 Molly C. McCloskey1
Molly C. McCloskey1 Victor Z. Zhang1,2
Victor Z. Zhang1,2 S. Danial Ahmad1Samuel Walker1Samantha S. Romanick1
S. Danial Ahmad1Samuel Walker1Samantha S. Romanick1 Hani A. Awad1,2,3
Hani A. Awad1,2,3 James L. McGrath1*
James L. McGrath1*
- 1Department of Biomedical Engineering, University of Rochester, Rochester, NY, United States
- 2Center for Musculoskeletal Research, University of Rochester Medical Center, Rochester, NY, United States
- 3Department of Orthopaedics, University of Rochester Medical Center, Rochester, NY, United States
The vascular system plays a critical role in the progression and resolution of inflammation. The contributions of the vascular endothelium to these processes, however, vary with tissue and disease state. Recently, tissue chip models have emerged as promising tools to understand human disease and for the development of personalized medicine approaches. Inclusion of a vascular component within these platforms is critical for properly evaluating most diseases, but many models to date use “generic” endothelial cells, which can preclude the identification of biomedically meaningful pathways and mechanisms. As the knowledge of vascular heterogeneity and immune cell trafficking throughout the body advances, tissue chip models should also advance to incorporate tissue-specific cells where possible. Here, we discuss the known heterogeneity of leukocyte trafficking in vascular beds of some commonly modeled tissues. We comment on the availability of different tissue-specific cell sources for endothelial cells and pericytes, with a focus on stem cell sources for the full realization of personalized medicine. We discuss sources available for the immune cells needed to model inflammatory processes and the findings of tissue chip models that have used the cells to studying transmigration.
Introduction
Infection triggers a series of events within a tissue, including tissue-resident immune cell polarization, endothelial activation, and immune cell recruitment. The endothelium plays a central role in this response by modulating blood flow, initiating the coagulation cascade, and assisting in immune cell transendothelial migration (TEM) from the blood into the infected tissue. Although this cascade is largely conserved, endothelial cells (ECs), and their underlying support cells, throughout the body have nuanced differences that enable them to best serve and protect the underlying tissue from disease. During the past few decades, great progress has been made in understanding this tissue-specificity of the vasculature, and within just the last few years several reviews have been published to help researchers apply this knowledge to in vitro model systems (1–6). In this review, we will briefly cover the most up-to-date knowledge of vascular heterogeneity of several key organs and discuss how we can reasonably apply this knowledge to human in vitro models in the context of immune cell migration. We will highlight key cell types that comprise the vasculature and immune system, covering possible cell sources, including stem cell differentiation protocols. As TEM occurs mostly within microvasculature, this will be our focus of discussion for EC heterogeneity and organ models.
The cardiovascular system is the earliest system to arise during embryonic development. As other organ systems form, they require oxygen and nutrients supplied by this early vascular network (7, 8). The process of de novo vessel formation, termed vasculogenesis, is initiated by formation of the vascular plexus from vascular progenitor cells that are derived from the mesoderm, known as angioblasts. Following EphrinB2/EphB4-mediated cell repulsion, angioblasts separate into arterial and venous territories and further differentiate into arterial and venous phenotypes (9, 10). This differentiation was initially thought to be flow driven, but several studies have conclusively demonstrated that these phenotypes arise prior to the heartbeat (9). Rather, nerve-derived signals promote arterial differentiation and alignment of blood vessels, and the cells are then further matured by exposure to higher blood pressure and flow. One key mediator in arterial vs. venous specification is vascular endothelial growth factor (VEGF). High levels of VEGF contribute to arterial specification, and lower levels to venous specification (11). From these initial blood vessels, the process of angiogenesis begins, in which new blood vessels branch from the existing ones. The sprouting vessels find each other, forming capillary networks which are reiteratively formed, remodeled, and pruned. For more detail on vasculogenesis and angiogenesis, we recommend a set of excellent reviews (9, 10, 12). Importantly, as the vasculature develops and matures, it recruits a variety of support cells, including pericytes (PC) and smooth muscle cells (SMC), for stabilization. Finally, as organs and tissues develop, the endothelium specializes itself to best serve the organ it vascularizes. Some EC specificity is mitotically stable, whereas other phenotypes arise from outside cues and are lost upon removal from the microenvironment (13–17).
While the mature vasculature was initially thought to serve as a passive barrier whose main function was to supply oxygen and nutrients to tissues, a host of studies over the last several decades have elucidated multiple roles of a very active endothelium. These roles are discussed in several excellent book chapters and review articles (7, 18–21). In healthy conditions the vasculature is largely quiescent; however, research suggests ECs play a role in controlling immune surveillance even in a quiescent state (22–24). Further, during inflammation ECs become activated, modulating blood flow and vascular permeability, creating new vasculature through angiogenesis, and promoting leukocyte TEM, primarily at post-capillary venules. To assist in TEM, ECs upregulate expression of leukocyte adhesion molecules (LAMs), such as selectins and cell adhesion molecules (CAMs) that interact with complimentary molecules on circulating immune cells to slow them down and guide entry into the tissue [reviewed in (25, 26)]. Generally, selectins enable rolling of immune cells across the endothelium to slow them down, and CAMs are used for firm arrest and then extravasation of the immune cell across the EC layer, with aid for extravasation from adhesion molecules such as platelet endothelial cell adhesion molecule-1 (PECAM-1). However, the expression of these molecules by ECs varies between tissues, and the mechanisms of migration are tissue-dependent (Figure 1) (18, 19, 27, 28). While EC heterogeneity is well-acknowledged, only recently have tissue-specific characteristics and functions been studied in detail, and only in a few organ systems, such as the brain, lung, kidney, and liver. Since immune cell interactions with endothelium depend on disease states and localization, and adhesive molecules serve as potential therapeutic targets, incorporating tissue-specific ECs in in vitro models of inflammation is important for the models to predict effective treatment strategies (29).
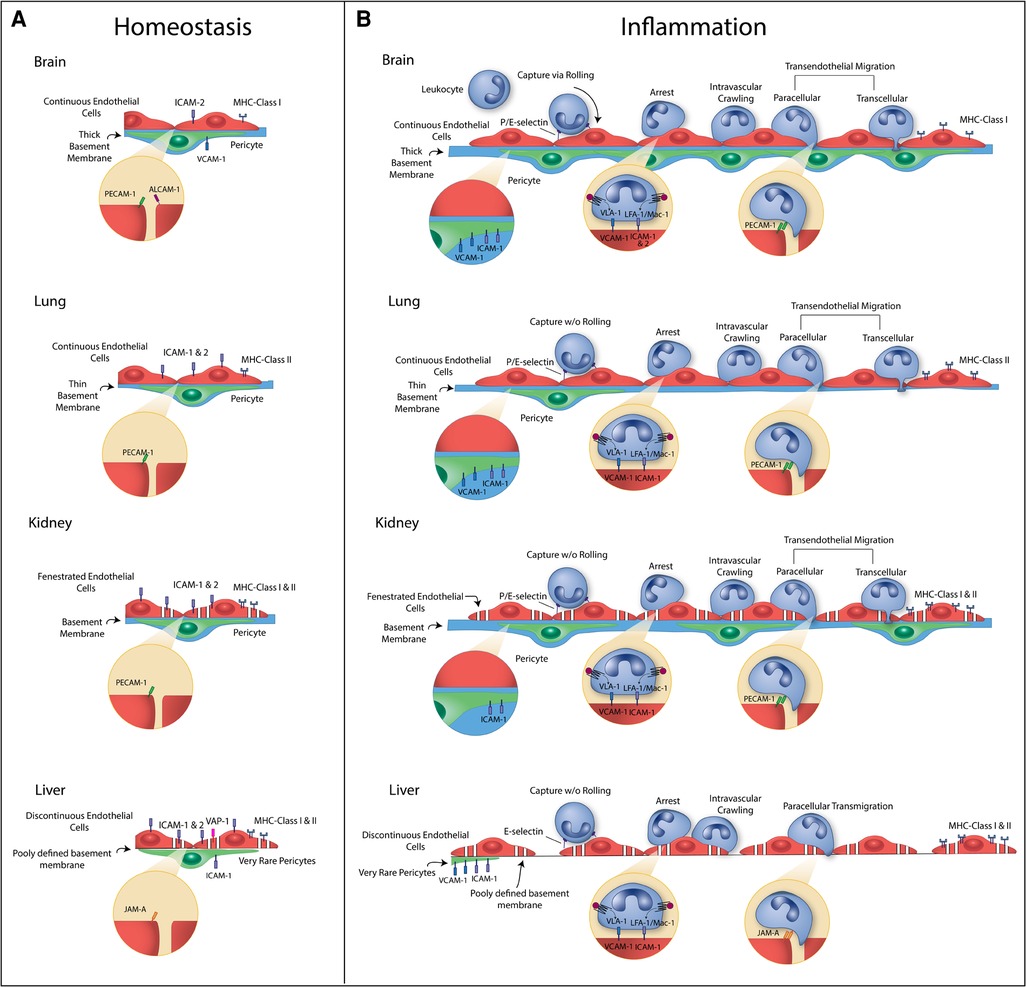
Figure 1. Summary of vascular heterogeneity during homeostasis (A) and inflammation (B). Heterogeneity illustrated includes endothelial cell (EC) type, EC to pericyte (PC) ratios, basement membrane (BM) characteristics, and leukocyte adhesion molecule (LAM) and major histocompatibility complex (MHC) expression. Brain: Brain microvessels have continuous ECs with a high EC : PC ratio and thick basement membrane. During homeostasis, there is very low expression of LAMs and MHC molecules. E-selectin and P-selectin are upregulated during inflammation and contribute to leukocyte rolling. ICAM-1, ICAM-2, and VCAM-1 are upregulated and contribute to leukocyte arrest and crawling. PECAM-1 at cell junctions assists in leukocyte transmigration (TEM), and ICAM-1 and VCAM-1 are upregulated on PCs to guide leukocytes through the BM into the tissue. MHC Class I molecules can be upregulated, which signal to T cells. Lung: Lung ECs at the air-blood barrier are continuous. There is a relatively low EC:PC ratio and thin basement membrane. During homeostasis, there is low expression of LAMs and MHC molecules. E-selectin and P-selectin can be upregulated during inflammation but do not contribute leukocyte rolling. ICAM-1 is upregulated and contributes to leukocyte sequestration and arrest. PECAM-1 at cell junctions assists in leukocyte TEM, and ICAM-1 and VCAM-1 are upregulated on PCs. MHC Class II molecules can be upregulated, which signal to T cells. Kidney: Kidney glomerulus capillaries have fenestrated ECs with a moderate EC:PC ratio and thick basement membrane. During homeostasis, there is constitutive expression of ICAM-1 and ICAM-2 and low expression of MHC molecules. E-selectin can be upregulated during inflammation, but leukocyte rolling is debated in the glomerulus. Glomerular ECs rely on platelets for P-selectin. ICAM-1 and VCAM-1 are upregulated and contribute to leukocyte arrest and crawling. PECAM-1 at cell junctions assists in leukocyte TEM, and ICAM-1 is upregulated on PCs to guide leukocytes through the BM into the tissue. MHC Class I and II molecules can be upregulated. Liver: Liver sinusoidal capillaries have discontinuous ECs with a low EC:PC ratio and poorly defined basement membrane. During homeostasis, there is expression of ICAM-1, ICAM-2, VAP-1 and MHC molecules. E-selectin can be upregulated in a subset of ECs during inflammation but do not contribute leukocyte rolling. ICAM-1, VCAM-1, and VAP-1 are upregulated and contribute to leukocyte arrest and crawling. JAM-A at cell junctions assists in leukocyte TEM, and ICAM-1 and VCAM-1 are upregulated on PCs. MHC Class II molecules can be upregulated.
