 Dominique Sanglard
Dominique Sanglard
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 15 March 2016
Sec. Infectious Agents and Disease
Volume 3 - 2016 | https://doi.org/10.3389/fmed.2016.00011
The use of antifungal drugs in the therapy of fungal diseases can lead to the development of antifungal resistance. Resistance has been described for virtually all antifungal agents in diverse pathogens, including Candida and Aspergillus species. The majority of resistance mechanisms have also been elucidated at the molecular level in these pathogens. Drug resistance genes and genome mutations have been identified. Therapeutic choices are limited for the control of fungal diseases, and it is tempting to combine several drugs to achieve better therapeutic efficacy. In the recent years, several novel resistance patterns have been observed, including antifungal resistance originating from environmental sources in Aspergillus fumigatus and the emergence of simultaneous resistance to different antifungal classes (multidrug resistance) in different Candida species. This review will summarize these current trends.
Progresses in the therapy of human diseases have increased the survival of critically ill patients or patients with impaired function of their immune system. As a consequence, risk factors accumulate and favor the progression of other diseases, such as infectious diseases. Among these diseases, invasive fungal infections in humans represent a significant proportion. The most frequent fungal pathogens are Candida, Aspergillus, Pneumocystis, and Cryptococcus spp. It is estimated that these fungal species cause at least 1.4 million deaths worldwide per year (1). Compared to other microbial pathogens causing bloodstream infections, Candida spp. are ranked fourth among the most common agents of bloodstream infections, after other common bacterial pathogens (2). Aspergillus infections are the most common microbial infections in hematopoietic stem cell transplant (HSCT) recipients (3). About 30–50% of invasive aspergillosis patients still die, and the mortality from candidemia also remains high at ~50% (4).
There are only four major classes of antifungal drugs available to treat invasive fungal infections. They include polyenes, pyrimidine analogs, echinocandins, and triazoles (5). A fifth class (allylamines) is also existing; however, compounds of this class (for example, terbinafine) are used only for treating superficial dermatophytic infections (6). Polyenes, such as amphotericin B (AmB), have the ability to bind ergosterol and act as a sterol “sponge,” thus destabilizing membrane functions (7). Ergosterol is a major sterol of fungal membranes and is required for maintaining cell membrane integrity. AmB may exert intrinsic toxic effects in humans; however, this negative effect can be avoided by using liposome formulations (5). Pyrimidine analogs, such as 5-fluorocytosine (5-FC), are metabolized by fungal cells into fluorinated pyrimidines, which destabilize nucleic acids (RNA, DNA) and therefore result in growth arrest. 5-FC is used mainly for the treatment of Cryptococcus spp. meningitis and in combination with AmB (8). Echinocandins block the catalytic subunit of the β-1,3 glucan synthase and thus inhibit cell wall biosynthesis (9). In medical practice, triazoles are still the most used antifungals. These compounds target a specific step in the biosynthesis of ergosterol that catalyzes lanosterol 14α-demethylation (5). Fluconazole is the major triazole in clinical settings, probably due to its high oral availability and tolerability by patients.
The physical measure that determines antifungal activity is the reduction of growth in vitro as compared to drug-untreated cells. Antifungal activity is usually measured with standard dilutions in liquid media, although solid surface agars with drug gradients can be used as well (10). Two major protocols are currently used, both originating from major antifungal susceptibility testing subcommittees (CLSI, Clinical Laboratory Standards Institute; EUCAST, European Committee on Antimicrobial Susceptibility Testing). The protocols yield so-called minimum inhibition concentration (MIC) values (given in microgram per milliliter) as measures of antifungal activity. Although these protocols differ in several technical aspects, the agreement between the two methods in terms of antifungal activities is generally high (Table 1) (11). If a collection of isolates from the same species (for example, Candida albicans) is tested for susceptibility with a single agent (fluconazole), the resulting MICs will be distributed in a Gaussian bell-shaped manner. Such distribution helps to identify isolates (non-wild type isolates) differing from the general population of wild type isolates (12). The distinction between the two groups can be made with the help of the so-called epidemiological cut off (ECOFF) values. The ECOFF value is defined as the upper limit of the wild type population and in general will encompass about 95–99% of a given population for a specific agent. The ECOFF helps to detect non-wild type isolates that can typically be assigned as resistant isolates and may exhibit specific antifungal resistance mechanisms (13). In vitro resistance (or microbiological resistance) may be predictive of in vivo resistance (or clinical resistance). In order to achieve this, several studies have established clinical breakpoints (CBPs) for specific agents and specific fungal pathogens using several clinical parameters, including in vivo drug pharmacokinetics, resistance mechanisms, and clinical response. With MICs above CBPs, the success of therapy with a given agent is limited. For example, CBPs for fluconazole and C. albicans are declared as 2 and 4 μg/ml by EUCAST and CLSI, respectively. In a study in which candidemia episodes were enrolled (217) and treated with fluconazole monotherapy, infection-related mortality was significantly increased in C. albicans episodes with an MIC ≥2 μg/ml compared with those below this MIC target (20.6 versus 4.9%) (14). These results support well the proposed fluconazole CBPs of both EUCAST and CLSI. Table 1 gives an overview of current CBP of five important Candida spp. and currently available antifungal agents.
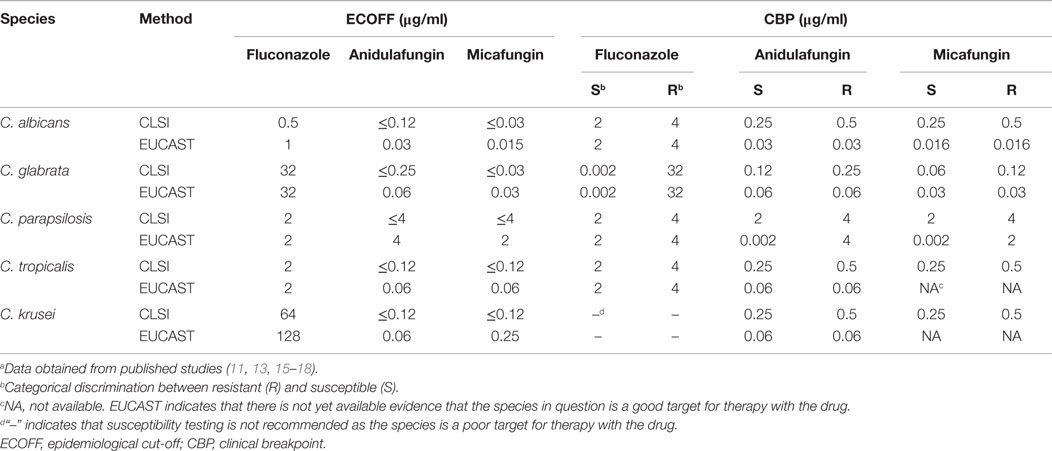
Table 1. ECOFF and CBP of different antifungal agents and fungal species.a
A number of fungal species are not perturbed by specific antifungal agents at any concentrations. The absence of drug activity in a species that was not pre-exposed to the tested agent is also known as intrinsic resistance. Taking as example the response of Candida and Aspergillus spp. to fluconazole, it is known that wild type C. albicans is susceptible to fluconazole, whereas Aspergillus fumigatus and C. krusei are intrinsically resistant to this drug (19). It is reported that Cryptococcus neoformans is resistant to echinocandins, even if the target of these drugs, a β-1,3 glucan synthase, is in vitro highly sensitive to these drugs (20). It is thought that echinocandin resistance is rather due to the high content of other sugar polymers (β-1,6 glucans) in Cr. neoformans, since their biosynthesis is not affected by echinocandins (21). Nevertheless, antifungal resistance can be acquired in vitro by drug exposure or during therapy. Antifungal resistance can measured by elevated MICs as compared to those of a wild type population. Acquired antifungal resistance has been reported virtually for all existing antifungal agents and major fungal pathogens (22).
