 Marek Cernoch1
Marek Cernoch1 Ondrej Viklicky1,2*
Ondrej Viklicky1,2*
- 1Transplant Laboratory, Transplant Center, Institute for Clinical and Experimental Medicine, Prague, Czechia
- 2Department of Nephrology, Transplant Center, Institute for Clinical and Experimental Medicine, Prague, Czechia
The complement system is considered to be an important part of innate immune system with a significant role in inflammation processes. The activation can occur through classical, alternative, or lectin pathway, resulting in the creation of anaphylatoxins C3a and C5a, possessing a vast spectrum of immune functions, and the assembly of terminal complement cascade, capable of direct cell lysis. The activation processes are tightly regulated; inappropriate activation of the complement cascade plays a significant role in many renal diseases including organ transplantation. Moreover, complement cascade is activated during ischemia/reperfusion injury processes and influences delayed graft function of kidney allografts. Interestingly, complement system has been found to play a role in both acute cellular and antibody-mediated rejections and thrombotic microangiopathy. Therefore, complement system may represent an interesting therapeutical target in kidney transplant pathologies.
Introduction
The complement system was, for a long time since its discovery, considered to be a mere support system for the innate immunity mechanisms, playing a role in bacterial lysis. Over the years, it became obvious that the complement is an important part of the immune system, and its functions are more various than previously assumed. It was shown to play an important role in various kidney-related diseases. Its participation was observed in various forms of glomerulonephritis, including those connected to systemic diseases, for example, systemic lupus erythematodes. The complement was also found to participate in other renal diseases and conditions, such as atypical hemolytic uremic syndrome (aHUS), IgA nephropathy, or diabetes mellitus-related renal complications. The complement system was also shown to be involved in many of the transplant-related pathologies. Complement proteins also play a role in the ischemia/reperfusion injury, the innate and adaptive alloimmune responses. Recently, many complement-targeting therapies have been suggested in organ transplantation.
Role of the Complement System
The complement system includes more than 40 proteins, both membrane-bound and soluble, including receptors and various regulatory proteins (1). The complement system is a fully integrated part of immune system, capable of cooperation with other parts of immune system (both native and adaptive) (2).
Components of the complement system function in cooperation when the complement is activated, resulting in a coordinated sequence of enzymatic activities and leading to the creation of components with effector function (3). The functions of complement system reach far beyond direct bacterial membrane lysis, which is performed by the membrane attack complex (MAC), a result of terminal complement cascade (4). The complement also participates in apoptotic cell removal (5), in a strictly controlled manner in order to prevent excessive complement activation which could lead to cell lysis and necrosis (6). Anaphylatoxins generated by C3 and C5 cleavage (i.e., C3a and C5a) are generally considered to have the pro-inflammatory effect and to possess a chemotactic capability, attracting and activating granulocytes, mast cells, and phagocytes (7). Complement components bound to the pathogen surface also opsonize the pathogen, marking it for phagocytosis (8).
The complement receptors were found to be able to influence B cell activation and the antibody production (9). Studies also showed that T cells are functioning in close cooperation with complement proteins, producing complement component endogenously. Conversely, complement anaphylatoxins C3a and C5a were shown to function as both costimulatory and survival signals to naive CD4+ T cells (10). The absence of this signaling was later found to have the effect of changing the cytokine production in CD4+ T cells, resulting in the creation of induced regulatory T cells (11).
Complement Activation Pathways
Complement activation can generally be achieved through one of three different pathways (Figure 1). All pathways require different stimuli to be triggered; the products of C5 cleavage and the formation of MAC are identical for all pathways (12). The classical pathway of the complement can be triggered by any structure that is recognized by C1q, the target recognition molecule for classical pathway. Mannose-binding lectin (MBL) and ficolins have the same function for lectin pathway (3). C1q can bind a wide variety of target structures, including antibodies IgM and IgG (certain isotypes), retroviral envelope proteins, or lipopolysaccharides, and is involved in immunological processes other than complement activation (13). MBL and ficolins are similar in structure to C1q to some extent; however, their C-terminal domains are different. These molecules are able to recognize and bind to saccharide patterns expressed on bacterial membranes and viruses (8). Lectin pathway can also be triggered by molecules found on the surface of dying cells (14).
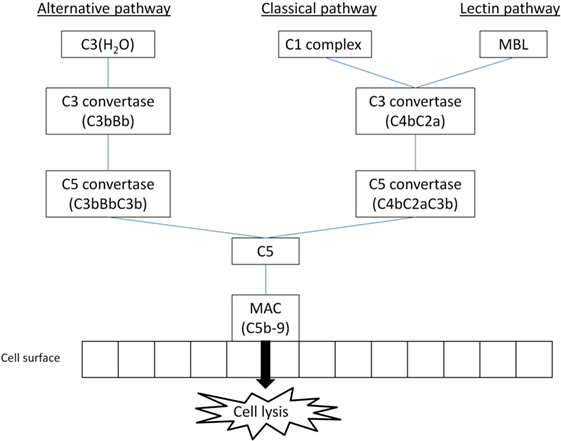
Figure 1. The simplified overview of main complement activation pathways (MBL, mannose-binding lectin; MAC, membrane attack complex); three main activation pathways are recognized in the complement system, leading to anaphylatoxin release and MAC formation: classical and lectin pathways are reliant on special recognition molecules to recognize a target, alternative pathway is triggered spontaneously with host cells protected by regulator molecules.
In the classical pathway, the binding of C1q to its target molecule is followed by the recruitment of C1r and C1s components, forming a C1 complex (15). The newly formed C1 complex interacts with C4 and C2 complement components, which are expressed in their inactive forms, cleaving them and subsequently forming C4bC2a, a molecule with C3 convertase function (12). In lectin pathway, the target recognition molecules induce conformational changes in MBL-associated serine proteases (MASPs). This is followed by the assembly of C3 convertase, C4bC2a, similar to classical pathway (12).
The alternative pathway is different in a sense that it has no dedicated target recognition molecule. Instead, there is a constant process of low-level spontaneous hydrolysis (so-called “tick-over”) of thioester bond in C3 component, changing it into active molecule (16). This molecule can bind Factor B and induce its cleavage by Factor D. The resulting complex C3(H2O)Bb acts as a C3 convertase and is capable of cleaving native C3 molecules to C3a and C3b fragments (17). In order to prevent damage to healthy host cells, complement regulators are present both in plasma and on cell membrane, inactivating C3b molecules that bind to the cell surface. Further studies revealed a significant role of plasma protein properdin. This protein is produced by activated neutrophils and was proved to prevent the rapid spontaneous destabilization of C3bBb complex (18). It can also bind to C3b and prevent its cleavage by complement regulators (19). Properdin is sometimes described as the only known positive regulator of the complement cascade (20).
The C3 convertases for all pathways have the same function—the cleavage of C3 into C3a and C3b fragments. The C3b fragment can subsequently bind to C3 convertase, turning it into C5 convertase. The C5 convertases promote the complement cascade by cleaving C5 into C5a and C5b fragments (12). The composition of the MAC, which is formed on pathogen membrane by the C5b fragment created by C5 convertases and subsequent recruitment of C6, C7, C8, and several (10–18) molecules C9, is known for longer time (21). Its precise molecular structure is, however, still being examined and described in more detail (22).
Earlier studies had uncovered evidence of various agents capable of inducing C5 component activation without triggering one of the standard complement pathways, in a C3-independent manner (23). Later studies highlighted the relationship between complement and the coagulation system (24, 25) and, more importantly, suggested that C5 component can be cleaved by thrombin, generating C5a in the absence of traditional C5 convertases (26, 27). Recent study, however, questioned these conclusions, suggesting that thrombin influences C5a fragment generation indirectly and it is in fact plasmin that acts as a major C5 cleavage mediator (28).
Anaphylatoxins and Their Function
Component anaphylatoxins, fragments C3a and C5a cleaved by their respective convertases, do not directly participate in the complement cascade progression. Instead, these small molecules are released and perform a number of immunomodulative functions. Anaphylatoxins possess significant pro-inflammatory functions, for example, leukocyte recruitment including neutrophils (29), mastocytes (30), and T lymphocytes (31), induction of oxygen radicals production in granulocytes (32, 33), induction of pro-inflammatory mediators production (34, 35), or blood vessel permeability increase (7). Apart from pro-inflammatory functions, studies showed that anaphylatoxins can also participate in tissue regeneration (36) and tissue fibrogenesis (37).
The complement activation is strongly associated with inflammatory processes. Recently, a new structure was identified in immune cells, a multiprotein complex called inflammasome. The inflammasome is important for proper induction and processing of IL-1β and IL-18, significant pro-inflammatory cytokines, via the activation of caspase-1 (38). Inflammasome assembly is induced by a variety of pattern recognition receptors, most notably Nod-like receptors, which also participate in its structure (39). Several studies focused on the link between complement activity and inflammasome activation, revealing that both C3a (40) and C5a (41, 42) anaphylatoxins are capable of influencing the IL-1 β production in immune cells. Newer research results have shown a novel mechanism of inflammasome activation in macrophages supported by MAC via the phagocytosis of opsonized particles (43). Since the complement system is an integral part of rejection mechanisms, it can be assumed that the complement-activated inflammasome formation could potentially influence the rejection processes, more research is, however, needed before any definitive conclusion is made.