As organ-specific functions of the vasculature develop from both stable epigenetic programming and environmental signals, careful thought must be given both to cell sources and the microenvironment these cells are cultured in when modeling a given tissue system. The development and application of tissue chip technology (a.k.a. “microphysiological systems” or “organ-on-chips”) provides researchers the opportunity to more faithfully recapitulate specific microenvironments for in vitro models. They do so by recreating both the structure and function of an organ or organ unit, often incorporating multiple key cell types and mechanical stimuli. They enable a reductionist approach to probe at disease mechanisms or make more accurate predictions of drug efficacy or toxicity prior to human clinical trials. By using patient-derived cells, tissue chips have the potential for personalized medicine. Several foundational studies have illustrated the ability of these models to accurately predict drug toxicity and drug and vaccine efficacy (30). More recently acknowledged has been the importance of vascularizing these tissue chip models. Several methods of vascularization have been developed, and we refer readers to these reviews for details on these techniques (3, 6). Further, different vascularization strategies and tissue chip configurations have been used to model a variety of critical organs (Figure 2). Many models thus far have relied heavily on the readily available and easy-to-culture human umbilical vein EC (HUVEC) line. However, given the importance of the endothelium in disease and the heterogeneity of endothelial cells and their inflammatory responses, tissue chip models should start to incorporate tissue specific ECs, as well as critical support cells, when possible.
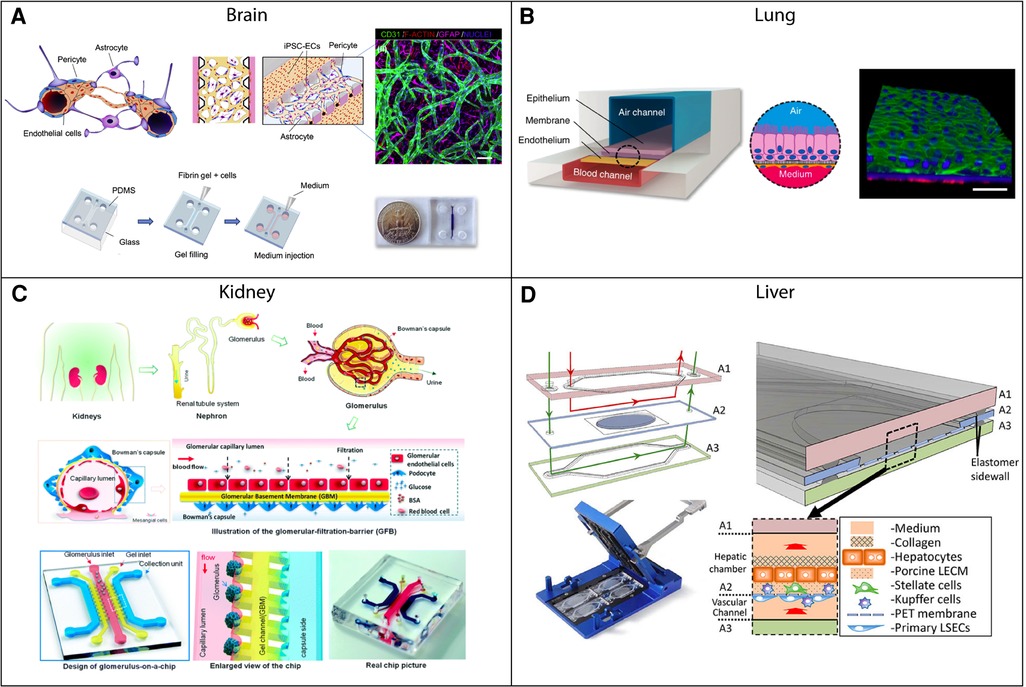
Figure 2. Examples of vascularized tissue chips for different organ systems. (A) Tissue chip example of the blood-brain barrier (BBB). The 3D-BBB contains a microvascular network that forms through a fibrin gel via vasculogenesis. Adapted from (31) with permission; copyright Elsevier Science & Technology Journals. (B) Small airway-on-a-chip example. The tissue chip contains blood and air channels separated by a porous membrane. Pulmonary microvascular endothelial cells line the blood channel and are exposed to fluid flow. Adapted from (32) with permission; Copyright © 2015, Nature Publishing Group, a division of Macmillan Publishers Limited (CC-BY). (C) Example of a glomerulus-on-a-chip. The chip contains capillary, gel, and collection channels, with glomerular endothelial cells cultured in the capillary channel and exposed to fluid flow. Adapted from (33) with permission from Royal Society of Chemistry. (D) Tissue chip example of the liver. Liver sinusoidal endothelial cells and Kupffer cells are cultured in a vascular chamber, which is separated from a hepatic chamber by a porous membrane. Both chambers have fluid flow rates that are representative of the flow experienced in vivo. Adapted from (34) with permission from the Royal Society of Chemistry.
Endothelial cells
Background
Endothelial cells are a heterogenous population that demonstrate high levels of organ-specific structure and function (summarized in Figure 1, Table 1), emerging from both epigenetic specification and environmental influences. EC heterogeneity was first recognized in the 1980s as a fundamental characteristic of the endothelium, appearing in the vasculature of the oldest extant vertebrate, the hagfish (35, 36). Differences between ECs include cell shape, junctional protein expression, endocytic and transcytosis levels and pathways, and glycocalyx composition. Further, ECs can be classified broadly into three categories: (1) fenestrated, (2) discontinuous, and (3) continuous (19, 37). Fenestrated cells are found in organs where filtration or secretion are primary functions of the vasculature, such as the kidney and liver. Similar to fenestrated endothelium, discontinuous endothelial cells have fenestrations that are larger in diameter and lack a diaphragm, appearing as gaps in the cell. Discontinuous ECs also have poorly developed basement membranes. The sinusoidal vascular beds of the liver are one example that contains discontinuous cells. In contrast, in the brain, brain microvascular ECs (BMECs) serve a critical role in protecting the tissue from fluctuations in blood composition and, as such, are continuous. Other continuous endothelial cells are found in the skin, lung, heart and muscle. Continuous ECs are often adhered to each other via tight junctions, in addition to adherens junctions, to limit passage of small molecules.
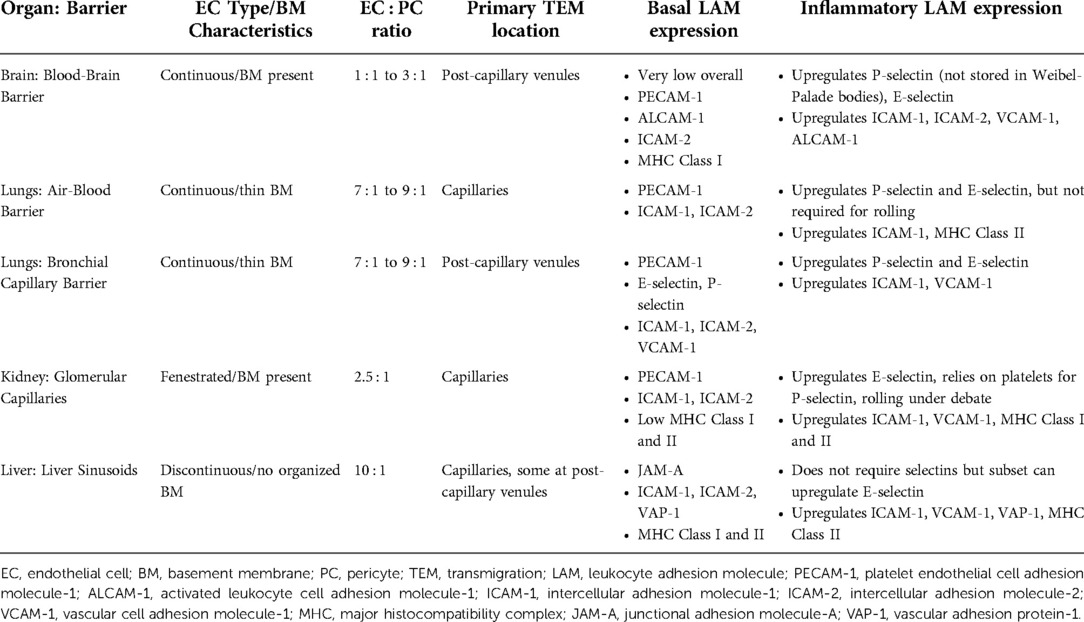
Table 1. Endothelial Cell characteristics at sites of leukocyte transmigration.
While the mechanisms driving vascular heterogeneity are still not fully understood, it is clear that some tissue specificity emerges from cell-intrinsic developmental pathways that are epigenetically regulated (12–14, 36). Certain genes persist for several passages of in vitro cell culture and are thus, mitotically stable (13, 18, 38). For example, in a study of lung EC cultures, the majority of studied protein persisted in culture, while several proteins expressed in vivo were lost (17). Microarray profiling and bulk and single cell RNA sequencing (scRNAseq) have identified transcription factors that regulate these tissue specific expression patterns. GATA Binding Protein 4 (GATA4) upregulates sinusoidal endothelial genes in hepatic endothelial cells (14, 39, 40), and mesenchyme homeobox 2 (MEOX2) and transcription factor 15 (TCF15) are critical regulators in heart ECs to mediate fatty acid uptake (41). Interestingly, new evidence from RiboTag transgenic mouse studies illustrate that tissue-specific ECs also express markers thought to be specific to other cell types within that tissue (27). For example, the authors demonstrated that brain ECs express canonical neuronal markers, and heart endothelium expresses markers thought to be restricted to cardiomyocytes. To exclude non-EC contamination, the authors crosschecked their data with two scRNAseq datasets and confirmed these findings.
Environmental cues clearly contribute to the unique phenotypes of endothelial cells along the regions of vasculature and in different tissues (15, 16). This is most clearly demonstrated in in vitro studies, where removal of ECs from their in vivo environment results in dedifferentiation or phenotypic drift of the primary cells. For example, when BMECs are isolated from the brain and cultured in vitro, they form more permeable barriers than seen in vivo. Addition of support cells, such as pericytes and/or astrocytes, or conditioned media from these cells, to the microenvironment can help recover some of the lost functionality (42). Other stimuli include contact-dependent cell communication and mechanical stimuli such as shear stress, cyclic strain, matrix stiffness, and curvature (6, 43). The mechanical stresses an EC encounters will differ depending on tissue location (12, 43). Further, ECs in certain tissues can be exposed to extreme environments, such as high oxygen across the blood-air barrier in the lungs and hypoxic conditions in the kidney (12, 36). Therefore, environmental factors driving particular EC phenotypes include paracrine signals, interactions with matrix, and mechanical factors. Any in vitro environment that fails to reconstitute these factors is likely to experience shortcomings as a tissue-specific model, however, tissue chip platforms help mitigate some of these shortcomings. For example, the dynamic in vitro blood–brain barrier (DIV-BBB) is designed in a tube structure to add the curvature to the ECs and fluid flow (44). Other vascularized chip models include post-capillary venule expansions (45), many incorporate physiologically-relevant hydrogels [reviewed in (46)], and most have fluid flow capabilities to mimic the shear stresses from blood flow experienced by endothelial cells (31, 34, 44, 47–54). Addition of these environmental cues improves endothelial cell permeability (55), viability (56), and cytoskeletal architecture (57). Incorporation of extracellular matrix (ECM) in tissue chip models helps enable cell attachment, guide cell function, and establish EC polarization (46). Further, tissue chips incorporating hydrogels or other ECM matrices (e.g., fibrin matrices) can be used for development of perfusable, physiological vascular networks via vasculogenesis and/or angiogenesis (31, 47, 48). Finally, the consequences of modulating the microenvironment can be thoroughly evaluated on tissue chip platforms. For example, disturbed flow experienced by ECs in atherosclerotic lesions has been investigated on a tissue chip platform. Wang and colleagues discovered the critical role of Yes-associated protein (YAP)/transcriptional coactivator with PDZ-binding motif (TAZ) activation in promoting the proinflammatory EC phenotype seen in these lesions (58). Therefore, tissue chips enable researchers to evaluate the role of environmental cues on EC function in conditions of health and disease.
