 Mehmet Coskun
Mehmet Coskun
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Med., 25 August 2014
Sec. Gastroenterology
Volume 1 - 2014 | https://doi.org/10.3389/fmed.2014.00024
The intestinal epithelium has a strategic position as a protective physical barrier to luminal microbiota and actively contributes to the mucosal immune system. This barrier is mainly formed by a monolayer of specialized intestinal epithelial cells (IECs) that are crucial in maintaining intestinal homeostasis. Therefore, dysregulation within the epithelial layer can increase intestinal permeability, lead to abnormalities in interactions between IECs and immune cells in underlying lamina propria, and disturb the intestinal immune homeostasis, all of which are linked to the clinical disease course of inflammatory bowel disease (IBD). Understanding the role of the intestinal epithelium in IBD pathogenesis might contribute to an improved knowledge of the inflammatory processes and the identification of potential therapeutic targets.
Inflammatory bowel disease (IBD) is characterized by a chronic idiopathic inflammation of the intestine and consists of two main forms, i.e., ulcerative colitis (UC) (1) and Crohn’s disease (CD) (2). The inflammation in CD can be transmural affecting any parts of the entire gastrointestinal tract, whereas UC is restricted to the mucosa of the colon. Although the etiology of IBD is largely unknown, it involves a complex interaction between genetic, luminal, and environmental factors that trigger an inappropriate mucosal immune response (Figure 1) (3–7). The importance of genetic susceptibility has over the past decades been established through genome-wide association studies, which have identified a total of 163 IBD-associated gene loci, most of which are associated with both CD and UC (110/163) – suggesting shared pathways in IBD pathogenesis despite differences in clinical phenotype – and 30 gene loci classified specifically for CD and 23 as UC specific (8). However, not all patients with genetic changes develop IBD. Therefore, other factors like environmental risk factors including diet, smoking, drugs, infections, geography, and stress have an important role in the pathogenesis of IBD (9). In particular, changes in the composition of the intestinal microbiota are likely the most important environmental factor in IBD by inducing an overactive immune response that harms the mucosal barrier (10–14). It is therefore not surprising that intestinal barrier dysfunction plays a key pathogenic role in IBD (15). Thus, IBD is a multifactorial disease driven by an exaggerated immune response to gut microbiota in a genetically susceptible host causing defects in epithelial barrier function and epithelial response to pathogens (Figure 1).
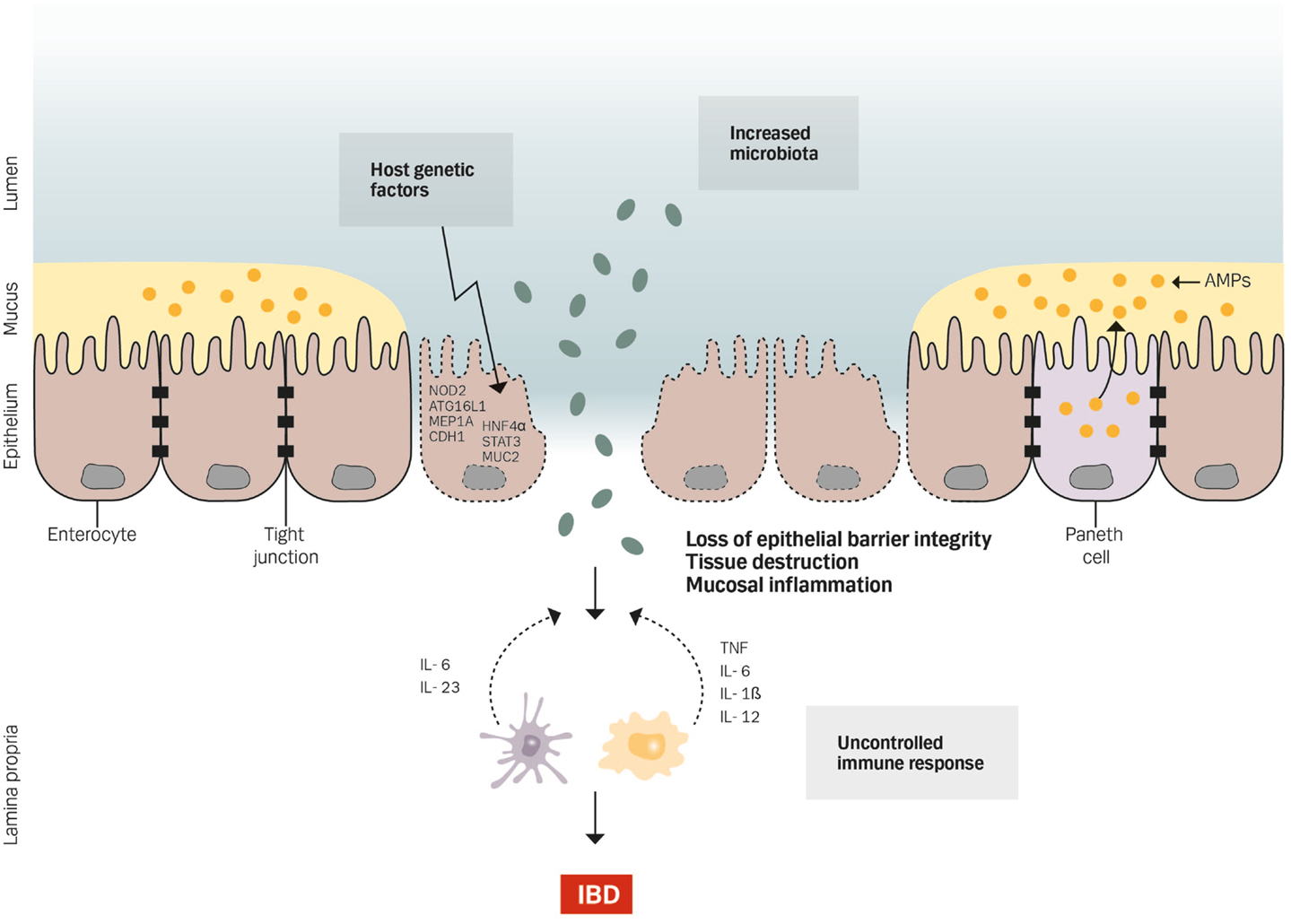
Figure 1. Genetics, gut microbiota, and an uncontrolled immune response cause defects in epithelial barrier function by affecting the barrier integrity, increasing tissue destruction and mucosal inflammation.
