 Jherna Balany
Jherna Balany Vineet Bhandari
Vineet Bhandari
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 21 December 2015
Sec. Pulmonary Medicine
Volume 2 - 2015 | https://doi.org/10.3389/fmed.2015.00090
This article is part of the Research Topic Bronchopulmonary Dysplasia: Past, Current and Future Pathophysiologic Concepts and Their Contribution to Understanding Lung Disease View all 14 articles
The concerted interaction of genetic and environmental factors acts on the preterm human immature lung with inflammation being the common denominator leading to the multifactorial origin of the most common chronic lung disease in infants – bronchopulmonary dysplasia (BPD). Adverse perinatal exposure to infection/inflammation with added insults like invasive mecha nical ventilation, exposure to hyperoxia, and sepsis causes persistent immune dysregulation. In this review article, we have attempted to analyze and consolidate current knowledge about the role played by persistent prenatal and postnatal inflammation in the pathogenesis of BPD. While some parameters of the early inflammatory response (neutrophils, cytokines, etc.) may not be detectable after days to weeks of exposure to noxious stimuli, they have already initiated the signaling pathways of the inflammatory process/immune cascade and have affected permanent defects structurally and functionally in the BPD lungs. Hence, translational research aimed at prevention/amelioration of BPD needs to focus on dampening the inflammatory response at an early stage to prevent the cascade of events leading to lung injury with impaired healing resulting in the pathologic pulmonary phenotype of alveolar simplification and dysregulated vascularization characteristic of BPD.
Bronchopulmonary dysplasia (BPD) is the most common chronic respiratory disease affecting infants wherein the developmental program of the lung is altered secondary to preterm birth of the baby (1). Lung development progresses in five distinct stages: embryonic, pseudoglandular, canalicular, saccular, and alveolar (2, 3). Human preterm babies who develop BPD are born in the late canalicular or early saccular stage of lung development. The late canalicular stage is characterized by development of the primitive alveoli and the alveolar capillary barrier, and the differentiation of type I and type II pneumocytes. The early saccular stage is marked by initiation of surfactant production, pulmonary vascularization, and enlargement of terminal airways (2–5). Unique to lung development is the fact that unlike other organs, the lungs complete their development after birth (up to 8 years of age) (6). Alveolar sacs are formed by secondary septation of alveolar ducts. With preterm birth, this programed development is disrupted, and in the setting of inflammation [whether it is due to infection, mechanical ventilation (MV), or hyperoxia] causes impaired alveolarization leading to BPD. We need to remember that while in sheep, baboons, and humans, the saccular stage occurs in utero; in rodent models, it begins at embryonic day 18 and continues through postnatal (PN) day 5 (4, 5).
In spite of many advances in neonatal medicine in the past few decades, like the introduction of better MV strategies and the use of surfactant and antenatal steroids, the incidence of BPD has not declined (7). The incidence of BPD in the United States is about 10,000–15,000 new cases each year out of which the majority of those affected have a birth weight <1250 g (8). Pulmonary and neurodevelopmental sequelae of this devastating disease extend even into adulthood (9).
Genetic (10) and environmental factors (pre- and/or postnatal sepsis, invasive MV, and hyperoxia) (1) act on the preterm human immature lung with inflammation being the common denominator in all these interactions leading to the multifactorial origins of this disease. As shown in Figure 1, it is postulated that adverse perinatal exposure/infection with added insults like invasive MV, exposure to hyperoxia, and sepsis causes persistent immune dysregulation. This on top of genetic susceptibility and prematurity leads to persistent inflammation leading to lung remodeling and evolution of BPD.
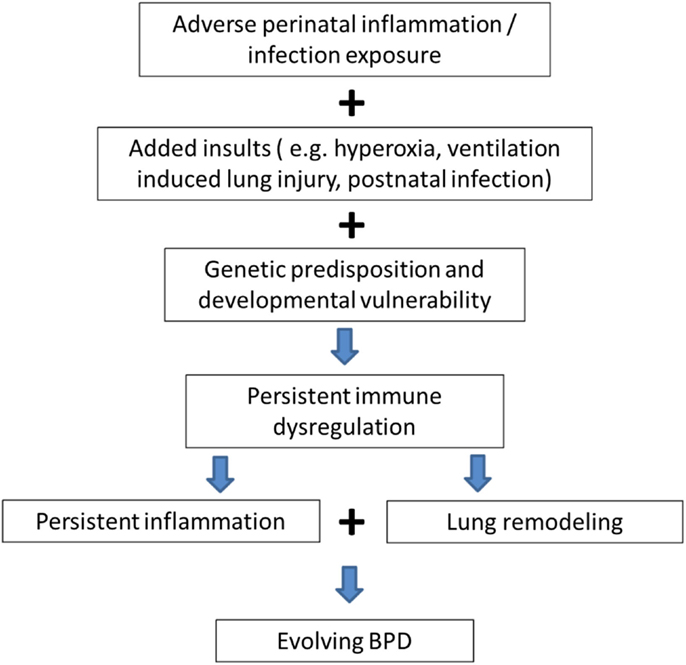
Figure 1. Genetic predisposition and persistent inflammation due to environmental factors (sepsis, invasive mechanical ventilation, and hyperoxia) acting on the foundation of immature lung underlie the pathogenesis of BPD.
In this review article, we have attempted to analyze and consolidate current knowledge about the role played by persistent prenatal and postnatal inflammation in the pathogenesis of BPD. We searched PubMed for articles limited to English language with the keywords: “Bronchopulmonary dysplasia or BPD,” “inflammation,” “chorioamnionitis,” “mechanical ventilation,” “hyperoxia,” “postnatal sepsis,” either individually or in combination. We focused on articles published over the last 10 years and used the most relevant ones for this review.
Bronchopulmonary dysplasia has been linked to the development of an inflammatory response that can occur in absence of clinical infection. Systemic fetal inflammatory response (11) and neonatal leukemoid reactions (12) have been implicated as a risk factor for BPD. Pulmonary inflammation in BPD is characterized by the presence of inflammatory cells like neutrophils and monocytes, pro-inflammatory cytokines, and other mediators, including soluble adhesion molecules.
The innate immunity and adaptive immunity reinforce each other and act in unison. Cells of the innate immune system secrete cytokines, which can prime lymphocytes thereby modulating adaptive immunity (13). Exposure to a specific antigen causes these primed lymphocytes to have a more rapid and intense immune response (14, 15). Naïve T cells express CD62L (L-selectin) (16). Upon activation, the T cells shed their surface CD62L molecules. In infants with BPD, the expression of the CD62L is decreased on these CD4+ T-cells thereby suggesting T cell activation. CD54 (intercellular adhesion molecule-1 or ICAM-1) is an adhesion molecule that mediates a co-stimulatory signal in T cell activation. CD54 expression is increased upon cell activation (17).
The premature lung is exposed to ongoing oxidative and cellular damage. Damaged lung tissue releases chemotactic factors and inflammatory cytokines, such as interleukin (IL)-1, IL-8 (CXCL-8), and tumor necrosis factor alpha (TNF-α). This leads to an influx of neutrophils and other inflammatory cells with increased release/production of additional pro-inflammatory cytokines (Table 1). Multiple studies have shown that IL-1β, IL-6, and IL-8 are elevated very early in the respiratory course of the human preterm population that ultimately develop BPD, and in tracheal aspirates of those with BPD. In contrast, decreased levels of IL-10 in serum and tracheal aspirates have been shown in studies of those infants who developed BPD. In addition to ILs, a large variety of other biomarkers have been detected and associated with the development of BPD in tracheal aspirates, as well as blood and urine samples of premature infants (9, 18). The ones that have been implicated in the animal models include inflammatory cytokines, matrix proteins, growth factors, and vascular factors (9, 18–33). Their role is illustrated in Table 1.