Complement Regulatory Proteins
To prevent the complement-mediated damage to healthy host cells caused by unchecked activation, regulatory proteins are expressed by the host cells on the cellular membranes or present in the plasma that can disrupt the function of convertases or their products (8). Some of the regulators are described as having decay-accelerating functions, which means they are able to directly influence and inhibit the structure and function of convertases. Membrane-bound decay-accelerating factor (DAF/CD55) causes the dissociation of Bb and C2a from their respective C3 convertases, while also preventing them from reassembling (44). Complement receptor 1 (CR1/CD35) also possesses decay-accelerating activity and is able to influence all three complement activation pathways through binding of C3b and C4b (45, 46) and functioning as a cofactor for Factor I, a protease capable of cleaving C3b and C4b components (47). The C4b-binding protein also works as a cofactor for Factor I, but is capable of targeting only C4b component and, therefore, plays a role only in classical and lectin pathways (48).
A number of regulators are aimed at alternative pathway products, since this pathway is triggered spontaneously and is, therefore, the most dangerous for host cells. Factor H plays a major role in the alternative pathway regulation; it functions as an inhibitor of Factor B binding to C3b, preventing the C3 convertase creation. Factor H was also shown to a have a role in C3b proteolytic cleavage as a cofactor for Factor I (49). Similarly, membrane cofactor protein (MCP/CD46) can act as a cofactor for Factor I, participating in proteolytic cleavage of both C3b and C4b (50).
Complement in Renal Ischemia/Reperfusion Injury
The process of blood flow restoration after a period of its lack is common in organ transplantation. The ischemic injury to the renal tissue is a result of complex sequence of events, including renin synthesis, the shutdown of aerobic metabolism, and the generation of oxygen radicals (51). Together with other mediators such as pro-inflammatory cytokine release and complement activation, the environment becomes distinctly pro-inflammatory; leukocyte migration and activation are supported by these mediators (52). The expression of MHC class II molecules is higher both on endothelial and tubular epithelial cells during ischemia, supporting the T-cell response (53).
Both adaptive and innate immunity mechanisms are enhanced during ischemia/reperfusion injury. In our previous study with three renal graft biopsies describing thoroughly the ischemia/reperfusion process, the expression levels of several genes associated with renal allograft injury, i.e., cell proliferation and apoptosis, were shown to be upregulated during reperfusion (54). The complement activation is a significant marker of ongoing renal ischemia/reperfusion injury and has an influence on inflammatory processes. The C3 component expression is upregulated in the renal allograft (55) and the levels of systemic C3adesArg fragment (carboxypeptidase-cleaved C3a) released into circulation are increased (56). Interestingly, the main source of C3 component participating in ischemia/reperfusion injury was shown to be the renal allograft, with only minor role attributed to recipient’s circulating C3 (57).
Similarly, multiple studies have inquired into the role of C5 component during ischemia/reperfusion processes. The inhibition of C5 was shown to have the strong protective effect against inflammatory response in renal allograft, reducing significantly the influx of neutrophiles and suggesting that the chemokine production is dependent on complement activation during ischemia/reperfusion injury (58). Further study has confirmed the importance of C5a/C5aR interaction for neutrophile infiltration, while also hinting at the existence of different, neutrophile-independent functions of C5aR (59). The expression of C5aR was also found to be upregulated in deceased donor grafts, and the increased expression levels were associated with cold ischemia time and graft function (60). A study comparing the effects of C3a and C5a on the pathogenesis of ischemia/reperfusion injury has described C5a as having more significant impact (61). Since C5 component was shown to influence T-cell differentiation toward Th1 type (62), it is possible that not only innate but also adaptive immune mechanisms are influenced by C5 and its cleavage product. Recent study of the effects of C5a on human kidney tissue revealed the increase in pro-inflammatory cytokine production (IL-1β, IL-6, and IL-8) by the renal tissue linked to C5a stimulation (63).
Research comparing genome expression in renal allografts from living and deceased donors revealed that complement components were expressed at significantly higher levels before reperfusion in allografts from deceased donors (64). These results seem to be in agreement with earlier study that described increased activation of complement and other inflammatory mediators in kidneys recovered from rats with diagnosed brain death (65). Damman et al. (66) found the evidence of increased induction of C3 component in renal allografts in donors with diagnosed brain death, which was associated with decreased allograft functionality. Similar results were obtained from an experiment with murine cardiac allografts (67). Further study analyzing the complement activation markers in deceased kidney donors described a relationship between donor plasma levels of MAC and the observed graft functionality, with higher MAC levels correlating with acute rejection incidence (68). Newer, abovementioned study described an increase in systemic C5a levels in plasma of donors with diagnosed brain death, compared to living donors, although no correlation with graft function was found; the expression rates of C5aR in allograft were also increased in comparison to living donors (63).
Further studies have shown that the complement activation and inflammation associated with the ischemia/reperfusion injury also leads to an increased antibody production, which might influence the probability of late allograft failure (69). The T cell infiltration of the allograft is also supported by the activated complement, providing another potential negative influence for long-term outcome (70).
The significance of IgM antibodies for complement activation during ischemia/reperfusion injury was also addressed by researchers. Originally, it was considered to be a proof of classical pathway activation (71) and these findings were supported by further studies (72). Newer experiments, however, came with evidence that natural IgM antibody binds to MBL and triggers the lectin pathway (73), which has been further confirmed (74).
The question of participating complement activation pathways has been the subject of many studies and is still not fully resolved. The matter is complicated by the fact that the complement activity in ischemia/reperfusion injury appears to differ significantly depending on both the organ selected for study and the model used (75). For instance, Zhou et al. (76) showed that C4-deficient mice were not protected against renal ischemia/reperfusion injury, suggesting that classical pathway does not significantly participate. These results were further supported by a study that showed that gastrointestinal ischemia/reperfusion injury in mice is not dependent on C1q, a key component for classical pathway activation (77). However, newer study proved the presence of C1q in murine intestinal ischemia/reperfusion injury, casting doubt on the previously published results (78). The same study also confirmed the role of lectin pathway, while failing to find evidence of alternative pathway activation, which was originally supposed to play an important role (79). Recently, Miwa et al. (80), based on their murine DAF−/−CD59−/− model, suggested that alternative pathway plays a significant role during early renal ischemia/reperfusion injury, with properdin having an important pathogenic role.
The role of lectin pathway in endothelium complement activation during oxidative stress was known for some time (81), and cytokeratin 1 was found to play a significant part in activation process on hypoxic endothelial cells (82). Subsequent experiments suggested that lectin pathway is connected to ischemia/reperfusion injury, with MBL deposition correlating with postischemic inflammation (83). Further studies stressed the importance of lectin pathway in renal (84) and gastrointestinal (77) ischemia/reperfusion injury in mice. Recent experiments suggested that lectin pathway indeed plays a significant role in ischemia/reperfusion injury, indicating that the activation of complement proceeds in a C4-independent manner, where MASP-2 plays a crucial role for complement activation, suggesting a possible novel lectin pathway activation alternative (85).
Based on the abovementioned experimental studies, recently several prospective trials in human kidney transplantation studies were initiated to investigate the effect of complement inhibition in the prevention of delayed graft function (NCT01919346 and NCT02145182). Anti-C5 monoclonal antibody (eculizumab) used as C5 complement component is widely implicated in renal ischemia/reperfusion injury. Experiments on animal models already showed a positive effect of terminal pathway blockade on graft function, suggesting a possible way of ischemia/reperfusion injury and delayed graft function prevention (86); however, very recent data from the PROTECT study (NCT02145182) on 286 kidney transplant recipients randomized to receive either eculizumab (1,200 mg prior reperfusion and 900 mg on first postoperative day) or placebo along with antithymocyte globulin failed to meet its primary end point (delayed graft function defined as dialysis within first posttransplant week), with the results from both arms not reaching statistical significance (87). Clearly, it is too premature to draw any other conclusion from the PROTECT study as secondary analysis has not been finished yet and no data were published beside initial press release.
Complement in Posttransplant Thrombotic Microangiopathy (TMA)
The complement system is also highly involved in the TMA characterized by microvascular thrombosis with thrombocytopenia, hemolytic anemia, and red blood cell fragmentation. In the renal allograft, either de novo or recurrent posttransplant TMA can appear (88). Many cases of recurrent TMA have been linked to complement-associated gene mutations, such as CFH, CFI, MCP, C3, and others; however, similar to aHUS, gene mutation confirmation is not necessary for diagnosis as the genetical evaluation is far too complex and cannot necessarily prove complement-associated gene mutation while complement gene risk haplotypes are frequently found in the population. TMA occurs in a wide range of diseases other than originally described thrombotic thrombocytopenic purpura and HUS, such as autoimmune diseases, malignant hypertension, HCV infection, vascular rejection, and endothelial damage due to immunosuppressant, such as cyclosporine A, tacrolimus, and sirolimus, toxicity (89, 90). Many of those trigger complement (over)activity. TMA seems to be not a rare histological finding in kidney graft biopsies performed early after transplantation (91). With the advent of Luminex technology allowing detection of donor specific anti-HLA antibodies, many of TMA cases were shown to be associated with antibody-mediated rejection and C4d positivity. In a significant proportion of patients, however, the cause of TMA remains unclear. In many cases, TMA is associated with progressive graft dysfunction and graft loss despite the plasma exchange and immunosuppressive therapy. It is likely that in most cases, unrecognized complement genetic mutations risk haplotypes might be responsible for TMA development and subsequent graft failure. It was shown that even TMA associated with AMR may be associated with otherwise “silent” CFH mutation (92). Therefore, a blockage of terminal complement activity with anti C5 monoclonal antibody, eculizumab, has been introduced as a treatment of choice in recurrent TMA in kidney transplantation (93–95). Interestingly, many centers have been using eculizumab off-label also for de novo posttransplant TMA; however, it is premature to draw any conclusion as there is a lack of prospective studies and information from registries is still missing. Moreover, economical aspects of such therapy slow down clinical research in this interesting area.