The intestinal epithelium forms the protective barrier and host defense against the harmful luminal microenvironment with selective permeability and absorption of nutrients. The epithelium is covered by a single-cell layer composed of different subtypes of specialized intestinal epithelial cells (IECs) including absorptive cells, goblet cells, enteroendocrine cells, Paneth cells, M cells, cup cells, and Tuft cells, all of which differentiate from epithelial stem cells (16–18). These subsets of IECs are functionally different and essential to maintain intestinal homeostasis by separating the intestinal lumen from the underlying lamina propria and by controlling the crosstalk between microbiota and subjacent immune cells. Thus, a dysregulation of the differentiation system for correct IEC formation has a crucial role in the pathogenesis of IBD (19). Indeed, several crucial genes for the differentiation of IECs have been demonstrated to become aberrantly expressed during IBD (20–22).
The epithelial monolayer is the main component of the epithelial barrier and its ability to act as a protective physical barrier is mediated by the formation of a web of tight junctions (TJs) that regulate the paracellular permeability and barrier integrity, production of mucus layer covering the luminal surface of the epithelium, and recognition of pathogens and production of antimicrobial peptides (AMPs) to ensure effective immunity (23). Hence, TJs seal the paracellular space between epithelial cells and separate the cell membrane into apical and basolateral domains, thus forming a physical barrier against foreign antigens. In fact, an altered expression and structural changes of the intestinal TJ proteins are closely associated with the development of IBD (24–26). Moreover, several pro-inflammatory cytokines, such as tumor necrosis factor (TNF)-α and interferon-γ, have been shown to increase TJ permeability and to induce apoptosis of IECs (27–29). This leads to the loss of epithelial barrier function and induces epithelial damage and ulcers that are present in mucosal inflammation. Indeed, a strong linkage has been established between abnormal intestinal permeability and mucosal inflammation in both CD and UC patients (30), which has been supported by several studies demonstrating a restored intestinal permeability in patients responding to anti-TNF therapy (31, 32). Moreover, among the various experimental mouse models of intestinal inflammation (33), two available models, namely, SAMP1/YitFc and C3H/HeJBir, develop spontaneous chronic inflammation due to defects in innate and adaptive immune responses. Thus, SAMP1/YitFc mice have epithelial dysfunction and increased permeability (34), whereas C3H/HeJBir mice are more likely to have dysregulated innate immune responses and bacterial clearance (35).