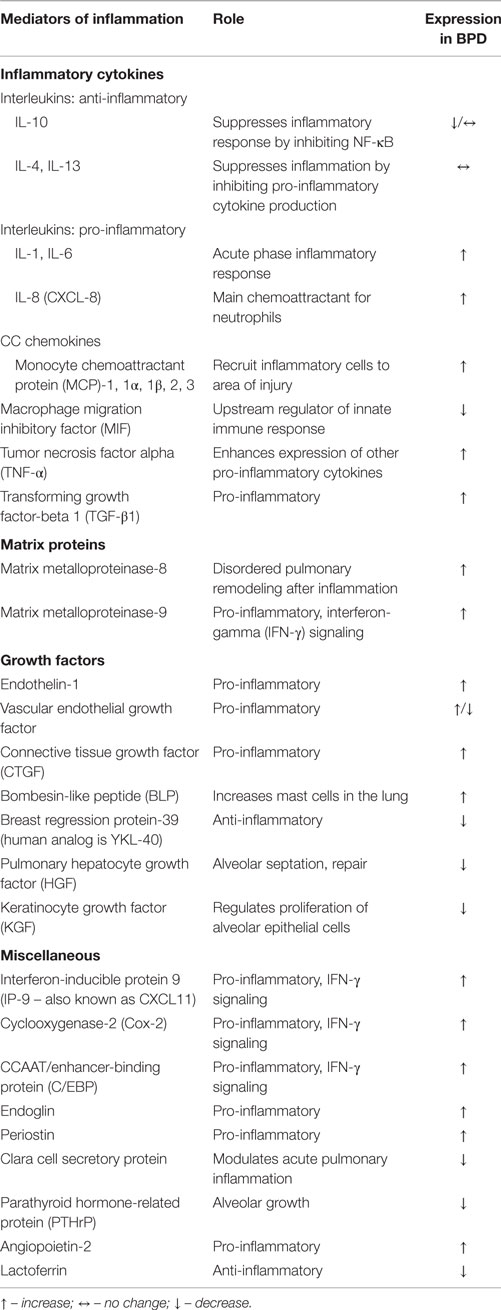
Table 1. Selected mediators of inflammation, their role, and corresponding expression in BPD.
We will now describe the major environmental factors that contribute to inflammation, and its persistence, in the pathogenesis of BPD. These include prenatal influences (chorioamnionitis) and postnatal influences, namely early- and late- onset sepsis, invasive MV, and hyperoxia.
As the name suggests, chorioamnionitis is inflammation of the chorion and amnion membranes of the placenta (34). Although commonly seen in clinical practice, chorioamnionitis is a complex syndrome associated with pregnancy leading to preterm deliveries (34). Chorioamnionitis has been classified as either histological or clinical. With histological chorioamnionitis, there is infiltration of polymorphonuclear leukocytes and other inflammatory cells like macrophages and T cells as seen microscopically (35–37). Clinical chorioamnionitis is evidenced by fever >37.5°C, uterine tenderness, foul smelling vaginal discharge, abdominal pain, maternal tachycardia with a heart rate >100 bpm, fetal tachycardia HR >160 bpm, and white blood cell (WBC) count >15,000/mm3 (38, 39).
It has been shown in in vitro studies that bacterial products like phospholipase A2, peptidoglycan polysaccharide, proteolytic enzymes, and endotoxins can initiate an inflammatory response. Inoculation of the amniotic cavity with E. coli lipopolysaccharide (LPS) or live Ureaplasma organisms has been shown to induce structural and functional fetal lung maturation (40–43). Antenatal lung inflammation impacts a variety of signaling pathway regulators like toll-like receptors 2 and 4 (TLR2 and TLR4), growth factors like TGF-β and CTGF, and mesenchymal structural proteins like bone morphogenetic protein-4 leading to vascular remodeling and alveolar simplification, which could be considered akin to a mild BPD phenotype (40–43). However, repetitive LPS exposure and/or chronic chorioamnionitis leads to immune tolerance and a dampened inflammatory response, which in turn allows the lungs to develop close to normal in experimental BPD animals (40–42).
Adverse perinatal outcomes are seen with intra-amniotic inflammation irrespective of the presence of intra-amniotic infection. Colonization per se, without inflammation is not associated with adverse outcomes (44). The severity of the adverse outcomes is directly related to the severity of the intra-amniotic inflammation (44). Maternal antibiotic use has been associated with decreased BPD (45).
To summarize, in experimental animals, antenatal inflammation causes lung maturation and some degrees of lung injury, which is modified by the not fully developed innate immune response, exposure to antenatal steroids, and noxious postnatal factors. Not surprisingly, given the variability in definition and impact of various confounding factors, the issue of antenatal inflammation causing BPD in human infants is controversial (42, 46–49). Chorioamnionitis increases the incidence of preterm birth, and if accompanied by lung inflammation could result in surfactant dysfunction allowing for prolonged exposure to supplemental oxygen and invasive MV (11, 48). This “multiple hit” of events could explain the propensity to BPD in such infants (48), though this has not been consistently shown (50). In addition, persistence and non-resolution of lung inflammation lead to BPD by inhibiting secondary septation, alveolarization and normal vascular development, and the compromised ability of the lungs to heal.
Preterm infants are more susceptible to infections since their immune defenses are not fully developed, have vulnerable skin barrier, and require multiple invasive procedures (51). Postnatal infection/inflammation could either be localized to the lung or could be systemic in origin. Chorioamnionitis increases the risk of early-onset neonatal sepsis, which sets off an inflammatory cascade (48). Also, it has been shown that late-onset sepsis induces a pro-inflammatory and pro-fibrotic response in the preterm lung predisposing it to BPD (51).
Local (intra-tracheal) exposure to LPS (bacterial endotoxin) or dsRNA (a marker of viral replication) in the neonatal rat led to acute cellular and cytokine inflammatory responses, which were associated with histologic features of impaired alveolar development (52, 53).
Neonatal mice injected with intraperitoneal LPS demonstrated reduced lung inflammation and apoptosis after 24 h as compared to adults, and this was associated with activation of the transcription factor, nuclear factor kappa B (NF-κB) (54). Inhibition of NF-κB resulted in increased cell death and alveolar simplification and disruption of angiogenesis via vascular growth factor (VEGF)-R2 (55). It has also been shown that using a targeted deletion of NF-κB signaling (using a lung epithelium-specific deletion of IKKβ – which is a known activating kinase upstream of NF-κB) in a mouse model results in alveolar hypoplasia with decreased VEGF expression (56). In addition, there was increased expression of CXCL-1, as well as its receptor CXCR2. Pre-treatment with CXCR2-neutralizing antibody was able to reverse the effects in the developing lung (53). In summary, exposure to either bacterial or viral agents in the rodent model led to features of inflammation, with pulmonary histology suggestive of BPD.
Inflammatory response secondary to viral infections in early post natal stages could be worth considering in the evolution of BPD. Increased neutrophil accumulation, increased expression of CXCL-1 and its receptor CXCR2, and decreased lung alveolarization have been seen with intra-tracheal delivery of viral pro-inflammatory dsRNA in 10-day-old mouse model (53).
Mechanical ventilation is a risk factor for the development of BPD in premature infants. Lung injury from MV results due to volutrauma, barotrauma, or atelectrauma (57).
When lungs are exposed to high tidal volumes, over distension leads to production of pro-inflammatory cytokines like IL-6, IL-8, and TNFα and reduced expression of anti-inflammatory cytokines like IL-10 (58). Even ventilation at low tidal volumes is deleterious because of the stretch injury it can induce by over-distending partially collapsed lungs. Sustained lung inflation (SLI) has been shown to increase levels of pro-inflammatory cytokines and BPD-like changes in the lungs of preterm lambs (59). There is great need to find non-invasive ventilation strategies for preterm neonates because even “gentle” invasive MV for a shorter duration can induce an inflammatory response (60).
In neonatal rats (7- to 14-day-old – in the alveolar phase of lung development), high tidal volume ventilation increased IL-6 mRNA and upregulated the TGF-β signaling molecule, CTGF mRNA, and protein expression compared to controls (61). In an 8-day-old rat ventilation model, high tidal volumes increased the neutrophilic and inflammatory cytokine mRNA and/or protein expression (IL-1β, IL-6, CXCL-1 and 2) response (62). In a 7-day-old rat model, exposure to MV for 24 h in room air led to cell cycle arrest (63), suggesting a harbinger to alveolar simplification, the pathologic hallmark of BPD.
In an invasive MV model in 2-week-old mice (well into the alveolar phase of lung development) for 1 h, IL-6 lung levels were increased in the high tidal volume ventilation group (64). Studies conducted in 2- to 6-day-old mice (late saccular to early alveolar phase of lung development) ventilated for 8–24 h with room air or 40% O2 revealed dysregulated elastin (ELN) assembly, a threefold to fivefold increase in cell death, TGF-β activation, and a decrease in VEGF-R2 expression (65, 66). Inhibiting lung elastase activity by using recombinant human elafin or genetically modified mice that expressed elafin in the vascular endothelium was protective of the lung injury (67, 68).
Early studies using a chronically ventilated (3–4 weeks) preterm lamb model of BPD showed evidence of non-uniform inflation patterns and impaired alveolar formation with an abnormal abundance of elastin (69). Inflammation was evident by the presence of inflammatory cells, namely alveolar macrophages, neutrophils, and mononuclear cells and edema (69). In this model, there was also reduced lung expression of growth factors that regulate alveolarization and differential alteration of matrix proteins that regulate ELN assembly (70). A non-invasive (nasal) ventilation approach preserved alveolar architecture (71) and had a positive effect on parathyroid hormone-related protein-peroxisome proliferator-activated receptor-gamma (PTHrP-PPARγ)-driven alveolar homeostatic epithelial–mesenchymal signaling in the preterm lamb model (72).
