 Jean Emmanuel Kahn
Jean Emmanuel Kahn Matthieu Groh
Matthieu Groh Guillaume Lefèvre
Guillaume Lefèvre- 1Service de Médecine Interne, Centre de Référence des Syndromes Hyperéosinophiliques-CEREO, Hôpital Foch, Université Versailles-Saint Quentin en Yvelines, Suresnes, France
- 2Service de Médecine Interne, Hôpital Saint Louis, Université Paris-Diderot, Paris, France
- 3Université de Lille, INSERM, CHU Lille, U995 – LIRIC – Lille Inflammation Research International Center, Institut d’Immunologie, Centre de Référence des Syndromes Hyperéosinophiliques-CEREO, Unité d’Immunologie Clinique, Lille, France
Hypereosinophilia (HE) is a heterogeneous condition that can be reported in various (namely inflammatory, allergic, infectious, or neoplastic) diseases with distinct pathophysiological pathways. In 1975, Chusid et al. published the first diagnostic criteria of hypereosinophilic syndromes (HES). Over the years, as both basic and clinical knowledge improved, several updates have been suggested, with a focus on better distinguishing isolated or asymptomatic eosinophilia from diseases with specific eosinophil-related organ damage. Moreover, underlying molecular and cellular mechanisms of eosinophilia gradually became the cornerstone of successive attempts to classify HE-related diseases. In 2011, the International Cooperative Working Group on Eosinophil Disorders criteria emerged from a multidisciplinary Working Conference on Eosinophil Disorders and Syndromes, and provided substantial contribution to the clarification of general concepts and definitions in the field of HE. Yet, owing to the low prevalence of HE/HES, to the numerous diseases encompassed in the spectrum of HE-related disorders (with sometimes overlapping phenotypes), many questions are left unanswered (e.g., the need to better standardize the use of modern molecular tools, or the clinical relevance of distinguishing different subtypes of idiopathic HES). Here, we review the current state of knowledge in the fields of classification and diagnosis criteria of HE-related diseases, with emphasis on the analysis of both strengths and weaknesses of present concepts and their usefulness in daily practice.
Introduction
The concept of “hypereosinophilic syndromes” (HES) was introduced by Hardy and Anderson in 1968 (1), and Chusid et al. later suggested in 1975 diagnostic criteria for HES (2). These readable and easy-to-use criteria comprised chronic (i.e., longer than 6 months) hypereosinophilia (HE) (i.e., above 1.5 × 109/L) with no identifiable cause, associated with clinical manifestations. Nowadays, given the various identified molecular mechanisms underpinning HE (e.g., T-cell-dependent IL-5 production, clonal abnormalities of the myeloid lineage) and the subsequent heterogeneity of diseases encompassed in the spectrum of HES, this first set of diagnostic criteria has become outdated. Hereafter, we will review the main classifications in HES, and discuss their strengths and potential pitfalls.
Current Classifications and Definitions of Eosinophilic Disorders
Initially, the concept of HES was commonly applied to patients with multi-organ damage (often involving the heart) and unexplained chronic HE above 1.5 × 109/L. Yet, different clinico-biological phenotypes were already observed in the first published series of patients with HES, suggesting that various underlying pathophysiological processes could be involved (2, 3). Hence, a subgroup of patients presented with features suggestive of myeloproliferative neoplasm (i.e., anemia, splenomegaly, myelofibrosis, etc.), which led to the concept of myeloproliferative (“leukemic” for Chusid) HES. Decades later, clonal abnormalities involving fusion transcripts (among which PDGFRA and PDGFRB genes) were identified in the same subgroup of patients, thereby validating ex post the initial hypothesis (4). Next, the lymphoid variant of HES was defined in another subgroup of patients with dermatologic manifestations that responded to corticosteroids and were originally classified as hypersensitivity (or non-malignant or allergic) HES but in which abnormal clonal T-cells (e.g., CD3−CD4+ T-cells) that produced eosinophilopoietins were later identified (5).
