 Daniele Corridoni
Daniele Corridoni Thomas Chapman
Thomas Chapman Tim Ambrose
Tim Ambrose Alison Simmons1,2
Alison Simmons1,2

94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 19 February 2018
Sec. Gastroenterology
Volume 5 - 2018 | https://doi.org/10.3389/fmed.2018.00032
This article is part of the Research Topic Intestinal Inflammation View all 6 articles
Activation of the innate immune system through pattern-recognition receptor (PRR) signaling plays a pivotal role in the early induction of host defense following exposure to pathogens. Loss of intestinal innate immune regulation leading aberrant immune responses has been implicated in the pathogenesis of inflammatory bowel disease (IBD). The precise role of PRRs in gut inflammation is not well understood, but considering their role as bacterial sensors and their genetic association with IBD, they likely contribute to dysregulated immune responses to the commensal microbiota. The purpose of this review is to evaluate the emerging functions of PRRs including their functional cross-talk, how they respond to mitochondrial damage, induce mitophagy or autophagy, and influence adaptive immune responses by interacting with the antigen presentation machinery. The review also summarizes some of the recent attempts to harness these pathways for therapeutic approaches in intestinal inflammation.
The innate immune system represents the first line of defense against pathogens, providing an immediate response to infection, and is conserved throughout evolution (1, 2). In vertebrates, the innate immune system also serves to prime the adaptive immune system, generating long-lasting immunological memory. The critical importance of the innate immune system is underlined by the observation that defects can be lethal to the host (2) or associated with inflammatory disorders, such as inflammatory bowel disease (IBD).
The innate immune system faces two key challenges. First, it is faced with a near constant barrage of microorganisms. Second, it must accurately and rapidly discriminate between non-infectious self and infectious pathogen. To achieve this, it relies upon a relatively small number of pattern-recognition receptors (PRRs) that recognize features common to many pathogens, known as pathogen-associated molecular patterns (2). Pathogen-associated molecular patterns are typically critical for survival of the pathogen and thus cannot be mutated by the pathogen to avoid detection; consequently, they are often invariant across entire classes of pathogens (3).
Pattern-recognition receptors are germline-encoded receptors that are expressed on the cell surface, in intracellular compartments such as the cytosol or endosomes, or secreted into serum or tissue fluid. PRRs are classified according to their function, localization, and ligand specificity. Two main classes of PRRs have been described in mammalian cells, namely membrane bound receptors, including toll-like receptors (TLRs) and C-type lectin receptors (CLRs), and cytoplasmic receptors, including nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), RIG-I-like receptors (RLRs), AIM2-like receptors, and the more recently identified cyclic GMP-AMP synthase (cGAS). PRRs often act in synergy allowing rapid amplification of the initial immune response. The varied location of PRRs provides the ability to detect pathogens invading a range of cellular compartments. Multiple PRRs are capable of recognizing a single microorganism, which may present a broad array of antigenic ligands, ensuring a robust immune response. PRRs also recognize host-derived endogenous ligands known as danger-associated molecular patterns. These endogenous danger response signals, such are uric acid and HMGB1, are released by stressed cells upon necrosis to promote an innate immune response through activation of PRRs (4, 5).
Toll-like receptors are the best-characterized PRRs. First identified in Drosophila (6), to date 11 separate receptors have been identified in humans, classified as TLR1–10, with human TLR11 believed to be a pseudogene (7). A further two TLRs, TLR12 and TLR13, have been described in mice, but are not found in humans. Although TLRs sense a broad range of ligands, derived from both exogenous microbial pathogens and host (damage-associated molecular patterns), they share a common structure. This consists of an extracellular ectodomain containing a variable number of leucine-rich repeat (LRR) motifs that mediate ligand binding, a single transmembrane helix, and an intracellular toll-like interleukin 1 receptor domain required for intracellular signaling (8). Dependent on which TLR is activated, specific adaptor molecules are recruited which can broadly be considered as MyD88 dependent or independent. It is this selectivity of adapter molecule recruitment that leads to the specificity of TLR signaling pathways and the subsequent inflammatory response. The role of TLR signaling in the pathogenesis of IBD, together with the potential for therapeutic modulation, is discussed in Section “Potential Therapeutic Targets Altering Signaling through TLRs.”
There is emerging evidence for the importance of another class of PRR, the CLR in IBD. CLRs comprise a large family of receptors that bind to carbohydrates via carbohydrate-recognition domains and appear of particular importance in mediating antifungal immunity, but are also able to recognize other pathogens including bacteria and protozoa (9). A polymorphism in the gene for Dectin-1 is linked to a severe form of ulcerative colitis, driven by an aberrant response to commensal intestinal fungi (10). Genetic variants in mannose-binding lectin (MBL) have been linked to Crohn’s disease (11) while mice deficient in MBL show increased susceptibility to experimental colitis (12). Macrophage galactose-type C-type lectin-1 and SIGN-R3 have also separately been linked to protective regulatory roles in murine models of colitis (13, 14). It is clear that CLRs may also act in synergy with TLRs, with a deficiency of both the CLR SIGN-R1 and TLR4 leading to reduced susceptibility to colitis in a murine model, with reduced responsiveness to the TLR4 ligand lipopolysaccharide (15).
The NLR family of proteins are cytosolic PRRs that sense a diverse range of microbial structures such as peptidoglycan and flagellin, and also endogenous danger signals, to trigger innate immune activation (16, 17). NLRs consist of three domains—an N-terminal protein interaction domain, a central NOD domain, and a C terminal LRR. The NLRs are divided into four subfamilies on the basis of their N-terminal effector domains: NLRA, acidic domain containing; NLRB, Baculovirus inhibitor of apoptosis protein repeat domain containing; NLRC, caspase recruitment domain (CARD) domain containing; NLRP, pyrin domain containing. NLRX represents other NLR proteins with no significant homology to the N-terminal domain of the other NLR subfamilies (18).
Functionally, the NLR family can be divided into further subgroups related to inflammasome assembly, autophagy, antigen presentation, signaling transduction, and transcription activation (19). The N-terminus effector domain that mediates protein–protein interactions is important in determining function, for example, the pyrin domain of the NLRP subfamily allows binding and activation of the caspase-1 inflammasome, while the CARD domain of the NLRC subfamily binds and activates receptor-interacting serine/threonine protein kinase 2 (RIPK2), activating downstream NF-κB, and MAPK signaling pathways (20, 21). The LRR domain is required for binding and detection of ligands and consists of leucine-rich amino acid strands forming a peptide loop.
The NOD domain, which has ATPase activity, is required for self-oligomerization of NLRs following binding of ligand to the LRR domain, likely facilitating binding and activation of downstream effector molecules via the N-terminus effector domain (22). The structural diversity of the LRR and N-terminus effector domains allows NLRs to interact with a wide array of ligands and binding partners, activating a broad range of signaling pathways. This breadth of role is reflected in the wide variety of human diseases that result from mutations in NLR-encoding genes (23).
Two related members of the NLR family, NOD1 and NOD2, are critical for the innate immune response to many bacterial infections. Both NOD1 and NOD2 respond to distinct structural motifs derived from intracellular peptidoglycan, a fragment of bacterial cell walls. NOD1 recognizes the dipeptide d-glutamyl-meso-diaminopimelic acid, present in all Gram-negative, and a limited number of Gram-positive bacteria such as Listeria spp. and Bacillus spp. (24, 25). By contrast, NOD2 recognizes muramyl dipeptide (MDP), the largest fraction of peptidoglycan consisting of one carbohydrate and two amino acids, present in all Gram-negative and Gram-positive bacteria (26). Activation of NOD1 and NOD2 signaling upon peptidoglycan sensing leads to their direct interaction with RIPK2 in a complex called “nodosome.” The nodosome formation leads to activation of pro-inflammatory and antimicrobial responses as discussed further in this review (27, 28). In 2001, NOD2 was found to be associated with Crohn’s disease and it remains one of the strongest genetic risk factors (29, 30). Given this evidence, the review of innate mechanisms relating to NLRs will focus on NOD2.
NOD2 is expressed in cells of the gastrointestinal tract, specifically Paneth cells, intestinal epithelial cells (IECs), stem cells, and stromal cells. It is also expressed in the hematopoietic compartment in monocyte-derived cells including dendritic cells (DCs) and macrophages. In IECs, membrane targeting of NOD2 is required for NF-κB activation by MDP (31). NOD2 also recruits the autophagy protein autophagy-related 16-like 1 (ATG16L1) to the plasma membrane at the site of bacterial entry to drive autophagy (32). It remains unclear where NOD2 engages ligand in other cellular locations, such as ligands derived from endosome resident bacteria including Salmonella enterica serovar Typhimurium (33).
NOD2 consists of a LRR domain, a central NOD domain, and an N-terminal effector domain that contains two CARDs in tandem. Extrapolation of the recently solved crystal structure of NLRC4 predicts that the NOD domain is followed by a proximal helical domain, a winged-helix domain, and a distal helical domain (34). The ADP-mediated interaction between the NOD domain and winged-helix help stabilize a closed, auto-inhibited conformation, with the LRR occluding these two domains and maintaining a monomeric state (27, 34).
The precise mechanisms by which MDP enters cells and activates NOD2 remains uncertain, but a number of entry routes have been described: (1) phagocytosis of bacteria and degradation into fragments including MDP, (2) shedding of peptidoglycan by invasive bacteria (35), (3) uptake of peptidoglycan fragments from bacteria-derived extracellular outer membrane vesicles (36), (4) endocytosis (37), and (5) transport across host membranes through bacterial secretion systems or channels (27, 38). The relative importance of these varied mechanisms is likely to differ depending on the host cell type. Taken together, these mechanisms suggest that NOD2 may be able to sense extracellular bacteria, significantly expanding upon its classically described role as a sensor of invading cytosolic bacteria (33). The ability of soluble MDP to stimulate NOD2 in vitro further supports the presence of routes of direct entry for the ligand (27).
Following MDP binding and conformational change of NOD2 to an open structure and homo-oligomerization, NOD2 recruits RIPK2 via interaction between their CARD domains (27, 39). The NOD2–RIPK2 signaling axis has been extensively studied and mapped. RIPK2 is essential for NOD2 signaling, as evidenced by the failure of RIPK2-deficient murine macrophages to respond to MDP (40). Engagement of RIPK2 can occur via NOD1 or NOD2 but not via TLR-mediated signaling pathways (33).
Although it was initially uncertain whether MDP binds directly to NOD2 once it has reached the cytosol, recent reports support a direct interaction but do not exclude the requirement for accessory molecules (41, 42). It is generally believed that recognition is mediated by the LRR domain, although a potential role for the NOD domain has also been suggested (42). A mechanism has been proposed whereby ATP binding to NOD2 leads to homo-oligomerization and enhances MDP binding and subsequent signal transduction (42). It is likely that the distal helical domain is required to mediate NOD2 conformational change to an open activated structure upon ligand binding (34). However, further structural data are required to confirm the mechanism of NOD2 activation.
The stable ubiquitination of RIPK2 leads to the recruitment and activation of the kinase TAK1 (27). This activated TAK1 complex interacts with the simultaneously bound IκB kinase (IKK) complex, ubiquitinating and degrading IKKγ, which allows IKKα and IKKβ to phosphorylate IκKBα (NF-κB inhibitor IκB) (38). Phosphorylated IκKBα is degraded, allowing the translocation of NF-κB to the nucleus and inflammatory gene transcription (27, 43). In addition to NF-κB pathways, NOD2 activation via RIPK2/TAK1 also activates the MAPK regulated extracellular ERK1/2, JNK, and p38 (39). NOD2 can also activate p38 and JNK via CARD9 (44).
More recently, it has been shown that NOD2 is able to respond to pathogen-associated molecular patterns other than MDP, most notably viral ssRNA (45). This results in a quite separate RIPK2 independent signaling pathway, mediated by the mitochondrial antivirus signaling protein (MAVS). Engagement of MAVS at the mitochondria leads to a signaling cascade involving interferon regulatory factor 3 (IRF3) that produces interferon (IFN)-β (45). It was shown in vitro that NOD2 drives type 1 IFN production in response to a range of viruses containing an ssRNA genome including respiratory syncytial virus and influenza virus (45). This was confirmed in vivo, with NOD2 knockout mice demonstrating an increased susceptibility to infection with respiratory syncytial virus or influenza, although increased susceptibility to viral infections has not yet been shown in humans expressing NOD2 polymorphisms (45).
Much remains to be understood about how NOD2 signaling pathways are regulated but it is clear that a complex system of protein–protein interactions underlies this. In addition to the actions of the E3 ligases described above, roles for TRAF 2, 4, and 5 have been described (43, 46). NOD2 signaling can also be fine-tuned by the removal of ubiquitin. An important example is the action of ovarian tumor deubiquitinase A20, which inhibits NOD2 signaling by regulating the MDP-induced ubiquitination of RIPK2 (47).
Additional negative regulators include Erbin that directly binds to NOD2 and inhibits MDP signaling (48), Rac1 GTPase (49) and RIG-I (50). By contrast, GRIM-19 (51), a cell death protein, and CARD9 (44) positively regulate NOD2 signaling. It appears that the cytoskeleton plays a key role in modulating NOD2 activity; in addition to cytoskeletal-related proteins, Erbin and Rac1 GTPase, the intermediate protein filament vimentin is also important (52). Intriguingly, three RHO GTPases, including Rac1 GTPase, that play important roles in cytoskeletal modeling, are apparently able to activate NOD2 in the absence of MDP, raising the possibility of NOD2 stimulation in the absence of a bacterial pathogen (33, 53).
A complex interplay between PRRs provides cross-regulation of innate immune receptor signaling and can both amplify and suppress the immune response (54). The intricacy of this interrelationship results from the varied array of PRR ligands, many of which may arise from the same pathogen, and the divergent signaling pathways they may simultaneously induce (54). Synergistic signaling results from NOD2 acting together with a number of PRR-mediated pathways including NOD1, TLR2, TLR3, TLR4, and TLR9 to boost production of a range of both pro- and anti-inflammatory cytokines (IL-6, IL-8, TNF, IL-1β, and IL-10) in antigen-presenting cells (APCs) (22, 55–57). The cross-talk between NOD2 and TLR2 remains the most well characterized to date. Their close relationship is not surprising given that both respond to ligands derived from the same bacterial component, peptidoglycan. Although the precise mechanisms of cross-regulation are not well understood, both NOD2 and TLR2 activate separate upstream signaling cascades to recruit the same MAPK and NF-κB pathways, which play a central role in cytokine production (54). NOD2 and TLR2 also collaborate in adaptive immune roles, and they have been shown to cooperatively regulate the functional maturation of DCs (58).
Mitochondria are highly active organelles that continuously relocate within the cell, undergoing fission, fusion, biogenesis, and mitophagy to maintain a pool suitably responsive to cellular demands. These processes are termed mitochondrial dynamics and control mitochondrial morphology, quality, abundance, and location, which are critical to the immune role of mitochondria (59). There is still much to understand about how the innate immune system regulates mitochondrial dynamics, but a recent study provides an interesting insight. Following recognition of bacteria by TLRs, macrophages were found to adapt their electron transport chain architecture by destabilizing complex I in a NLRP3 and reactive oxygen species (ROS)-dependent manner (60). This resulted in enhanced mitochondrial respiration from complex I and II and increased mitochondrial reactive oxygen species (mROS) and may regulate IL-1β and IL-10 production.
Mitochondria act as a platform for innate immune signaling, with key innate immune effectors assembling on the outer mitochondrial membrane (59, 61). One of the first proteins discovered was MAVS, a key adapter protein for the RLR signaling pathway that responds to viral infections (62). On viral infection and RLR triggering, MAVS is targeted to the outer membrane by a C-terminal transmembrane domain. Although it remains unclear precisely why MAVS must be recruited to the mitochondria, localization here is essential to mediate downstream signaling via NF-κB and IRF3 to regulate type-1 IFN production (61, 62). As described earlier, NOD2 can also engage MAVS in response to viral ssRNA triggering a similar IFN cascade (45). MAVS activation is additionally regulated by mitochondrial dynamics (63) and mROS (64) (Figure 1D). A further illustration of the intersecting nature of mitochondrial signaling pathways is the recent discovery of the regulation of the NLRP3 inflammasome by MAVS (65). Another mitochondrial membrane protein, cardiolipin, also controls NLRP3 inflammasome activation in an mROS-independent manner (66).