There are variable accounts on the frequency at which antifungal resistance occurs in hospitalized patients. The epidemiology of invasive fungal infections and associated resistance is based on data collected by sentinel and population-based surveillance programs. Here, we will review some data available for major fungal pathogens, including C. albicans, C. glabrata, A. fumigatus, and Cr. neoformans. Rates of resistance that are calculated from available data depend on the values that are used as CBP for given agents as was reported recently (23). The CBP recommended by the CLSI and EUCAST committees used to be very divergent for specific agents (for example, for fluconazole in C. albicans), but now tend to be more harmonized (Table 1). Given these divergences, it is sometimes difficult to make comparisons between old and more recent epidemiological studies (23). In any case, antifungal resistance rates in C. albicans are in general low. In a study from two different areas between 2008 and 2011, resistance to agents, such as fluconazole (CBP: ≥64 μg/ml) or echinocandins (CBP: ≥4 μg/ml), ranged between 1 and 2% in bloodstream isolates (24). Resistance rates in C. glabrata are higher than in C. albicans. According to data available in the ARTEMIS Antifungal Surveillance Program, C. glabrata increased as a cause of invasive candidiasis from 18% of all BSI isolates in 1992–2001 to 25% in 2001–2007. Fluconazole resistance rates in C. glabrata increased over the same period from 9 to 14% (CBP: ≥64 μg/ml) (25, 26). Resistance of C. glabrata to the class of echinocandins also reaches significant proportions. It was reported that, within a 10-year survey (2001–2010) in an US hospital (Duke University Hospital), echinocandin resistance rate increased from 4.9 to 12.3% (27). Similar trends are reported in Europe, although resistance rates range between 1 and 4% (28).
Antifungal resistance in A. fumigatus from clinical origin is mainly reported for the class of azoles, including itraconazole, voriconazole, and posaconazole. Rates of resistance are geographically variable. In general, resistance occurs when MIC values are above the ECOFF. These values are 1 μg/ml for itraconazole and voriconazole and 0.25 μg/ml for posaconazole (9). For example, resistance reached levels varying between 6 and 27% in the UK between 1997 and 2009, while it is up to 8% and only 0.6% in the Netherlands and USA, respectively (29).
Cryptococcus neoformans antifungal resistance is known for fluconazole (MIC ≥ 16 μg/ml). Fluconazole resistance has been described mostly in AIDS patients suffering cryptococcal meningitis. Although resistance rates were up to 28% in the early 90s (1990–1994), which corresponds to a period before the introduction of highly active anti-retroviral therapy (HAART), the current trends are around 1% (30, 31).
Mechanisms of antifungal resistance have been resolved at the molecular level for most antifungal agents and fungal pathogens. In principle, these mechanisms fall into distinct categories, including (1) decrease of effective drug concentration, (2) drug target alterations, and (3) metabolic bypasses. The major features for each of these principles are summarized below (Figure 1).
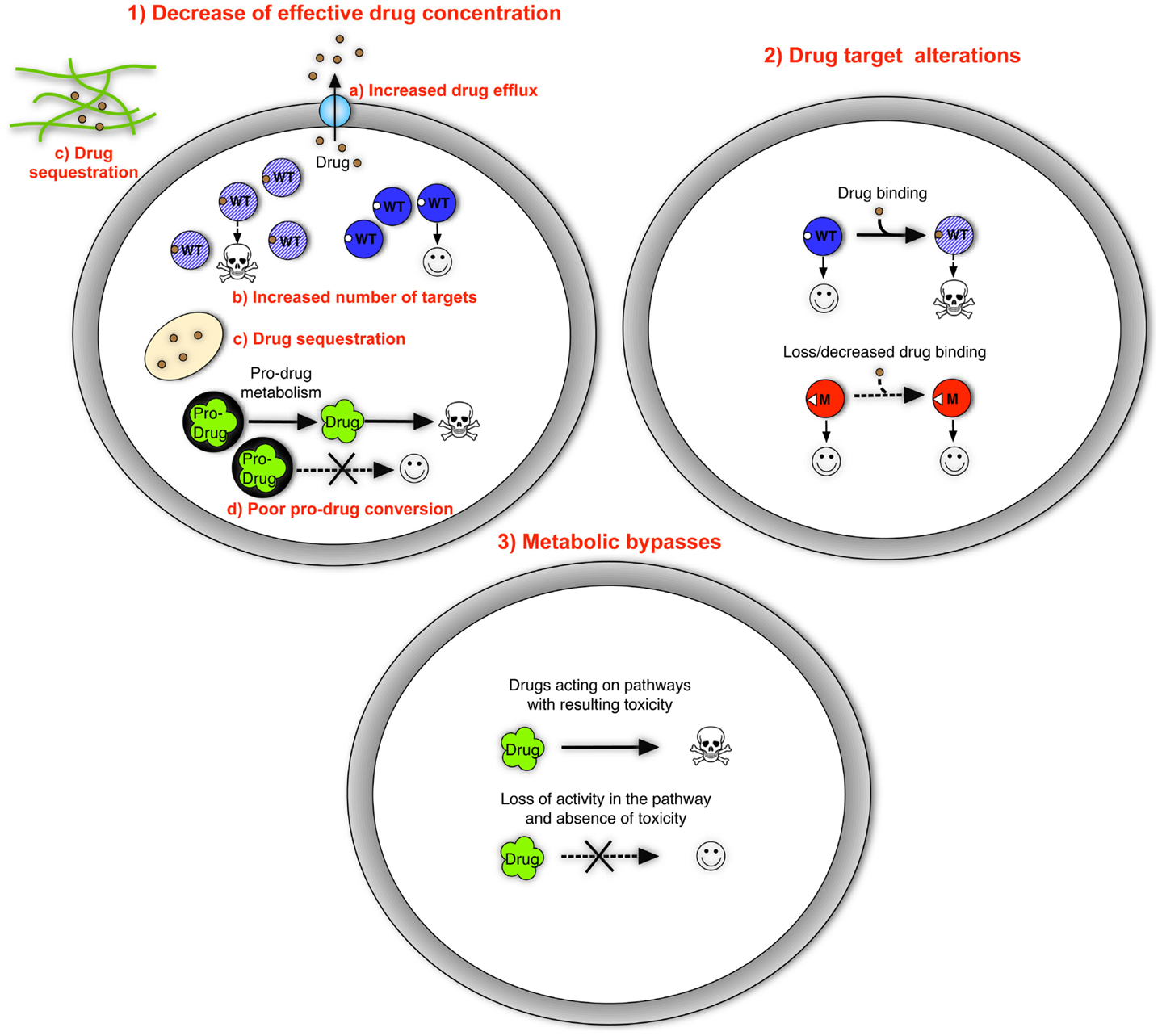
Figure 1. The three basic resistance mechanisms to antifungal drugs. They include (as listed in the text) (1) decrease of effective drug concentration with specific mechanisms including increased drug efflux, increased number of targets, drug sequestration of extracellular and intracellular origins, and poor pro-drug conversion; (2) drug target alterations; and (3) metabolic bypasses. Genome mutations are generally responsible for these three basic principles. Drug sequestration can be mediated by the formation of matrix polymers in biofilms, a state of cells that is not dependent on the occurrence of genome mutations. Wild type proteins are represented by blue circles catalyzing cellular functions; blue-shaded circles represent proteins blocked by drugs in which cellular functions are blocked causing decreased growth or death. Mutant proteins are represented by red circles. Drugs are represented with different symbols. Symbols: WT, wild type; M, mutant.
(1) Decreased effective drug concentrations can be achieved itself by several distinct mechanisms:
(a) The drug intracellular concentrations can be decreased by active efflux.