In the early 2000s, numerous expert classifications embedded the above-defined concepts of molecularly defined myeloproliferative-HES, lymphoid HES, and idiopathic HES (when no molecular or immunological abnormality can be found) (6, 7). Yet, less than two decades later, these classifications already seem outdated due to recent major breakthroughs in molecular biology. Currently, the two main—and partially redundant—classifications are the one proposed by the WHO (which covers only primary/neoplastic HES) (8) and the one proposed by the International Cooperative Working Group on Eosinophil Disorders (ICOG-EO) in 2011 (9). The ICOG-EO (an international and multidisciplinary panel of experts) agreed on unifying terminologies and criteria, and suggested a classification that delineates various forms of HE and HES (including primary and secondary variants) based on specific hematologic and immunologic conditions.
Definitions of Eosinophilia and HE
International Cooperative Working Group on Eosinophil Disorders first provided basic definitions of what should be considered as HE (Table 1). The cut-off of 1.5 × 109/L was chosen to differentiate HE (>1.5 × 109/L) from “eosinophilia” (between 0.5 and 1.5 × 109/L). The duration of 1 month of blood HE (instead of the 6 month delay comprised in Chusid criteria) was retained as sufficient and indeed makes sense from a clinical viewpoint, considering that life-threatening organ involvement is frequent in HES. Importantly, the latter criteria also include tissue eosinophilia in the field of HE-related disorders, thereby highlighting the fact that discrepancies between blood and tissue eosinophilia (e.g., marked tissue eosinophilia without blood eosinophilia or HE) can be reported in some eosinophilic disorders (e.g., eosinophilic esophagitis or acute eosinophilic pneumonia). Hence, the pathologist’s definition of tissue HE includes more than 20% of eosinophils in bone marrow sections, and/or (for other tissues) extensive tissue infiltration by eosinophils, and/or marked deposition of eosinophils granule proteins (Table 1).

Table 1. Summary of the ICOG-EO’s definitions of eosinophilic disorders.
Definition of HES
The ICOG-EO defined HES as blood HE or tissue HE associated with HE-related organ damage (precluding the absence of an alternative diagnosis for the organ dysfunction) (Table 1). As compared with the Chusid criteria, this definition comprises a causal link between the observed tissue HE and organ damage, the probability of which is considered sufficient in presence of the following specific histological findings: (1) fibrosis or (2) thrombosis or (3) cutaneous eosinophilia (with erythema or angioedema or pruritus or eczema or ulceration) or (4) peripheral or central neurologic defect.
Definition of Neoplastic HE/HES (HEN/SHEN)
Hypereosinophilia or HES are considered as neoplastic (or clonal or primary, HEN/SHEN) when an underlying myeloid/lymphoid/stem cell neoplasm with HE and rearrangement of PDGFRA, PDGFRB, FGFR or with PCM1-JAK2 translocation is identified.
HEN/HESN also encompasses the broad spectrum of other WHO-defined myeloid neoplasms with associated eosinophilia, such as BCR-ABL1+ chronic myeloid leukemia, JAK2-mutated myeloproliferative neoplasms, KIT D816V+ systemic mastocytosis, acute myeloid leukemia (AML) associated with CBFβ fusion gene, myelodysplastic syndromes associated with HE, and other WHO-defined myeloid neoplasms with HE (10).
Finally, HEN/SHEN also includes a last subgroup of patients classified as chronic eosinophilic leukemia not otherwise specified, which should remain an exclusion diagnosis defined by (1) the exclusion of all genetically predisposed conditions described previously, (2) the absence of molecular or cytological features of AML, and (3) the presence of a non-specific clonal cytogenetic or molecular abnormality (i.e., TET2, ASXL1, IDH2, SF3B1) or blast cells >2% in the blood and >5% in the bone marrow.
