 Verónica Urdaneta
Verónica Urdaneta Josep Casadesús
Josep Casadesús
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 03 October 2017
Sec. Gastroenterology
Volume 4 - 2017 | https://doi.org/10.3389/fmed.2017.00163
Bile salts and bacteria have intricate relationships. The composition of the intestinal pool of bile salts is shaped by bacterial metabolism. In turn, bile salts play a role in intestinal homeostasis by controlling the size and the composition of the intestinal microbiota. As a consequence, alteration of the microbiome–bile salt homeostasis can play a role in hepatic and gastrointestinal pathological conditions. Intestinal bacteria use bile salts as environmental signals and in certain cases as nutrients and electron acceptors. However, bile salts are antibacterial compounds that disrupt bacterial membranes, denature proteins, chelate iron and calcium, cause oxidative damage to DNA, and control the expression of eukaryotic genes involved in host defense and immunity. Bacterial species adapted to the mammalian gut are able to endure the antibacterial activities of bile salts by multiple physiological adjustments that include remodeling of the cell envelope and activation of efflux systems and stress responses. Resistance to bile salts permits that certain bile-resistant pathogens can colonize the hepatobiliary tract, and an outstanding example is the chronic infection of the gall bladder by Salmonella enterica. A better understanding of the interactions between bacteria and bile salts may inspire novel therapeutic strategies for gastrointestinal and hepatobiliary diseases that involve microbiome alteration, as well as novel schemes against bacterial infections.
Bile salts are produced in the liver from cholesterol, specifically in pericentral hepatocytes, and their daily production is approximately 350 mg (1). Seventeen enzymes convert cholesterol into bile acids, which are transformed into bile salts by the association with Na+ or K+ ions. There are four types of bile salts: primary and secondary, conjugated, and non-conjugated (2, 3). Conjugation involves the formation of an amide bond with either taurine or glycine.
Primary bile salts are the immediate products of cholesterol degradation. Variations in the pool of primary bile salts occur among vertebrates: for instance, in humans and rats, the primary bile salts are cholate and chenodeoxycholate while in mice they are cholate and muricholate (2). Intestinal bacteria transform primary bile salts into secondary bile salts by removal of the hydroxyl group at C7. In humans, secondary bile salts are deoxycholate (DOC, from cholate) and litocholate (from chenodeoxycholate) (4, 5). The hydroxyl groups of bile salts protrude in the same direction, a feature that is partly responsible for their detergent activity because it confers high-water solubility and generates delimited hydrophilic and hydrophobic regions (6).
Intestinal anaerobes play a major role in bile salt metabolism (7, 8), and the main transformations are as follows:
(i) Hydrolysis of the amide bond between the glycine or the taurine conjugated to the steroid nucleus. This reaction, called deconjugation, makes bile salts available as substrates for further modifications by the intestinal microbiota and is, therefore, crucial in bile biotransformation (9–11). Deconjugation is catalyzed by bacterial enzymes known as bile salt hydrolases (BSH), which are widespread in the bacterial world and include Gram-positive intestinal species like Lactobacillus (12–16), Enteroccocus (17, 18), Bifidobacterium (19–21), and Clostridium (22). BSH activity has also been reported in the commensal, Gram-negative Bacteroides spp. (23), and in the Archaea domain, specifically in species of the intestinal microbiome, such as Methanobrevibacter smithii and Methanosphera stadmanae (23). The high levels of identity found between BSH of different domains suggest horizontal gene transfer (23). Additionally, BSH genes show high redundancy in the gut ecosystem, and the number of BSH paralogs varies from strain to strain; for instance, in some serovars of Lactobacillus plantarum four different functional BSH genes have been described (13, 24). A potential selective value of BSH activity is enhancement of bile tolerance, thus facilitating survival in the gut (23, 25). Furthermore, bile salts serve as acceptors of electrons generated by fermentation while glycine and taurine can be used as sources of carbon and nitrogen (6).
(ii) 7α/β-dehydroxylation converts primary bile salts into secondary bile salts. Examples are 7α-dehydroxylation of cholate and chenodeoxycholate yielding deoxycholate and lithocholate, respectively, and 7β-dehydroxylation of ursodeoxycholate yielding lithocholate (11). These biotransformations occur in the human colon, and deoxycholate and lithocholate are the predominant bile salts in human feces (7, 26). A pre-requisite for these transformations is deconjugation because 7α/β-dehydroxylation occurs in free bile salts (27). Unlike BSH activity, only a small number of bacterial species belonging to the class Clostridia have 7α/β-dehydroxylation activity (28). Transformation of primary into secondary bile salts requires transport of free primary bile salts into the bacterial cell, which is carried out by the proton-dependent bile acid transporter BaiG (29). Once inside the cell, a series of reactions occur, beginning with ligation of the bile salt to CoA in a Mg2+- and ATP-dependent reaction catalyzed by CoA ligase (30). The bile salt-Coa thioester is then oxidized at the 3-hydroxy group by a 3α-hydroxysteroid dehydrogenase (31). After oxidation, NAD-dependent flavoproteins synthesize 3-dehydro-Δ4-chenodeoxycholate or 3-dehydro-Δ4-cholate and 3-dehydro-Δ4-ursodeoxycholate, respectively (31, 32), making the bile salt chemically labile for irreversible 7α-dehydration (7). The enzymes involved in further steps of this pathway have not yet been identified; they may include oxidoreductases that catalyze reduction to secondary bile salts (7). A potential advantage for 7α/β-dehydroxylating bacteria might be favorable niche competition upon exclusion of microorganisms sensitive to secondary bile salts (7); additionally, production of reduced NADPH might be energetically useful by providing proton motive force (9).
(iii) Numerous enteric especies (e.g., Clostridium, Peptostreptococcus, Bacteroides, Eubacterium, and Escherichia coli) can perform oxidation and epimerization of hydroxy groups at the positions C3, C7, and C12 of bile salts, generating isobile (β-hidroxy) salts. Examples are 3-oxocholanoate and isocholate; 7-oxocholanoate and 7-epicholate; and 12-oxocholanoate and 12-epicholate (11). Oxidation and epimerization are catalyzed by hydroxysteroid dehydrogenase. Epimerization is a reversible stereochemical change from α to β configuration or vice versa, with the formation of a stable oxo-bile salt intermediate (7). These modified bile salts (epimers and isobile salts) are usually recycled to the liver and repaired before rejoining bile (9).
Some of these biotransformations contribute to the salvage of bile salts that escape active transport in the distal ileum during enterohepatic circulation. Particularly, deconjugation and 7α-dehydroxylation increases hydrophobicity and Pka of bile salts, facilitating their recovery by passive transport in the colon epithelium (7).
In the absence of microbial transformations, the diversity of the bile salt pool decreases (33). The intestinal microbiota has an active role in the regulation of bile salt synthesis: bacterial metabolism decreases the level of taurine-conjugated muricholic acid, a farnesoid X receptor (FXR) antagonist that inhibits FXR signaling in the intestine. FXR signaling reduces the expression of cholesterol 7α-hydrolase (CYP7a1), a rate-limiting enzyme in bile acid synthesis; and as a consequence, primary bile acid synthesis is reduced. Hence, the gut microbiota regulates not only secondary bile salt metabolism but also primary bile salt synthesis by alleviating FXR inhibition (34).
Figure 1 depicts the process of synthesis of the most abundant bile salts present in human bile and their circulation trough the hepatic, biliary, and digestive systems, as well as their chemical structure and physicochemical properties.
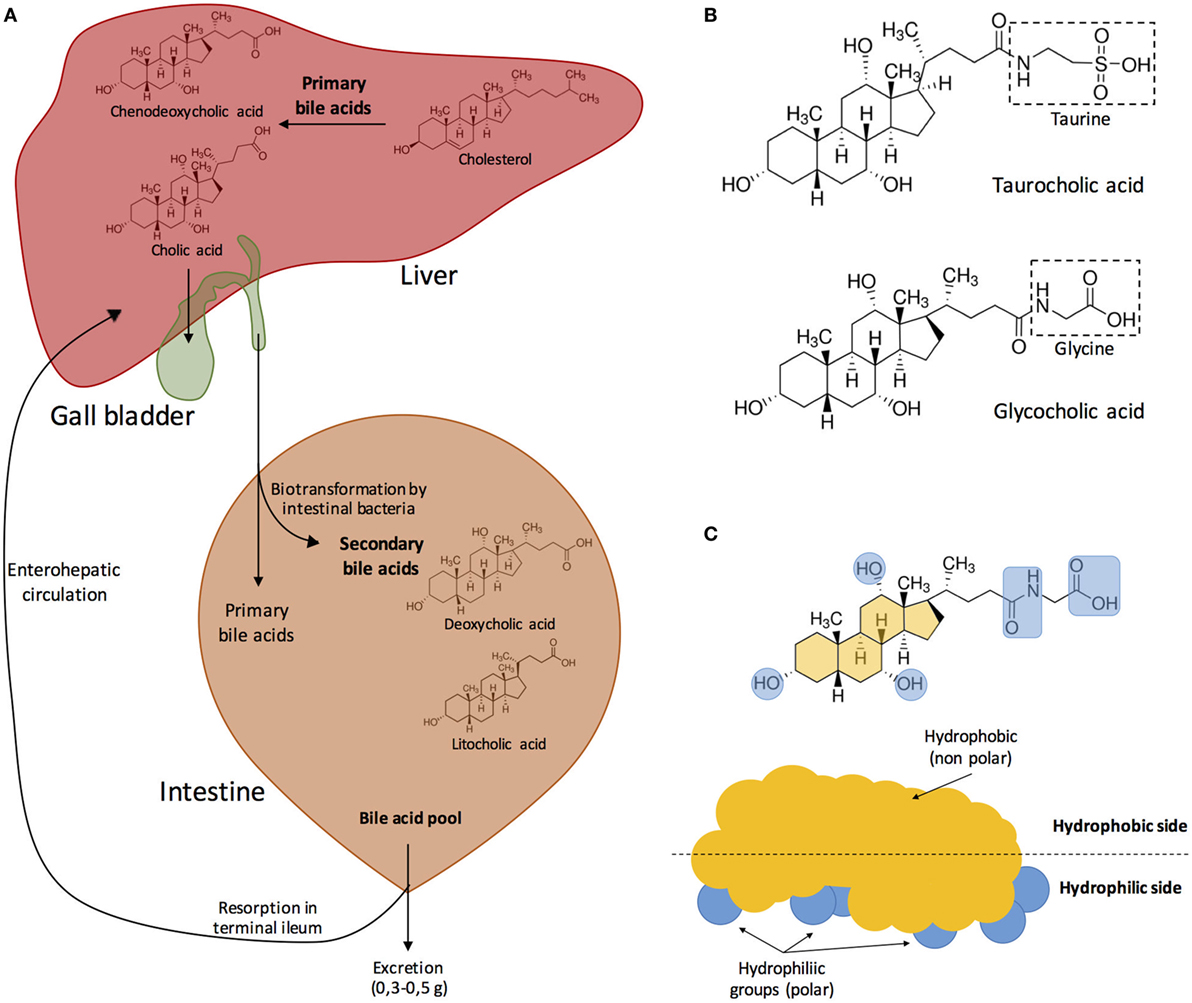
Figure 1. (A) Synthesis of the major bile acids of human bile and circulation in the hepatic, biliary, and digestive systems. Bile acids are transformed into bile salts by the association with Na+ or K+ ions. Primary bile acids are stored in the gall bladder. After food ingestion, bile released into the small intestine contains 5–15 g of bile acids. In the intestine, bile acids are modified by the effect of intestinal microbiota. Both primary and secondary bile acids are reabsorbed in the terminal ileum into the blood stream. Modified from Fontana et al. (3). (B) Bile acids can be conjugated with either glycine or taurine. In this example, cholic acid becomes either taurocholic acid or glycocholic acid after conjugation with taurine or glycine, respectively. (C) Bile acids are amphipathic, and their hydrophobic side associates with the surface of lipid droplets while the polar groups interact with water creating a stable emulsion of small, mixed micelles. Modified from Hofmann (5) and Vander et al. (35).