To support the epithelial barrier, a mucus layer covers the single layer of IECs (Figure 1), and the critical role of the mucus layer in microbiota sequestration has been demonstrated by animal studies with Muc2-deficient mice (36) – the producer of the main mucus protein secreted by goblet cells – and by reduced goblet cell numbers and depleted mucus secretion in IBD patients (37). In mice, Muc2 deficiency results in a diminished mucus layer, elevated levels of pro-inflammatory cytokines, and development of spontaneous colitis. Moreover, the intestinal epithelial barrier is supported by the production of AMPs – mainly secreted by Paneth cells. These AMPs contribute to the mucus layer by preventing bacteria from reaching the epithelial surface or interact with the underlying immune system (Figure 1). Thus, the role of AMPs has been implicated in mucosal homeostasis and in the pathogenesis of several conditions including IBD (38–40). Indeed, a defective expression of AMPs has been revealed in patients with CD (41–43).
Apart from forming a tight protective barrier, IECs are actively involved in the innate immune response as many epithelial cells have pattern recognition receptors such as Toll-like receptors on the cell surface and nucleotide-binding oligomerization domain (NOD)-like receptors in the cytoplasm that are essential in sensing bacterial products and initiating the immune response through the pro-inflammatory transcription factor nuclear factor-κB to maintain homeostasis (18). Therefore, any defects in these IECs-related processes might trigger mucosal inflammation.
Genes within IBD-associated genetic loci highlight the importance of epithelial barrier defects (44). While the functional role of many loci or single-nucleotide polymorphisms is incompletely understood (8), several of the IBD-susceptibility genes (few examples are discussed below) are associated with different aspects of epithelial functions. Moreover, experimental mouse models of intestinal inflammation have been extensively used to investigate the importance of components of a healthy intestinal barrier function [reviewed in detail in Ref. (33)].
The nuclear transcription factor, hepatocyte nuclear factor 4α (HNF4α), encoded by HNF4α, regulates the expression of several components involved in epithelial TJs and intestinal permeability. HNF4α is down-regulated in patients with IBD and, moreover, mice with IEC-specific conditional knockout of Hnf4α are more susceptible to chemically induced colitis (20), indicating that HNF4α is crucial for the barrier function of the intestinal mucosa. Another example is the CDH1 gene that encodes for E-cadherin, a cell adhesion molecule expressed in the epithelium, that is important for key morphogenetic processes such as cell growth, epithelial differentiation, and proliferation (45). Hence, loss or mislocalization of E-cadherin is involved in the pathogensis of IBD by increasing epithelial permeability (46).
Moreover, several polymorphisms in the meprin 1A (MEP1A) gene have been associated with UC (47). This gene encodes the α subunit of meprins found as a secreted form or as a membrane-bound form at the brush-border membrane in association with the transmembrane β subunit in IECs where their main function is to cleave diverse substrates such as laminins, TJ proteins, and cytokines (48–51). The expression of MEP1A is decreased in patients with active UC and experimental mice models with Mep1A-deficiency are more susceptible to chemically induced colitis (22, 52).