Complement and T-Cell-Mediated Rejection
While the complement system plays an important role in the innate immunity mechanisms, its role in the adaptive immunity functions is also significant. It is becoming apparent that the complement plays a significant role in T-cell-mediated rejection, influencing the proliferation and activity of T-cells participating in rejection (Table 1). The mechanisms of complement involvement are not yet fully understood, however, they are a subject of focused research. Despite the fact that majority of C3 complement component is expressed in liver, C3 can also be expressed locally in other tissues, including kidneys (96). Pratt et al. (97) showed that in the absence of locally synthesized C3 component, the acute rejection of the murine renal allograft was not as powerful and the T-cell response was significantly regulated. More recent study confirmed that T-cell alloresponse is affected by both C3a and C5a anaphylatoxins produced locally, by dendritic cells (98). Further experiments on both human and murine cells showed that the blockage of C3a and C5a interactions with their respective receptors in T-cells leads to regulatory T cells induction and stabilization (99).

Table 1. Complement function in transplantation.
An important role is also played by C3a and C5a receptors (C3aR and C5aR), which are expressed on numerous types of immune cells. Gueler et al. (100) described an increase in C5aR expression on infiltrating cells in renal biopsies in patients with acute cellular rejection, as compared to patients with histologically normal biopsies. Subsequent experiments with murine renal allografts showed the similar pattern and the use of C5aR antagonist pretreatment led to increased graft survival and the decrease in monocyte/macrophage infiltration. In the same year, another study revealed the connection between C3aR on the surface of dendritic cells and the attenuation of T-cell stimulation by cyclic adenosine monophosphate level alteration (101). Experiments performed on murine cells revealed that signaling via C3aR and C5aR on natural regulatory T-cells attenuates the FoxP3 expression, leading to diminished suppressive function and, subsequently, enhanced pro-inflammatory T-cell response (102).
As mentioned above, the complement can also influence the creation of regulatory T-cells (11). Recent study showed that in C3-deficient mice, the T-cell-mediated rejection of skin grafts was attenuated by the reduction of Th1/Th17 (i.e., pro-inflammatory) T-lymphocyte activity while upregulating the regulatory T-cell expression (103). Another study reported correlation of regulatory T-cell count and serum C4 levels in patients with systemic lupus erythematosus; according to the results of following in vitro experiment, the C4 component is able to influence the T-cell differentiation in favor of regulatory T-cells in the presence of dendritic cells (104). Similar effect was shown for C1q component, where macrophages that ingested C1q-opsonized apoptotic cells demonstrated the ability to affect the levels of Th1/Th17 T-cell proliferation, resulting in increased tendency toward regulatory T-cells (105).
The exact role of regulators of complement activation during T-cell-mediated rejection is not yet fully understood. Earlier studies showed that murine DAF homolog acts as a regulator of T-cell immune response in mice (106) and that DAF expressed on antigen-presenting cells has the ability to affect the T-cell differentiation by regulating the C5a component production (62). The experiments on murine cardiac allografts from DAF-deficient donors showed that DAF is able to regulate T-cell activity and to affect T-cell-mediated rejection (107). This effect was later confirmed in a study of graft-versus-host disease (108).
Recent study revealed the decrease in mRNA expression of CD59 and Crry (murine regulator of complement, functionally similar to MCP) in rat allografts compared to control group with syngeneic grafts. The same study also compared levels of MCP in patients’ clinical data and found a significant difference in 5-year renal graft survival, where the grafts of patients expressing high levels of MCP showed higher survival rates (109). MCP was also shown to be able to regulate the T-cell immune response by affecting the cytokine production of Th1 cells (110).
We recently described, in clinical settings, that T-cell-mediated acute rejection can occur even in patients with full C5 blockade while using eculizumab, suggesting that the role of C5 is not critical for T-cell-mediated rejection or, alternatively, other mechanisms are able to replace C5 functionally in such a case (111).
Complement and Antibody-Mediated Rejection
While the main role of complement system during T-cell-mediated rejection is to regulate activity and differentiation of participating T-cells, in antibody-mediated rejection, the complement participates in more direct manner and its components can be found directly at the site of the antibody binding (112). Although the classical pathway is considered to be the primary mechanism of complement system activation during antibody-mediated rejection, experiments have also shown that the influence of MBL levels was found to be associated with rejection severity (113). Another study suggested connection between lectin pathway of activation and C4d deposition in kidney allografts, hinting at possible role of lectin pathway activated by H-ficolin in antibody-mediated rejection (114). The complement fragment C4d, created by C4 cleavage, has a specific significance for antibody-mediated rejection. Although being an inactive component, it remains bound at the site of C4 cleavage, acting as a marker of complement activation (115). The immunohistological assessments of renal allograft biopsy samples revealed that a deposition of C4d in interstitial capillaries can be linked to worse allograft outcomes and was suggested to be a marker of humoral response to the allograft (116). Further studies confirmed the importance of C4d as an independent predictive factor of long-term graft survival (117, 118). The presence of C4d deposits in allograft was included in Banff’ classification of antibody-mediated rejection as a criterium for positive identification in 2001, and in 2007 update the C4d scoring was further specified as a criterium for the identification of chronic antibody-mediated rejection (119, 120). Later studies, however, hinted at the existence of another, C4d-negative form of antibody-mediated rejection. A study comparing the protocol kidney biopsies from renal transplant recipients reached the conclusion that C4d presence cannot be considered an evidence of chronic antibody-mediated rejection (121), while the results of a study focused on endothelial gene expression in allografts suggested that only about 40% of renal transplants undergoing chronic antibody-mediated rejection are C4d-positive (122). Based on the results from studies questioning the usability of C4d positivity as criterium for chronic antibody-mediated rejection, the Banff classification was revised during the 2013 Banff Conference. The previous immunohistological evidence required for classification was changed into a requirement of the evidence of the antibody interaction with the endothelium. This new classification category includes C4d positivity, among other markers (120).
The classical pathway presents the dominant mechanism of complement activation during antibody-mediated rejection (123). The antibody production was found to be directly influenced through complement receptors CR1 and CR2 expressed on the surface of follicular dendritic cells (124), while antigens with C3d component attached were able to react more efficiently with B-cells via CR2, lowering significantly the amount of antigen needed to induce the antibody production (125). IgG and IgM antibodies bound on graft endothelium are capable of interacting directly with C1q, leading to the classical pathway activation of the complement, anaphylatoxin release, and MAC formation (126). In recent study, a novel view on complement activity in allograft injury was revealed; it was shown that the antibodies bound to the vascular endothelium lead to complement activation and the formation of MAC, which can subsequently trigger a non-canonical NF-κB signaling, resulting in pro-inflammatory reaction and T-cell response enhancement instead of cell lysis (127). While some studies showed the correlation between the presence of donor-specific antibodies (DSA) with complement-binding capability and decreased graft survival with higher rate of antibody-mediated rejection (128, 129), the results from different studies revealed no predictive value for these DSA concerning graft survival or rejection rates (130). Recent research uncovered the evidence of existence of polyreactive antibodies, capable of reacting to multiple different antigens, including HLA molecules (131); this discovery opposes the traditional view of antibody specificity to single target. The possible presence of these antibodies has been suggested to be related to the increased levels of autoantibody response in patients with chronic antibody-mediated rejection described earlier (132). Further study confirmed both the presence and the development of polyreactive antibodies in kidney transplants with chronic antibody-mediated rejection and also their ability to activate complement via classical pathway, contributing to C4d deposition in allograft tissue (133).
Several studies suggested a significant role of terminal complement activation during acute antibody-mediated rejection (134). Stegall et al. (135) later suggested that the role of complement in chronic antibody-mediated rejection could be, in contrast to its acute form, more oriented toward the chemotaxis and inflammation stimulation function. Therefore, terminal complement blockade was suggested to be an efficacious therapy in DSA-positive kidney transplantation. However, prospective randomized trial (NCT01399593) was ceased due to the lack of convincing efficacy in antibody-mediated rejection reduction, despite numerical difference between study and control groups (136). One of the possible explanations for these results is the involvement of complement-independent mechanisms of antibody-mediated rejection. Regarding this aspect, a study on mice revealed a significant role of NK-cells in chronic antibody-mediated rejection of heart transplants, since the depletion of these cells lowered the frequency and severity of rejection in allografts regardless of complement activation (137). These results tend to support conclusions of earlier study of transcripts expressed in renal biopsies obtained from DSA-positive patients. NK-cell activity was put in association with antibody-mediated rejection, regardless of C4d positivity (138). Further experiments confirmed the association of NK-cell transcripts with cases of antibody-mediated rejection and DSA positivity and also revealed NK-cell transcripts in some cases of early T-cell-mediated rejection, suggesting the possible participation of NKT-cells (139). The results of recent study on CCR5-deficient mouse model of renal transplant rejection provide further data supporting the link between antibody-mediated graft injury and failure and the activity of the NK-cells (140). Legris et al. (141) suggested that further study of peripheral NK-cell activity may provide additional data concerning the complement-independent antibody-mediated rejection mechanisms linked to NK-cells.