The human body is a complex ecosystem where the number of commensal bacterial cells are roughly as abundant as “human” cells (36). Particularly, the gut microbiome has one of the highest bacterial densities in nature (1012 bacteria/g feces, wet weight) (7, 37), and may be viewed as a dynamic community that has co-evolved with their host to facilitate digestion and absorption of complex food components (38). In this symbiotic process, the host must control bacterial colonization of the small intestine since bacterial overgrowth can lead to deficient absorption of nutrients due to bacterial competition with the host.
The factors that can induce changes in the gut microbiome include transit time, abundance of proteolytic enzymes, antimicrobial peptides, diet, age, antibiotic use, disease, and bile (7, 39, 40). Several studies have reported the effect of bile salts on gut microbial communities. According to Kakiyama et al. (40), there is a connection between liver health, bile composition, and gut microbiome structure: as an example, patients with cirrhosis exhibit lower levels of fecal bile salts, which reflects a drop in conversion of primary to secondary bile salts. This decrease may be correlated with an alteration of the gut microbiome upon overgrowth of enteric bacteria (potential pathogens) and decreased abundance of 7α-dehydroxylating Gram-positives like Lachnospiraceae, Ruminococcaceae, and Blautia. On the other hand, the amount of bile released into the intestine can alter gut colonization: a low level of bile salts favors proliferation of Gram-negative bacteria (including pathogens), while high levels of bile salts favors the proliferation of Gram-positive bacteria and reduction of the Gram-negative Bacteroides (41).
High-fat diets, characteristic of Western populations, also affect the structure of the intestinal microbiome by altering the bile salt pool (42, 43). Experiments carried out with an animal model for inflammatory bowel disease (interleukin-10-deficient mice), have shown that the gut microbiome community is altered when the animals are fed with a high saturated fat diet. In particular, the authors detected proliferation of the sulfite-reducing pathobiont Bilophila wadswortia (42), a bacterial species that is difficult to detect in healthy organisms but is associated with appendicitis and intestinal inflammation (44, 45). A diet high in saturated fat favors taurine conjugation of bile salts over glycine conjugation, increasing organic sulfur availability for B. wadswortia (43).
Bile participates in maintaining the intestinal homeostasis as an activator of the FXR and of the Vitamin D nuclear receptor (VDR) (45–48). FXR is a transcription factor from the nuclear receptor family and functions as the main sensor of intracellular bile levels. It is most abundant in tissues exposed to bile salts like the liver and the intestine (49, 50) and modulates intestinal innate immunity (49). The role of FXR in antibacterial defense was inferred from the observation that mice with obstructed bile ducts suffered from microbial proliferation followed by invasion of the epithelium; these symptoms were alleviated by either a synthetic FXR ligand or by bile salts (46, 47, 51). FXR activates genes involved in enteric protection and inhibition of bacterial overgrowth like the angiogenin gene Ang1 and the nitric oxide synthase gene iNos (46, 50). Furthermore, bile salts can induce the synthesis of cathelicidin in biliary epithelial cells. Cathelicidins are antimicrobial peptides that prevent bacterial infections in vivo (51) including those caused by pathogenic E. coli (52) and are involved in innate immunity (53). Bile salts induce the extracellular protein kinase (ERK 1/2) pathway which in turn activates the VDR receptor, resulting in cathelicidin synthesis (48).
Because bile salts control the structure of the intestinal microbiome and the microbiome regulates the composition and size of the bile salt pool, alteration of the microbiome–bile salt homeostasis can have multiple pathological consequences. In cirrhotic patients, for instance, a shrinking bile salt pool may alter the intestinal microbiome by increasing the size of bacterial populations that produce proinflammatory molecules, which trigger a feedback loop as inflammation downregulates bile acid synthesis in the liver (54, 55). As cirrhosis progresses, decreased concentrations of bile salts in the small intestine permit bacterial overgrowth, which many contribute to cirrhosis complications like intestinal endotoxemia and hepatic encephalopathy (54, 56). Microbiome-induced alteration of the bile salt pool may also play a role in non-alcoholic fatty liver disease by impairing the activity of bile salt receptors and bile salt transporters (57).
Bile salt metabolism and signaling is also impaired in cholestasis, which causes accumulation of bile salts in the liver with concomitant hepatocyte injury and inflammation. Patients with chronic cholestasis may be at higher risk of developing hepatocellular and bile duct cancer (58). This hypothesis is consistent with the observation that bile salts can promote cell proliferation by activating mitogenic pathways in the hepatobiliary tract (59).
The composition of the intestinal microbiota may also play a role in progression of colorectal cancer (58), and alteration of the composition of bile salt pool may be indirectly involved in this condition by favoring intestinal colonization by Firmicutes and Proteobacteria with concomitant decrease of Bacteriodetes (60).
In the stomach, alterations such as diet and drug use may favor the colonization of pathogens like Helicobacter pylori (61), which promotes mucosal inflammation of the gastric epithelium and has been identified as the strongest risk factor for gastric adenocarcinoma (62). In the particular case of adenocarcinomas associated with H. pylori-related proximal atrophic gastritis, an increase in concentration of bile salts in the distal stomach may prompt migration of the pathogen to the proximal stomach (63).
Other Helicobacter species have also been isolated from patients with biliary diseases like cholelithiasis, cholecystitis, gall bladder polyp, and gall bladder and biliary tract cancers (64–66).
The expression of certain bacterial loci is regulated by bile salts, which may serve as signals that identify the intestinal environment. For instance, Shigella invasion genes and certain Salmonella genes belonging to the PhoPQ regulon, which controls multiple virulence traits, are upregulated in the presence of bile salts (67–69). In contrast, expression of the Salmonella enterica pathogenicity island 1 (SPI-1), which encodes a type 3 secretion system necessary for invasion of the ileal epithelium, is inhibited by bile salts (70). This repression may prevent synthesis of invasion proteins in intestinal environments that are not appropriate for invasion. In Vibrio cholerae, bile salts activate transcription of genes involved in virulence and biofilm formation (71, 72). Changes in gene expression and/or protein synthesis in the presence of bile have been also described in the Gram-negative Campylobacter jejuni (73) and in the Gram-positives Enterococcus faecalis (74) and Listeria monocytogenes (75).
The molecular mechanisms involved in transcriptional regulation by bile salts are known in certain cases. In Vibrio spp., bile salts bind specific receptors in the bacterial surface, activating signal transduction pathways that modulate gene expression patterns. For instance, the inner membrane proteins VtrA and VtrC of Vibrio parahaemolyticus are part of a bile salt-sensitive signal transduction system, and binding of bile salts to VtrC activates expression of type 3 secretion genes (76). Inner membrane proteins also control virulence gene expression in V. cholerae in response to bile salts: the TcpP/TcpH and ToxR/ToxS pairs constitute bile salt-sensitive signal transduction systems that control transcription of toxT (77, 78). In turn, ToxR is a transcriptional activator of genes encoding the cholera toxin and the toxin co-regulated pilus (79). In S. enterica, repression of invasion in the presence of bile salts involves postranscriptional destabilization of the transcription factor HilD (80).
Bile salts also control pathogenesis of Clostridium difficile, a Gram-positive spore-forming anaerobe (81). C. difficile vegetative cells produce enterotoxins (e.g., TccA and TcdB) that cause inflammation and diarrhea. This process is controlled by the cspBAC locus (82). Taurocholate, a conjugated primary bile salt, activates germination of the spores through interaction with the spore receptor CspC (83). Interestingly, the secondary bile salt deoxycholate can also promote spore germination but inhibits vegetative growth (84). On the other hand, chenodeoxycholate, an unconjugated primary bile salt, inhibits spore germination and is a competitive inhibitor of taurocholate (85). Mice treated with broad spectrum antibiotics show increased susceptibility to C. difficile infection if their pools of secondary bile salts are diminished (86). These observations suggest that the type of bile salt predominant in the medium serves as an environmental signal to either remain dormant or trigger spore germination.