“Generic” endothelial cells
The first isolated human endothelial cells came from umbilical veins (HUVECs). HUVECs are easy to access and culture, express many key EC markers, junctional proteins and inflammatory proteins, and have served as a robust line for many scientific discoveries (6). Because of these conveniences, HUVECs are often used as a “generic” EC in models of various tissues (Table 2). As one example, HUVECs were incorporated into a liver biochip to identify a potential new biomarker for sepsis, CAAP48, which is found in higher concentrations in sepsis patients and appears to contribute to liver dysfunction during sepsis (59). In this study, HUVECs were cultured with differentiated HepaRG hepatocytes in MOTiF biochips. While immune cell migration was not directly evaluated, Blaurock-Möller and colleagues did measure release of soluble intercellular adhesion molecule-1 (sICAM-1) and vascular cell adhesion molecule-1 (sVCAM-1) after treatment with CAAP48. Both are shed during sepsis and contribute to leukocyte migration. The study found that CAAP48 led to increases in both sICAM-1 and sVCAM-1 and may contribute to the uncontrolled inflammatory response during sepsis. Our lab has also utilized HUVECs to create a microvascular mimetic tissue chip model, termed (µSiM-MVM). HUVECs were cultured on ultrathin, nanoporous, optically-transparent membranes to monitor neutrophil transmigration. One study found that neutrophil TEM may cause small, local increases in EC layer permeability (60), while the other probed at directional stimulation and its effects on TEM (61). In the latter study, ICAM-1 reorganization was visualized on the EC surface following tissue-side, or abluminal, stimulation, likely in order to capture neutrophils and guide extravasation to the underlying tissue.

Table 2. Endothelial cell sources for tissue models.
Despite the conveniences of HUVECs, an obvious limitation of this cell line is that they are derived from the macrovasculature, which have mechanical, structural, and functional differences compared to the microvasculature (38). In a comparison of LAM expression kinetics between HUVECs, glomerular endothelial cells, and dermal microvascular endothelial cells, each cell type had significant differences in response to tumor necrosis factor-α (TNFα) and interferon gamma (IFNγ) stimulation (Figure 3) (28). Another recent study from our lab demonstrated differential barrier responses of HUVECs compared to human pulmonary microvascular endothelial cells in response to barrier modulating molecule, sphingosine-1-phosphate (Figure 3) (68). Both studies highlight the limitations of this model for representing tissue-specific vascular barriers. Further, immune cell trafficking and most tissue-specific heterogeneity occurs at the level of the microvasculature (22, 25, 26). A common alternative to HUVECs are commercially-available human microvascular endothelial cells (hMVEC) (Table 2). hMVECs can come from several sources but are commonly derived from the dermis. This cell line was used in a tissue chip model designed to study mechanisms of neutrophil transmigration (62). By creating chemotactic gradients, the group was able to model in vivo-like neutrophil TEM across the hMVEC layer and through a collagen gel to the “wound” chamber. They discovered that in the absence of an endothelial layer, neutrophils did not migrate as far, indicating EC-neutrophil interactions assist in neutrophil migration to the wound site. While there are likely benefits to using hMVEC over HUVEC to model the microvasculature, neither cell type fully recapitulates the tissue-specificity desired for tissue chip models.
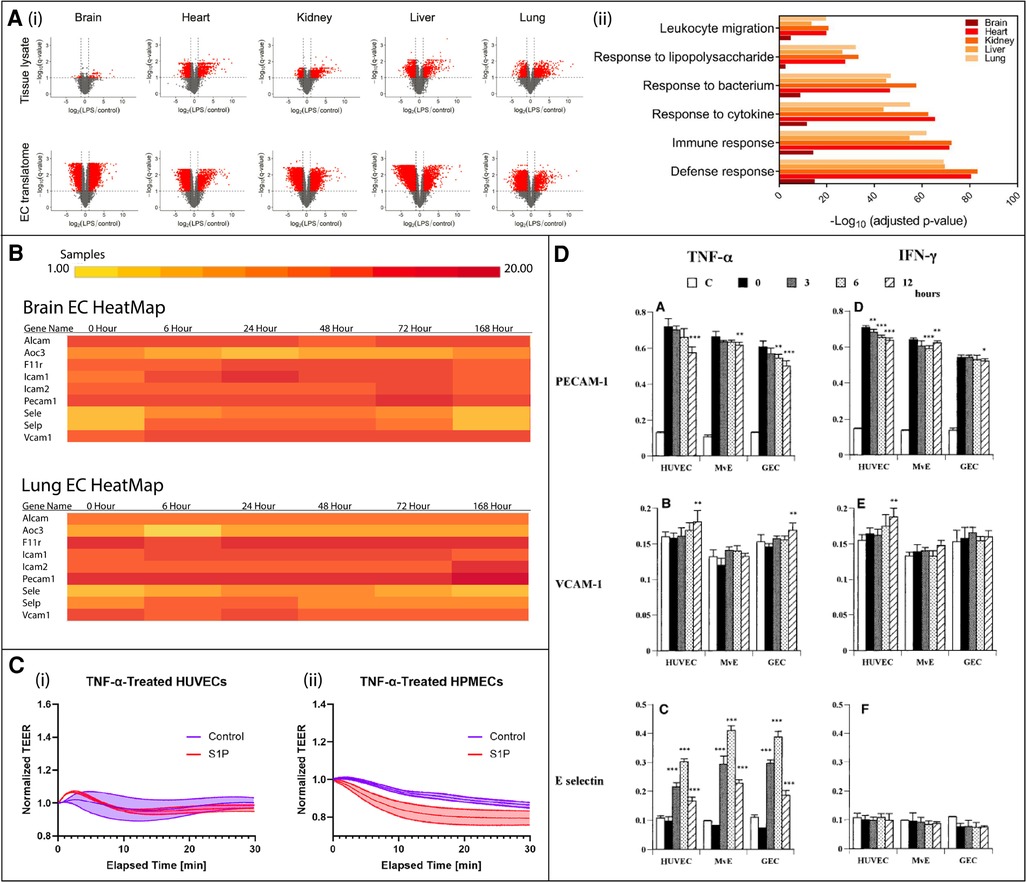
Figure 3. In vivo murine and in vitro human studies illustrating endothelial cell (EC) heterogeneity in response to inflammatory stimuli. (A) In vivo murine EC-translating ribosome affinity purification (TRAP) study highlights tissue heterogeneity as demonstrated by variable upregulated and downregulated transcriptomes and translatomes following LPS exposure via volcano plots (i) and GO analysis (ii). Leukocyte migration responses vary by tissue. Adapted from (15), used under Creative Commons PNAS license. (B) In vivo RiboTag transgenic mouse study shows differentially expressed genes in brain and lung ECs in response to LPS. The time course response varies between tissues. Created at http://www.rehmanlab.org/ribo from open access data generated by (27), under Creative Commons Attribution License (CC BY 4.0). (C) In vitro comparison of the response of two human cell lines, human umbilical vein ECs (HUVECs) and human pulmonary microvascular ECs (HPMECs), to TNFα stimulation with or without EC permeability regulator sphingosine-1-phosphate (S1P). While HUVECs are desensitized to TNFα following S1P exposure (i), HPMEC barriers are further disrupted (ii). Adapted from (68), used under Creative Commons CC BY-NC-ND license. (D) In vitro comparison of the response of three human cell lines, HUVECs, glomerular endothelial cells (GEC), and dermal microvascular endothelial cells (MvE), to TNFα and IFNγ stimulation. The kinetics of the expression of leukocyte adhesion molecules, PECAM-1, VCAM-1, and E-selectin, significantly differs between cell sources. Adapted from (28) with permission from Elsevier; Copyright © 2001 Academic Press.
Another alternative “generic” EC source is blood outgrowth ECs (BOECs) (Table 2). BOECs are endothelial progenitors isolated from the blood using density gradient isolation and can be matured in culture to create patient-specific EC models (69–72). Their gene expression is similar to HUVECs, and they are responsive to shear stress and cytokine stimulation (73). Recent studies have incorporated BOECs into tissue chip platforms. For example, Mathur and colleagues developed an arteriole “vessel-chip” using BOECs, which upregulated ICAM-1 after TNFα stimulation (74). The vessel-chips were also supportive of platelet adhesion and increased small molecule permeability upon TNFα treatment. In another study led by Mathur, BOECs were isolated from patients with sickle cell disease (SCD) and cultured in microfluidic vessel-chips (49). Both SCD patients exhibited upregulation of E- and P-selectin, as well as more moderate upregulation of ICAM-1 and VCAM-1. While neither of these studies tested immune cell migration, given BOEC's expression of cell adhesion molecules, these studies are feasible. While BOECs are a promising, patient-specific EC source, there has been variability in the success rate of BOEC cultures, with some individuals appearing unable to support BOEC isolation and culture (70, 75). Future studies would be needed to optimize this technique.
Another patient-specific “generic” EC source is human induced pluripotent stem cell (hiPSC)-derived ECs. Several protocols have been developed that produce CD31+, CD34+, and/or von Willebrand factor (vWF)+ endothelial cells, which have been described in prior reviews (Table 2) (76–78). Most protocols transition the cells initially through a mesodermal lineage using an embryoid body approach, 2D approach, co-culture or 3D culture techniques. Following these steps, a variety of methods have been developed to drive cells towards an endothelial phenotype. While many EC differentiation strategies are limited by low yield and require a purification step via magnetic-activated cell sorting (MACS), more efficient methods have recently been developed using bone morphogenetic protein 4 (BMP4), transforming growth factor-β (TGF-β), or transcription factor approaches (76, 77, 79). Several of the methods for generating hiPSC-ECs have been evaluated for their inflammatory response phenotypes. For example, Adams and colleagues’ embryoid body technique produces cells which express E-selectin, ICAM-1, and VCAM-1 in a time-dependent manner when stimulated with proinflammatory cytokines, TNFα and interleukin-1β (IL-1β), and bacterial antigen, lipopolysaccharide (LPS) (80). Further, neutrophil and T cell transmigration was comparable to HUVECs. Another embryoid body approach to generate hiPSC-ECs cultured these cells in a microfluidic device and found increased monocyte adhesion with TNFα stimulation (50).
Despite recent advances, hiPSC-derived ECs appear to be developmentally immature compared to primary ECs (81). In a side-by-side comparison of CD31+ and CD34+ hiPSC-ECs generated using Orlova et al.'s method (82), HUVECs, and human dermal blood ECs (HDMECs), the inflammatory responses of each cell differed significantly (81). While all cells upregulated some adhesion molecules, such as ICAM-1, in response to TNFα, hiPSC-derived ECs lacked upregulation of VCAM-1. hiPSC-derived ECs also promoted less immune cell adhesion compared to HUVECs. Interestingly, CD31+ hiPSC-derived ECs formed tighter barriers and had a more robust inflammatory responses compared to CD34+ hiPSC-derived ECs. It would be beneficial to have similar comparisons for other stem-cell derived ECs, as understanding their inflammatory phenotypes could help researchers decide which method to employ for particular models and/or hypotheses. In addition, while baseline characterization of inflammatory responses is critical, technical challenges have hindered validation of the inflammatory response in the context of specific tissue microenvironments. It is not unreasonable to think that developmentally immature ECs may progress towards a more mature phenotype when cultured within a tissue chip environment containing critical support cells and environmental signals. In fact, a recent BMEC-like cell differentiation protocol which differentiates hiPSCs into endothelial progenitor cells and sorts for CD31+ cells before specifying a brain microvascular EC-like phenotype, also did not see upregulation of VCAM-1 on BMEC-like cells grown as a monoculture (83), similar to the results of CD31+ ECs generated via the Orlova method. However, VCAM-1 upregulation was rescued by coculture with smooth muscle-like cells (SMLCs) or SMLC-conditioned medium. Future studies, therefore, could characterize “generic” ECs cultured within tissue chips for further differentiation and mature inflammatory phenotypes.