It is known that drug resistance can be mediated by the activity of several efflux transport systems, including ATP-binding cassette (ABC) transporters and transporters of the major facilitator superfamily (MFS). The analysis of fungal pathogen genomes has identified varying numbers of ABC transporters and MFS transporters with different topologies. C. albicans is predicted to contain 28 ABC proteins and 96 potential MFS transporters (32–34), whereas C. glabrata has at least 18 ABC transporters (35) and 33 MFS transporters (deduced from http://www.ebi.ac.uk/interpro/entry/IPR011701/taxonomy). Larger numbers of ABC and MFS proteins are found in A. fumigatus (45 and 275, respectively) and Cr. neoformans (29 and 192, respectively) (36, 37) (data available at http://www.membranetransport.org). ABC transporters are arranged in different subfamilies; however, they all contain membrane spanning domains and use ATP hydrolysis for drug transport. MFS transporters are transmembrane proteins, which use the electrochemical proton-motive force to mediate drug efflux. MFS are involved in multidrug resistance (MDR) (MFS–MDR transporters) function as proton antiporters and are classified into two groups: the drug:H+ antiporter-1 DHA1 family and the drug:H+ antiporter-2 DHA2 family (32, 38).
Fungal ABC transporters have been arranged into several classes; however, only ABC transporters of the pleiotropic drug resistance (PDR) class are relevant for antifungal drug resistance. In C. albicans, the PDR class comprises the major transporters involved in azole resistance, including CDR1 (for Candida drug resistance) and CDR2, and also other transporters not shown yet to be involved in antifungal resistance (CDR3, CDR4, CDR11, and SNQ2). Basically, the upregulation of both CDR1 and CDR2 mediates azole resistance by enhanced drug efflux and reduces azole accumulation in some C. albicans clinical strains (39). Several other ABC transporters known to be involved in azole resistance by their upregulation are the C. glabrata CgCDR1, CgCDR2, CgSNQ2 genes, and AFR1 from Cr. neoformans (40). In A. fumigatus, the association between azole resistance and transporter upregulation is less clear. The ABC transporter atrF was shown as upregulated in an azole-resistant clinical isolate; however, this could not be firmly attributed as a cause of resistance (41). AfuMDR4 was strongly upregulated in several itraconazole-resistant laboratory-derived mutants (42). Transcriptional profiling revealed transporter genes whose expression was induced in response to voriconazole (43). These included five ABC transporter genes (designated abcA–E) and three MFS transporter genes (designated mfsA–C). The abcA gene, renamed cdr1B, is the only known transporter gene with a direct role in azole resistance in A. fumigatus (44). AfuMDR3, a MFS transporter in A. fumigatus, was found as upregulated in a collection of itraconazole-resistant laboratory-derived mutants; however, its participation in azole resistance of clinical isolates remains elusive (42). MFS involved in the development of azole resistance in clinical isolates are restricted to MDR1 from C. albicans and C. dubliniensis. MDR1 is upregulated in specific strains, which results in enhanced azole efflux (45, 46). FLU1 (for Fluconazole resistance) from C. albicans is another MFS, and heterologous expression in S. cerevisiae revealed that it served as a fluconazole efflux transporter (47). Until now, however, no studies have shown the participation of FLU1 in azole resistance in clinical isolates.
Upregulation of ABC and MFS transporters is mediated by specific regulators in resistant fungal pathogens. In C. albicans, CDR1 and CDR2 are known to be regulated by a zinc cluster finger transcriptional regulator called TAC1 and MDR1 by another regulator called MRR1 (48, 49). Mutations (gain-of-function or GOF mutations) in these regulators have been described, and they confer an hyperactivation state that does not require additional stimulation, thus explaining the inherent high expression levels of the transporters in drug-resistant isolates (50, 51). Other transcriptional regulators of drug transporters relevant to azole resistance, such as PDR1, have been described in C. glabrata (52, 53).
(b) The drug target is overexpressed.
By increasing the number of drug targets, the effective drug concentration needs to be also increased to saturate all target molecules, which results in drug resistance. For example, ERG11 upregulation has been associated with azole resistance in C. albicans. This transcriptional regulation is mediated by a zinc cluster finger transcription factor called UPC2. As in the case of other drug resistance transcriptional regulators, GOF mutations in UPC2 have been described and result in upregulation of various genes, among which is ERG11 (54). Upregulation of Cyp51A is also known in azole-resistant A. fumigatus isolates; however, the upregulation is mediated by duplication of 34- and 42-bp elements (trans-regulation) in the Cyp51A promoter. This duplication is associated with specific Cyp51A mutations (L98H, Y121F/T289A) (55). These combined mutation signatures are enriched in azole-resistant A. fumigatus isolates originating from the environment that probably arose from the use of azoles in the agriculture (29).
(c) The drug is sequestered in extra- or intracellular compartments.
Fungal pathogens have the ability to sequester drugs within extracellular compartments. Several fungal pathogens, including Candida and Aspergillus spp., are able to form biofilms in specific growth conditions (56). Biofilms are multicellular structures in which cells form a dense network that is covered by the so-called matrix. The matrix is composed of different elements in C. albicans biofilms, including several cell wall polymers (57). Biofilm formation is known to be associated with resistance to several drugs, including azoles, polyenes, and pyrimidine analogs (58). Interestingly, recent data showed that the matrix participates to this process by its capacity to sequester antifungal agents. This process has been clearly documented for fluconazole (57, 59) and was suggested for AmB in C. albicans (60).
Much less is known in drug sequestration in intracellular compartments. A single report document the accumulation in C. albicans of fluconazole in organelles that were described as vesicular vacuoles. Whether or not this type of mechanism could occur in other isolates remains unknown (61).
(d) A pro-drug is poorly converted to an active drug.
Poor drug metabolization as a principle of antifungal resistance is also observed when 5-FC resistance occurs. 5-FC is a pro-drug, which is metabolized by cells into fluorinated pyrimidine analogs, thus inhibiting nucleic acid and protein biosynthesis. After import into cells, cytosine deaminase converts 5-FC into 5-fluorouridine (5-FU) and therefore the deficiency of this step decreases further processing and toxicity of the drug. Mutations in cytosine deaminase in C. albicans (FCA1) (62) and C. glabrata (FCY1) have been reported, resulting in 5FC resistance (63, 64).
(2) Drug target alterations have been reported for at least two classes of antifungal agents, including azoles and echinocandins. The targets of these two drugs are a 14α-lanosterol demethylase and a β-1,3 glucan synthase, respectively. Lanosterol demethylase is encoded by ERG11 in C. albicans and Cyp51A and Cyp51B in A. fumigatus. Mutations in ERG11 resulting in non-synonymous amino acid substitutions that are present in azole-resistant C. albicans isolates are numerous and were shown to decrease the affinity of the target to azoles (65). The effects of ERG11 mutations have different outcomes on azole MICs that depend on structural features of azole drugs. Although most known mutations decrease affinity to fluconazole, they have only a moderate effect on posaconazole affinity (66). In many cases, simultaneous ERG11 mutations can be present on the same ERG11 allele and be accompanied by drug transport modifications, thus resulting in azole-resistant isolates with high MIC values against azoles (for example, fluconazole MIC > 128 μg/ml) (67). Mutations in lanosterol demethylase genes from azole-resistant A. fumigatus isolates have been only reported in Cyp51A until now. Single Cyp51A mutations are sufficient to confer high level resistance to azoles in this species (68). As in the case of ERG11, Cyp51A mutations have different impact on MICs that depend on the azole structure (69).
Decreased affinity to the target is also known for echinocandins. β-1,3 glucan synthases are encoded by FKS genes in different fungal species. Up to now, echinocandin resistance has been attributed to specific mutations leading to amino acid substitutions in two different regions of these genes (Hot spot 1 and 2 or HS1 and HS2). FKS1 mutations have been reported in these two regions (HS1: region 640–650 and HS2: 1345–1365) in clinical isolates of C. albicans (70). Equivalent mutations in the HS1 of FKS2 (an homolog to FKS1) of C. glabrata and FKS1 of Candida lusitaniae (71, 72), C. tropicalis, and C. krusei (73) have been reported. Some Candida species (the Candida parapsilosis family, including C. parapsilosis, C. orthopsilosis, and C. metapsilosis) exhibit intrinsic low susceptibilities to echinocandins. FKS1 genes in these species contain a natural polymorphism (P660A at the 3′-extremity of HS1) enabling decreased affinity of the β-1,3 glucan synthase to echinocandins. However, this natural FKS1 polymorphism of these species has less impact than those acquired by mutations, since these Candida species still respond to echinocandin therapy (74).