Complement Targeting Therapies in Kidney Transplantation
Complement system plays a significant role in most cases of antibody-mediated rejection. Therefore, its components can be considered as potential therapeutic targets in attempts to prevent or treat such type of the rejection. Recently, the inhibition of several complement components has shown certain results in preventing the antibody-mediated rejection.
C1 inhibitor is a serine protease inhibitor that is capable of irreversibly blocking not only C1 complex components (142), as the name suggests, but also MASP-1 and MASP-2 (143). It is, therefore, an effective inhibitor of both classical and lectin pathway activation. Several studies with C1 inhibitor were already published. Earlier study on non-human primate model showed a potential for inhibition of acute antibody-mediated rejection with C1-inhibitor (144). Very recently, a double-blind study assessed the safety of C1-inhibitor in kidney transplant recipients and suggested the positive effect of this treatment regarding the C1q+HLA antibody levels reduction and antibody-mediated rejection occurrence (145). The possible use of complement inhibitor based on viral coat proteins is also a subject of ongoing research (146, 147).
Eculizumab is a humanized monoclonal antibody capable of blocking the cleavage of C5 complement component, thus preventing the release of anaphylatoxin C5a and the assembly of MAC (148). The C5 component participates in all three main pathways of complement activation; therefore, the blockade of its cleavage can have a positive influence on various medical conditions regardless of the active pathway. Eculizumab was shown to be an effective treatment for paroxysmal nocturnal hemoglobinuria (149) and aHUS including its recurrence in kidney transplantation (93, 150). Since the complement system is a prominent factor during antibody-mediated rejection, the effects of eculizumab were studied with regard to this condition as well. Stegall et al. (134) studied the effects of terminal complement inhibition on patients with a positive crossmatch and concluded that eculizumab is capable of decreasing the incidence of acute antibody-mediated rejection. Some success in treating the acute antibody-mediated rejection in renal allograft was also reported in several case reports (151–153). Extended follow-up study, however, showed that eculizumab treatment has no effect on chronic antibody-mediated rejection in recipients with significant amount of DSA (154). Moreover, there are known cases of C4d-negative antibody-mediated rejection where the use of eculizumab was ineffective (155). Similarly, the existence of IgM-mediated acute antibody-mediated rejection was described in patients treated with eculizumab (156). Therefore, C5 inhibition might be efficacious in the treatment of acute antibody-mediated rejection at least in some cases; however, these effects are lost in preventing chronic rejection development.
Conclusion
The complement system is an intricate system of proteins participating in a wide range of processes connected to immune system. It possesses a significant influence and it is carefully regulated. The precise involvement of the complement system in the rejection processes is not yet fully understood, but it is becoming clear that it plays an important role in both T-cell-mediated and antibody-mediated organ rejection. Lately, new evidence is emerging suggesting that new, complement-independent mechanisms participate in transplant rejection as well. Recent uncertainty in particular complement proteins targeting therapies in kidney transplantation reflects the complexity and phylogenetical conservation of complement system.
Author Contributions
MC wrote the paper. OV designed and wrote the paper.
Conflict of Interest Statement
OV received a speaker fee from Alexion. MC declares no conflict of interest.
Funding
The study was supported by Ministry of Health of the Czech Republic, grant nr. 15-26865A, all rights reserved, and MH CZ DRO (“Institute for Clinical and Experimental Medicine—IKEM, IN 00023001”).
References
1. Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: role in immunity. Front Immunol (2015) 6:257. doi: 10.3389/fimmu.2015.00257
2. Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res (2010) 20(1):34–50. doi:10.1038/cr.2009.139
3. Bohlson SS, O’Conner SD, Hulsebus HJ, Ho MM, Fraser DA. Complement, c1q, and c1q-related molecules regulate macrophage polarization. Front Immunol (2014) 5:402. doi:10.3389/fimmu.2014.00402
4. Bubeck D. The making of a macromolecular machine: assembly of the membrane attack complex. Biochemistry (2014) 53(12):1908–15. doi:10.1021/bi500157z
5. Ogden CA, deCathelineau A, Hoffmann PR, Bratton D, Ghebrehiwet B, Fadok VA, et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med (2001) 194(6):781–95. doi:10.1084/jem.194.6.781
6. Trouw LA, Bengtsson AA, Gelderman KA, Dahlbäck B, Sturfelt G, Blom AM. C4b-binding protein and factor H compensate for the loss of membrane-bound complement inhibitors to protect apoptotic cells against excessive complement attack. J Biol Chem (2007) 282(39):28540–8. doi:10.1074/jbc.M704354200
7. Klos A, Tenner AJ, Johswich KO, Ager RR, Reis ES, Köhl J. The role of the anaphylatoxins in health and disease. Mol Immunol (2009) 46(14):2753–66. doi:10.1016/j.molimm.2009.04.027
8. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I – molecular mechanisms of activation and regulation. Front Immunol (2015) 6:262. doi:10.3389/fimmu.2015.00262
9. Molina H, Holers VM, Li B, Fung Y, Mariathasan S, Goellner J, et al. Markedly impaired humoral immune response in mice deficient in complement receptors 1 and 2. Proc Natl Acad Sci U S A (1996) 93(8):3357–61. doi:10.1073/pnas.93.8.3357
10. Strainic MG, Liu J, Huang D, An F, Lalli PN, Mugim N, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity (2008) 28(3):425–35. doi:10.1016/j.immuni.2008.02.001
11. Strainic MG, Shevach EM, An F, Lin F, Medof ME. Absence of signaling into CD4? cells via C3aR and C5aR enables autoinductive TGF-β1 signaling and induction of Foxp3? regulatory T cells. Nat Immunol (2013) 14(2):162–71. doi:10.1038/ni.2499
12. Sarma JV, Ward PA. The complement system. Cell Tissue Res (2011) 343(1):227–35. doi:10.1007/s00441-010-1034-0
13. Kishore U, Ghai R, Greenhough TJ, Shrive AK, Bonifati DM, Gadjeva MG, et al. Structural and functional anatomy of the globular domain of complement protein C1q. Immunol Lett (2004) 95(2):113–28. doi:10.1016/j.imlet.2004.06.015
14. Nauta AJ, Raaschou-Jensen N, Roos A, Daha MR, Madsen HO, Borrias-Essers MC, et al. Mannose-binding lectin engagement with late apoptotic and necrotic cells. Eur J Immunol (2003) 33(10):2853–63. doi:10.1002/eji.200323888
15. Girija UV, Gingras AR, Marshall JE, Panchal R, Sheikh MA, Gál P, et al. Structural basis of the C1q/C1s interaction and its central role in assembly of the C1 complex of complement activation. Proc Natl Acad Sci U S A (2013) 110(34):13916–20. doi:10.1073/pnas.1311113110
16. Pangburn MK, Schreiber RD, Müller-Eberhard HJ. Formation of the initial C3 convertase of the alternative complement pathway. Acquisition of C3b-like activities by spontaneous hydrolysis of the putative thioester in native C3. J Exp Med (1981) 154(3):856–67. doi:10.1084/jem.154.3.856
17. Li K, Gor J, Perkins SJ. Self-association and domain rearrangements between complement C3 and C3u provide insight into the activation mechanism of C3. Biochem J (2010) 431(1):63–72. doi:10.1042/BJ20100759
18. Fearon DT, Austen KF. Properdin: binding to C3b and stabilization of the C3b-dependent C3 convertase. J Exp Med (1975) 142(4):856–63. doi:10.1084/jem.142.4.856
19. Kemper C, Atkinson JP, Hourcade DE. Properdin: emerging roles of a pattern-recognition molecule. Annu Rev Immunol (2010) 28:131–55. doi:10.1146/annurev-immunol-030409-101250
20. Lesher AM, Nilsson B, Song WC. Properdin in complement activation and tissue injury. Mol Immunol (2013) 56(3):191–8. doi:10.1016/j.molimm.2013.06.002
21. Podack ER. Molecular composition of the tubular structure of the membrane attack complex of complement. J Biol Chem (1984) 259(13):8641–7.