Brain
The capillary network within the brain is the tightest in the body. The blood-brain barrier contains continuous endothelial cells zipped together by tight junction molecules, in contact with pericytes embedded within a basement membrane and nearby astrocyte endfeet. These cells have very low rates of transcytosis and upregulate expression of several transporters, such as glucose transporter GLUT1 and ABC transporter MDR1, to supply the underlying brain tissue with necessary nutrients (84). RiboTagEC murine data indicates that a large portion of enriched genes in BMECs are involved in transport, including ion, acid, and neurotransmitter transport (27). Breakdown of this barrier is a common feature of several cognitive dysfunctions, including Alzheimer's disease (AD), Parkinson's Disease, and Multiple Sclerosis (MS) (85–88). Traditionally thought of as “immune privileged” the blood-brain barrier does allow immune cell migration during inflammation, as well as low level entry of T cells in healthy conditions for surveillance purposes (89–91). This process is aided by constitutive expression major histocompatibility complex (MHC) class I molecules by BMECs. However, expression of other adhesion molecules during homeostasis is lower than peripheral ECs (92). The brain endothelium responds to inflammatory stimuli with upregulation of LAMs, allowing immune cells to cross the BBB using the traditional mechanisms of rolling, arrest, and TEM (27, 89, 93). P-selectin, however, is not stored in Weibel-Palade bodies on brain endothelial cells, leading to delayed surface expression and slowed recruitment of leukocytes during inflammation (94). Interestingly, different regions of the brain express varying levels of adhesion molecules, and thus, immune cell migration differs between brain regions (90). Further separating BMEC inflammatory responses from other tissue-specific ECs, brain endothelium appears to upregulate, rather than downregulate, more of its ribosome associated transcripts in response to LPS (Figure 3) (15). The consequences of this increase in actively translated mRNAs, however, have not been studied further.
Given that the BBB is implicated in numerous diseases and is a critical target for drug delivery, many tissue chip models of this barrier have been developed (65, 95–99). Because the unique properties of BMECs are long recognized and well-studied, these models rarely settle for generic endothelial cells and the topic of BMEC source has been reviewed in depth (100–102). The most common cells lines incorporated into in vitro tissue chip models have been human primary cells and stem cell-derived brain ECs (Table 2). Human primary brain ECs are difficult to obtain, and immortalized lines, such as hCMEC/D3, are notorious for dedifferentiation in culture and weaker barrier properties compared to in vivo barriers. However, incorporation of primary cells into tissue chips systems, in particularly under flow conditions and in coculture with pericytes and/or astrocytes, has been shown to improve these phenotypes (42, 103). Commercially available primary human brain ECs were incorporated into the DIV-BBB within hollow fibers containing a mixture of 0.2 to 0.5 µm pores and ≈2 to 4 µm pores to allow for immune cell migration across chambers (44). As the fibers were designed to mimic the diameter of the microvasculature, the platform serves as a potential model to directly study immune cell transmigration mechanisms. The group demonstrated that monocytes crossed the barrier during flow cessation followed by reperfusion, and the barrier experienced a biphasic opening, as seen in prior in vivo studies. In another model, primary human BMECs were cultured in a blood-brain barrier-on-a-chip (B3C) in vascular channels, with 3 µm pores for leukocyte migration (65). The set up enabled real-time analysis of neutrophil migration, discovering that protein kinase C-delta inhibits interaction of neutrophils with endothelial cells, reducing both adhesion and migration. Therefore, primary human brain ECs provide a facile alternative for those interested in studying immune cell migration mechanisms despite their weak barrier properties compared to their in vivo counterparts.
One of the first human alternatives to primary cells were umbilical cord blood (UBC)-derived CD34+ ECs, which obtain BBB-like properties when cocultured with pericytes (104, 105). Mossu and colleagues utilized these cells in a microfluidic cerebrovascular barrier model (µSiM-CVB) to study T-cell TEM under flow (63). Upon confirmation that the cells expressed ICAM-1, ICAM-2, and VCAM-1 upon TNFα stimulation, the tissue chip platform was used to image all steps of the TEM cascade using live cell imaging. However, this cell source is not sufficiently BBB-specific, and thus the field has turned its efforts to developing hiPSC-based BMECs.
Differentiation protocols for BMECs have undergone several iterations, including an early protocol that produced hybrid endothelial/epithelial cell phenotypes (102, 106–108). Despite these challenges, stem cells continue to be the cell of choice for many BBB tissue chips (Table 2). BMEC differentiation generally starts similar to other EC differentiation protocols, transitioning through a mesodermal state. The first hiPSC-based protocol developed co-differentiated endothelial cells and neurons, following the embryonic developmental process (109). Future iterations transitioned from undefined to defined medium and introduced retinoic acid to improve barrier properties (110). While the cells were well characterized for their brain-like phenotypes in terms of transporters and tight barrier properties, it was soon discovered that they lacked several key EC characteristics, including expression of selectins, ICAM-2 and VCAM-1, as well as expressing several epithelial cell markers (83, 102). Recently, a few responses have emerged. Lu and colleagues produced phenotypic endothelial cells (rECs) by overexpression of EC-specific transcription factors, however, the cells have not yet been cultured to produce a brain-specific phenotype. They are confirmed to increase E-selectin expression upon TNFα stimulation (111). Another novel protocol was produced by Nishihara and colleagues, which uses an endothelial progenitor cell differentiation method before specifying a brain-like phenotype through an extended endothelial cell culture method (EECM) with brain-based supplement B27 and human fibroblast growth factor 2 (83). Cells produced with this method (known as EECM-BMEC-like cells) express key inflammatory molecules. Excitingly, EECM-BMEC-like cells derived from MS patients demonstrated increased expression of both ICAM-1 and VCAM-1 at baseline and upon proinflammatory stimulation compared to healthy controls. The model was further confirmed to mimic other key MS phenotypes, including disrupted tight junctions and increased interactions with immune cells, and opens the door for testing new drugs and therapeutic approaches (112). Further, our group recently incorporated EECM-BMEC-like cells into a tissue chip platform known as the modular-µSiM (m-µSiM) (66). m-µSiM culture was confirmed to mimic the baseline and inflammatory phenotypes of EECM-BMEC-like cells cultured in traditional culture plates and Transwell systems, and supported studies of both neutrophil and T cell TEM. While the new protocols do not achieve the barrier tightness of some previous BMEC differentiation methods, they represent valuable alternatives for groups interested in studying disease mechanisms involving immune cell migration.
Lung
Microvascular endothelial cells of the lung can be broadly classified as pulmonary or bronchial and are another example of continuous ECs. The site of gas exchange occurs at the alveolar-capillary interface, or air-blood barrier, within the pulmonary circulation, where ECs and epithelial cells are separated only by a thin basement membrane (18). Alveolar capillary ECs have several defining factors, separating them from other ECs. For example, the entire population of alveolar capillary ECs express angiotensin I–converting enzyme, in comparison to only 10% of systemic ECs (18). Recent studies have subclassified alveolar capillary ECs into “aerocytes” (aCap) and “general” capillary (gCap) (113). The main function of aerocytes is gas exchange and leukocyte migration, whereas gCaps serve roles in vasomotor tone, capillary homeostasis, and repair. Interestingly, aerocytes lack expression of major constituents of Weibel-Palade bodies specific to ECs, such as vWF, P-selectin, and endothelin 1 (EDN-1). The thinnest of lung capillaries, where gas exchange takes place, are where Weibel-Palade bodies are known to be absent (114, 115). They do, however, express other common EC markers, such as PECAM-1 and CD34 (13, 18).
There are unique aspects of the air-blood barrier that make it a site of high leukocyte transmigration. Transmigration does not appear to involve selectin mediated rolling and occurs in the small pulmonary capillaries, instead of post-capillary venules (18, 116, 117). Neutrophil deformation leads to sequestering in the capillaries, and then LAMs such as ICAM-1 keep the neutrophils in place. In addition, P-selectin can contribute to leukocyte sequestration, and upregulation of P-selectin and E-selectin are critical markers of lung inflammation (29, 118). While lung ECs significantly upregulate genes related to leukocyte adhesion and migration (Figure 3), T cell activation, and regulation of immune system processes in response to LPS, at least in mice, this response is delayed compared to the responses of brain and heart ECs (27). Additionally, Car4-high ECs have recently been identified in mice using single cell RNA sequencing (119). Car4-high ECs express high levels of Car4, CD34, and VEGF receptors, and localization and proliferation is enriched at sites of influenza-induced lung injury in the alveolus. Car4-high ECs are primed to receive VEGFA signals from damaged alveolar type I epithelial cells for repair and regeneration, as it has been suggested that Car4-high ECs may play a role in vasculogenesis. Bronchial capillary ECs, on the other hand, are less well studied and therefore poorly understood. Although less leukocyte TEM occurs in the bronchial compared to pulmonary capillaries, bronchial ECs constitutively express E-selectin and P-selectin (18, 120). This is likely due to constant exposure to antigens. They can upregulates selectins, ICAM-1 and VCAM-1 under inflammation (117). Future studies characterizing these cells will be critical, as they are implicated in several diseases, including chronic obstructive pulmonary disease (COPD) and SARS-CoV-2 (32, 121).
Most of the discussion around cell sources for lung tissue chips has focused on lung epithelial cells, however a few recent reviews have discussed EC sources in in vitro lung models (113, 122, 123). Endothelial cells used in tissue chip models thus far have largely been HUVECs and primary human lung microvascular ECs (HMVEC-L, LMVEC, HLVEC, or HPMEC; Table 2) (32, 51, 67, 123–125). Primary lung ECs are available commercially and often obtained during biopsies. As with most primary human cells, they risk having a diseased phenotype, may be pooled and therefore genetically heterogenous, are slow-growing, and have a limited passages (122–125). In addition, commercial sources are now specifying that the available lung microvascular ECs are a mix of lymphatic and vascular ECs, as it is extremely challenging to distinguish between the two cell types (32). Despite these limitations, they upregulate E-selectin, ICAM-1, and VCAM-1 in response to various proinflammatory stimuli (123). Primary lung microvascular ECs have been used in lung tissue chip models of both alveolar-capillary interface and bronchial capillaries (32, 51, 67). The seminal tissue chip study was a lung-on-chip model, incorporating alveolar epithelial cells with HLVECs. They observed upregulation of ICAM-1 in response to TNFα stimulation, along with secretion of cytokines by HLVECs (51). This tissue chip was later modified to model a human “small airway-on-a-chip” to study asthma and COPD (32). The model contained commercially-available healthy control and COPD donor primary human airway epithelial cells in one chamber and commercial human lung microvascular endothelial cells in the opposite chamber. The epithelial cells were first differentiated into a bronchiolar epithelium, composed of the many cell types found within that layer. The model was confirmed to mimic drug responses in vivo in terms of neutrophil recruitment and LAM expression after stimulation with viral mimic poly(I:C) or LPS. When testing a drug with known limited activity in COPD patients, there was no change in neutrophil adhesion or expression of E-selectin, ICAM-1, or VCAM-1 upon drug administration. However, an experimental drug reduced expression of all three molecules, corresponding to reduced neutrophil adhesion to the endothelial layer. Importantly, this was only observed in the flow conditions of the lung tissue chip, and not in a Transwell™ model, highlighting the importance of mechanical stimuli to EC function. Another bronchiole lung tissue chip by Barkal and colleagues consisting of primary human bronchial epithelial cells, pulmonary fibroblasts, and LMVECs was used to characterize the bronchiole inflammatory response to fungal infection (67). The group analyzed polymorphonuclear leukocyte (PMN) migration across the vasculature and toward the Aspergillus fumigatus (A. fumigatus) hyphae. There was increased PMN transmigration in infected chips compared to controls, and even greater migration in a ΔlaeA mutant model. ΔlaeA mutants are known to be less virulent, likely due to reduced production of molecules used to evade the host immune system. This work indicates that greater PMN recruitment may aid in the clearance ΔlaeA mutants over wildtype A. fumigatus.
