 Martin Laurence
Martin Laurence Mark Asquith
Mark Asquith James T. Rosenbaum
James T. Rosenbaum

95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Med. , 05 April 2018
Sec. Rheumatology
Volume 5 - 2018 | https://doi.org/10.3389/fmed.2018.00080
Spondyloarthritis is a common type of arthritis which affects mostly adults. It consists of idiopathic chronic inflammation of the spine, joints, eyes, skin, gut, and prostate. Inflammation is often asymptomatic, especially in the gut and prostate. The HLA-B*27 allele group, which presents intracellular peptides to CD8+ T cells, is by far the strongest risk factor for spondyloarthritis. The precise mechanisms and antigens remain unknown. In 1959, Catterall and King advanced a novel hypothesis explaining the etiology of spondyloarthritis: an as-yet-unrecognized sexually acquired microbe would be causing all spondyloarthritis types, including acute anterior uveitis. Recent studies suggest an unrecognized sexually acquired fungal infection may be involved in prostate cancer and perhaps multiple sclerosis. This warrants reanalyzing the Catterall–King hypothesis based on the current literature. In the last decade, many links between spondyloarthritis and fungal infections have been found. Antibodies against the fungal cell wall component mannan are elevated in spondyloarthritis. Functional polymorphisms in genes regulating the innate immune response against fungi have been associated with spondyloarthritis (CARD9 and IL23R). Psoriasis and inflammatory bowel disease, two common comorbidities of spondyloarthritis, are both strongly associated with fungi. Evidence reviewed here lends credence to the Catterall–King hypothesis and implicates a common fungal etiology in prostate cancer, benign prostatic hyperplasia, multiple sclerosis, psoriasis, inflammatory bowel disease, and spondyloarthritis. However, the evidence available at this time is insufficient to definitely confirm this hypothesis. Future studies investigating the microbiome in relation to these conditions should screen specimens for fungi in addition to bacteria. Future clinical studies of spondyloarthritis should consider antifungals which are effective in psoriasis and multiple sclerosis, such as dimethyl fumarate and nystatin.
More than four decades ago, major histocompatibility complex (MHC) class I allele group HLA-B*27 was identified as a potent risk factor for developing spondyloarthritis (1, 2). Despite the strength of this association, mechanisms accounting for the link between HLA-B*27 and spondyloarthritis remain a mystery. The bacterial microbiome has attracted a great deal of attention as a possible explanation for this link (3). While we acknowledge the importance of bacteria, we describe a likely role for fungi and suggest that the prostate may be an important locale harboring microbes etiologically related to spondyloarthritis.
Recent studies have linked the immune response against fungi with prostate cancer (4) and multiple sclerosis (5). Sexual risk factors of prostate cancer (6–8) and multiple sclerosis (9–12) suggest that there may exist an as-yet-unrecognized sexually transmitted infection (STI) etiologically involved in these two diseases (4, 5, 13, 14). Epidemiological evidence suggests this elusive STI may be a fungal infection (15).
Many forms of spondyloarthritis have links with fungal infections, including ankylosing spondylitis (AS) (16), acute anterior uveitis (AAU) (16), Crohn’s disease (17, 18), and psoriasis (19, 20). One form of spondyloarthritis, reactive arthritis (ReA), has clear sexual risk factors—though causal associations with known STIs remain uncertain (21, 22). ReA and other spondyloarthritides are rare in children, and onset typically occurs in young adulthood (23, 24), mirroring the occurrence of STIs (25). In 1959, Catterall and King postulated a common sexually acquired infectious etiology for all spondyloarthritis types, whose primary focus in men is the prostate (26, 27).
In this article, we review the evidence which allowed Catterall and King to postulate their hypothesis, and related studies published in the six intervening decades. We then review the links between fungi and spondyloarthritis, in part to determine if the sexually acquired prostatic infection sought by Catterall and King (28) could be the same as the putative sexually acquired fungal infection suspected of causing prostate cancer (4, 15).
Between 1818 and 1948 many case reports were published describing a syndrome affecting men, consisting mainly of the simultaneous inflammation of the urethra, eyes, and joints (29). This syndrome was initially called Reiter’s disease. It is now called ReA due to the genital and enteric infections which often immediately precede onset. The classic eye inflammation in ReA is bilateral conjunctivitis on the external surface of the eye and less frequently unilateral AAU (30).
After studying 126 cases, Harkness published a comprehensive review of ReA (29). He made several key observations (Table 1). Harkness extended the definition of ReA to include incomplete cases in which either eye or joint inflammation was absent, and to similar cases in women where the main genital symptom was cervicitis. Harkness noted that generally no microbe could be observed or cultured to account for urethritis or arthritis symptoms, and that the presence of Neisseria gonorrhoeae in a subset of patients—previously thought to be causative—seemed to be coincidental. Some idiopathic urethritis cases in his series were likely caused by infections that were not as well characterized at the time, such as Chlamydia trachomatis (serology was positive in 15% of his cases) and Mycoplasmataceae species. Idiopathic urethritis remains frequent today: its prevalence is about 13% in healthy American men (31), and no infectious agent can be found in about half of American men presenting with urethritis at an STI clinic (32). Similarly, no causative microbe can be found in about half of sexually acquired ReA cases (22).

Table 1. Nine key observations in Harkness’ 1949 review of ReA.
Harkness advanced a four-part hypothesis to explain the etiology of ReA: (a) a single as-yet-unrecognized infectious agent is necessary in all ReA cases; (b) some relapses are caused by non-sexual “additional factors” that reactivate the infectious agent; (c) the infectious agent cannot be cleared by the immune system in at least some cases and remains in a latent state between attacks; and (d) enteric microbes are not directly causative, but rather unleash the single causative infectious agent.
The association between inflammation of the prostate—usually defined as a high concentration of leukocytes in expressed prostatic secretion obtained by prostate massage—and non-genital symptoms was firmly established by Visher in 1929 (41). Visher tested the expressed prostatic secretion of 500 consecutive young men admitted for any reason to the Veterans’ Hospital in Waukesha, Wisconsin (41). 87 men (17%) had a high prostate leukocyte concentration (41). Of these 87 men, 36 men had radiographs taken of their spine and sacroiliac joints, of which 20 showed signs of AS (55%) (41). This greatly exceeded the expected rate, though control radiographs were not used.
In 1958, a Swedish group (42) and a British group (43) published independent studies which replicated the association between prostate, spine, and sacroiliac inflammation reported by Visher. Both studies reported prostatic inflammation in a third of controls (22/66 and 28/85, respectively) and in nearly all AS patients (71/73 and 45/54, respectively) (42, 43). ReA cases were also included in these two studies, and a strong association with prostatic inflammation was found again (34/40 and 56/59, respectively) (42, 43). Many other studies reported high rates of prostatic inflammation in AS and ReA patients (44–47), though they did not include controls, so their results are difficult to interpret (48).
By 1957, it was recognized that ReA often occurred in an incomplete form (29). King suspected that idiopathic AAU, a common comorbidity of ReA and AS, was part of the same syndrome even when it occurred alone. King thus predicted that prostatic inflammation would be associated with isolated AAU (49), as previously reported in small case series (50–54). To formally demonstrate this association, King asked Catterall to perform a prospective study of all new male uveitis cases from May 1957 to December 1958 at the Institute of Ophthalmology in London (49). Catterall’s study confirmed that AAU was strongly associated with prostatic inflammation as compared to controls and to other patients in the series (Table 2) (27, 28).
Based on these prostatic inflammation association studies, Catterall and King advanced a three-part hypothesis (26, 28): (a) isolated AAU, ReA, and AS are part of the same syndrome; (b) an as-yet-unidentified genital infection is necessary for this syndrome; and (c) this infection is generally sexually acquired. The main marker of this genital infection in men was considered to be prostatic inflammation, as determined by counting the number of leukocytes in expressed prostatic secretion. Unfortunately, this was a low specificity marker because the vast majority of men with prostatic inflammation never develop the syndrome (43). This was thought to be due to rare genetic predispositions which were necessary for inflammation to occur outside the genital area (43). No assay available at the time managed to detect the putative infection (48). Four years later, one of King’s coworkers stated (55): “The trouble is that we have not yet identified the organism.”
The association between prostatic inflammation and isolated AAU was not considered biologically plausible, and Catterall’s results were deemed unlikely (56, 57). All studies associating prostatic inflammation with isolated AAU, ReA, or AS used semi-quantitative leukocyte per high-powered field methods, which could have been biased by improper blinding during analysis or sample collection, yielding a spurious association.
To test the association between prostatic inflammation and isolated AAU with the least possible bias and highest accuracy, Dark and Morton used total ejaculate rather than expressed prostatic secretion (eliminating sample collection bias), used cell counting chambers rather than plain microscope slides (a fully quantitative counting method), blinded their analysis (eliminating observer bias), and excluded men with a history of urethritis or ReA (eliminating bias toward sexually acquired cases) (57). Their results were unequivocal: the association between genital inflammation and isolated AAU is real (57).
The HLA-B*27 allele group is the strongest genetic risk factor for spondyloarthritis. Its association with AS exceeds odds ratios of 40 in Caucasian populations (58–60), and it is also strongly associated with ReA (36) and isolated AAU (61). Table 3 lists conditions associated with the HLA-B*27 allele group. Though hypotheses other than antigen presentation have been proposed, the association of ERAP1 polymorphisms with AS in HLA-B*27 carriers strongly suggests major histocompatibility complex (MHC) class I antigen presentation to CD8+ T cells is part of the causative pathway leading to spondyloarthritis (62). The discovery of HLA-B*27 and its association with isolated AAU, ReA, and AS gave much credence to the Catterall–King hypothesis by confirming that these conditions shared an underlying immune mechanism (56). It also provided evidence that the putative etiological infectious agent was intracellular and that a genetically determined immune response caused symptoms (43).
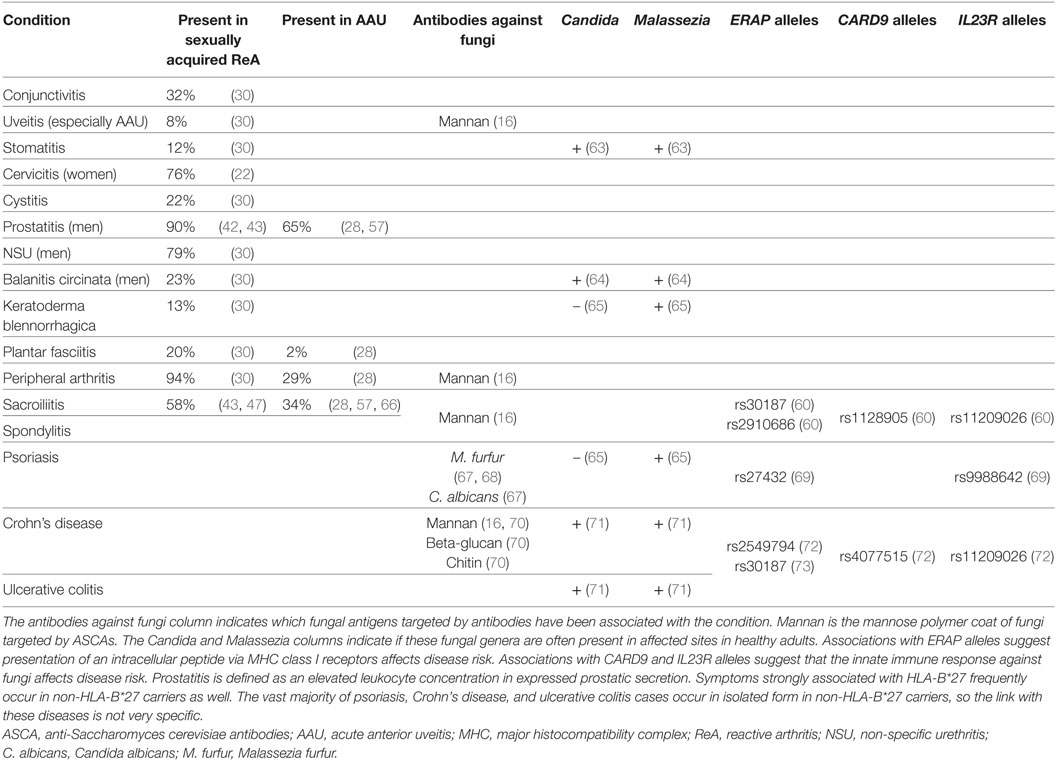
Table 3. Conditions associated with HLA-B*27 spondyloarthritides.
The expression of HLA-B in various tissues and cell types was recently measured by the Human Protein Atlas project (74). HLA-B exhibited highly variable expression in different tissues and surprisingly was either undetected or negligibly expressed in a number of tissues including hepatocytes, myocytes, and soft tissues. HLA-B was highly expressed in the secretory epithelial cells of the prostate (although less in the cervix), glandular cells of the small intestine and colon, as well as the skin. Its expression was not measured in the eyes or joints, though the strong expression of HLA-B*27 in synovial lining cells of AS patients was reported by an older study (75). HLA-B*27 presentation of intracellular antigens is thus consistent with HLA-B expression in all spondyloarthritis sites tested.