22. Serna M, Giles JL, Morgan BP, Bubeck D. Structural basis of complement membrane attack complex formation. Nat Commun (2016) 7:10587. doi:10.1038/ncomms10587
23. Vogt W, Zimmermann B, Hesse D, Nolte R. Activation of the fifth component of human complement, C5, without cleavage, by methionine oxidizing agents. Mol Immunol (1992) 29(2):251–6. doi:10.1016/0161-5890(92)90106-8
24. Distelmaier K, Adlbrecht C, Jakowitsch J, Winkler S, Dunkler D, Gerner C, et al. Local complement activation triggers neutrophil recruitment to the site of thrombus formation in acute myocardial infarction. Thromb Haemost (2009) 102(3):564–72. doi:10.1160/TH09-02-0103
25. Howes JM, Richardson VR, Smith KA, Schroeder V, Somani R, Shore A, et al. Complement C3 is a novel plasma clot component with anti-fibrinolytic properties. Diab Vasc Dis Res (2012) 9(3):216–25. doi:10.1177/1479164111432788
26. Huber-Lang M, Younkin EM, Sarma JV, Riedemann N, McGuire SR, Lu KT, et al. Generation of C5a by phagocytic cells. Am J Pathol (2002) 161(5):1849–59. doi:10.1016/S0002-9440(10)64461-6
27. Auger JL, Haasken S, Binstadt BA. Autoantibody-mediated arthritis in the absence of C3 and activating Fcγ receptors: C5 is activated by the coagulation cascade. Arthritis Res Ther (2012) 14(6):R269. doi:10.1186/ar4117
28. Foley JH, Walton BL, Aleman MM, O’Byrne AM, Lei V, Harrasser M, et al. Complement activation in arterial and venous thrombosis is mediated by plasmin. EBioMedicine (2016) 5:175–82. doi:10.1016/j.ebiom.2016.02.011
29. Ehrengruber MU, Geiser T, Deranleau DA. Activation of human neutrophils by C3a and C5A. Comparison of the effects on shape changes, chemotaxis, secretion, and respiratory burst. FEBS Lett (1994) 346(2–3):181–4. doi:10.1016/0014-5793(94)00463-3
30. Hartmann K, Henz BM, Krüger-Krasagakes S, Köhl J, Burger R, Guhl S, et al. C3a and C5a stimulate chemotaxis of human mast cells. Blood (1997) 89(8):2863–70.
31. Nataf S, Davoust N, Ames RS, Barnum SR. Human T cells express the C5a receptor and are chemoattracted to C5a. J Immunol (1999) 162(7):4018–23.
32. Elsner J, Oppermann M, Czech W, Dobos G, Schöpf E, Norgauer J, et al. C3a activates reactive oxygen radical species production and intracellular calcium transients in human eosinophils. Eur J Immunol (1994) 24(3):518–22. doi:10.1002/eji.1830240304
33. Elsner J, Oppermann M, Czech W, Kapp A. C3a activates the respiratory burst in human polymorphonuclear neutrophilic leukocytes via pertussis toxin-sensitive G-proteins. Blood (1994) 83(11):3324–31.
34. el-Lati SG, Dahinden CA, Church MK. Complement peptides C3a- and C5a-induced mediator release from dissociated human skin mast cells. J Invest Dermatol (1994) 102(5):803–6. doi:10.1111/1523-1747.ep12378589
35. Fischer WH, Jagels MA, Hugli TE. Regulation of IL-6 synthesis in human peripheral blood mononuclear cells by C3a and C3a(desArg). J Immunol (1999) 162(1):453–9.
36. Strey CW, Markiewski M, Mastellos D, Tudoran R, Spruce LA, Greenbaum LE, et al. The proinflammatory mediators C3a and C5a are essential for liver regeneration. J Exp Med (2003) 198(6):913–23. doi:10.1084/jem.20030374
37. Silasi-Mansat R, Zhu H, Georgescu C, Popescu N, Keshari RS, Peer G, et al. Complement inhibition decreases early fibrogenic events in the lung of septic baboons. J Cell Mol Med (2015) 19(11):2549–63. doi:10.1111/jcmm.12667
38. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10(2):417–26. doi:10.1016/S1097-2765(02)00599-3
39. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140(6):821–32. doi:10.1016/j.cell.2010.01.040
40. Asgari E, Le Friec G, Yamamoto H, Perucha E, Sacks SS, Köhl J, et al. C3a modulates IL-1β secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood (2013) 122(20):3473–81. doi:10.1182/blood-2013-05-502229
41. An LL, Mehta P, Xu L, Turman S, Reimer T, Naiman B, et al. Complement C5a potentiates uric acid crystal-induced IL-1β production. Eur J Immunol (2014) 44(12):3669–79. doi:10.1002/eji.201444560
42. Samstad EO, Niyonzima N, Nymo S, Aune MH, Ryan L, Bakke SS, et al. Cholesterol crystals induce complement-dependent inflammasome activation and cytokine release. J Immunol (2014) 192(6):2837–45. doi:10.4049/jimmunol.1302484
43. Suresh R, Chandrasekaran P, Sutterwala FS, Mosser DM. Complement-mediated ’bystander’ damage initiates host NLRP3 inflammasome activation. J Cell Sci (2016) 129(9):1928–39. doi:10.1242/jcs.179291
44. Fujita T, Inoue T, Ogawa K, Iida K, Tamura N. The mechanism of action of decay-accelerating factor (DAF). DAF inhibits the assembly of C3 convertases by dissociating C2a and Bb. J Exp Med (1987) 166(5):1221–8. doi:10.1084/jem.166.5.1221
45. Krych-Goldberg M, Hauhart RE, Subramanian VB, Yurcisin BM II, Crimmins DL, Hourcade DE, et al. Decay accelerating activity of complement receptor type 1 (CD35). Two active sites are required for dissociating C5 convertases. J Biol Chem (1999) 274(44):31160–8. doi:10.1074/jbc.274.44.31160
46. Hourcade DE, Mitchell L, Kuttner-Kondo LA, Atkinson JP, Medof ME. Decay-accelerating factor (DAF), complement receptor 1 (CR1), and factor H dissociate the complement AP C3 convertase (C3bBb) via sites on the type A domain of Bb. J Biol Chem (2002) 277(2):1107–12. doi:10.1074/jbc.M109322200
47. Java A, Liszewski MK, Hourcade DE, Zhang F, Atkinson JP. Role of complement receptor 1 (CR1; CD35) on epithelial cells: a model for understanding complement-mediated damage in the kidney. Mol Immunol (2015) 67(2 Pt B):584–95. doi:10.1016/j.molimm.2015.07.016
48. Blom AM. Structural and functional studies of complement inhibitor C4b-binding protein. Biochem Soc Trans (2002) 30(Pt 6):978–82. doi:10.1042/bst0300978
49. Perkins SJ, Nan R, Li K, Khan S, Miller A. Complement factor H-ligand interactions: self-association, multivalency and dissociation constants. Immunobiology (2012) 217(2):281–97. doi:10.1016/j.imbio.2011.10.003
50. Seya T, Turner JR, Atkinson JP. Purification and characterization of a membrane protein (gp45-70) that is a cofactor for cleavage of C3b and C4b. J Exp Med (1986) 163(4):837–55. doi:10.1084/jem.163.4.837
51. Siedlecki A, Irish W, Brennan DC. Delayed graft function in the kidney transplant. Am J Transplant (2011) 11(11):2279–96. doi:10.1111/j.1600-6143.2011.03754.x
52. Arumugam TV, Shiels IA, Woodruff TM, Granger DN, Taylor SM. The role of the complement system in ischemia-reperfusion injury. Shock (2004) 21(5):401–9. doi:10.1097/00024382-200405000-00002
53. Bohmova R, Viklicky O. Renal ischemia–reperfusion injury: an inescapable event affecting kidney transplantation outcome. Folia Microbiol (Praha) (2001) 46(4):267–76. doi:10.1007/BF02815613
54. Wohlfahrtova M, Brabcova I, Zelezny F, Balaz P, Janousek L, Honsova E, et al. Tubular atrophy and low netrin-1 gene expression are associated with delayed kidney allograft function. Transplantation (2014) 97(2):176–83. doi:10.1097/TP.0b013e3182a95d04
55. Serinsöz E, Bock O, Gwinner W, Schwarz A, Haller H, Kreipe H, et al. Local complement C3 expression is upregulated in humoral and cellular rejection of renal allografts. Am J Transplant (2005) 5(6):1490–4. doi:10.1111/j.1600-6143.2005.00873.x
56. Thurman JM, Royer PA, Ljubanovic D, Dursun B, Lenderink AM, Edelstein CL, et al. Treatment with an inhibitory monoclonal antibody to mouse factor B protects mice from induction of apoptosis and renal ischemia/reperfusion injury. J Am Soc Nephrol (2006) 17(3):707–15. doi:10.1681/ASN.2005070698
57. Farrar CA, Zhou W, Lin T, Sacks SH. Local extravascular pool of C3 is a determinant of postischemic acute renal failure. FASEB J (2006) 20(2):217–26. doi:10.1096/fj.05-4747com