(3) Metabolic bypasses occur when given metabolic pathways are perturbed by loss or strong decrease of specific functions. Metabolic bypass can be compared to compensatory mechanisms in which cells divert the toxic effects exerted by some antifungal agents. For example, resistance to azoles can be mediated by loss-of-function mutations in the gene ERG3 that encodes a sterol Δ5,6 desaturase. If active, the gene product converts 14α-methylated sterols that arise from azole exposure into a toxic 3,6-diol derivative (75). Fungi unable to produce this metabolite acquire azole resistance. Several studies have reported ERG3 loss-of-function mutations to account for azole resistance (76–79). Due to a deficiency in ergosterol biosynthesis, these isolates can be, however, less competitive than wild type isolates in conditions encountered in the host. As a result of loss-of-function of ERG3 in specific mutants, ergosterol is absent from cell membranes. This way, the mutants escape the toxic effect of AmB, which normally acts as a “sponge” for ergosterol to rapidly destabilize membrane functions (7). Several other mutations in the ergosterol biosynthesis pathway (ERG6, ERG24, and ERG2) lead to the same effect and have also a compensatory effect (80–82).
A mutation in the gene FUR1 encoding uracil phosphoribosyltransferase decreases the conversion of 5-FU, which is produced from 5-FC deamination (see above), into a toxic metabolite (5-FC monophosphate). Thus, the toxic effect of 5-FC cannot be exerted (83).
Azole antifungal agents are not only widely used in medicine but they also largely contribute to crop protection in agriculture and are used to preserve materials from fungal decay (84). Therefore, A. fumigatus, as a ubiquitous fungus, is likely to come into contact in the environment with the same substance class that is used in medicine. A first report on azole resistance from environmental isolates was published in 2007 from the Netherlands (85). In this study, the authors were able to identify a mutation in the azole target Cyp51A (a L98H substitution), which was associated with a 34-bp tandem repeat (TR34) in the gene promoter. Interestingly, the same mutation was recovered from patient samples, strongly suggesting that the Cyp51A L98H/TR34 mutation was acquired from environmental isolates. This mutation results in resistance to all medical azoles (pan-azole resistance). One argument that is crucial to support environmental acquisition of azole resistance is that between 64 and 71% of patients with IA due to an azole-resistant A. fumigatus isolate had never received azole treatment before (86). There are concerns that azole resistance could become a global public health threat, since fungal spores can disperse easily by circulating air flows across long distances (84). Currently, environmental resistance is documented in several other European and Asian countries and America (29, 87, 88). Other Cyp51A mutations than the L98H/TR34 are now also reported from environmental isolates, including TR46/Y121F/T289A (89), as well as others (G54A and M220I) that were until now exclusively recovered from clinical isolates (90). These data suggest that systematic surveillance programs should be initiated worldwide. The use of azoles in the environment will be difficult to restrict, unless scientists raise better public and political awareness on this problem.
Antifungal resistance has been observed in most occasions as a process involving resistance to single classes of drugs. Within the same class, several different agents can exist. Examples are for the classes of azoles (fluconazole, itraconazole, voriconazole, posaconazole, and isavuconazole) and echinocandins (caspofungin, micafungin, and anidulafungin).
Specific resistance mechanisms can result in cross-resistance to several drugs of the same class. It is known that the expression of ABC transporters (i.e., CDR1 or CgCDR1) mediate cross-resistance to all azoles used in medicine (66). Likewise, specific FKS1 mutations in C. albicans (F641S, S645Y) yield cross-resistance to all echinocandins (91).
Multidrug resistance is the simultaneous resistance to at least two different classes of antifungal agents. In the recent years, reports documenting cases of MDR in fungal pathogens have been published. We will give here an overview of the latest trends in the emergence of MDR.
Many fungal infections are treated with different antifungal agent classes, including azoles and polyenes. MDR between these two classes could be mediated expectedly by different genomic mutations; however, it has been reported that it is sufficient to harbor only loss-of-function mutations in ERG3 to result in simultaneous MDR against azoles and AmB. Such mutations have been reported in C. albicans (79, 92–94) and C. dubliniensis (95). Other ERG gene defects may also confer MDR to both drug classes, such as the loss-of-function mutation in ERG2 observed in C. albicans (82).
Nevertheless, some specific isolates may show simultaneous mutations in several genes as a cause of MDR. This was reported in C. tropicalis by ERG3/ERG11 loss-of-function mutations (77, 82) and in C. albicans by ERG11/ERG5 mutations (92).
Echinocandins are being increasingly used for the therapy of fungal infections, especially those caused by Candida spp. Resistance to echinocandins logically appeared soon after its introduction in medicine in 2005 (96). A first report of MDR to caspofungin and azoles in C. glabrata isolated from blood cultures was made in 2010 after caspofungin therapy (97). Resistance mechanisms were combining mutations in the β-1,3 glucan synthase FKS2 (S663P) and ABC transporters upregulation. Closely related isolates became resistant to 5-FC after therapy with this drug; however, it was still susceptible to the two other drugs. These isolates exhibited a non-synonymous mutation (G190D) in FUR1, which probably accounted to decrease 5-FC toxicity.
The current trends show that the highest proportion of resistant isolates is from the species C. glabrata (70). Other observations were made recently on MDR with both azoles and echinocandins in C. glabrata. Among echinocandin-resistant isolates sampled between 2008 and 2013 in two US surveillance hospital sites, 36% were also resistant to azoles (98). Similar observations were reported in another US site between 2005 and 2013, in which 10.3% C. glabrata isolates from cancer patients were resistant to caspofungin and from which about 60% had a MDR phenotype with azoles (99). The data of this study also indicated that caspofungin exposure alone could induce MDR without azole exposure. Here, resistance mechanisms were not systematically investigated in these isolates; however, they are likely to involve FKS1/FKS2 and PDR1 mutations. This MDR pattern is therefore of concern, especially when considering that very few therapeutic alternatives are available.
Combining resistance for more than two drug classes is not a frequent observation in clinical isolates. However, a few cases have emerged recently and highlight the capacity of specific pathogens to adapt to strong antifungal selective pressure.
A recent case illustrated the evolution of MDR in C. albicans sequential isolates taken from a patient at different sites (oropharynx, esophagus, feces, and colon) treated over time (100). The isolates were related to each other as confirmed by genotyping methods. The evolution of drug resistance followed the course of drug treatments. Fluconazole treatment induced first a GOF mutation in TAC1 with corresponding azole resistance (MIC fluconazole >16 μg/ml). Caspo- and anidulafungin treatment resulted in resistance (MIC caspofungin >32 μg/ml) with a corresponding FKS1 mutation (S645P). Lastly, AmB treatment established AmB resistance (MIC > 32 μg/ml) with a loss-of-function mutation in ERG2. All three mutations were conserved in the final MDR strain (100). MDR evolution took place within a time lapse of 5 years.
Another example originates from a C. lusitaniae infection in a young immunocompromised patient with severe enterocolitis and visceral adenoviral disease (71). C. lusitaniae isolates were recovered from blood cultures and stools over a period of 3 months. Very early at onset of therapy, fully susceptible isolates were recovered. Caspofungin regimen resulted in detection of resistance (MIC = 4 μg/ml) with a corresponding FKS1 mutation in HS1 (S638Y). This resistance coincided with AMB resistance, although not administered simultaneously. The therapy was continued by fluconazole from which resistance rapidly emerged (MIC = 32 μg/ml). Fluconazole resistance could be associated with upregulation of a MFS transporter (MFS7) but was also accompanied by 5-FC resistance, even if no 5-FC was administered. Combination therapy with caspofungin and voriconazole was next attempted, and isolates with simultaneous resistance to caspofungin, fluconazole, and 5-FC were detected. These isolates exhibited another FKS1 mutation (S631Y) and MFS7 upregulation. This study highlighted a very dynamic property of C. lusitaniae, which responded quickly to antifungal exposure. In this specific type of abdominal disease, it is believed that a reservoir of fungal cells was present with mixed MDR genotypes, and, depending on the drug treatment regimen, dominant populations could emerge (71).