Definition of Reactive HE/HES
Reactive HE and HES (HER and HESR) aggregate all conditions (e.g., parasitic infections, adverse drug reactions, inflammatory, or neoplastic diseases) in which eosinophils are considered as non-clonal and are thought to be driven by Th-2 (mainly IL-5) cytokines. Importantly, although it may seem counterintuitive at first sight, ICOG-EO classification emphasizes the fact that HER and HESR (with reactive eosinophils) can be observed in neoplastic diseases in which the clonal cells (T-cells, Reed-Steinberg cells, carcinomatous cells, mast-cells, etc.) are the main sources of IL-5 and other eosinophilopoietins. Hence, the lymphocytic variant of HES (an indolent T-cell lymphoproliferative disease) is, therefore, classified as a subtype of HESR.
Definition of Idiopathic HES
When a patient fulfills the criteria of HES but does not comply with the definitions of both HESN and HESR, the diagnosis of idiopathic HES (HESI) can be retained. Interestingly, in main expert centers in HES, more than half of HES patients are classified as HESI, while 10–20% of patients each belong to HESN and HESR categories (11).
Definition of HE of Undetermined Significance
Patients with isolated blood HE but without organ dysfunction and who will remain completely asymptomatic over time are not that uncommon. Hence, after that an initial comprehensive evaluation excludes HEN and HER, the ICOG-EO classification suggests that such patients should be classified as HE of undetermined significance (HEUS). This newly defined subgroup has major clinical implications since recent data strongly suggests that such patients carry a good prognosis and should only be closely followed without treatment (12).
Old and New Criteria for the Classification of Neoplastic HE/HES
In the 2005 and 2010 classifications of HE/HES, experts brought to the forefront “good old fashioned” features suggestive of a myeloid neoplasm (i.e., hepatosplenomegaly, increased serum vitamin B12 or tryptase, anemia, thrombocytopenia, myelofibrosis) as criteria for “myeloproliferative-HES,” even in the absence of a molecularly proven HESN (6, 7). With the exception of the blast cell count, the ICOG-EO and WHO classifications have nearly completely excluded these patients—which are now classified as HESI—from the field of HESN. This distinction of patients with presumed myeloid neoplasm is clinically relevant as response to different treatment options differs (less response to steroids, more likely to respond to imatinib). Hitherto, such patients carry a guarded prognosis [the latter being closer to HESN than that of other HESI patients (13, 14)] and, as patients with HESN, may require treatment with tyrosine kinase (TK) inhibitors, cytotoxic drugs, or even bone marrow transplantation. Hence, the current distinction between patients with molecularly defined HESN from those with similar clinical features but without any identifiable mutation is questionable, and it seems desirable that further updates of HE/HES classifications should individualize these myeloproliferative-HES patients as a specific subgroup even in the absence of an identified mutation.
Next, due to the development of new sequencing methods and tools in malignant hematology [especially the next-generation sequencing (NGS)], the field of neoplastic HES has considerably evolved. Since the FIP1L1–PDGFRA fusion transcript gene was discovered in 2003, the list of genetically defined HE has regularly been implemented over the years and now comprises 72 distinct entities consisting mostly of TK fusion genes (10).
The identification of numerous mutations in myeloproliferative disorders and myelodysplastic syndrome raise the question whether these new mutations should be included in further classifications of HES. Two recent studies report NGS results in two cohorts of 98 and 51 patients with HEUS and/or HES (15, 16). Interestingly, such mutations (including ASXL1, TET2, SETBP1, CSFR3, and SF3B1) were identified in 11 and 28% of patients, respectively, suggesting that a significant proportion of patients otherwise classified as HEUS and/or HESUS might rather belong to the HEN and HESN subgroups. Yet, none of these studies provided convincing elements demonstrating the transforming capacity of these mutations, suggesting that they may not be the true driving mutation for HES/HEUS. In addition, the impact of NGS-identified mutations on survival remains an open question, a poorer prognosis in patients with NGS-mutations being suggested in a single study (16). Hence, NGS seems to be a highly powerful tool to identify molecular defects in HE/HES. Yet, large prospective registries are needed in order to evaluate its potential usefulness in daily practice regarding patients’ treatment and prognosis, before this tool be included as a new criterion for HEN or HESN.