Given the success of various lung-on-chip models using primary human microvascular cells, as well as the ability to obtain these samples directly from patient populations, there appears to be little drive to produce stem-cell derived lung ECs. To the best of our knowledge, currently, the only in vitro models that have explored hiPSC-ECs in lungs have been for tissue regeneration purposes (Table 2) (126). Due to the immature cell phenotype, they fell short in several parameters compared to both HUVECs and HMVEC-Ls. Regardless, with the advantages of personalized medicine and the limitations of primary lung microvascular ECs, including lymphatic EC contamination, it may be beneficial for the field to start exploring these options.
Kidney
Filtration within the kidney occurs in the glomerulus, which contains fenestrated endothelial cells, a basement membrane, and podocytes. Like most endothelium, glomerular ECs (GECs, also GEnC) arise from the mesoderm. However, their development occurs mainly through vasculogenesis rather than in combination with angiogenesis (127). GECs produce a robust glycocalyx and have fenestrations which are 60–80 nm in diameter, allowing selective filtration based on size and charge (18). Similar to lung ECs, GECs express normal levels of PECAM-1 and CD34, but lower levels of vWF (13, 18). Barrier function is, in part, controlled by paracrine signaling from podocytes, which wrap their foot processes around the ECs (11). While many tissue chip studies focus on filtration functions of GECs, leukocyte infiltration occurs during glomerulus inflammation and has been implicated as the cause of damage in several diseases, including diabetic nephropathy, lupus nephritis, and sepsis (128–131). Immune cell migration in the kidney is unique, as the glomerulus is a capillary-based structure and lacks post-capillary venules. However, glomerular capillaries, similar to pulmonary capillaries, are supportive of leukocyte TEM (132, 133). Further, unlike most endothelium, which stores P-selectin in Weibel-Palade bodies, GECs do not express significant amounts of P-selectin on their surface. Thus, the adhesion of leukocytes through P-selectin/P-selectin glycoprotein ligand-1 (PSGL-1) interactions in the glomerulus first requires the adhesion of platelets, an often neglected component of tissue chip models (134). GECs also constitutively express ICAM-1, and can further upregulate ICAM-1, along with E-selectin, VCAM-1, and MHC Class I and II proteins during inflammation (28, 29, 135). Despite expression of selectins on GECs, immune cell migration does not always appear to start with rolling in the glomerulus, though this has been debated (29, 136, 137).
Due to challenges in isolating and culturing primary human GECs, in vitro studies have often relied on mouse or rat cells (138). However, recent improvements have led to commercial availability of primary human GECs, as well as protocols for isolation of GECs from patient samples, both of which have been implemented in glomerulus tissue chip models (Table 2) (52, 53, 139). There are even commercial sources of primary human glomerular microvascular endothelial cells which have been functionally assayed for inducible expression of E-selectin (140). As with most primary cells, however, commercial and self-isolated GECs have limited lifespans. One tissue chip model was able to culture primary GECs obtained from kidneys that were “non-suitable for transplantation” - primarily from infant patients whose cause of death were not related to the kidney (141). The cells retained several key properties of GECs, including presence of fenestrations. It is likely, however, that most labs do not have access to these materials. To our knowledge, none of these models have studied immune cell trafficking into the glomerulus.
To date, there are no GEC-specific differentiation protocols, and hiPSC-derived EC incorporation into glomerulus tissue chips is limited. However, Roye and colleagues developed a personalized glomerulus tissue chip using stem cell-derived epithelium and vascular endothelium (viEC) (Table 2) (54). The group utilized a protocol by Atchison et al. that follows the same initial transition through the mesoderm, and then specifies into viEC through VEGF-A and a cyclic adenosine monophosphate (cAMP) booster (64). Cells were then sorted for CD31+/VE-cadherin+ expression. While these cells are not differentiated into a tissue-specific phenotype, co-culture with hiPSC-derived podocytes demonstrated mature functions of the glomerulus, along with a disease phenotype when treated with a nephrotoxic drug. Future studies will need to analyze the immune response of the ECs in this culture. As the first glomerulus-on-chip was developed under a decade ago, it is not surprising that studies thus far have focused on filtration properties. Hopefully as the field develops, immune cell infiltration will be incorporated into these disease models to gather a more complete picture of disease progression and identification of therapeutic targets.
Liver
The capillaries of the liver are contained within its sinusoids. Blood arrives here from two vascular systems, the arterial and portal vasculature (18). The endothelial cells in the sinusoids are discontinuous, with their primary function being filtration. They lack an organized basement membrane and function in partner with tissue resident macrophages, called Kupffer cells, to remove wastes, ECs doing so mainly through endocytosis (11, 18). Liver sinusoidal endothelial cells (LSECs) represent a heterogenous population, with distinct phenotypes in different zones of the liver, as each zone is its own unique microenvironment (142). As the liver is a key immune organ, LSECs can present antigens to both CD4+ and CD8+ T cells via MHC Class I and II molecules [reviewed in (143, 144)]. Interactions between LSECs and T cells can decrease T cell activation, preventing autoimmune dysfunction throughout the body. Traditional transmigration is not always necessary for immune cell surveillance. In some instances, T cells can pass through the LSEC gaps and directly contact hepatocytes, or contact hepatocytes without crossing the LSEC barrier by contacting the hepatocyte protrusions through the fenestrations (145). Leukocytes are recruited and infiltrate the liver sinusoids in several disease condition, including hepatitis and sepsis (29). While the liver has post-capillary venules, leukocyte migration primarily occurs in the sinusoidal capillaries and does not require selectins (29, 146). However, expression of E-selectin by a small percentage of LSECs was detected in Kupffer cell (resident liver macrophages)-depleted mice and serves to recruit monocytes that replenish the Kupffer cell population (147). Another unique aspect of liver sinusoidal TEM is that junctional adhesion molecule-A (JAM-A), rather than PECAM-1, is necessary for transmigration (148, 149). LSECs also express baseline levels of ICAM-1 and vascular adhesion protein-1 (VAP-1). These, along with VCAM-1 and MHC Class II molecules, are upregulated in response to proinflammatory stimulation (146, 150, 151). Finally, LSECs can lose their fenestrations upon tissue damage (152). The complex and unique features of the liver sinusoids make in vitro modeling for immune cell TEM particularly challenging.
Similar to brain endothelial cells, LSECs rapidly dedifferentiate in culture, which can be partially salvaged by coculture with relevant cell types, most often hepatocytes (152, 153). Still, they are difficult to cryopreserve, and some commercially available sinusoidal ECs are derived from large vessels rather than the microvasculature (154). Commonly used alternatives have been HUVECs and human foreskin endothelium cells (HMEC-1), both of which improve hepatocyte function (155). However, neither of these cell types are discontinuous, and they lack liver-specific receptors. In fact, even immortalized LSEC line, TMNK-1, lacks fenestrations and have a chronically activated phenotype (Table 2) (155). One commercial source of LSECs is a mix of human liver derived ECs (HLECs) and reports containing up to 80% LSECs (Table 2). This source was used to develop a “Continuously Zonated and Vascularized Human Liver Acinus Microphysiological System” (vLAMPS) (34). The group designed their chip system to mimic the structure of the liver sinusoid, containing hepatocytes, Kupffer cells, and stellate cells in a hepatic chamber and LSECs in the vascular channel. The liver derived ECs formed a continuous layer with fenestrations averaging 170 nm pore diameters and demonstrated upregulation of ICAM-1 under inflammatory stimuli [1 μg/ml LPS + 15 nM epidermal growth factor (EGF) + 10 ng/ml TGF-β]. Further, fluorescently labelled PMNs could be tracked migrating across the LSEC layer and into the hepatic chamber.
Stem cell-derived LSEC protocols are still in their infancy, with the first protocol published in 2017 (Table 2) (156). Following the developmental pathway, LSEC progenitors were first generated by induction of mesodermal cells and selection of FLK1+CD31+CD34+ cells. Interestingly, this population already expressed several LSEC-specific genes. FLK1+CD31+CD34+ cells were expanded and matured by TGFβ inhibition and culture in hypoxic conditions and a LSEC signature was confirmed by looking at expression of cell-specific markers. A later publication used an embryoid body approach to induce mesodermal cells and, from there, generated arterial and venous ECs (157). Gage et al. also used TGFβ inhibition and culture in hypoxic conditions to mature the arterial and venous ECs into LSECs, but also included cAMP agonism. They discovered that the venous-like cells more readily adopted a LSEC phenotype, as evidenced by molecular, structural, and functional features, including fenestrations and scavenger capabilities. Neither the Koui et al. or Gage et al. protocols tested the inflammatory response of hiPSC-derived LSECs and to our knowledge have not been incorporated into tissue chip models. Other potential avenues for generating LSECs could utilize using newly identified transcription factors that determine LSEC fate, such as C-MAF, GATA4, and MEIS2 (39), PU.1 (encoded by the SPI1 gene) (158), and c-Maf (159).
Musculoskeletal system
The musculoskeletal system encompasses numerous tissues with varying types and levels of vascularization. As tissue chip models of musculoskeletal tissues become more sophisticated, it is important to consider the accurate representation of vascular barriers and their roles within these tissues (160). Tissues such as muscles (161), bone (162), synovium (163), and menisci (164) are vascularized while other tissues such as tendons and ligaments (165, 166), intervertebral discs (167), and cartilage (163, 168) generally have very little or no vascularization. In these “avascular” tissues, vascularity is typically an important feature of disease states, thereby motivating the need for accurate “pathological” vascular barriers in in vitro models. While there are many remaining unknowns, specific EC characteristics have been identified for certain tissues.
In muscle, ECs have been shown to function in partnership with myogenic cells especially during early development. Muscle ECs secrete growth factors including insulin-like growth factor 1(IGF-1), hepatocyte growth factor (HGF), basic fibroblast growth factor (bFGF), platelet-derived growth factor-BB (PDGF-BB), and VEGF to induce myogenesis, while responding to angiogenic signals from myogenic cells (161). A tissue chip model by Osaki and colleagues recapitulated this crosstalk using light sensitive channelrhodopsin-2 (ChR2) C2C12 skeletal muscle cells (C2C12-ChR2) and HUVECs to create muscle and vascular structures in a device, which could be optically stimulated to contract (169). The group demonstrated that HUVECs responded to angiopoietin-1 secreted by muscle cells to stimulate angiogenesis. Additionally, HUVEC coculture upregulated myogenesis in muscle cells, thereby improving muscle contraction via angiopoetin-1/neuregulin-1 signaling.