NOD2, encoded by CARD15, as the first genetic susceptibility locus for CD is an intracellular receptor that recognizes bacterial muramyl dipeptide and induces autophagy and bacterial clearance. NOD2 has, therefore, a central role in innate immune activation of epithelial cells attributed in part to Paneth cells. Autophagy is an important participant in the defense against intracellular invading pathogens and the NOD2-directed autophagy is dependent on, among others, the autophagy-related 16-like 1 (ATG16L1) protein, which is encoded by ATG16L1 (53). Thus, carrying the CD-associated loss-of-function NOD2 and/or ATG16L1 risk variants displays an augmented inflammatory status (54) with a defective autophagy induction, bacterial clearance, and antigen presentation. As a consequence, the immune response is diminished, and luminal bacteria may invade the intestinal mucosa and trigger inflammation (55). However, neither partial nor complete loss of function of Atg16l1 or Nod2 leads to spontaneous intestinal inflammation (56, 57). Interestingly, specific deletion of the gene encoding X-box-binding protein 1 (Xbp1) – a transcription factor central to the unfolded protein response in the setting of endoplasmic reticulum (ER) stress – in IECs results in ER stress and Paneth cell impairment (58). Moreover, a recent study demonstrated that ER stress induced by IECs-specific Xbp1 deletion was compensated by autophagy responses in Paneth cells. However, deletion of the autophagy-related gene, Atg16l1 or Atg7, in addition to Xbp1 developed severe spontaneous CD-like transmural ileitis (59). Thus, both ER stress and autophagy seem to be crucial regulatory mechanisms in intestinal homeostasis further underlying the importance of Paneth cells in intestinal inflammation, and this has indeed been supported by a recent study by Deuring et al. (60). In this study, the authors have shown that patients with CD, homozygous, or heterozygous for the ATG16L1 risk allele, are associated with ER stress in Paneth cells (60).
A large number of mammalian cytokines modulate intracellular signaling by inducing the mitogen-activated protein kinase pathway (61), as well as the Janus kinase/signal transducer and activator of transcription (STAT) pathway (62). STATs are transcription factors that orchestrate an appropriate cellular response through target gene expression, and STAT1, STAT3, and STAT4 have been reported as IBD-related susceptibility genes (8). In particular, animal studies have revealed the crucial role of an intact Stat3 in intestinal homeostasis and its protective role as IEC-specific Stat3-deficient mice are highly susceptible to chemically induced colitis (63).
Altogether, given the importance of many transcription factors, adhesion molecules, and immunological factors in the maintenance of the integrity of the epithelial barrier and regulating the homeostasis of epithelial cells, it is likely that subtle defects in epithelial gene function or expression may contribute to IBD pathogenesis.
The medical management of IBD includes glucocorticoids, immunomodulators (64), and anti-TNF-α biological agents (65, 66) as well as inhibitors of other molecular pathways (67). These drugs block key molecules that are involved in the induction and maintenance of inflammation in several signaling pathways, all of which have different contributions. However, no treatment strategy is curative or free of side effects. Hence, despite increased knowledge on its pathophysiology and improvements in medical therapy, about one-third of IBD patients need surgery due to an inadequate response or treatment failure to conventional therapy. Thus, achieving mucosal healing, preventing disease relapse, and avoiding complications are among the major goals in the management of patients with IBD (68–71). It is therefore likely that the mechanisms that drive mucosal inflammation differ between patients and that the inflammatory cascades are much more complex than initially thought.
Considering the culminating evidence that IECs are central to the maintenance of intestinal homeostasis and intestinal epithelium dysfunction is associated with IBD pathogenesis, potential IEC-based therapeutics will remain a crucial field of interest for IBD therapy. In fact, there are several indications that IECs are central in the response to TNF-α (72), and more importantly, IECs are a target of TNF-α inhibitors (73, 74).