Reactive HES: A Disorder with Bystander HE or a True Reactive HES?
Many pathologic conditions can induce reactive blood and/or tissue eosinophilia (HER), due to the overproduction of eosinophilopoietic cytokines, mostly IL-5. Yet, in many of these situations, skin and organ damage seem to be due to an autoimmune process (e.g., bullous pemphigoid), a malignant disease (e.g., cutaneous lymphoma, histiocytosis, mastocytosis) or to the massive tissue infiltration by IgG4+ plasma cells (e.g., IgG4-related disorders) rather than to eosinophilic tissue infiltration. From a pathological viewpoint, substantial effort (including assessment of extracellular deposition of eosinophil granule proteins by immunohistostaining) has been made by the ICOG-EO classification to define eosinophilic tissue infiltration. Yet, these laboratory techniques are often neither standardized nor performed routinely, and not used as a diagnostic tool in daily practice.
On the other side, in some patients with solid cancer, lymphoma or helminthiasis, a pronounced blood and tissue eosinophilia may occur in organs other than those affected by the underlying disease. In such situations, a true eosinophilic endomyocardial fibrosis due to eosinophil toxicity—as well as many other organ involvements—have been reported. Hence, physicians must be aware that the same disease may induce a bystander blood and/or tissue HE without meaningful consequences related to eosinophils, or a true reactive HESR.
According to the clinical context, the initial workup of an unexplained HE/HES must include broad investigations in order to rule out with certainty an underlying disease likely to favor HER/HESR. The choice of keeping HESR as part of the ICOG-EO’s nosology has clinical implications: (1) HESI is an exclusion diagnosis which supposes that all etiologies of HESR must be excluded and (2) the treatment of the underlying cause may reverse HER, but in case of organ damage and/or dysfunction attributable to tissue HE (i.e., HESR), corticosteroids may be considered from the outset in addition to the treatment of the underlying disease.
Unmet Needs in the Diagnosis of the Lymphoid Variant of HES
HESL, a subtype of HESR, is a chronic clonal indolent T-cell lymphoproliferative disorder in which mature peripheral T-cells secrete high amounts of IL-5, leading to the polyclonal expansion of eosinophils. Hence, to some extent, HESL can be considered as the archetype of Th-2 driven eosinophilic disorders (17). Patients can be aymptomatic or poorly symptomatic for years, with HE being the sole manifestation of the indolent T-cell proliferative disorder (18). Diagnosing HESL is important for three reasons: (1) its treatment can differ from that of HESI, notably because of frequent corticosteroid dependency requiring additional corticosteroid-sparing-treatments, (2) clonal T-cells that are found in blood and tissues of HESL patients can be mistaken for aggressive T-cell lymphoma, and patients wrongly treated as such with chemotherapy, and (3) authentic T-cell lymphomas (e.g., angioimmunoblastic T-cell lymphomas) may occur during disease course of these patients, who should be closely monitored (18–20).
Diagnosing HESL usually requires the detection of an abnormal T-cell subset in the peripheral blood. Experts agree that three main subsets of HESL must be systematically searched by flow cytometry: CD3−CD4+TCRab−, CD3+CD4+CD7−, and CD3+CD4−CD8−TCRab+ (21). Although lacking specificity, further confirmation of HESL is supported by a clonal TCR rearrangement.
Yet, a clear definition of what should (or should not) be diagnosed as HESL is lacking in all current classifications. Pertinently, should a cutoff of absolute or relative counts of such abnormal T-cells be defined? Is the demonstration of their ability to produce high levels of IL-5 necessary? Is a clonal TCR rearrangement necessary or sufficient to define HESL? Could other abnormal subsets of blood cells (e.g., type 2 innate lymphoid cells) define HESL? Last, several biological biomarkers such as IL-5, CCL-17/TARC, IgE, which demonstrated in many studies potential utility in their ability to distinguish between various subtypes of HES (18, 22) could also be added as additional diagnostic criteria in future classifications.