In enterohemorrhagic E. coli O157:H7, bile reduces the expression of genes of the enterocyte effacement (LEE) pathogenicity island (87). When the concentration of bile decreases in downstream regions of the intestine, the LEE locus is activated. LEE expression induces attaching and effacing pathogenesis, and ultimately results in loss of the intestinal epithelial barrier (88). This pathology, which occurs specifically in the large intestine, provides another example of bile salt-mediated intestinal signaling (87).
Bile inhibits bacterial growth (69). In patients with primary cirrhosis, where biliary tract sterility is disrupted (89), the administration of bile salts decreases endotoxin accumulation in biliary epithelial cells (90). The inhibitory effect of bile salts on bacterial growth can be also observed under laboratory conditions, and appears to be the consequence of multiple injuries caused by salts to the bacterial cell.
Bile acids are surface active, amphipathic molecules, and their detergent activity damages cell membranes. Not surprisingly, many bile-sensitive mutants of both Gram-negative and Gram-positive bacteria carry mutations that impair membrane integrity. Likewise, electron microscopy studies have described a shrunken phenotype in Propionibacterium freudenreichii cells exposed to bile (91). Enzymatic assays in E. coli, Clostridium perfringens, and Lactobacillus acidophilus have shown that bile causes leakage of intracellular material (92, 93). Factors that influence the severity of membrane disruption are as follows:
(i) Concentration of bile, high concentrations will dissolve membrane lipids, causing leakage and cell death (94). Low concentrations of bile may have more subtle effects on membrane fluidity and permeability by altering membrane-bound proteins or increasing trans-membrane divalent cation flux. Low levels of bile can also alter the hydrophobicity of the cell surface (92, 93, 95, 96).
(ii) Type and structure of bile salts. Bile salts conjugated with taurine or glycine are fully ionized at physiological pH and for this reason they remain in the outer hemi-leaflet of the membrane; on the contrary, unconjugated bile salts passively cross membranes and enter the cell (97–99).
(iii) Membrane architecture and composition. Changes in lipopolysaccharide (LPS), membrane electric charge, hydrophobicity, lipid fluidity, and fatty acid composition alter bile resistance levels in multiple bacterial species (e.g., E. coli, L. monocytogenes, and L. acidophilus) (100–103).
Upon entry into the bacterial cell, bile salts cause nucleic acid damage. In E. coli, sodium chenodeoxycholate and sodium deoxycholate activate the SOS response (104). Increased transcription of the SoxRS regulon genes osmY and micF suggests that DNA oxidative damage may occur upon exposure to bile salts (105, 106). In S. enterica, bile increases the frequency of nucleotide substitutions, frameshifts, and chromosomal rearrangements (107), and the mutational spectrum of bile suggests that one primary lesion may be oxidative damage of cytosine (108). Bile salts also induce curing of the Salmonella virulence plasmid (109), a feature common among DNA damaging agents (110).
The detergent activity of bile salts can alter the conformation of proteins. Not surprisingly, synthesis of chaperones DnaKJ and GroESL (91, 111), which assist in proper folding of proteins, increases in the presence of bile salts.
Bile salts are able to chelate iron and calcium. In the case of iron, the primary bile acids taurocholic and glycocholic can form soluble Fe2+–bile salt complexes. This binding increases intestinal iron uptake (112). Because both the host and the microbiota require iron for fundamental cellular processes, bile salts may withhold iron from microorganisms, limiting their growth (113).
Calcium ions (Ca2+) can bind to micelles of bile salts conjugated with either taurine or glycine. This binding reduces Ca+2 precipitation, thereby decreasing the contribution of calcium to the formation of gallstones (114). Calcium is also a signal involved in motility, cell cycle and cell division, control of gene expression, and chemotaxis (115, 116). Hence, shortage of Ca2+ upon bile salt chelation may also limit bacterial proliferation.
The ambivalent nature of bile salts as signals of the host environment and as antibacterial agents requires that intestinal bacteria can cope with bile-induced injuries. Not surprisingly, bacterial species adapted to the mammalian intestine are resistant to bile salts, a trait exploited for the design of selective microbiological media such as the MacConkey agar used in the identification of genera of the family Enterobacteriaceae.
Addition of ox bile or individual bile salts to microbiological media is also a strategy to study resistance to bile salts under laboratory conditions. Using this reductionist approach, genetic and biochemical analyses have identified cell functions and mechanisms involved in bile resistance in a number of species including the model organisms E. coli and S. enterica (25, 117, 118) (Table 1). An overall conclusion from these studies is that resistance to bile involves multiple cell functions and mechanisms.
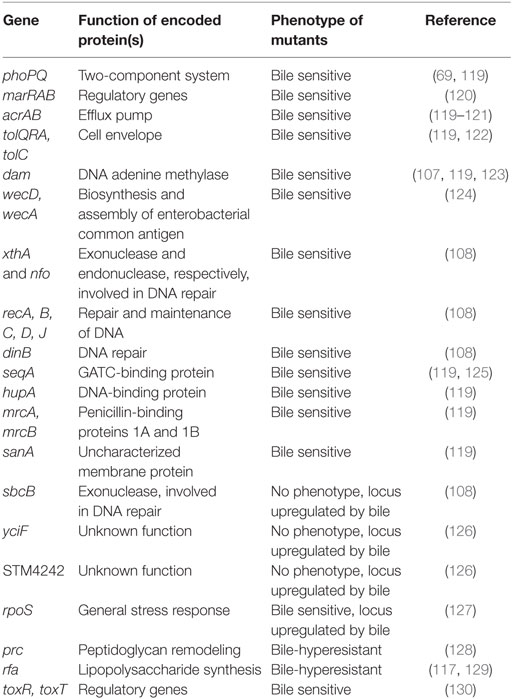
Table 1. Genetic loci that contribute to bile resistance in enteric bacteria.
The cell envelope of Gram-negative bacteria contains three layers: the cytoplasmic (inner) membrane, the peptidoglycan cell wall, and the outer membrane. The outer membrane is asymmetrical: its inner leaflet consists mainly of phospholipids while the outer leaflet is almost entirely composed of a glycolipid known as LPS (131). Loss of the O-antigen in the LPS results in decreased resistance to bile (117, 129); on the contrary, very long O-antigen chains increase bile resistance (132). The relevance of the LPS structure in bile resistance is further supported by the observation that S. enterica mutants hyper-resistant to bile often carry mutations in LPS transport genes (127).
Another cell envelope component that contributes to bile resistance is the enterobacterial common antigen (ECA), a family-specific glycolipid present in the outer leaflet of the outer membrane. In S. enterica, mutations in genes involved in ECA synthesis cause bile sensitivity (124).
Bile salts also induce peptidoglycan remodeling, and remodeling increases bile resistance. Growth of S. enterica in the presence of a sublethal concentration of DOC is accompanied by a reduction in the amount of Braun lipoprotein (Lpp) anchored to peptidoglycan (133). Because Lpp-containing muropeptides provide covalent linkage between the outer membrane and the peptidoglycan layer, reduction of this union may increase flexibility in the cell envelope, perhaps altering outer-membrane fluidity. Growth of S. enterica in the presence of DOC is also associated with a decrease in 3–3 crosslinks between the sugar components of peptidoglycan (N-acetylmuramic acid and N-acetylglucosamine), suggesting that low crosslinking may increase bile resistance (133).
Even though the bacterial envelope provides a barrier that reduces bile salt uptake, bile salts can enter the cell by diffusion or by passage through porins like OmpF. As a consequence, active efflux is necessary to reduce their concentration inside the cell (134). Among the efflux systems found in enterobacterial species, AcrAB–TolC is the best characterized (135–138). It comprises the outer-membrane protein channel TolC, the proton force-dependent transporter AcrB located in the inner membrane, and the periplasmic protein AcrA, which aids in efflux by bridging the TolC and AcrA integral membrane proteins (139). The AcrAB–TolC efflux pump is able to transport a diverse array of compounds with little chemical similarity (140), and is essential for bile resistance (120, 121, 141, 142). The genes encoding the AcrAB–TolC multidrug efflux system are under the control of a transcriptional regulator known as RamA. In turn, transcription of the ramA gene is activated by bile salts, mainly by relieving transcriptional repression exerted by the RamR protein (138, 143).
DNA adenine methylase (Dam–) mutants of S. enterica are bile sensitive (123, 144), and genetic analysis unveils the involvement of Dam-directed mismatch repair (107): mutations in any of the mismatch repair genes mutH, mutL, or mutS suppress bile sensitivity in dam mutants, providing evidence that bile sensitivity is caused by MutHLS activity. Salmonella MutHLS− mutants are not sensitive to bile, indicating that bile-induced DNA damage can be repaired by mechanisms other than Dam-dependent mismatch repair. In Dam− mutants, however, lack of DNA strand discrimination causes DNA strand breakage when the MutHLS systems deal with bile-induced lesions (107).
Surveys of bile sensitivity among S. enterica DNA repair mutants have revealed that base excision repair (BER), SOS-associated DNA repair, and recombinational repair by the RecBCD enzyme are required to cope with bile-induced DNA damage (108). In contrast, nucleotide excision repair (NER) is dispensable. The observation that bile resistance requires BER but not NER suggests that bile-induced DNA lesions are unlikely to be bulky, thus providing indirect support for the occurrence of oxidative damage (107, 108).
Several lines of evidence suggest that bile salts may impair DNA replication in Salmonella: (i) dinB mutations confer bile sensitivity, suggesting that SOS-associated translesion DNA synthesis may be required to overcome bile-induced DNA damage; (ii) RecB–, RecC–, and RecA− RecD− double mutants are also bile sensitive, indicating that survival to bile may require RecB-dependent homologous recombination (108). A tentative scenario is that primary lesions (e.g., oxidized cytosine moieties) may trigger BER. As a consequence, BER exonucleases will produce DNA strand breaks as an intermediate step in the DNA repair process. Furthermore, as indicated above, DNA strand breaks can also be formed as a consequence of MutHLS activity. These DNA strand breaks may impair progression of DNA replication forks, inducing the SOS response (see below); as a consequence, DinB-mediated translesion DNA synthesis may occur (108). It is also conceivable that bile-induced lesions could directly block DNA replication, thus inducing the SOS response in a direct fashion. In such a scenario, the need of homologous recombination mediated by the RecBCD enzyme complex might reflect the occurrence of stalled DNA replication forks (108).