Due to the complexity of signaling within the intestinal epithelium, monocultures of intestinal cells have until now limited their applicability as disease modeling. However, given the proper culture conditions, primary IEC cultures (organoids) can be maintained long-term in vitro and are therefore an invaluable new tool for research of more physiological relevance (75). Within the next several years, further studies and improved knowledge of the role of IECs in IBD will undoubtedly contribute to a better understanding of the pathogenesis of IBD, and will provide more insights into the potential and efficacy of IECs-based therapeutic opportunities and their application.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Ordás I, Eckmann L, Talamini M, Baumgart DC, Sandborn WJ. Ulcerative colitis. Lancet (2012) 380:1606–19. doi: 10.1016/S0140-6736(12)60150-0
2. Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet (2012) 380:1590–605. doi:10.1016/S0140-6736(12)60026-9
3. Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol (2010) 28:573–621. doi:10.1146/annurev-immunol-030409-101225
4. Sartor RB. Mechanisms of disease: pathogenesis of Crohn’s disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol (2006) 3:390–407. doi:10.1038/ncpgasthep0528
5. Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet (2010) 42:1118–25. doi:10.1038/ng.717
6. Anderson CA, Boucher G, Lees CW, Franke A, D’Amato M, Taylor KD, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet (2011) 43:246–52. doi:10.1038/ng.764
7. Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature (2011) 474:307–17. doi:10.1038/nature10209
8. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature (2012) 491:119–24. doi:10.1038/nature11582
9. Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med (2009) 361:2066–78. doi:10.1056/NEJMra0804647
10. Frank DN, Robertson CE, Hamm CM, Kpadeh Z, Zhang T, Chen H, et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis (2011) 17:179–84. doi:10.1002/ibd.21339
11. Nagalingam NA, Lynch SV. Role of the microbiota in inflammatory bowel diseases. Inflamm Bowel Dis (2012) 18:968–84. doi:10.1002/ibd.21866
12. Manichanh C, Borruel N, Casellas F, Guarner F. The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol (2012) 9:599–608. doi:10.1038/nrgastro.2012.152
13. Brown EM, Sadarangani M, Finlay BB. The role of the immune system in governing host-microbe interactions in the intestine. Nat Immunol (2013) 14:660–7. doi:10.1038/ni.2611
14. Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology (2014) 146:1489–99. doi:10.1053/j.gastro.2014.02.009
15. Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature (2011) 474:298–306. doi:10.1038/nature10208
16. van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol (2009) 71:241–60. doi:10.1146/annurev.physiol.010908.163145
17. Noah TK, Donahue B, Shroyer NF. Intestinal development and differentiation. Exp Cell Res (2011) 317:2702–10. doi:10.1016/j.yexcr.2011.09.006
18. Dupaul-Chicoine J, Dagenais M, Saleh M. Crosstalk between the intestinal microbiota and the innate immune system in intestinal homeostasis and inflammatory bowel disease. Inflamm Bowel Dis (2013) 19:2227–37. doi:10.1097/MIB.0b013e31828dcac7
19. Koch S, Nusrat A. The life and death of epithelia during inflammation: lessons learned from the gut. Annu Rev Pathol (2012) 7:35–60. doi:10.1146/annurev-pathol-011811-120905
20. Ahn SH, Shah YM, Inoue J, Morimura K, Kim I, Yim S, et al. Hepatocyte nuclear factor 4alpha in the intestinal epithelial cells protects against inflammatory bowel disease. Inflamm Bowel Dis (2008) 14:908–20. doi:10.1002/ibd.20413
21. Zheng X, Tsuchiya K, Okamoto R, Iwasaki M, Kano Y, Sakamoto N, et al. Suppression of hath1 gene expression directly regulated by hes1 via notch signaling is associated with goblet cell depletion in ulcerative colitis. Inflamm Bowel Dis (2011) 17:2251–60. doi:10.1002/ibd.21611