58. De Vries B, Matthijsen RA, Wolfs TG, Van Bijnen AA, Heeringa P, Buurman WA. Inhibition of complement factor C5 protects against renal ischemia-reperfusion injury: inhibition of late apoptosis and inflammation. Transplantation (2003) 75(3):375–82. doi:10.1097/01.TP.0000044455.05584.2A
59. de Vries B, Köhl J, Leclercq WK, Wolfs TG, van Bijnen AA, Heeringa P, et al. Complement factor C5a mediates renal ischemia-reperfusion injury independent from neutrophils. J Immunol (2003) 170(7):3883–9. doi:10.4049/jimmunol.170.7.3883
60. Lewis AG, Köhl G, Ma Q, Devarajan P, Köhl J. Pharmacological targeting of C5a receptors during organ preservation improves kidney graft survival. Clin Exp Immunol (2008) 153(1):117–26. doi:10.1111/j.1365-2249.2008.03678.x
61. Peng Q, Li K, Smyth LA, Xing G, Wang N, Meader L, et al. C3a and C5a promote renal ischemia-reperfusion injury. J Am Soc Nephrol (2012) 23(9):1474–85. doi:10.1681/ASN.2011111072
62. Lalli PN, Strainic MG, Lin F, Medof ME, Heeger PS. Decay accelerating factor can control T cell differentiation into IFN-gamma-producing effector cells via regulating local C5a-induced IL-12 production. J Immunol (2007) 179(9):5793–802. doi:10.4049/jimmunol.179.9.5793
63. van Werkhoven MB, Damman J, van Dijk MC, Daha MR, de Jong IJ, Leliveld A, et al. Complement mediated renal inflammation induced by donor brain death: role of renal C5a-C5aR interaction. Am J Transplant (2013) 13(4):875–82. doi:10.1111/ajt.12130
64. Naesens M, Li L, Ying L, Sansanwal P, Sigdel TK, Hsieh HC, et al. Expression of complement components differs between kidney allografts from living and deceased donors. J Am Soc Nephrol (2009) 20(8):1839–51. doi:10.1681/ASN.2008111145
65. Kusaka M, Pratschke J, Wilhelm MJ, Ziai F, Zandi-Nejad K, Mackenzie HS, et al. Activation of inflammatory mediators in rat renal isografts by donor brain death. Transplantation (2000) 69(3):405–10. doi:10.1097/00007890-200002150-00017
66. Damman J, Nijboer WN, Schuurs TA, Leuvenink HG, Morariu AM, Tullius SG, et al. Local renal complement C3 induction by donor brain death is associated with reduced renal allograft function after transplantation. Nephrol Dial Transplant (2011) 26(7):2345–54. doi:10.1093/ndt/gfq717
67. Atkinson C, Floerchinger B, Qiao F, Casey S, Williamson T, Moseley E, et al. Donor brain death exacerbates complement-dependent ischemia/reperfusion injury in transplanted hearts. Circulation (2013) 127(12):1290–9. doi:10.1161/CIRCULATIONAHA.112.000784
68. Damman J, Seelen MA, Moers C, Daha MR, Rahmel A, Leuvenink HG, et al. Systemic complement activation in deceased donors is associated with acute rejection after renal transplantation in the recipient. Transplantation (2011) 92(2):163–9. doi:10.1097/TP.0b013e318222c9a0
69. Fuquay R, Renner B, Kulik L, McCullough JW, Amura C, Strassheim D, et al. Renal ischemia-reperfusion injury amplifies the humoral immune response. J Am Soc Nephrol (2013) 24(7):1063–72. doi:10.1681/ASN.2012060560
70. Raedler H, Vieyra MB, Leisman S, Lakhani P, Kwan W, Yang M, et al. Anti-complement component C5 mAb synergizes with CTLA4Ig to inhibit alloreactive T cells and prolong cardiac allograft survival in mice. Am J Transplant (2011) 11(7):1397–406. doi:10.1111/j.1600-6143.2011.03561.x
71. Williams JP, Pechet TT, Weiser MR, Reid R, Kobzik L, Moore FD Jr, et al. Intestinal reperfusion injury is mediated by IgM and complement. J Appl Physiol (1999) 86(3):938–42.
72. Zhang M, Austen WG Jr, Chiu I, Alicot EM, Hung R, Ma M, et al. Identification of a specific self-reactive IgM antibody that initiates intestinal ischemia/reperfusion injury. Proc Natl Acad Sci U S A (2004) 101(11):3886–91. doi:10.1073/pnas.0400347101
73. Zhang M, Takahashi K, Alicot EM, Vorup-Jensen T, Kessler B, Thiel S, et al. Activation of the lectin pathway by natural IgM in a model of ischemia/reperfusion injury. J Immunol (2006) 177(7):4727–34. doi:10.4049/jimmunol.177.7.4727
74. Busche MN, Pavlov V, Takahashi K, Stahl GL. Myocardial ischemia and reperfusion injury is dependent on both IgM and mannose-binding lectin. Am J Physiol Heart Circ Physiol (2009) 297(5):H1853–9. doi:10.1152/ajpheart.00049.2009
75. Diepenhorst GM, van Gulik TM, Hack CE. Complement-mediated ischemia-reperfusion injury: lessons learned from animal and clinical studies. Ann Surg (2009) 249(6):889–99. doi:10.1097/SLA.0b013e3181a38f45
76. Zhou W, Farrar CA, Abe K, Pratt JR, Marsh JE, Wang Y, et al. Predominant role for C5b-9 in renal ischemia/reperfusion injury. J Clin Invest (2000) 105(10):1363–71. doi:10.1172/JCI8621
77. Hart ML, Ceonzo KA, Shaffer LA, Takahashi K, Rother RP, Reenstra WR, et al. Gastrointestinal ischemia-reperfusion injury is lectin complement pathway dependent without involving C1q. J Immunol (2005) 174(10):6373–80. doi:10.4049/jimmunol.174.10.6373
78. Lee H, Green DJ, Lai L, Hou YJ, Jensenius JC, Liu D, et al. Early complement factors in the local tissue immunocomplex generated during intestinal ischemia/reperfusion injury. Mol Immunol (2010) 47(5):972–81. doi:10.1016/j.molimm.2009.11.022
79. Stahl GL, Xu Y, Hao L, Miller M, Buras JA, Fung M, et al. Role for the alternative complement pathway in ischemia/reperfusion injury. Am J Pathol (2003) 162(2):449–55. doi:10.1016/S0002-9440(10)63839-4
80. Miwa T, Sato S, Gullipalli D, Nangaku M, Song WC. Blocking properdin, the alternative pathway, and anaphylatoxin receptors ameliorates renal ischemia-reperfusion injury in decay-accelerating factor and CD59 double-knockout mice. J Immunol (2013) 190(7):3552–9. doi:10.4049/jimmunol.1202275
81. Collard CD, Väkevä A, Morrissey MA, Agah A, Rollins SA, Reenstra WR, et al. Complement activation after oxidative stress: role of the lectin complement pathway. Am J Pathol (2000) 156(5):1549–56. doi:10.1016/S0002-9440(10)65026-2
82. Collard CD, Montalto MC, Reenstra WR, Buras JA, Stahl GL. Endothelial oxidative stress activates the lectin complement pathway: role of cytokeratin 1. Am J Pathol (2001) 159(3):1045–54. doi:10.1016/S0002-9440(10)61779-8
83. de Vries B, Walter SJ, Peutz-Kootstra CJ, Wolfs TG, van Heurn LW, Buurman WA. The mannose-binding lectin-pathway is involved in complement activation in the course of renal ischemia-reperfusion injury. Am J Pathol (2004) 165(5):1677–88. doi:10.1016/S0002-9440(10)63424-4
84. Møller-Kristensen M, Wang W, Ruseva M, Thiel S, Nielsen S, Takahashi K, et al. Mannan-binding lectin recognizes structures on ischaemic reperfused mouse kidneys and is implicated in tissue injury. Scand J Immunol (2005) 61(5):426–34. doi:10.1111/j.1365-3083.2005.01591.x
85. Asgari E, Farrar CA, Lynch N, Ali YM, Roscher S, Stover C, et al. Mannan-binding lectin-associated serine protease 2 is critical for the development of renal ischemia reperfusion injury and mediates tissue injury in the absence of complement C4. FASEB J (2014) 28(9):3996–4003. doi:10.1096/fj.13-246306
86. Yu ZX, Qi S, Lasaro MA, Bouchard K, Dow C, Moore K, et al. Targeting complement pathways during cold ischemia and reperfusion prevents delayed graft function. Am J Transplant (2016) 16(9):2589–97. doi:10.1111/ajt.13797
87. Business Wire. Alexion Announces Top-Line Results from Phase 2/3 PROTECT Study of Eculizumab (Soliris®) for the Prevention of Delayed Graft Function (DGF) After Kidney Transplantation. (2017). Available from: http://www.businesswire.com/news/home/20161221005838/en/Alexion-Announces-Top-Line-Results-Phase-23-PROTECT
88. Noris M, Remuzzi G. Thrombotic microangiopathy after kidney transplantation. Am J Transplant (2010) 10(7):1517–23. doi:10.1111/j.1600-6143.2010.03156.x
89. Ardalan MR. Review of thrombotic microangiopathy (TMA), and post-renal transplant TMA. Saudi J Kidney Dis Transpl (2006) 17(2):235–44.
