). 57:7031–41 doi: 10.1021/jm500749d
96. Mackman N. Tissue-specific hemostasis in mice. Arterioscler Thromb Vasc Biol. 25:2273–81. doi: 10.1161/01.ATV.0000183884.06371.52
97. Singh I, Smith A, Vanzieleghem B, Collen D, Burnand K, Saint-Remy JM, et al. Antithrombotic effects of controlled inhibition of factor VIII with a partially inhibitory human monoclonal antibody in a murine vena cava thrombosis model. Blood (2002) 99:3235–40. doi: 10.1182/blood.V99.9.3235
Keywords: GAS6, TYRO3, AXL, MERTK, platelet activation, signaling pathways
Citation: Law LA, Graham DK, Di Paola J and Branchford BR (2018) GAS6/TAM Pathway Signaling in Hemostasis and Thrombosis. Front. Med. 5:137. doi: 10.3389/fmed.2018.00137
Received: 12 February 2018; Accepted: 23 April 2018;
Published: 09 May 2018.
Edited by:
Evi X. Stavrou, Case Western Reserve University, United StatesCopyright © 2018 Law, Graham, Di Paola and Branchford. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brian R. Branchford, YnJpYW4uYnJhbmNoZm9yZEB1Y2RlbnZlci5lZHU=