22. Coskun M, Olsen AK, Holm TL, Kvist PH, Nielsen OH, Riis LB, et al. TNF-alpha-induced down-regulation of CDX2 suppresses MEP1A expression in colitis. Biochim Biophys Acta (2012) 1822:843–51. doi:10.1016/j.bbadis.2012.01.012
23. Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol (2014) 14:141–53. doi:10.1038/nri3608
24. Zeissig S, Bürgel N, Günzel D, Richter J, Mankertz J, Wahnschaffe U, et al. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut (2007) 56:61–72. doi:10.1136/gut.2006.094375
25. Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology (2005) 129:550–64. doi:10.1016/j.gastro.2005.05.002
26. Schmitz H, Barmeyer C, Fromm M, Runkel N, Foss HD, Bentzel CJ, et al. Altered tight junction structure contributes to the impaired epithelial barrier function in ulcerative colitis. Gastroenterology (1999) 116:301–9. doi:10.1016/S0016-5085(99)70126-5
27. Marini M, Bamias G, Rivera-Nieves J, Moskaluk CA, Hoang SB, Ross WG, et al. TNF-alpha neutralization ameliorates the severity of murine Crohn’s-like ileitis by abrogation of intestinal epithelial cell apoptosis. Proc Natl Acad Sci U S A (2003) 100:8366–71. doi:10.1073/pnas.1432897100
28. Su L, Nalle SC, Shen L, Turner ES, Singh G, Breskin LA, et al. TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterology (2013) 145:407–15. doi:10.1053/j.gastro.2013.04.011
29. Nava P, Koch S, Laukoetter MG, Lee WY, Kolegraff K, Capaldo CT, et al. Interferon-gamma regulates intestinal epithelial homeostasis through converging beta-catenin signaling pathways. Immunity (2010) 32:392–402. doi:10.1016/j.immuni.2010.03.001
30. Mankertz J, Schulzke JD. Altered permeability in inflammatory bowel disease: pathophysiology and clinical implications. Curr Opin Gastroenterol (2007) 23:379–83. doi:10.1097/MOG.0b013e32816aa392
31. Suenaert P, Bulteel V, Lemmens L, Noman M, Geypens B, Van Assche G, et al. Anti-tumor necrosis factor treatment restores the gut barrier in Crohn’s disease. Am J Gastroenterol (2002) 97:2000–4. doi:10.1111/j.1572-0241.2002.05914.x
32. Toedter G, Li K, Sague S, Ma K, Marano C, Macoritto M, et al. Genes associated with intestinal permeability in ulcerative colitis: changes in expression following infliximab therapy. Inflamm Bowel Dis (2012) 18:1399–410. doi:10.1002/ibd.22853
33. Pastorelli L, De Salvo C, Mercado JR, Vecchi M, Pizarro TT. Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: lessons learned from animal models and human genetics. Front Immunol (2013) 4:280. doi:10.3389/fimmu.2013.00280
34. Pizarro TT, Pastorelli L, Bamias G, Garg RR, Reuter BK, Mercado JR, et al. SAMP1/YitFc mouse strain: a spontaneous model of Crohn’s disease-like ileitis. Inflamm Bowel Dis (2011) 17:2566–84. doi:10.1002/ibd.21638
35. Sundberg JP, Elson CO, Bedigian H, Birkenmeier EH. Spontaneous, heritable colitis in a new substrain of C3H/HeJ mice. Gastroenterology (1994) 107:1726–35.
36. Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology (2006) 131:117–29. doi:10.1053/j.gastro.2006.04.020
37. Boltin D, Perets TT, Vilkin A, Niv Y. Mucin function in inflammatory bowel disease: an update. J Clin Gastroenterol (2013) 47:106–11. doi:10.1097/MCG.0b013e3182688e73
38. Wehkamp J, Stange EF. Paneth’s disease. J Crohns Colitis (2010) 4:523–31. doi:10.1016/j.crohns.2010.05.010
39. Muniz LR, Knosp C, Yeretssian G. Intestinal antimicrobial peptides during homeostasis, infection, and disease. Front Immunol (2012) 3:310. doi:10.3389/fimmu.2012.00310
40. Ostaff MJ, Stange EF, Wehkamp J. Antimicrobial peptides and gut microbiota in homeostasis and pathology. EMBO Mol Med (2013) 5:1465–83. doi:10.1002/emmm.201201773
41. Arijs I, De Hertogh G, Lemaire K, Quintens R, Van Lommel L, Van Steen K, et al. Mucosal gene expression of antimicrobial peptides in inflammatory bowel disease before and after first infliximab treatment. PLoS One (2009) 4:e7984. doi:10.1371/journal.pone.0007984