90. Benz K, Amann K. Thrombotic microangiopathy: new insights. Curr Opin Nephrol Hypertens (2010) 19(3):242–7. doi:10.1097/MNH.0b013e3283378f25
91. Ponticelli C. De novo thrombotic microangiopathy. An underrated complication of renal transplantation. Clin Nephrol (2007) 67(6):335–40. doi:10.5414/CNP67335
92. Broeders EN, Stordeur P, Rorive S, Dahan K. A ’silent’, new polymorphism of factor H and apparent de novo atypical haemolytic uraemic syndrome after kidney transplantation. BMJ Case Rep (2014). doi:10.1136/bcr-2014-207630
93. Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med (2013) 368(23):2169–81. doi:10.1056/NEJMoa1208981
94. Licht C, Greenbaum LA, Muus P, Babu S, Bedrosian CL, Cohen DJ, et al. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int (2015) 87(5):1061–73. doi:10.1038/ki.2014.423
95. Zuber J, Le Quintrec M, Krid S, Bertoye C, Gueutin V, Lahoche A, et al. Eculizumab for atypical hemolytic uremic syndrome recurrence in renal transplantation. Am J Transplant (2012) 12(12):3337–54. doi:10.1111/j.1600-6143.2012.04252.x
96. Passwell J, Schreiner GF, Non-aka M, Beuscher HU, Colten HR. Local extrahepatic expression of complement genes C3, factor B, C2, and C4 is increased in murine lupus nephritis. J Clin Invest (1988) 82(5):1676–84. doi:10.1172/JCI113780
97. Pratt JR, Basheer SA, Sacks SH. Local synthesis of complement component C3 regulates acute renal transplant rejection. Nat Med (2002) 8(6):582–7. doi:10.1038/nm0602-582
98. Cravedi P, Leventhal J, Lakhani P, Ward SC, Donovan MJ, Heeger PS. Immune cell-derived C3a and C5a costimulate human T cell alloimmunity. Am J Transplant (2013) 13(10):2530–9. doi:10.1111/ajt.12405
99. van der Touw W, Cravedi P, Kwan WH, Paz-Artal E, Merad M, Heeger PS. Cutting edge: receptors for C3a and C5a modulate stability of alloantigen-reactive induced regulatory T cells. J Immunol (2013) 190(12):5921–5. doi:10.4049/jimmunol.1300847
100. Gueler F, Rong S, Gwinner W, Mengel M, Bröcker V, Schön S, et al. Complement 5a receptor inhibition improves renal allograft survival. J Am Soc Nephrol (2008) 19(12):2302–12. doi:10.1681/ASN.2007111267
101. Li K, Anderson KJ, Peng Q, Noble A, Lu B, Kelly AP, et al. Cyclic AMP plays a critical role in C3a-receptor-mediated regulation of dendritic cells in antigen uptake and T-cell stimulation. Blood (2008) 112(13):5084–94. doi:10.1182/blood-2008-05-156646
102. Kwan WH, van der Touw W, Paz-Artal E, Li MO, Heeger PS. Signaling through C5a receptor and C3a receptor diminishes function of murine natural regulatory T cells. J Exp Med (2013) 210(2):257–68. doi:10.1084/jem.20121525
103. Zheng QY, Liang SJ, Li GQ, Lv YB, Li Y, Tang M, et al. Complement component 3 deficiency prolongs MHC-II disparate skin allograft survival by increasing the CD4(+) CD25(+) regulatory T cells population. Sci Rep (2016) 6:33489. doi:10.1038/srep33489
104. Cheng HB, Chen RY, Wu JP, Chen L, Liang YH, Pan HF, et al. Complement C4 induces regulatory T cells differentiation through dendritic cell in systemic lupus erythematosus. Cell Biosci (2015) 5:73. doi:10.1186/s13578-015-0052-8
105. Clarke EV, Weist BM, Walsh CM, Tenner AJ. Complement protein C1q bound to apoptotic cells suppresses human macrophage and dendritic cell-mediated Th17 and Th1 T cell subset proliferation. J Leukoc Biol (2015) 97(1):147–60. doi:10.1189/jlb.3A0614-278R
106. Liu J, Miwa T, Hilliard B, Chen Y, Lambris JD, Wells AD, et al. The complement inhibitory protein DAF (CD55) suppresses T cell immunity in vivo. J Exp Med (2005) 201(4):567–77. doi:10.1084/jem.20040863
107. Pavlov V, Raedler H, Yuan S, Leisman S, Kwan WH, Lalli PN, et al. Donor deficiency of decay-accelerating factor accelerates murine T cell-mediated cardiac allograft rejection. J Immunol (2008) 181(7):4580–9. doi:10.4049/jimmunol.181.7.4580
108. Kwan WH, Hashimoto D, Paz-Artal E, Ostrow K, Greter M, Raedler H, et al. Antigen-presenting cell-derived complement modulates graft-versus-host disease. J Clin Invest (2012) 122(6):2234–8. doi:10.1172/JCI61019
109. Yamanaka K, Kakuta Y, Miyagawa S, Nakazawa S, Kato T, Abe T, et al. Depression of complement regulatory factors in rat and human renal grafts is associated with the progress of acute T-cell mediated rejection. PLoS One (2016) 11(2):e0148881. doi:10.1371/journal.pone.0148881
110. Cardone J, Le Friec G, Vantourout P, Roberts A, Fuchs A, Jackson I, et al. Complement regulator CD46 temporally regulates cytokine production by conventional and unconventional T cells. Nat Immunol (2010) 11(9):862–71. doi:10.1038/ni.1917
111. Parikova A, Fronek JP, Viklicky O. Living-donor kidney transplantation for atypical haemolytic uremic syndrome with pre-emptive eculizumab use. Transpl Int (2015) 28(3):366–9. doi:10.1111/tri.12440
112. Farrar CA, Sacks SH. Mechanisms of rejection: role of complement. Curr Opin Organ Transplant (2014) 19(1):8–13. doi:10.1097/MOT.0000000000000037
113. Berger SP, Roos A, Mallat MJ, Fujita T, de Fijter JW, Daha MR. Association between mannose-binding lectin levels and graft survival in kidney transplantation. Am J Transplant (2005) 5(6):1361–6. doi:10.1111/j.1600-6143.2005.00841.x
114. Imai N, Nishi S, Alchi B, Ueno M, Fukase S, Arakawa M, et al. Immunohistochemical evidence of activated lectin pathway in kidney allografts with peritubular capillary C4d deposition. Nephrol Dial Transplant (2006) 21(9):2589–95. doi:10.1093/ndt/gfl210
115. Zwirner J, Felber E, Herzog V, Riethmüller G, Feucht HE. Classical pathway of complement activation in normal and diseased human glomeruli. Kidney Int (1989) 36(6):1069–77. doi:10.1038/ki.1989.302
116. Feucht HE, Schneeberger H, Hillebrand G, Burkhardt K, Weiss M, Riethmüller G, et al. Capillary deposition of C4d complement fragment and early renal graft loss. Kidney Int (1993) 43(6):1333–8. doi:10.1038/ki.1993.187
117. Lederer SR, Kluth-Pepper B, Schneeberger H, Albert E, Land W, Feucht HE. Impact of humoral alloreactivity early after transplantation on the long-term survival of renal allografts. Kidney Int (2001) 59(1):334–41. doi:10.1046/j.1523-1755.2001.00495.x
118. Herzenberg AM, Gill JS, Djurdjev O, Magil AB. C4d deposition in acute rejection: an independent long-term prognostic factor. J Am Soc Nephrol (2002) 13(1):234–41.
119. Solez K, Colvin RB, Racusen LC, Haas M, Sis B, Mengel M, et al. Banff 07 classification of renal allograft pathology: updates and future directions. Am J Transplant (2008) 8(4):753–60. doi:10.1111/j.1600-6143.2008.02159.x
120. Hass M. An updated Banff schema for diagnosis of antibody-mediated rejection in renal allografts. Curr Opin Organ Transplant (2014) 19(3):315–22. doi:10.1097/MOT.0000000000000072
121. Miura M, Ogawa Y, Kubota KC, Harada H, Shimoda N, Ono T, et al. Donor-specific antibody in chronic rejection is associated with glomerulopathy, thickening of peritubular capillary basement membrane, but not C4d deposition. Clin Transplant (2007) 21:8–12. doi:10.1111/j.1399-0012.2007.00710.x
122. Sis B, Jhangri GS, Bunnag S, Allanach K, Kaplan B, Halloran PF. Endothelial gene expression in kidney transplants with alloantibody indicates antibody-mediated damage despite lack of C4d staining. Am J Transplant (2009) 9(10):2312–23. doi:10.1111/j.1600-6143.2009.02761.x
123. Sheen JH, Heeger PS. Effects of complement activation on allograft injury. Curr Opin Organ Transplant (2015) 20(4):468–75. doi:10.1097/MOT.0000000000000216
124. Fang Y, Xu C, Fu YX, Holers VM, Molina H. Expression of complement receptors 1 and 2 on follicular dendritic cells is necessary for the generation of a strong antigen-specific IgG response. J Immunol (1998) 160(11):5273–9.