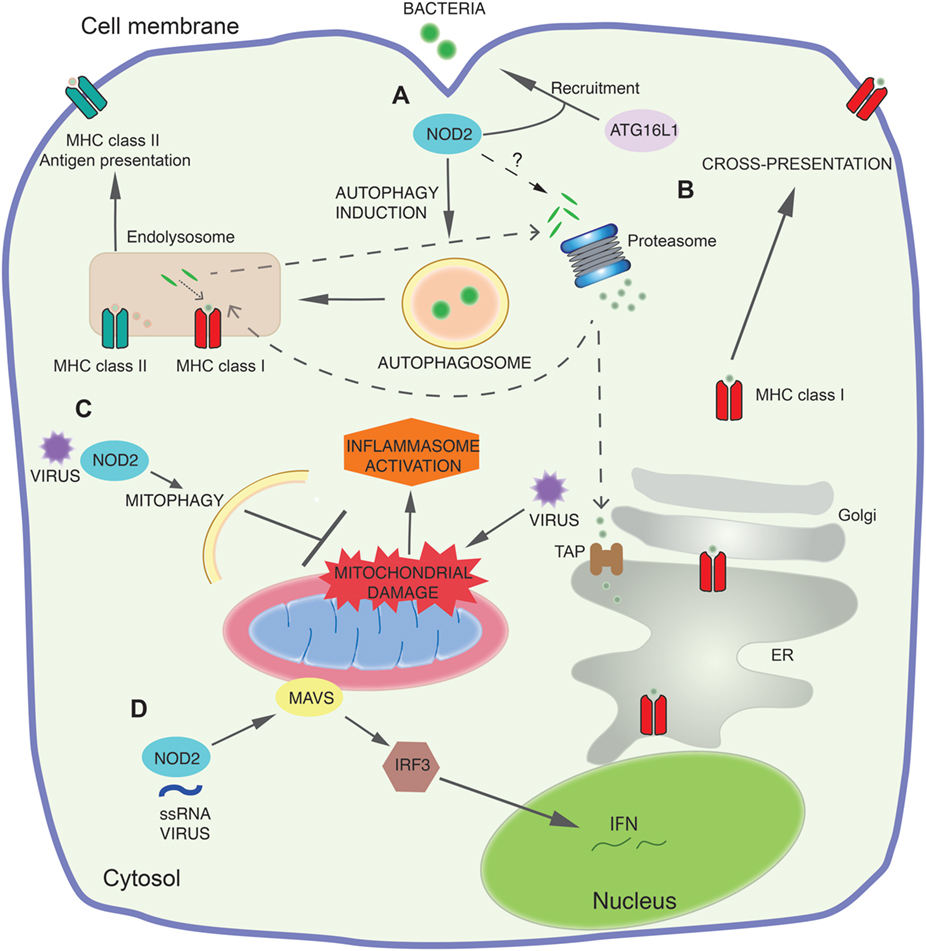
Figure 1. NOD2 signaling pathways. (A) At sites of bacterial entry to the cell cytosol, NOD2 recruits the autophagy protein autophagy-related 16-like 1 (ATG16L1) to the plasma membrane and together they direct autophagy of the invading bacteria. This leads to both direct bacterial killing and loading of the antigens to major histocompatibility complex class II (MHC II). (B) Early NOD2 engagement by components of bacteria increases cross-presentation for MHC class I-dependent antigen presentation. Antigens destined for cross-presentation can have different routes: they can be transported from the endocytic vesicles to the cytosol for proteasomal degradation and transported into the endoplasmic reticulum (ER) or back into the endosomal compartment for loading onto MHC class I. The molecular mechanisms by which NOD2 controls these pathways and proteasome function are still not defined. (C) NOD2 also functions as a viral pattern-recognition receptor and modulates immune response to viral infection. NOD2 drives RIPK2-mediated mitophagy of damaged mitochondria following viral invasion of the cytosol. This negatively regulates the NLRP3 inflammasome response by limiting the release of mitochondrial reactive oxygen species and other mitochondrial damage-associated molecular patterns following the mitochondrial damage caused by viral infection. (D) NOD2 recognizes viral ssRNA to trigger mitochondrial antivirus signaling protein (MAVS) mediated activation of interferon regulatory factor 3 (IRF3), leading to production of the antiviral interferon (IFN) response.
Given the ancient bacterial roots of mitochondria, it is unsurprising that mitochondrial injury releases compounds carrying bacterial molecular motifs, sensed as danger-associated molecular patterns, to trigger an immune response (67). Mitochondrial DNA (mtDNA), which shares hypomethylated CpG motifs with bacterial DNA, is one such danger-associated molecular pattern, which can be sensed by the innate immune receptor for CpG DNA, TLR9 (67). TLR9 activation leads to NF-κB and MAPK inflammatory cascades and can also signal through IRF7 to enhance type-1 IFN responses (68). mtDNA is also an important endogenous agonist for NLRP3 inflammasome (69), and in macrophages has been shown to activate the NLRC4 (70) and AIM2 (absent in melanoma 2) inflammasomes (69). Finally, mtDNA can also trigger pro-inflammatory cytokine responses via the cGAS PRR (71, 72). mtDNA-dependent inflammatory pathology has been described in a diverse range of diseases including heart failure, atherosclerosis, rheumatoid arthritis, and liver disease, and following bacterial infection (68).
It is essential that cells are able to efficiently eliminate damaged mitochondria and their danger-associated molecular patterns to prevent aberrant inflammation, and here autophagy plays a vital role. Mitophagy inhibits NLRP3 inflammasome activation by clearing damaged mitochondria and limiting the release of both mROS and mtDNA (69, 73). Mice deficient in the autophagy protein LC3B showed enhanced caspase-1-dependent cytokines in sepsis models (69). As described earlier, following viral infection the NOD2–RIPK2 axis also acts to increase mitophagy and limit inflammasome activation (Figure 1C). These studies underline the importance of mitophagy in responding to the mitochondrial damage that occurs during an immune response, thereby preventing hyperactivation of the immune system.
While it is clear that mitochondria are of central importance in immunity, their role in Crohn’s disease is not well understood. A number of studies suggest the presence of mitochondrial dysfunction in IBD. Enterocytes isolated from patients with IBD have abnormal mitochondrial morphology (74), and murine models of colitis result in similar mitochondrial changes in IECs (75). There is evidence of elevated ROS, although the source was not clarified, and impaired mitochondrial membrane potential in patients with active Crohn’s, which was found to improve on disease remission (76). In pediatric Crohn’s disease patients, proteomic analysis of mucosal biopsies suggested a marked downregulation of mitochondrial proteins, including components of the mitochondrial respiratory chain (77). It has been suggested that IBD is a state of energy deficiency, with impaired mitochondrial β-oxidation of fatty acids implicated (78). This theory is supported by the findings that, mutations in SLC22A5 (OCTN2), which encodes a carnitine transporter, are associated with Crohn’s disease (79), with carnitine essential for fatty acid oxidation. Mice lacking OCTN2 develop spontaneous intestinal inflammation and atrophy (80). Conversely, mice carrying mutations that increase mitochondrial oxidative phosphorylation activity and ATP generation are protected from chemical models of colitis (81). It is clear that more research is needed to elucidate the contribution of mitochondrial dysfunction to Crohn’s disease, but the potential intersection with processes such as autophagy and inflammasome activation is intriguing.
A distinct role for innate immunity is the induction of a form of autophagy, macroautophagy (32, 82), a highly conserved mechanism for the bulk degradation of cellular contents. Macroautophagy is distinguished from the two other primary types of autophagy in mammalian cells, microautophagy and chaperone-mediated autophagy, by the formation of a double membrane bound phagophore and autophagosome (83). In its most basic form, autophagy represents a cellular adaptation to starvation and allows the non-specific breakdown of a cell’s own constituents to recycle nutrients and balance biosynthetic pathways (84, 85). This form of autophagy, known as non-selective autophagy, is also induced by cellular stress. However, autophagy can also be utilized by cells in a more precisely targeted process (selective autophagy) for various indications, and indeed is not restricted to “self-constituents” as it can target invading pathogens (86). This important host defense against pathogens, termed xenophagy, can target a range of invasive bacteria including S. enterica serovar Typhimurium, Listeria monocytogenes, Shigella flexneri, and Mycobacterium tuberculosis, as well as bacteria internalized by phagocytosis (87). Both NOD2 and TLRs can mediate xenophagy and this influences loading of microbial antigens to late endosomal compartments, where substrates are then degraded by lysosomal hydrolases. During antigen presentation, foreign proteins are captured by autophagosomes and are delivered to major histocompatibility complex (MHC) class II-processing and -loading compartments (Figure 1A).
Following induction of autophagy, the core pathway of macroautophagy begins with the nucleation of a double-membrane phagophore. The origin of the phagophore is a source of considerable debate, with a role proposed for numerous membrane compartments including the endoplasmic reticulum (ER) and the mitochondria (88, 89). This step and the subsequent extension of the phagophore into an autophagosome that engulfs the targeted cytoplasmic components requires the autophagy ubiquitin-like protein LC3 (the mammalian homolog of ATG8) (90). To date, over 40 proteins have been identified as important for macroautophagy, primarily the autophagy-related genes first mapped in yeast (91) with mammalian homologs subsequently described. Broadly, they can be grouped according the stage of autophagy at which they function—(1) induction, (2) nucleation and expansion, (3) fusion, and (4) degradation and efflux (90). A number of these proteins are involved in the processing of LC3, underlining its importance. The ATG4 protease first cleaves the inactive pro-form of LC3 at its C-terminus to generate the cytosolic LC3-I. LC3-I is then conjugated to phosphatidylethanolamine in the phagophore membrane by ATG3 and 7, producing the lipidated LC3-II, which initiates the formation and maturation of the autophagosome. A critical role of LC3 is to recruit other autophagy proteins to the autophagosome, including the ULK1 kinase (90, 92) and ATG13 (90). This typically requires an LC3-interacting region (LIR), characterized by a WXXI/L sequence, with tryptophan (W) beginning the sequence, any two amino acids (XX) following, and either isoleucine (I) or leucine (L) two residues downstream (93). A number of LIR containing autophagy proteins also contain an ubiquitin-binding domain, allowing recruitment of the aforementioned ubiquitinated targets to the autophagosome. Important examples in xenophagy are p62 (SQSTM1) (94), NDP52 (95), NBR1 (96), and optineurin (OPTN) (97). In addition, and vital to the concept of xenophagy, an LC3-decorated single membrane phagosome may also become sequestered within an autophagosome (98). This type of xenophagy is restricted to phagocytic cells (macrophages, DCs, and neutrophils).
During the process of autophagosome maturation, LC3-II is deconjugated by ATG4 and thus released from the outer membrane. By contrast, the GATE-16/γ-aminobutyric acid receptor-associated family (GABARAPs) appear important for the maturation of the autophagosome (99). Following closure and maturation, the autophagosome progresses toward fusion with the lysosome. The autophagosome may fuse with a late endosome forming an amphisome (100), which subsequently fuses with a lysosome to generate an autolysosome. Alternatively, an autophagosome may fuse directly with a lysosome, and this should also be termed an autolysosome (101). By contrast, when a phagosome is incorporated into an autophagosome and fuses with a lysosome, this should be distinguished as an autophagolysosome (101). The term autophagolysosome also describes the fusion of an LC-decorated phagosome with a lysosome (102), a process independent of autophagy termed LC3-mediated phagocytosis, but which shares a number of overlapping features with autophagy.
Three protein families are key to the process of autophagosome–lysosome fusion. Rab GTPases localize to the membranes of both structures, recruiting membrane-tethering complexes that bridge the autophagosome to the lysosome. Soluble N-ethylmaleimide-sensitive factor attachment protein receptors then drive the fusion of the opposing lipid bilayer membranes (103). In addition, GABARAPs also regulate modulate autophagosome–lysosome fusion by regulating the generation of phosphatidylinositol-4-phosphate (104). Once fused, lysosomal hydrolases digest the contents of the autophagolysosome.
A cascade of intracellular pathways have evolved to respond to protein misfolding—this is known as the UPR (105). The combined action of three ER transmembrane stress sensors is responsible for UPR activation: inositol-requiring enzyme 1α (IRE1α), PKR-like ER kinase (PERK), and activating transcription factor 6α (ATF6α). Usually, the luminal domains of these proteins are inactive through association with binding immunoglobulin protein (BiP). However, BiP has higher affinity for misfolded proteins. Therefore when misfolded proteins accumulate, BiP dissociates from the stress sensors activating downstream signaling cascades (106). These signaling pathways lead primarily to a reduction in the quantity of proteins that enter the ER. Also, through increased transcription of ERAD- and autophagy-related proteins, misfolded proteins are eliminated. Finally, the ER expands, and the capacity to refold proteins is increased (107–110).
Dimerization of IRE1α, followed by release from BiP, initiates splicing of a single mRNA encoding X-box-binding protein 1 (XBP1) (111, 112). This generates XBP1s, an activator of transcription, which induces the transcription of chaperones and protein-folding enzymes resident in the ER. Together, these increase ER size and function (113). Dissociation of BiP from PERK allows PERK homodimerization and autophosphorylation to activate the cytoplasmic kinase domain. Activated PERK attenuates global translation of mRNA by inhibiting eIF2-TC to enable cells to temporarily manage ER stress (114, 115). Finally, following BiP dissociation, ATF6α relocates to the Golgi apparatus. Here, it is processed by site 1 and 2 proteases (S1P and S2P) producing a p50 fragment. This fragment translocates to the nucleus and induces gene expression related to proteins that increase overall ER capacity, the ability to refold proteins, and activates the ERAD pathway (116–118).
Inflammation represents a critical factor in the induction of the UPR. Immune cells are highly sensitive to environmental factors which induce ER stress. Protein-folding demand is markedly increased following pathogen exposure (105). ER stress and TLR signaling are linked—TLR signaling in macrophages induces ER stress and ER stress acts to increase the response to TLR signaling. The TLR 2 and 4 ligands, Pam3CSK4 and LPS, induce IRE1α activation. IRE1α-induced XBP1 splicing in response to TLR ligation affects pro-inflammatory cytokine production, such as the production of IL-6, TNF, and IFNβ. Increased IL-1β production results from the activation of glycogen synthase 3β (GSK3β) which is IRE1α dependent. Furthermore, GSK3β inhibits ongoing splicing of XBP1, attenuating TNF transcription thereby altering the inflammatory response (119, 120).
Dysregulation in UPR signaling have been associated with various complex inflammatory diseases, including IBD. Among the risk genes associated with IBD identified by genome-wide association studies (GWAS), some encode for proteostatic proteins including Orosomucoid-like 3 (ORMDL3) or anterior gradient 2 (AGR2) (121–124). Paneth cells are abnormally located in the ileum of Agr2-deficient mice who also exhibit reduced mucin 2 (MUC2) expression in goblet cells. These mice also develop spontaneous ileo-colitis and an activated UPR (125). Mice with a knockout of XBP1 specifically in IECs (Xbp1ΔIEC) exhibit signs of ER stress. They are more susceptible to chemical colitis induced by dextran sulfate sodium (DSS) and develop a spontaneous ileitis (126). If these mice also lack the autophagy gene ATG16L1 in IECs they develop a Crohn-like transmural spontaneous enteritis (127). Importantly, patients carrying the Crohn’s risk allele ATG16L1 (T300A) have evidence of ER stress in Paneth cells (128).
Activation of the innate immune signaling pathways through PRRs discussed above provides immediate detection of microbial presence and viability, which is necessary to determine a successful activation of naïve T cells and generate appropriate effector responses. Activation of PRRs leads to a significant change in the phenotype of APCs, which is characterized by enhanced expression of costimulatory molecules and increased secretion of pro-inflammatory cytokines (129). In addition, signals derived from PRRs in DCs determine whether the antigen presentation machinery leads to activation or cross-tolerance of T cells, depending on whether or not DCs are exposed to PRR ligands and on the length of this exposure (130). This contributes, in physiological conditions, to minimize the risk of generating pro-inflammatory responses to self-antigens, to maximize T-cell priming against microbial antigens during the initial phase of DC maturation, and to temporally control MHC class I and II antigen presentation and prevent excessive priming during chronic phases of pathogen handling. Consequently, dysregulation of these mechanisms may be highly relevant in the pathogenesis of IBD.
NOD2 recruits the autophagy protein ATG16L1 to the plasma membrane at the site of bacterial entry to direct autophagy. This facilitates bacterial trafficking to the autophagosome through a signaling pathway independent of RIPK2. NOD2 signaling via RIPK2 also upregulates the autophagosome formation and increases autophagic flux, further potentiating autophagy. This is required for the generation of MHC class II antigen-specific CD4+ T cell responses (82) (Figure 1A). Importantly, DCs expressing the Crohn’s disease NOD2 and ATG16L1 variants have reduced autophagic response and MHC class II antigen presentation in response to MDP. Decreased macroautophagy due to NOD2 and ATG16L1 mutations may impair innate resistance to invading bacteria and, thereby, trigger inflammation as a result of increased antigenic load or lead to insufficient tolerance induction against commensals in the gut and trigger Crohn’s disease (82).
In the context of NOD2-dependent generation of MHC class II antigen-specific CD4+ T cell responses, several studies also suggest that NOD2 signaling influences CD4-specific adaptive immune responses. The induction of IL-17 by bacteria-primed DCs is not through the TGF-β–IL-6 pathway in naïve human Th cells. Instead, Th17 cells develop from memory Th cells (27). In addition, MDP programs DCs to increase IL-23 and IL-1 production which orchestrates Th17-mediated immunity in humans. In line with this, expression of IL-17 following MDP stimulation is impaired in DCs derived from Crohn’s disease patients with polymorphisms in the NOD2 gene. This can be attributed to loss of NOD2/TLR synergy on production of IL-1α and IL-1β, and IL-23 (131, 132).
It is important that the synergistic effects of NOD2 with other TLR signaling pathways inducing effector cytokines such as IL-23 are tightly regulated so that homeostasis can be restored at the termination of an immune response. MicroRNAs (miRNAs) are important regulators of gene expression whose main function is to repress target mRNA levels including regulators of innate immune responses by targeting key signaling proteins and cytokines (133–137). Our group has previously demonstrated that NOD2 can regulate induction of the miRNA family 29a, 29b, and 29c and induces this family alone or additively with TLR2 or TLR5 in DCs. miRNA-29 downregulates IL-12p40/IL-23 and attenuates Th17 CD4+ T cell responses; however, Crohn’s disease DCs expressing associated NOD2 variants are incapable of inducing miR-29 following NOD2 triggering. Therefore, NOD2 has an immunoregulatory function in human DCs of the miR-29 family, and a loss of miR-29 induction in Crohn’s DCs might contribute to the abnormal elevation of IL-23 observed in inflamed lesions during this disease (132).