Enteric microbial epidemics and sexual risk factors have both been convincingly associated with the onset of ReA (76). Proving this link was relatively easy due to the short lag of a few weeks between enteric/genital symptoms and ReA symptoms. The list of confirmed or suspected precipitating infectious agents is very long, especially for enteric ReA (76–78). The relative incidence of enteric and genital ReA cases is not firmly established. The largest prospective community-based study, performed in Oslo (Norway) between 1988 and 1990, found that half of the cases were idiopathic and the remaining cases were evenly split between enteric and sexually acquired types (36). A smaller study published 10 years earlier reported a similar distribution (37). Another small study published 10 years later found a similar fraction of idiopathic cases, but a much lower fraction of sexually acquired cases as defined by C. trachomatis seropositivity (38).
Though various infections have been considered as possible triggers for isolated AAU and AS, fewer studies have investigated this link as compared to ReA. The onset of AS symptoms is gradual, so retracing infectious triggers which occurred years earlier is difficult.
One of the best controlled studies of enteric ReA was conducted in 1962 on an American Navy ship after a sudden outbreak of Shigella-induced dysentery (40). Because the outbreak timing was circumscribed and the ship was at sea during the following months, all ReA cases coinciding with this outbreak could be traced (40). Out of a population of 1,276 male crew members, 602 proven cases of dysentery occurred, of which nine developed ReA (1.5%) (40). The sequence of symptoms varied between individuals, with six out of nine showing the complete triad and with urethritis preceding other symptoms in most cases (40). This matches ReA presentation described in sexually acquired cases (29, 33). Less well-controlled community-based outbreak studies also support enteric triggers for ReA, and have implicated many enteric infections beyond Shigella (76, 77). These infections include not only bacteria such as Salmonella, Campylobacter, Yersinia, Clostridium difficile, and Escherichia coli but also protists such as Giardia lamblia (79). Idiopathic enteric symptoms, where no plausible causative infection can be found, are also common in ReA (79). There is no obvious pattern linking these infections other than enteric inflammation.
Unlike enteric infections, genital infections like Candida albicans and STIs do not occur in large confined epidemics, making associations with ReA more difficult to prove. Prior to large prospective studies, sporadic ReA cases seemed concentrated in men who consulted for urethritis symptoms resembling gonorrhea and shortly thereafter developed inflammation of the eyes and joints (33). Sexually acquired ReA cases were thus initially (wrongly) deemed to be post-gonorrheal polyarthritis (47).
Sporadic ReA cases were mainly seen by urologists and venereologists, who considered urethritis a necessary symptom. In 1933, Harkness realized that a majority of urethral discharge cases were of non-gonococcal origin (80). The search for genital infections which could explain idiopathic urethritis and ReA began in earnest after World War II, and the presence of C. trachomatis (29), Ureaplasma urealyticum (29), Mycoplasma hominis (29), and Trichomonas vaginalis (26) was quickly demonstrated in some cases. However, even after accounting for these new genital infections, most urethritis cases remain unexplained (22, 31, 32).
C. trachomatis became widely recognized as an STI in the 1970s (81) and is currently the genital infection most convincingly associated with ReA (21, 82). The strongest evidence of such an association can be found in case–control studies that measure either serological markers of past exposure to C. trachomatis or the presence of C. trachomatis itself in the genital tract (22, 36, 37, 83). The largest prospective community-based study cultured C. trachomatis in 25 of 112 ReA cases (22%) which occurred in Oslo (Norway) between 1988 and 1990 (36). The largest serological study analyzed 323 ReA cases referred to the Diagnostic-Research Centre of Sexually Transmitted Diseases in Bialystok (Poland) between 2001 and 2012, finding C. trachomatis IgG seropositivity in 70 cases (22%) (83). Both studies considered these rates to be much higher than those of control populations.
Due to the lack of natural experiments such as confined epidemics, it is difficult to demonstrate that C. trachomatis directly causes a subset of ReA cases and is not acting as a surrogate for another genital infection—as occurred with N. gonorrhoeae before it. This was well understood in 1968, when early reports of C. trachomatis in ReA were met with skepticism because C. trachomatis rates in ReA series were similar to those seen in STI clinics (84), suggesting it was acting as a surrogate for sexual activity and was not directly involved (85). Exposure to C. trachomatis cannot be demonstrated in a majority of sexually acquired cases (22, 82), let alone in all types of ReA cases combined (36, 38, 83), and two prospective STI clinic studies have confirmed that C. trachomatis is not associated with ReA in high STI risk populations (86, 87).
N. gonorrhoeae and C. trachomatis can be excluded as a cause of most ReA cases, even in STI clinic series (22, 29, 82, 84, 86, 87). Their association with ReA could be due to these two STIs acting as surrogates for an as-yet-unrecognized sexually acquired infection, as proposed by Catterall and King, or it could be due to a direct etiological role in a minority of sexually acquired ReA cases. N. gonorrhoeae and C. trachomatis are both associated with recent changes in sexual partners (88) and thus to genital exposure to a new set of microbes. In contrast with other STIs, their clearance by the immune system within a few months (89) makes them excellent markers of a recent change in sexual partners. If known STIs are acting as surrogates for an as-yet-unrecognized sexually acquired infection which causes ReA, then the strongest associations would be expected to be found with N. gonorrhoeae and C. trachomatis.
Infectious triggers of AS and AAU have not been as widely studied as in ReA. The studies which have been run are small. Demonstrating associations with infections occurring a month before ReA onset is easy in comparison, whereas it often takes a decade for AS to be recognized (24): triggering infections will be hard to identify because causative microbes may have been cleared and seropositivity may have been lost in the interim. For example, C. trachomatis IgG seropositivity is lost within 6 years when using microimmunofluorescence assays (90).
Antibodies against Klebsiella have been associated with AS in many studies (91), and Klebsiella stool cultures have been associated with disease activity in AS (92) and AAU (93), though Klebsiella cannot be found in most patients (94). Antibodies against peptidoglycan, a common component of bacterial cell walls, have been associated with AS (95) and other spondyloarthritis types (96).
Two small studies found high rates of C. trachomatis in male AS patients, respectively, using cell culture (15/31) (97) and IgG/IgA solid phase enzyme immunoassay (20/32) (98). The selective reporting of C. trachomatis in only a subset of AS patients in the first study and the lack of comparison to a control group in both studies means these results are difficult to interpret. A study of genital infections in women with AS reported a similarly high C. trachomatis rate detected by cell culture (15/32), which was significantly higher than the rate measured in controls (5/33) (99). However, a similar study performed in men with AS using sensitive molecular methods found much lower rates of C. trachomatis (1/32), which were indistinguishable from controls (3/120) (98); note that this study found high rates of C. trachomatis antibodies in these AS patients (20/32) (98), as reported earlier in this paragraph. Finally, two small C. trachomatis serological studies of AS patients of both sexes did not find a statistically significant association, perhaps due to low power (100, 101).
Studies analyzing spondyloarthritis phenotypes reported an increased risk of AAU in C. trachomatis seropositive patients (OR = 7.0, 95% CI: 1.1–44.1) (98) and Saccharomyces cerevisiae seropositive patients (OR = 4.36, 95% CI: 1.08–17.64) (16). Anti-Saccharomyces cerevisiae antibodies (ASCAs) bind to the mannose polymer (mannan) coat of all fungi: such antibodies can be generated in response to any fungal infection. ASCAs were also associated with peripheral arthritis (OR = 3.78, 95% CI: 1.57–9.15) (16) and inflammatory bowel disease (OR = 3.43, 95% CI: 1.15–10.20) (16). Spondyloarthritis and AS have been linked to ASCAs in many studies (16, 102–105), though the association is not as strong as in Crohn’s disease (70, 106).
Two small serological studies did not find an association between AAU and C. trachomatis when comparing with age-matched controls, nor did they find an association with any enteric infection suspected of causing ReA (107, 108). Neither study measured ASCAs.
Strong consistent associations between AS/AAU and infections suspected of causing ReA are lacking. The moderate association between STI seropositivity and AS/AAU implied by the Catterall–King hypothesis cannot be confirmed nor refuted by current studies: higher powered studies are warranted.
Ankylosing spondylitis is more common and has a more predictable course than ReA and AAU, making it easier to study from an epidemiological point of view. ReA and AAU typically occur in attacks lasting a few weeks or months, whereas AS is characterized by long-term inflammation of the sacroiliac joints and spine.
Ankylosing spondylitis risk before puberty is very low (23, 24). Its risk of onset increases sharply around the age of 15 years, peaks during young adulthood (age: 18–29 years), and tapers off exponentially over the following 30 years (23, 24). This distribution suggests environmental factors necessary for AS (if any) affect mainly adults, not children.
The observation that many HLA-B*27 carriers do not develop AS suggests that additional genetic and environmental factors contribute to the disease. AS monozygotic twin concordance does not reach 100% (109), strongly supporting the existence of environmental factors. Because the monozygotic twin concordance observed in AS exceeds 50% (109), environmental factors required for triggering the disease in genetically susceptible individuals must be ubiquitous (110). This is also consistent with the tapering off of AS onset risk after the age of 30 years, since by that age most individuals would presumably have been exposed to any environmental trigger and have already developed symptoms.
Sexually acquired infections match well with the age at onset of AS because their incidence is very low in children and peaks in young adulthood (25). However, no known sexually acquired infections other than all human papillomavirus (HPV) types combined are present in over half of the population. For STIs which can be cleared by the immune system, peak prevalence occurs between 18 and 24 years of age (89). STI prevalence in Americans of this age group was estimated to be 53.8% for HPV (all types), 3.9% for herpes simplex virus type 2, 1.6% for C. trachomatis, 0.9% for T. vaginalis, and 0.3% for N. gonorrhoeae (89). C. trachomatis and N. gonorrhoeae are both well-established risk factors of ReA; other STIs in this list have not been widely studied in ReA because they do not often cause urethritis. Lifetime risk of sexual exposure to HPV (all types) in men is estimated to be 91% (111), so this infection reaches a high enough fraction of the population to be able to cause AS through a hit-and-run mechanism. In contrast, it appears unlikely that well over half the male population could be exposed to either C. trachomatis or N. gonorrhoeae, though formally demonstrating this is difficult based on currently published studies (89).
Studies of the age at first attack of ReA and AAU are much smaller than those related to AS. AAU is rare in children, and the highest risk of onset occurs in young adults (66, 112, 113). Sexually acquired ReA generally does not occur in children due to a lack of sexual activity, and its rate peaks in young adults (34, 35, 114); this distribution is very similar to that of AS. Enteric ReA has a more even age distribution within adults (36) and is also rare in children (115–117). At first blush, the paucity of enteric ReA cases in children is difficult to explain.
The largest sexually acquired ReA cohort study demonstrated that relapses sometimes coincide with genital or enteric infections but in many cases seem unprovoked (33, 39). Most of these relapses were attributed to flare-ups rather than to repeated exposure to triggering microbes (39). Urethritis is often the first symptom in enteric ReA, suggesting enteric infections are acting as one of many possible cofactors that can trigger flare-ups of a latent genital infection. This could explain why a wide range of enteric infections are associated with ReA, and why enteric ReA is rare in children (115–117) despite ample exposure to enteric microbes (115).
The association between ASCAs and spondyloarthritis (16, 102–105) warrants analyzing links between fungal infections and conditions listed in Table 3. In a recent study, Maillet et al demonstrated that ASCAs are more strongly associated with peripheral symptoms (uveitis, arthritis, and inflammatory bowel disease) than axial symptoms (spondylitis and sacroiliitis), and with the absence of HLA-B*27 alleles (16). ASCAs are a biomarker of CD4+ T cell recognition of fungal mannoproteins, suggesting the recognition of fungi by CD4+ T cells may play an important role in peripheral spondyloarthritis symptoms, and in cases where antigen presentation to CD8+ T cells is less efficient due to the absence of HLA-B*27 alleles. CD4+ T cell recognition of Malassezia antigens resulting in a Th1 response has been reported in psoriasis (20), though it has not been studied in spondyloarthritis.
Rare homozygous mutations in either CARD9 or in the IL-17/IL-23 pathway cause chronic mucocutaneous candidiasis by impairing the immune response against fungi (118–120). Similarly, IL-17 inhibitors increase candidiasis risk (121). Genome wide association studies have linked prevalent functional CARD9 and IL23R polymorphisms to AS and associated conditions (Table 3). These genes are involved in the inflammatory cascade downstream of phagocyte recognition of fungal cell wall components beta-glucan (mainly through Dectin-1) and mannan (mainly through Dectin-2) (110, 122, 123). These are the two primary antigens leading to phagocytosis of fungi such as Malassezia furfur (124) and Cryptococcus neoformans (125). These links with fungi suggest that the immune response against fungal antigens may be an important component of spondyloarthritis, and interventions targeting cytokines associated with fungal infections—for example, the anti-IL-17A drug secukinumab (126)—may improve symptoms.
ASCAs are strongly associated with uveitis in spondyloarthritis patients (16), but antibodies against various bacteria suspected of causing ReA are not (107, 108). Similarly, circulating CD4+ T cells in uveitis patients are more sensitive than controls to fungal antigens (C. albicans) after a 24-h antigen exposure period (127), whereas sensitivity to bacterial antigens (Staphylococcus aureus, Clostridium tetani, and Mycobacterium tuberculosis) and protist antigens (Toxoplasma gondii) are similar in both groups (127). Intermediate uveitis is associated with multiple sclerosis onset (128) and with the HLA-DRB1*1501 allele (129), which are both associated with the immune response to fungi (5). A small study reported that oral dimethyl fumarate, a fungicidal compound known to be effective in multiple sclerosis and psoriasis, improved chronic idiopathic uveitis symptoms in four patients (130).