A consequence of the use of antifungal agents in the therapy of fungal diseases is to face antifungal resistance in fungi. The extent of the problem is variable and depends on the type of fungus, the type of antifungal agent and on the geographical location of hospitals. In the recent years, however, reports on novel resistance profiles have appeared, and one of the most problematic is the development of MDR. It seems that, up to now, MDR occurs mostly in the species C. glabrata, especially since the introduction of echinocandins in the clinic. The reasons behind MDR in this pathogen are still unclear. Since C. glabrata harbors a haploid genome, single genetic events are sufficient to express phenotypes, which are less the case for diploid organisms (for example, C. albicans). One other reason is that the genome context of C. glabrata may facilitate the occurrence of genetic events. Very recent data from the laboratory of D. Perlin suggest that some C. glabrata exhibit much higher mutations rates than others (hypermutator phenotype) (101). With the appearance of these novel resistance profiles, alternative therapeutic approaches are required and novel antifungal agents need to be identified.
The author confirms being the sole contributor of this work and approved it for publication.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
DS is supported by grants from the Swiss National Research Foundation No. 31003A_146936 and CRSII3_141848. DS is thankful to Sara Vaz and Alix Coste for critical reading.
1. Brown GD, Denning DW, Gow NAR, Levitz SM, Netea MG, White TC. Hidden killers: human fungal infections. Sci Transl Med (2012) 4:165rv13. doi: 10.1126/scitranslmed.3004404
2. Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis (2004) 39:309–17. doi:10.1086/421946
3. Kontoyiannis DP, Marr KA, Park BJ, Alexander BD, Anaissie EJ, Walsh TJ, et al. Prospective surveillance for invasive fungal infections in hematopoietic stem cell transplant recipients, 2001-2006: overview of the transplant-associated infection surveillance network (TRANSNET) database. Clin Infect Dis (2010) 50:1091–100. doi:10.1086/651263
4. Denning DW, Bromley MJ. Infectious disease. How to bolster the antifungal pipeline. Science (2015) 347:1414–6. doi:10.1126/science.aaa6097
5. Sanglard D, Odds FC. Resistance of Candida species to antifungal agents: molecular mechanisms and clinical consequences. Lancet Infect Dis (2002) 2:73–85. doi:10.1016/s1473-3099(02)00181-0
6. Jessup CJ, Ghannoum MA, Ryder NS. An evaluation of the in vitro activity of terbinafine. Med Mycol (2000) 38:155–9. doi:10.1080/mmy.38.2.155.159
7. Anderson TM, Clay MC, Cioffi AG, Diaz KA, Hisao GS, Tuttle MD, et al. Amphotericin forms an extramembranous and fungicidal sterol sponge. Nat Chem Biol (2014) 10:400–6. doi:10.1038/nchembio.1496
8. Nussbaum JC, Jackson A, Namarika D, Phulusa J, Kenala J, Kanyemba C, et al. Combination flucytosine and high-dose fluconazole compared with fluconazole monotherapy for the treatment of cryptococcal meningitis: a randomized trial in Malawi. Clin Infect Dis (2010) 50:338–44. doi:10.1086/649861
9. Arendrup MC. Update on antifungal resistance in Aspergillus and Candida. Clin Microbiol Infect (2014) 20(Suppl 6):42–8. doi:10.1111/1469-0691.12513
10. Pfaller MA, Diekema DJ. Progress in antifungal susceptibility testing of Candida spp. using clinical and laboratory standards institute broth microdilution methods, 2010-2012. J Clin Microbiol (2012) 50:2846–56. doi:10.1128/JCM.00937-12
11. Pfaller MA, Castanheira M, Messer SA, Rhomberg PR, Jones RN. Comparison of EUCAST and CLSI broth microdilution methods for the susceptibility testing of 10 systemically active antifungal agents when tested against Candida spp. Diagn Microbiol Infect Dis (2014) 79:198–204. doi:10.1016/j.diagmicrobio.2014.03.004
12. Delarze E, Sanglard D. Defining the frontiers between antifungal resistance, tolerance and the concept of persistence. Drug Resist Updat (2015) 23:12–9. doi:10.1016/j.drup.2015.10.001
13. Arendrup MC, Cuenca-Estrella M, Lass-Flörl C, Hope WW. Breakpoints for antifungal agents: an update from EUCAST focussing on echinocandins against Candida spp. and triazoles against Aspergillus spp. Drug Resist Updat (2013) 16:81–95. doi:10.1016/j.drup.2014.01.001
14. van Hal SJ, Chen SC-A, Sorrell TC, Ellis DH, Slavin M, Marriott DM. Support for the EUCAST and revised CLSI fluconazole clinical breakpoints by Sensititre® YeastOne® for Candida albicans: a prospective observational cohort study. J Antimicrob Chemother (2014) 69:2210–4. doi:10.1093/jac/dku124
15. Arendrup MC, Cuenca-Estrella M, Lass-Flörl C, Hope WW; The European Committee on Antimicrobial Susceptibility Testing-Subcommittee on Antifungal Susceptibility Testing (EUCAST-AFST). EUCAST technical note on Candida and micafungin, anidulafungin and fluconazole. Mycoses (2014) 57:377–9. doi:10.1111/myc.12170
16. Maubon D, Garnaud C, Calandra T, Sanglard D, Cornet M. Resistance of Candida spp. to antifungal drugs in the ICU: where are we now? Intensive Care Med (2014) 40:1–15. doi:10.1007/s00134-014-3404-7
17. Pfaller MA, Castanheira M, Diekema DJ, Messer SA, Moet GJ, Jones RN. Comparison of European Committee on Antimicrobial Susceptibility Testing (EUCAST) and Etest methods with the CLSI broth microdilution method for echinocandin susceptibility testing of Candida species. J Clin Microbiol (2010) 48:1592–9. doi:10.1128/JCM.02445-09
18. EUCAST-AFST. EUCAST technical note on fluconazole. Clin Microbiol Infect (2008) 14:193–5. doi:10.1111/j.1469-0691.2007.01899.x
19. Pfaller MA. Antifungal drug resistance: mechanisms, epidemiology, and consequences for treatment. Am J Med (2012) 125:S3–13. doi:10.1016/j.amjmed.2011.11.001
20. Maligie MA, Selitrennikoff CP. Cryptococcus neoformans resistance to echinocandins: (1,3)β-glucan synthase activity is sensitive to echinocandins. Antimicrob Agents Chemother (2005) 49:2851–6. doi:10.1128/AAC.49.7.2851-2856.2005
21. Feldmesser M, Kress Y, Mednick A, Casadevall A. The effect of the echinocandin analogue caspofungin on cell wall glucan synthesis by Cryptococcus neoformans. J Infect Dis (2000) 182:1791–5. doi:10.1086/317614
22. Perlin DS, Shor E, Zhao Y. Update on antifungal drug resistance. Curr Clin Micro Rpt (2015) 2:84–95. doi:10.1007/s40588-015-0015-1
23. Fothergill AW, Sutton DA, McCarthy DI, Wiederhold NP. Impact of new antifungal breakpoints on antifungal resistance in Candida species. J Clin Microbiol (2014) 52:994–7. doi:10.1128/JCM.03044-13
24. Cleveland AA, Farley MM, Harrison LH, Stein B, Hollick R, Lockhart SR, et al. Changes in incidence and antifungal drug resistance in candidemia: results from population-based laboratory surveillance in Atlanta and Baltimore, 2008-2011. Clin Infect Dis (2012) 55:1352–61. doi:10.1093/cid/cis697