Similarly, ECs in bone serve several important roles in development and maintaining homeostasis of surrounding tissue. Vascularization in bone includes the Haversian system of canals, which allow blood vessels to supply cortical bone, and the vasculature of the periosteum surrounding bones. Studies investigating bone ECs have identified many subpopulations that can influence osteogenesis and the formation of new blood vessels (170, 171). Additionally, ECs in the bone marrow have important traits that maintain the hematopoietic environment for production of blood cells. For example, the two main types of blood vessels found in bone marrow, arteries and sinusoids, have been shown to differentially regulate hematopoiesis. Factors including EC morphology, cell signaling, and barrier integrity influence the maintenance of stem cell or leukocyte populations (172). Bone marrow ECs have shown greater ability to induce hematopoietic progenitor cell adhesion and migration when compared to HUVECs or lung ECs. They also exhibit lower expression of vWF and constitutive expression of adhesion molecules such as VCAM-1 and E-selectin compared to other EC populations (173). Chou and colleagues created a bone marrow tissue chip using CD34+ and marrow-derived stromal cells to model the periarterial, perisinusoidal, mesenchymal, and osteoblastic hematopoietic niches of bone marrow (174). HUVECs were incorporated into a vascular channel. Their model was able to recapitulate pathological features, such as hematopoietic dysfunction and neutrophil maturation abnormality, when constructed using cells from patients with Shwachman-Diamond syndrome.
Tendons and ligaments together form a large category of tissue that is often considered avascular. In healthy mature tissue, vascularization is sparse and mostly limited to supportive sheath structures that provide organization, such as the endotenon or epitenon, and interfaces with muscle and bone (165, 166). While the “healthy” level of vascularization may vary among tendons and ligaments in different parts of the body, neovascularization is a common feature seen after acute injury and long-term pathological states such as tendinopathy (175). The notion that these tissues are avascular can be misleading, and it is important that in vitro models carefully consider the role of vasculature for their designs, particularly in disease models. Knowledge of tendon-specific ECs is still limited when compared to other musculoskeletal tissues. One tendon study has shown that resident EC populations may form a “blood-tendon barrier” that maintains a tendon stem cell niche which may have implications for tendon healing and regeneration. Further, mouse tendon vasculature was shown to form tighter barriers than cardiac vasculature, but not as tight as those found in the blood-brain barrier (176).
The endothelial cells discussed here represent some of the diversity of EC populations found throughout musculoskeletal tissues. Some ECs, such as those in bone, are better characterized than others, but all play important roles in maintaining their surrounding tissue and tissue-specific functions. Importantly, increased vascular proliferation is common in sites of injury and disease, suggesting that the notion of avascular tissues is misleading. Therefore, ECs are an important component for researchers to consider when creating in vitro models of musculoskeletal tissues. Most existing musculoskeletal tissue chips with vasculature components utilize HUVECs, and other sources for musculoskeletal tissue ECs are rare (160). However, accurate representation of musculoskeletal EC function will facilitate more physiologically relevant tissue systems, particularly in disease and injury models. hiPSC-derived ECs are beginning to emerge in musculoskeletal models (177), and as hiPSC techniques become more sophisticated and more tissue chip researchers move toward patient-specific models, it is likely that hiPSC-derived ECs will become increasingly incorporated into these models. Further research is needed for developing protocols to generate musculoskeletal-specific endothelial cells.
Pericytes
Background
Perivascular cells are support cells that line the vasculature and serve a variety of roles in both homeostasis and tissue repair (178). The three main types of perivascular cells are smooth muscle cells (SMCs), pericytes (PCs), and fibroblasts (FBs). SMCs are predominantly found along larger vessels, whereas PCs surround smaller blood vessels, including capillaries and post-capillary venules (3, 179). Cells with intermediate phenotypes are found along midsize vessels. FBs are found throughout much of the vasculature, mainly on arterioles and veins but not within capillaries (179). They are important regulators of extracellular matrix deposition and tissue remodeling after injury, along with important roles in guiding angiogenesis. As this review focuses on the microvasculature and immune cell trafficking, we will only discuss PC heterogeneity and immune functions.
Pericytes have multiple developmental lineages. While most PCs, like ECs, arise from the mesoderm, PCs of the brain and retina have a neural crest lineage (180). Mature pericytes are known to embed themselves within the basement membrane of blood vessels, where they communicate with ECs via gap junctions, adherens junctions, and soluble signals (180, 181). EC to PC ratio varies in different organs, with the highest coverage found in the brain and retina and lowest in skeletal muscle (Figure 1) (180, 182). Along with other mural cells, PCs aid with angiogenesis and vessel stabilization. They also regulate blood flow and have various roles in immunological defense and response to inflammation (180, 183). They are generally characterized by vessel location and morphology, which is generally stellate but varies along the vasculature and between tissues (184). Defining pericytes and other mural cells has been difficult, due to overlapping markers, but recent studies suggest inwardly rectifying potassium channel, KCNJ8, and ABC transporter, ABCC9, may be good candidates (179, 180). Traditionally, however, PCs have been identified by expression of PDGFRβ and/or NG2 (178, 180, 184).
Following TEM across the EC layer, migrating immune cells must also breach the basement membrane and pericyte layer to reach extravascular tissue. This process has been reviewed in detail prior (26, 185, 186). The resistance created by these final two layers depends on the tissue and their coverage. Pericytes have multiple roles in assisting transmigrating leukocytes during inflammation, including secretion of cytokines, remodeling and degradation of the basement membrane, and upregulation of adhesion molecules, such as ICAM-1, to guide migrating immune cells after they cross the EC barrier (187–190). Expression of LAMs, at least in some part, appears to be tissue-dependent (Figure 1, Table 3) (186). Neutrophils preferentially migrate across “hotspots,” crawling across PCs to regions with gaps in coverage or high ICAM-1 expression and regions that have low deposition of some basement membrane components (185, 189). PCs can directly interact with transmigrating immune cells before they enter the extravascular tissue. These interactions include signals that promote differentiation into immune cell subtypes (26). PCs can also damper immune responses by signaling to ECs to reduce neutrophil migration and can negatively regulate T cell responses (192–194).
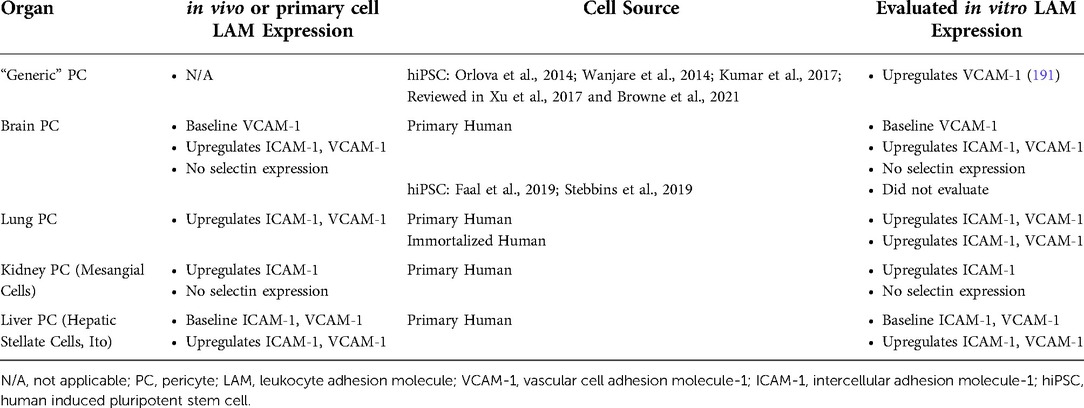
Table 3. Pericyte sources for tissue models.
Pericyte sources
Pericytes can be isolated from several tissues throughout the body, including the placenta, brain, heart, glomerulus, skeletal muscle and, most recently, lung (Table 3) (178, 186, 195). The characteristics of PCs from different sources vary (178). For example, brain-derived pericytes have a more elongated morphology compared to the rounded and compact pericytes found in the kidney glomerulus, also known as mesangial cells (184). Some functions appear to be tissue-specific as well. For instance, liver pericytes contribute to Vitamin A metabolism (178). In terms of immune cell migration, brain-derived PCs can upregulate both ICAM-1 and VCAM-1 upon proinflammatory stimulation and human glomerular pericytes can upregulate ICAM, but neither express selectins (26, 196–198). E-selectin was, however, expressed on human dermal pericytes derived from skin biopsies (26). Liver PCs express baseline levels of ICAM-1 and VCAM-1, both of which can be upregulated in response to TNFα (199). Both primary and immortalized human lung PCs show comparable expression of ICAM-1 and VCAM-1 in in vitro culture (195).
Several protocols for generating stem-cell derived pericytes have emerged in recent years. Given the variability in cell origin, some protocols transition through the mesoderm and others through the neuroectoderm. There are methods that co-differentiate ECs and PCs by generating mesodermal cells and sorting these cells to further specify and expand the EC and PC populations (Table 3) (82). Suppressing the TGF-β pathway is common in order to drive cells towards a PC-specific, rather than SMC, phenotype (82, 191, 200, 201). The characterization of the generated pericytes differs by protocol and continues to develop as the knowledge of PC and SMC phenotypes has grown. Reviews on these protocols and the phenotypic characterization of the cells generated have been previously published (3, 202). To the best of our knowledge, the only tissue-specific pericyte protocols available are two publications from 2019, both of which produce brain pericyte-like cells (Table 3). One publication by Stebbins and colleagues transitions the cells through a neural crest intermediate, while the other by Faal and colleagues presents two methods, one method transitioning cells through the mesoderm and the other via neural crest (200, 203). Until recently PCs have largely been ignored in in vitro models, likely due to the heterogeneity of this cell type and difficulty in identification of pericytes over other perivascular cells. However, several vascular barrier chips [reviewed in (4)] and BBB tissue chips (31, 204–206) have incorporated pericytes. Thus far, none of these tissue chip models have been used to study immune cell trafficking. However, one BBB model did measure adhesion molecule, VCAM-1, in hiPSC-derived pericyte-like mural cells (iMCs) differentiated from an Alzheimer's disease “APOE4/4 risk” line and an “APOE4/4-risk edited to APOE3/3-non-risk” hiPSC line (204). They discovered iMCs derived from the AD-risk cell line had increased basal VCAM-1 expression compared to the non-risk line. It is likely we will see future tissue chip studies incorporating PCs from various sources as their importance in disease continues to be established.
Immune cells
Background
While this review has covered the major vascular contributors to leukocyte trafficking, the sourcing of immune cells is also critical. During inflammation, an acute response is initiated by innate immune cells, first transmigrating neutrophils and then monocytes. This can turn into chronic inflammation, which involves both innate and acquired immune cells, including several types of T cells and B cells. Leukocytes migrating from the circulation into tissue can activate tissue resident immune cells such as macrophages. Discussion of the subsets of each of these immune cells is beyond the scope of this review but are detailed in many excellent books and reviews (207–209). The actions of immune cells can either lead to resolution or exacerbation of inflammation, and their inclusion into tissue chip models of disease are critical. Below, we will discuss a few subsets of immune cells and their potential cell sources.
Neutrophils
Neutrophils, or polymorphonuclear leukocytes (PMNs), are the most abundant white blood cell in the human body and comprise between 50%–70% of the circulating leukocyte population in healthy adults (210, 211). They are rapidly produced in the bone marrow (>1011 cells/day) via differentiation of hematopoietic stem cells (HSCs), the cell type that gives rise to all blood cells, towards granulocyte-macrophage progenitors (GMPs), ultimately resulting in mature PMNs (210, 212–214). The exact factors that promote HSC differentiation towards a terminal PMN lineage are poorly understood, and a growing body of evidence suggests that PMNs are a heterogenous population of cells with robust plasticity and polarized phenotypes, rather than a homogenous one (214, 215). This understanding emerges from multiple characterizations, which are reviewed elsewhere (210, 216–218). Circulating PMNs are thought to be short lived cells with a general half-life of 7 h postproduction from the bone marrow (219). However, this characterization is now contradicted by recent evidence suggesting that PMNs survive for longer in the body with the capability of recirculating back into vasculature and bone marrow (220, 221). Regardless, PMNs typically function by migrating from post-capillary venules towards sites of inflammation in tissue via the adhesion cascade (221, 222). Upon tissue infiltration, PMNs will chemotactically migrate towards pathogens and destroy them through phagocytosis or the expulsion of DNA to form neutrophil extracellular traps (NETs) (222, 223). PMNs are also known to engage in a complex cross-talk with elements of the adaptive immune system, modulating immune effector function responses to infections (224).