It has been seen in preterm fetal sheep that there is increased expression of early response gene-1 (Egr-1) as well as pro- and anti-inflammatory cytokines and dynamic changes in heat shock protein 70 (HSP70) (57). This stretch injury also increases expression of granulocyte/macrophage colony-stimulating factor mRNA leading to maturation of lung monocytes to alveolar macrophages (57). Induction of surfactant proteins A, B, and C mRNA is also increased (57). More recently, even short-term stretch injury (15 min) secondary to invasive MV in preterm fetal sheep led to increased levels of pro-inflammatory cytokines, IL-1β, IL-6, monocyte chemoattractant protein (MCP)-1, and MCP-2 mRNA by 1 h (57). This was accompanied by increased presence of inflammatory cells in the bronchoalveolar lavage fluid (BALF) with initial increases in neutrophils and monocytes by 1 h and a transition to macrophages by 24 h (57).
The preterm ventilated baboon model of BPD (delivered at 125 days – at 68% of gestation) showed evidence of alveolar hypoplasia and dysmorphic vasculature, akin to that seen in human BPD (73). Importantly, there were significant elevations of TNF-α, IL-6, IL-8 levels, but not of IL-1β and IL-10, in tracheal aspirate fluids at various times during the period of ventilator support, supporting a role for inflammation (73). In addition, increased matrix metalloproteinase-9 (MMP-9) levels were associated with lung inflammation and edema seen in this invasive ventilation model (74). Alteration of VEGF was also noted in the lungs of various baboon models (75, 76). Bombesin is a 14-amino acid peptide, initially detected in amphibian skin, but immunoreactive studies have shown the presence of bombesin-like peptide (BLP) in multiple organ systems in mammals (77). In the lung, BLP have been shown to be released by pulmonary neuroendocrine cells (77). BLP blockade improved alveolar septation and angiogenesis in the preterm baboon models (78, 79).
In the 125-day baboon model, treatment with early nasal continuous positive airway pressure (NCPAP) for 28 days led to a pulmonary phenotype similar to 156 days gestational control lungs, suggesting that this non-invasive approach could minimize lung injury (80). In the same model, delayed extubation (till 5 days) versus early extubation to NCPAP at 24 h led to significantly increased BALF IL-6, IL-8, MCP-1, macrophage inflammatory protein-1 alpha (MIP-1α), and growth-regulated oncogene-alpha (GRO-α) in the delayed NCPAP group (81).
Some epidemiological studies showed that replacing invasive MV with NCPAP was associated with BPD reduction (82). No reduction in the incidence of BPD or mortality in the NCPAP group was seen in the COIN study that randomized infants born at 25–28 weeks to receive either NCPAP or intubation with MV in the delivery room (83). The INSURE (IN: intubation, SUR: surfactant, E: extubation) technique has been shown to reduce the need for MV and incidence of BPD (84). Non-invasive ventilation strategies like nasal intermittent-positive pressure ventilation (NIPPV) not only reduce the need for intubation within the first 48–72 h of life, but also have been associated with decreased mortality and/or BPD and hence is a feasible option for the newborn (85–87), though additional studies are required (88). The optimal mode of non-invasive ventilation (for example: type of NCPAP, maximum level of NCPAP, synchronized or non-synchronized method of nasal ventilation), selection of the best nasal interface (short-prongs or mask), and choice of ventilator need to be determined, and this information would be helpful in management of the disease.
To summarize, while the lamb/sheep/baboon ventilation models are in the saccular stage (akin to the human premature babies who are at most risk for BPD at birth), the rat/mouse ventilation models are in the alveolar phase of lung development. However, it is quite obvious that mechanical stretch injury generates an inflammatory response (mostly neutrophils, IL-1β, IL-6, CXCL-1/-2, TGF-β signaling), along with alterations in matrix proteins (ELN, MMP-9) and VEGF. In addition, there is increased cell death and cell cycle arrest. Thus, it appears that an initial inflammatory cascade triggers the signaling of additional molecular mediators that lead to dysregulated vascularization and impaired alveolarization. Interestingly, non-invasive (nasal) ventilation approaches were protective of these responses. Thus, prolonged invasive MV sets off a persistent cascade of inflammatory response that in the setting of hyperoxia takes the “multiple hit” pathway of leading to BPD.
Many studies have documented the injurious effects of perinatal supplemental oxygen on lung development. Target levels of O2 in extremely low birth weight (ELBW) have been studied extensively. The morphologic changes of human BPD resemble hyperoxic lung injury in newborn animals (73). Prolonged exposure to hyperoxia in the neonatal mouse for 14 days or longer results in a phenotype of “old” BPD (89, 90). Exposure to hyperoxia in the critical saccular stage of lung development replicates human BPD, with effects that are dose-dependent on the fraction of inspired oxygen (FiO2) concentration; the effects last lifelong with increased susceptibility to respiratory tract infections (91–95). Acute lung injury caused by hyperoxia (Figure 2) occurs secondary to an inflammatory response, which causes destruction of the alveolar–capillary barrier, vascular leak, influx of inflammatory mediators, pulmonary edema, and ultimately cell death (96). With continued exposure to hyperoxia this inflammatory response and pulmonary edema improve initially but chronic pulmonary inflammation ensues in the following weeks (97). At the cellular level, alveolar or interstitial macrophages express early response cytokines when exposed to hyperoxia, which in turn attract inflammatory cells to the lungs (19).
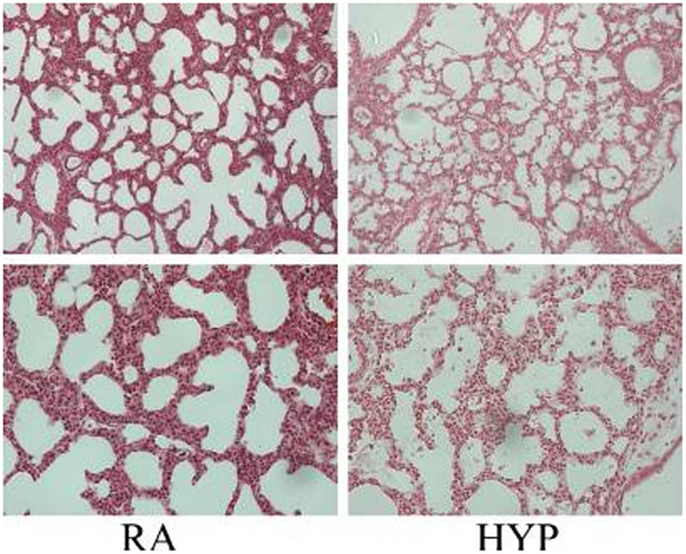
Figure 2. Photomicrographs (×10, upper panel; ×20 lower panel; hematoxylin and eosin stain) of neonatal lung injury noted in newborn mice at postnatal day 2, after 100% O2 exposure since birth. Note the alveolar exudates and presence of inflammatory cells in the hyperoxia-exposed lungs compared with litter-mate controls in room air. RA, room air; HYP, hyperoxia [with permission from Semin Fetal Neonatal Med (2010) 15(4):223–9].
It has been shown that there exists a dose-dependent effect of hyperoxia on severity of BPD in the murine model. Mice exposed to varying concentrations of oxygen ranging from 40 to 100% at PN days 1–4 had more severe disease at higher concentrations of oxygen (92). An oxygen dose-dependent inflammatory response to influenza-A viral infection in adult mice that had been exposed to hyperoxia as neonates has been reported (95). Furthermore, this response was dependent upon the cumulative exposure to oxygen (98).
The specific role of individual inflammatory molecular mediators in the pathogenesis of BPD has been particularly well illustrated by utilizing lung-targeted overexpressing transgenic models, in room air, resulting in pulmonary phenotypes reminiscent of human BPD. These include IL-1β (99, 100) and IFN-γ (25, 91). In the case of IL-1β transgenic mice, absence of the beta6 integrin subunit was protective of the BPD phenotype (101). Interestingly, inhibition of cyclooxygenase-2 (Cox-2) ameliorated the BPD phenotype in the hyperoxia-induced as well as the IFN-γ lung overexpressing transgenic mouse model in room air. A recent paper has reported that increased Cox-2 activity may contribute to proinflammatory responses in hyperoxia-exposed developing mouse lungs (102).