25. Pfaller MA, Messer SA, Boyken L, Tendolkar S, Hollis RJ, Diekema DJ. Variation in susceptibility of bloodstream isolates of Candida glabrata to fluconazole according to patient age and geographic location. J Clin Microbiol (2003) 41:2176–9. doi:10.1128/JCM.41.5.2176-2179.2003
26. Pfaller MA, Messer SA, Hollis RJ, Boyken L, Tendolkar S, Kroeger J, et al. Variation in susceptibility of bloodstream isolates of Candida glabrata to fluconazole according to patient age and geographic location in the United States in 2001 to 2007. J Clin Microbiol (2009) 47:3185–90. doi:10.1128/JCM.00946-09
27. Alexander BD, Johnson MD, Pfeiffer CD, Jimenez-Ortigosa C, Catania J, Booker R, et al. Increasing echinocandin resistance in Candida glabrata: clinical failure correlates with presence of FKS mutations and elevated minimum inhibitory concentrations. Clin Infect Dis (2013) 56:1724–32. doi:10.1093/cid/cit136
28. Arendrup MC, Perlin DS. Echinocandin resistance: an emerging clinical problem? Curr Opin Infect Dis (2014) 27:484–92. doi:10.1097/QCO.0000000000000111
29. Vermeulen E, Lagrou K, Verweij PE. Azole resistance in Aspergillus fumigatus: a growing public health concern. Curr Opin Infect Dis (2013) 26:493–500. doi:10.1097/QCO.0000000000000005
30. Aller AI, Martin-Mazuelos E, Lozano F, Gomez-Mateos J, Steele-Moore L, Holloway WJ, et al. Correlation of fluconazole MICs with clinical outcome in cryptococcal infection. Antimicrob Agents Chemother (2000) 44:1544–8. doi:10.1128/AAC.44.6.1544-1548.2000
31. Pfaller MA, Castanheira M, Diekema DJ, Messer SA, Jones RN. Wild-type MIC distributions and epidemiologic cutoff values for fluconazole, posaconazole, and voriconazole when testing Cryptococcus neoformans as determined by the CLSI broth microdilution method. Diagn Microbiol Infect Dis (2011) 71:252–9. doi:10.1016/j.diagmicrobio.2011.07.007
32. Gaur M, Puri N, Manoharlal R, Rai V, Mukhopadhayay G, Choudhury D, et al. MFS transportome of the human pathogenic yeast Candida albicans. BMC Genomics (2008) 9:579. doi:10.1186/1471-2164-9-579
33. Paulsen IT, Sliwinski MK, Nelissen B, Goffeau A, Saier MH. Unified inventory of established and putative transporters encoded within the complete genome of Saccharomyces cerevisiae. FEBS Lett (1998) 430:116–25. doi:10.1016/S0014-5793(98)00629-2
34. Prasad R, Goffeau A. Yeast ATP-binding cassette transporters conferring multidrug resistance. Annu Rev Microbiol (2012) 66:39–63. doi:10.1146/annurev-micro-092611-150111
35. Gbelska Y, Krijger J-J, Breunig KD. Evolution of gene families: the multidrug resistance transporter genes in five related yeast species. FEMS Yeast Res (2006) 6:345–55. doi:10.1111/j.1567-1364.2006.00058.x
36. Kovalchuk A, Driessen AJM. Phylogenetic analysis of fungal ABC transporters. BMC Genomics (2010) 11:177. doi:10.1186/1471-2164-11-177
37. Lamping E, Baret PV, Holmes AR, Monk BC, Goffeau A, Cannon RD. ScienceDirect – fungal genetics and biology: fungal PDR transporters: phylogeny, topology, motifs and function. Fungal Genet Biol (2010) 47:127–42. doi:10.1016/j.fgb.2009.10.007
38. Costa C, Dias PJ, Sá-Correia I, Teixeira MC. MFS multidrug transporters in pathogenic fungi: do they have real clinical impact? Front Physiol (2014) 5:197–197. doi:10.3389/fphys.2014.00197
39. Sanglard D, Coste A, Ferrari S. Antifungal drug resistance mechanisms in fungal pathogens from the perspective of transcriptional gene regulation. FEMS Yeast Res (2009) 9:1029–50. doi:10.1111/j.1567-1364.2009.00578.x
40. Coleman JJ, Mylonakis E. Efflux in fungi: la pièce de résistance. PLoS Pathog (2009) 5:e1000486. doi:10.1371/journal.ppat.1000486.t002
41. Slaven JW, Anderson MJ, Sanglard D, Dixon GK, Bille J, Roberts IS, et al. Increased expression of a novel Aspergillus fumigatus ABC transporter gene, atrF, in the presence of itraconazole in an itraconazole resistant clinical isolate. Fungal Genet Biol (2002) 36:199–206. doi:10.1016/S1087-1845(02)00016-6
42. Nascimento AM, Goldman GH, Park S, Marras SAE, Delmas G, Oza U, et al. Multiple resistance mechanisms among Aspergillus fumigatus mutants with high-level resistance to itraconazole. Antimicrob Agents Chemother (2003) 47:1719–26. doi:10.1128/aac.47.5.1719-1726.2003
43. da Silva Ferreira ME, Malavazi I, Savoldi M, Brakhage AA, Goldman MH, Kim HS, et al. Transcriptome analysis of Aspergillus fumigatus exposed to voriconazole. Curr Genet (2006) 50:32–44. doi:10.1007/s00294-006-0073-2
44. Fraczek MG, Bromley M, Buied A, Moore CB, Rajendran R, Rautemaa R, et al. The cdr1B efflux transporter is associated with non-cyp51a-mediated itraconazole resistance in Aspergillus fumigatus. J Antimicrob Chemother (2013) 68:1486–96. doi:10.1093/jac/dkt075
45. Lamping E, Monk BC, Niimi K, Holmes AR, Tsao S, Tanabe K, et al. Characterization of three classes of membrane proteins involved in fungal azole resistance by functional hyperexpression in Saccharomyces cerevisiae. Eukaryot Cell (2007) 6:1150–65. doi:10.1128/EC.00091-07
46. Sanglard D, Kuchler K, Ischer F, Pagani JL, Monod M, Bille J. Mechanisms of resistance to azole antifungal agents in Candida albicans isolates from AIDS patients involve specific multidrug transporters. Antimicrob Agents Chemother (1995) 39:2378–86. doi:10.1128/aac.39.11.2378
47. Calabrese D, Bille J, Sanglard, D. A novel multidrug efflux transporter gene of the major facilitator superfamily from Candida albicans (FLU1) conferring resistance to fluconazole. Microbiology (2000) 146(Pt 11):2743–54. doi:10.1099/00221287-146-11-2743
48. Coste AT, Karababa M, Ischer F, Bille J, Sanglard D. TAC1, transcriptional activator of CDR genes, is a new transcription factor involved in the regulation of Candida albicans ABC transporters CDR1 and CDR2. Eukaryot Cell (2004) 3:1639–52. doi:10.1128/EC.3.6.1639-1652.2004
49. Morschhäuser J, Barker KS, Liu TT, BlaB-Warmuth J, Homayouni R, Rogers PD. The transcription factor Mrr1p controls expression of the MDR1 efflux pump and mediates multidrug resistance in Candida albicans. PLoS Pathog (2007) 3:e164. doi:10.1371/journal.ppat.0030164
50. Coste A, Turner V, Ischer F, Morschhäuser J, Forche A, Selmecki A, et al. A mutation in Tac1p, a transcription factor regulating CDR1 and CDR2, is coupled with loss of heterozygosity at chromosome 5 to mediate antifungal resistance in Candida albicans. Genetics (2006) 172:2139–56. doi:10.1534/genetics.105.054767
51. Dunkel N, Blass J, Rogers PD, Morschhäuser J. Mutations in the multi-drug resistance regulator MRR1, followed by loss of heterozygosity, are the main cause of MDR1 overexpression in fluconazole-resistant Candida albicans strains. Mol Microbiol (2008) 69:827–40. doi:10.1111/j.1365-2958.2008.06309.x
52. Ferrari S, Ischer F, Calabrese D, Posteraro B, Sanguinetti M, Fadda G, et al. Gain of function mutations in CgPDR1 of Candida glabrata not only mediate antifungal resistance but also enhance virulence. PLoS Pathog (2009) 5:e1000268. doi:10.1371/journal.ppat.1000268