Though most cases of uveitis are considered idiopathic, some cases can be attributed to infections: bacteria (especially M. tuberculosis, C. trachomatis, Treponema pallidum, and Borrelia burgdorferi), viruses (especially herpes viruses), protists (especially T. gondii), and fungi (especially C. albicans) can cause uveitis (131). In a very large study of uveitis etiology, infections were reported in 13% of anterior uveitis, 7% of intermediate uveitis, 40% of posterior uveitis, and 43% of panuveitis cases (131). The abovementioned studies indicate that increased immune sensitivity to fungal antigens may be an important risk factor of idiopathic uveitis, especially in association with spondyloarthritis and multiple sclerosis.
About 10% of spondyloarthritis patients also have inflammatory bowel disease (Crohn’s disease or ulcerative colitis) (16, 24), a rate significantly higher than that of the general population (<1%) (132). Enteric inflammation can be found in about 60% of spondyloarthritis patients, though it is usually subclinical (133). Functional CARD9 and IL23R alleles are also associated with isolated Crohn’s disease and ulcerative colitis (Table 3), suggesting the immune response against fungi may play an important role here as well.
The recognition of fungal antigens is accentuated in human peripheral blood mononuclear cells (PBMC) from Crohn’s disease patients as compared to controls (134, 135); this was attributed to increased expression of Dectin-1, Dectin-2, and the mannose receptor (135). A similar study focused on bacterial antigens found that CD4+ T cell activation through antigen presentation by PBMC was elevated in inflammatory bowel disease patients as compared to controls when exposed to either C. albicans or nine bacterial species (136). Because CD4+ T cells recognize antigenic peptides which are more species specific than sugars recognized by monocytes, this study should be repeated with a wider panel of medically important fungi including Malassezia and Cryptococcus species. These two genera are taxonomically very distant from Candida and are expected to share few antigenic peptides despite sharing antigenic sugars such as beta-glucan and mannan. CD4+ T cell recognition of Candida and Malassezia antigens can be very different, as demonstrated in psoriasis (20). Crohn’s disease is strongly associated with antibodies against conserved fungal antigen sugars (especially mannan, but also beta-glucan and chitin) (70), and recent gut microbiome studies suggest a fungal etiology (17, 18, 137, 138).
Three recent studies using vedolizumab in Crohn’s disease and ulcerative colitis patients reported that this intervention triggered peripheral arthritis, sacroiliitis, or psoriasis in about 10% of cases (139–141); these are the most common symptoms associated with spondyloarthritis (Table 3). Vedolizumab blocks the α4β7 integrin homing receptor, preventing lymphocytes originating from the gut from returning there during recirculation (141). It is thus plausible that following activation due to antigen exposure in the gut, stray lymphocytes subsequently trigger inflammation upon reaching the peripheral joints, sacroiliac joints, and skin (142, 143) by recognizing a similar or identical antigen in these sites (144). Since Crohn’s disease is strongly associated with an immune response against fungi, these may be fungal antigens.
The distribution of the age at onset of isolated AS (24) is very similar to that of AS with inflammatory bowel disease (24) and to that of isolated Crohn’s disease (145, 146): risk is low before puberty, increases sharply around the age of 15 years, peaks during young adulthood (age: 18–29 years), and tapers off over the following 30 years (146). Though Crohn’s disease is not as strongly associated with an MHC class I allele as AS, associations with ERAP alleles and interactions between ERAP alleles and MHC class I alleles have also been reported in Crohn’s disease (72, 73). It is thus plausible that exposure to the same sexually acquired intracellular fungal infection is necessary for both spondyloarthritis and isolated Crohn’s disease, and that genetic predisposition determines which symptoms appear following exposure to this environmental factor.
Approximately 15–35% of spondyloarthritis patients also have psoriasis (16, 24), which is somewhat higher than the rate measured in the general population (<10%) (147). Many studies have associated psoriasis with fungi. Candida colonization of the gut (19, 148) and antibodies against C. albicans (67) and M. furfur (67, 68) are associated with psoriasis. The application of lysed M. furfur cells to the skin of psoriasis patients induces psoriasis-like lesions much more frequently than in controls (149). Psoriasis patients’ PBMC readily produce a Th1 response when incubated with M. furfur antigens (20), whereas the same response does not occur in controls, nor when using C. albicans or Trichophyton rubrum antigens (20). The chitin binding protein YLK-40 is associated with psoriasis and is a marker of disease severity (150); chitin is a highly conserved fungal antigen which is not present in bacteria or in human cells. Finally, many fungicidal compounds have been shown to reduce psoriasis symptoms (151–158), though such drugs can also induce psoriasis flare-ups (159)—such flare-ups have been tentatively attributed to a Jarisch–Herxheimer reaction (154, 156).
Though the incidence of psoriasis peaks at about the same age as AS (24), incidence in children and older adults is much higher in psoriasis (160). The age at onset of isolated AS and AS with psoriasis is nearly identical (24). This suggests that psoriasis may be a heterogeneous disease, of which only a subset of cases share a common etiology with AS (161). A recent study of ERAP alleles in psoriasis supports this hypothesis: ERAP alleles are only associated with psoriasis onset in adolescents and young adults (162), suggesting that the same sexually acquired intracellular fungal infection may be necessary for both spondyloarthritis and this subset of psoriasis cases.
The efficacy of oral nystatin in reducing psoriasis symptoms (154–157) [oral nystatin is not absorbed and thus limited to killing fungi in the gut (163, 164)], the expression of αEβ7 integrin by CD8+ T cells in psoriatic lesions (165) (αEβ7 integrin is a marker of mucosal origin), and the triggering of psoriasis by vedolizumab (140, 141) suggest that some psoriasis cases may be caused by lymphocytes originating in the gut. The many links with fungi described earlier and the associations between ERAP alleles and psoriasis (162) suggest that similar intracellular fungal antigens are present in the gut and on the skin, providing a common antigenic target for CD8+ T cells migrating from the gut to the skin. The only fungal genus currently known to be highly prevalent on the human skin is Malassezia (65), present in both healthy skin and psoriatic lesions (166). Several groups have proposed that a loss of immune tolerance to Malassezia may cause some psoriasis cases (149, 151, 153, 167, 168). Malassezia are found within skin keratinocytes (169) and have recently been detected in the gut (71, 138, 170).
The presence of idiopathic prostatic inflammation affecting men has been recognized for a very long time, both in isolation and in association with spondyloarthritis (41, 43, 48). The association between prostatic inflammation and spondyloarthritis, combined with other genital symptoms and sexual risk factors in ReA, suggests that a chronic genital infection may reach the prostate and cause these conditions (26, 27).
Fungal infections of the prostate are considered rare in immune-competent individuals (171), and few studies have investigated possible fungal etiologies in prostate disease (4). The recent discovery of an abundant fungicidal protein in the prostate (172) indicates that a fungal infection reaches this site, hence necessitating such an antimicrobial protein. This protein is called either prostate secretory protein 94 (PSP94) or beta-microseminoprotein, and is encoded by the MSMB gene. High PSP94 concentration in the prostate protects men from prostate cancer in a dose-dependent manner (173–176), suggesting prostatic fungi may be an important etiological component of prostate cancer (4). PSP94 concentration interacts with sexual risk factors in affecting prostate cancer risk, suggesting this fungal infection may be sexually acquired (15). Sexual risk factors have been widely studied in prostate cancer and are well recognized (6–8), though no specific causative infection has been strongly associated with prostate cancer (177). Due to inhibition by calcium ions (172), PSP94’s fungicidal activity within the prostate is restricted to the cytosol of secretory epithelial cells, suggesting that targeted fungi must be intracellular and must invade these cells (4).
Interestingly, an aberrant truncated form of PSP94 lacking the fungicidal region is strongly associated with benign prostatic hyperplasia (BPH) (178, 179), and prostate epithelial cells in BPH tissue do not stain for PSP94 in histological studies, as opposed to healthy tissue (180, 181). This suggests that the cytotoxic T cell immune response against secretory epithelial cells observed in BPH (182) may be triggered by the loss of PSP94-mediated immunity against a ubiquitous (183) intracellular fungal infection.
Histological studies have found melanin inclusions within secretory epithelial cells of the prostate targeted by CD8+ T cells (184–186), though a fungal infection was not considered as a possible explanation. Fungi synthesize melanin as a defense mechanism when exposed to fungicides (187, 188), so an intracellular fungus exposed to PSP94 within these cells could explain the presence of melanin.
PSP94 is also present in glandular epithelial cells of the cervix (189) [the main genital inflammation site of ReA in women (22)] and colon (189, 190) (one of the main inflammatory bowel disease inflammation sites). It is not present in the skin (190). Its presence in the eyes and joints has not been tested.
PSP94’s ortholog in pigs has conserved its fungicidal activity (172) despite rapid evolution (191) resulting in changes to half of the amino acids in the protein (192). Humans and pigs shared an ancestor about 90 million years ago, suggesting that PSP94 is coevolving with fungal infections in both species, and PSP94’s fungicidal activity confers a selective advantage to host fitness (193).
PSP94’s conserved fungicidal properties suggest that rodents may also be infected with fungal species targeted by this protein. In 1956, Pearson noticed that the injection of Freund’s adjuvant (lysed Mycobacterium suspended in oil and water) into the footpad of Wistar and Long-Evans rats resulted in symptoms resembling ReA: lymphocyte-mediated inflammation of the joints, genitals, skin, and eyes ensued 10–16 days later (194, 195). Lipidic Mycobacterium extracts readily induced arthritis as well, suggesting that the adjuvant’s causative antigens were not proteins or peptides (195). It was later shown that alpha beta T cells were the main mediators of inflammation (196). Because alpha beta T cells directly recognize peptides rather than lipids, offending antigens may have already been present at the site of adjuvant administration. Host and commensal microbe antigens were considered the most probable target of these T cells, both in the footpad and in the many other sites of inflammation (195). Such sites appeared to be sterile by cell culture (195), and no self-antigens were strongly associated with this condition (197): the underlying mechanisms in this animal model remain unresolved (3). Likewise, the antigenic target in the widely used HLA-B27/β2m transgenic rat model of spondyloarthritis remains to be identified. It has been demonstrated that development of genital tract inflammation precedes and is required for the development of arthritic symptoms in these animals (198). We therefore hypothesize that a fungal infection at this site could be a contributing agent to the ensuing pathology.
Reactive arthritis-like symptoms can be induced in genetically predisposed rodents through exposure to various fungal stimuli: C. albicans (199), beta-glucan (110, 200), and mannan (110, 123). Dectin-2, CARD9, and IL-23 seem to be important in these animal models because knocking out Dectin-2 or CARD9 or blocking IL-23 reduces symptoms (110, 123).
An animal model of arthritis based on the injection of Streptococcus pyogenes cell wall antigens into the knee joint of mice demonstrated that a minute quantity of C. albicans added to this injection skewed the T cell immune response toward Th17, markedly increasing the level of inflammation (201). Interestingly, C. albicans alone, even in large quantities, caused little inflammation (201). This suggests that the immune response to fungal infections is muted unless inflammatory cofactors such as bacterial antigens are also present, giving a plausible explanation for Pearson’s animal model of ReA: Mycobacterium antigens may have stimulated an immune response against an elusive fungal infection already present in the rat footpad and other sites of inflammation. For example, Mycobacterium glycolipid trehalose dimycolate upregulates the expression of Mincle (202), and Mincle was recently shown to be a key receptor in an animal model of Mycobacterium-induced uveitis (203). Mincle has a very high affinity to alpha-mannose, an antigen found in Malassezia but not in other medically important fungal species (204). A very recent study reported that alpha-mannan induces uveitis in an animal model with an efficiency similar to Mycobacterium antigens (123). These findings suggest antigenic challenges may increase phagocytic activity directed against Malassezia, leading to activation of alpha beta T cells which recognize Malassezia peptides.
The hypothesis of an adaptive immune response against a fungal infection proposed in the previous paragraph is quite speculative and would be on more solid ground if there existed a disseminated ubiquitous fungal infection in an animal. One such infection was discovered in 2012 in mealworms (205): this fungus-like eukaryote is vertically transmitted to all individuals and is present throughout the mealworm body, though it is concentrated in the genitals and is sexually transmitted as well (205). To the best of our knowledge, no fungal infection with similar properties has been found in a rodent or other mammal. If such a fungal infection existed in humans, it could explain why a heterogeneous set of inflammatory stimuli can cause ReA: each stimulus can trigger the loss of immune tolerance to this disseminated fungal infection by acting as an immunological adjuvant which provokes an adaptive immune response against fungal antigens. This would explain why fungal colonization and fungal antigens are effective at breaking immune tolerance. If this elusive fungal infection was mainly sexually acquired in humans, this would explain why children and young teens are at very low risk of conditions associated with HLA-B*27, and why ReA has manifest sexual risk factors and genital symptoms. Finally, a fungal etiology would explain why functional polymorphisms in genes which are part of the immune response against fungi are associated with conditions listed in Table 3.