NOD2 activation by MDP in mice generates specific Th2-type immune responses. Costimulation with TLR agonists promotes the priming of not only Th1 and Th2 but also Th17 responses (138, 139). In this context, direct triggering of NOD2 by MDP, generates a specific Th2-type immune response and stromal NOD2 expression is needed to prime effector CD4+ Th2 responses. In addition, NOD2-dependent stimulation induces OX40 ligand, necessary for Th2 immunity (140, 141). A more recent study has investigated the effect of Nod2 deletion in a spontaneous mouse model of chronic intestinal inflammation, SAMP1/YitFc, characterized by a progressive cobblestone CD-like ileitis that develops in the absence of chemical, genetic, or immunological manipulation (142). Nod2 deletion in SAMP1/YitFc mice was associated with inhibition of the Th2 cytokines IL-4, IL-5, and IL-13 whereas no effect was observed in Th1 cytokine expression, including TNF-α and IFN-γ. In addition, the role of Th2 effector signaling pathways were also affected by NOD2 deletion, with decreased phosphorylated-STAT6 and GATA3 in the gut mucosa of SAMP × NOD2−/− mice. These effects were observed in the presence of the same bacterial flora, which suggests that changes in the bacterial community are not associated with the effects of NOD2 in ameliorating intestinal inflammation in SAMP1/YitFc mice, and that NOD2 regulates Th2 responses in the intestine independent of acute dysbiosis (142).
With the availability of new drug compounds that target inhibition of NOD2 and RIPK2 signaling, potential pharmacological inhibition of NOD2 signaling may be a reasonable therapeutic strategy to prevent Th2-driven intestinal inflammation and other CD4 T cell responses.
Cross-presentation represents a critical mechanism for priming adaptive immune responses against exogenous antigens derived from microbial pathogens or tumors (143), which are presented by MHC class I molecules (144). During cross-presentation, the establishment of CD8+ T-cell-mediated responses is dictated by DCs whose function is to acquire exogenous antigens and direct the formation of a complex between the MHC-I peptide and a cognate TCR leading to activation and proliferation of antigen-specific CD8+ T cells. Different pathways and subcellular locations regulating cross-presentation have been described, including the vacuolar and phagocytic pathways (143). Engagement of the vacuolar pathway implicates the degradation of antigens by endosomal or phagosomal proteases (i.e., cathepsin S) and the resultant peptides are loaded onto MHC class I molecules (145). By contrast, during activation of the phagocytic pathway, internalized antigens from endosomes or phagosomes are exported to the cytosol where they are degraded by the proteasome. The resultant peptides are then transported back into the phagosome and loaded onto MHC-I (144, 146) or, instead, peptides are transported via TAP into the ER, for loading onto ER-resident heavy chain-B2m complexes (147) (Figure 1B). It has been indicated in several studies that PRRs signaling influence on multiple processes associated with cross-presentation. Thus, any dysfunction of these mechanisms may lead to abnormal activation of CD8+ T cells responses associated with inflammation.
There is temporal control of MHC-I antigen presentation by the innate immune system, with initial PRRs engagement promoting its efficiency during the early or acute phase of microbial exposure and with prolonged stimulation leading to mechanisms to prevent excessive priming during chronic phases of pathogen handling. Following a short stimulation of TLR4 by LPS in DCs, antigen translocation from the phagosome to the cytosol is increased. During this phase, Blander and colleagues have also recently shown that the recruitment to phagosomes of MHC class I molecules, stored in endosomal recycling compartments (ERCs), is enhanced by TLR4 stimulation (148). In fact, LPS stimulation leads to IKK2-dependent phosphorylation of phagosome-associated SNAP-23 (synaptosome-associated protein of 23 kDa) promoting fusion between ERCs and phagosomes. This mechanism acts to deliver to phagosomes enough numbers of MHC-I molecules once TLRs senses microbial components to increase cross-presentation and priming of CD8+ T cells (148, 149).
During the intermediate phase of DC maturation followed by 3–16 h of TLR engagement, the efficacy of cross-presentation is still increased (150, 151). During this phase, DCs exhibit increased endocytosis, proteasomal and TAP activity, delayed phagosomal degradation, and decreased acidification (mediated by a decrease in recruitment of lysosomal proteases to phagosomes). RAB34-dependent peri-nuclear clustering of lysosomes and reduced shift of phagosomes along microtubules prevents their fusion, with the end result of increasing the efficiency of cross-presentation (152, 153).
During the late stage of DCs stimulation by TLR agonists (24–40 h), there is a markedly reduced efficiency of cross-presentation (150). This is likely due to decreased antigen uptake or antigen export to the cytosol (154, 155).
Given the presence of NLRs at sites that are in close proximity to phagosomal and endosomal membranes containing high levels of bacterial components, engagement of their signaling pathways may play a critical role in interacting with the MHC class I antigen presentation machinery. However, there are only few studies investigating how NLR activity affects cross-presentation (156).
NLRP3 inflammasome and caspase-1 activation modulate phagosome activity by causing locally restricted modification of the proteins associated to the organelle, which results in induction of the microbicidal activity (157). In fact, phagosome-associated caspase-1 can control the activity of NADPH oxidase NOX2 inducing changes on pH of the vacuole. In line with this, NLRP3-deficient and caspase-1-deficient cells fail to induce phagosome acidification in response to microbial infection (157). Since cross-presentation of phagocytosed antigens to CD8+ T cells occurs primarily from a non-acidified phagosome, activation of the inflammasome negatively impact cross-presentation by controlling the pH of phagosomes, which accelerates degradation of antigens.
Early stimulation of both NOD1 and NOD2 signaling alone or together with other PRRs enhances cross-presentation while during the late phase DC maturation their stimulation lead to decreased cross-presentation. NOD1 and NOD2 activation by peptidoglycan in DCs increase cross-presentation (Figure 1A) via upregulation of intracellular components, such as TAP, SEC61, and calnexin, which are essential for MHC class I-dependent antigen presentation and enhanced antigen-specific CD8+ T cells. During this process, NOD/RIPK2-mediated signals might mimic the TLR4–MyD88 signals necessary to induce recruitment of TAP to the early endosomes, which is an important step for enhancing cross-presentation of soluble antigens (158). By contrast, peptidoglycan pre-treatment of DCs led to progressive inhibition of cross-presentation over time, with decreased cross-presentation after 12 h and complete inhibition after 18 h (159). This demonstrates that maturation of DCs by NOD1 and NOD2 engagement, prior to antigen encounter negatively modulates cross-presentation. Therefore, NOD-dependent signaling pathways temporally control MHC class I antigen presentation and may prevent excessive priming during chronic phases of pathogen handling. This may be highly relevant in the context of chronic intestinal inflammation in which loss of function associated polymorphisms in the NOD2 gene are associated with increased susceptibility to Crohn’s disease. As increasing evidence suggests that CD8+ T cells may play an earlier role in IBD development than the CD4+ T cells (160), whether dysregulated NOD2 signaling may affect CD8+ T cell responses by aberrant priming via the cross-presentation pathway remains to be investigated.
NOD1, polymorphisms of which are associated with human IBD (161), is constitutively expressed in IECs and has been shown to activate NF-κB by pathogens in particular those which have developed methods to bypass sensing by TLRs (162). Furthermore, NOD1 is required for innate immune responses in human IECs to Campylobacter jejuni infection and transient knockdown of NOD1 increases bacterial invasion (163). However, NOD1−/− mice do not exhibit any pathological differences to wild-type in a Salmonella-induced colitis model, whereas NOD1−/−NOD2−/− double knockout resulted in milder colitis (164). Contrary to this, NOD1 deficiency has been shown to result in increased colitis-associated colonic cancer in a chemical colitis model (165), which may, in part, reflect differences in colitis modeling. Together, this suggests that augmentation rather than complete inhibition of NOD1 may derive benefit in intestinal inflammation. There are currently no preclinical or clinical studies modulating NOD1 signaling directly although a number of inhibitors have been developed, including Noditinib-1 (ML130) and ML146, to enable further study of this pathway (166–169).
The function of NLRs in intestinal inflammation is of particular interest given the strong association between NOD2 polymorphisms [R702W, G908R, and 1007fs (3020insC) (29, 30)] and Crohn’s disease. Patients with a NOD2 variant are more likely to have disease affecting the terminal ileum, exhibit a fibrostenosing phenotype, and to require surgical intervention (170). Interestingly, there are no differences in phenotype between homozygotes or compound heterozygotes for the three polymorphisms listed. Approximately half of patients with Crohn’s disease have at least one mutation in NOD2 (170) and importantly not everyone in the wider population with NOD2 variants develop intestinal inflammation (171, 172) highlighting the complexities of this disease and the non-essential role of NOD2 in disease pathogenesis. Unlike NOD1, NOD2 is expressed predominantly in myeloid cells (173). However, a role for recognition of intracellular bacteria in IECs, which is lost in 3020insC mutants, has been demonstrated (174).
Given that NOD2 polymorphisms are generally thought to lead to loss of function, it is intriguing as to why these might lead to an increased risk of intestinal inflammation. It is unclear as to whether repair of NOD2 signaling defects might represent a therapeutic strategy for patients expressing Crohn’s disease-associated polymorphisms (175, 176). An alternative to targeting the receptors directly is modulating the function of downstream signaling mediators such as RIPK2. RIPK2 inhibitors have shown benefit in inflammatory disease including in the SAMP1/YitFc model of Crohn’s disease-like ileitis (177–179) although there are no clinical studies as yet in intestinal inflammation.
Polymorphisms in the NLRP3 gene are associated with Crohn’s disease (180) although the effect size of this may be small (181). Mice deficient in NLRP3 are more susceptible to chemical colitis through lack of IL-18 (182) but, in contrast, excessive IL-1β and IL-18 production is seen in human IBD tissues suggesting activation of NLRP3 may mediate chronic intestinal inflammation [discussed in Ref. (123)]. To this end, inhibitors of the NLRP3 inflammasome such as Astragalus polysaccharide (183, 184), INF39 (an acrylate derivative) (185), and levornidazole (186) have been shown in experimental models to improve colitis. The peptide SPA4 has anti-inflammatory actions through indirect suppression of the NLRP3 inflammasome by interacting with TLR4 (187) although this has not yet been used in colitis models. However, the plant flavonoid alpinetin does improve DSS colitis through a similar mechanism of modulation of the NLRP3 and TLR4 pathways (188). Recently, a novel inhibitor of NLRP3, CY-09, was identified which binds to the NACHT domain of the complex to inhibit activation in murine macrophages and human PBMCs, and in a relevant murine model of NLRP3-associated inflammation (189). Ongoing study is required to elucidate whether this compound is of benefit in intestinal inflammation. Interfering with inflammasome-related cytokines may hold promise for the management of human IBD. A UK-based, multicentre, randomized, double-blind, placebo-controlled trial of the IL-1 receptor antagonist Anakinra alongside corticosteroids in acute severe colitis is planned.1 A comprehensive review of the role of the NLRP3 inflammasome in intestinal health and disease has recently been published (190).
TLR2 has been shown to be essential for tight junction function in the intestine through MyD88 and PI3K/Akt (191). In this study, mice deficient for TLR2 or MyD88 demonstrated tight junction dysfunction when challenged with DSS while oral treatment with a TLR2 ligand, Pam3CSK4, improved barrier function and reduced colonic inflammation. Another group has shown that deletion of TLR2 in the multidrug resistance model of colitis resulted in more aggressive disease (192). The commensal microbiota may yield benefits for modulating inflammation through TLR2. Polysaccharide A (PSA) is produced by the mammalian intestinal commensal Bacteroides fragilis. PSA acts via TLR2 on APCs to modulate Th1/Th2 balance (193) and via plasmacytoid DCs to induce regulatory T cells (194). In a Helicobacter hepaticus model of colitis, co-colonization with B. fragilis resulted in a milder disease (195). Similar effects were seen with purified PSA alone (195). Oral PSA was of benefit in a 2,4,6-trinitrobenzenesulphonic acid (TNBS) chemical colitis model through increased production of the anti-inflammatory cytokine IL-10 (195). These effects on IL-10 levels have been replicated in germ-free mice challenged with DSS. In those mice which had prior administration of B. fragilis, this resulted in improved histology, reduced mortality, reduced pro-inflammatory TNF-α mRNA, and increased IL-10 mRNA (196). Surprisingly, despite the improved colitis, a significant increase in colonic pro-inflammatory IL-17 mRNA was seen in the Bacteroides group. A subsequent study by the same group confirmed the essential role of TLR2 in the modulation of colitis by B. fragilis. TLR2−/− mice did not gain appreciable benefit from Bacteroides administration as measured by colon length and histological scores, and these mice also fail to upregulate IL-10 transcription following bacterial administration (197). Similar to the findings for PSA and B. fragilis, curli fibers (amyloid fibers in enteric biofilms) are recognized by TLR2, result in IL-10 production, and ameliorate TNBS-induced colitis (198).
Other avenues have corroborated the role of TLR2 in mediating inflammation. The TLR2 agonists lipoarabinomannan and lipoteichoic acid reduced indomethacin-induced murine ileitis via modulation of TLR4 pathways on macrophages and effects on leukocyte migration (199). The lipoxin A4 agonist, BML-111, alters the expression of TLR2 and 4 in a murine cecal ligation/puncture model of sepsis with improvements in pro-inflammatory IL-6 and TNF-α production (200). In addition to directly acting agonists, interfering with TLR2 dimerization has been shown to ameliorate DSS colitis through effects on monocyte activation (201) and to improve Pam3CSK4-induced hepatic inflammation (202). VB-201, an oxidized phospholipid mimic which is orally available, binds to TLR2 and CD14 to limit downstream inflammatory pathways and has beneficial effects on atherosclerosis (203) and experimental autoimmune encephalomyelitis (204). In addition to directly modulating TLR signaling pathways, effects are mediated through inhibition of monocyte migration (205). A phase 2 study of VB-201 in mild-to-moderate ulcerative colitis has been completed but the results have not yet been published (NCT01839214). A humanized IgG4-monoclonal anti-TLR2 antibody (OPN-305) is effective in a porcine model of cardiac ischemia–reperfusion injury (206) and has been shown to be well tolerated and effective in reducing IL-6 production from peripheral whole blood in a phase 1, randomized, double-blind, placebo-controlled clinical trial (207).
TLR3, which responds to viral double-stranded RNA and is the only TLR not to signal through MyD88 (208), is expressed in human IBD tissues and stimulates the production of the antimicrobial peptide lipocalin-2 in the HT29 colonic epithelial cell line (209). Activation of TLR3 using poly(I:C) has been shown to ameliorate DSS colitis (210) through maintenance of epithelial barrier integrity (211). The enteric virome and an intact TLR3 signaling pathway is important in maintaining intestinal health. Treatment of wild-type mice with antiviral agents before administration of DSS results in worsened colitis, similar to TLR3−/−TLR7−/− double knockout, and this is probably mediated through effects on IFNβ production by plasmacytoid DCs (212). Preconditioning of human umbilical cord-derived mesenchymal stem cells with poly(I:C) enhanced the immunosuppressive effects in both TNBS (213) and DSS (214) models of colitis. Although not yet used in human IBD, the TLR3 agonist rintatolimod has benefits in chronic fatigue syndrome/myalgic encephalomyelitis (215, 216), a disease of uncertain etiology but with evidence of viral triggers to the disease and impaired natural killer cell function (217).
Poly(I:C) is rapidly hydrolyzed in serum but can be stabilized with poly-l-lysine and carboxymethylcellulose (polyICLC) to resist this (218). PolyICLC is under evaluation as part of cancer therapy as a vaccine adjunct and may have roles in modulating cross-presentation of antigens by APCs to naïve CD8+ lymphocytes [reviewed in Ref. (219)]. Although not yet used in models of intestinal inflammation, polyICLC is protective in models of infections caused by influenza (220) or Dengue virus (221), murine cryptococcosis (222), and has been shown to enhance T cell responses in the lung of non-human primates when coadministered with anti-CD40 (223). An alternative to PolyICLC is PIKA, a stabilized double-stranded RNA, which has been shown to promote the maturation of DCs (224) but has been studied less extensively.
In addition to the studies on SPA4 (187), alpinetin (188), and BML-111 (200) detailed above, other lines of enquiry suggest that modulation of TLR4 signaling may yield benefit in the management of intestinal inflammation. Unlike studies on TLR3 where enhanced signaling is effective the evidence with TLR4 is predominantly in blocking signaling. An in vitro study using SPA4 demonstrated a reduction in NF-κB-dependent cytokine production, migration, and invasion of the SW480 colonic cancer cell line suggesting that interfering with TLR4 signaling may be of benefit in colitis-associated cancer (225). To this end, transgenic mice with constitutively activated TLR4 are more susceptible to both DSS colitis and colitis-induced neoplasia (226) and the TLR4 antagonist 1A6 inhibits neoplasia in this model (226). Previously, it had been shown that 1A6 improved DSS colitis, but not the adoptive transfer model of colitis, although adverse effects on mucosal healing were noted (227). An alternative TLR4 antagonist, CRX-526, blocks the ability of LPS, the natural ligand for TLR4 (208), to induce pro-inflammatory cytokines and improves both DSS and multidrug resistance gene 1a-deficient models of colitis (228).
Infection of rhesus macaques with Shigella dysenteriae caused colitis, which was inhibited by oral administration of a small, non-absorbable polypropyletherimine dendrimer glucosamine (229), which acts to inhibit LPS signaling through TLR4–MD2 (230, 231).
The small molecule, TAK-242 (resatorvid), binds to the intracellular domain of TLR4 to inhibit signaling by interfering with the interaction between TLR4 and its adaptor molecules to inhibit NF-κB activation and interleukin-1 receptor-associated kinase (232). Two phase 3 clinical trials of resatorvid in severe sepsis have been performed (NCT00143611 and NCT00633477). Unfortunately, neither trial has been published nor the product was discontinued by the company leaving open the question as to its efficacy in intestinal inflammation.