There is increased expression of IL-1α mRNA in neonatal mice exposed to hyperoxia (89). Lung mRNA for IL-1β also increases in neonatal mice exposed to hyperoxia (103). Transgenic IL-1β overexpression in lung epithelium resulted in BPD phenotype in neonatal mice (100). In hyperoxia-exposed newborn rabbits, the pattern of IL-1β rise and fall matches the rise and fall of histologic inflammation (104). However, in the immature baboon model of BPD, no such pattern between IL-1β levels and inflammation was seen in the tracheal aspirates (73). CINC-1 in premature rat lungs (105) and newborn rabbits (104) exposed to hyperoxia was upregulated. Also, IL-8 levels in tracheal aspirates of the premature baboon model of BPD have been shown to be increased (73).
The lungs of hyperoxia-exposed neonatal mice had no change in IL-10 mRNA expression (103). Also tracheal aspirates of baboon model of BPD show no difference in IL-10 levels (73). IL-1β, IL-6, and IL-8 are pro-inflammatory cytokines and are elevated very early in the course of BPD.
Typically viewed as pro-inflammatory, these cytokines have been shown to be elevated very early in the respiratory course of the human preterm population that ultimately develops BPD (20). Studies have found that serum and tracheal aspirate IL-10 levels were decreased in those infants who developed BPD (20).
A variety of potential therapeutic agents have been used in hyperoxia-exposed mice models that have been shown to decrease inflammation and/or attenuate other parameters of lung injury/BPD phenotype. These include rosiglitazone (106, 107), hepatocyte growth factor (HGF) (108), B-naphthoflavone (109), arginyl-glutamine as well as docosahexaenoic acid (110), and a combination of vitamin A and retinoic acid (111). Treatment with human amnion epithelial cells attenuated some parameters of hyperoxia-induced inflammatory lung injury (mRNA expression of IL-1α, IL-6, TGF-β, platelet-derived growth factor-beta or PDGF-β, mean linear intercept, and septal crest density), but not other aspects, for example, alveolar airspace volume, collagen content, or leukocyte infiltration in neonatal mice (112).
To summarize, while variable initiation and duration of exposure to hyperoxia animal models have been reported as models of human BPD, exposure to hyperoxia for a relatively short (PN1–4) duration in mice, which is at the critical saccular stage of lung development, can result in an inflammatory response sufficient to create the BPD pulmonary phenotype. This can be recapitulated using transgenic mice models of the inflammatory mediators, but kept in room air. Importantly, exposure to 0.4, 0.6, >0.8 FiO2 can mimic mild, moderate, and severe BPD, respectively. A vast array of therapeutic agents has been reported to be effective in improving alveolar and/or vascular architecture of the hyperoxia-exposed neonatal lung in lambs, rats, and mice.
While hyperoxia exposure is a good starting point for testing the efficacy of potential therapeutic agents, it is important to be able to delineate the responsible molecule/signaling pathway in developmentally appropriate room air models and confirm the results in preventing/ameliorating the BPD phenotype. This would avoid the confounding variable of hyperoxia-induced alterations in multiple other molecular mediators, allowing delineation of targeted molecules in specific signaling pathways for maximal potential therapeutic relevance. Among the inflammatory mediators of hyperoxia-induced lung injury that can mimic the BPD phenotype in room air, the well-defined ones are IL-1β, TGF-β1, CTGF, IFN-γ, and MIF. It would be important to attempt to translate some of the newer targets in specific signaling pathways that have been recently reported, for example, inhibition of Cox-2 (91, 102) as a potential therapeutic option for prevention/amelioration of BPD.
It is important to highlight the fact that for BPD to occur, it requires the known environmental factors to be exposed to the immature lung for a sustained duration, resulting in persistent inflammation. For the chorioamnionitis rodent models, the exposure to LPS is over a few days in the late canalicular/early saccular stage of lung development. For the relative short duration of exposure to invasive MV and hyperoxia in rodent models, 1 postnatal day in the saccular stage of lung development is equivalent to 3–4 weeks in a human preterm infant. Obviously, the larger animal models (sheep/lamb/baboon) also need few days to weeks of injury to develop the pulmonary phenotype of BPD. While some parameters of the early inflammatory response (neutrophils, cytokines such as IL-1, TNFα) may not be detectable after days to weeks of exposure to noxious stimuli, they have already initiated the signaling pathways of the inflammatory process/immune cascade and have affected permanent defects structurally and functionally in the BPD lungs. This is borne out by the facts that the pathologic appearance of large simplified alveoli is permanent following just the first 4 PN days of hyperoxia exposure in mice models (93). Furthermore, these mice have increased mortality when exposed to viral infectious challenge as adults (98, 113). In concordance, preterm neonates with BPD have anatomical and functional pulmonary deficits well into childhood and as adults (114–116). There is some clinical evidence that early interruption of the initial inflammatory response could result in amelioration and potential reversal of these effects (117).
It is important to remember that while in vitro studies are helpful in figuring out the mechanistic significance of a signaling pathway, these are usually conducted with cell lines or freshly isolated single cells of a particular phenotype. Thus, the results of such studies may not accurately reflect the in vivo situation of interaction with the multiple cell types found in the lung. In addition, while the significantly different responses between adult and neonatal lungs to the postnatal factors discussed here – invasive ventilation (118–121), local/systemic sepsis (52, 54, 122–124), and hyperoxia (19, 125–127) – are well established, it is also important to be cognizant of the stages of lung development when comparing animal data for relevance to humans. This is best exemplified by studies that highlight the differential responses in the various stages of lung development (mostly, saccular vs. alveolar) in animal models (99). Furthermore, the degree and duration of exposure to the noxious stimulus (hyperoxia, for example) in the animal models needs to be appropriate in order to attempt to extrapolate the data to humans. For example, a prolonged exposure to hyperoxia from birth to 2 weeks in the mouse, i.e., almost to the end of alveolarization is akin to exposing a preterm neonate to the same to at least up to 2 years of age.
To conclude, it is the preterm lung in the late canalicular/saccular phase of development that is most predisposed to BPD, when exposed to the pre- and postnatal factors. Inflammation and then its persistence in the preterm lung – whether initiated by prenatal factors like chorioamnionitis or whether propagated postnatally with the use of high FiO2 and invasive MV or sepsis – culminates in BPD. Hence, translational research needs to be aimed at decreasing chorioamnionitis and finding better strategies for early non-invasive MV and optimum use of oxygen for the immature preterm lung for dampening the inflammatory response.
JB wrote the initial draft. VB did substantial re-organization and editing of the manuscript. Both authors have approved the final version of the manuscript as submitted.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funded by NIH grant HL 085103 and The Hartwell Foundation.