The hypotheses put forward by Harkness in 1949 to explain the etiology of ReA (29), and expanded by Catterall and King in 1959 to include prostatic inflammation, AS, and isolated AAU (26, 28), have remained consistent with the scientific literature published since. The evidence reviewed here supports the existence of an as-yet-unrecognized genital infection which is a necessary etiological factor in spondyloarthritis. While the existence of an as-yet-unrecognized genital infection may seem to be far-fetched at first, such a hypothesis has been proposed to explain the sexual risk factors of multiple sclerosis (9, 14) and prostate cancer (7, 8, 13) based on evidence completely unrelated to spondyloarthritis (Table 4). Recent studies have implicated fungal infections in multiple sclerosis (5, 206–208) and prostate cancer (4, 15, 172), and these infections appear to be intracellular in both cases (Table 4) (4, 209, 210).

Table 4. Main observations supporting a common sexually acquired intracellular fungal infection in spondyloarthritis, prostate disease, and multiple sclerosis.
A commonly held view is that ReA-causing inflammation is aseptic, and chronic inflammation in the joints, spine, and eyes is due to autoimmunity (75). Associations with ERAP alleles and the wide variety of MHC class I alleles present in HLA-B*27-negative patients suffering from conditions listed in Table 3 suggest that many epitopes can be involved in the disease process. Thus, the hypothesis of molecular mimicry-induced autoimmunity triggered by an intracellular infection confined to the genitals cannot easily explain non-genital symptoms, unless this infection mimicked a wide range of human antigens (this seems unlikely). This suggests that the putative infection is not confined to the genitals, but rather spreads throughout the body and provides antigens necessary for inflammation in all affected sites, as proposed by Visher (41). This hypothesis is plausible because the only known microbial STI in humans which cannot be cleared by the immune system also spreads from the genitals and causes a wide variety of seemingly unrelated symptoms in infected sites: these symptoms are known as secondary and tertiary syphilis, and are caused by the bacterium T. pallidum (211).
The many links between fungi and conditions listed in Table 3 reviewed here suggest that a ubiquitous intracellular fungus, which usually reaches the genitals of adolescents and young adults through sexual activity, is a necessary etiological factor in these conditions. In a majority of individuals, this infection would remain asymptomatic or subclinical because immune tolerance is maintained. How and where immune tolerance is lost would be mostly genetically determined (43); this would explain why symptoms vary widely between infected individuals and why different conditions in Table 3 are associated with different genes. For example, HLA-B*27 molecules would have a high affinity to an antigenic protein in or on this fungus (Figure 1), frequently leading to a loss of immune tolerance in the sacroiliac joints and spine through the detection of this fungus in these sites. When immune tolerance is marginal, extraneous factors such as unrelated enteric or genital microbes would cause exacerbation of symptoms or flare-ups by acting as immunological adjuvants. These adjuvants are likely bacteria and unrelated fungi which infect or colonize mucosal surfaces, pushing lymphocytes in these sites to recognize the intracellular fungus, clonally expand, recirculate, enter non-mucosal tissue, and cause inflammation by detecting the intracellular fungus in such tissue.
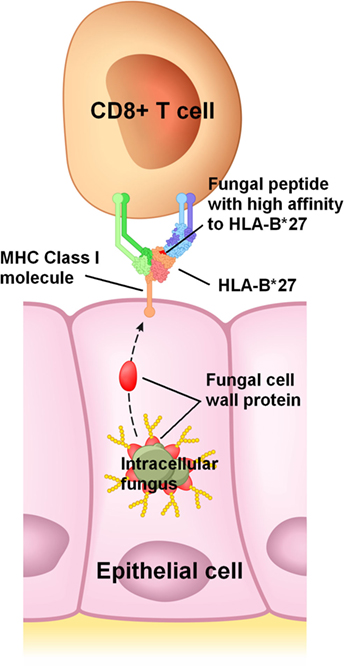
Figure 1. Proposed mechanism for HLA-B*27 in spondyloarthritis. HLA-B*27 would efficiently bind to a peptide from an abundant protein present in or on an intracellular fungus, and then present this peptide to CD8+ T cells on the infected host cell’s surface. In this example, a peptide from a fungal cell wall mannoprotein is presented to a CD8+ T cell. Cell wall mannoproteins are good antigen candidates due to their abundance, though presentation of peptides from other fungal proteins is also plausible.
The updated Catterall–King hypothesis proposed here has important implications for future studies. The microbiome of the prostate and cervix should be included in studies of conditions listed in Table 3. The many links with fungal infections described here highlight the limitations of 16S consensus microbiome techniques which can only detect bacteria. Techniques such as deep sequencing of total RNA (212–214) or DNA (166), while more expensive, should be used because they can detect microbes from the entire tree of life (including fungi). Recent studies have shown that many as-yet-unrecognized infections are present in humans (166, 215), so the existence of a novel microbe present in the genitals and in other sites is plausible. For example, molecular studies of oral (216) and genital (217) fungi in healthy individuals found many novel species. It would be interesting to know if fungal colonization of the gut is a risk factor of spondyloarthritis, as demonstrated in Crohn’s disease (17, 18) and psoriasis (19, 148), and if antifungal compounds such as dimethyl fumarate improve the course of spondyloarthritis, as demonstrated in psoriasis (158), psoriatic arthritis (218, 219), and multiple sclerosis (220).
The possible association between Malassezia and psoriasis (20, 149, 151, 153, 167, 168) suggests that particular attention should be given to Malassezia species in spondyloarthritis. Malassezia are ubiquitous facultative intracellular fungi which are difficult to detect. Because of their unique requirement for lipids, they do not grow in commonly used fungal culture media (221). Their DNA is difficult to extract (222), possibly because of their uniquely thick cell wall (221). ITS1 and ITS2, two of the most commonly used primers for fungal consensus PCR, have, respectively, two and one mismatches with Malassezia sequences, leading to underrepresentation in PCR products. Recent reports suggest that Malassezia are not limited to the skin and are present in the mouth (63), nose (223), gut (71, 138, 170), breast (224), brain (225, 226), and lung (227).
Malassezia have the right properties to be able to cause spondyloarthritis: they are ubiquitous, intracellular, present on the skin (65, 166), glans penis (64), mouth (63), and gut (71, 138, 170). Malassezia have beta-endorphin receptors which stimulate the secretion of lipases necessary for their growth (228–230), and high beta-endorphin levels are found in the prostate (231–233). Malassezia have been strongly associated with granulomatous Crohn’s disease in a pediatric biopsy study (OR = 25.2, 95% CI: 2.45–259, P = 0.0025) (138) and with Dectin-1 SNPs in an inflammatory bowel disease study (18). Reads from unidentified Malasseziales were reported in several recent studies of the skin (166, 234) and gut (18), suggesting some species and strains remain to be discovered. To the best of our knowledge, Malassezia’s presence in the joints, eyes, and prostate has not been tested.
Because Malassezia are common on the human skin, they can be inadvertently inserted in clinical specimens, causing spurious findings. Much care must be taken to ensure that these fungi are actually present in sampled sites. Though our preliminary microbiome results suggest that they are present in the prostate, excluding contamination beyond all doubt has proven to be challenging. As opposed to most infection types, Malassezia are ubiquitous and are part of the normal human microbiome, making their association with disease very difficult to prove. For example, distinguishing chronic inflammation caused by autoimmunity against human antigens and immunity against ubiquitous commensal microbe antigens on the psoriatic skin is not trivial, as healthy sites and psoriatic lesions have about the same microbiome (166). Microbes present in more sterile sites such as the joints, eyes, or prostate could provide important clues as to which species may be involved in spondyloarthritis.
Since the evidence available at this time is insufficient to definitely confirm the Catterall–King hypothesis, microbiome studies similar to that performed by Kellermayer et al (138) should be performed to test this hypothesis.
First draft was written by ML after discussion with JR and MA. All the authors critiqued and revised the draft.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
JR receives support from the William and Mary Bauman Foundation, the Stan and Madelle Rosenfeld Family Trust, the Rheumatology Research Foundation, and Research to Prevent Blindness. Mark Asquith is a Jane Bruckel Scholar supported by the Spondylitis Association of America.
1. Schlosstein L, Terasaki PI, Bluestone R, Pearson CM. High association of an HL-A antigen, W27, with ankylosing spondylitis. N Engl J Med (1973) 288(14):704–6. doi:10.1056/NEJM197304052881403
2. Brewerton D, Hart F, Nicholls A, Caffrey M, James D, Sturrock R. Ankylosing spondylitis and HL-A 27. Lancet (1973) 301(7809):904–7. doi:10.1016/S0140-6736(73)91360-3
3. Rosenbaum JT. Why HLA-B27? My thirty-year quest the Friedenwald lecture. Invest Ophthalmol Vis Sci (2011) 52(10):7712–5. doi:10.1167/iovs.11-8247
4. Sutcliffe S, De Marzo AM, Sfanos KS, Laurence M. MSMB variation and prostate cancer risk: clues towards a possible fungal etiology. Prostate (2014) 74(6):569–78. doi:10.1002/pros.22778
5. Benito-Leon J, Laurence M. The role of fungi in the etiology of multiple sclerosis. Front Neurosci (2017) 8:535. doi:10.3389/fneur.2017.00535
6. Rosenblatt KA, Wicklund KG, Stanford JL. Sexual factors and the risk of prostate cancer. Am J Epidemiol (2001) 153(12):1152–8. doi:10.1093/aje/153.12.1152
7. Dennis LK, Dawson DV. Meta-analysis of measures of sexual activity and prostate cancer. Epidemiology (2002) 13(1):72–9. doi:10.1097/00001648-200201000-00012
8. Taylor ML, Mainous AG III, Wells BJ. Prostate cancer and sexually transmitted diseases: a meta-analysis. Fam Med (2005) 37(7):506–12.
9. Hawkes CH, Giovannoni G, Keir G, Cunnington M, Thompson EJ. Seroprevalence of herpes simplex virus type 2 in multiple sclerosis. Acta Neurol Scand (2006) 114(6):363–7. doi:10.1111/j.1600-0404.2006.00677.x
10. Ferrante P, Castellani P, Barbi M, Bergamini F. The Italian Cooperative Multiple Sclerosis case-control study: preliminary results on viral antibodies. Ital J Neurol Sci (1987) 6:45–50.
11. Wandinger K, Jabs W, Siekhaus A, Bubel S, Trillenberg P, Wagner H, et al. Association between clinical disease activity and Epstein-Barr virus reactivation in MS. Neurology (2000) 55(2):178–84. doi:10.1212/WNL.55.2.178
12. Catalano LW. Herpesvirus hominis antibody in multiple sclerosis and amyotrophic lateral sclerosis. Neurology (1972) 22(5):473–473. doi:10.1212/WNL.22.5.473
13. Strickler HD, Goedert JJ. Sexual behavior and evidence for an infectious cause of prostate cancer. Epidemiol Rev (2001) 23(1):144–51. doi:10.1093/oxfordjournals.epirev.a000781
14. Hawkes CH. Is multiple sclerosis a sexually transmitted infection? J Neurol Neurosurg Psychiatry (2002) 73(4):439–43. doi:10.1136/jnnp.73.4.439
15. Stott-Miller M, Wright JL, Stanford JL. MSMB gene variant alters the association between prostate cancer and number of sexual partners. Prostate (2013) 73(16):1803–9. doi:10.1002/pros.22719
16. Maillet J, Ottaviani S, Tubach F, Roy C, Nicaise-Rolland P, Palazzo E, et al. Anti-Saccharomyces cerevisiae antibodies (ASCA) in spondyloarthritis: prevalence and associated phenotype. Joint Bone Spine (2016) 83(6):665–8. doi:10.1016/j.jbspin.2015.10.011
17. Hoarau G, Mukherjee P, Gower-Rousseau C, Hager C, Chandra J, Retuerto M, et al. Bacteriome and mycobiome interactions underscore microbial dysbiosis in familial Crohn’s disease. mBio (2016) 7(5):e1250–1216. doi:10.1128/mBio.01250-16
18. Sokol H, Leducq V, Aschard H, Pham H-P, Jegou S, Landman C, et al. Fungal microbiota dysbiosis in IBD. Gut (2016) 66(6):1039–48. doi:10.1136/gutjnl-2015-310746
19. Waldman A, Gilhar A, Duek L, Berdicevsky I. Incidence of Candida in psoriasis – a study on the fungal flora of psoriatic patients. Mycoses (2001) 44(3–4):77–81. doi:10.1046/j.1439-0507.2001.00608.x
20. Kanda N, Tani K, Enomoto U, Nakai K, Watanabe S. The skin fungus-induced Th1-and Th2-related cytokine, chemokine and prostaglandin E2 production in peripheral blood mononuclear cells from patients with atopic dermatitis and psoriasis vulgaris. Clin Exp Allergy (2002) 32(8):1243–50. doi:10.1046/j.1365-2745.2002.01459.x
21. Taylor-Robinson D, Keat A. Observations on Chlamydia trachomatis and other microbes in reactive arthritis. Int J STD AIDS (2015) 26(3):139–44. doi:10.1177/0956462414533319
22. Butrimiene I, Ranceva J, Griskevicius A. Potential triggering infections of reactive arthritis. Scand J Rheumatol (2006) 35(6):459–62. doi:10.1080/03009740600906750
23. Ciurea A, Scherer A, Weber U, Neuenschwander R, Tamborrini G, Exer P, et al. Age at symptom onset in ankylosing spondylitis: is there a gender difference? Ann Rheum Dis (2014) 73(10):1908–10. doi:10.1136/annrheumdis-2014-205613
24. Feldtkeller E, Khan MA, van der Heijde D, van der Linden S, Braun J. Age at disease onset and diagnosis delay in HLA-B27 negative vs. positive patients with ankylosing spondylitis. Rheumatol Int (2003) 23(2):61–6. doi:10.1007/s00296-002-0237-4
25. PHE. Sexually Transmitted Infections and Chlamydia Screening in England, 2015. HPR 10(22) Advance Access report (2016). Available from: www.psp94.com/phe_sti_2015.pdf (Accessed: December 7, 2016).