Alkaline phosphatase has been shown to detoxify LPS by dephosphorylation of the lipid A component (233–235). The potential benefit of alkaline phosphatase in inflammation was demonstrated in mice and piglets challenged with LPS (236) and in zebrafish was shown to be important in mucosal immunity to gut microbiota (237). In patients with IBD (both ulcerative colitis and Crohn’s disease), there is reduced expression of alkaline phosphatase in both the inflamed and non-inflamed epithelium (238). Furthermore, oral administration of acid-resistant alkaline phosphatase (to prevent degradation in the stomach) to rats undergoing DSS challenge ameliorated colitis (238). An uncontrolled phase 2 trial of intraduodenal alkaline phosphatase in patients with ulcerative colitis demonstrated reductions in C-reactive protein, fecal calprotectin, and clinical activity indices with no particular safety concerns identified (239). Thus, modification of TLR4 signaling through detoxifying its primary ligand is promising in intestinal inflammation, and this has been the subject of a recent review article (240).
Contrary to the beneficial effects of the pharmacological inhibitors on TLR4-induced inflammation, there is some evidence, related to the pediatric condition necrotizing enterocolitis, that TLR4 signaling may be essential for controlling inflammation. Use of probiotic-conditioned media from Bifidobacterium longum subsp infantis prevents IL-6 induction in immature enterocytes but requires an intact TLR4 signaling pathway through TLR4–IRAK1–AP1 (241). Therefore, TLR4 inhibition may not be of benefit in all types of intestinal inflammation. A phase 2 clinical trial of Bifidobacterium infantis 35624 in maintenance of remission in ulcerative colitis was registered in 2007 but the outcome is not known (NCT00510978). However, a different strain of this bacterium (strain 24737) is a component of the probiotic VSL#3 which has been shown in a recent systematic review to be of benefit in inducing remission in ulcerative colitis (242).
Altering TLR4 expression has also shown promise for understanding and modulating intestinal inflammation. The colons of mice deficient in corticotropin-releasing factor express less TLR4 mRNA and develop a more severe colitis in response to DSS (243). The flavonoid Baicalin improves colitis to a similar degree as mesalazine with a reduction in colonic TLR4 as measured by immunohistochemistry and a reduction in NF-κB-dependent pro-inflammatory cytokine production (244). TLR4 expression is reduced in enteric glial cells by the endocannabinoid-related lipid ligand palmitoylethanolamine, in a PPARα-dependent manner, to inhibit NF-κB activation, which is associated with a reduction in severity of DSS colitis (245).
Although no clinical studies of TLR4 modulation in intestinal inflammation have yet been performed, the TLR4 antagonist eritoran tetrasodium (E5564) has been used in a phase 3, randomized, double-blind, placebo-controlled clinical trial of sepsis (246). Although there was no benefit of eritoran in mortality rates from severe sepsis, there were no differences in adverse events between groups and this may pave the way for further studies of TLR4 modulation for other conditions.
The role of TLR5 in intestinal inflammation is less clear. TLR5−/− mice develop a spontaneous colitis (247) and mice treated with purified Salmonella-derived flagellin as a TLR5 agonist are protected from Clostridium difficile colitis (248). Furthermore, the TLR5 agonist CBLB502 has antioxidant actions and scavenges oxygen free radicals (249) through which it may exert anti-inflammatory properties. However, flagellin enemas have been shown to exacerbate established DSS colitis (250) although in a non-TLR5-dependent manner (251), highlighting the need for further studies.
The TLR7 agonist imiquimod, administered orally, induces type-1 IFN responses in the colonic mucosa to ameliorate DSS colitis and has effects in vitro on antimicrobial peptide production (252). A further study has confirmed a similar benefit in the TNBS model of colitis extending the mechanism of action to regulation of regulatory T cells (253). The development of 2′-O-methyl modifications to RNA has been used to develop TLR7 antagonists (254, 255), which may improve understanding of the signaling pathway in intestinal inflammation.
TLR7, along with TLR8 and 9, is an endosomal PRR (256). Inhibition of both TLR7 and 9 with IMO-3100, an oligonucleotide antagonist, or of TLR7, 8 and 9 with IMO-8400, resulted in a decrease in IL17A expression in an IL-23-dependent murine model of skin inflammation (257). Similar effects have been seen in human peripheral blood mononuclear cells (258) and, specifically for IMO-8400, on the induction of psoriatic lesions by inhibiting Th1/17 cytokines (259). Short-term treatment of patients with psoriasis in a phase 2a clinical trial yielded clinical benefit without significant adverse events (260). It is plausible therefore that this approach may yield benefit in Crohn’s disease which is also associated with IL-12/23 dysfunction (261).
TLR9 detects bacterial immunostimulatory DNA sequences. Synthetic DNA oligonucleotides (ODNs) ameliorate colitis whether induced by DSS, hapten, or in the IL10−/− mouse (262). However, it appears that the effects of ODNs are dependent on time of administration relative to induction of colitis. ODNs given therapeutically after colitis has developed worsen disease whereas when given prophylactically there is a reduction in inflammation thought possible due to a tolerance effects on IFNγ or on increasing IL-10 production (263). The same group went on to show that intestinal inflammation following DSS challenge is reduced in TLR9−/− mice compared with wild-type and that administration of adenoviral ODNs (which blocks the effects of DNA sequences) to wild-type mice with established colitis results in amelioration of disease (264). The findings were replicated in the severe combined immunodeficiency disease transfer model of colitis and in the IL10−/− mouse and the authors conclude that DNA sequences from gut microbes perpetuate chronic inflammation through TLR9 and that adenoviral ODNs may be of benefit in intestinal inflammation (264). In vitro studies of the c41 ODN, from Pseudomonas aeruginosa, in a murine macrophage cell line and in human monocytes demonstrates that this sequence binds to TLR9 without triggering downstream cascades but prevents other ODNs from binding and so has uses as a TLR9 antagonist (265). A subsequent study demonstrated more diverse actions of c41 on TLR activation suggesting wider effects than simply TLR9 (266). Similar to c41, the antimalarial drug chloroquine has diverse effects on TLR signaling but particularly via TLR9 to alleviate murine colitis (267).
Unlike many of the TLRs, TLR9 modulation has been studied clinically in patients with ulcerative colitis using a TLR9 agonist, DIMS0150 (also known as cobitolimod/Kappaproct). A single dose instilled to the mucosa of the transverse or descending colon at colonoscopy resulted in 43% clinical response at week 1, 71% at week 4, and 86% at week 12 (268). Remission rates were slightly lower than response but similarly increased to week 12. Clinical scores were mirrored by endoscopic improvement. Note was made of improved steroid sensitivity in patients treated with DIMS0150. An analysis of steroid-response genes identified from the pilot study (CD163, TSP-1, IL-1RII) were validated in a prospective cohort of patients treated with rectal placebo or DIMS0150 demonstrating utility of this gene panel in identifying patients most likely to benefit from TLR9 agonist therapy (269). Unfortunately, a larger randomized, double-blind, placebo-controlled trial of 131 patients with ulcerative colitis failed to reach its primary endpoint to demonstrate benefit of DIMS0150 over placebo at week 12 although some benefits were seen at earlier timepoints including on mucosal healing at week 4 (270). The treatment was not associated with serious adverse events and, as the authors conclude, further trials are merited. A second trial is currently in the recruitment stages (NCT03178669).
Similar to DIMS0150, a synthetic oligonucleotide acting as a TLR9 agonist, BL-7040 (previously known as Monarsen and EN101), which can be administered orally may also have potential in the management of human IBD. Originally designed as a therapy for myasthenia gravis due to its effects on acetylcholinesterase transcripts (271), BL-7040 was shown to be an activator of TLR9 signaling to increase levels of indoleamine and IFNα (272). It has subsequently been demonstrated that this compound induces miRNA changes to activate the alternative pathway of NF-κB activation through TLR9 (273). These preclinical findings have been extended to a phase 2a trial in ulcerative colitis, which confirmed the safety of BL-7040 in intestinal inflammation and demonstrated an improvement in colitis in half of the patients who completed the full study protocol (274).
Modulation of signaling cascades downstream of TLRs themselves have also shown promise for intestinal inflammation. The MyD88 inhibitor TJ-M2010-5 improved azoxymethane/DSS colitis and consequent colitis-associated colonic cancer (275). Nur77 is a transcription factor which is associated, from GWAS, with IBD and interacts with TRAF6 to interfere with TLR–IL1-R signaling to inhibit NF-κB cytokine production (276). Cytosporone B is an agonist of Nur77 and ameliorates DSS colitis (276).
Apilimod inhibits the lipid kinase activity of phosphatidylinositol-3-phosphate-5-kinase (PIKfyve) resulting in inhibition of IL-12/23p40 (277). Although initially promising, a phase 2 trial of apilimod in 220 patients with active Crohn’s disease failed to show benefit over placebo, although was well tolerated (278). A novel PIKfyve inhibitor, APY0201, administered orally ameliorated intestinal inflammation in the IL10−/− cell transfer model and showed effects in vitro on IL-12/23 production in macrophages (279). A more in depth understanding of the PIKfyve pathway may yet yield opportunities for reducing intestinal inflammation.
The narrow spectrum kinase inhibitor TOP1288 targets p38, Src, Lck, and Syk and has been shown to have anti-inflammatory effects relevant for colitis both in vitro and in vivo (280, 281). Recently, a clinical trial of six patients with active ulcerative colitis despite oral mesalazine was conducted with TOP1288 (282). The drug had minimal systemic bioavailability, was well tolerated and demonstrated promising reductions in colonic IL-6 and 8. Other therapies involving modulation of innate immune cell function under evaluation for intestinal inflammation include laquinimod which exerts at least some effects through effects on antigen presentation (283, 284). A phase 2 clinical trial of laquinimod in Crohn’s disease was safe and demonstrated benefit on clinical remission especially at lower doses (285).
Pattern-recognition receptor-mediated control of innate immunity has a fundamental role in both mounting immune defense and maintaining intestinal homeostasis. In this review, we have summarized emerging key functions of this receptor class that, if dysregulated due to functional or genetic defects, may be responsible for the induction of intestinal inflammation such as IBD. Attempts are being made to manipulate PRR-directed pathways for therapeutic purposes (summarized in Table 1 and Figure 2). Although the number of clinical trials that have been performed using this approach is currently small there are a number of compounds in the preclinical evaluation stage. The results from trials performed to date have not been greatly successful in IBD. It is likely a better molecular understanding of PRR biology within human intestinal cells and in vivo models is required to harness the full potential of this approach clinically.
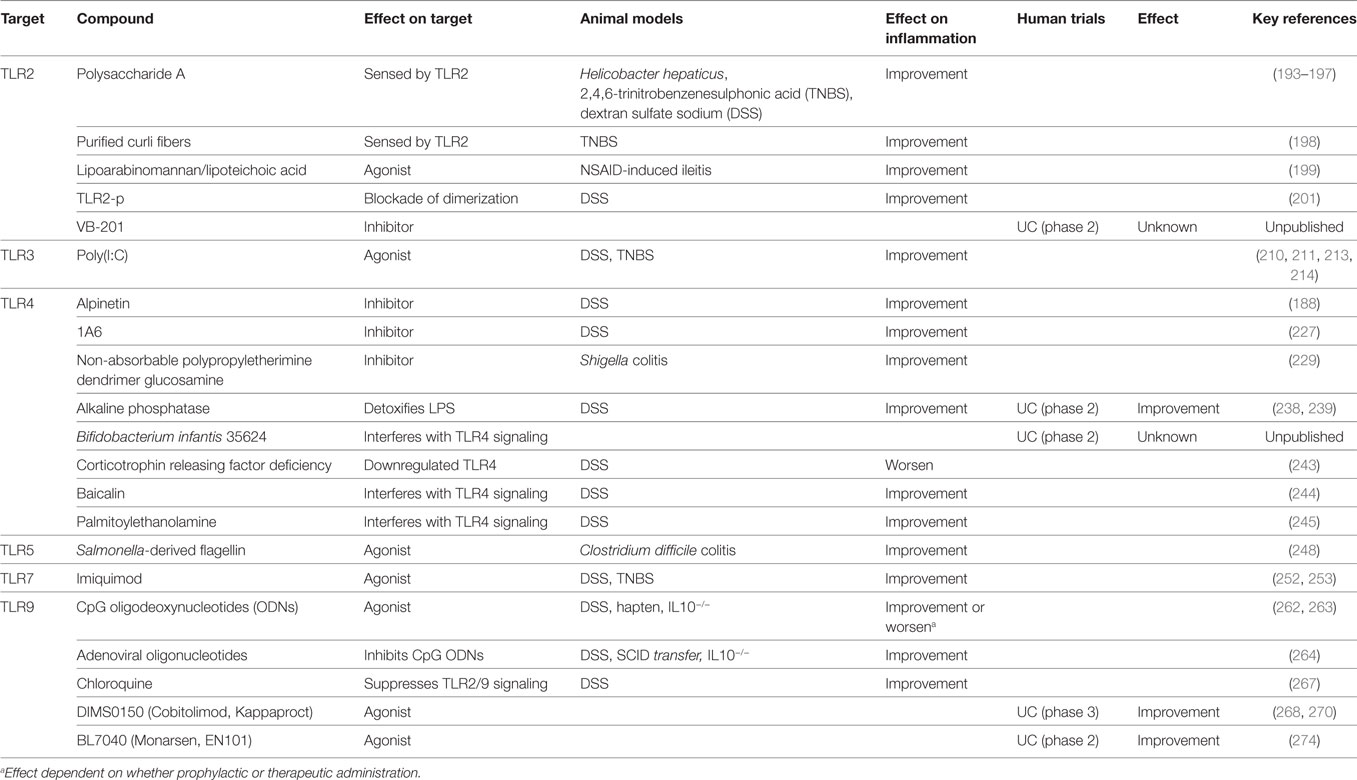
Table 1. A summary of compounds targeting Toll-like receptors to modulate animal and/or human intestinal inflammation.
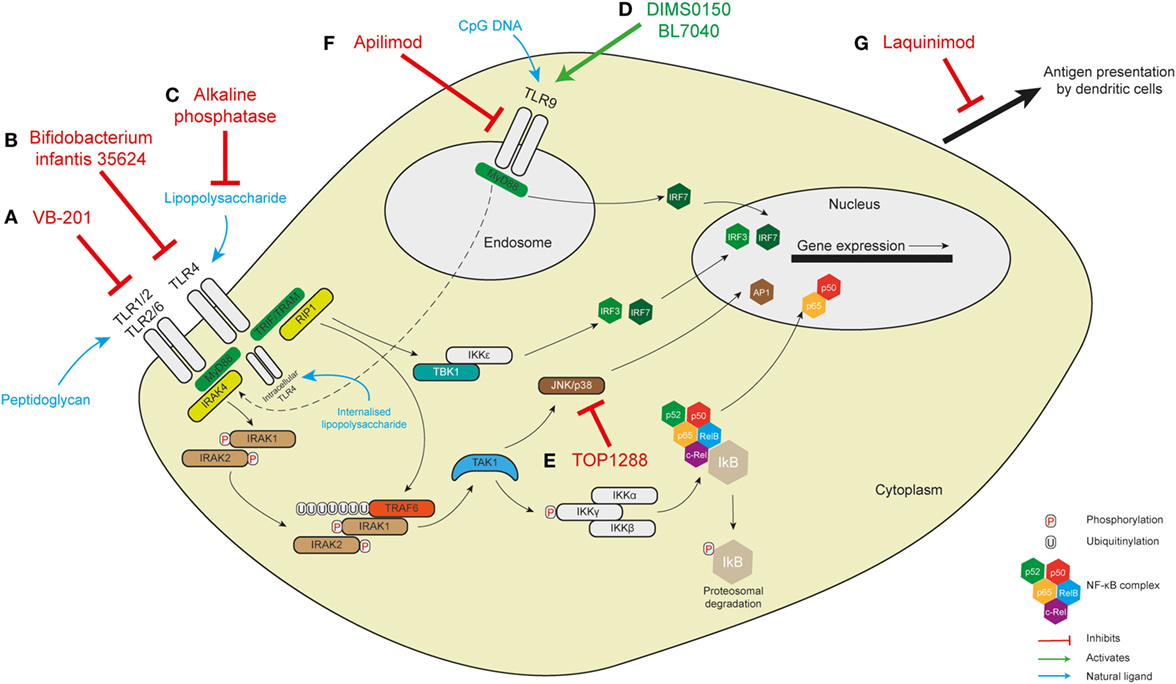
Figure 2. Targeting pattern-recognition receptor signaling in human intestinal inflammation. (A) The small molecule VB-201 interferes with downstream signaling from TLR2 and has been trialed in ulcerative colitis. (B) Similarly, Bifidobacterium infantis 35624, which probably acts to interfere with the TLR4 signaling pathway, has been trialed in maintaining remission in ulcerative colitis. The results of these two trials are not known. TLR4 is found on the cell membrane in immune cells but in intestinal epithelial cells has been demonstrated intracellularly where it responds to internalized lipopolysaccharide. (C) Alkaline phosphatase detoxifies LPS to inhibit TLR4 signaling and has been shown to be of benefit in ulcerative colitis. It is not known whether it might interfere with intracellular TLR4. Similar benefits have been seen with the TLR9 agonists DIMS0150 and BL7040 (D). (E) TOP1288, a narrow spectrum kinase inhibitor, has effects on p38, Src, Lck, and Syk to reduce colonic IL-6 and -8 in ulcerative colitis. (F) The exact role of apilimod in innate immune sensing is not well understood but there are effects on endosomal maturation and TLR9 sensing—this was not, however, effective in a trial in Crohn’s disease. (G) Laquinimod exerts some effects on antigen presentation by dendritic cells with some improvement in remission in Crohn’s disease.