Given the multiple injuries caused by bile salts to the bacterial cell, it is not surprising that stress regulons are induced in the presence of bile. The relevance of the DNA damage responsive SOS regulon, mentioned already in the previous section, is supported by the observation that LexA(Ind–) mutants, which are unable to induce the SOS response, are bile sensitive (108). Somehow surprisingly, the oxidative damage-responsive OxyR and SoxRS regulons, which are also activated by bile salts, are not essential for bile resistance (108). A tentative explanation is that redundance may exist in the stress responses of the bacterial cell so that certain functions can be backed up by others.
The RpoS-dependent general stress response is essential for bile resistance, and RpoS− mutants are bile sensitive. In S. enterica, an interesting feature of the RpoS response is inhibition of conjugation in the presence of bile, which involves posttranscriptional control of rpoS mRNA and ricI mRNAs by the small regulatory RNA RprA (145). The ricI gene encodes a cytoplasmic membrane protein that inhibits plasmid transfer by direct interaction with the conjugation apparatus protein TraV. RpoS− mutants of S. enterica are avirulent in the mouse model of typhoid (146, 147), and it seems reasonable to hypothesize that one cause of avirulence may be bile sensitivity.
Transcriptomic analysis has identified additional, RpoS-independent stress-inducible genes that increase their expression in the presence of DOC (cspD, uspA, aphC, etc.) (127). Although the contribution of these loci to bile resistance remains to be established, the number and variety of stress functions activated by bile salts supports the view that multiple stress responses contribute to bile resistance. Some such responses appear to be essential while others are not.
Salmonella stands out among bile-resistant bacterial genera because of its ability to colonize the hepatobiliary tract causing chronic infection. The current taxonomy defines six subspecies of S. enterica, which are in turn classified into serovars. The majority of serovars belong to subspecies enterica (148), which colonizes warm-blooded vertebrates (149) and accounts for 99% of human infections by Salmonella (150). Serovars belonging to subsp. enterica differ in host specificity and in the type of disease they produce. Some serovars are host-restricted, while others can infect a wide variety of animal hosts (151).
Figure 2 depicts the biology of Salmonella infections in humans. All diseases start upon invasion of the intestinal epithelium, often through M cells. Translocation across the intestinal epithelium is mediated by the virulence-associated type 3 secretion system encoded by Salmonella pathogenicity island 1 (SPI-1) (152), and invasion allows the bacteria to reach lymphocytes B and T below Peyer patches (153). Once the epithelium is crossed, S. enterica can produce three main types of infection: gastroenteritis, systemic infection, and asymptomatic chronic carriage.
(i) Gastroenteritis, a self-limited infection of the terminal ileum and colon which is the most common Salmonella infection worldwide, with more that 90 million cases per year (158). Gastroenteritis is produced by typhoidal serovars, especially Typhimuriun and Enteritidis. A localized inflammatory response induces infiltration of polymorphonuclear leukocytes, which helps to contain bacterial dissemination beyond the intestinal submucosa. Secretion of fluids and electrolytes in the small and large intestines produces diarrhea.
(ii) Systemic infection is produced by Salmonella serotypes that invade intestinal macrophages and disseminate inside the organism trough the lymphatic system, permitting colonization of internal organs such as the liver, the spleen, the bone marrow, and the gall bladder (Figure 2). In humans, typhoid and paratyphoid fever are caused by serotypes Typhi and Paratyphi, respectively. These infections are associated with high morbidity and mortality rates (159). Typhoid fever is estimated to cause over 20 million illnesses and over 200 thousand deaths worldwide, while the number of cases of paratyphoid fever is estimated over 5 million (160).
(iii) Chronic carriage. A fraction of individuals recovering from typhoid fever become asymptomatic, life-long carriers of S. Typhi. Non-typhoidal Salmonella serovars can also cause persistent infections, either associated with cholecystitis or asymptomatic, although the duration of carriage is usually limited to several months. The mesenteric lymph nodes, the liver, and the gall bladder appear to be the main Salmonella reservoirs during chronic carriage (161).
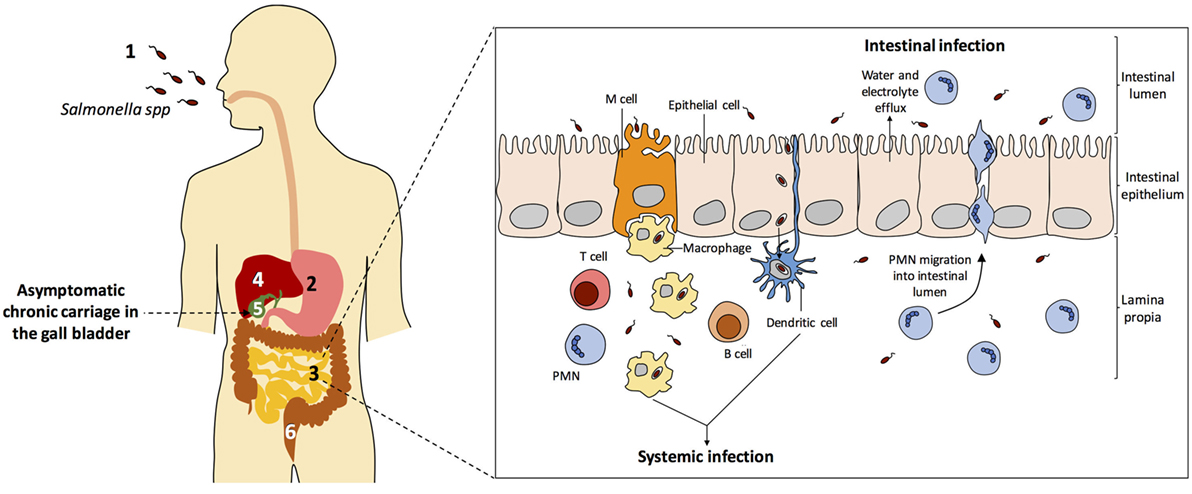
Figure 2. Salmonella enterica infection pathogenesis. (1) Salmonella infection starts with the ingestion of contaminated food or water. (2) Salmonellae invade intestinal epithelial cells and migrate to the lamina propria. (3) Two types of infection can occur: gastroenteritis and systemic infection. (4) During systemic dissemination, Salmonella colonizes the liver, the spleen, and the bone marrow. (5) From the liver, Salmonella reaches the gall bladder and can cause chronic carriage. (6) Salmonella carriers shed bacteria into the small intestine upon secretion of bile, and feces contain Salmonella cells. Inspired from Ref. (154–157).
Colonization of the gall bladder by Salmonella in asymptomatic carriers permits shedding of Salmonella cells into the intestine upon gall bladder contraction, with concomitant release of bile. From the small intestine, the bacteria travel downwards in the gut, ultimately being released with feces (153, 155). Aside from its epidemiological implications, chronic Salmonella carriage may predispose to gall bladder cancer, mainly as a consequence of chronic cholecystitis (162–164).
The bacterial lifestyle in the gall bladder is one of the less known aspects of Salmonella biology and a fascinating scientific conundrum. How is it possible that the high concentration of bile salts present in the gall bladder permits Salmonella survival? How is it possible that Salmonella survival in such a harsh environment can last for a lifetime? Answers to these questions are not simple; survival of Salmonella in the gall bladder appears to involve several adaptive strategies, which may be perhaps simultaneously adopted by subpopulations as a bet hedging strategy.
(i) A fraction of the Salmonella population may escape from bile salts by invasion of the gall bladder epithelium in a SPI-1-dependent manner, followed by replication in a vacuole (165). In this situation, extrusion of infected epithelial cells and release of Salmonella cells into the lumen has been observed. According to Gonzalez-Escobedo and Gunn (166), this mechanism could be important in maintaining the chronic carrier state and dissemination, because the bacteria released could either re-infect the epithelium or be shed into the medium.
(ii) Gallstones may play a major role in chronic infection. Using a murine model of typhoid carriage, John Gunn and co-workers have provided evidence that S. Typhimurium can form biofilms on the surface of cholesterol gallstones. The biofilm matrix provides high resistance to antimicrobial agents (167, 168), thereby explaining why antibiotic therapy is ineffective in carriers of S. enterica serovar Typhi who suffer from gallstone formation (169). This view is supported by the fact that S. typhi cells are detected on gallstones from human typhoid carriers (169).
(iii) Planktonic Salmonella cells can multiply in the gall bladder lumen, presumably using phospholipids as carbon and energy sources (170). How these unprotected cells endure the bactericidal activities of bile remains unknown. A tentative speculation is that activation of bile-responsive stress responses may generate cell lineages in which resistance to bile is maintained by feedback loops that are transmissible through cell division. In addition, bile-resistant mutants may appear, especially during longtime colonization as bile salts are mutagenic (108).
The composition of the bile salt pool is shaped by bacterial metabolism, and bile salts are used as physiological signals by both bacteria and hepatic cells. This entangled relationship is made even more complex by the fact that bile salts are antibacterial agents. A better understanding of the contribution of the bile salt pool to gastrointestinal and hepatobiliary homeostasis may inspire novel therapeutic strategies for conditions that involve microbiome alteration (e.g., cirrhosis, fatty liver disease, cholestasis, colorectal cancer, and certain types of Helicobacter-associated cancer). In turn, knowledge of mechanisms of bile resistance in intestinal pathogens may stimulate novel schemes to combat infectious diseases. For instance, eradication of Salmonella Typhi from the gall bladder of chronic carriers by procedures other than cholecystectomy might reduce chronic carriage of typhoid, a public health problem aggravated by global travel.
VU: literature search, writing, and drawing of figures. JC: literature search and writing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This study was supported by grant BIO2013-44220-R from the Spanish Ministerio de Economía y Competitividad (MINECO) and the European Regional Fund. VU is a postdoctoral researcher with a contract from the Consejería de Innovación, Ciencia y Empresa, Junta de Andalucía, Spain.