Idiopathic HES: A Multifaceted Disease with Many Overlapping Conditions
Should Multi-Organ and Single-Organ Diseases Be Distinguished?
According to current definitions, all patients with blood HE and organ damage with significant eosinophilic infiltration could be classified as HES, whatever the number of organs involved.
Yet, the ICOG-EO classification also makes a clear distinction between HES and several other organ-restricted conditions with HE (e.g., eosinophilic cystitis, eosinophilic esophagitis, eosinophilic gastroenteritis, eosinophilic pneumonia, dermatologic conditions associated with HE, etc.) that by definition only affect a single organ during the entire course of the disease (the main suggested reason being that the causative role of eosinophils in organ damage is unclear) (9). Nevertheless, some patients with HESL, FIP1L1–PDGFRA HESN or HESI may only have single-organ involvement at disease onset, with further organ involvement only occurring during follow-up (22). Furthermore, the single-organ damage also depends on the way the clinician looks at the patient: in chronic eosinophilic pneumonia, many patients have sinonasal polyposis. Should they be classified as single-organ disease or HES? Hence, such separation at diagnosis between eosinophilic single-organ diseases and HES appears questionable. In our mind, most patients with eosinophilic single-organ disorders demonstrated by histopathology should be investigated and subsequently followed as HES, even though some of these patients will indeed subsequently never develop multi-organ HES.
Should Outcome Profiles of HEI/HESI Be Distinguished?
In daily practice, HEI/HESI treating physician are confronted with various patterns of disease courses. First, some patients will present a single flare of variable duration and will recover (spontaneously or after corticosteroids tapering and discontinuation) without subsequent relapse (Figure 1A). Of note, according to previous classifications of HES disorders (6, 7), these latter patients with less than 6 months of disease duration would not have been classified as HES per se. Next, other patients periodically relapse during follow-up, with a variable delay in between flares (that can be weeks, months or years in some cases, and whose severity may also fluctuate overtime). Hence, a pragmatic approach to treating such patients could be short courses of corticosteroids but without long-term therapy (Figure 1B). Notably, both outcome profiles have been reported in various single-organ eosinophil disorders [e.g., acute eosinophilic pneumonia, eosinophilic gastroenteritis (23), episodic angioedema with HE (Gleich’s syndrome) (24)] as well as in HESI. Last, a third set of patients will present, usually in the context of corticosteroid dependency, chronic persistent disease requiring second-line treatments (Figure 1C).
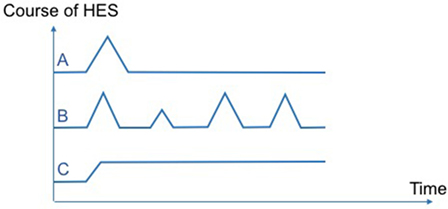
Figure 1. Various patterns of disease courses observed in hypereosinophilic syndromes (HES). Pattern (A): single flare without subsequent relapse. Pattern (B): several relapses with intervals of complete remission. Pattern (C): chronic persistent disease.
Underlying mechanisms involved in eosinophilia are likely to differ between the three disease patterns described here above. Hence, a single flare of HES without subsequent relapse (pattern A) strongly suggests temporary exposure to an extrinsic trigger (e.g., drug-induced eosinophilia for the disease). Conversely, recurrent or chronic persistent HEI/HESI (patterns B and C) suggest an intrinsic dysfunction of eosinophil regulation and/or a persistent unidentified underlying cause. By analogy with multiple sclerosis, it is advisable that these different patterns of disease courses be incorporated into further updates of disease classifications in an effort to homogenize inclusion criteria in clinical trials and to individualize patient care.