All authors contributed equally to the conception and overview of the manuscript content. DC, TC, and TA contributed equally to the writing of the manuscript with AS providing intellectual content and oversight. All authors edited and approved the final version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the UK MRC and by a Wellcome Investigator Award (AS), an NIHR Research Professorship (AS), a Crohn’s and Colitis UK Medical Research Award (DC), and a Wellcome Clinical Training Fellowship (TC). We acknowledge Saara Chapman for assisting with the design of Figure 1, and the support of the Oxford NIHR Biomedical Research Centre.
DAMP, damage-associated molecular pattern; DCs, dendritic cells; DSS, dextran sulfate sodium; IBD, inflammatory bowel disease; IEC, intestinal epithelial cell; mtDNA, mitochondrial DNA; MDP, muramyl dipeptide; mROS, mitochondrial reactive oxygen species; NLR, NOD-like receptor; PRR, pattern-recognition receptor; TLR, toll-like receptor; TNBS, 2,4,6-trinitrobenzenesulphonic acid.
1. Kimbrell DA, Beutler B. The evolution and genetics of innate immunity. Nat Rev Genet (2001) 2(4):256–67. doi:10.1038/35066006
2. Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol (2002) 20:197–216. doi:10.1146/annurev.immunol.20.083001.084359
3. Janeway CA Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol (1989) 54(Pt 1):1–13. doi:10.1101/SQB.1989.054.01.003
4. Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science (1999) 285(5425):248–51. doi:10.1126/science.285.5425.248
5. Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature (2003) 425(6957):516–21. doi:10.1038/nature01991
6. Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/toll/cactus controls the potent antifungal response in Drosophila adults. Cell (1996) 86(6):973–83. doi:10.1016/S0092-8674(00)80172-5
7. Ishii KJ, Koyama S, Nakagawa A, Coban C, Akira S. Host innate immune receptors and beyond: making sense of microbial infections. Cell Host Microbe (2008) 3(6):352–63. doi:10.1016/j.chom.2008.05.003
8. Botos I, Segal DM, Davies DR. The structural biology of toll-like receptors. Structure (2011) 19(4):447–59. doi:10.1016/j.str.2011.02.004
9. Hoving JC, Wilson GJ, Brown GD. Signalling C-type lectin receptors, microbial recognition and immunity. Cell Microbiol (2014) 16(2):185–94. doi:10.1111/cmi.12249
10. Iliev ID, Funari VA, Taylor KD, Nguyen Q, Reyes CN, Strom SP, et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science (2012) 336(6086):1314–7. doi:10.1126/science.1221789
11. Seibold F, Konrad A, Flogerzi B, Seibold-Schmid B, Arni S, Juliger S, et al. Genetic variants of the mannan-binding lectin are associated with immune reactivity to mannans in Crohn’s disease. Gastroenterology (2004) 127(4):1076–84. doi:10.1053/j.gastro.2004.07.056
12. Muller S, Schaffer T, Flogerzi B, Seibold-Schmid B, Schnider J, Takahashi K, et al. Mannan-binding lectin deficiency results in unusual antibody production and excessive experimental colitis in response to mannose-expressing mild gut pathogens. Gut (2010) 59(11):1493–500. doi:10.1136/gut.2010.208348
13. Saba K, Denda-Nagai K, Irimura T. A C-type lectin MGL1/CD301a plays an anti-inflammatory role in murine experimental colitis. Am J Pathol (2009) 174(1):144–52. doi:10.2353/ajpath.2009.080235
14. Eriksson M, Johannssen T, von Smolinski D, Gruber AD, Seeberger PH, Lepenies B. The C-type lectin receptor SIGNR3 binds to fungi present in commensal microbiota and influences immune regulation in experimental colitis. Front Immunol (2013) 4:196. doi:10.3389/fimmu.2013.00196
15. Saunders SP, Barlow JL, Walsh CM, Bellsoi A, Smith P, McKenzie AN, et al. C-type lectin SIGN-R1 has a role in experimental colitis and responsiveness to lipopolysaccharide. J Immunol (2010) 184(5):2627–37. doi:10.4049/jimmunol.0901970
16. Inohara N, Chamaillard M, McDonald C, Nunez G. NOD-LRR proteins: role in host-microbial interactions and inflammatory disease. Annu Rev Biochem (2005) 74:355–83. doi:10.1146/annurev.biochem.74.082803.133347
17. Magalhaes JG, Sorbara MT, Girardin SE, Philpott DJ. What is new with Nods? Curr Opin Immunol (2011) 23(1):29–34. doi:10.1016/j.coi.2010.12.003
18. Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, et al. The NLR gene family: a standard nomenclature. Immunity (2008) 28(3):285–7. doi:10.1016/j.immuni.2008.02.005
19. Fritz JH, Kufer TA. Editorial: NLR-protein functions in immunity. Front Immunol (2015) 6:306. doi:10.3389/fimmu.2015.00306
20. Kufer TA, Sansonetti PJ. NLR functions beyond pathogen recognition. Nat Immunol (2011) 12(2):121–8. doi:10.1038/ni.1985
21. Maekawa T, Kufer TA, Schulze-Lefert P. NLR functions in plant and animal immune systems: so far and yet so close. Nat Immunol (2011) 12(9):817–26. doi:10.1038/ni.2083
22. Strober W, Murray PJ, Kitani A, Watanabe T. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat Rev Immunol (2006) 6(1):9–20. doi:10.1038/nri1747
23. Zhong Y, Kinio A, Saleh M. Functions of NOD-like receptors in human diseases. Front Immunol (2013) 4:333. doi:10.3389/fimmu.2013.00333
24. Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L, et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol (2003) 4(7):702–7. doi:10.1038/ni945
25. Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science (2003) 300(5625):1584–7. doi:10.1126/science.1084677
26. Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem (2003) 278(11):8869–72. doi:10.1074/jbc.C200651200
27. Caruso R, Warner N, Inohara N, Nunez G. NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity (2014) 41(6):898–908. doi:10.1016/j.immuni.2014.12.010
28. Keestra AM, Baumler AJ. Detection of enteric pathogens by the nodosome. Trends Immunol (2014) 35(3):123–30. doi:10.1016/j.it.2013.10.009
29. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature (2001) 411(6837):599–603. doi:10.1038/35079107
30. Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature (2001) 411(6837):603–6. doi:10.1038/35079114
31. Barnich N, Aguirre JE, Reinecker HC, Xavier R, Podolsky DK. Membrane recruitment of NOD2 in intestinal epithelial cells is essential for nuclear factor-{kappa}B activation in muramyl dipeptide recognition. J Cell Biol (2005) 170(1):21–6. doi:10.1083/jcb.200502153
32. Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol (2010) 11(1):55–62. doi:10.1038/ni.1823
33. Philpott DJ, Sorbara MT, Robertson SJ, Croitoru K, Girardin SE. NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol (2014) 14(1):9–23. doi:10.1038/nri3565
34. Hu Z, Yan C, Liu P, Huang Z, Ma R, Zhang C, et al. Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science (2013) 341(6142):172–5. doi:10.1126/science.1236381
35. Nigro G, Fazio LL, Martino MC, Rossi G, Tattoli I, Liparoti V, et al. Muramylpeptide shedding modulates cell sensing of Shigella flexneri. Cell Microbiol (2008) 10(3):682–95. doi:10.1111/j.1462-5822.2007.01075.x
36. Bielig H, Rompikuntal PK, Dongre M, Zurek B, Lindmark B, Ramstedt M, et al. NOD-like receptor activation by outer membrane vesicles from Vibrio cholerae non-O1 non-O139 strains is modulated by the quorum-sensing regulator HapR. Infect Immun (2011) 79(4):1418–27. doi:10.1128/IAI.00754-10
37. Nakamura N, Lill JR, Phung Q, Jiang Z, Bakalarski C, de Maziere A, et al. Endosomes are specialized platforms for bacterial sensing and NOD2 signalling. Nature (2014) 509(7499):240–4. doi:10.1038/nature13133
38. Boyle JP, Parkhouse R, Monie TP. Insights into the molecular basis of the NOD2 signalling pathway. Open Biol (2014) 4(12):140178. doi:10.1098/rsob.140178
39. Kobayashi K, Inohara N, Hernandez LD, Galan JE, Nunez G, Janeway CA, et al. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature (2002) 416(6877):194–9. doi:10.1038/416194a
40. Park JH, Kim YG, McDonald C, Kanneganti TD, Hasegawa M, Body-Malapel M, et al. RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J Immunol (2007) 178(4):2380–6. doi:10.4049/jimmunol.178.4.2380
41. Grimes CL, Ariyananda Lde Z, Melnyk JE, O’Shea EK. The innate immune protein Nod2 binds directly to MDP, a bacterial cell wall fragment. J Am Chem Soc (2012) 134(33):13535–7. doi:10.1021/ja303883c
42. Mo J, Boyle JP, Howard CB, Monie TP, Davis BK, Duncan JA. Pathogen sensing by nucleotide-binding oligomerization domain-containing protein 2 (NOD2) is mediated by direct binding to muramyl dipeptide and ATP. J Biol Chem (2012) 287(27):23057–67. doi:10.1074/jbc.M112.344283
43. Hasegawa M, Fujimoto Y, Lucas PC, Nakano H, Fukase K, Nunez G, et al. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-kappaB activation. EMBO J (2008) 27(2):373–83. doi:10.1038/sj.emboj.7601962
44. Hsu YM, Zhang Y, You Y, Wang D, Li H, Duramad O, et al. The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat Immunol (2007) 8(2):198–205. doi:10.1038/ni1426
45. Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol (2009) 10(10):1073–80. doi:10.1038/ni.1782
46. Marinis JM, Homer CR, McDonald C, Abbott DW. A novel motif in the Crohn’s disease susceptibility protein, NOD2, allows TRAF4 to down-regulate innate immune responses. J Biol Chem (2011) 286(3):1938–50. doi:10.1074/jbc.M110.189308
47. Hitotsumatsu O, Ahmad RC, Tavares R, Wang M, Philpott D, Turer EE, et al. The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity (2008) 28(3):381–90. doi:10.1016/j.immuni.2008.02.002
48. McDonald C, Chen FF, Ollendorff V, Ogura Y, Marchetto S, Lecine P, et al. A role for Erbin in the regulation of Nod2-dependent NF-kappaB signaling. J Biol Chem (2005) 280(48):40301–9. doi:10.1074/jbc.M508538200
49. Eitel J, Krull M, Hocke AC, N’Guessan PD, Zahlten J, Schmeck B, et al. Beta-PIX and Rac1 GTPase mediate trafficking and negative regulation of NOD2. J Immunol (2008) 181(4):2664–71. doi:10.4049/jimmunol.181.4.2664
50. Morosky SA, Zhu J, Mukherjee A, Sarkar SN, Coyne CB. Retinoic acid-induced gene-I (RIG-I) associates with nucleotide-binding oligomerization domain-2 (NOD2) to negatively regulate inflammatory signaling. J Biol Chem (2011) 286(32):28574–83. doi:10.1074/jbc.M111.227942
51. Barnich N, Hisamatsu T, Aguirre JE, Xavier R, Reinecker HC, Podolsky DK. GRIM-19 interacts with nucleotide oligomerization domain 2 and serves as downstream effector of anti-bacterial function in intestinal epithelial cells. J Biol Chem (2005) 280(19):19021–6. doi:10.1074/jbc.M413776200
52. Stevens C, Henderson P, Nimmo ER, Soares DC, Dogan B, Simpson KW, et al. The intermediate filament protein, vimentin, is a regulator of NOD2 activity. Gut (2013) 62(5):695–707. doi:10.1136/gutjnl-2011-301775
53. Zhao Y, Alonso C, Ballester I, Song JH, Chang SY, Guleng B, et al. Control of NOD2 and Rip2-dependent innate immune activation by GEF-H1. Inflamm Bowel Dis (2012) 18(4):603–12. doi:10.1002/ibd.21851
54. Cao X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat Rev Immunol (2016) 16(1):35–50. doi:10.1038/nri.2015.8
55. Ferwerda G, Girardin SE, Kullberg BJ, Le Bourhis L, de Jong DJ, Langenberg DM, et al. NOD2 and toll-like receptors are nonredundant recognition systems of Mycobacterium tuberculosis. PLoS Pathog (2005) 1(3):e34. doi:10.1371/journal.ppat.0010034
56. Netea MG, Ferwerda G, de Jong DJ, Jansen T, Jacobs L, Kramer M, et al. Nucleotide-binding oligomerization domain-2 modulates specific TLR pathways for the induction of cytokine release. J Immunol (2005) 174(10):6518–23. doi:10.4049/jimmunol.174.10.6518
57. Tada H, Aiba S, Shibata K, Ohteki T, Takada H. Synergistic effect of Nod1 and Nod2 agonists with toll-like receptor agonists on human dendritic cells to generate interleukin-12 and T helper type 1 cells. Infect Immun (2005) 73(12):7967–76. doi:10.1128/IAI.73.12.7967-7976.2005
58. Ghorpade DS, Kaveri SV, Bayry J, Balaji KN. Cooperative regulation of NOTCH1 protein-phosphatidylinositol 3-kinase (PI3K) signaling by NOD1, NOD2, and TLR2 receptors renders enhanced refractoriness to transforming growth factor-beta (TGF-beta)- or cytotoxic T-lymphocyte antigen 4 (CTLA-4)-mediated impairment of human dendritic cell maturation. J Biol Chem (2011) 286(36):31347–60. doi:10.1074/jbc.M111.232413
59. Mills EL, Kelly B, O’Neill LAJ. Mitochondria are the powerhouses of immunity. Nat Immunol (2017) 18(5):488–98. doi:10.1038/ni.3704
60. Garaude J, Acin-Perez R, Martinez-Cano S, Enamorado M, Ugolini M, Nistal-Villan E, et al. Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat Immunol (2016) 17(9):1037–45. doi:10.1038/ni.3509
61. Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity (2015) 42(3):406–17. doi:10.1016/j.immuni.2015.02.002
62. Seth RB, Sun LJ, Ea CK, Chen ZJJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappa B and IRF3. Cell (2005) 122(5):669–82. doi:10.1016/j.cell.2005.08.012
63. Castanier C, Garcin D, Vazquez A, Arnoult D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep (2010) 11(2):133–8. doi:10.1038/embor.2009.258
64. Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci U S A (2009) 106(8):2770–5. doi:10.1073/pnas.0807694106
65. Park S, Juliana C, Hong S, Datta P, Hwang I, Fernandes-Alnemri T, et al. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J Immunol (2013) 191(8):4358–66. doi:10.4049/jimmunol.1301170
66. Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity (2013) 39(2):311–23. doi:10.1016/j.immuni.2013.08.001
67. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464(7285):104–7. doi:10.1038/nature08780
68. West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol (2017) 17(6):363–75. doi:10.1038/nri.2017.21
69. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol (2011) 12(3):222–30. doi:10.1038/ni.1980
70. Jabir MS, Hopkins L, Ritchie ND, Ullah I, Bayes HK, Li D, et al. Mitochondrial damage contributes to Pseudomonas aeruginosa activation of the inflammasome and is downregulated by autophagy. Autophagy (2015) 11(1):166–82. doi:10.4161/15548627.2014.981915
71. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature (2015) 520(7548):553–7. doi:10.1038/nature14156
72. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med (2016) 22(2):146–53. doi:10.1038/nm.4027
73. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469(7329):221–5. doi:10.1038/nature09663
74. Delpre G, Avidor I, Steinherz R, Kadish U, Ben-Bassat M. Ultrastructural abnormalities in endoscopically and histologically normal and involved colon in ulcerative colitis. Am J Gastroenterol (1989) 84(9):1038–46.