42. Wehkamp J, Harder J, Weichenthal M, Mueller O, Herrlinger KR, Fellermann K, et al. Inducible and constitutive beta-defensins are differentially expressed in Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis (2003) 9:215–23. doi:10.1097/00054725-200307000-00001
43. Wehkamp J, Schmid M, Fellermann K, Stange EF. Defensin deficiency, intestinal microbes, and the clinical phenotypes of Crohn’s disease. J Leukoc Biol (2005) 77:460–5. doi:10.1189/jlb.0904543
44. Mokry M, Middendorp S, Wiegerinck CL, Witte M, Teunissen H, Meddens CA, et al. Many inflammatory bowel disease risk loci include regions that regulate gene expression in immune cells and the intestinal epithelium. Gastroenterology (2013) 146:1040–7. doi:10.1053/j.gastro.2013.12.003
45. van Roy F, Berx G. The cell-cell adhesion molecule E-cadherin. Cell Mol Life Sci (2008) 65:3756–88. doi:10.1007/s00018-008-8281-1
46. Muise AM, Walters TD, Glowacka WK, Griffiths AM, Ngan BY, Lan H, et al. Polymorphisms in E-cadherin (CDH1) result in a mis-localised cytoplasmic protein that is associated with Crohn’s disease. Gut (2009) 58:1121–7. doi:10.1136/gut.2008.175117
47. Banerjee S, Oneda B, Yap LM, Jewell DP, Matters GL, Fitzpatrick LR, et al. MEP1A allele for meprin A metalloprotease is a susceptibility gene for inflammatory bowel disease. Mucosal Immunol (2009) 2:220–31. doi:10.1038/mi.2009.3
48. Bertenshaw GP, Turk BE, Hubbard SJ, Matters GL, Bylander JE, Crisman JM, et al. Marked differences between metalloproteases meprin A and B in substrate and peptide bond specificity. J Biol Chem (2001) 276:13248–55. doi:10.1074/jbc.M011414200
49. Kruse MN, Becker C, Lottaz D, Köhler D, Yiallouros I, Krell HW, et al. Human meprin alpha and beta homo-oligomers: cleavage of basement membrane proteins and sensitivity to metalloprotease inhibitors. Biochem J (2004) 378:383–9. doi:10.1042/BJ20031163
50. Broder C, Becker-Pauly C. The metalloproteases meprin alpha and meprin beta: unique enzymes in inflammation, neurodegeneration, cancer and fibrosis. Biochem J (2013) 450:253–64. doi:10.1042/BJ20121751
51. Bao J, Yura RE, Matters GL, Bradley SG, Shi P, Tian F, et al. Meprin A impairs epithelial barrier function, enhances monocyte migration, and cleaves the tight junction protein occludin. Am J Physiol Renal Physiol (2013) 305:F714–26. doi:10.1152/ajprenal.00179.2012
52. Banerjee S, Jin G, Bradley SG, Matters GL, Gailey RD, Crisman JM, et al. Balance of meprin A and B in mice affects the progression of experimental inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol (2011) 300:G273–82. doi:10.1152/ajpgi.00504.2009
53. Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhães JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol (2010) 11:55–62. doi:10.1038/ni.1823
54. Hoefkens E, Nys K, John JM, Van Steen K, Arijs I, Van der Goten J, et al. Genetic association and functional role of Crohn disease risk alleles involved in microbial sensing, autophagy, and endoplasmic reticulum (ER) stress. Autophagy (2013) 9:2046–55. doi:10.4161/auto.26337
55. Goto Y, Kiyono H. Epithelial barrier: an interface for the cross-communication between gut flora and immune system. Immunol Rev (2012) 245:147–63. doi:10.1111/j.1600-065X.2011.01078.x
56. Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature (2008) 456:259–63. doi:10.1038/nature07416
57. Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nuñez G, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science (2005) 307:731–4. doi:10.1126/science.1104911
58. Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell (2008) 134:743–56. doi:10.1016/j.cell.2008.07.021
59. Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Böck J, Martinez-Naves E, et al. Paneth cells as a site of origin for intestinal inflammation. Nature (2013) 503:272–6. doi:10.1038/nature12599