26. King A. Non-gonococcal urethritis and trichomoniasis in the male. Urol Int (1959) 9(3–6):127–45. doi:10.1159/000277449
27. Catterall R, Perkins ES. Uveitis and urogenital disease in the male. Br J Ophthalmol (1961) 45(2):109. doi:10.1136/bjo.45.2.109
28. Catterall R. Significance of non-specific genital infection in uveitis and arthritis. Lancet (1961) 278(7205):739–42. doi:10.1016/S0140-6736(61)90688-2
30. Csonka G. Reiter’s syndrome. Ergebnisse der inneren Medizin und Kinderheilkunde. Berlin, Heidelberg: Springer (1965). p. 125–89. doi:10.1007/978-3-642-94912-8_4
31. Gillespie CW, Manhart LE, Lowens MS, Golden MR. Asymptomatic urethritis is common and is associated with characteristics that suggest sexually transmitted etiology. Sex Transm Dis (2013) 40(3):271–4. doi:10.1097/OLQ.0b013e31827c9e42
32. Wetmore CM, Manhart LE, Lowens MS, Golden MR, Whittington WL, Xet-Mull AM, et al. Demographic, behavioral, and clinical characteristics of men with nongonococcal urethritis differ by etiology: a case-comparison study. Sex Transm Dis (2011) 38(3):180–6. doi:10.1097/OLQ.0b013e3182040de9
33. Csonka GW. The course of Reiter’s syndrome. Br Med J (1958) 1(5079):1088. doi:10.1136/bmj.1.5079.1088
34. Hall WH, Finegold S. A study of 23 cases of Reiter’s syndrome. Ann Intern Med (1953) 38(3):533–50. doi:10.7326/0003-4819-38-3-533
35. Ford DK, Rasmussen G. Relationships between genitourinary infection and complicating arthritis. Arthritis Rheumatol (1964) 7(3):220–7. doi:10.1002/art.1780070305
36. Kvien T, Glennås A, Melby K, Granfors K, Andrup O, Karstensen B, et al. Reactive arthritis: incidence, triggering agents and clinical presentation. J Rheumatol (1994) 21(1):115–22.
37. Valtonen V, Leirisalo M, Pentikäinen P, Räsänen T, Seppälä I, Larinkari U, et al. Triggering infections in reactive arthritis. Ann Rheum Dis (1985) 44(6):399–405. doi:10.1136/ard.44.6.399
38. Söderlin MK, Kautiainen H, Puolakkainen M, Hedman K, Söderlund-Venermo M, Skogh T, et al. Infections preceding early arthritis in southern Sweden: a prospective population-based study. J Rheumatol (2003) 30(3):459–64.
39. Csonka G. Recurrent attacks in Reiter’s disease. Arthritis Rheumatol (1960) 3(2):164–9. doi:10.1002/art.1780030208
40. Noer HR. An experimental epidemic of Reiter’s syndrome. JAMA (1966) 198(7):693–8. doi:10.1001/jama.198.7.693
41. Visher J. Chronic prostatitis: its role in etiology of sacroiliac and spinal arthritis. Med J (1929) 130:214–5.
42. Domeij B, Giertz G, Olhagen B, Romanus R. Genitourinary focus in rheumatic disorder in the male. Acta Chir Scand (1958) 115(1–2):1.
43. Mason RM, Murray RS, Oates JK, Young AC. Prostatitis and ankylosing spondylitis. Br Med J (1958) 1(5073):748–51. doi:10.1136/bmj.1.5073.748
44. Weinberger H, Dienes L, Bauer W. Clinical features and bacteriological studies in Reiter’s syndrome. Rheumatic Diseases, American Rheumatism Association. Philadelphia, PA: WB Saunders Co. (1952). p. 73–7.
45. Romanus R. Pelvo-spondylitis ossificans in the male and genitourinary infection. Acta Med Scand (1953) 145(S280):178–241. doi:10.1111/j.0954-6820.1953.tb17256.x
46. Oates J. Incidence of genital infection in male patients with ankylosing spondylitis. Br J Vener Dis (1959) 35(2):89.
47. Olhagen B. Chronic uro-polyarthritis in the male. Acta Med Scand (1960) 168(4):339–45. doi:10.1111/j.0954-6820.1960.tb13455.x
48. Anonymous. Editorial: ankylosing spondylitis and urogenital infection. Br Med J (1960) 1(5176):865–6. doi:10.1136/bmj.1.5176.865
49. Catterall R. Incidence of chronic genital infection in male patients with uveitis: a preliminary report. Br J Vener Dis (1958) 34(4):254.
50. Benedict WL, Von Lackum WH, Nickel AA. The pelvic organs as foci of infection in inflammatory diseases of the eye. Trans Am Ophthalmol Soc (1926) 24:145.
51. Von Lackum WH. Clinical and experimental data on prostatic infection. J Urol (1927) 18(3):293–306. doi:10.1016/S0022-5347(17)73283-1
52. Zentmayer W. The prostate as a remote focus of infection in ocular inflammations. J Am Med Assoc (1926) 87(15):1172–6. doi:10.1001/jama.1926.02680150006002
53. Pelouze P. Ophthalmologic importance of focal infective prostatitis. Arch Ophthalmol (1932) 7(3):372–7. doi:10.1001/archopht.1932.00820100026004
54. Kretschmer H, Berkey H, Heckel M, Ockuly E. Chronic prostatitis: a critical review of 1,000 cases. Ill Med J (1937) 71:151–61.
55. Wright V, Reed WB. The link between Reiter’s syndrome and psoriatic arthritis. Ann Rheum Dis (1964) 23(1):12. doi:10.1136/ard.23.1.12
56. Perkins E. Uveitis in B27-related disease. Ann Rheum Dis (1979) 38(Suppl 1):92. doi:10.1136/ard.38.Suppl_1.92
57. Dark A, Morton R. Acute anterior uveitis in men. Association with chronic prostatitis. Br J Ophthalmol (1968) 52(12):907. doi:10.1136/bjo.52.12.907
58. Costantino F, Talpin A, Said-Nahal R, Goldberg M, Henny J, Chiocchia G, et al. Prevalence of spondyloarthritis in reference to HLA-B27 in the French population: results of the GAZEL cohort. Ann Rheum Dis (2015) 74(4):689–93. doi:10.1136/annrheumdis-2013-204436
59. Brown MA, Pile KD, Kennedy LG, Calin A, Darke C, Bell J, et al. HLA class I associations of ankylosing spondylitis in the white population in the United Kingdom. Ann Rheum Dis (1996) 55(4):268–70. doi:10.1136/ard.55.4.268
60. I. G. o. A. S. Consortium. Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat Genet (2013) 45(7):730–8. doi:10.1038/ng.2667
61. Linssen A, Rothova A, Valkenburg H, Dekker-Saeys A, Luyendijk L, Kijlstra A, et al. The lifetime cumulative incidence of acute anterior uveitis in a normal population and its relation to ankylosing spondylitis and histocompatibility antigen HLA-B27. Invest Ophthalmol Vis Sci (1991) 32(9):2568–78.
62. Evans DM, Spencer CC, Pointon JJ, Su Z, Harvey D, Kochan G, et al. Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility. Nat Genet (2011) 43(8):761–7. doi:10.1038/ng.873
63. Dupuy AK, David MS, Li L, Heider TN, Peterson JD, Montano EA, et al. Redefining the human oral mycobiome with improved practices in amplicon-based taxonomy: discovery of Malassezia as a prominent commensal. PLoS One (2014) 9(3):e90899. doi:10.1371/journal.pone.0090899
64. Mayser P, Schütz M, Schuppe HC, Jung A, Schill WB. Frequency and spectrum of Malassezia yeasts in the area of the prepuce and glans penis. BJU Int (2001) 88(6):554–8. doi:10.1046/j.1464-410X.2001.02375.x
65. Findley K, Oh J, Yang J, Conlan S, Deming C, Meyer JA, et al. Topographic diversity of fungal and bacterial communities in human skin. Nature (2013) 498:367–70. doi:10.1038/nature12171
66. Haarr M. Rheumatic iridocyclitis. Acta Ophthalmol (1960) 38(1):37–45. doi:10.1111/j.1755-3768.1960.tb00176.x
67. Liang Y, Wen H, Xiao R. Serum levels of antibodies for IgG, IgA, and IgM against the fungi antigen in psoriasis vulgaris. Bull Hunan Med Univ (2003) 28(6):638–40.
68. Squiquera L, Galimberti R, Morelli L, Plotkin L, Milicich R, Kowalckzuk A, et al. Antibodies to proteins from Pityrosporum ovale in the sera from patients with psoriasis. Clin Exp Dermatol (1994) 19(4):289–93. doi:10.1111/j.1365-2230.1994.tb01197.x
69. Tsoi LC, Spain SL, Knight J, Ellinghaus E, Stuart PE, Capon F, et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet (2012) 44(12):1341–8. doi:10.1038/ng.2467
70. Dotan I, Fishman S, Dgani Y, Schwartz M, Karban A, Lerner A, et al. Antibodies against laminaribioside and chitobioside are novel serologic markers in Crohn’s disease. Gastroenterology (2006) 131(2):366–78. doi:10.1053/j.gastro.2006.04.030
71. Suhr MJ, Banjara N, Hallen-Adams HE. Sequence-based methods for detecting and evaluating the human gut mycobiome. Lett Appl Microbiol (2016) 62(3):209–15. doi:10.1111/lam.12539
72. Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet (2010) 42(12):1118–25. doi:10.1038/ng.717
73. Castro-Santos P, Moro-García MA, Marcos-Fernández R, Alonso-Arias R, Díaz-Peña R. ERAP1 and HLA-C interaction in inflammatory bowel disease in the Spanish population. Innate Immun (2017) 23(5):476–81. doi:10.1177/1753425917716527
74. Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-based map of the human proteome. Science (2015) 347(6220):1260419. doi:10.1126/science.1260419
75. Husby G, Tsuchiya N, Schwimmbeck PL, Keat A, Pahle JA, Oldstone M, et al. Cross-reactive epitope with Klebsiella pneumoniae nitrogenase in articular tissue of HLA–B27+ patients with ankylosing spondylitis. Arthritis Rheumatol (1989) 32(4):437–45. doi:10.1002/anr.1780320413
76. Hannu T. Reactive arthritis. Best Pract Res Clin Rheumatol (2011) 25(3):347–57. doi:10.1016/j.berh.2011.01.018
77. Hughes RA, Keat AC. Reiter’s syndrome and reactive arthritis: a current view. Semin Arthritis Rheum (1994) 23:190–210. doi:10.1016/0049-0172(94)90075-2
78. Sibilia J, Limbach FX. Reactive arthritis or chronic infectious arthritis? Ann Rheum Dis (2002) 61(7):580–7. doi:10.1136/ard.61.7.580
79. Uotila T, Antonen J, Laine J, Kujansuu E, Haapala A, Lumio J, et al. Reactive arthritis in a population exposed to an extensive waterborne gastroenteritis outbreak after sewage contamination in Pirkanmaa, Finland. Scand J Rheumatol (2011) 40(5):358–62. doi:10.3109/03009742.2011.562533
81. Thompson SE, Washington AE. Epidemiology of sexually transmitted Chlamydia trachomatis infections. Epidemiol Rev (1983) 5:96. doi:10.1093/oxfordjournals.epirev.a036266
82. Keat A, Thomas BJ, Taylor-Robinson D. Chlamydial infection in the aetiology of arthritis. Br Med Bull (1983) 39(2):168–74. doi:10.1093/oxfordjournals.bmb.a071811
83. Ostaszewska-Puchalska I, Zdrodowska-Stefanow B, Kuryliszyn-Moskal A, Bułhak-Kozioł V, Sokołowska M. Incidence of Chlamydia trachomatis infection in patients with reactive arthritis. Reumatologia (2015) 53(2):69. doi:10.5114/reum.2015.51505
84. Kinsella T, Norton W, Ziff M. Complement-fixing antibodies to bedsonia organisms in Reiter’s syndrome and ankylosing spondylitis. Ann Rheum Dis (1968) 27(3):241. doi:10.1136/ard.27.3.241
85. Ford D. Non-gonococcal urethritis and Reiter’s syndrome: personal experience with etiological studies during 15 years. Can Med Assoc J (1968) 99(18):900.