75. Rodenburg W, Keijer J, Kramer E, Vink C, van der Meer R, Bovee-Oudenhoven IM. Impaired barrier function by dietary fructo-oligosaccharides (FOS) in rats is accompanied by increased colonic mitochondrial gene expression. BMC Genomics (2008) 9:144. doi:10.1186/1471-2164-9-144
76. Beltran B, Nos P, Dasi F, Iborra M, Bastida G, Martinez M, et al. Mitochondrial dysfunction, persistent oxidative damage, and catalase inhibition in immune cells of naive and treated Crohn’s disease. Inflamm Bowel Dis (2010) 16(1):76–86. doi:10.1002/ibd.21027
77. Mottawea W, Chiang CK, Muhlbauer M, Starr AE, Butcher J, Abujamel T, et al. Altered intestinal microbiota-host mitochondria crosstalk in new onset Crohn’s disease. Nat Commun (2016) 7:13419. doi:10.1038/ncomms13419
78. Novak EA, Mollen KP. Mitochondrial dysfunction in inflammatory bowel disease. Front Cell Dev Biol (2015) 3:62. doi:10.3389/fcell.2015.00062
79. Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet (2008) 40(8):955–62. doi:10.1038/ng.175
80. Shekhawat PS, Srinivas SR, Matern D, Bennett MJ, Boriack R, George V, et al. Spontaneous development of intestinal and colonic atrophy and inflammation in the carnitine-deficient jvs (OCTN2(-/-)) mice. Mol Genet Metab (2007) 92(4):315–24. doi:10.1016/j.ymgme.2007.08.002
81. Bar F, Bochmann W, Widok A, Von Medem K, Pagel R, Hirose M, et al. Mitochondrial gene polymorphisms that protect mice from colitis. Gastroenterology (2013) 145(5):1055. doi:10.1053/j.gastro.2013.07.015
82. Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med (2010) 16(1):90–7. doi:10.1038/nm.2069
83. Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res (2014) 24(1):24–41. doi:10.1038/cr.2013.168
84. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature (2008) 451(7182):1069–75. doi:10.1038/nature06639
85. Lassen KG, Xavier RJ. Genetic control of autophagy underlies pathogenesis of inflammatory bowel disease. Mucosal Immunol (2017) 10(3):589–97. doi:10.1038/mi.2017.18
86. Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe (2009) 5(6):527–49. doi:10.1016/j.chom.2009.05.016
87. Shibutani ST, Saitoh T, Nowag H, Munz C, Yoshimori T. Autophagy and autophagy-related proteins in the immune system. Nat Immunol (2015) 16(10):1014–24. doi:10.1038/ni.3273
88. Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell (2010) 141(4):656–67. doi:10.1016/j.cell.2010.04.009
89. Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol (2010) 12(9):831–5. doi:10.1038/ncb0910-831
90. Klionsky DJ, Schulman BA. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat Struct Mol Biol (2014) 21(4):336–45. doi:10.1038/nsmb.2787
91. Klionsky DJ, Cregg JM, Dunn WA Jr, Emr SD, Sakai Y, Sandoval IV, et al. A unified nomenclature for yeast autophagy-related genes. Dev Cell (2003) 5(4):539–45. doi:10.1016/S1534-5807(03)00296-X
92. Kraft C, Kijanska M, Kalie E, Siergiejuk E, Lee SS, Semplicio G, et al. Binding of the Atg1/ULK1 kinase to the ubiquitin-like protein Atg8 regulates autophagy. EMBO J (2012) 31(18):3691–703. doi:10.1038/emboj.2012.225
93. Noda NN, Kumeta H, Nakatogawa H, Satoo K, Adachi W, Ishii J, et al. Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells (2008) 13(12):1211–8. doi:10.1111/j.1365-2443.2008.01238.x
94. Ponpuak M, Davis AS, Roberts EA, Delgado MA, Dinkins C, Zhao Z, et al. Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity (2010) 32(3):329–41. doi:10.1016/j.immuni.2010.02.009
95. Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol (2009) 10(11):1215–21. doi:10.1038/ni.1800
96. Mostowy S, Sancho-Shimizu V, Hamon MA, Simeone R, Brosch R, Johansen T, et al. p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J Biol Chem (2011) 286(30):26987–95. doi:10.1074/jbc.M111.223610
97. Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science (2011) 333(6039):228–33. doi:10.1126/science.1205405
98. Amer AO, Swanson MS. Autophagy is an immediate macrophage response to Legionella pneumophila. Cell Microbiol (2005) 7(6):765–78. doi:10.1111/j.1462-5822.2005.00509.x
99. Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J (2010) 29(11):1792–802. doi:10.1038/emboj.2010.74
100. Berg TO, Fengsrud M, Stromhaug PE, Berg T, Seglen PO. Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J Biol Chem (1998) 273(34):21883–92. doi:10.1074/jbc.273.34.21883
101. Klionsky DJ, Eskelinen EL, Deretic V. Autophagosomes, phagosomes, autolysosomes, phagolysosomes, autophagolysosomes… wait, I’m confused. Autophagy (2014) 10(4):549–51. doi:10.4161/auto.28448
102. Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A (2011) 108(42):17396–401. doi:10.1073/pnas.1113421108
103. Nakamura S, Yoshimori T. New insights into autophagosome-lysosome fusion. J Cell Sci (2017) 130(7):1209–16. doi:10.1242/jcs.196352
104. Wang H, Sun HQ, Zhu X, Zhang L, Albanesi J, Levine B, et al. GABARAPs regulate PI4P-dependent autophagosome:lysosome fusion. Proc Natl Acad Sci U S A (2015) 112(22):7015–20. doi:10.1073/pnas.1507263112
105. Grootjans J, Kaser A, Kaufman RJ, Blumberg RS. The unfolded protein response in immunity and inflammation. Nat Rev Immunol (2016) 16(8):469–84. doi:10.1038/nri.2016.62
106. Pincus D, Chevalier MW, Aragon T, van Anken E, Vidal SE, El-Samad H, et al. BiP binding to the ER-stress sensor Ire1 tunes the homeostatic behavior of the unfolded protein response. PLoS Biol (2010) 8(7):e1000415. doi:10.1371/journal.pbio.1000415
107. Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature (1999) 397(6716):271–4. doi:10.1038/16729
108. Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell (2000) 6(5):1099–108. doi:10.1016/S1097-2765(00)00108-8
109. Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, et al. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell (2001) 7(6):1165–76. doi:10.1016/S1097-2765(01)00265-9
110. Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol (2003) 23(21):7448–59. doi:10.1128/MCB.23.21.7448-7459.2003
111. Ishii A, Kawai T, Tekura K, Oshida H, Nakayama J. A convenient method for the generation of a disulfur monoxide equivalent and its reaction with diazoalkanes to yield dithiirane 1-oxides this work was supported by a Grant-in-Aid for Scientific Research (No. 90193242) from the Ministry of Education, Science, Sports, and Culture, Japan. Angew Chem Int Ed Engl (2001) 40(10):1924–6. doi:10.1002/1521-3773(20010518)40:10<1924::AID-ANIE1924>3.0.CO;2-F
112. Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, et al. The unfolded protein response signals through high-order assembly of Ire1. Nature (2009) 457(7230):687–93. doi:10.1038/nature07661
113. Hassler JR, Scheuner DL, Wang S, Han J, Kodali VK, Li P, et al. The IRE1alpha/XBP1s pathway is essential for the glucose response and protection of beta cells. PLoS Biol (2015) 13(10):e1002277. doi:10.1371/journal.pbio.1002277
114. Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, et al. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature (2011) 473(7348):528–31. doi:10.1038/nature09968
115. Volmer R, van der Ploeg K, Ron D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci U S A (2013) 110(12):4628–33. doi:10.1073/pnas.1217611110
116. Talloczy Z, Jiang W, Virgin HWT, Leib DA, Scheuner D, Kaufman RJ, et al. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A (2002) 99(1):190–5. doi:10.1073/pnas.012485299
117. Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ (2007) 14(2):230–9. doi:10.1038/sj.cdd.4401984
118. Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J, et al. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell (2007) 13(3):351–64. doi:10.1016/j.devcel.2007.07.005
119. Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol (2010) 11(5):411–8. doi:10.1038/ni.1857
120. Kim S, Joe Y, Kim HJ, Kim YS, Jeong SO, Pae HO, et al. Endoplasmic reticulum stress-induced IRE1alpha activation mediates cross-talk of GSK-3beta and XBP-1 to regulate inflammatory cytokine production. J Immunol (2015) 194(9):4498–506. doi:10.4049/jimmunol.1401399
121. Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet (2009) 41(6):703–7. doi:10.1038/ng.381
122. Van Limbergen J, Wilson DC, Satsangi J. The genetics of Crohn’s disease. Annu Rev Genomics Hum Genet (2009) 10:89–116. doi:10.1146/annurev-genom-082908-150013
123. Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol (2010) 28:573–621. doi:10.1146/annurev-immunol-030409-101225
124. McGovern DP, Gardet A, Torkvist L, Goyette P, Essers J, Taylor KD, et al. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat Genet (2010) 42(4):332–7. doi:10.1038/ng.549
125. Zhao F, Edwards R, Dizon D, Afrasiabi K, Mastroianni JR, Geyfman M, et al. Disruption of Paneth and goblet cell homeostasis and increased endoplasmic reticulum stress in Agr2-/- mice. Dev Biol (2010) 338(2):270–9. doi:10.1016/j.ydbio.2009.12.008
126. Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell (2008) 134(5):743–56. doi:10.1016/j.cell.2008.07.021
127. Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Bock J, Martinez-Naves E, et al. Paneth cells as a site of origin for intestinal inflammation. Nature (2013) 503(7475):272–6. doi:10.1038/nature12599
128. Deuring JJ, Fuhler GM, Konstantinov SR, Peppelenbosch MP, Kuipers EJ, de Haar C, et al. Genomic ATG16L1 risk allele-restricted Paneth cell ER stress in quiescent Crohn’s disease. Gut (2014) 63(7):1081–91. doi:10.1136/gutjnl-2012-303527
129. Reis e Sousa C. Dendritic cells in a mature age. Nat Rev Immunol (2006) 6(6):476–83. doi:10.1038/nri1845
130. Heath WR, Carbone FR. Cross-presentation, dendritic cells, tolerance and immunity. Annu Rev Immunol (2001) 19:47–64. doi:10.1146/annurev.immunol.19.1.47
131. van Beelen AJ, Zelinkova Z, Taanman-Kueter EW, Muller FJ, Hommes DW, Zaat SA, et al. Stimulation of the intracellular bacterial sensor NOD2 programs dendritic cells to promote interleukin-17 production in human memory T cells. Immunity (2007) 27(4):660–9. doi:10.1016/j.immuni.2007.08.013
132. Brain O, Owens BM, Pichulik T, Allan P, Khatamzas E, Leslie A, et al. The intracellular sensor NOD2 induces microRNA-29 expression in human dendritic cells to limit IL-23 release. Immunity (2013) 39(3):521–36. doi:10.1016/j.immuni.2013.08.035
133. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A (2006) 103(33):12481–6. doi:10.1073/pnas.0605298103
134. Tili E, Michaille JJ, Cimino A, Costinean S, Dumitru CD, Adair B, et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol (2007) 179(8):5082–9. doi:10.4049/jimmunol.179.8.5082
135. Bazzoni F, Rossato M, Fabbri M, Gaudiosi D, Mirolo M, Mori L, et al. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc Natl Acad Sci U S A (2009) 106(13):5282–7. doi:10.1073/pnas.0810909106
136. Ceppi M, Pereira PM, Dunand-Sauthier I, Barras E, Reith W, Santos MA, et al. MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proc Natl Acad Sci U S A (2009) 106(8):2735–40. doi:10.1073/pnas.0811073106
137. Sheedy FJ, Palsson-McDermott E, Hennessy EJ, Martin C, O’Leary JJ, Ruan Q, et al. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat Immunol (2010) 11(2):141–7. doi:10.1038/ni.1828
138. Fritz JH, Le Bourhis L, Sellge G, Magalhaes JG, Fsihi H, Kufer TA, et al. Nod1-mediated innate immune recognition of peptidoglycan contributes to the onset of adaptive immunity. Immunity (2007) 26(4):445–59. doi:10.1016/j.immuni.2007.03.009
139. Magalhaes JG, Fritz JH, Le Bourhis L, Sellge G, Travassos LH, Selvanantham T, et al. Nod2-dependent Th2 polarization of antigen-specific immunity. J Immunol (2008) 181(11):7925–35. doi:10.4049/jimmunol.181.11.7925
140. Duan W, Mehta AK, Magalhaes JG, Ziegler SF, Dong C, Philpott DJ, et al. Innate signals from Nod2 block respiratory tolerance and program T(H)2-driven allergic inflammation. J Allergy Clin Immunol (2010) 126(6):1284–1293.e1210. doi:10.1016/j.jaci.2010.09.021
141. Magalhaes JG, Lee J, Geddes K, Rubino S, Philpott DJ, Girardin SE. Essential role of Rip2 in the modulation of innate and adaptive immunity triggered by Nod1 and Nod2 ligands. Eur J Immunol (2011) 41(5):1445–55. doi:10.1002/eji.201040827
142. Corridoni D, Rodriguez-Palacios A, Di Stefano G, Di Martino L, Antonopoulos DA, Chang EB, et al. Genetic deletion of the bacterial sensor NOD2 improves murine Crohn’s disease-like ileitis independent of functional dysbiosis. Mucosal Immunol (2017) 10(4):971–82. doi:10.1038/mi.2016.98
143. Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol (2012) 12(8):557–69. doi:10.1038/nri3254
144. Burgdorf S, Scholz C, Kautz A, Tampe R, Kurts C. Spatial and mechanistic separation of cross-presentation and endogenous antigen presentation. Nat Immunol (2008) 9(5):558–66. doi:10.1038/ni.1601
145. Shen L, Sigal LJ, Boes M, Rock KL. Important role of cathepsin S in generating peptides for TAP-independent MHC class I crosspresentation in vivo. Immunity (2004) 21(2):155–65. doi:10.1016/j.immuni.2004.07.004
146. Houde M, Bertholet S, Gagnon E, Brunet S, Goyette G, Laplante A, et al. Phagosomes are competent organelles for antigen cross-presentation. Nature (2003) 425(6956):402–6. doi:10.1038/nature01912
147. Kovacsovics-Bankowski M, Rock KL. A phagosome-to-cytosol pathway for exogenous antigens presented on MHC class I molecules. Science (1995) 267(5195):243–6. doi:10.1126/science.7809629
148. Nair-Gupta P, Baccarini A, Tung N, Seyffer F, Florey O, Huang Y, et al. TLR signals induce phagosomal MHC-I delivery from the endosomal recycling compartment to allow cross-presentation. Cell (2014) 158(3):506–21. doi:10.1016/j.cell.2014.04.054
149. Blander JM. The comings and goings of MHC class I molecules herald a new dawn in cross-presentation. Immunol Rev (2016) 272(1):65–79. doi:10.1111/imr.12428
150. Gil-Torregrosa BC, Lennon-Dumenil AM, Kessler B, Guermonprez P, Ploegh HL, Fruci D, et al. Control of cross-presentation during dendritic cell maturation. Eur J Immunol (2004) 34(2):398–407. doi:10.1002/eji.200324508
151. Alloatti A, Kotsias F, Magalhaes JG, Amigorena S. Dendritic cell maturation and cross-presentation: timing matters! Immunol Rev (2016) 272(1):97–108. doi:10.1111/imr.12432
152. Wang T, Hong W. Interorganellar regulation of lysosome positioning by the Golgi apparatus through Rab34 interaction with Rab-interacting lysosomal protein. Mol Biol Cell (2002) 13(12):4317–32. doi:10.1091/mbc.E02-05-0280
153. Kasmapour B, Gronow A, Bleck CK, Hong W, Gutierrez MG. Size-dependent mechanism of cargo sorting during lysosome-phagosome fusion is controlled by Rab34. Proc Natl Acad Sci U S A (2012) 109(50):20485–90. doi:10.1073/pnas.1206811109
154. Wilson NS, Behrens GM, Lundie RJ, Smith CM, Waithman J, Young L, et al. Systemic activation of dendritic cells by toll-like receptor ligands or malaria infection impairs cross-presentation and antiviral immunity. Nat Immunol (2006) 7(2):165–72. doi:10.1038/ni1300
155. Weck MM, Grunebach F, Werth D, Sinzger C, Bringmann A, Brossart P. TLR ligands differentially affect uptake and presentation of cellular antigens. Blood (2007) 109(9):3890–4. doi:10.1182/blood-2006-04-015719
156. Corridoni D, Simmons A. Innate immune receptors for cross-presentation: the expanding role of NLRs. Mol Immunol (2017). doi:10.1016/j.molimm.2017.11.028
157. Sokolovska A, Becker CE, Ip WK, Rathinam VA, Brudner M, Paquette N, et al. Activation of caspase-1 by the NLRP3 inflammasome regulates the NADPH oxidase NOX2 to control phagosome function. Nat Immunol (2013) 14(6):543–53. doi:10.1038/ni.2595
158. Asano J, Tada H, Onai N, Sato T, Horie Y, Fujimoto Y, et al. Nucleotide oligomerization binding domain-like receptor signaling enhances dendritic cell-mediated cross-priming in vivo. J Immunol (2010) 184(2):736–45. doi:10.4049/jimmunol.0900726
159. Wagner CS, Cresswell P. TLR and nucleotide-binding oligomerization domain-like receptor signals differentially regulate exogenous antigen presentation. J Immunol (2012) 188(2):686–93. doi:10.4049/jimmunol.1102214
160. Lee JC, Lyons PA, McKinney EF, Sowerby JM, Carr EJ, Bredin F, et al. Gene expression profiling of CD8+ T cells predicts prognosis in patients with Crohn disease and ulcerative colitis. J Clin Invest (2011) 121(10):4170–9. doi:10.1172/JCI59255
161. McGovern DP, Hysi P, Ahmad T, van Heel DA, Moffatt MF, Carey A, et al. Association between a complex insertion/deletion polymorphism in NOD1 (CARD4) and susceptibility to inflammatory bowel disease. Hum Mol Genet (2005) 14(10):1245–50. doi:10.1093/hmg/ddi135
162. Kim JG, Lee SJ, Kagnoff MF. Nod1 is an essential signal transducer in intestinal epithelial cells infected with bacteria that avoid recognition by toll-like receptors. Infect Immun (2004) 72(3):1487–95. doi:10.1128/IAI.72.3.1487-1495.2004
163. Zilbauer M, Dorrell N, Elmi A, Lindley KJ, Schuller S, Jones HE, et al. A major role for intestinal epithelial nucleotide oligomerization domain 1 (NOD1) in eliciting host bactericidal immune responses to Campylobacter jejuni. Cell Microbiol (2007) 9(10):2404–16. doi:10.1111/j.1462-5822.2007.00969.x
164. Geddes K, Rubino S, Streutker C, Cho JH, Magalhaes JG, Le Bourhis L, et al. Nod1 and Nod2 regulation of inflammation in the Salmonella colitis model. Infect Immun (2010) 78(12):5107–15. doi:10.1128/IAI.00759-10
165. Chen GY, Shaw MH, Redondo G, Nunez G. The innate immune receptor Nod1 protects the intestine from inflammation-induced tumorigenesis. Cancer Res (2008) 68(24):10060–7. doi:10.1158/0008-5472.CAN-08-2061
166. Magnuson G, Khan P, Yuan H, Brown B, Divlianska DB, Stonich D, et al. High throughput screening assays for NOD1 inhibitors – probe 2. Probe Reports from the NIH Molecular Libraries Program. Bethesda, MD (2010).