PMN migratory function in response to inflammatory stimuli or environmental conditions is frequently modeled on tissue chip platforms (61, 225–227). Because of their short lifespan, PMNs are most commonly isolated from whole blood via density gradient separation and used immediately (Table 4). In fact, all tissue chip models utilizing PMNs described in this report thus far, have used freshly isolated PMNs. Cells obtained from donors provide high physiological relevance, however, they are subject to high degrees of biological variability and supply constraints. Cryopreserved purified human PMNs are also commercially available from biological supply vendors, however a recent study by Avci and colleagues demonstrated that PMNs exposed to a 9-week freeze-thaw cycle exhibited reduced chemotactic and polarization capability compared to freshly isolated cells when exposed to a potent PMN chemoattractant (Table 4) (233). This physiological limitation, in addition to the costs associated with purchasing frozen cells, makes isolating PMNs directly from donors the cheaper, and more common, of the two options.

Table 4. Leukocyte sources for tissue models.
Beyond acquisition from human donors, PMN-like cells can be created through differentiation protocols featuring promyelocytic cell lines, such as HL-60, or hiPSCs (Table 4) (234, 235). In one tissue chip study, PMN-like cells were produced by culturing human promyelocytic leukemia cells in complete media supplemented with 1.5% dimethyl sulfoxide (DMSO) for 4–5 days (227). This study did not include an EC layer, as the group was focused on developing a reductionist approach to study electrotaxis. Studies have demonstrated, however, that PMN-like cells derived from the HL-60 promyelocytic cell line have modified or diminished PMN effector functions, such as NET production and antimicrobial properties when compared to primary blood-derived PMNs (236, 237). As an alternative, a recent protocol produces PMNs from hiPSCs by transfection with ETV2 mRNA, a hematoendothelial programmer (238). hiPSC-derived PMNs generated by this method, however, have a decreased chemotactic response when compared to donor PMNs and contain a population of immature cells which lack phagocytic activity (238). These cells do, however, exhibit similar levels of reactive oxygen species production. To date, hiPSC-derived PMNs have not been incorporated into tissue chip models, likely due to the novelty of these protocols and the low cost of freshly isolating PMNs from blood.
Monocytes
Monocytes are another major type of circulating leukocyte and can further differentiate into macrophages and dendritic cells (DCs). Together, these cell types play important roles in the innate immune system. Monocytes are derived from bone marrow where they originate from HSCs. Their precursors differentiate into monoblasts and then promonocytes, which finally divide into monocytes. After maturation, monocytes remain in the bone marrow for less than a day before entering circulation, where they can stay for up to 3 days before migrating into organs and tissues (239, 240). Monocytes can differentiate into macrophages and DCs, which can undergo further changes to serve more specific functions (241). For example, macrophages can polarize into pro- and anti-inflammatory phenotypes such as M1 and M2 macrophages, respectively. They are distinct from tissue-resident macrophages and DCs, which are unique to their tissue and may even have different developmental origins. The classification of monocytes, macrophages, and DCs is complex, and this diversity makes the use of monocytes and their derivatives in in vitro disease models complicated (240, 241).
Careful selection of monocyte source and/or differentiation method is needed for the creation of relevant in vitro models. Most methods identify monocytes using the monocyte/macrophage marker CD14. Similar to neutrophils, monocytes are most commonly freshly isolated from the blood, in this case selecting for peripheral blood mononuclear cells (PBMCs) (Table 4). However, they can also be obtained from commercially available immortalized monocyte-like cell lines (MCLCs), such as THP-1 and U-937 cells (Table 4). Subtle differences exist between freshly isolated and commercial monocytes. While commercial lines are well characterized and validated through gene expression and cytokine profiles, there are differences in their phenotypic effect when responding to inflammation and with functional measures such as migration (242, 243). One tissue chip study demonstrated some of these differences by comparing the responses of freshly isolated PBMCs and THP-1 cells in an inflamed liver sinusoidal organoid biochip (228). The model incorporated HUVECs and HepaRG hepatocytes, and upon stimulation with LPS, HUVECs upregulated ICAM-1 and VCAM-1. This enabled monocytes from both sources to transmigrate across the HUVEC layer, which, surprisingly, seemed to reduce the inflammatory status of the HUVECs, as measured by lower ICAM-1 and VCAM-1 levels compared to monocyte-free conditions. This is in contrast to data indicating monocyte migration further activates ECs but appears to be due to a shift to M2 polarization of macrophages upon monocyte migration. While most results were comparable between the two monocyte sources, the group did find a difference in cytokine secretion, with lower secretion by THP-1 monocytes, consistent with previous studies comparing monocyte responses to LPS. In another study, Sharifi and colleagues also used both PBMCs and THP-1 cells in their foreign body response-on-a-chip (FBROC) (229). The FBROC combined monocytes and Ti microbeads in a device separated by a vascular barrier composed of HUVECs. When monocyte chemoattractant protein-1 (MCP-1) was added to the microbead compartment, monocyte-endothelial cell interactions were activated, and THP-1 cells transmigrated into the Ti compartment. Here, they differentiated further into a pro-inflammatory M1 phenotype indicating recognition of the Ti microbeads as a foreign body. PBMCs isolated from three patient donors had varying polarization of monocytes to M1 and M2 phenotypes as indicated by different CD80/CD206 expression ratios. This highlights the potential for using PBMCs in patient-specific models of disease which is not possible with commercial MCLCs.
Monocytes derived from hiPSCs have become increasingly common, and numerous protocol exist, most of which follow a similar framework of differentiating hiPSCs to mesoderm lineage, followed by hematopoietic progenitors and finally CD14+ monocytes (Table 4) (244–247). These protocols have been shown to robustly produce CD14+ monocytes that have similar physiology to monocytes obtained from other sources. A recent study published by Ronaldson-Bouchard and colleagues utilized a commercially available monocyte differentiation kit to produce patient-specific hiPSC-derived monocytes for their multi-organ chip (230). The study demonstrated that monocytes maintained the classical CD16− CD14+ phenotype over 4 weeks circulating in the device and were confined in the vascular channels under uninjured conditions. When cryo-injury of a heart tissue compartment was induced, monocytes were able to selectively extravasate across the vascular layer into the injured compartment while not entering the healthy compartments. As the tissue chip community continues to develop patient-specific models of disease, it is likely that hiPSC-derived monocytes will become increasingly common moving forward.
T cells
T (thymus-dependent) and B (bone marrow-dependent) lymphocytes contribute to the acquired, or adaptive, immune response. More specifically, this class represents the only cell types that can recognize and respond to specific antigens (248). The development of both cell types initiates from common lymphoid progenitors (CLPs), which themselves derive from multipotent HSCs. T and B lymphocyte developmental pathways share common mechanisms including the rearrangement of antigen-receptor genes, testing for successful rearrangement, and the assembly of a heterodimeric antigen receptor (249). Additionally, both T and B cell differentiation is guided by environment-specific signaling conveyed by thymic epithelial cells and bone marrow stromal cells, respectively. The established repertoire of mature T cells is long-lived and potentially self-renewing, while the repertoire of mature B cells is comprised of short-lived cells and requires replenishment from the bone marrow (250). The developmental process of lymphocytes is a complicated process, and a detailed discussion is beyond the scope of this summary but can be found elsewhere in literature (248–250). We will focus on T lymphocytes in this review, as these cells have been studied extensively using tissue chip models.
The T lymphocyte family includes two lineages distinguished by their T-cell receptor (TCR), the majority αβ TCR lineage and the minority γδ TCR lineage. The γδ lineage is less studied, and these cells are believed to be involved in processes more closely associated with the innate immune response (249). The αβ lineage undergoes further development in the thymus and is subject to developmental checkpoints known as positive and negative selection; these mechanisms ensure emerging T cells recognize self-MHC molecules yet exhibit self-tolerance (250). Additionally, the αβ lineage is comprised of sublineages distinguished by the presence of co-receptors CD4 and CD8. Functionally, these sublineages are unique as CD4 and CD8 T cells recognize and bind to MHC class II and MHC class I molecules, respectively. After recognizing specific pathogen-associated peptides in secondary lymphoid organs, T cells become activated and modify expression of adhesion molecules (251). This allows T cells to travel to sites of infection and carry out effector functions. Broadly put, cytotoxic CD8 T cells (CTLs) are involved in the direct killing of cells compromised with intracellular pathogens while effector CD4 T cells assist with the removal of extracellular pathogens and provide cytokine signaling support (250). Effector CD4 T cells can differentiate into various subtypes dependent on the local chemical environment. The TH1 (T Helper 1), TH2, and TH17 subtypes orchestrate the elimination of distinct pathogen classes while the TFH (T follicular helper) subtype promotes B cell responses within lymph nodes. Lastly, the Treg (regulatory T cell) subtype dampens the immune response and inhibits the effector functions of other subtypes (249).
The study of T lymphocytes using tissue chip models primarily focuses on their activation (252–255), trafficking (including TEM) (231, 232, 256–258) and interactions with target (usually tumor) cells (259–262). Here, we focus on interactions between T cells and ECs in the context of TEM. Very thorough reviews that cover the abovementioned topics have been written and include information relevant to other immune cells as well (208, 263–266). Traditionally, PBMCs are used as the source of T lymphocytes in tissue chips (231, 257, 258), although others have incorporated immortalized Jurkat T cells (Table 4) (232, 256). In addition, a number of approaches have been utilized for isolation of tumor infiltrating lymphocytes from human tissues utilizing enzymatic digestion, mechanical digestion, or both (267–269). Depending on the downstream application, negative selection of T cells can be utilized to prevent activation (270). Despite the fact that primary and Jurkat T cells are often used interchangeably, there are documented differences, namely, distinct actin organization and dynamics (271). Jurkat T cells lack negative regulators of TCR signaling and are impaired in their ability to receive costimulatory signals (272, 273). Additionally, a comparative analysis of post-stimulation transcription profiles revealed a substantial subset of genes was differentially expressed among primary and Jurkat T cells (274). PBMCs are isolated from blood samples using a standard density gradient centrifugation method. Beyond this, it is common to utilize an isolation kit that works on the principle of negative selection. de Haan et al. constructed an endothelium-on-a-chip model that was used to study freshly isolated T cell dynamics in response to chemokine gradients and pro-inflammatory cytokines (231). Culturing immortalized human dermal microvascular endothelial cells (HMEC-1) with stimulated T cells resulted in a near 3-fold increase of ICAM-1 expression, as well as an increased frequency of TEM relative to unstimulated cells. Interestingly, stimulated T cell TEM was mediated by a C-X-C motif chemokine ligand 12 (CXCL12) gradient but did not depend on TNF-α HMEC-1 pretreatment. It was hypothesized that perfusion of stimulated T cells established an inflammatory environment through secretion of cytokines, and this environment was not further developed through TNF-α pretreatment. Park and colleagues also aimed to study T cell-endothelium interactions and optimized a microfluidic system using stimulated Jurkat T cells (232). They reported a lack of binding between stimulated T cells and unstimulated HUVECs. This is contrary to the findings described by de Haan et al. that suggest stimulated T cells not only bind to unstimulated ECs but can subsequently extravasate. This could be due to experimental differences (rocking perfusion vs. flow, endothelium geometry, timescales examined) but may also reflect inherent features of the cell types selected (HMEC-1 vs. HUVEC and primary T cells vs. Jurkat T cells).