125. Dempsey PW, Allison ME, Akkaraju S, Goodnow CC, Fearon DT. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science (1996) 271(5247):348–50. doi:10.1126/science.271.5247.348
126. Akiyoshi T, Hirohashi T, Alessandrini A, Chase CM, Farkash EA, Neal Smith R, et al. Role of complement and NK cells in antibody mediated rejection. Hum Immunol (2012) 73(12):1226–32. doi:10.1016/j.humimm.2012.07.330
127. Jane-Wit D, Manes TD, Yi T, Qin L, Clark P, Kirkiles-Smith NC, et al. Alloantibody and complement promote T cell-mediated cardiac allograft vasculopathy through non-canonical nuclear factor-κB signaling in endothelial cells. Circulation (2013) 128(23):2504–16. doi:10.1161/CIRCULATIONAHA.113.002972
128. Yabu JM, Higgins JP, Chen G, Sequeira F, Busque S, Tyan DB. C1q-fixing human leukocyte antigen antibodies are specific for predicting transplant glomerulopathy and late graft failure after kidney transplantation. Transplantation (2011) 91(3):342–7. doi:10.1097/TP.0b013e318203fd26
129. Loupy A, Lefaucheur C, Vernerey D, Prugger C, Duong van Huyen JP, Mooney N, et al. Complement-binding anti-HLA antibodies and kidney-allograft survival. N Engl J Med (2013) 369(13):1215–26. doi:10.1056/NEJMoa1302506
130. Crespo M, Torio A, Mas V, Redondo D, Pérez-Sáez MJ, Mir M, et al. Clinical relevance of pretransplant anti-HLA donor-specific antibodies: does C1q-fixation matter? Transpl Immunol (2013) 29(1–4):28–33. doi:10.1016/j.trim.2013.07.002
131. Porcheray F, DeVito J, Helou Y, Dargon I, Fraser JW, Nobecourt P, et al. Expansion of polyreactive B cells cross-reactive to HLA and self in the blood of a patient with kidney graft rejection. Am J Transplant (2012) 12(8):2088–97. doi:10.1111/j.1600-6143.2012.04053.x
132. Porcheray F, DeVito J, Yeap BY, Xue L, Dargon I, Paine R, et al. Chronic humoral rejection of human kidney allografts associates with broad autoantibody responses. Transplantation (2010) 89(10):1239–46. doi:10.1097/TP.0b013e3181d72091
133. Porcheray F, Fraser JW, Gao B, McColl A, DeVito J, Dargon I, et al. Polyreactive antibodies developing amidst humoral rejection of human kidney grafts bind apoptotic cells and activate complement. Am J Transplant (2013) 13(10):2590–600. doi:10.1111/ajt.12394
134. Stegall MD, Diwan T, Raghavaiah S, Cornell LD, Burns J, Dean PG, et al. Terminal complement inhibition decreases antibody-mediated rejection in sensitized renal transplant recipients. Am J Transplant (2011) 11(11):2405–13. doi:10.1111/j.1600-6143.2011.03757.x
135. Stegall MD, Chedid MF, Cornell LD. The role of complement in antibody-mediated rejection in kidney transplantation. Nat Rev Nephrol (2012) 8(11):670–8. doi:10.1038/nrneph.2012.212
136. Alexion Pharmaceuticals Inc. Alexion Provides Update on Phase 2 Clinical Trial with Eculizumab in Antibody Mediated Rejection (AMR) in Living-Donor Kidney Transplant Recipients. (2017). Available from: http://news.alexionpharma.com/press-release/company-news/alexion-provides-update-phase-2-clinical-trial-eculizumab-antibody-mediat
137. Hirohashi T, Chase CM, Della Pelle P, Sebastian D, Alessandrini A, Madsen JC, et al. A novel pathway of chronic allograft rejection mediated by NK cells and alloantibody. Am J Transplant (2012) 12(2):313–21. doi:10.1111/j.1600-6143.2011.03836.x
138. Hidalgo LG, Sis B, Sellares J, Campbell PM, Mengel M, Einecke G, et al. NK cell transcripts and NK cells in kidney biopsies from patients with donor-specific antibodies: evidence for NK cell involvement in antibody-mediated rejection. Am J Transplant (2010) 10(8):1812–22. doi:10.1111/j.1600-6143.2010.03201.x
139. Hidalgo LG, Sellares J, Sis B, Mengel M, Chang J, Halloran PF. Interpreting NK cell transcripts versus T cell transcripts in renal transplant biopsies. Am J Transplant (2012) 12(5):1180–91. doi:10.1111/j.1600-6143.2011.03970.x
140. Kohei N, Tanaka T, Tanabe K, Masumori N, Dvorina N, Valujskikh A, et al. Natural killer cells play a critical role in mediating inflammation and graft failure during antibody-mediated rejection of kidney allografts. Kidney Int (2016) 89(6):1293–306. doi:10.1016/j.kint.2016.02.030
141. Legris T, Picard C, Todorova D, Lyonnet L, Laporte C, Dumoulin C, et al. Antibody-dependent NK cell activation is associated with late kidney allograft dysfunction and the complement-independent alloreactive potential of donor-specific antibodies. Front Immunol (2016) 7:288. doi:10.3389/fimmu.2016.00288
142. Davis AE III, Mejia P, Lu F. Biological activities of C1 inhibitor. Mol Immunol (2008) 45(16):4057–63. doi:10.1016/j.molimm.2008.06.028
143. Paréj K, Dobó J, Závodszky P, Gál P. The control of the complement lectin pathway activation revisited: both C1-inhibitor and antithrombin are likely physiological inhibitors, while α2-macroglobulin is not. Mol Immunol (2013) 54(3–4):415–22. doi:10.1016/j.molimm.2013.01.009
144. Tillou X, Poirier N, Le Bas-Bernardet S, Hervouet J, Minault D, Renaudin K, et al. Recombinant human C1-inhibitor prevents acute antibody-mediated rejection in alloimmunized baboons. Kidney Int (2010) 78(2):152–9. doi:10.1038/ki.2010.75
145. Vo AA, Zeevi A, Choi J, Cisneros K, Toyoda M, Kahwaji J, et al. A phase I/II placebo-controlled trial of C1-inhibitor for prevention of antibody-mediated rejection in HLA sensitized patients. Transplantation (2015) 99(2):299–308. doi:10.1097/TP.0000000000000592
146. Mauriello CT, Pallera HK, Sharp JA, Woltmann JL Jr, Qian S, Hair PS, et al. A novel peptide inhibitor of classical and lectin complement activation including ABO incompatibility. Mol Immunol (2013) 53(1–2):132–9. doi:10.1016/j.molimm.2012.07.012
147. Sharp JA, Hair PS, Pallera HK, Kumar PS, Mauriello CT, Nyalwidhe JO, et al. Peptide inhibitor of complement C1 (PIC1) rapidly inhibits complement activation after intravascular injection in rats. PLoS One (2015) 10(7):e0132446. doi:10.1371/journal.pone.0132446
148. Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol (2007) 25(11):1256–64. doi:10.1038/nbt1207-1488c
149. Hillmen P, Young NS, Schubert J, Brodsky RA, Socié G, Muus P, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med (2006) 355(12):1233–43. doi:10.1056/NEJMoa061648
150. Fakhouri F, Hourmant M, Campistol JM, Cataland SR, Espinosa M, Gaber AO, et al. Terminal complement inhibitor eculizumab in adult patients with atypical hemolytic uremic syndrome: a single-arm, open-label trial. Am J Kidney Dis (2016) 68(1):84–93. doi:10.1053/j.ajkd.2015.12.034
151. Stewart ZA, Collins TE, Schlueter AJ, Raife TI, Holanda DG, Nair R, et al. Case report: eculizumab rescue of severe accelerated antibody-mediated rejection after ABO-incompatible kidney transplant. Transplant Proc (2012) 44(10):3033–6. doi:10.1016/j.transproceed.2012.03.053
152. Chehade H, Rotman S, Matter M, Girardin E, Aubert V, Pascual M. Eculizumab to treat antibody-mediated rejection in a 7-year-old kidney transplant recipient. Pediatrics (2015) 135(2):e551–5. doi:10.1542/peds.2014-2275
153. Tran D, Boucher A, Collette S, Payette A, Royal V, Senécal L. Eculizumab for the treatment of severe antibody-mediated rejection: a case report and review of the literature. Case Rep Transplant (2016) 2016:9874261. doi:10.1155/2016/9874261
154. Cornell LD, Schinstock CA, Gandhi MJ, Kremers WK, Stegall MD. Positive crossmatch kidney transplant recipients treated with eculizumab: outcomes beyond 1 year. Am J Transplant (2015) 15(5):1293–302. doi:10.1111/ajt.13168
155. Burbach M, Suberbielle C, Brochériou I, Ridel C, Mesnard L, Dahan K, et al. Report of the inefficacy of eculizumab in two cases of severe antibody-mediated rejection of renal grafts. Transplantation (2014) 98(10):1056–9. doi:10.1097/TP.0000000000000184
Keywords: complement, kidney transplantation, antibody-mediated rejection, ischemia/reperfusion injury, anticomplement therapy
Citation: Cernoch M and Viklicky O (2017) Complement in Kidney Transplantation. Front. Med. 4:66. doi: 10.3389/fmed.2017.00066
Received: 27 January 2017; Accepted: 09 May 2017;
Published: 30 May 2017
Edited by:
Ignacio Anegon, University of Nantes, FranceReviewed by:
Marcelo Hill, Institut Pasteur de Montevideo, UruguayMarian Klinger, Medical University, Poland
Copyright: © 2017 Cernoch and Viklicky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ondrej Viklicky, b25kcmVqLnZpa2xpY2t5JiN4MDAwNDA7aWtlbS5jeg==