1. Hofmann AF, Hagey LR. Bile acids: chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol Life Sci (2008) 65:2461–83. doi:10.1007/s00018-008-7568-6
2. Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem (2003) 72:137–74. doi:10.1146/annurev.biochem.72.121801.161712
3. Fontana J, Trnka J, Mada P, Ivák P, Lavríková P, Nováková L, et al. Functions of Cells and Human Body. Prague: Karlovy University (2012).
4. Monte MJ, Marin JJG, Antelo A, Vazquez-Tato J. Bile acids: chemistry, physiology, and pathophysiology. World J Gastroenterol (2009) 15:804–16. doi:10.3748/wjg.15.804
6. Philipp B. Bacterial degradation of bile salts. Appl Microbiol Biotechnol (2011) 89:903–15. doi:10.1007/s00253-010-2998-0
7. Ridlon JM, Kang D-J, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res (2006) 47:241–59. doi:10.1194/jlr.R500013-JLR200
8. Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, et al. Host-gut microbiota metabolic interactions. Science (2012) 108:1262–8. doi:10.1126/science.1223813
9. Long SL, Gahan CGM, Joyce SA. Interactions between gut bacteria and bile in health and disease. Mol Aspects Med (2017) 5:1–12. doi:10.1016/j.mam.2017.06.002
10. Begley M, Hill C, Gahan CGM. Bile salt hydrolase activity in probiotics. Appl Environ Microbiol (2006) 72:1729–38. doi:10.1128/AEM.72.3.1729
11. Ridlon JM, Harris SC, Bhowmik S, Kang DJ, Hylemon PB. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes (2016) 7:22–39. doi:10.1080/19490976.2015.1127483
12. Elkins CA, Moser SA, Savage DC. Genes encoding bile salt hydrolases and conjugated bile salt transporters in Lactobacillus johnsonii 100-100 and other Lactobacillus species. Microbiology (2001) 147:3403–12. doi:10.1099/00221287-147-12-3403
13. Ren J, Sun K, Wu Z, Yao J, Guo B. All 4 bile salt hydrolase proteins are responsible for the hydrolysis activity in Lactobacillus plantarum ST-III. J Food Sci (2011) 76:622–8. doi:10.1111/j.1750-3841.2011.02431.x
14. Chae JP, Valeriano VD, Kim GB, Kang DK. Molecular cloning, characterization and comparison of bile salt hydrolases from Lactobacillus johnsonii PF01. J Appl Microbiol (2013) 114:121–33. doi:10.1111/jam.12027
15. Gu XC, Luo XG, Wang CX, Ma DY, Wang Y, He YY, et al. Cloning and analysis of bile salt hydrolase genes from Lactobacillus plantarum CGMCC No. 8198. Biotechnol Lett (2014) 36:975–83. doi:10.1007/s10529-013-1434-9
16. Jayashree S, Pooja S, Pushpanathan M, Rajendhran J, Gunasekaran P. Identification and characterization of bile salt hydrolase genes from the genome of Lactobacillus fermentum MTCC 8711. Appl Biochem Biotechnol (2014) 174:855–66. doi:10.1007/s12010-014-1118-5
17. Franz CM, Specht I, Haberer P, Holzapfel WH. Bile salt hydrolase activity of Enterococci isolated from food: screening and quantitative determination. J Food Prot (2001) 64:725–9. doi:10.4315/0362-028X-64.5.725
18. Wijaya A, Hermann A, Abriouel H, Specht I, Yousif NMK, Holzapfel WH, et al. Cloning of the bile salt hydrolase (bsh) gene from Enterococcus faecium FAIR-E 345 and chromosomal location of bsh genes in food enterococci. J Food Prot (2004) 67:2772–8. doi:10.4315/0362-028X-67.12.2772
19. Grill JP, Schneider F, Crociani J, Kim GB. Purification and characterization of two types of bile salt hydrolase from Bifidobacterium spp. Appl Environ Microbiol (1995) 61:2577–82.
20. Kim G-B, Yi S-H, Lee BH. Purification and characterization of three different types of bile salt hydrolases from Bifidobacterium strains. J Dairy Sci (2004) 87:258–66. doi:10.3168/jds.S0022-0302(04)73164-1
21. Tanaka H, Hashiba H, Kok J, Mierau I. Bile salt hydrolase of Bifidobacterium longum – biochemical and genetic characterization. Appl Environ Microbiol (2000) 66:2502–12. doi:10.1128/AEM.66.6.2502-2512.2000
22. Rossocha M, Schultz-Heienbrok R, Von Moeller H, Coleman JP, Saenger W. Conjugated bile acid hydrolase is a tetrameric N-terminal thiol hydrolase with specific recognition of its cholyl but not of its tauryl product. Biochemistry (2005) 44:5739–48. doi:10.1021/bi0473206
23. Jones BV, Begley M, Hill C, Gahan CGM, Marchesi JR. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc Natl Acad Sci U S A (2008) 105:13580–5. doi:10.1073/pnas.0804437105
24. Lambert JM, Bongers RS, De Vos WM, Kleerebezem M. Functional analysis of four bile salt hydrolase and penicillin acylase family members in Lactobacillus plantarum WCFS1. Appl Environ Microbiol (2008) 74:4719–26. doi:10.1128/AEM.00137-08
25. Begley M, Sleator RD, Gahan CG, Hill C. Contribution of the three bile-associated loci, bsh, pva, and btlB, to gastrointestinal persistence and bile tolerance of Listeria monocytogenes. Infect Immun (2005) 73:894–904. doi:10.1128/IAI.73.2.894
26. Hylemon PB, Harder J. Biotransformation of monoterpenes, bile acids, and other isoprenoids in anaerobic ecosystems. FEMS Microbiol Rev (1998) 22:475–88. doi:10.1016/S0168-6445(98)00027-8
27. Batta AK, Salen G, Arora R, Shefer S, Batta M, Person A. Side chain conjugation prevents bacterial 7-dehydroxylation of bile acids. J Biol Chem (1990) 265:10925–8.
28. Wells JE, Williams KB, Whitehead TR, Heuman DM, Hylemon PB. Development and application of a polymerase chain reaction assay for the detection and enumeration of bile acid 7α-dehydroxylating bacteria in human feces. Clin Chim Acta (2003) 331:127–34. doi:10.1016/S0009-8981(03)00115-3
29. Mallonee DH, Hylemon PB. Sequencing and expression of a gene encoding a bile acid transporter from Eubacterium sp. strain VPI 12708. J Bacteriol (1996) 178:7053–8. doi:10.1128/jb.178.24.7053-7058.1996
30. Ridlon JM, Hylemon PB. Identification and characterization of two bile acid coenzyme A transferases from Clostridium scindens, a bile acid 7-dehydroxylating intestinal bacterium. J Lipid Res (2012) 53:66–76. doi:10.1194/jlr.M020313
31. Ridlon JM, Kang DJ, Hylemon PB. Isolation and characterization of a bile acid inducible 7α-dehydroxylating operon in Clostridium hylemonae TN271. Anaerobe (2010) 16:137–46. doi:10.1016/j.anaerobe.2009.05.004
32. Kang D-J, Ridlon JM, Moore DR II, Barnes S, Hylemon PB. Clostridium scindens baiCD and baiH genes encode stereo-specific 7α/7β-hydroxy-3-oxo-Δ4-cholenoic acid oxidoreductases. Biochim Biophys Acta (2008) 1781:16–25. doi:10.1016/j.bbalip.2007.10.008
33. Swann JR, Want EJ, Geier FM, Spagou K, Wilson ID, Sidaway JE, et al. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc Natl Acad Sci U S A (2011) 108:4523–30. doi:10.1073/pnas.1006734107
34. Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab (2013) 17:225–35. doi:10.1016/j.cmet.2013.01.003
35. Vander A, Sherman J, Luciano D. Human Physiology: The Mechanisms of Body Function. 8th ed. Boston: McGraw-Hill (2001).
36. Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. bioRxiv (2016) 14(8):e1002533. doi:10.1371/journal.pbio.1002533
37. Whitman WB, Coleman DC, Wiebe WJ. Prokaryotes: the unseen majority. Proc Natl Acad Sci U S A (1998) 95:6578–83. doi:10.1073/pnas.95.12.6578
38. Montagner A, Korecka A, Polizzi A, Lippi Y, Blum Y, Canlet C, et al. Hepatic circadian clock oscillators and nuclear receptors integrate microbiome-derived signals. Sci Rep (2016) 6:20127. doi:10.1038/srep20127
39. Ridlon J, Kang D, Hylemon P, Bajaj J. Bile acids and the gut microbiome. Curr Opin Gastroenterol (2014) 30:332–8. doi:10.1097/MOG.0000000000000057
40. Kakiyama G, Pandak WM, Gillevet PM, Hylemon PB, Heuman DM, Daita K, et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J Hepatol (2013) 58:949–55. doi:10.1016/j.jhep.2013.01.003
41. Bajaj JS, Hylemon PB, Ridlon JM, Heuman DM, Daita K, White MB, et al. Colonic mucosal microbiome differs from stool microbiome in cirrhosis and hepatic encephalopathy and is linked to cognition and inflammation. AJP Gastrointest Liver Physiol (2012) 303:G675–85. doi:10.1152/ajpgi.00152.2012
42. Devkota S, Wang Y, Musch M. Dietary fat-induced taurocholic acid production promotes pathobiont and colitis in IL-10-/-mice. Nature (2012) 487:1–9. doi:10.1038/nature11225
43. Baron EJ, Summanen P, Downes J, Roberts MC, Wexler HFS. Bilophila wadsworthia, a unique gram-negative anaerobic rod recovered from appendicitis specimens and human faeces. J Gen Microbiol (1989) 135:3405–11.