167. Correa RG, Khan PM, Askari N, Zhai D, Gerlic M, Brown B, et al. Discovery and characterization of 2-aminobenzimidazole derivatives as selective NOD1 inhibitors. Chem Biol (2011) 18(7):825–32. doi:10.1016/j.chembiol.2011.06.009
168. Khan PM, Correa RG, Divlianska DB, Peddibhotla S, Sessions EH, Magnuson G, et al. Identification of inhibitors of NOD1-induced nuclear factor-kappaB activation. ACS Med Chem Lett (2011) 2(10):780–5. doi:10.1021/ml200158b
169. Jakopin Z. Nucleotide-binding oligomerization domain (NOD) inhibitors: a rational approach toward inhibition of NOD signaling pathway. J Med Chem (2014) 57(16):6897–918. doi:10.1021/jm401841p
170. Lesage S, Zouali H, Cezard JP, Colombel JF, Belaiche J, Almer S, et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet (2002) 70(4):845–57. doi:10.1086/339432
171. Hampe J, Cuthbert A, Croucher PJ, Mirza MM, Mascheretti S, Fisher S, et al. Association between insertion mutation in NOD2 gene and Crohn’s disease in German and British populations. Lancet (2001) 357(9272):1925–8. doi:10.1016/S0140-6736(00)05063-7
172. Linde K, Boor PP, Houwing-Duistermaat JJ, Kuipers EJ, Wilson JH, de Rooij FW. Card15 and Crohn’s disease: healthy homozygous carriers of the 3020insC frameshift mutation. Am J Gastroenterol (2003) 98(3):613–7. doi:10.1111/j.1572-0241.2003.07287.x
173. Gutierrez O, Pipaon C, Inohara N, Fontalba A, Ogura Y, Prosper F, et al. Induction of Nod2 in myelomonocytic and intestinal epithelial cells via nuclear factor-kappa B activation. J Biol Chem (2002) 277(44):41701–5. doi:10.1074/jbc.M206473200
174. Hisamatsu T, Suzuki M, Reinecker HC, Nadeau WJ, McCormick BA, Podolsky DK. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology (2003) 124(4):993–1000. doi:10.1053/gast.2003.50153
175. Bielig H, Velder J, Saiai A, Menning M, Meemboor S, Kalka-Moll W, et al. Anti-inflammatory arene – chromium complexes acting as specific inhibitors of NOD2 signalling. ChemMedChem (2010) 5(12):2065–71. doi:10.1002/cmdc.201000320
176. Rickard DJ, Sehon CA, Kasparcova V, Kallal LA, Zeng X, Montoute MN, et al. Identification of benzimidazole diamides as selective inhibitors of the nucleotide-binding oligomerization domain 2 (NOD2) signaling pathway. PLoS One (2013) 8(8):e69619. doi:10.1371/journal.pone.0069619
177. Tigno-Aranjuez JT, Benderitter P, Rombouts F, Deroose F, Bai X, Mattioli B, et al. In vivo inhibition of RIPK2 kinase alleviates inflammatory disease. J Biol Chem (2014) 289(43):29651–64. doi:10.1074/jbc.M114.591388
178. Canning P, Ruan Q, Schwerd T, Hrdinka M, Maki JL, Saleh D, et al. Inflammatory signaling by NOD-RIPK2 is inhibited by clinically relevant type II kinase inhibitors. Chem Biol (2015) 22(9):1174–84. doi:10.1016/j.chembiol.2015.07.017
179. Nachbur U, Stafford CA, Bankovacki A, Zhan YF, Lindqvist LM, Fiil BK, et al. A RIPK2 inhibitor delays NOD signalling events yet prevents inflammatory cytokine production. Nat Commun (2015) 6:6442. doi:10.1038/ncomms7442
180. Villani AC, Lemire M, Fortin G, Louis E, Silverberg MS, Collette C, et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat Genet (2009) 41(1):71–6. doi:10.1038/ng.285
181. Lewis GJ, Massey DC, Zhang H, Bredin F, Tremelling M, Lee JC, et al. Genetic association between NLRP3 variants and Crohn’s disease does not replicate in a large UK panel. Inflamm Bowel Dis (2011) 17(6):1387–91. doi:10.1002/ibd.21499
182. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity (2010) 32(3):379–91. doi:10.1016/j.immuni.2010.03.003
183. Lv J, Zhang Y, Tian Z, Liu F, Shi Y, Liu Y, et al. Astragalus polysaccharides protect against dextran sulfate sodium-induced colitis by inhibiting NF-kappacapital VE, Cyrillic activation. Int J Biol Macromol (2017) 98:723–9. doi:10.1016/j.ijbiomac.2017.02.024
184. Tian Z, Liu Y, Yang B, Zhang J, He H, Ge H, et al. Astagalus polysaccharide attenuates murine colitis through inhibiton of the NLRP3 inflammasome. Planta Med (2017) 83(1–02):70–7. doi:10.1055/s-0042-108589
185. Cocco M, Pellegrini C, Martinez-Banaclocha H, Giorgis M, Marini E, Costale A, et al. Development of an acrylate derivative targeting the NLRP3 inflammasome for the treatment of inflammatory bowel disease. J Med Chem (2017) 60(9):3656–71. doi:10.1021/acs.jmedchem.6b01624
186. Wang X, Wang S, Hu C, Chen W, Shen Y, Wu X, et al. A new pharmacological effect of levornidazole: inhibition of NLRP3 inflammasome activation. Biochem Pharmacol (2015) 97(2):178–88. doi:10.1016/j.bcp.2015.06.030
187. Ramani V, Awasthi S. Toll-like receptor 4-interacting SPA4 peptide suppresses the NLRP3 inflammasome in response to LPS and ATP stimuli. J Leukoc Biol (2015) 98(6):1037–48. doi:10.1189/jlb.3A1114-570R
188. He X, Wei Z, Wang J, Kou J, Liu W, Fu Y, et al. Alpinetin attenuates inflammatory responses by suppressing TLR4 and NLRP3 signaling pathways in DSS-induced acute colitis. Sci Rep (2016) 6:28370. doi:10.1038/srep28370
189. Jiang H, He H, Chen Y, Huang W, Cheng J, Ye J, et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med (2017) 214(11):3219–38. doi:10.1084/jem.20171419
190. Pellegrini C, Antonioli L, Lopez-Castejon G, Blandizzi C, Fornai M. Canonical and non-canonical activation of NLRP3 inflammasome at the crossroad between immune tolerance and intestinal inflammation. Front Immunol (2017) 8:36. doi:10.3389/fimmu.2017.00036
191. Cario E, Gerken G, Podolsky DK. Toll-like receptor 2 controls mucosal inflammation by regulating epithelial barrier function. Gastroenterology (2007) 132(4):1359–74. doi:10.1053/j.gastro.2007.02.056
192. Ey B, Eyking A, Klepak M, Salzman NH, Gothert JR, Runzi M, et al. Loss of TLR2 worsens spontaneous colitis in MDR1A deficiency through commensally induced pyroptosis. J Immunol (2013) 190(11):5676–88. doi:10.4049/jimmunol.1201592
193. Wang Q, McLoughlin RM, Cobb BA, Charrel-Dennis M, Zaleski KJ, Golenbock D, et al. A bacterial carbohydrate links innate and adaptive responses through toll-like receptor 2. J Exp Med (2006) 203(13):2853–63. doi:10.1084/jem.20062008
194. Dasgupta S, Erturk-Hasdemir D, Ochoa-Reparaz J, Reinecker HC, Kasper DL. Plasmacytoid dendritic cells mediate anti-inflammatory responses to a gut commensal molecule via both innate and adaptive mechanisms. Cell Host Microbe (2014) 15(4):413–23. doi:10.1016/j.chom.2014.03.006
195. Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature (2008) 453(7195):620–5. doi:10.1038/nature07008
196. Chiu CC, Ching YH, Wang YC, Liu JY, Li YP, Huang YT, et al. Monocolonization of germ-free mice with Bacteroides fragilis protects against dextran sulfate sodium-induced acute colitis. Biomed Res Int (2014) 2014:675786. doi:10.1155/2014/675786
197. Chang YC, Ching YH, Chiu CC, Liu JY, Hung SW, Huang WC, et al. TLR2 and interleukin-10 are involved in Bacteroides fragilis-mediated prevention of DSS-induced colitis in gnotobiotic mice. PLoS One (2017) 12(7):e0180025. doi:10.1371/journal.pone.0180025
198. Oppong GO, Rapsinski GJ, Tursi SA, Biesecker SG, Klein-Szanto AJ, Goulian M, et al. Biofilm-associated bacterial amyloids dampen inflammation in the gut: oral treatment with curli fibres reduces the severity of hapten-induced colitis in mice. NPJ Biofilms Microbiomes (2015) 1:15019. doi:10.1038/npjbiofilms.2015.19
199. Narimatsu K, Higashiyama M, Kurihara C, Takajo T, Maruta K, Yasutake Y, et al. Toll-like receptor (TLR) 2 agonists ameliorate indomethacin-induced murine ileitis by suppressing the TLR4 signaling. J Gastroenterol Hepatol (2015) 30(11):1610–7. doi:10.1111/jgh.12980
200. Liu H, Liu Z, Zhao S, Sun C, Yang M. Effect of BML-111 on the intestinal mucosal barrier in sepsis and its mechanism of action. Mol Med Rep (2015) 12(2):3101–6. doi:10.3892/mmr.2015.3746
201. Shmuel-Galia L, Aychek T, Fink A, Porat Z, Zarmi B, Bernshtein B, et al. Neutralization of pro-inflammatory monocytes by targeting TLR2 dimerization ameliorates colitis. EMBO J (2016) 35(6):685–98. doi:10.15252/embj.201592649
202. Mistry P, Laird MH, Schwarz RS, Greene S, Dyson T, Snyder GA, et al. Inhibition of TLR2 signaling by small molecule inhibitors targeting a pocket within the TLR2 TIR domain. Proc Natl Acad Sci U S A (2015) 112(17):5455–60. doi:10.1073/pnas.1422576112
203. Mendel I, Feige E, Yacov N, Salem Y, Levi I, Propheta-Meiran O, et al. VB-201, an oxidized phospholipid small molecule, inhibits CD14- and toll-like receptor-2-dependent innate cell activation and constrains atherosclerosis. Clin Exp Immunol (2014) 175(1):126–37. doi:10.1111/cei.12212
204. Mendel I, Shoham A, Propheta-Meiran O, Ishai E, Halperin G, Feige E, et al. A Lecinoxoid, an oxidized phospholipid small molecule, constrains CNS autoimmune disease. J Neuroimmunol (2010) 226(1–2):126–35. doi:10.1016/j.jneuroim.2010.06.011
205. Feige E, Yacov N, Salem Y, Levi I, Mendel I, Propheta-Meiran O, et al. Inhibition of monocyte chemotaxis by VB-201, a small molecule lecinoxoid, hinders atherosclerosis development in ApoE(-/-) mice. Atherosclerosis (2013) 229(2):430–9. doi:10.1016/j.atherosclerosis.2013.06.005
206. Arslan F, Houtgraaf JH, Keogh B, Kazemi K, de Jong R, McCormack WJ, et al. Treatment with OPN-305, a humanized anti-toll-like receptor-2 antibody, reduces myocardial ischemia/reperfusion injury in pigs. Circ Cardiovasc Interv (2012) 5(2):279–87. doi:10.1161/CIRCINTERVENTIONS.111.967596
207. Reilly M, Miller RM, Thomson MH, Patris V, Ryle P, McLoughlin L, et al. Randomized, double-blind, placebo-controlled, dose-escalating phase I, healthy subjects study of intravenous OPN-305, a humanized anti-TLR2 antibody. Clin Pharmacol Ther (2013) 94(5):593–600. doi:10.1038/clpt.2013.150
208. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell (2006) 124(4):783–801. doi:10.1016/j.cell.2006.02.015
209. Ostvik AE, Granlund AV, Torp SH, Flatberg A, Beisvag V, Waldum HL, et al. Expression of toll-like receptor-3 is enhanced in active inflammatory bowel disease and mediates the excessive release of lipocalin 2. Clin Exp Immunol (2013) 173(3):502–11. doi:10.1111/cei.12136
210. Vijay-Kumar M, Wu H, Aitken J, Kolachala VL, Neish AS, Sitaraman SV, et al. Activation of toll-like receptor 3 protects against DSS-induced acute colitis. Inflamm Bowel Dis (2007) 13(7):856–64. doi:10.1002/ibd.20142
211. Zhao HW, Yue YH, Han H, Chen XL, Lu YG, Zheng JM, et al. Effect of toll-like receptor 3 agonist poly I:C on intestinal mucosa and epithelial barrier function in mouse models of acute colitis. World J Gastroenterol (2017) 23(6):999–1009. doi:10.3748/wjg.v23.i6.999
212. Yang JY, Kim MS, Kim E, Cheon JH, Lee YS, Kim Y, et al. Enteric viruses ameliorate gut inflammation via toll-like receptor 3 and toll-like receptor 7-mediated interferon-beta production. Immunity (2016) 44(4):889–900. doi:10.1016/j.immuni.2016.03.009
213. Qiu Y, Guo J, Mao R, Chao K, Chen BL, He Y, et al. TLR3 preconditioning enhances the therapeutic efficacy of umbilical cord mesenchymal stem cells in TNBS-induced colitis via the TLR3-Jagged-1-Notch-1 pathway. Mucosal Immunol (2017) 10(3):727–42. doi:10.1038/mi.2016.78
214. Fuenzalida P, Kurte M, Fernandez-O’ryan C, Ibanez C, Gauthier-Abeliuk M, Vega-Letter AM, et al. Toll-like receptor 3 pre-conditioning increases the therapeutic efficacy of umbilical cord mesenchymal stromal cells in a dextran sulfate sodium-induced colitis model. Cytotherapy (2016) 18(5):630–41. doi:10.1016/j.jcyt.2016.02.002
215. Strayer DR, Carter WA, Stouch BC, Stevens SR, Bateman L, Cimoch PJ, et al. A double-blind, placebo-controlled, randomized, clinical trial of the TLR-3 agonist rintatolimod in severe cases of chronic fatigue syndrome. PLoS One (2012) 7(3):e31334. doi:10.1371/journal.pone.0031334
216. Mitchell WM. Efficacy of rintatolimod in the treatment of chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME). Expert Rev Clin Pharmacol (2016) 9(6):755–70. doi:10.1586/17512433.2016.1172960
217. Navaneetharaja N, Griffiths V, Wileman T, Carding SR. A role for the intestinal microbiota and virome in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS)? J Clin Med (2016) 5(6):E55. doi:10.3390/jcm5060055
218. Levy HB, Baer G, Baron S, Buckler CE, Gibbs CJ, Iadarola MJ, et al. A modified polyriboinosinic-polyribocytidylic acid complex that induces interferon in primates. J Infect Dis (1975) 132(4):434–9. doi:10.1093/infdis/132.4.434
219. Ammi R, De Waele J, Willemen Y, Van Brussel I, Schrijvers DM, Lion E, et al. Poly(I:C) as cancer vaccine adjuvant: knocking on the door of medical breakthroughs. Pharmacol Ther (2015) 146:120–31. doi:10.1016/j.pharmthera.2014.09.010
220. Wong JP, Christopher ME, Viswanathan S, Dai X, Salazar AM, Sun LQ, et al. Antiviral role of toll-like receptor-3 agonists against seasonal and avian influenza viruses. Curr Pharm Des (2009) 15(11):1269–74. doi:10.2174/138161209787846775
221. Hu Y, Hu Y, Sun L, Wong J, Wang M. Antiviral effects of liposome-encapsulated PolyICLC against dengue virus in a mouse model. Biochem Biophys Res Commun (2016) 478(2):913–8. doi:10.1016/j.bbrc.2016.08.050
222. Sionov E, Mayer-Barber KD, Chang YC, Kauffman KD, Eckhaus MA, Salazar AM, et al. Type I IFN induction via poly-ICLC protects mice against cryptococcosis. PLoS Pathog (2015) 11(8):e1005040. doi:10.1371/journal.ppat.1005040
223. Thompson EA, Liang F, Lindgren G, Sandgren KJ, Quinn KM, Darrah PA, et al. Human anti-CD40 antibody and poly IC:LC adjuvant combination induces potent T cell responses in the lung of nonhuman primates. J Immunol (2015) 195(3):1015–24. doi:10.4049/jimmunol.1500078
224. Lau YF, Tang LH, Ooi EE. A TLR3 ligand that exhibits potent inhibition of influenza virus replication and has strong adjuvant activity has the potential for dual applications in an influenza pandemic. Vaccine (2009) 27(9):1354–64. doi:10.1016/j.vaccine.2008.12.048
225. Rakhesh M, Cate M, Vijay R, Shrikant A, Shanjana A. A TLR4-interacting peptide inhibits lipopolysaccharide-stimulated inflammatory responses, migration and invasion of colon cancer SW480 cells. Oncoimmunology (2012) 1(9):1495–506. doi:10.4161/onci.22089
226. Fukata M, Shang L, Santaolalla R, Sotolongo J, Pastorini C, Espana C, et al. Constitutive activation of epithelial TLR4 augments inflammatory responses to mucosal injury and drives colitis-associated tumorigenesis. Inflamm Bowel Dis (2011) 17(7):1464–73. doi:10.1002/ibd.21527
227. Ungaro R, Fukata M, Hsu D, Hernandez Y, Breglio K, Chen A, et al. A novel toll-like receptor 4 antagonist antibody ameliorates inflammation but impairs mucosal healing in murine colitis. Am J Physiol Gastrointest Liver Physiol (2009) 296(6):G1167–79. doi:10.1152/ajpgi.90496.2008
228. Fort MM, Mozaffarian A, Stover AG, Correia Jda S, Johnson DA, Crane RT, et al. A synthetic TLR4 antagonist has anti-inflammatory effects in two murine models of inflammatory bowel disease. J Immunol (2005) 174(10):6416–23. doi:10.4049/jimmunol.174.10.6416
229. Islam D, Lombardini E, Ruamsap N, Imerbsin R, Khantapura P, Teo I, et al. Controlling the cytokine storm in severe bacterial diarrhoea with an oral toll-like receptor 4 antagonist. Immunology (2016) 147(2):178–89. doi:10.1111/imm.12549
230. Shaunak S, Thomas S, Gianasi E, Godwin A, Jones E, Teo I, et al. Polyvalent dendrimer glucosamine conjugates prevent scar tissue formation. Nat Biotechnol (2004) 22(8):977–84. doi:10.1038/nbt995
231. Barata TS, Teo I, Brocchini S, Zloh M, Shaunak S. Partially glycosylated dendrimers block MD-2 and prevent TLR4-MD-2-LPS complex mediated cytokine responses. PLoS Comput Biol (2011) 7(6):e1002095. doi:10.1371/journal.pcbi.1002095
232. Matsunaga N, Tsuchimori N, Matsumoto T, Ii M. TAK-242 (resatorvid), a small-molecule inhibitor of toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol Pharmacol (2011) 79(1):34–41. doi:10.1124/mol.110.068064
233. Poelstra K, Bakker WW, Klok PA, Hardonk MJ, Meijer DK. A physiologic function for alkaline phosphatase: endotoxin detoxification. Lab Invest (1997) 76(3):319–27.