1. Bhandari V. Postnatal inflammation in the pathogenesis of bronchopulmonary dysplasia. Birth Defects Res A Clin Mol Teratol (2014) 100(3):189–201. doi: 10.1002/bdra.23220
2. Kotecha S. Lung growth: implications for the newborn infant. Arch Dis Child Fetal Neonatal Ed (2000) 82(1):F69–74. doi:10.1136/fn.82.1.F69
3. Maeda Y, Dave V, Whitsett JA. Transcriptional control of lung morphogenesis. Physiol Rev (2007) 87(1):219–44. doi:10.1152/physrev.00028.2006
4. Joshi S, Kotecha S. Lung growth and development. Early Hum Dev (2007) 83(12):789–94. doi:10.1016/j.earlhumdev.2007.09.007
5. Kramer EL, Deutsch GH, Sartor MA, Hardie WD, Ikegami M, Korfhagen TR, et al. Perinatal increases in TGF-{alpha} disrupt the saccular phase of lung morphogenesis and cause remodeling: microarray analysis. Am J Physiol Lung Cell Mol Physiol (2007) 293(2):L314–27. doi:10.1152/ajplung.00354.2006
6. Berger J, Bhandari V. Animal models of bronchopulmonary dysplasia. The term mouse models. Am J Physiol Lung Cell Mol Physiol (2014) 307(12):L936–47. doi:10.1152/ajplung.00159.2014
7. Smith VC, Zupancic JA, McCormick MC, Croen LA, Greene J, Escobar GJ, et al. Trends in severe bronchopulmonary dysplasia rates between 1994 and 2002. J Pediatr (2005) 146(4):469–73. doi:10.1016/j.jpeds.2004.12.023
8. Bhandari A, Bhandari V. “New” bronchopulmonary dysplasia: a clinical review. Clin Pulm Med (2011) 18(3):137–43. doi:10.1097/CPM.0b013e318218a071
9. Bhandari A, Bhandari V. Pitfalls, problems, and progress in bronchopulmonary dysplasia. Pediatrics (2009) 123(6):1562–73. doi:10.1542/peds.2008-1962
10. Bhandari V, Gruen JR. The genetics of bronchopulmonary dysplasia. Semin Perinatol (2006) 30(4):185–91. doi:10.1053/j.semperi.2006.05.005
11. Hofer N, Kothari R, Morris N, Muller W, Resch B. The fetal inflammatory response syndrome is a risk factor for morbidity in preterm neonates. Am J Obstet Gynecol (2013) 209(6):e1–11. doi:10.1016/j.ajog.2013.08.030
12. Zanardo V, Savio V, Giacomin C, Rinaldi A, Marzari F, Chiarelli S. Relationship between neonatal leukemoid reaction and bronchopulmonary dysplasia in low-birth-weight infants: a cross-sectional study. Am J Perinatol (2002) 19(7):379–86. doi:10.1055/s-2002-35612
13. Hoebe K, Janssen E, Beutler B. The interface between innate and adaptive immunity. Nat Immunol (2004) 5(10):971–4. doi:10.1038/ni1004-971
14. Vivier E, Malissen B. Innate and adaptive immunity: specificities and signaling hierarchies revisited. Nat Immunol (2005) 6(1):17–21. doi:10.1038/ni1153
15. Lanzavecchia A, Sallusto F. From synapses to immunological memory: the role of sustained T cell stimulation. Curr Opin Immunol (2000) 12(1):92–8. doi:10.1016/S0952-7915(99)00056-4
16. Hannet I, Erkeller-Yuksel F, Lydyard P, Deneys V, DeBruyere M. Developmental and maturational changes in human blood lymphocyte subpopulations. Immunol Today (1992) 13(6):215–218. doi:10.1016/0167-5699(92)90157-3
17. Springer TA. Adhesion receptors of the immune system. Nature (1990) 346(6283):425–34. doi:10.1038/346425a0
18. Bhandari A, Bhandari V. Biomarkers in bronchopulmonary dysplasia. Paediatr Respir Rev (2013) 14(3):173–9. doi:10.1016/j.prrv.2013.02.008
19. Bhandari V, Elias JA. Cytokines in tolerance to hyperoxia-induced injury in the developing and adult lung. Free Radic Biol Med (2006) 41(1):4–18. doi:10.1016/j.freeradbiomed.2006.01.027
20. Thompson A, Bhandari V. Pulmonary biomarkers of bronchopulmonary dysplasia. Biomark Insights (2008) 3:361–73.
21. Alapati D, Rong M, Chen S, Hehre D, Rodriguez MM, Lipson KE, et al. Connective tissue growth factor antibody therapy attenuates hyperoxia-induced lung injury in neonatal rats. Am J Respir Cell Mol Biol (2011) 45(6):1169–77. doi:10.1165/rcmb.2011-0023OC
22. Bozyk PD, Bentley JK, Popova AP, Anyanwu AC, Linn MD, Goldsmith AM, et al. Neonatal periostin knockout mice are protected from hyperoxia-induced alveolar simplification. PLoS One (2012) 7(2):e31336. doi:10.1371/journal.pone.0031336
23. De Paepe ME, Patel C, Tsai A, Gundavarapu S, Mao Q. Endoglin (CD105) up-regulation in pulmonary microvasculature of ventilated preterm infants. Am J Respir Crit Care Med (2008) 178(2):180–7. doi:10.1164/rccm.200608-1240OC
24. Choo-Wing R, Syed MA, Harijith A, Bowen B, Pryhuber G, Janér C, et al. Hyperoxia and interferon-γ–induced injury in developing lungs occur via cyclooxygenase-2 and the endoplasmic reticulum stress–dependent pathway. Am J Respir Cell Mol Biol (2013) 48(6):749–57. doi:10.1165/rcmb.2012-0381OC
25. Harijith A, Choo-Wing R, Cataltepe S, Yasumatsu R, Aghai ZH, Janer J, et al. A role for matrix metalloproteinase 9 in IFNgamma-mediated injury in developing lungs: relevance to bronchopulmonary dysplasia. Am J Respir Cell Mol Biol (2011) 44(5):621–30. doi:10.1165/rcmb.2010-0058OC
26. Bhatt AJ, Pryhuber GS, Huyck H, Watkins RH, Metlay LA, Maniscalco WM. Disrupted pulmonary vasculature and decreased vascular endothelial growth factor, Flt-1, and TIE-2 in human infants dying with bronchopulmonary dysplasia. Am J Respir Crit Care Med (2001) 164(10 Pt 1):1971–80. doi:10.1164/ajrccm.164.10.2101140
27. Lassus P, Turanlahti M, Heikkila P, Andersson LC, Nupponen I, Sarnesto A, et al. Pulmonary vascular endothelial growth factor and Flt-1 in fetuses, in acute and chronic lung disease, and in persistent pulmonary hypertension of the newborn. Am J Respir Crit Care Med (2001) 164(10 Pt 1):1981–7. doi:10.1164/ajrccm.164.10.2012036
28. Meller S, Bhandari V. VEGF levels in humans and animal models with RDS and BPD: temporal relationships. Exp Lung Res (2012) 38(4):192–203. doi:10.3109/01902148.2012.663454
29. Subramaniam M, Sugiyama K, Coy DH, Kong Y, Miller YE, Weller PF, et al. Bombesin-like peptides and mast cell responses: relevance to bronchopulmonary dysplasia? Am J Respir Crit Care Med (2003) 168(5):601–11. doi:10.1164/rccm.200212-1434OC
30. Bhattacharya S, Go D, Krenitsky DL, Huyck HL, Solleti SK, Lunger VA, et al. Genome-wide transcriptional profiling reveals connective tissue mast cell accumulation in bronchopulmonary dysplasia. Am J Respir Crit Care Med (2012) 186(4):349–58. doi:10.1164/rccm.201203-0406OC
31. Sohn MH, Kang MJ, Matsuura H, Bhandari V, Chen NY, Lee CG, et al. The chitinase-like proteins breast regression protein-39 and YKL-40 regulate hyperoxia-induced acute lung injury. Am J Respir Crit Care Med (2010) 182(7):918–28. doi:10.1164/rccm.200912-1793OC
32. Bhandari V, Choo-Wing R, Lee CG, Zhu Z, Nedrelow JH, Chupp GL, et al. Hyperoxia causes angiopoietin 2-mediated acute lung injury and necrotic cell death. Nat Med (2006) 12(11):1286–93. doi:10.1038/nm1494
33. Aghai ZH, Faqiri S, Saslow JG, Nakhla T, Farhath S, Kumar A, et al. Angiopoietin 2 concentrations in infants developing bronchopulmonary dysplasia: attenuation by dexamethasone. J Perinatol (2008) 28(2):149–55. doi:10.1038/sj.jp.7211886
34. Menon R, Taylor RN, Fortunato SJ. Chorioamnionitis – a complex pathophysiologic syndrome. Placenta (2010) 31(2):113–20. doi:10.1016/j.placenta.2009.11.012
35. Pankuch GA, Appelbaum PC, Lorenz RP, Botti JJ, Schachter J, Naeye RL. Placental microbiology and histology and the pathogenesis of chorioamnionitis. Obstet Gynecol (1984) 64(6):802–6.
36. Duff P, Sanders R, Gibbs RS. The course of labor in term patients with chorioamnionitis. Am J Obstet Gynecol (1983) 147(4):391–5.