44. Loubinoux J, Bronowicki JP, Pereira IAC, Mougenel JL, Faou AE. Sulfate-reducing bacteria in human feces and their association with inflammatory bowel diseases. FEMS Microbiol Ecol (2002) 40:107–12. doi:10.1111/j.1574-6941.2002.tb00942.x
45. Forman BM, Goode E, Chen J, Oro AE, Bradley DJ, Perlmann T, et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell (1995) 81:687–93. doi:10.1016/0092-8674(95)90530-8
46. Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, et al. Identification of a nuclear receptor for bile acids. Science (1999) 284:1362–5. doi:10.1126/science.284.5418.1362
47. Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, et al. Vitamin D receptor as an intestinal bile acid sensor. Science (2002) 296:1313–6. doi:10.1126/science.1070477
48. D’Aldebert E, Biyeyeme Bi Mve MJ, Mergey M, Wendum D, Firrincieli D, Coilly A, et al. Bile salts control the antimicrobial peptide cathelicidin through nuclear receptors in the human biliary epithelium. Gastroenterology (2009) 136:1435–43. doi:10.1053/j.gastro.2008.12.040
49. Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol (2009) 183:6251–61. doi:10.4049/jimmunol.0803978
50. Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A (2006) 103:3920–5. doi:10.1073/pnas.0509592103
51. Chromek M, Slamová Z, Bergman P, Kovács L, Podracká L, Ehrén I, et al. The antimicrobial peptide cathelicidin protects the urinary tract against invasive bacterial infection. Nat Med (2006) 12:636–41. doi:10.1038/nm1407
52. Dorschner RA, Pestonjamasp VK, Tamakuwala S, Ohtake T, Rudisill J, Nizet V, et al. Cutaneous injury induces the release of cathelicidin anti-microbial peptides active against group A streptococcus. J Invest Dermatol (2001) 117:91–7. doi:10.1046/j.1523-1747.2001.01340.x
53. Zanetti M. Cathelicidins, multifunctional peptides of the innate immunity. J Leukoc Biol (2003) 75:39–48. doi:10.1189/jlb.0403147
54. Ridlon JM, Alves JM, Hylemon PB, Bajaj JS. Cirrhosis, bile acids and gut microbiota. Gut Microbes (2013) 4:382–7. doi:10.4161/gmic.25723
55. Kakiyama G, Hylemon PB, Zhou H, Pandak WM, Heuman DM, Kang DJ, et al. Colonic inflammation and secondary bile acids in alcoholic cirrhosis. Am J Physiol Gastrointest Liver Physiol (2014) 306:G929–37. doi:10.1152/ajpgi.00315.2013
56. Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Gut microbiota, cirrhosis, and alcohol regulate bile acid metabolism in the gut. Dig Dis (2015) 33:338–45. doi:10.1159/000371678
57. Arab JP, Karpen SJ, Dawson PA, Arrese M, Trauner M. Bile acids and nonalcoholic fatty liver disease: molecular insights and therapeutic perspectives. Hepatology (2017) 65:350–62. doi:10.1002/hep.28709
58. Li T, Apte U. Bile acid metabolism and signaling in cholestasis, inflammation and cancer. Adv Pharmacol (2015) 74:263–302. doi:10.1016/bs.apha.2015.04.003
59. Fan M, Wang X, Xu G, Yan Q, Huang W. Bile acid signaling and liver regeneration. Biochim Biophys Acta (2014) 1849:196–200. doi:10.1016/j.bbagrm.2014.05.021
60. Fukui H. Gut microbiota and host reaction in liver diseases. Microorganisms (2015) 3:759–91. doi:10.3390/microorganisms3040759
61. Nardone G, Compare D. The human gastric microbiota: is it time to rethink the pathogenesis of stomach diseases? United Eur Gastroenterol J (2015) 3:255–60. doi:10.1177/2050640614566846
62. Wroblewski LE, Peek RM. Helicobacter pylori in gastric carcinogenesis: mechanisms. Gastroenterol Clin North Am (2013) 42:285–98. doi:10.1016/j.gtc.2013.01.006
63. Mukaisho KI, Nakayama T, Hagiwara T, Hattori T, Sugihara H. Two distinct etiologies of gastric cardia adenocarcinoma: interactions among pH, Helicobacter pylori, and bile acids. Front Microbiol (2015) 6:1–7. doi:10.3389/fmicb.2015.00412
64. de Martel C, Plummer M, Parsonnet J, van Doorn LJ, Franceschi S. Helicobacter species in cancers of the gallbladder and extrahepatic biliary tract. Br J Cancer (2009) 100:194–9. doi:10.1038/sj.bjc.6604780
65. Hamada T, Yokota K, Ayada K, Hirai K, Kamada T, Haruma K, et al. Detection of Helicobacter hepaticus in human bile samples of patients with biliary disease. Helicobacter (2009) 14:545–51. doi:10.1111/j.1523-5378.2009.00729.x
66. Sabbaghian MS, Ranaudo J, Zeng L, Alongi AP, Perez-Perez G, Shamamian P. Identification of Helicobacter spp. in bile and gallbladder tissue of patients with symptomatic gallbladder disease. HPB (2010) 12:129–33. doi:10.1111/j.1477-2574.2009.00148.x
67. Pope LM, Reed KE, Payne SM. Increased protein secretion and adherence to HeLa cells by Shigella spp. following growth in the presence of bile salts. Infect Immun (1995) 63:3642–8.
68. Faherty CS, Redman JC, Rasko DA, Barry EM, Nataro JP. Shigella flexneri effectors OspE1 and OspE2 mediate induced adherence to the colonic epithelium following bile salts exposure. Mol Microbiol (2012) 85(1):107–21. doi:10.1111/j.1365-2958.2012.08092.x
69. van Velkinburgh JC, Gunn JS. PhoP-PhoQ-regulated loci are required for enhanced bile resistance in Salmonella spp. Infect Immun (1999) 67:1614–22.
70. Prouty AM, Gunn JS. Salmonella enterica serovar Typhimurium invasion is repressed in the presence of bile. Infect Immun (2000) 68:6763–9. doi:10.1128/IAI.68.12.6763-6769.2000
71. Gupta S, Chowdhury R. Bile affects production of virulence factors and motility of Vibrio cholerae. Infect Immun (1997) 65:1131–4.
72. Hung DT, Zhu J, Sturtevant D, Mekalanos JJ. Bile acids stimulate biofilm formation in Vibrio cholerae. Mol Microbiol (2006) 59:193–201. doi:10.1016/j.jallcom.2005.11.011
73. Malik-Kale P, Parker CT, Konkel ME. Culture of Campylobacter jejuni with sodium deoxycholate induces virulence gene expression. J Bacteriol (2008) 190:2286–97. doi:10.1128/JB.01736-07
74. Solheim M, Aakra AF, Vebø H, Snipen L, Nes IF. Transcriptional responses of Enterococcus faecalis V583 to bovine bile and sodium dodecyl sulfate. Appl Environ Microbiol (2007) 73:5767–74. doi:10.1128/AEM.00651-07
75. Quillin SJ, Schwartz KT, Leber JH. The novel Listeria monocytogenes bile sensor BrtA controls expression of the cholic acid efflux pump MdrT. Mol Microbiol (2011) 81:129–42. doi:10.1111/j.1365-2958.2011.07683.x
76. Li P, Rivera-Cancel G, Kinch LN, Salomon D, Tomchick DR, Grishin NV, et al. Bile salt receptor complex activates a pathogenic type III secretion system. Elife (2016) 5:1–26. doi:10.7554/eLife.15718
77. Schuhmacher DA, Klose KE. Environmental signals modulate ToxT-dependent virulence factor expression in Vibrio cholerae. J Bacteriol (1999) 181:1508–14.
78. Provenzano D, Klose KE. Altered expression of the ToxR-regulated porins OmpU and OmpT diminishes Vibrio cholerae bile resistance, virulence factor expression, and intestinal colonization. Proc Natl Acad Sci U S A (2000) 97:10220–4. doi:10.1073/pnas.170219997
79. DiRita VJ, Parsot C, Jander G, Mekalanos JJ. Regulatory cascade controls virulence in Vibrio cholerae. Proc Natl Acad Sci U S A (1991) 88:5403–7. doi:10.1073/pnas.88.12.5403
80. Eade CR, Hung CC, Bullard B, Gonzalez-Escobedo G, Gunn JS, Altier C. Bile acids function synergistically to repress invasion gene expression in Salmonella by destabilizing the invasion regulator HilD. Infect Immun (2016) 84:2198–208. doi:10.1128/IAI.00177-16
81. Bartlett JG. The new epidemic of Clostridium difficile-associated enteric disease. An Intern Med (2006) 145:758–64. doi:10.7326/0003-4819-145-10-200611210-00008
82. Abt MC, McKenney PT, Pamer EG. Clostridium difficile colitis: pathogenesis and host defence. Nat Rev Microbiol (2016) 14:609–20. doi:10.1038/nrmicro.2016.108
83. Francis MB, Allen CA, Shrestha R, Sorg JA. Bile acid recognition by the Clostridium difficile germinant receptor, CspC, is important for establishing infection. PLoS Pathog (2013) 9:e1003356. doi:10.1371/journal.ppat.1003356
84. Wilson KH. Efficiency of various bile salt preparations for stimulation of Clostridium difficile spore germination. J Clin Microbiol (1983) 18:1017–9.