60. Deuring JJ, Fuhler GM, Konstantinov SR, Peppelenbosch MP, Kuipers EJ, de Haar C, et al. Genomic ATG16L1 risk allele-restricted Paneth cell ER stress in quiescent Crohn’s disease. Gut (2014) 63:1081–91. doi:10.1136/gutjnl-2012-303527
61. Coskun M, Olsen J, Seidelin JB, Nielsen OH. MAP kinases in inflammatory bowel disease. Clin Chim Acta (2011) 412:513–20. doi:10.1016/j.cca.2010.12.020
62. Coskun M, Salem M, Pedersen J, Nielsen OH. Involvement of JAK/STAT signaling in the pathogenesis of inflammatory bowel disease. Pharmacol Res (2013) 76C:1–8. doi:10.1016/j.phrs.2013.06.007
63. Willson TA, Jurickova I, Collins M, Denson LA. Deletion of intestinal epithelial cell STAT3 promotes T-lymphocyte STAT3 activation and chronic colitis following acute dextran sodium sulfate injury in mice. Inflamm Bowel Dis (2013) 19:512–25. doi:10.1097/MIB.0b013e31828028ad
64. Nielsen OH, Coskun M, Steenholdt C, Rogler G. The role and advances of immunomodulator therapy for inflammatory bowel disease. Expert Rev Gastroenterol Hepatol (2014). doi:10.1586/17474124.2014.945914
65. Nielsen OH, Ainsworth MA. Tumor necrosis factor inhibitors for inflammatory bowel disease. N Engl J Med (2013) 369:754–62. doi:10.1056/NEJMct1209614
66. Nielsen OH, Ainsworth MA. Which biological agents are most appropriate for ulcerative colitis? Ann Intern Med (2014) 160:733–4. doi:10.7326/M14-0605
67. Pedersen J, Coskun M, Soendergaard C, Salem M, Nielsen OH. Inflammatory pathways of importance for management of inflammatory bowel disease. World J Gastroenterol (2014) 20:64–77. doi:10.3748/wjg.v20.i1.64
68. Seidelin JB, Coskun M, Nielsen OH. Mucosal healing in ulcerative colitis: pathophysiology and pharmacology. Adv Clin Chem (2013) 59:101–23. doi:10.1016/B978-0-12-405211-6.00004-8
69. Neurath MF, Travis SP. Mucosal healing in inflammatory bowel diseases: a systematic review. Gut (2012) 61:1619–35. doi:10.1136/gutjnl-2012-302830
70. De Cruz P, Kamm MA, Prideaux L, Allen PB, Moore G. Mucosal healing in Crohn’s disease: a systematic review. Inflamm Bowel Dis (2013) 19:429–44. doi:10.1002/ibd.22977
71. Vaughn BP, Shah S, Cheifetz AS. The role of mucosal healing in the treatment of patients with inflammatory bowel disease. Curr Treat Options Gastroenterol (2014) 12:103–17. doi:10.1007/s11938-013-0008-1
72. Bamias G, Dahman MI, Arseneau KO, Guanzon M, Gruska D, Pizarro TT, et al. Intestinal-specific TNFalpha overexpression induces Crohn’s-like ileitis in mice. PLoS One (2013) 8:e72594. doi:10.1371/journal.pone.0072594
73. Hassan C, Ierardi E, Burattini O, De Francesco V, Zullo A, Stoppino G, et al. Tumour necrosis factor alpha down-regulation parallels inflammatory regression in ulcerative colitis patients treated with infliximab. Dig Liver Dis (2007) 39:811–7. doi:10.1016/j.dld.2007.06.003
74. Fischer A, Gluth M, Pape UF, Wiedenmann B, Theuring F, Baumgart DC. Adalimumab prevents barrier dysfunction and antagonizes distinct effects of TNF on tight junction proteins and signaling pathways in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol (2013) 304:G970–9. doi:10.1152/ajpgi.00183.2012
Keywords: barrier dysfunction, Crohn’s disease, inflammatory bowel disease, intestinal epithelium, tight junctions, ulcerative colitis
Citation: Coskun M (2014) Intestinal epithelium in inflammatory bowel disease. Front. Med. 1:24. doi: 10.3389/fmed.2014.00024
Received: 28 May 2014; Paper pending published: 14 July 2014;
Accepted: 11 August 2014; Published online: 25 August 2014.
Edited by:
Giovanni Cammarota, Catholic University, ItalyReviewed by:
Luca Pastorelli, University of Milan, ItalyCopyright: © 2014 Coskun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mehmet Coskun, Department of Gastroenterology, Medical Section 54 O3, Herlev Hospital, University of Copenhagen, Herlev Ringvej 75, Herlev DK-2730, Denmark e-mail:bWVobWV0LmNvc2t1bkByZWdpb25oLmRr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.