37. Redline RW, Heller D, Keating S, Kingdom J. Placental diagnostic criteria and clinical correlation – a workshop report. Placenta (2005) 26(Suppl A):S114–7. doi:10.1016/j.placenta.2005.02.009
38. Miyazaki K, Furuhashi M, Matsuo K, Minami K, Yoshida K, Kuno N, et al. Impact of subclinical chorioamnionitis on maternal and neonatal outcomes. Acta Obstet Gynecol Scand (2007) 86(2):191–7. doi:10.1080/00016340601022793
39. Gibbs RS, Blanco JD, St Clair PJ, Castaneda YS. Quantitative bacteriology of amniotic fluid from women with clinical intraamniotic infection at term. J Infect Dis (1982) 145(1):1–8. doi:10.1093/infdis/145.1.1
40. Viscardi RM. Perinatal inflammation and lung injury. Semin Fetal Neonatal Med (2012) 17(1):30–5. doi:10.1016/j.siny.2011.08.002
41. Kramer BW, Kallapur S, Newnham J, Jobe AH. Prenatal inflammation and lung development. Semin Fetal Neonatal Med (2009) 14(1):2–7. doi:10.1016/j.siny.2008.08.011
42. Kunzmann S, Collins JJ, Kuypers E, Kramer BW. Thrown off balance: the effect of antenatal inflammation on the developing lung and immune system. Am J Obstet Gynecol (2013) 208(6):429–37. doi:10.1016/j.ajog.2013.01.008
43. Kallapur SG, Kramer BW, Jobe AH. Ureaplasma and BPD. Semin Perinatol (2013) 37(2):94–101. doi:10.1053/j.semperi.2013.01.005
44. Combs CA, Gravett M, Garite TJ, Hickok DE, Lapidus J, Porreco R, et al. Amniotic fluid infection, inflammation, and colonization in preterm labor with intact membranes. Am J Obstet Gynecol (2014) 210(2):e1–15. doi:10.1016/j.ajog.2013.11.032
45. Eriksson L, Haglund B, Odlind V, Altman M, Kieler H. Prenatal inflammatory risk factors for development of bronchopulmonary dysplasia. Pediatr Pulmonol (2014) 49(7):665–72. doi:10.1002/ppul.22881
46. Been JV, Zimmermann LJ. Histological chorioamnionitis and respiratory outcome in preterm infants. Arch Dis Child Fetal Neonatal Ed (2009) 94(3):F218–25. doi:10.1136/adc.2008.150458
47. Thomas W, Speer CP. Chorioamnionitis: important risk factor or innocent bystander for neonatal outcome? Neonatology (2011) 99(3):177–87. doi:10.1159/000320170
48. Thomas W, Speer CP. Chorioamnionitis is essential in the evolution of bronchopulmonary dysplasia – the case in favour. Paediatr Respir Rev (2014) 15(1):49–52. doi:10.1016/j.prrv.2013.09.004
49. Plakkal N, Soraisham AS, Trevenen C, Freiheit EA, Sauve R. Histological chorioamnionitis and bronchopulmonary dysplasia: a retrospective cohort study. J Perinatol (2013) 33(6):441–5. doi:10.1038/jp.2012.154
50. Nasef N, Shabaan AE, Schurr P, Iaboni D, Choudhury J, Church P, et al. Effect of clinical and histological chorioamnionitis on the outcome of preterm infants. Am J Perinatol (2013) 30(1):59–68. doi:10.1055/s-0032-1321501
51. Shah J, Jefferies AL, Yoon EW, Lee SK, Shah PS, Canadian Neonatal Network. Risk factors and outcomes of late-onset bacterial sepsis in preterm neonates born at <32 weeks’ gestation. Am J Perinatol (2015) 32(7):675–82. doi:10.1055/s-0034-1393936
52. Franco ML, Waszak P, Banalec G, Levame M, Lafuma C, Harf A, et al. LPS-induced lung injury in neonatal rats: changes in gelatinase activities and consequences on lung growth. Am J Physiol Lung Cell Mol Physiol (2002) 282(3):L491–500. doi:10.1152/ajplung.00140.2001
53. Londhe VA, Belperio JA, Keane MP, Burdick MD, Xue YY, Strieter RM. CXCR2/CXCR2 ligand biological axis impairs alveologenesis during dsRNA-induced lung inflammation in mice. Pediatr Res (2005) 58(5):919–26. doi:10.1203/01.PDR.0000181377.78061.3E
54. Alvira CM, Abate A, Yang G, Dennery PA, Rabinovitch M. Nuclear factor-kappaB activation in neonatal mouse lung protects against lipopolysaccharide-induced inflammation. Am J Respir Crit Care Med (2007) 175(8):805–15. doi:10.1164/rccm.200608-1162OC
55. Iosef C, Alastalo TP, Hou Y, Chen C, Adams ES, Lyu SC, et al. Inhibiting NF-kappaB in the developing lung disrupts angiogenesis and alveolarization. Am J Physiol Lung Cell Mol Physiol (2012) 302(10):L1023–36. doi:10.1152/ajplung.00230.2011
56. Londhe VA, Maisonet TM, Lopez B, Jeng JM, Xiao J, Li C, et al. Conditional deletion of epithelial IKKbeta impairs alveolar formation through apoptosis and decreased VEGF expression during early mouse lung morphogenesis. Respir Res (2011) 12:134. doi:10.1186/1465-9921-12-134
57. Hillman NH, Polglase GR, Pillow JJ, Saito M, Kallapur SG, Jobe AH. Inflammation and lung maturation from stretch injury in preterm fetal sheep. Am J Physiol Lung Cell Mol Physiol (2011) 300(2):L232–41. doi:10.1152/ajplung.00294.2010
58. Carvalho CG, Silveira RC, Procianoy RS. Ventilator-induced lung injury in preterm infants. Rev Bras Ter Intensiva (2013) 25(4):319–26. doi:10.5935/0103-507X.20130054
59. Hillman NH, Kemp MW, Noble PB, Kallapur SG, Jobe AH. Sustained inflation at birth did not protect preterm fetal sheep from lung injury. Am J Physiol Lung Cell Mol Physiol (2013) 305(6):L446–53. doi:10.1152/ajplung.00162.2013
60. Allison BJ, Crossley KJ, Flecknoe SJ, Davis PG, Morley CJ, Harding R, et al. Ventilation of the very immature lung in utero induces injury and BPD-like changes in lung structure in fetal sheep. Pediatr Res (2008) 64(4):387–92. doi:10.1203/PDR.0b013e318181e05e
61. Wu S, Capasso L, Lessa A, Peng J, Kasisomayajula K, Rodriguez M, et al. High tidal volume ventilation activates Smad2 and upregulates expression of connective tissue growth factor in newborn rat lung. Pediatr Res (2008) 63(3):245–50. doi:10.1203/PDR.0b013e318163a8cc
62. Kroon AA, Wang J, Huang Z, Cao L, Kuliszewski M, Post M. Inflammatory response to oxygen and endotoxin in newborn rat lung ventilated with low tidal volume. Pediatr Res (2010) 68(1):63–9. doi:10.1203/00006450-201011001-00120
63. Kroon AA, Wang J, Kavanagh BP, Huang Z, Kuliszewski M, van Goudoever JB, et al. Prolonged mechanical ventilation induces cell cycle arrest in newborn rat lung. PLoS One (2011) 6(2):e16910. doi:10.1371/journal.pone.0016910
64. Cannizzaro V, Zosky GR, Hantos Z, Turner DJ, Sly PD. High tidal volume ventilation in infant mice. Respir Physiol Neurobiol (2008) 162(1):93–9. doi:10.1016/j.resp.2008.04.010
65. Bland RD, Ertsey R, Mokres LM, Xu L, Jacobson BE, Jiang S, et al. Mechanical ventilation uncouples synthesis and assembly of elastin and increases apoptosis in lungs of newborn mice. Prelude to defective alveolar septation during lung development? Am J Physiol Lung Cell Mol Physiol (2008) 294(1):L3–14. doi:10.1152/ajplung.00362.2007
66. Mokres LM, Parai K, Hilgendorff A, Ertsey R, Alvira CM, Rabinovitch M, et al. Prolonged mechanical ventilation with air induces apoptosis and causes failure of alveolar septation and angiogenesis in lungs of newborn mice. Am J Physiol Lung Cell Mol Physiol (2010) 298(1):L23–35. doi:10.1152/ajplung.00251.2009
67. Hilgendorff A, Parai K, Ertsey R, Jain N, Navarro EF, Peterson JL, et al. Inhibiting lung elastase activity enables lung growth in mechanically ventilated newborn mice. Am J Respir Crit Care Med (2011) 184(5):537–46. doi:10.1164/rccm.201012-2010OC
68. Hilgendorff A, Parai K, Ertsey R, Juliana Rey-Parra G, Thebaud B, Tamosiuniene R, et al. Neonatal mice genetically modified to express the elastase inhibitor elafin are protected against the adverse effects of mechanical ventilation on lung growth. Am J Physiol Lung Cell Mol Physiol (2012) 303(3):L215–27. doi:10.1152/ajplung.00405.2011