86. Keat A, Maini R, Nkwazi G, Pegrum G, Ridgway G, Scott J. Role of Chlamydia trachomatis and HLA-B27 in sexually acquired reactive arthritis. Br Med J (1978) 1(6113):605–7. doi:10.1136/bmj.1.6113.605
87. Rich E, Hook EW, Alarcón GS, Moreland LW. Reactive arthritis in patients attending an urban sexually transmitted diseases clinic. Arthritis Rheumatol (1996) 39(7):1172–7. doi:10.1002/art.1780390715
88. Van Duynhoven Y, Van De Laar M, Schop W, Mouton J, Van der Meijden W, Sprenger M. Different demographic and sexual correlates for chlamydial infection and gonorrhoea in Rotterdam. Int J Epidemiol (1997) 26(6):1373–85. doi:10.1093/ije/26.6.1373
89. Satterwhite CL, Torrone E, Meites E, Dunne EF, Mahajan R, Ocfemia MCB, et al. Sexually transmitted infections among US women and men: prevalence and incidence estimates, 2008. Sex Transm Dis (2013) 40(3):187–93. doi:10.1097/OLQ.0b013e318286bb53
90. Clad A, Freidank H, Kunze M, Schnoeckel U, Hofmeier S, Flecken U, et al. Detection of seroconversion and persistence of Chlamydia trachomatis antibodies in five different serological tests. Eur J Clin Microbiol Infect Dis (2000) 19(12):932–7. doi:10.1007/s100960000397
91. Rashid T, Ebringer A. Ankylosing spondylitis is linked to Klebsiella – the evidence. Clin Rheumatol (2007) 26(6):858–64. doi:10.1007/s10067-006-0488-7
92. Ebringer R, Cawdell D, Cowling P, Ebringer A. Sequential studies in ankylosing spondylitis. Association of Klebsiella pneumoniae with active disease. Ann Rheum Dis (1978) 37(2):146–51. doi:10.1136/ard.37.2.146
93. White L, McCoy R, Tait B, Ebringer R. A search for gram-negative enteric micro-organisms in acute anterior uveitis: association of klebsiella with recent onset of disease, HLA-B27, and B7 CREG. Br J Ophthalmol (1984) 68(10):750–5. doi:10.1136/bjo.68.10.750
94. Smith G, Blackwell C, Nuki G. Faecal flora in spondyloarthropathy. Br J Rheumatol (1997) 36(8):850–4. doi:10.1093/rheumatology/36.8.850
95. Park H, Schumacher H, Zeiger A, Rosenbaum J. Antibodies to peptidoglycan in patients with spondylarthritis: a clue to disease aetiology? Ann Rheum Dis (1984) 43(5):725–8. doi:10.1136/ard.43.5.725
96. Rahman M, Ahmed S, Schumacher H, Zeiger A. High levels of antipeptidoglycan antibodies in psoriatic and other seronegative arthritides. J Rheumatol (1990) 17(5):621–5.
97. Schiefer HG, Weidner W, Krauss H, Gerhardt U, Schmidt KL. Rheumatoid factor-negative arthritis, especially ankylosing spondylitis, and infections of the male urogenital tract. Zentralbl Bakteriol Mikrobiol Hyg A (1983) 255(4):511–7.
98. Van der Paardt M, van Denderen J, van den Brule A, Morré S, van der Horst-Bruinsma I, Bezemer P, et al. Prevalence of Chlamydia trachomatis in urine of male patients with ankylosing spondylitis is not increased. Ann Rheum Dis (2000) 59(4):300–2. doi:10.1136/ard.59.4.300
99. Lange U, Berliner M, Ludwig M, Schiefer H, Teichmann J, Weidner W, et al. Ankylosing spondylitis and infections of the female urogenital tract. Rheumatol Int (1998) 17(5):181–4. doi:10.1007/s002960050031
100. Csango P, Upsahl M, Romberg Ø, Kornstad L, Sarov I. Chlamydia trachomatis serology in ankylosing spondylitis. Clin Rheumatol (1987) 6(3):384–90. doi:10.1007/BF02206837
101. Savolainen E, Kettunen A, Närvänen A, Kautiainen H, Kärkkäinen U, Luosujärvi R, et al. Prevalence of antibodies against Chlamydia trachomatis and incidence of C. trachomatis-induced reactive arthritis in an early arthritis series in Finland in 2000. Scand J Rheumatol (2009) 38(5):353–6. doi:10.1080/03009740902769559
102. Hoffman I, Demetter P, Peeters M, De Vos M, Mielants H, Veys EM, et al. Anti-Saccharomyces cerevisiae IgA antibodies are raised in ankylosing spondylitis and undifferentiated spondyloarthropathy. Ann Rheum Dis (2003) 62(5):455–9. doi:10.1136/ard.62.5.455
103. Török H-P, Glas J, Gruber R, Brumberger V, Strasser C, Kellner H, et al. Inflammatory bowel disease-specific autoantibodies in HLA-B27-associated spondyloarthropathies: increased prevalence of ASCA and pANCA. Digestion (2004) 70(1):49–54. doi:10.1159/000080081
104. Aydin S, Atagunduz P, Temel M, Bicakcigil M, Tasan D, Direskeneli H. Anti-Saccharomyces cerevisiae antibodies (ASCA) in spondyloarthropathies: a reassessment. Rheumatology (2008) 47(2):142–4. doi:10.1093/rheumatology/kem324
105. Mundwiler ML, Mei L, Landers CJ, Reveille JD, Targan S, Weisman MH. Inflammatory bowel disease serologies in ankylosing spondylitis patients: a pilot study. Arthritis Res Ther (2009) 11(6):R177. doi:10.1186/ar2866
106. Riente L, Chimenti D, Pratesi F, Delle Sedie A, Tommasi S, Tommasi C, et al. Antibodies to tissue transglutaminase and Saccharomyces cerevisiae in ankylosing spondylitis and psoriatic arthritis. J Rheumatol (2004) 31(5):920–4.
107. Huhtinen M, Laasila K, Granfors K, Puolakkainen M, Seppälä I, Laasonen L, et al. Infectious background of patients with a history of acute anterior uveitis. Ann Rheum Dis (2002) 61(11):1012–6. doi:10.1136/ard.61.11.1012
108. Onal S, Kazokoglu H, Incili B, Demiralp EE, Yavuz S. Prevalence and levels of serum antibodies to gram negative microorganisms in Turkish patients with HLA-B27 positive acute anterior uveitis and controls. Ocul Immunol Inflamm (2006) 14(5):293–9. doi:10.1080/09273940600977241
109. Brown MA, Kennedy LG, MacGregor AJ, Darke C, Duncan E, Shatford JL, et al. Susceptibility to ankylosing spondylitis in twins: the role of genes, HLA, and the environment. Arthritis Rheum (1997) 40(10):1823–8. doi:10.1002/art.1780401015
110. Ruutu M, Thomas G, Steck R, Degli-Esposti MA, Zinkernagel MS, Alexander K, et al. β-glucan triggers spondylarthritis and Crohn’s disease–like ileitis in SKG mice. Arthritis Rheum (2012) 64(7):2211–22. doi:10.1002/art.34423
111. Chesson HW, Dunne EF, Hariri S, Markowitz LE. The estimated lifetime probability of acquiring human papillomavirus in the United States. Sex Transm Dis (2014) 41(11):660–4. doi:10.1097/OLQ.0000000000000193
112. Van Metre TE, Brown WH, Knox DL, Maumenee AE. The relation between nongranulomatous uveitis and arthritis. J Allergy (1965) 36(2):158–74. doi:10.1016/0021-8707(65)90164-4
113. Huhtinen M, Karma A. HLA-B27 typing in the categorisation of uveitis in a HLA-B27 rich population. Br J Ophthalmol (2000) 84(4):413–6. doi:10.1136/bjo.84.4.413
114. Popert A, Gill A, Laird S. A prospective study of Reiter’s syndrome: an interim report on the first 82 cases. Br J Vener Dis (1964) 40(3):160.
115. Eastmond C. Gram-negative bacteria and B27 disease. Rheumatology (1983) 22(Suppl_2):67–74. doi:10.1093/rheumatology/XXII.suppl_2.67
116. Hannu T, Mattila L, Siitonen A, Leirisalo-Repo M. Reactive arthritis attributable to Shigella infection: a clinical and epidemiological nationwide study. Ann Rheum Dis (2005) 64(4):594–8. doi:10.1136/ard.2004.027524
117. Rudwaleit M, Richter S, Braun J, Sieper J. Low incidence of reactive arthritis in children following a salmonella outbreak. Ann Rheum Dis (2001) 60(11):1055–7. doi:10.1136/ard.60.11.1055
118. Smeekens SP, Plantinga TS, van de Veerdonk FL, Heinhuis B, Hoischen A, Joosten LA, et al. STAT1 hyperphosphorylation and defective IL12R/IL23R signaling underlie defective immunity in autosomal dominant chronic mucocutaneous candidiasis. PLoS One (2011) 6(12):e29248. doi:10.1371/journal.pone.0029248
119. Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science (2011) 332(6025):65–8. doi:10.1126/science.1200439
120. Glocker E-O, Hennigs A, Nabavi M, Schäffer AA, Woellner C, Salzer U, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med (2009) 361(18):1727–35. doi:10.1056/NEJMoa0810719
121. Saunte D, Mrowietz U, Puig L, Zachariae C. Candida infections in patients with psoriasis and psoriatic arthritis treated with interleukin-17 inhibitors and their practical management. Br J Dermatol (2017) 177(1):47–62. doi:10.1111/bjd.15015
122. Romani L. Immunity to fungal infections. Nat Rev Immunol (2011) 11(4):275–88. doi:10.1038/nri2939
123. Brown BR, Lee EJ, Snow PE, Vance EE, Iwakura Y, Ohno N, et al. Fungal-derived cues promote ocular autoimmunity through a Dectin-2/Card9-mediated mechanism. Clin Exp Immunol (2017) 190(3):293–303. doi:10.1111/cei.13021
124. Suzuki T, Ohno N, Ohshima Y, Yadomae T. Soluble mannan and β-glucan inhibit the uptake of Malassezia furfur by human monocytic cell line, THP-1. FEMS Immunol Med Microbiol (1998) 21(3):223–30. doi:10.1111/j.1574-695X.1998.tb01169.x
125. Cross C, Bancroft G. Ingestion of acapsular Cryptococcus neoformans occurs via mannose and beta-glucan receptors, resulting in cytokine production and increased phagocytosis of the encapsulated form. Infect Immun (1995) 63(7):2604–11.