Antineutrophil Cytoplasm Antibody (ANCA)-Negative Eosinophil Granulomatosis with Polyangiitis (EGPA) and HESI: The Diagnostic (and Therapeutic) Dilemma
Eosinophil granulomatosis with polyangiitis is defined in the 2012 International Chapel Hill Consensus Conference on the Nomenclature of Vasculitides as an eosinophil-rich and necrotizing granulomatous inflammation often involving the respiratory tract, with necrotizing vasculitis predominantly affecting small to medium vessels, and associated with asthma and eosinophilia (25). EGPA differs from other ANCA-associated vasculitides (AAV) by the constant presence of asthma, blood and tissue eosinophilia, and the low prevalence of ANCA positivity, which are detected in only 20–40% of patients (26). Next, the phenotype of ANCA-positive and ANCA-negative EGPA patients differ, with ANCA-negative patients having less vasculitic manifestations (purpura, peripheral neuropathy, glomerulonephritis, scleritis) but more frequent cardiomyopathy (often mimicking that observed in HES) (27). Considering that ≈40% of patients with asthma, HE above 1.5 G/L and at least another systemic manifestation had neither symptoms of vasculitis nor ANCA, a recent European Respiratory Society-endorsed Taskforce on EGPA suggested that this subgroup of patients be considered to have hypereosinophilic asthma with systemic (non-vasculitic) manifestations (HASM) rather than genuine EGPA (28). The results of a negative trial testing azathioprine versus placebo in low-risk EGPA (29) and the recent MIRRA study demonstrating the benefit of mepolizumab in EGPA (30) both confirm the overlap between ANCA-negative EGPA and HES. Given the fact that clinical and biological profiles of ANCA-negative EGPA and HES overlap markedly (31), it seems appropriate to consider a diagnosis of HES in patients with HASM should be mentioned in further updates of both AAV and HES classifications (32).
Conclusion
The concept of HES has evolved considerably since the first classification by Chusid in 1975, and the recent ICOG-EO classification has successfully embedded most of the field’s recent breakthroughs. These include, albeit not exclusively, the identification of numerous TK fusion genes, the concept of HESR (among which HESL), and the need for a clear histopathological definition of eosinophilic tissue infiltration. Moreover, this classification puts an end to many longstanding issues in the HE/HES domain and is a useful tool for the physician in daily care, allowing for better classification of patients between single-organ disease, HESN, HESR, HESI, and HEUS (a condition that does not require therapy). Nevertheless, due to the lack of large prospective cohorts of HE/HES patients, one major limitation of the ICOG-EO classification is that it is mainly expert based and, thus, remains low-evidenced. Pertinently, many points (e.g., the need for a clear definition of HESL; how to treat patients with a myeloproliferative phenotype but for whom a clonal mutation is not (yet) evidenced; improving the diagnostic workup of patients suspected of ANCA-negative EGPA versus HESI) are open for improvement and should be the starting point of future HE/HES-targeted research. Hence, the implementation of international multicentric registries is awaited in order to improve current classifications of HE/HE and subsequently patient care.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
The handling editor declared a past supervisory role with one of the authors GL.
References
1. Hardy WR, Anderson RE. The hypereosinophilic syndromes. Ann Intern Med (1968) 68(6):1220–9. doi:10.7326/0003-4819-68-6-1220