69. Albertine KH, Jones GP, Starcher BC, Bohnsack JF, Davis PL, Cho SC, et al. Chronic lung injury in preterm lambs. Disordered respiratory tract development. Am J Respir Crit Care Med (1999) 159(3):945–58. doi:10.1164/ajrccm.159.3.9804027
70. Bland RD, Xu L, Ertsey R, Rabinovitch M, Albertine KH, Wynn KA, et al. Dysregulation of pulmonary elastin synthesis and assembly in preterm lambs with chronic lung disease. Am J Physiol Lung Cell Mol Physiol (2007) 292(6):L1370–84. doi:10.1152/ajplung.00367.2006
71. Reyburn B, Li M, Metcalfe DB, Kroll NJ, Alvord J, Wint A, et al. Nasal ventilation alters mesenchymal cell turnover and improves alveolarization in preterm lambs. Am J Respir Crit Care Med (2008) 178(4):407–18. doi:10.1164/rccm.200802-359OC
72. Rehan VK, Fong J, Lee R, Sakurai R, Wang ZM, Dahl MJ, et al. Mechanism of reduced lung injury by high-frequency nasal ventilation in a preterm lamb model of neonatal chronic lung disease. Pediatr Res (2011) 70(5):462–6. doi:10.1038/pr.2011.687
73. Coalson JJ, Winter VT, Siler-Khodr T, Yoder BA. Neonatal chronic lung disease in extremely immature baboons. Am J Respir Crit Care Med (1999) 160(4):1333–46. doi:10.1164/ajrccm.160.4.9810071
74. Tambunting F, Beharry KD, Hartleroad J, Waltzman J, Stavitsky Y, Modanlou HD. Increased lung matrix metalloproteinase-9 levels in extremely premature baboons with bronchopulmonary dysplasia. Pediatr Pulmonol (2005) 39(1):5–14. doi:10.1002/ppul.20135
75. Maniscalco WM, Watkins RH, Pryhuber GS, Bhatt A, Shea C, Huyck H. Angiogenic factors and alveolar vasculature: development and alterations by injury in very premature baboons. Am J Physiol Lung Cell Mol Physiol (2002) 282(4):L811–23. doi:10.1152/ajplung.00325.2001
76. Tambunting F, Beharry KD, Waltzman J, Modanlou HD. Impaired lung vascular endothelial growth factor in extremely premature baboons developing bronchopulmonary dysplasia/chronic lung disease. J Investig Med (2005) 53(5):253–62. doi:10.2310/6650.2005.53508
77. Ganter MT, Pittet JF. Bombesin-like peptides: modulators of inflammation in acute lung injury? Am J Respir Crit Care Med (2006) 173(1):1–2. doi:10.1164/rccm.2510002
78. Sunday ME, Yoder BA, Cuttitta F, Haley KJ, Emanuel RL. Bombesin-like peptide mediates lung injury in a baboon model of bronchopulmonary dysplasia. J Clin Invest (1998) 102(3):584–94. doi:10.1172/JCI2329
79. Subramaniam M, Bausch C, Twomey A, Andreeva S, Yoder BA, Chang L, et al. Bombesin-like peptides modulate alveolarization and angiogenesis in bronchopulmonary dysplasia. Am J Respir Crit Care Med (2007) 176(9):902–12. doi:10.1164/rccm.200611-1734OC
80. Thomson MA, Yoder BA, Winter VT, Martin H, Catland D, Siler-Khodr TM, et al. Treatment of immature baboons for 28 days with early nasal continuous positive airway pressure. Am J Respir Crit Care Med (2004) 169(9):1054–62. doi:10.1164/rccm.200309-1276OC
81. Thomson MA, Yoder BA, Winter VT, Giavedoni L, Chang LY, Coalson JJ. Delayed extubation to nasal continuous positive airway pressure in the immature baboon model of bronchopulmonary dysplasia: lung clinical and pathological findings. Pediatrics (2006) 118(5):2038–50. doi:10.1542/peds.2006-0622
82. Aly H, Milner JD, Patel K, El-Mohandes AA. Does the experience with the use of nasal continuous positive airway pressure improve over time in extremely low birth weight infants? Pediatrics (2004) 114(3):697–702. doi:10.1542/peds.2003-0572-L
83. Morley CJ, Davis PG, Doyle LW, Brion LP, Hascoet JM, Carlin JB. Nasal CPAP or intubation at birth for very preterm infants. N Engl J Med (2008) 358(7):700–8. doi:10.1056/NEJMoa072788
84. Stevens TP, Harrington EW, Blennow M, Soll RF. Early surfactant administration with brief ventilation vs. selective surfactant and continued mechanical ventilation for preterm infants with or at risk for respiratory distress syndrome. Cochrane Database Syst Rev (2007) 4:CD003063.
85. Bhandari V. The potential of non-invasive ventilation to decrease BPD. Semin Perinatol (2013) 37(2):108–14. doi:10.1053/j.semperi.2013.01.007
86. Mehta P, Berger J, Bucholz E, Bhandari V. Factors affecting nasal intermittent positive pressure ventilation failure and impact on bronchopulmonary dysplasia in neonates. J Perinatol (2014) 34(10):754–60. doi:10.1038/jp.2014.100
87. Jasani B, Nanavati R, Kabra N, Rajdeo S, Bhandari V. Comparison of non-synchronized nasal intermittent positive pressure ventilation versus nasal continuous positive airway pressure as post-extubation respiratory support in preterm infants with respiratory distress syndrome: a randomized controlled trial. J Matern Fetal Neonatal Med (2015) 10:1–6. doi:10.3109/14767058.2015.1059809
88. Kirpalani H, Millar D, Lemyre B, Yoder BA, Chiu A, Roberts RS. A trial comparing noninvasive ventilation strategies in preterm infants. N Engl J Med (2013) 369(7):611–20. doi:10.1056/NEJMoa1214533
89. Warner BB, Stuart LA, Papes RA, Wispe JR. Functional and pathological effects of prolonged hyperoxia in neonatal mice. Am J Physiol (1998) 275(1 Pt 1):L110–7.
90. Zhang X, Wang H, Shi Y, Peng W, Zhang S, Zhang W, et al. Role of bone marrow-derived mesenchymal stem cells in the prevention of hyperoxia-induced lung injury in newborn mice. Cell Biol Int (2012) 36(6):589–94. doi:10.1042/CBI20110447
91. Choo-Wing R, Syed MA, Harijith A, Bowen B, Pryhuber G, Janer C, et al. Hyperoxia and interferon-gamma-induced injury in developing lungs occur via cyclooxygenase-2 and the endoplasmic reticulum stress-dependent pathway. Am J Respir Cell Mol Biol (2013) 48(6):749–57. doi:10.1165/rcmb.2012-0381OC
92. Yee M, Chess PR, McGrath-Morrow SA, Wang Z, Gelein R, Zhou R, et al. Neonatal oxygen adversely affects lung function in adult mice without altering surfactant composition or activity. Am J Physiol Lung Cell Mol Physiol (2009) 297(4):L641–9. doi:10.1152/ajplung.00023.2009
93. O’Reilly MA, Marr SH, Yee M, McGrath-Morrow SA, Lawrence BP. Neonatal hyperoxia enhances the inflammatory response in adult mice infected with influenza A virus. Am J Respir Crit Care Med (2008) 177(10):1103–10. doi:10.1164/rccm.200712-1839OC
94. Li Z, Choo-Wing R, Sun H, Sureshbabu A, Sakurai R, Rehan VK, et al. A potential role of the JNK pathway in hyperoxia-induced cell death, myofibroblast transdifferentiation and TGF-beta1-mediated injury in the developing murine lung. BMC Cell Biol (2011) 12:54. doi:10.1186/1471-2121-12-54
95. Buczynski BW, Yee M, Paige Lawrence B, O’Reilly MA. Lung development and the host response to influenza A virus are altered by different doses of neonatal oxygen in mice. Am J Physiol Lung Cell Mol Physiol (2012) 302(10):L1078–87. doi:10.1152/ajplung.00026.2012
96. Thickett DR, Armstrong L, Christie SJ, Millar AB. Vascular endothelial growth factor may contribute to increased vascular permeability in acute respiratory distress syndrome. Am J Respir Crit Care Med (2001) 164(9):1601–5. doi:10.1164/ajrccm.164.9.2011071
97. Ben-Ari J, Makhoul IR, Dorio RJ, Buckley S, Warburton D, Walker SM. Cytokine response during hyperoxia: sequential production of pulmonary tumor necrosis factor and interleukin-6 in neonatal rats. Isr Med Assoc J (2000) 2(5):365–9.