234. Poelstra K, Bakker WW, Klok PA, Kamps JA, Hardonk MJ, Meijer DK. Dephosphorylation of endotoxin by alkaline phosphatase in vivo. Am J Pathol (1997) 151(4):1163–9.
235. Bentala H, Verweij WR, Huizinga-Van der Vlag A, van Loenen-Weemaes AM, Meijer DK, Poelstra K. Removal of phosphate from lipid A as a strategy to detoxify lipopolysaccharide. Shock (2002) 18(6):561–6. doi:10.1097/00024382-200212000-00013
236. Beumer C, Wulferink M, Raaben W, Fiechter D, Brands R, Seinen W. Calf intestinal alkaline phosphatase, a novel therapeutic drug for lipopolysaccharide (LPS)-mediated diseases, attenuates LPS toxicity in mice and piglets. J Pharmacol Exp Ther (2003) 307(2):737–44. doi:10.1124/jpet.103.056606
237. Bates JM, Akerlund J, Mittge E, Guillemin K. Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe (2007) 2(6):371–82. doi:10.1016/j.chom.2007.10.010
238. Tuin A, Poelstra K, de Jager-Krikken A, Bok L, Raaben W, Velders MP, et al. Role of alkaline phosphatase in colitis in man and rats. Gut (2009) 58(3):379–87. doi:10.1136/gut.2007.128868
239. Lukas M, Drastich P, Konecny M, Gionchetti P, Urban O, Cantoni F, et al. Exogenous alkaline phosphatase for the treatment of patients with moderate to severe ulcerative colitis. Inflamm Bowel Dis (2010) 16(7):1180–6. doi:10.1002/ibd.21161
240. Bilski J, Mazur-Bialy A, Wojcik D, Zahradnik-Bilska J, Brzozowski B, Magierowski M, et al. The role of intestinal alkaline phosphatase in inflammatory disorders of gastrointestinal tract. Mediators Inflamm (2017) 2017:9074601. doi:10.1155/2017/9074601
241. Meng D, Zhu W, Ganguli K, Shi HN, Walker WA. Anti-inflammatory effects of Bifidobacterium longum subsp infantis secretions on fetal human enterocytes are mediated by TLR-4 receptors. Am J Physiol Gastrointest Liver Physiol (2016) 311(4):G744–53. doi:10.1152/ajpgi.00090.2016
242. Derwa Y, Gracie DJ, Hamlin PJ, Ford AC. Systematic review with meta-analysis: the efficacy of probiotics in inflammatory bowel disease. Aliment Pharmacol Ther (2017) 46(4):389–400. doi:10.1111/apt.14203
243. Chaniotou Z, Giannogonas P, Theoharis S, Teli T, Gay J, Savidge T, et al. Corticotropin-releasing factor regulates TLR4 expression in the colon and protects mice from colitis. Gastroenterology (2010) 139(6):2083–92. doi:10.1053/j.gastro.2010.08.024
244. Feng J, Guo C, Zhu Y, Pang L, Yang Z, Zou Y, et al. Baicalin down regulates the expression of TLR4 and NFkB-p65 in colon tissue in mice with colitis induced by dextran sulfate sodium. Int J Clin Exp Med (2014) 7(11):4063–72.
245. Esposito G, Capoccia E, Turco F, Palumbo I, Lu J, Steardo A, et al. Palmitoylethanolamide improves colon inflammation through an enteric glia/toll like receptor 4-dependent PPAR-alpha activation. Gut (2014) 63(8):1300–12. doi:10.1136/gutjnl-2013-305005
246. Opal SM, Laterre PF, Francois B, LaRosa SP, Angus DC, Mira JP, et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA (2013) 309(11):1154–62. doi:10.1001/jama.2013.2194
247. Vijay-Kumar M, Sanders CJ, Taylor RT, Kumar A, Aitken JD, Sitaraman SV, et al. Deletion of TLR5 results in spontaneous colitis in mice. J Clin Invest (2007) 117(12):3909–21. doi:10.1172/JCI33084
248. Jarchum I, Liu M, Lipuma L, Pamer EG. Toll-like receptor 5 stimulation protects mice from acute Clostridium difficile colitis. Infect Immun (2011) 79(4):1498–503. doi:10.1128/IAI.01196-10
249. Li W, Ge C, Yang L, Wang R, Lu Y, Gao Y, et al. CBLB502, an agonist of toll-like receptor 5, has antioxidant and scavenging free radicals activities in vitro. Int J Biol Macromol (2016) 82:97–103. doi:10.1016/j.ijbiomac.2015.10.033
250. Choi YJ, Im E, Chung HK, Pothoulakis C, Rhee SH. TRIF mediates toll-like receptor 5-induced signaling in intestinal epithelial cells. J Biol Chem (2010) 285(48):37570–8. doi:10.1074/jbc.M110.158394
251. Ivison SM, Himmel ME, Hardenberg G, Wark PA, Kifayet A, Levings MK, et al. TLR5 is not required for flagellin-mediated exacerbation of DSS colitis. Inflamm Bowel Dis (2010) 16(3):401–9. doi:10.1002/ibd.21097
252. Sainathan SK, Bishnupuri KS, Aden K, Luo Q, Houchen CW, Anant S, et al. Toll-like receptor-7 ligand imiquimod induces type I interferon and antimicrobial peptides to ameliorate dextran sodium sulfate-induced acute colitis. Inflamm Bowel Dis (2012) 18(5):955–67. doi:10.1002/ibd.21867
253. Suzuki R, Katakura K, Fujiwara T, Gunji N, Watanabe H, Ohira H. Imiquimod-induced CCR9 ameliorates murine TNBS colitis. Fukushima J Med Sci (2016) 62(2):90–100. doi:10.5387/fms.2015-28
254. Robbins M, Judge A, Liang L, McClintock K, Yaworski E, MacLachlan I. 2’-O-methyl-modified RNAs act as TLR7 antagonists. Mol Ther (2007) 15(9):1663–9. doi:10.1038/sj.mt.6300240
255. Sioud M. Development of TLR7/8 small RNA antagonists. Methods Mol Biol (2010) 629:387–94. doi:10.1007/978-1-60761-657-3_25
256. Gay NJ, Symmons MF, Gangloff M, Bryant CE. Assembly and localization of toll-like receptor signalling complexes. Nat Rev Immunol (2014) 14(8):546–58. doi:10.1038/nri3713
257. Suarez-Farinas M, Arbeit R, Jiang W, Ortenzio FS, Sullivan T, Krueger JG. Suppression of molecular inflammatory pathways by toll-like receptor 7, 8, and 9 antagonists in a model of IL-23-induced skin inflammation. PLoS One (2013) 8(12):e84634. doi:10.1371/journal.pone.0084634
258. Jiang WW, Bhagat L, Yu D, Kandimalla ER, Agrawal S. IMO-3100, an antagonist of toll-like receptors 7 and 9, modulates gene expression and regulatory networks induced by ligands. J Immunol (2009) 182(1 Suppl):48.25.
259. Jiang WV, Zhu FG, Yu D, Kandimalla ER, La Monica N, Agrawal S. A selective inhibitor of endosomal toll-like receptors, IMO-8400, suppresses activation of multiple cytokines, Th17 response and inflammasome activation. Arthritis Rheum (2012) 64(10):S461–461.
260. Balak DM, van Doorn MB, Arbeit RD, Rijneveld R, Klaassen E, Sullivan T, et al. IMO-8400, a toll-like receptor 7, 8, and 9 antagonist, demonstrates clinical activity in a phase 2a, randomized, placebo-controlled trial in patients with moderate-to-severe plaque psoriasis. Clin Immunol (2017) 174:63–72. doi:10.1016/j.clim.2016.09.015
261. Simon EG, Ghosh S, Iacucci M, Moran GW. Ustekinumab for the treatment of Crohn’s disease: can it find its niche? Therap Adv Gastroenterol (2016) 9(1):26–36. doi:10.1177/1756283X15618130
262. Rachmilewitz D, Karmeli F, Takabayashi K, Hayashi T, Leider-Trejo L, Lee J, et al. Immunostimulatory DNA ameliorates experimental and spontaneous murine colitis. Gastroenterology (2002) 122(5):1428–41. doi:10.1053/gast.2002.32994
263. Obermeier F, Dunger N, Strauch UG, Grunwald N, Herfarth H, Scholmerich J, et al. Contrasting activity of cytosin-guanosin dinucleotide oligonucleotides in mice with experimental colitis. Clin Exp Immunol (2003) 134(2):217–24. doi:10.1046/j.1365-2249.2003.02288.x
264. Obermeier F, Dunger N, Strauch UG, Hofmann C, Bleich A, Grunwald N, et al. CpG motifs of bacterial DNA essentially contribute to the perpetuation of chronic intestinal inflammation. Gastroenterology (2005) 129(3):913–27. doi:10.1053/j.gastro.2005.06.061
265. Li Y, Cao H, Wang N, Xiang Y, Lu Y, Zhao K, et al. A novel antagonist of TLR9 blocking all classes of immunostimulatory CpG-ODNs. Vaccine (2011) 29(11):2193–8. doi:10.1016/j.vaccine.2010.10.042
266. Liu W, Yang X, Wang N, Fan S, Zhu Y, Zheng X, et al. Multiple immunosuppressive effects of CpG-c41 on intracellular TLR-mediated inflammation. Mediators Inflamm (2017) 2017:6541729. doi:10.1155/2017/6541729
267. Nagar J, Ranade S, Kamath V, Singh S, Karunanithi P, Subramani S, et al. Therapeutic potential of chloroquine in a murine model of inflammatory bowel disease. Int Immunopharmacol (2014) 21(2):328–35. doi:10.1016/j.intimp.2014.05.005
268. Musch E, Lutfi T, von Stein P, Zargari A, Admyre C, Malek M, et al. Topical treatment with the toll-like receptor agonist DIMS0150 has potential for lasting relief of symptoms in patients with chronic active ulcerative colitis by restoring glucocorticoid sensitivity. Inflamm Bowel Dis (2013) 19(2):283–92. doi:10.1002/ibd.23019
269. Kuznetsov NV, Zargari A, Gielen AW, von Stein OD, Musch E, Befrits R, et al. Biomarkers can predict potential clinical responders to DIMS0150 a toll-like receptor 9 agonist in ulcerative colitis patients. BMC Gastroenterol (2014) 14:79. doi:10.1186/1471-230X-14-79
270. Atreya R, Bloom S, Scaldaferri F, Gerardi V, Admyre C, Karlsson A, et al. Clinical effects of a topically applied toll-like receptor 9 agonist in active moderate-to-severe ulcerative colitis. J Crohns Colitis (2016) 10(11):1294–302. doi:10.1093/ecco-jcc/jjw103
271. Sussman JD, Argov Z, McKee D, Hazum E, Brawer S, Soreq H. Antisense treatment for myasthenia gravis: experience with monarsen. Ann N Y Acad Sci (2008) 1132:283–90. doi:10.1196/annals.1405.022
272. Gilboa-Geffen A, Wolf Y, Hanin G, Melamed-Book N, Pick M, Bennett ER, et al. Activation of the alternative NFkappaB pathway improves disease symptoms in a model of Sjogren’s syndrome. PLoS One (2011) 6(12):e28727. doi:10.1371/journal.pone.0028727
273. Nadorp B, Soreq H. Gut feeling: microRNA discriminators of the intestinal TLR9-cholinergic links. Int Immunopharmacol (2015) 29(1):8–14. doi:10.1016/j.intimp.2015.04.058
274. Dotan I, Levy-Nissenbaum E, Chowers Y, Fich A, Israeli E, Adar T, et al. Ameliorating active ulcerative colitis via an orally available toll-like receptor-9 modifier: a prospective open-label, multicenter phase II trial. Dig Dis Sci (2016) 61(11):3246–54. doi:10.1007/s10620-016-4276-1
275. Xie L, Jiang FC, Zhang LM, He WT, Liu JH, Li MQ, et al. Targeting of MyD88 homodimerization by novel synthetic inhibitor TJ-M2010-5 in preventing colitis-associated colorectal cancer. J Natl Cancer Inst (2016) 108(4):djv364. doi:10.1093/jnci/djv364
276. Wu H, Li XM, Wang JR, Gan WJ, Jiang FQ, Liu Y, et al. NUR77 exerts a protective effect against inflammatory bowel disease by negatively regulating the TRAF6/TLR-IL-1R signalling axis. J Pathol (2016) 238(3):457–69. doi:10.1002/path.4670
277. Cai X, Xu Y, Cheung AK, Tomlinson RC, Alcazar-Roman A, Murphy L, et al. PIKfyve, a class III PI kinase, is the target of the small molecular IL-12/IL-23 inhibitor apilimod and a player in toll-like receptor signaling. Chem Biol (2013) 20(7):912–21. doi:10.1016/j.chembiol.2013.05.010
278. Sands BE, Jacobson EW, Sylwestrowicz T, Younes Z, Dryden G, Fedorak R, et al. Randomized, double-blind, placebo-controlled trial of the oral interleukin-12/23 inhibitor apilimod mesylate for treatment of active Crohn’s disease. Inflamm Bowel Dis (2010) 16(7):1209–18. doi:10.1002/ibd.21159
279. Andou A, Yamamoto T, Agung E, Seki Y, Nishio H, Shuto M, et al. A novel orally active phosphatidylinositol 3-phosphate 5-kinase (pikfyve) inhibitor ameliorates mouse colitis by inhibition of IL-12 and IL-23 production from macrophages. Gastroenterology (2012) 142(5):S864–864. doi:10.1016/S0016-5085(12)63351-4
280. Foster M, Biancheri P, Fyfe M, MacDonald T, Rowley A, Sirohi S, et al. The pharmacological profile of TOP1288, a narrow spectrum kinase inhibitor in clinical development as an anti-inflammatory treatment for ulcerative colitis. J Crohns Colitis (2017) 11:S95–95. doi:10.1093/ecco-jcc/jjx002.158
281. Solanke Y, Foster M, Fyfe M, Rowley A, Sirohi S, Webber S, et al. The narrow spectrum kinase inhibitor TOP1288 demonstrates potent anti-inflammatory effects in a T cell adoptive transfer colitis model through a topical mode of action. J Crohns Colitis (2017) 11:S108–9. doi:10.1093/ecco-jcc/jjx002.188
282. Taylor M, Duggal A, Rowley A, Foster M, Sirohi S, Solanke Y, et al. A first clinical trial of a novel narrow spectrum kinase inhibitor TOP1288 in patients with ulcerative colitis. J Crohns Colitis (2017) 11:S429–30. doi:10.1093/ecco-jcc/jjx002.808
283. Gurevich M, Gritzman T, Orbach R, Tuller T, Feldman A, Achiron A. Laquinimod suppress antigen presentation in relapsing-remitting multiple sclerosis: in-vitro high-throughput gene expression study. J Neuroimmunol (2010) 221(1–2):87–94. doi:10.1016/j.jneuroim.2010.02.010
284. Jolivel V, Luessi F, Masri J, Kraus SH, Hubo M, Poisa-Beiro L, et al. Modulation of dendritic cell properties by laquinimod as a mechanism for modulating multiple sclerosis. Brain (2013) 136(Pt 4):1048–66. doi:10.1093/brain/awt023
Keywords: innate immunity, pattern-recognition receptors, toll-like receptors, NOD-like receptors, inflammatory bowel disease
Citation: Corridoni D, Chapman T, Ambrose T and Simmons A (2018) Emerging Mechanisms of Innate Immunity and Their Translational Potential in Inflammatory Bowel Disease. Front. Med. 5:32. doi: 10.3389/fmed.2018.00032
Received: 11 December 2017; Accepted: 30 January 2018;
Published: 19 February 2018
Edited by:
Luca Pastorelli, Università degli Studi di Milano, ItalyReviewed by:
Elias Gounaris, Northwestern University, United StatesCopyright: © 2018 Corridoni, Chapman, Ambrose and Simmons. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniele Corridoni, ZGFuaWVsZS5jb3JyaWRvbmlAbmRtLm94LmFjLnVr
†Co-first authors.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.