2. Chusid MJ, Dale DC, West BC, Wolff SM. The hypereosinophilic syndrome: analysis of fourteen cases with review of the literature. Medicine (Baltimore) (1975) 54(1):1–27. doi:10.1097/00005792-197501000-00001
4. Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med (2003) 348(13):1201–14. doi:10.1056/NEJMoa025217
5. Cogan E, Schandene L, Crusiaux A, Cochaux P, Velu T, Goldman M. Brief report: clonal proliferation of type 2 helper T cells in a man with the hypereosinophilic syndrome. N Engl J Med (1994) 330(8):535–8. doi:10.1056/NEJM199402243300804
6. Klion AD, Bochner BS, Gleich GJ, Nutman TB, Rothenberg ME, Simon HU, et al. Approaches to the treatment of hypereosinophilic syndromes: a workshop summary report. J Allergy Clin Immunol (2006) 117(6):1292–302. doi:10.1016/j.jaci.2006.02.042
7. Simon HU, Rothenberg ME, Bochner BS, Weller PF, Wardlaw AJ, Wechsler ME, et al. Refining the definition of hypereosinophilic syndrome. J Allergy Clin Immunol (2010) 126(1):45–9. doi:10.1016/j.jaci.2010.03.042
8. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood (2016) 127(20):2391–405. doi:10.1182/blood-2016-03-643544
9. Valent P, Klion AD, Horny HP, Roufosse F, Gotlib J, Weller PF, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol (2012) 130(3):607–12.e9. doi:10.1016/j.jaci.2012.02.019
10. Reiter A, Gotlib J. Myeloid neoplasms with eosinophilia. Blood (2017) 129(6):704–14. doi:10.1182/blood-2016-10-695973
11. Klion AD. How I treat hypereosinophilic syndromes. Blood (2015) 126(9):1069–77. doi:10.1182/blood-2014-11-551614
12. Chen YY, Khoury P, Ware JM, Holland-Thomas NC, Stoddard JL, Gurprasad S, et al. Marked and persistent eosinophilia in the absence of clinical manifestations. J Allergy Clin Immunol (2014) 133(4):1195–202. doi:10.1016/j.jaci.2013.06.037
13. Lefebvre C, Bletry O, Degoulet P, Guillevin L, Bentata-Pessayre M, Le Thi Huong D, et al. [Prognostic factors of hypereosinophilic syndrome. Study of 40 cases]. Ann Med Interne (Paris) (1989) 140(4):253–7.
14. Helbig G, Soja A, Bartkowska-Chrobok A, Kyrcz-Krzemien S. Chronic eosinophilic leukemia-not otherwise specified has a poor prognosis with unresponsiveness to conventional treatment and high risk of acute transformation. Am J Hematol (2012) 87(6):643–5. doi:10.1002/ajh.23193
15. Pardanani A, Lasho T, Barraco D, Patnaik M, Elala Y, Tefferi A. Next generation sequencing of myeloid neoplasms with eosinophilia harboring the FIP1L1-PDGFRA mutation. Am J Hematol (2016) 91(3):E10–1. doi:10.1002/ajh.24273
16. Wang SA, Tam W, Tsai AG, Arber DA, Hasserjian RP, Geyer JT, et al. Targeted next-generation sequencing identifies a subset of idiopathic hypereosinophilic syndrome with features similar to chronic eosinophilic leukemia, not otherwise specified. Mod Pathol (2016) 29(8):854–64. doi:10.1038/modpathol.2016.75
17. Roufosse F, Cogan E, Goldman M. Lymphocytic variant hypereosinophilic syndromes. Immunol Allergy Clin North Am (2007) 27(3):389–413. doi:10.1016/j.iac.2007.07.002
18. Lefevre G, Copin MC, Staumont-Salle D, Avenel-Audran M, Aubert H, Taieb A, et al. The lymphoid variant of hypereosinophilic syndrome: study of 21 patients with CD3-CD4+ aberrant T-cell phenotype. Medicine (Baltimore) (2014) 93(17):255–66. doi:10.1097/md.0000000000000088
19. Lefevre G, Copin MC, Roumier C, Aubert H, Avenel-Audran M, Grardel N, et al. CD3-CD4+ lymphoid variant of hypereosinophilic syndrome: nodal and extranodal histopathological and immunophenotypic features of a peripheral indolent clonal T-cell lymphoproliferative disorder. Haematologica (2015) 100(8):1086–95. doi:10.3324/haematol.2014.118042
20. Roufosse F, de Leval L, van Krieken H, van Deuren M. Lymphocytic variant hypereosinophilic syndrome progressing to angioimmunoblastic T-cell lymphoma. Leuk Lymphoma (2015) 56(6):1891–4. doi:10.3109/10428194.2014.976823