126. Baeten D, Baraliakos X, Braun J, Sieper J, Emery P, Van Der Heijde D, et al. Anti-interleukin-17A monoclonal antibody secukinumab in treatment of ankylosing spondylitis: a randomised, double-blind, placebo-controlled trial. Lancet (2013) 382(9906):1705–13 doi:10.1016/S0140-6736(13)61134-4
127. Cozon GJN, Mbitikon-Kobo FM, Fatoohi F, Spire M, Grange J-D, Kodjikian L. Abnormal cellular reactivity to microbial antigens in patients with uveitis. Invest Ophthalmol Vis Sci (2008) 49(6):2526–30. doi:10.1167/iovs.07-1454
128. Messenger W, Hildebrandt L, Mackensen F, Suhler E, Becker M, Rosenbaum JT.Characterisation of uveitis in association with multiple sclerosis. Br J Ophthalmol (2015) 99(2):205–9. doi:10.1136/bjophthalmol-2014-305518
129. Tang WM, Pulido JS, Eckels DD, Han DP, Mieler WF, Pierce K. The association of HLA-DR15 and intermediate uveitis. Am J Ophthalmol (1997) 123(1):70–5. doi:10.1016/S0002-9394(14)70994-8
130. Heinz C, Heiligenhaus A. Improvement of noninfectious uveitis with fumaric acid esters: results of a pilot study. Arch Ophthalmol (2007) 125(4):569–71. doi:10.1001/archopht.125.4.569
131. Barisani-Asenbauer T, Maca SM, Mejdoubi L, Emminger W, Machold K, Auer H. Uveitis-a rare disease often associated with systemic diseases and infections-a systematic review of 2619 patients. Orphanet J Rare Dis (2012) 7(1):57. doi:10.1186/1750-1172-7-57
132. Cosnes J, Gower–Rousseau C, Seksik P, Cortot A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology (2011) 140(6):1785–1794. doi:10.1053/j.gastro.2011.01.055
133. Van Praet L, Van den Bosch FE, Jacques P, Carron P, Jans L, Colman R, et al. Microscopic gut inflammation in axial spondyloarthritis: a multiparametric predictive model. Ann Rheum Dis (2013) 72(3):414–7. doi:10.1136/annrheumdis-2012-202135
134. Jyonouchi H, Geng L, Cushing-Ruby A, Monteiro IM. Aberrant responses to TLR agonists in pediatric IBD patients; the possible association with increased production of Th1/Th17 cytokines in response to candida, a luminal antigen. Pediatr Allergy Immunol (2010) 21(4p2):e747–55. doi:10.1111/j.1399-3038.2009.00923.x
135. Baram L, Cohen-Kedar S, Spektor L, Elad H, Guzner-Gur H, Dotan I. Differential stimulation of peripheral blood mononuclear cells in Crohn’s disease by fungal glycans. J Gastroenterol Hepatol (2014) 29(12):1976–84. doi:10.1111/jgh.12701
136. Hegazy AN, West NR, Stubbington MJT, Wendt E, Suijker KIM, Datsi A, et al. Circulating and tissue-resident CD4+ T cells with reactivity to intestinal microbiota are abundant in healthy individuals and function is altered during inflammation. Gastroenterology (2017) 153(5):1320–37.e16. doi:10.1053/j.gastro.2017.07.047
137. Gerard R, Sendid B, Colombel J-F, Poulain D, Jouault T. An immunological link between Candida albicans colonization and Crohn’s disease. Crit Rev Microbiol (2015) 41(2):135–9. doi:10.3109/1040841X.2013.810587
138. Kellermayer R, Mir SA, Nagy-Szakal D, Cox SB, Dowd SE, Kaplan JL, et al. Microbiota separation and C-reactive protein elevation in treatment naïve pediatric granulomatous Crohn disease. J Pediatric Gastroenterol Nutr (2012) 55(3):243–50. doi:10.1097/MPG.0b013e3182617c16
139. Varkas G, Thevissen K, De Brabanter G, Van Praet L, Czul-Gurdian F, Cypers H, et al. An induction or flare of arthritis and/or sacroiliitis by vedolizumab in inflammatory bowel disease: a case series. Ann Rheum Dis (2017) 76(5):878–81. doi:10.1136/annrheumdis-2016-210233
140. Wendling D, Sondag M, Verhoeven F, Vuitton L, Koch S, Prati C. Arthritis occurrence or reactivation under Vedolizumab treatment for inflammatory bowel disease. A four cases report. Joint Bone Spine (2017) 85(2):255–6. doi:10.1016/j.jbspin.2017.01.012
141. Tadbiri S, Peyrin-Biroulet L, Serrero M, Filippi J, Pariente B, Roblin X, et al. Impact of vedolizumab therapy on extra-intestinal manifestations in patients with inflammatory bowel disease: a multicentre cohort study nested in the OBSERV-IBD cohort. Aliment Pharmacol Ther (2017) 47(4):485–93. doi:10.1111/apt.14419
142. Salmi M, Jalkanen S. Human leukocyte subpopulations from inflamed gut bind to joint vasculature using distinct sets of adhesion molecules. J Immunol (2001) 166(7):4650–7. doi:10.4049/jimmunol.166.7.4650
143. Ciccia F, Guggino G, Rizzo A, Saieva L, Peralta S, Giardina A, et al. Type 3 innate lymphoid cells producing IL-17 and IL-22 are expanded in the gut, in the peripheral blood, synovial fluid and bone marrow of patients with ankylosing spondylitis. Ann Rheum Dis (2015) 74(9):1739–47. doi:10.1136/annrheumdis-2014-206323
144. Salmi M, Jalkanen S. Endothelial ligands and homing of mucosal leukocytes in extraintestinal manifestations of IBD. Inflamm Bowel Dis (1998) 4(2):149–56. doi:10.1097/00054725-199805000-00026
145. Chouraki V, Savoye G, Dauchet L, Vernier-Massouille G, Dupas J-L, Merle V, et al. The changing pattern of Crohn’s disease incidence in northern France: a continuing increase in the 10- to 19-year-old age bracket (1988–2007). Aliment Pharmacol Ther (2011) 33(10):1133–42. doi:10.1111/j.1365-2036.2011.04628.x
146. Yao T, Matsui T, Hiwatashi N. Crohn’s disease in Japan. Dis Colon Rectum (2000) 43:S85–93. doi:10.1007/BF02237231
147. Parisi R, Symmons DP, Griffiths CE, Ashcroft DM. Global epidemiology of psoriasis: a systematic review of incidence and prevalence. J Invest Dermatol (2013) 133(2):377–85. doi:10.1038/jid.2012.339
148. Sepahi S, Riahi-Zanjani B, Ghorani A. The role of Candida albicans in the pathogenesis of Psoriasis vulgaris: systematic literature review. Rev Clin Med (2016) 3:122–7. doi:10.22038/rcm.2016.6485
149. Lober CW, Belew PW, Rosenberg EW, Bale G. Patch tests with killed sonicated microflora in patients with psoriasis. Arch Dermatol (1982) 118(5):322–5. doi:10.1001/archderm.1982.01650170036019
150. Imai Y, Tsuda T, Aochi S, Futatsugi-Yumikura S, Sakaguchi Y, Nakagawa N, et al. YKL-40 (chitinase 3-like-1) as a biomarker for psoriasis vulgaris and pustular psoriasis. J Dermatol Sci (2011) 64(1):75–7. doi:10.1016/j.jdermsci.2011.06.012
151. Faergemann J. Treatment of Sebopsoriasis with Itraconazole: Behandlung von Sebopsoriasis mit Itraconazole. Mycoses (1985) 28(12):612–8. doi:10.1111/j.1439-0507.1985.tb02094.x
152. Rosenberg EW, Belew PW. Improvement of psoriasis of the scalp with ketoconazole. Arch Dermatol (1982) 118(6):370–1. doi:10.1001/archderm.118.6.370
153. Farr P, Marks J, Krause L, Shuster S. Response of scalp psoriasis to oral ketoconazole. Lancet (1985) 326(8461):921–2. doi:10.1016/S0140-6736(85)90853-0
154. Crutcher N, Rosenberg EW, Belew PW, Skinner RB Jr, Eaglstein NF, Baker SM. Oral nystatin in the treatment of psoriasis. Arch Dermatol (1984) 120(4):435–6. doi:10.1001/archderm.120.4.435a
155. Ganor S. Treatment of early psoriasis lesions with a oral amphotericin B or nystatin. Int J Dermatol (1988) 27(6):420–420. doi:10.1111/j.1365-4362.1988.tb02398.x
156. Buslau M, Hänel H, Holzmann H. The significance of yeasts in seborrheic eczema. Hautarzt (1989) 40(10):611–3.
157. Ascioglu O, Soyuer U, Aktas E. Improvement of psoriasis with oral nystatin. In: Tümbay E, Seeliger HPR, Anǧ Ö, editors. Candida and Candidamycosis. Boston, MA: Springer (1991). p. 279–81. doi:10.1007/978-1-4684-5910-4_48
158. Balak D, Fallah Arani S, Hajdarbegovic E, Hagemans C, Bramer W, Thio H, et al. Efficacy, effectiveness and safety of fumaric acid esters in the treatment of psoriasis: a systematic review of randomized and observational studies. Br J Dermatol (2016) 175(2):250–62. doi:10.1111/bjd.14500
159. Chiu H-Y, Chang W-L, Tsai T-F, Tsai Y-W, Shiu M-N. Risk of psoriasis following terbinafine or itraconazole treatment for onychomycosis: a population-based case-control comparative study. Drug Safety (2018) 41(3):285–95. doi:10.1007/s40264-017-0614-2
160. Swanbeck G, Inerot A, Martinsson T, Wahlström J, Enerbäck C, Enlund F, et al. Age at onset and different types of psoriasis. Br J Dermatol (1995) 133(5):768–73. doi:10.1111/j.1365-2133.1995.tb02753.x
161. Rahman P, Schentag CT, Gladman DD. Immunogenetic profile of patients with psoriatic arthritis varies according to the age at onset of psoriasis. Arthritis Rheumatol (1999) 42(4):818–23. doi:10.1002/1529-0131(199904)42:4<818::AID-ANR30>3.0.CO;2-5
162. Lysell J, Padyukov L, Kockum I, Nikamo P, Ståhle M. Genetic association with ERAP1 in psoriasis is confined to disease onset after puberty and not dependent on HLA-C* 06. J Invest Dermatol (2013) 133(2):411–7. doi:10.1038/jid.2012.280
163. Adachi A, Horikawa T, Ichihashi M, Takashima T, Komura A. Role of Candida allergen in atopic dermatitis and efficacy of oral therapy with various antifungal agents. Allergy (1999) 48(7):719–25.
164. Larson JL, Wallace TL, Tyl RW, Marr MC, Myers CB, Cossum PA. The reproductive and developmental toxicity of the antifungal drug Nyotran® (liposomal nystatin) in rats and rabbits. Toxicol Sci (2000) 53(2):421–9. doi:10.1093/toxsci/53.2.421
165. Pauls K, Schön M, Kubitza RC, Homey B, Wiesenborn A, Lehmann P, et al. Role of integrin α E (CD103) β 7 for tissue-specific epidermal localization of CD8+ T lymphocytes. J Invest Dermatol (2001) 117(3):569–75. doi:10.1046/j.0022-202x.2001.01481.x
166. Tett A, Pasolli E, Farina S, Truong DT, Asnicar F, Zolfo M, et al. Unexplored diversity and strain-level structure of the skin microbiome associated with psoriasis. NPJ Biofilms Microbiomes (2017) 3:1–12. doi:10.1038/s41522-017-0022-5
167. Gomez-Moyano E, Crespo-Erchiga V, Martínez-Pilar L, Diaz DG, Martínez-García S, Navarro ML, et al. Do Malassezia species play a role in exacerbation of scalp psoriasis? J Mycol Med (2014) 24(2):87–92. doi:10.1016/j.mycmed.2013.10.007
168. Fry L, Baker BS. Triggering psoriasis: the role of infections and medications. Clin Dermatol (2007) 25(6):606–15. doi:10.1016/j.clindermatol.2007.08.015
169. Pierard J, Dockx P. The ultrastructure of tinea versicolor and Malassezia furfur. Int J Dermatol (1972) 11(2):116–24. doi:10.1111/j.1365-4362.1972.tb01735.x
170. Hallen-Adams HE, Kachman SD, Kim J, Legge RM, Martínez I. Fungi inhabiting the healthy human gastrointestinal tract: a diverse and dynamic community. Fungal Ecol (2015) 15:9–17. doi:10.1016/j.funeco.2015.01.006
171. de la Rosette JJ, Hubregtse MR, Meuleman EJ, Stolk-Engelaar MV, Debruyne FM. Diagnosis and treatment of 409 patients with prostatitis syndromes. Urology (1993) 41(4):301–7. doi:10.1016/0090-4295(93)90584-W
172. Edstrom Hagerwall AM, Rydengard V, Fernlund P, Morgelin M, Baumgarten M, Cole AM, et al. Beta-microseminoprotein endows post coital seminal plasma with potent candidacidal activity by a calcium- and pH-dependent mechanism. PLoS Pathog (2012) 8(4):e1002625. doi:10.1371/journal.ppat.1002625
173. Eeles RA, Kote-Jarai Z, Giles GG, Olama AA, Guy M, Jugurnauth SK, et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet (2008) 40(3):316–21. doi:10.1038/ng.90
174. Kote-Jarai Z, Easton DF, Stanford JL, Ostrander EA, Schleutker J, Ingles SA, et al. Multiple novel prostate cancer predisposition loci confirmed by an international study: the PRACTICAL Consortium. Cancer Epidemiol Biomarkers Prev (2008) 17(8):2052–61. doi:10.1158/1055-9965.EPI-08-0317
175. Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, Orr N, et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet (2008) 40(3):310–5. doi:10.1038/ng.91
176. Xu X, Valtonen-Andre C, Savblom C, Hallden C, Lilja H, Klein RJ. Polymorphisms at the microseminoprotein-beta locus associated with physiologic variation in beta-microseminoprotein and prostate-specific antigen levels. Cancer Epidemiol Biomarkers Prev (2010) 19(8):2035–42. doi:10.1158/1055-9965.EPI-10-0431
177. Hrbacek J, Urban M, Hamsikova E, Tachezy R, Heracek J. Thirty years of research on infection and prostate cancer: no conclusive evidence for a link. A systematic review. Urol Oncol (2012) 31(7):951–65. doi:10.1016/j.urolonc.2012.01.013
178. Xu K, Wang X, Ling MT, Lee DT, Fan T, Chan FL, et al. Identification of a specifically expressed modified form of novel PSP-94 protein in the secretion of benign prostatic hyperplasia. Electrophoresis (2003) 24(7–8):1311–8. doi:10.1002/elps.200390167
179. Tsai Y, Harrison H, Lee C, Daufeldt J, Oliver L, Grayhack J. Systematic characterization of human prostatic fluid proteins with two-dimensional electrophoresis. Clin Chem (1984) 30(12):2026–30.
180. Doctor VM, Sheth AR, Simha MM, Arbatti NJ, Aaveri JP, Sheth NA. Studies on immunocytochemical localization of inhibin-like material in human prostatic tissue: comparison of its distribution in normal, benign and malignant prostates. Br J Cancer (1986) 53(4):547–54. doi:10.1038/bjc.1986.85
181. Sathe VS, Sheth NA, Phadke MA, Sheth AR, Zaveri JP. Biosynthesis and localization of inhibin in human prostate. Prostate (1987) 10(1):33–43. doi:10.1002/pros.2990100107
182. Kramer G, Marberger M. Could inflammation be a key component in the progression of benign prostatic hyperplasia? Curr Opin Urol (2006) 16(1):25–9. doi:10.1097/01.mou.0000193368.91823.1b
183. Guess HA, Arrighi HM, Metter EJ, Fozard JL. Cumulative prevalence of prostatism matches the autopsy prevalence of benign prostatic hyperplasia. Prostate (1990) 17(3):241–6. doi:10.1002/pros.2990170308
184. Seman G, Gallager HS, Johnson DE. Melanin-like pigment in the human prostate. Prostate (1982) 3(1):59–72. doi:10.1002/pros.2990030109
185. Guillan R, Zelman S. The incidence and probable origin of melanin in the prostate. J Urol (1970) 104(1):151–3. doi:10.1016/S0022-5347(17)61689-6
186. Maksem JA, Johenning PW, Galang CF. Prostatitis and aspiration biopsy cytology of prostate. Urology (1988) 32(3):263–8. doi:10.1016/0090-4295(88)90398-6
187. Gómez BL, Nosanchuk JD. Melanin and fungi. Curr Opin Infect Dis (2003) 16(2):91–6. doi:10.1097/00001432-200304000-00005
188. Fernandes C, Prados-Rosales R, Silva BM, Nakouzi-Naranjo A, Zuzarte M, Chatterjee S, et al. Activation of melanin synthesis in Alternaria infectoria by antifungal drugs. Antimicrob Agents Chemother (2016) 60(3):1646–55. doi:10.1128/AAC.02190-15
189. Weiber H, Andersson C, Murne A, Rannevik G, Lindstrom C, Lilja H, et al. Beta microseminoprotein is not a prostate-specific protein. Its identification in mucous glands and secretions. Am J Pathol (1990) 137(3):593–603.