98. Maduekwe ET, Buczynski BW, Yee M, Rangasamy T, Stevens TP, Lawrence BP, et al. Cumulative neonatal oxygen exposure predicts response of adult mice infected with influenza A virus. Pediatr Pulmonol (2014) 50(3):222–30. doi:10.1002/ppul.23063
99. Backstrom E, Hogmalm A, Lappalainen U, Bry K. Developmental stage is a major determinant of lung injury in a murine model of bronchopulmonary dysplasia. Pediatr Res (2011) 69(4):312–8. doi:10.1203/PDR.0b013e31820bcb2a
100. Bry K, Whitsett JA, Lappalainen U. IL-1beta disrupts postnatal lung morphogenesis in the mouse. Am J Respir Cell Mol Biol (2007) 36(1):32–42. doi:10.1165/rcmb.2006-0116OC
101. Hogmalm A, Sheppard D, Lappalainen U, Bry K. beta6 Integrin subunit deficiency alleviates lung injury in a mouse model of bronchopulmonary dysplasia. Am J Respir Cell Mol Biol (2010) 43(1):88–98. doi:10.1165/rcmb.2008-0480OC
102. Britt RD Jr, Velten M, Tipple TE, Nelin LD, Rogers LK. Cyclooxygenase-2 in newborn hyperoxic lung injury. Free Radic Biol Med (2013) 61:502–11. doi:10.1016/j.freeradbiomed.2013.04.012
103. Johnston CJ, Wright TW, Reed CK, Finkelstein JN. Comparison of adult and newborn pulmonary cytokine mRNA expression after hyperoxia. Exp Lung Res (1997) 23(6):537–52. doi:10.3109/01902149709039242
104. D’Angio CT, LoMonaco MB, Chaudhry SA, Paxhia A, Ryan RM. Discordant pulmonary proinflammatory cytokine expression during acute hyperoxia in the newborn rabbit. Exp Lung Res (1999) 25(5):443–65. doi:10.1080/019021499270187
105. Wagenaar GT, ter Horst SA, van Gastelen MA, Leijser LM, Mauad T, van der Velden PA, et al. Gene expression profile and histopathology of experimental bronchopulmonary dysplasia induced by prolonged oxidative stress. Free Radic Biol Med (2004) 36(6):782–801. doi:10.1016/j.freeradbiomed.2003.12.007
106. Dasgupta C, Sakurai R, Wang Y, Guo P, Ambalavanan N, Torday JS, et al. Hyperoxia-induced neonatal rat lung injury involves activation of TGF-{beta} and Wnt signaling and is protected by rosiglitazone. Am J Physiol Lung Cell Mol Physiol (2009) 296(6):L1031–41. doi:10.1152/ajplung.90392.2008
107. Takeda K, Okamoto M, de Langhe S, Dill E, Armstrong M, Reisdorf N, et al. Peroxisome proliferator-activated receptor-g agonist treatment increases septation and angiogenesis and decreases airway hyperresponsiveness in a model of experimental neonatal chronic lung disease. Anat Rec (Hoboken) (2009) 292(7):1045–61. doi:10.1002/ar.20921
108. Ohki Y, Mayuzumi H, Tokuyama K, Yoshizawa Y, Arakawa H, Mochizuki H, et al. Hepatocyte growth factor treatment improves alveolarization in a newborn murine model of bronchopulmonary dysplasia. Neonatology (2009) 95(4):332–8. doi:10.1159/000187651
109. Couroucli XI, Liang YH, Jiang W, Wang L, Barrios R, Yang P, et al. Prenatal administration of the cytochrome P4501A inducer, Beta-naphthoflavone (BNF), attenuates hyperoxic lung injury in newborn mice: implications for bronchopulmonary dysplasia (BPD) in premature infants. Toxicol Appl Pharmacol (2011) 256(2):83–94. doi:10.1016/j.taap.2011.06.018
110. Ma L, Li N, Liu X, Shaw L, Li Calzi S, Grant MB, et al. Arginyl-glutamine dipeptide or docosahexaenoic acid attenuate hyperoxia-induced lung injury in neonatal mice. Nutrition (2012) 28(11–12):1186–91. doi:10.1016/j.nut.2012.04.001
111. James ML, Ross AC, Nicola T, Steele C, Ambalavanan N. VARA attenuates hyperoxia-induced impaired alveolar development and lung function in newborn mice. Am J Physiol Lung Cell Mol Physiol (2013) 304(11):L803–12. doi:10.1152/ajplung.00257.2012
112. Vosdoganes P, Lim R, Koulaeva E, Chan ST, Acharya R, Moss TJ, et al. Human amnion epithelial cells modulate hyperoxia-induced neonatal lung injury in mice. Cytotherapy (2013) 15(8):1021–9. doi:10.1016/j.jcyt.2013.03.004
113. Reilly EC, Martin KC, Jin GB, Yee M, O’Reilly MA, Lawrence BP. Neonatal hyperoxia leads to persistent alterations in NK responses to influenza A virus infection. Am J Physiol Lung Cell Mol Physiol (2015) 308(1):L76–85. doi:10.1152/ajplung.00233.2014
114. Bhandari A, McGrath-Morrow S. Long-term pulmonary outcomes of patients with bronchopulmonary dysplasia. Semin Perinatol (2013) 37(2):132–7. doi:10.1053/j.semperi.2013.01.010
115. Islam JY, Keller RL, Aschner JL, Hartert TV, Moore PE. Understanding the short- and long-term respiratory outcomes of prematurity and bronchopulmonary dysplasia. Am J Respir Crit Care Med (2015) 192(2):134–56. doi:10.1164/rccm.201412-2142PP
116. Saarenpaa HK, Tikanmaki M, Sipola-Leppanen M, Hovi P, Wehkalampi K, Siltanen M, et al. Lung function in very low birth weight adults. Pediatrics (2015) 136(4):642–50. doi:10.1542/peds.2014-2651
117. Berger J, Mehta P, Bucholz E, Dziura J, Bhandari V. Impact of early extubation and reintubation on the incidence of bronchopulmonary dysplasia in neonates. Am J Perinatol (2014) 31(12):1063–72. doi:10.1055/s-0034-1371702
118. Naik AS, Kallapur SG, Bachurski CJ, Jobe AH, Michna J, Kramer BW, et al. Effects of ventilation with different positive end-expiratory pressures on cytokine expression in the preterm lamb lung. Am J Respir Crit Care Med (2001) 164(3):494–8. doi:10.1164/ajrccm.164.3.2010127
119. Ikegami M, Moss TJ, Kallapur SG, Mulrooney N, Kramer BW, Nitsos I, et al. Minimal lung and systemic responses to TNF-alpha in preterm sheep. Am J Physiol Lung Cell Mol Physiol (2003) 285(1):L121–9. doi:10.1152/ajplung.00393.2002
120. Copland IB, Martinez F, Kavanagh BP, Engelberts D, McKerlie C, Belik J, et al. High tidal volume ventilation causes different inflammatory responses in newborn versus adult lung. Am J Respir Crit Care Med (2004) 169(6):739–48. doi:10.1164/rccm.200310-1417OC
121. Kornecki A, Tsuchida S, Ondiveeran HK, Engelberts D, Frndova H, Tanswell AK, et al. Lung development and susceptibility to ventilator-induced lung injury. Am J Respir Crit Care Med (2005) 171(7):743–52. doi:10.1164/rccm.200408-1053OC
122. Lee PT, Holt PG, McWilliam AS. Role of alveolar macrophages in innate immunity in neonates: evidence for selective lipopolysaccharide binding protein production by rat neonatal alveolar macrophages. Am J Respir Cell Mol Biol (2000) 23(5):652–61. doi:10.1165/ajrcmb.23.5.4016
123. Lee PT, Holt PG, McWilliam AS. Ontogeny of rat pulmonary alveolar macrophage function: evidence for a selective deficiency in il-10 and nitric oxide production by newborn alveolar macrophages. Cytokine (2001) 15(1):53–7. doi:10.1006/cyto.2001.0894
124. Martin TR, Ruzinski JT, Wilson CB, Skerrett SJ. Effects of endotoxin in the lungs of neonatal rats: age-dependent impairment of the inflammatory response. J Infect Dis (1995) 171(1):134–44. doi:10.1093/infdis/171.1.134
125. Choo-Wing R, Nedrelow JH, Homer RJ, Elias JA, Bhandari V. Developmental differences in the responses of IL-6 and IL-13 transgenic mice exposed to hyperoxia. Am J Physiol Lung Cell Mol Physiol (2007) 293(1):L142–50. doi:10.1152/ajplung.00434.2006
126. Bhandari V, Choo-Wing R, Lee CG, Yusuf K, Nedrelow JH, Ambalavanan N, et al. Developmental regulation of NO-mediated VEGF-induced effects in the lung. Am J Respir Cell Mol Biol (2008) 39(4):420–30. doi:10.1165/rcmb.2007-0024OC
Keywords: premature newborn, chronic lung disease, cytokines, sepsis, hyperoxia, mechanical ventilation
Citation: Balany J and Bhandari V (2015) Understanding the Impact of Infection, Inflammation, and Their Persistence in the Pathogenesis of Bronchopulmonary Dysplasia. Front. Med. 2:90. doi: 10.3389/fmed.2015.00090
Received: 05 October 2015; Accepted: 03 December 2015;
Published: 21 December 2015
Edited by:
Anne Hilgendorff, Helmholtz Zentrum München, GermanyReviewed by:
Mattias Svensson, Karolinska Institutet, SwedenCopyright: © 2015 Balany and Bhandari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vineet Bhandari, dmluZWV0LmJoYW5kYXJpQGRyZXhlbG1lZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.