21. Roufosse F. Hypereosinophilic syndrome variants: diagnostic and therapeutic considerations. Haematologica (2009) 94(9):1188–93. doi:10.3324/haematol.2009.010421
22. Ogbogu PU, Bochner BS, Butterfield JH, Gleich GJ, Huss-Marp J, Kahn JE, et al. Hypereosinophilic syndrome: a multicenter, retrospective analysis of clinical characteristics and response to therapy. J Allergy Clin Immunol (2009) 124(6):1319–25.e3. doi:10.1016/j.jaci.2009.09.022
23. Pineton de Chambrun G, Gonzalez F, Canva JY, Gonzalez S, Houssin L, Desreumaux P, et al. Natural history of eosinophilic gastroenteritis. Clin Gastroenterol Hepatol (2011) 9(11):950–6.e1. doi:10.1016/j.cgh.2011.07.017
24. Khoury P, Herold J, Alpaugh A, Dinerman E, Holland-Thomas N, Stoddard J, et al. Episodic angioedema with eosinophilia (Gleich syndrome) is a multilineage cell cycling disorder. Haematologica (2015) 100(3):300–7. doi:10.3324/haematol.2013.091264
25. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum (2013) 65(1):1–11. doi:10.1002/art.37715
26. Sable-Fourtassou R, Cohen P, Mahr A, Pagnoux C, Mouthon L, Jayne D, et al. Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome. Ann Intern Med (2005) 143(9):632–8. doi:10.7326/0003-4819-143-9-200511010-00006
27. Comarmond C, Pagnoux C, Khellaf M, Cordier JF, Hamidou M, Viallard JF, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): clinical characteristics and long-term followup of the 383 patients enrolled in the French Vasculitis Study Group cohort. Arthritis Rheum (2013) 65(1):270–81. doi:10.1002/art.37721
28. Cottin V, Bel E, Bottero P, Dalhoff K, Humbert M, Lazor R, et al. Revisiting the systemic vasculitis in eosinophilic granulomatosis with polyangiitis (Churg-Strauss): a study of 157 patients by the Groupe d’Etudes et de Recherche sur les Maladies Orphelines Pulmonaires and the European Respiratory Society Taskforce on eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Autoimmun Rev (2017) 16(1):1–9. doi:10.1016/j.autrev.2016.09.018
29. Puechal X, Pagnoux C, Baron G, Quemeneur T, Neel A, Agard C, et al. Adding azathioprine to remission-induction glucocorticoids for eosinophilic granulomatosis with polyangiitis, microscopic polyangiitis or polyarteritis nodosa without poor prognosis factors a randomized-controlled trial. Arthritis Rheumatol (2017) 6(11):2175–86. doi:10.1002/art.40205
30. Wechsler ME, Akuthota P, Jayne D, Khoury P, Klion A, Langford CA, et al. Mepolizumab or placebo for eosinophilic granulomatosis with polyangiitis. N Engl J Med (2017) 376(20):1921–32. doi:10.1056/NEJMoa1702079
31. Khoury P, Zagallo P, Talar-Williams C, Santos CS, Dinerman E, Holland NC, et al. Serum biomarkers are similar in churg-strauss syndrome and hypereosinophilic syndrome. Allergy (2012) 67(9):1149–56. doi:10.1111/j.1398-9995.2012.02873.x
Keywords: hypereosinophilic syndrome, hypereosinophilia, classification, eosinophilic granulomatosis with polyangiitis, eosinophilic disorders
Citation: Kahn JE, Groh M and Lefèvre G (2017) (A Critical Appraisal of) Classification of Hypereosinophilic Disorders. Front. Med. 4:216. doi: 10.3389/fmed.2017.00216
Received: 05 September 2017; Accepted: 16 November 2017;
Published: 05 December 2017
Edited by:
Florence Emmanuelle Roufosse, Free University of Brussels, BelgiumReviewed by:
Gerald Joseph Gleich, University of Utah, United StatesOwen McCarty, Oregon Health & Science University, United States
Peter F. Weller, Harvard Medical School, United States
Copyright: © 2017 Kahn, Groh and Lefèvre. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean Emmanuel Kahn, amUua2FobkBob3BpdGFsLWZvY2gub3Jn