53. Vermitsky J-P, Earhart KD, Smith WL, Homayouni R, Edlind TD, Rogers PD. Pdr1 regulates multidrug resistance in Candida glabrata: gene disruption and genome-wide expression studies. Mol Microbiol (2006) 61:704–22. doi:10.1111/j.1365-2958.2006.05235.x
54. Dunkel N, Liu TT, Barker KS, Homayouni R, Morschhäuser J, Rogers PD. A gain-of-function mutation in the transcription factor Upc2p causes upregulation of ergosterol biosynthesis genes and increased fluconazole resistance in a clinical Candida albicans isolate. Eukaryot Cell (2008) 7:1180–90. doi:10.1128/EC.00103-08
55. Snelders E, Melchers WJG, Verweij PE. Azole resistance in Aspergillus fumigatus: a new challenge in the management of invasive aspergillosis? Future Microbiol (2011) 6:335–47. doi:10.2217/fmb.11.4
56. Ramage G, Saville SP, Thomas DP, Lopez-Ribot JL. Candida biofilms: an update. Eukaryot Cell (2005) 4:633–8. doi:10.1128/EC.4.4.633-638.2005
57. Mitchell KF, Zarnowski R, Sanchez H, Edward JA, Reinicke EL, Nett JE, et al. Community participation in biofilm matrix assembly and function. Proc Natl Acad Sci U S A (2015) 112(13):4092–7. doi:10.1073/pnas.1421437112
58. Desai JV, Mitchell AP, Andes DR. Fungal biofilms, drug resistance, and recurrent infection. Cold Spring Harb Perspect Med (2014) 4:a019729–019729. doi:10.1101/cshperspect.a019729
59. Bonhomme J, d’Enfert C. Candida albicans biofilms: building a heterogeneous, drug-tolerant environment. Curr Opin Microbiol (2013) 16:398–403. doi:10.1016/j.mib.2013.03.007
60. Vediyappan G, Rossignol T, d’Enfert C. Interaction of Candida albicans biofilms with antifungals: transcriptional response and binding of antifungals to beta-glucans. Antimicrob Agents Chemother (2010) 54:2096–111. doi:10.1128/AAC.01638-09
61. Maebashi K, Kudoh M, Nishiyama Y, Makimura K, Uchida K, Mori T, et al. A novel mechanism of fluconazole resistance associated with fluconazole sequestration in Candida albicans isolates from a myelofibrosis patient. Microbiol Immunol (2002) 46:317–26. doi:10.1111/j.1348-0421.2002.tb02702.x
62. Hope WW, Tabernero L, Denning DW, Anderson MJ. Molecular mechanisms of primary resistance to flucytosine in Candida albicans. Antimicrob Agents Chemother (2004) 48:4377–86. doi:10.1128/AAC.48.11.4377-4386.2004
63. Edlind TD, Katiyar SK. Mutational analysis of flucytosine resistance in Candida glabrata. Antimicrob Agents Chemother (2010) 54:4733–8. doi:10.1128/AAC.00605-10
64. Vandeputte P, Pineau L, Larcher G, Noël T, Brèthes D, Chabasse D, et al. Molecular mechanisms of resistance to 5-fluorocytosine in laboratory mutants of Candida glabrata. Mycopathologia (2011) 171:11–21. doi:10.1007/s11046-010-9342-1
65. Lamb D, Kelly D, White T, Kelly S. The R467K amino acid substitution in Candida albicans sterol 14alpha-demethylase causes drug resistance through reduced affinity. Antimicrob Agents Chemother (2000) 44:63–7. doi:10.1128/aac.44.1.63-67.2000
66. Sanglard D, Coste AT. Activity of isavuconazole and other azoles against Candida clinical isolates and yeast model systems with known azole resistance mechanisms. Antimicrob Agents Chemother (2015) 60:229–38. doi:10.1128/AAC.02157-15
67. Morio F, Loge C, Besse B, Hennequin C, Le Pape P. Screening for amino acid substitutions in the Candida albicans Erg11 protein of azole-susceptible and azole-resistant clinical isolates: new substitutions and a review of the literature. Diagn Microbiol Infect Dis (2010) 66:373–84. doi:10.1016/j.diagmicrobio.2009.11.006
68. Diaz-Guerra TM, Mellado E, Cuenca-Estrella M, Rodriguez-Tudela JL. A point mutation in the 14alpha-sterol demethylase gene cyp51A contributes to itraconazole resistance in Aspergillus fumigatus. Antimicrob Agents Chemother (2003) 47:1120–4. doi:10.1128/AAC.47.3.1120-1124.2003
69. Garcia-Effron G, Mellado E, Gomez-Lopez A, Alcazar-Fuoli L, Cuenca-Estrella M, Rodríguez-Tudela JL. Differences in interactions between azole drugs related to modifications in the 14-alpha sterol demethylase gene (cyp51A) of Aspergillus fumigatus. Antimicrob Agents Chemother (2005) 49:2119–21. doi:10.1128/AAC.49.5.2119-2121.2005
70. Perlin DS. Echinocandin resistance in Candida. Clin Infect Dis (2015) 61:S612–7. doi:10.1093/cid/civ791
71. Asner SA, Giulieri S, Diezi M, Marchetti O, Sanglard D. Acquired multidrug antifungal resistance in Candida lusitaniae during therapy. Antimicrob Agents Chemother (2015) 59. doi:10.1128/AAC.02204-15
72. Desnos-Ollivier M, Moquet O, Chouaki T, Guerin AM, Dromer F. Development of echinocandin resistance in Clavispora lusitaniae during caspofungin treatment. J Clin Microbiol (2011) 49:2304–6. doi:10.1128/JCM.00325-11
73. Desnos-Ollivier M, Bretagne S, Raoux D, Hoinard D, Dromer F, Dannaoui E, et al. Mutations in the fks1 gene in Candida albicans, C. tropicalis, and C. krusei correlate with elevated caspofungin MICs uncovered in AM3 medium using the method of the European Committee on Antibiotic Susceptibility Testing. Antimicrob Agents Chemother (2008) 52:3092–8. doi:10.1128/AAC.00088-08
74. Garcia-Effron G, Katiyar SK, Park S, Edlind TD, Perlin DS. A naturally occurring proline-to-alanine amino acid change in Fks1p in Candida parapsilosis, Candida orthopsilosis, and Candida metapsilosis accounts for reduced echinocandin susceptibility. Antimicrob Agents Chemother (2008) 52:2305–12. doi:10.1128/AAC.00262-08
75. Kelly S, Lamb D, Corran A, Baldwin B, Kelly D. Mode of action and resistance to azole antifungals associated with the formation of 14alpha-methylergosta-8,24(28)-dien-3ß,6a-diol. Biochem Biophys Res Commun (1995) 207:910–5. doi:10.1006/bbrc.1995.1272
76. Chau AS, Gurnani M, Hawkinson R, Laverdiere M, Cacciapuoti A, McNicholas PM. Inactivation of sterol delta5,6-desaturase attenuates virulence in Candida albicans. Antimicrob Agents Chemother (2005) 49:3646–51. doi:10.1128/AAC.49.9.3646-3651.2005
77. Eddouzi J, Parker JE, Vale-Silva LA, Coste A, Ischer F, Kelly S, et al. Molecular mechanisms of drug resistance in clinical Candida species isolated from Tunisian hospitals. Antimicrob Agents Chemother (2013) 57:3182–93. doi:10.1128/AAC.00555-13
78. Martel CM, Parker JE, Bader O, Weig M, Gross U, Warrilow AGS, et al. Identification and characterization of four azole-resistant erg3 mutants of Candida albicans. Antimicrob Agents Chemother (2010) 54:4527–33. doi:10.1128/AAC.00348-10
79. Morio F, Pagniez F, Lacroix C, Miegeville M, Le Pape P. Amino acid substitutions in the Candida albicans sterol Δ5,6-desaturase (Erg3p) confer azole resistance: characterization of two novel mutants with impaired virulence. J Antimicrob Chemother (2012) 67:2131–8. doi:10.1093/jac/dks186
80. Jensen-Pergakes KL, Kennedy MA, Lees ND, Barbuch R, Koegel C, Bard M. Sequencing, disruption, and characterization of the Candida albicans sterol methyltransferase (ERG6) gene: drug susceptibility studies in erg6 mutants. Antimicrob Agents Chemother (1998) 42:1160–7.