85. Sorg JA, Sonenshein AL. Inhibiting the initiation of Clostridium difficile spore germination using analogs of chenodeoxycholic acid, a bile acid. J Bacteriol (2010) 192:4983–90. doi:10.1128/JB.00610-10
86. Theriot CM, Bowman AA, Young VB. Antibiotic-induced alterations of the gut microbiota alter secondary bile acid production and allow for Clostridium difficile spore germination and outgrowth in the large intestine. mSphere (2015) 1:e45–15. doi:10.1128/mSphere.00045-15
87. Hamner S, McInnerney K, Williamson K, Franklin MJ, Ford TE. Bile salts affect expression of Escherichia coli O157:H7 genes for virulence and iron acquisition, and promote growth under iron limiting conditions. PLoS One (2013) 8:e74647. doi:10.1371/journal.pone.0074647
88. Garmendia J, Frankel G, Crepin VF. Enteropathogenic and enterohemorrhagic Escherichia coli infections: translocation, translocation, translocation. Infect Immun (2005) 73:2573–85. doi:10.1128/IAI.73.5.2573
89. Hopf U, Stemerowicz R, Rodloff A, Galanos C, Möller B, Lobeck H, et al. Relation between Escherichia coli R(Rough)-forms in gut, lipid A in liver, and primary biliary cirrhosis. Lancet (1989) 334:1419–22. doi:10.1016/S0140-6736(89)92034-5
90. Sasatomi K, Noguchi K, Sakisaka S, Sata M, Tanikawa K. Abnormal accumulation of endotoxin in biliary epithelial cells in primary biliary cirrhosis and primary sclerosing cholangitis. J Hepatol (1998) 29:409–16. doi:10.1016/S0168-8278(98)80058-5
91. Leverrier P, Dimova D, Pichereau V, Auffray Y, Boyaval P, Jan G. Susceptibility and adaptive response to bile salts in Propionibacterium freudenreichii: physiological and proteomic analysis. Appl Environ Microbiol (2003) 69:3809–18. doi:10.1128/AEM.69.7.3809
92. Fujisawa T, Mori M. Influence of bile salts on β-glucuronidase activity of intestinal bacteria. Lett Appl Microbiol (1996) 22:271–4. doi:10.1111/j.1472-765X.1996.tb01159.x
93. Noh DO, Gilliland SE. Influence of bile on cellular integrity and beta-galactosidase activity of Lactobacillus acidophilus. J Dairy Sci (1993) 76:1253–9. doi:10.3168/jds.S0022-0302(93)77454-8
94. Coleman R, Lowe PJ, Billington D. Membrane lipid composition and susceptibility to bile salt damage. Biochim Biophys Acta (1980) 599:294–300. doi:10.1016/0005-2736(80)90075-9
95. Gómez Zavaglia A, Kociubinski G, Pérez P, Disalvo E, De Antoni G. Effect of bile on the lipid composition and surface properties of bifidobacteria. J Appl Microbiol (2002) 93:794–9. doi:10.1046/j.1365-2672.2002.01747.x
96. Záratea G, Gonzaleza S, Chaia AP, Oliver G. Effect of bile on the β-galactosidase activity of dairy propionibacteria. Lait (2000) 80:267–76. doi:10.1051/lait:2000125
97. Hofmann M, Schumann C, Zimmer G, Henzel K, Locher U, Leuschner U. LUV’s lipid composition modulates diffusion of bile acids. Chem Phys Lipids (2001) 110:165–71. doi:10.1016/S0009-3084(01)00131-1
98. Cabral DJ, Small DM, Lilly HS, Hamilton JA. Transbilayer movement of bile acids in model membranes. Biochemistry (1987) 26:1801–4. doi:10.1021/bi00381a002
99. Schubert R, Jaroni H, Schoelmerich J, Schmidt KH. Studies on the mechanism of bile salt-induced liposomal membrane damage. Digestion (1983) 28:181–90. doi:10.1159/000198984
100. Chou C, Cheng S. Recovery of low-temperature stressed E. coli O157:H7 and its susceptibility to crystal violet, bile salt, sodium chloride and ethanol. Int J Food Microbiol (2000) 61:127–36. doi:10.1016/S0168-1605(00)00266-X
101. Chou LS, Weimer B. Isolation and characterization of acid- and bile-tolerant isolates from strains of Lactobacillus acidophilus. J Dairy Sci (1999) 82:23–31. doi:10.3168/jds.S0022-0302(99)75204-5
102. King T, Ferenci T, Szabo EA. The effect of growth atmosphere on the ability of Listeria monocytogenes to survive exposure to acid, proteolytic enzymes and bile salts. Int J Food Microbiol (2003) 84:133–43. doi:10.1016/S0168-1605(02)00404-X
103. Fernández Murga ML, Bernik D, Font de Valdez G, Disalvo AE. Permeability and stability properties of membranes formed by lipids extracted from Lactobacillus acidophilus grown at different temperatures. Arch Biochem Biophys (1999) 364:115–21. doi:10.1006/abbi.1998.1093
104. Kandell RL, Bernstein C. Bile salt/acid induction of DNA damage in bacterial and mammalian cells: implications for colon cancer. Nutr Cancer (1991) 16:227–38. doi:10.1080/01635589109514161
105. Bernstein C, Bernstein H, Payne CM, Beard SE, Schneider J. Bile salt activation of stress response promoters in Escherichia coli. Curr Microbiol (1999) 39:68–72. doi:10.1007/s002849900420
106. Chou JH, Greenberg JT, Demple B. Posttranscriptional repression of Escherichia coli OmpF protein in response to redox stress: positive control of the micF antisense RNA by the soxRS locus. J Bacteriol (1993) 175:1026–31. doi:10.1128/jb.175.4.1026-1031.1993
107. Prieto AI, Ramos-Morales F, Casadesús J. Bile-induced DNA damage in Salmonella enterica. Genetics (2004) 168:1787–94. doi:10.1534/genetics.104.031062
108. Prieto AI, Ramos-Morales F, Casadesús J. Repair of DNA damage induced by bile salts in Salmonella enterica. Genetics (2006) 174:575–84. doi:10.1534/genetics.106.060889
109. García-Quintanilla M, Ramos-Morales F, Casadesús J. Conjugal transfer of the Salmonella enterica virulence plasmid in the mouse intestine. J Bacteriol (2008) 190:1922–7. doi:10.1128/JB.01626-07
110. Willetts NS. The elimination of Flac+ from Escherichia coli by mutagenic agents. Biochem Biophys Res Commun (1967) 27:112–7. doi:10.1016/S0006-291X(67)80048-2
111. Flahaut S, Frere J, Boutibonnes P, Auffray Y. Comparison of the bile salts and sodium dodecyl sulfate stress responses in Enterococcus faecalis. Appl Environ Microbiol (1996) 62:2416–20.
112. Sanyal AJ, Hirsch JI, Moore EW. Premicellar taurocholate enhances calcium uptake from all regions of rat small intestine. Gastroenterology (1994) 106:866–74. doi:10.1016/0016-5085(94)90744-7
113. Symeonidis A, Marangos M. Iron and microbial growth. Insight Control Infect Dis Glob Scenar (2012) 16:289–330. doi:10.5772/34760
114. Rajagopalan N, Lindenbaum S. The binding of Ca2+ to taurine and glycine-conjugated bile salt micelles. Biochim Biophys Acta (1982) 711:66–74. doi:10.1016/0005-2760(82)90010-8
115. Michiels J, Xi C, Verhaert J, Vanderleyden J. The functions of Ca2+ in bacteria: a role for EF-hand proteins? Trends Microbiol (2002) 10:87–93. doi:10.1016/S0966-842X(01)02284-3
116. Dominguez DC. Calcium signalling in bacteria. Mol Microbiol (2004) 54:291–7. doi:10.1111/j.1365-2958.2004.04276.x
117. Gunn JS. Mechanisms of bacterial resistance and response to bile. Microbes Infect (2000) 2:907–13. doi:10.1016/S1286-4579(00)00392-0
118. Casadesús J, Hernández SB, Cota I, Ramos-Morales F. Of bacteria and bile. In: Maloy S, Hughes KT, Casadesús J, editors. The Lure of Bacterial Genetics. Washington, DC: American Society of Microbiology (2010). p. 153–62.