190. Ohkubo I, Tada T, Ochiai Y, Ueyama H, Eimoto T, Sasaki M. Human seminal plasma beta-microseminoprotein: its purification, characterization, and immunohistochemical localization. Int J Biochem Cell Biol (1995) 27(6):603–11. doi:10.1016/1357-2725(95)00021-G
191. Nolet S, St-Louis D, Mbikay M, Chretien M. Rapid evolution of prostatic protein PSP94 suggested by sequence divergence between rhesus monkey and human cDNAs. Genomics (1991) 9(4):775–7. doi:10.1016/0888-7543(91)90375-O
192. Ghasriani H, Teilum K, Johnsson Y, Fernlund P, Drakenberg T. Solution structures of human and porcine beta-microseminoprotein. J Mol Biol (2006) 362(3):502–15. doi:10.1016/j.jmb.2006.07.029
193. Clark NL, Swanson WJ. Pervasive adaptive evolution in primate seminal proteins. PLoS Genet (2005) 1(3):e35. doi:10.1371/journal.pgen.0010035
194. Pearson CM. Development of arthritis, periarthritis and periostitis in rats given adjuvants. Proc Soc Exp Biol Med (1956) 91(1):95–101. doi:10.3181/00379727-91-22179
195. Pearson CM, Wood FD. Studies of polyarthritis and other lesions induced in rats by injection of mycobacterial adjuvant. I. General clinical and pathologic characteristics and some modifying factors. Arthritis Rheumatol (1959) 2(5):440–59. doi:10.1002/1529-0131(195910)2:5<440::AID-ART1780020510>3.0.CO;2-N
196. Taurog JD, Sandberg GP, Mahowald ML. The cellular basis of adjuvant arthritis: II. Characterization of the cells mediating passive transfer. Cell Immunol (1983) 80(1):198–204. doi:10.1016/0008-8749(83)90106-5
197. Waksman BH, Pearson CM, Sharp JT. Studies of arthritis and other lesions induced in rats by injection of mycobacterial adjuvant. J Immunol (1960) 85(4):403–17.
198. Taurog JD, Rival C, van Duivenvoorde LM, Satumtira N, Dorris ML, Sun M, et al. Autoimmune epididymoorchitis is essential to the pathogenesis of male-specific spondylarthritis in HLA–B27–transgenic rats. Arthritis Rheumatol (2012) 64(8):2518–28. doi:10.1002/art.34480
199. Sonoyama K, Miki A, Sugita R, Goto H, Nakata M, Yamaguchi N. Gut colonization by Candida albicans aggravates inflammation in the gut and extra-gut tissues in mice. Med Mycol (2011) 49(3):237–47. doi:10.3109/13693786.2010.511284
200. Hida S, Miura NN, Adachi Y, Ohno N. Cell wall β-glucan derived from Candida albicans acts as a trigger for autoimmune arthritis in SKG mice. Biol Pharm Bull (2007) 30(8):1589–92. doi:10.1248/bpb.30.1589
201. Marijnissen RJ, Koenders MI, Van de Veerdonk FL, Dulos J, Netea MG, Boots AM, et al. Exposure to Candida albicans polarizes a T-cell driven arthritis model towards Th17 responses, resulting in a more destructive arthritis. PLoS One (2012) 7(6):e38889. doi:10.1371/journal.pone.0038889
202. Miyake Y, Toyonaga K, Mori D, Kakuta S, Hoshino Y, Oyamada A, et al. C-type lectin MCL is an FcRγ-coupled receptor that mediates the adjuvanticity of mycobacterial cord factor. Immunity (2013) 38(5):1050–62. doi:10.1016/j.immuni.2013.03.010
203. Lee EJ, Brown BR, Vance EE, Snow PE, Silver PB, Heinrichs D, et al. Mincle activation and the Syk/Card9 signaling axis are central to the development of autoimmune disease of the eye. J Immunol (2016) 196(7):3148–58. doi:10.4049/jimmunol.1502355
204. Yamasaki S, Matsumoto M, Takeuchi O, Matsuzawa T, Ishikawa E, Sakuma M, et al. C-type lectin Mincle is an activating receptor for pathogenic fungus, Malassezia. Proc Natl Acad Sci U S A (2009) 106(6):1897–902. doi:10.1073/pnas.0805177106
205. Lord JC, Hartzer KL, Kambhampati S. A nuptially transmitted ichthyosporean symbiont of Tenebrio molitor (Coleoptera: Tenebrionidae). J Eukaryot Microbiol (2012) 59(3):246–50. doi:10.1111/j.1550-7408.2012.00617.x
206. Ramos M, Pisa D, Molina S, Rábano A, Juarranz Á, Carrasco L. Fungal infection in patients with multiple sclerosis. Open Mycol J (2008) 2(1):22–8. doi:10.2174/1874437000802010022
207. Benito-Leon J, Pisa D, Alonso R, Calleja P, Diaz-Sanchez M, Carrasco L. Association between multiple sclerosis and Candida species: evidence from a case-control study. Eur J Clin Microbiol Infect Dis (2010) 29(9):1139–45. doi:10.1007/s10096-010-0979-y
208. Pisa D, Alonso R, Jimenez-Jimenez FJ, Carrasco L. Fungal infection in cerebrospinal fluid from some patients with multiple sclerosis. Eur J Clin Microbiol Infect Dis (2013) 32(6):795–801. doi:10.1007/s10096-012-1810-8
209. Guerini FR, Cagliani R, Forni D, Agliardi C, Caputo D, Cassinotti A, et al. A functional variant in ERAP1 predisposes to multiple sclerosis. PLoS One (2012) 7(1):e29931. doi:10.1371/journal.pone.0029931
210. Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, et al. Clonal expansions of CD8+ T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med (2000) 192(3):393–404. doi:10.1084/jem.192.3.393
211. Hook EW III, Marra CM. Acquired syphilis in adults. N Engl J Med (1992) 326(16):1060–9. doi:10.1056/NEJM199204163261606
212. Palacios G, Druce J, Du L, Tran T, Birch C, Briese T, et al. A new arenavirus in a cluster of fatal transplant-associated diseases. N Engl J Med (2008) 358(10):991–8. doi:10.1056/NEJMoa073785
213. Lipkin WI. Microbe hunting. Microbiol Mol Biol Rev (2010) 74(3):363–77. doi:10.1128/MMBR.00007-10
214. Cottier F, Srinivasan KG, Yurieva M, Liao W, Poidinger M, Zolezzi F, et al. Advantages of meta-total RNA sequencing (MeTRS) over shotgun metagenomics and amplicon-based sequencing in the profiling of complex microbial communities. NPJ Biofilms Microbiomes (2018) 4(1):2. doi:10.1038/s41522-017-0046-x
215. Kowarsky M, Camunas-Soler J, Kertesz M, De Vlaminck I, Koh W, Pan W, et al. Numerous uncharacterized and highly divergent microbes which colonize humans are revealed by circulating cell-free DNA. Proc Natl Acad Sci U S A (2017) 114(36):9623–8. doi:10.1073/pnas.1707009114
216. Ghannoum MA, Jurevic RJ, Mukherjee PK, Cui F, Sikaroodi M, Naqvi A, et al. Characterization of the oral fungal microbiome (mycobiome) in healthy individuals. PLoS Pathog (2010) 6(1):e1000713. doi:10.1371/journal.ppat.1000713
217. Guo R, Zheng N, Lu H, Yin H, Yao J, Chen Y. Increased diversity of fungal flora in the vagina of patients with recurrent vaginal candidiasis and allergic rhinitis. Microb Ecol (2012) 64(4):918–27. doi:10.1007/s00248-012-0084-0
218. Peeters A, Dukmans B, Van der Schroeff J. Fumaric acid therapy for psoriatic arthritis. A randomized, double-blind, placebo-controlled study. Rheumatology (1992) 31(7):502–4. doi:10.1093/rheumatology/31.7.502
219. Altmeyer PJ, Mattlies U, Pawlak F, Hoffmann K, Frosch PJ, Ruppert P, et al. Antipsoriatic effect of fumaric acid derivatives: results of a multicenter double-blind study in 100 patients. J Am Acad Dermatol (1994) 30(6):977–81. doi:10.1016/S0190-9622(94)70121-0
220. Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med (2012) 367(12):1098–107. doi:10.1056/NEJMoa1114287
221. Ashbee HR, Evans EGV. Immunology of diseases associated with Malassezia species. Clin Microbiol Rev (2002) 15(1):21–57. doi:10.1128/CMR.15.1.21-57.2002
222. Vesty A, Biswas K, Taylor MW, Gear K, Douglas RG. Evaluating the impact of DNA extraction method on the representation of human oral bacterial and fungal communities. PLoS One (2017) 12(1):e0169877. doi:10.1371/journal.pone.0169877
223. Cleland EJ, Bassioni A, Boase S, Dowd S, Vreugde S, Wormald PJ. The fungal microbiome in chronic rhinosinusitis: richness, diversity, postoperative changes and patient outcomes. Int Forum Allergy Rhinol (2014) 4(4):259–65. doi:10.1002/alr.21297
224. Boix-Amorós A, Martinez-Costa C, Querol A, Collado MC, Mira A. Multiple approaches detect the presence of fungi in human breastmilk samples from healthy mothers. Sci Rep (2017) 7:13016. doi:10.1038/s41598-017-13270-x
225. Alonso R, Pisa D, Marina AI, Morato E, Rábano A, Rodal I, et al. Evidence for fungal infection in cerebrospinal fluid and brain tissue from patients with amyotrophic lateral sclerosis. Int J Biol Sci (2015) 11(5):546. doi:10.7150/ijbs.11084
226. Alonso R, Pisa D, Marina AI, Morato E, Rábano A, Carrasco L. Fungal infection in patients with Alzheimer’s disease. J Alzheimers Dis (2014) 41(1):301–11. doi:10.3233/JAD-132681
227. van Woerden HC, Gregory C, Brown R, Marchesi JR, Hoogendoorn B, Matthews IP. Differences in fungi present in induced sputum samples from asthma patients and non-atopic controls: a community based case control study. BMC Infect Dis (2013) 13(1):69. doi:10.1186/1471-2334-13-69
228. Cafarchia C, Dell’Aquila M, Capelli G, Minoia P, Otranto D. Role of β-endorphin on phospholipase production in Malassezia pachydermatis in dogs: new insights into the pathogenesis of this yeast. Med Mycol (2007) 45(1):11–5. doi:10.1080/13693780600962718
229. Cafarchia C, Dell’Aquila M, Traversa D, Albrizio M, Guaricci A, De Santis T, et al. Expression of the µ-opioid receptor on Malassezia pachydermatis and its effect in modulating phospholipase production. Med Mycol (2010) 48(1):73–8. doi:10.3109/13693780902718347
230. Honnavar P, Chakrabarti A, Prasad GS, Singh P, Dogra S, Rudramurthy SM. β-Endorphin enhances the phospholipase activity of the dandruff causing fungi Malassezia globosa and Malassezia restricta. Med Mycol (2017) 55(2):150–4. doi:10.1093/mmy/myw058
231. Davidson A, Vermesh M, Paulson RJ, Graczykowski JW, Lobo RA. Presence of immunoreactive β-endorphin and calcitonin in human seminal plasma, and their relation to sperm physiology. Fertil Steril (1989) 51(5):878–80. doi:10.1016/S0015-0282(16)60684-2
232. Shahed AR, Shoskes DA. Correlation of β-endorphin and prostaglandin E2 levels in prostatic fluid of patients with chronic prostatitis with diagnosis and treatment response. J Urol (2001) 166(5):1738–41. doi:10.1016/S0022-5347(05)65664-9
233. Quartuccio M. Immunoreactive beta-endorphin in the genital tract and seminal plasma of the bull. Reprod Domest Anim (2004) 39(4):264. doi:10.1111/J.1439-0531.2004.498_1.X
Keywords: spondyloarthritis, acute anterior uveitis, reactive arthritis, ankylosing spondylitis, fungal infections
Citation: Laurence M, Asquith M and Rosenbaum JT (2018) Spondyloarthritis, Acute Anterior Uveitis, and Fungi: Updating the Catterall–King Hypothesis. Front. Med. 5:80. doi: 10.3389/fmed.2018.00080
Received: 16 November 2017; Accepted: 09 March 2018;
Published: 05 April 2018
Edited by:
Xu Huji, Second Military Medical University, ChinaReviewed by:
Lars Vereecke, Ghent University, BelgiumCopyright: © 2018 Laurence, Asquith and Rosenbaum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin Laurence, bWxhdXJlbmNlQHNoaXBzaGF3LmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.