81. Jia N, Arthington-Skaggs B, Lee W, Pierson CA, Lees ND, Eckstein J, et al. Candida albicans sterol C-14 reductase, encoded by the ERG24 gene, as a potential antifungal target site. Antimicrob Agents Chemother (2002) 46:947–57. doi:10.1128/AAC.46.4.947-957.2002
82. Vincent BM, Lancaster AK, Scherz-Shouval R, Whitesell L, Lindquist S. Fitness trade-offs restrict the evolution of resistance to amphotericin B. PLoS Biol (2013) 11:e1001692. doi:10.1371/journal.pbio.1001692
83. Dodgson AR, Dodgson KJ, Pujol C, Pfaller MA, Soll DR. Clade-specific flucytosine resistance is due to a single nucleotide change in the FUR1 gene of Candida albicans. Antimicrob Agents Chemother (2004) 48:2223–7. doi:10.1128/AAC.48.6.2223-2227.2004
84. Verweij PE, Kema GHJ, Zwaan B, Melchers WJG. Triazole fungicides and the selection of resistance to medical triazoles in the opportunistic mould Aspergillus fumigatus. Pest Manag Sci (2013) 69:165–70. doi:10.1002/ps.3390
85. Snelders E, van der Lee HAL, Kuijpers J, Rijs AJMM, Varga J, Samson RA, et al. Emergence of azole resistance in Aspergillus fumigatus and spread of a single resistance mechanism. PLoS Med (2008) 5:e219. doi:10.1371/journal.pmed.0050219
86. van der Linden JWM, Camps SMT, Kampinga GA, Arends JPA, Debets-Ossenkopp YJ, Haas PJA, et al. Aspergillosis due to voriconazole highly resistant Aspergillus fumigatus and recovery of genetically related resistant isolates from domiciles. Clin Infect Dis (2013) 57:513–20. doi:10.1093/cid/cit320
87. van der Linden JWM, Arendrup MC, Warris A, Lagrou K, Pelloux H, Hauser PM, et al. Prospective multicenter international surveillance of azole resistance in Aspergillus fumigatus. Emerg Infect Dis (2015) 21:1041–4. doi:10.3201/eid2106.140717
88. Wiederhold NP, Gil VG, Gutierrez F, Lindner JR, Albataineh MT, Mccarthy DI, et al. First Detection of TR34 L98H and TR46 Y121F T289A Cyp51 Mutations Aspergillus fumigatus isolates in the United States. J Clin Microbiol (2016) 54:168–71. doi:10.1128/JCM.02478-15
89. Snelders E, Camps SMT, Karawajczyk A, Schaftenaar G, Kema GHJ, van der Lee HA, et al. Triazole fungicides can induce cross-resistance to medical triazoles in Aspergillus fumigatus. PLoS One (2012) 7:e31801. doi:10.1371/journal.pone.0031801
90. Bader O, Tünnermann J, Dudakova A, Tangwattanachuleeporn M, Weig M, Gross U. Environmental isolates of azole-resistant Aspergillus fumigatus in Germany. Antimicrob Agents Chemother (2015) 59:4356–9. doi:10.1128/AAC.00100-15
91. Perlin DS. Current perspectives on echinocandin class drugs. Future Microbiol (2011) 6:441–57. doi:10.2217/fmb.11.19
92. Martel CM, Parker JE, Bader O, Weig M, Gross U, Warrilow AGS, et al. A clinical isolate of Candida albicans with mutations in ERG11 (encoding sterol 14alpha-demethylase) and ERG5 (encoding C22 desaturase) is cross resistant to azoles and amphotericin B. Antimicrob Agents Chemother (2010) 54:3578–83. doi:10.1128/AAC.00303-10
93. Sanglard D, Ischer F, Parkinson T, Falconer D, Bille J. Candida albicans mutations in the ergosterol biosynthetic pathway and resistance to several antifungal agents. Antimicrob Agents Chemother (2003) 47:2404–12. doi:10.1128/aac.47.8.2404-2412.2003
94. Vale-Silva LA, Coste AT, Ischer F, Parker JE, Kelly SL, Pinto E, et al. Azole resistance by loss of function of the sterol δ5,6-desaturase gene (ERG3) in Candida albicans does not necessarily decrease virulence. Antimicrob Agents Chemother (2012) 56:1960–8. doi:10.1128/AAC.05720-11
95. Pinjon E, Moran GP, Jackson CJ, Kelly SL, Sanglard D, Coleman DC, et al. Molecular mechanisms of itraconazole resistance in Candida dubliniensis. Antimicrob Agents Chemother (2003) 47:2424–37. doi:10.1128/aac.47.8.2424-2437.2003
96. Park S, Kelly R, Kahn JN, Robles J, Hsu M-J, Register E, et al. Specific substitutions in the echinocandin target Fks1p account for reduced susceptibility of rare laboratory and clinical Candida sp. isolates. Antimicrob Agents Chemother (2005) 49:3264–73. doi:10.1128/AAC.49.8.3264-3273.2005
97. Chapeland-Leclerc F, Hennequin C, Papon N, Noël T, Girard A, Socié G, et al. Acquisition of flucytosine, azole, and caspofungin resistance in Candida glabrata bloodstream isolates serially obtained from a hematopoietic stem cell transplant recipient. Antimicrob Agents Chemother (2010) 54:1360–2. doi:10.1128/AAC.01138-09
98. Pham CD, Iqbal N, Bolden CB, Kuykendall RJ, Harrison LH, Farley MM, et al. Role of FKS mutations in Candida glabrata: MIC values, echinocandin resistance, and multidrug resistance. Antimicrob Agents Chemother (2014) 58:4690–6. doi:10.1128/AAC.03255-14
99. Farmakiotis D, Tarrand JJ, Kontoyiannis DP. Drug-resistant Candida glabrata infection in cancer patients. Emerg Infect Dis (2014) 20:1833–40. doi:10.3201/eid2011.140685
100. Jensen RH, Astvad KMT, Silva LV, Sanglard D, Jorgensen R, Nielsen KF, et al. Stepwise emergence of azole, echinocandin and amphotericin B multidrug resistance in vivo in Candida albicans orchestrated by multiple genetic alterations. J Antimicrob Chemother (2015) 70:1–5. doi:10.1093/jac/dkv140
Keywords: antifungals, drug resistance, Candida, Aspergillus
Citation: Sanglard D (2016) Emerging Threats in Antifungal-Resistant Fungal Pathogens. Front. Med. 3:11. doi: 10.3389/fmed.2016.00011
Received: 16 January 2016; Accepted: 03 March 2016;
Published: 15 March 2016
Edited by:
Yuji Morita, Aichi Gakuin University, JapanReviewed by:
Miguel Cacho Teixeira, University of Lisbon, PortugalCopyright: © 2016 Sanglard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dominique Sanglard, ZG9taW5pcXVlLnNhbmdsYXJkQGNodXYuY2g=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.