119. Langridge GC, Phan MD, Turner DJ, Perkins TT, Parts L, Haase J, et al. Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Res (2009) 19:2308–16. doi:10.1101/gr.097097.109
120. Prouty AM, Brodsky IE, Falkow S, Gunn JS. Bile-salt-mediated induction of antimicrobial and bile resistance in Salmonella typhimurium. Microbiology (2004) 150:775–83. doi:10.1099/mic.0.26769-0
121. Lacroix FJC, Cloeckaert A, Grépinet O, Pinault C, Popoff MY, Waxin H, et al. Salmonella typhimurium acrB-like gene: identification and role in resistance to biliary salts and detergents and in murine infection. FEMS Microbiol Lett (1996) 135:161–7. doi:10.1016/0378-1097(95)00443-2
122. Prouty AM, Van Velkinburgh JC, Gunn JS. Salmonella enterica serovar typhimurium resistance to bile: Identification and characterization of the tolQRA cluster. J Bacteriol (2002) 184:1270–6. doi:10.1128/JB.184.5.1270
123. Heithoff DM, Enioutina EY, Daynes RA, Sinsheimer RL, Low DA, Mahan MJ. Salmonella DNA adenine methylase mutants confer cross-protective immunity. Infect Immun (2001) 69:6725–30. doi:10.1128/IAI.69.11.6725-6730.2001
124. Ramos-Morales F, Prieto AI, Beuzon CR, Holden DW, Casadesús J. Role for Salmonella enterica enterobacterial common antigen in bile resistance and virulence. J Bacteriol (2003) 185:5328–32. doi:10.1128/JB.185.17.5328
125. Prieto AI, Jakomin M, Segura I, Pucciarelli MG, Ramos-Morales F, García-Del Portillo F, et al. The GATC-binding protein SeqA is required for bile resistance and virulence in Salmonella enterica serovar Typhimurium. J Bacteriol (2007) 189:8496–502. doi:10.1128/JB.01156-07
126. Prouty AM, Brodsky IE, Manos J, Belas R, Falkow S, Gunn JS. Transcriptional regulation of Salmonella enterica serovar Typhimurium genes by bile. FEMS Immunol Med Microbiol (2004) 41:177–85. doi:10.1016/j.femsim.2004.03.002
127. Hernández SB, Cota I, Ducret A, Aussel L, Casadesús J. Adaptation and preadaptation of Salmonella enterica to bile. PLoS Genet (2012) 8:e1002459. doi:10.1371/journal.pgen.1002459
128. Hernández SB, Ayala JA, Rico-Pérez G, García-del Portillo F, Casadesús J. Increased bile resistance in Salmonella enterica mutants lacking Prc periplasmic protease. Int Microbiol (2013) 16:87–92. doi:10.2436/20.1501.01.183
129. Picken R, Beacham I. Bacteriophage-resistant mutants of Escherichia coli K12. Location of receptors within the lipopolysaccharide. J Gen Microbiol (1977) 102:305–18. doi:10.1099/00221287-102-2-319
130. Provenzano D, Schuhmacher DA, Barker JL, Klose KE. The virulence regulatory protein ToxR mediates enhanced bile resistance in Vibrio cholerae and other pathogenic Vibrio species. Infect Immun (2000) 68:1491–7. doi:10.1128/IAI.68.3.1491-1497
131. Silhavy TJ, Kahne D, Walker S. The bacterial cell envelope. Cold Spring Harb Perspect Biol (2010) 2:1–17. doi:10.1101/cshperspect.a000414
132. Crawford RW, Keestra AM, Winter SE, Xavier MN, Tsolis RM, Tolstikov V, et al. Very long O-antigen chains enhance fitness during Salmonella-induced colitis by increasing bile resistance. PLoS Pathog (2012) 8:e1002918. doi:10.1371/journal.ppat.1002918
133. Hernández SB, Cava F, Pucciarelli MG, García-del Portillo F, de Pedro MA, Casadesús J. Bile-induced peptidoglycan remodelling in Salmonella enterica. Environ Microbiol (2015) 17:1081–9. doi:10.1111/1462-2920.12491
134. Thanassi DG, Cheng LW, Nikaido H. Active efflux of bile salts by Escherichia coli. J Bacteriol (1997) 179:2512–8. doi:10.1128/jb.179.8.2512-2518.1997
135. Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, Hearst JE. Genes acrA and acrB encode a stress-induced efflux system of Escherichia coli. Mol Microbiol (1995) 16:45–55. doi:10.1111/j.1365-2958.1995.tb02390.x
136. Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, Hearst JE. Molecular cloning and characterization of acrA and acrE genes of Escherichia coli. J Bacteriol (1993) 175:6299–313. doi:10.1128/jb.175.19.6299-6313.1993
137. Zgurskaya HI, Nikaido H. Bypassing the periplasm: reconstitution of the AcrAB multidrug efflux pump of Escherichia coli. Proc Natl Acad Sci U S A (1999) 96:7190–5. doi:10.1073/pnas.96.13.7190
138. Nikaido E, Yamaguchi A, Nishino K. AcrAB multidrug efflux pump regulation in Salmonella enterica serovar Typhimurium by RamA in response to environmental signals. J Biol Chem (2008) 283:24245–53. doi:10.1074/jbc.M804544200
139. Pos KM. Drug transport mechanism of the AcrB efflux pump. Biochim Biophys Acta (2009) 1794:782–93. doi:10.1016/j.bbapap.2008.12.015
140. Murakami S, Nakashima R, Yamashita E, Yamaguchi A. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature (2002) 419:587–93. doi:10.1038/nature01050
141. Nikaido H, Basina M, Nguyen V, Emiko Y, Rosenberg EY. Multidrug efflux pump AcrAB of Salmonella typhimurium excretes only those β-lactam antibiotics containing lipophilic side chains. J Bacteriol (1998) 180:4686–92.
142. Nishino K, Latifi T, Groisman EA. Virulence and drug resistance roles of multidrug efflux systems of Salmonella enterica serovar Typhimurium. Mol Microbiol (2006) 59:126–41. doi:10.1111/j.1365-2958.2005.04940.x
143. Baucheron S, Nishino K, Monchaux I, Canepa S, Maurel MC, Coste F, et al. Bile-mediated activation of the acrAB and tolC multidrug efflux genes occurs mainly through transcriptional derepression of ramA in Salmonella enterica serovar Typhimurium. J Antimicrob Chemother (2014) 69:2400–6. doi:10.1093/jac/dku140
144. Cano DA, Pucciarelli MG, Garcia-Del Portillo F, Casadesús J. Role of the RecBCD recombination pathway in Salmonella virulence. J Bacteriol (2002) 184:592–5. doi:10.1128/JB.184.2.592-595.2002
145. Papenfort K, Espinosa E, Casadesús J, Vogel J. Small RNA-based feedforward loop with AND-gate logic regulates extrachromosomal DNA transfer in Salmonella. Proc Natl Acad Sci U S A (2015) 112:E4772–81. doi:10.1073/pnas.1507825112
146. Fang FC, Libby SJ, Buchmeier NA, Loewen PC, Switala J, Harwood J, et al. The alternative sigma factor KatF (RpoS) regulates Salmonella virulence. Proc Natl Acad Sci U S A (1992) 89:11978–82. doi:10.1073/pnas.89.24.11978
147. Chen C, Buchmeier NA, Libby S, Fang FC, Krause M, Guiney DG. Central regulatory role for the RpoS sigma factor in expression of Salmonella dublin plasmid virulence genes. J Bacteriol (1995) 177:5303–9. doi:10.1128/jb.177.18.5303-5309.1995
148. Popoff MY, Bockemühl J, Gheesling LL. Supplement 2002 (no. 46) to the Kauffmann-White scheme. Res Microbiol (2004) 155:568–70. doi:10.1016/j.resmic.2004.04.005
149. McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, Courtney L, et al. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature (2001) 413:852–6. doi:10.1038/35101614
150. EFSA, ECDC. The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2014. EFSA J (2015) 13:1–191. doi:10.2903/j.efsa.2015.4329
151. Bäumler A, Fang FC. Host specificity of bacterial pathogens. Cold Spring Harb Perspect Med (2013) 3:1–20. doi:10.1101/cshperspect.a010041
152. Zhou D, Galán J. Salmonella entry into host cells: the work in concert of type III secreted effector proteins. Microbes Infect (2001) 3:1293–8. doi:10.1016/S1286-4579(01)01489-7
153. Haraga A, Ohlson MB, Miller SI. Salmonellae interplay with host cells. Nat Rev Microbiol (2008) 6:53–66. doi:10.1038/nrmicro1788
154. Sansonetti PJ. War and peace at mucosal surfaces. Nat Rev Immunol (2004) 4:953–64. doi:10.1038/nri1499
155. Tischler AD, Mckinney JD. Contrasting persistence strategies in Salmonella and Mycobacterium. Curr Opin Microbiol (2010) 4:93–9. doi:10.1016/j.mib.2009.12.007
156. Fournier BM, Parkos CA. The role of neutrophils during intestinal inflammation. Mucosal Immunol (2012) 5:354–66. doi:10.1038/mi.2012.24
157. Nell S, Suerbaum S, Josenhans C. The impact of the microbiota on the pathogenesis of IBD: lessons from mouse infection models. Nat Rev Microbiol (2010) 8:564–77. doi:10.1038/nrmicro2403
158. Majowicz SE, Musto J, Scallan E, Angulo FJ, Kirk M, O’Brien SJ, et al. The global burden of nontyphoidal Salmonella gastroenteritis. Clin Infect Dis (2010) 50:882–9. doi:10.1086/650733
159. Hardy A. Salmonella: a continuing problem. Postgrad Med J (2004) 80:541–5. doi:10.1136/pgmj.2003.016584
160. Crump JA, Luby SP, Mintz ED. The global burden of typhoid fever. Bull World Health Organ (2004) 82:346–53. doi:10.1590/S0042-96862004000500008
161. Watson KG, Holden DW. Dynamics of growth and dissemination of Salmonella in vivo. Cell Microbiol (2010) 12:1389–97. doi:10.1111/j.1462-5822.2010.01511.x
162. Dutta U, Garg PK, Kumar R, Tandon RK. Typhoid carriers among patients with gallstones are at increased risk for carcinoma of the gallbladder. Am J Gastroenterol (2000) 95:784–7. doi:10.1111/j.1572-0241.2000.01860.x
163. Shukla VK, Singh H, Pandey M, Upadhyay SK, Nath G. Carcinoma of the gallbladder-is it a sequel of typhoid? Dig Dis Sci (2000) 45:900–3. doi:10.1023/A:1005564822630
164. Kumar S. Infection as a risk factor for gallbladder cancer. J Surg Oncol (2006) 93:633–9. doi:10.1002/jso.20530
165. Menendez A, Arena ET, Guttman JA, Thorson L, Vallance BA, Vogl W, et al. Salmonella infection of gallbladder epithelial cells drives local inflammation and injury in a model of acute typhoid fever. J Infect Dis (2009) 200:1703–13. doi:10.1086/646608
166. Gonzalez-Escobedo G, Gunn JS. Gallbladder epithelium as a niche for chronic Salmonella carriage. Infect Immun (2013) 81:2920–30. doi:10.1128/IAI.00258-13
167. Crawford RW, Gibson DL, Kay WW, Gunn JS. Identification of a bile-induced exopolysaccharide required for Salmonella biofilm formation on gallstone surfaces. Infect Immun (2008) 76:5341–9. doi:10.1128/IAI.00786-08
168. Prouty AM, Schwesinger WH, Gunn JS. Biofilm formation and interaction with the surfaces of gallstones by Salmonella spp. Infect Immun (2002) 70:2640–9. doi:10.1128/IAI.70.5.2640
169. Crawford RW, Rosales-Reyes R, Ramírez-Aguilar ML, Chapa-Azuela O, Alpuche-Aranda C, Gunn JS. Gallstones play a significant role in Salmonella spp. gallbladder colonization and carriage. Proc Natl Acad Sci U S A (2010) 107:4353–8. doi:10.1073/pnas.1000862107
Keywords: bile salts, gall bladder, intestinal microbiome, gene regulation, resistance to bile, Salmonella, chronic infection
Citation: Urdaneta V and Casadesús J (2017) Interactions between Bacteria and Bile Salts in the Gastrointestinal and Hepatobiliary Tracts. Front. Med. 4:163. doi: 10.3389/fmed.2017.00163
Received: 02 August 2017; Accepted: 19 September 2017;
Published: 03 October 2017
Edited by:
Samy A. Azer, King Saud University, Saudi Arabia; University of Melbourne, AustraliaReviewed by:
Taro Takami, Yamaguchi University, JapanCopyright: © 2017 Urdaneta and Casadesús. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Josep Casadesús, Y2FzYWRlc3VzQHVzLmVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.