Protocols to generate T cells from hiPSCs have been researched extensively in the field of cancer treatment (Table 4) (275–279). These protocols are used for cell-based immunotherapy in which hiPSC methods allow for the cloning and expansion of tumor antigen-specific T cells. Essentially, tumor-specific CTLs are isolated, reprogrammed to generate hiPSCs that inherit rearranged TCR genes, expanded, and finally differentiated to yield large numbers of functionally mature CTLs (277, 279). The protocols found in literature vary but differentiation is carried out with hematopoietic/lymphopoietic cytokine cocktail treatment and co-culture with bone marrow-derived stromal cells that express the Notch ligand Delta-like 1 (OP9-DL1). Conventional methods generate a class of CTLs that is roughly 100-fold less competent compared to primary CTL counterparts, however, subtle alterations made to differentiation protocols can generate an alternative CTL class that provides comparable antigen-specific cytotoxicity relative to primary CTLs (276). Further fine tuning of protocols has resulted in enhanced hiPSC-derived CTL stability and specificity through deletion of recombination activating gene 2, thereby avoiding unwanted TCR rearrangements (280). To our knowledge hiPSC-derived T cells have not been studied in tissue chip systems, however, this endeavor would aid in the study of patient-specific T cell mechanisms as well as designing personalized treatment regimens for a broad range of diseases.
Conclusions
The vasculature plays a central role in disease development and resolution. As tissue chip systems become more widespread for disease modeling and personalized medicine, it is important that a vascular component is included when possible. This vascular component should include relevant cell types, including endothelial cells and pericytes, for studying the impact of immune cell migration in the context of disease and inflammation. To properly target immune cell migration for therapies, careful consideration should be given to the sourcing of these cells. It is increasingly clear that mechanisms of leukocyte entry into different tissues varies, and cells from different sources can more or less faithfully recapitulate these critical phenotypes. In certain cases, “generic” endothelial cells may be sufficient, given the robustness of cell lines such as HUVECs and the ability of ECs to adopt tissue-specific phenotypes through cell-cell signaling. However, in order for personalized medicine to realize its full potential, it may be necessary to utilize tissue-specific primary cells or stem-cell derived endothelial cells, pericytes, and/or immune cells. This, of course, poses several challenges in terms of time, cost, and the general newness of many differentiation protocols. Further, generating a fully isogenic multicellular model requires careful timing of several differentiation protocols when cryopreservation is not an option. Currently, many hiPSC-derived endothelial cell methods generate immature ECs or fail to fully recapitulate the necessary immune phenotypes. This will improve with careful research and more studies characterizing tissue-specific vascular cells during inflammation. It is our hope that researchers interested in studying immune cell migration in tissue chip models will find this review helpful for identifying cell sources which express the proper leukocyte adhesion molecules for the tissue of interest.
Author contributions
MM conceptualized, wrote, and edited the manuscript. VZ, DA, SW, SR, and HA contributed writing to the manuscript. JM conceptualized and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
MM, DA, and JM were partially supported by the National Institutes of Health (NIH) award numbers UG3TR003281 and R61HL154249. DA was also partially by supported Arnold and Mabel Beckman Foundation 2020LSM-31. SR was supported by NIH under award number T32 HL066988, Multidisciplinary Training in Pulmonary Research. HA and VZ was supported by the National Center for Advancing Translational Sciences (NCATS) and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) of the NIH under award number UG3TR003281. VZ is a trainee in the Medical Scientist Training Program funded by NIH T32 GM007356. The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Science or NIH.
Acknowledgments
The authors acknowledge A. Salminen and J. Flax for their insights and feedback regarding the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Nguyen J, Lin YY, Gerecht S. The next generation of endothelial differentiation: tissue-specific ECs. Cell Stem Cell. (2021) 28(7):1188–204. doi: 10.1016/j.stem.2021.05.002
2. Hennigs JK, Matuszcak C, Trepel M, Korbelin J. Vascular endothelial cells: heterogeneity and targeting approaches. Cells. (2021) 10(10):2712. doi: 10.3390/cells10102712
3. Browne S, Gill EL, Schultheiss P, Goswami I, Healy KE. Stem cell-based vascularization of microphysiological systems. Stem Cell Rep. (2021) 16(9):2058–75. doi: 10.1016/j.stemcr.2021.03.015
4. Cochrane A, Albers HJ, Passier R, Mummery CL, van den Berg A, Orlova VV, et al. Advanced in vitro models of vascular biology: human induced pluripotent stem cells and organ-on-chip technology. Adv Drug Deliv Rev. (2019) 140:68–77. doi: 10.1016/j.addr.2018.06.007
5. Wang X, Sun Q, Pei J. Microfluidic-Based 3D engineered microvascular networks and their applications in vascularized microtumor models. Micromachines (Basel). (2018) 9(10):493. doi: 10.3390/mi9100493
6. Ewald ML, Chen YH, Lee AP, Hughes CCW. Correction: the vascular niche in next generation microphysiological systems. Lab Chip. (2021) 21(18):3615–6. doi: 10.1039/D1LC90093E
7. Feletou M. The endothelium: Part 1: Multiple functions of the endothelial cells-focus on endothelium-derived vasoactive mediators. San Rafael, CA: Integrated Systems Physiology: from Molecule to Function to Disease (2011).
8. Risau W, Flamme I. Vasculogenesis. Annu Rev Cell Dev Biol. (1995) 11:73–91. doi: 10.1146/annurev.cb.11.110195.000445
9. Jain RK. Molecular regulation of vessel maturation. Nat Med. (2003) 9(6):685–93. doi: 10.1038/nm0603-685
10. Niklason L, Dai G. Arterial venous differentiation for vascular bioengineering. Annu Rev Biomed Eng. (2018) 20:431–47. doi: 10.1146/annurev-bioeng-062117-121231
11. Augustin HG, Koh GY. Organotypic vasculature: from descriptive heterogeneity to functional pathophysiology. Science. (2017) 357(6353):eaal2379. doi: 10.1126/science.aal2379
12. Potente M, Makinen T. Vascular heterogeneity and specialization in development and disease. Nat Rev Mol Cell Biol. (2017) 18(8):477–94. doi: 10.1038/nrm.2017.36
13. Marcu R, Choi YJ, Xue J, Fortin CL, Wang Y, Nagao RJ, et al. Human organ-specific endothelial cell heterogeneity. iScience. (2018) 4:20–35. doi: 10.1016/j.isci.2018.05.003
14. Sabbagh MF, Heng JS, Luo C, Castanon RG, Nery JR, Rattner A, et al. Transcriptional and epigenomic landscapes of CNS and non-CNS vascular endothelial cells. Elife. (2018) 7:e36187. doi: 10.7554/eLife.36187
15. Cleuren ACA, van der Ent MA, Jiang H, Hunker KL, Yee A, Siemieniak DR, et al. The in vivo endothelial cell translatome is highly heterogeneous across vascular beds. Proc Natl Acad Sci U S A. (2019) 116(47):23618–24. doi: 10.1073/pnas.1912409116
16. Nolan DJ, Ginsberg M, Israely E, Palikuqi B, Poulos MG, James D, et al. Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev Cell. (2013) 26(2):204–19. doi: 10.1016/j.devcel.2013.06.017
17. Durr E, Yu J, Krasinska KM, Carver LA, Yates JR, Testa JE, et al. Direct proteomic mapping of the lung microvascular endothelial cell surface in vivo and in cell culture. Nat Biotechnol. (2004) 22(8):985–92. doi: 10.1038/nbt993
18. Aird WC. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ Res. (2007) 100(2):174–90. doi: 10.1161/01.RES.0000255690.03436.ae
19. Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. (2007) 100(2):158–73. doi: 10.1161/01.RES.0000255691.76142.4a
20. Aird WC. Endothelial cell heterogeneity. Cold Spring Harb Perspect Med. (2012) 2(1):a006429. doi: 10.1101/cshperspect.a006429
21. Pober JS, Sessa WC. Inflammation and the blood microvascular system. Cold Spring Harb Perspect Biol. (2014) 7(1):a016345. doi: 10.1101/cshperspect.a016345
22. Kalucka J, de Rooij L, Goveia J, Rohlenova K, Dumas SJ, Meta E, et al. Single-cell transcriptome atlas of murine endothelial cells. Cell. (2020) 180(4):764–79.e20. doi: 10.1016/j.cell.2020.01.015
23. Mai J, Virtue A, Shen J, Wang H, Yang XF. An evolving new paradigm: endothelial cells–conditional innate immune cells. J Hematol Oncol. (2013) 6:61. doi: 10.1186/1756-8722-6-61
24. Young MR. Endothelial cells in the eyes of an immunologist. Cancer Immunol Immunother. (2012) 61(10):1609–16. doi: 10.1007/s00262-012-1335-0
25. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. (2007) 7(9):678–89. doi: 10.1038/nri2156
26. Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity. (2014) 41(5):694–707. doi: 10.1016/j.immuni.2014.10.008
27. Jambusaria A, Hong Z, Zhang L, Srivastava S, Jana A, Toth PT, et al. Endothelial heterogeneity across distinct vascular beds during homeostasis and inflammation. Elife. (2020) 9:e51413. doi: 10.7554/eLife.51413
28. Murakami S, Morioka T, Nakagawa Y, Suzuki Y, Arakawa M, Oite T. Expression of adhesion molecules by cultured human glomerular endothelial cells in response to cytokines: comparison to human umbilical vein and dermal microvascular endothelial cells. Microvasc Res. (2001) 62(3):383–91. doi: 10.1006/mvre.2001.2356
29. Hickey MJ, Westhorpe CL. Imaging inflammatory leukocyte recruitment in kidney, lung and liver–challenges to the multi-step paradigm. Immunol Cell Biol. (2013) 91(4):281–9. doi: 10.1038/icb.2012.83
30. Ingber DE. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat Rev Genet. (2022) 23:467–91. doi: 10.1038/s41576-022-00466-9
31. Campisi M, Shin Y, Osaki T, Hajal C, Chiono V, Kamm RD. 3D self-organized microvascular model of the human blood-brain barrier with endothelial cells, pericytes and astrocytes. Biomaterials. (2018) 180:117–29. doi: 10.1016/j.biomaterials.2018.07.014
32. Benam KH, Villenave R, Lucchesi C, Varone A, Hubeau C, Lee HH, et al. Small airway-on-a-chip enables analysis of human lung inflammation and drug responses in vitro. Nat Methods. (2016) 13(2):151–7. doi: 10.1038/nmeth.3697
33. Wang L, Tao T, Su W, Yu H, Yu Y, Qin J. A disease model of diabetic nephropathy in a glomerulus-on-a-chip microdevice. Lab Chip. (2017) 17(10):1749–60. doi: 10.1039/C7LC00134G
34. Li X, George SM, Vernetti L, Gough AH, Taylor DL. A glass-based, continuously zonated and vascularized human liver acinus microphysiological system (vLAMPS) designed for experimental modeling of diseases and ADME/TOX. Lab Chip. (2018) 18(17):2614–31. doi: 10.1039/C8LC00418H
35. Auerbach R. Vascular endothelial cell differentiation: organ-specificity and selective affinities as the basis for developing anti-cancer strategies. Int J Radiat Biol. (1991) 60(1–2):1–10. doi: 10.1080/09553009114551401
36. Minami T, Aird WC. Endothelial cell gene regulation. Trends Cardiovasc Med. (2005) 15(5):174–84. doi: 10.1016/j.tcm.2005.06.002
37. Bennett HS, Luft JH, Hampton JC. Morphological classifications of vertebrate blood capillaries. Am J Physiol. (1959) 196(2):381–90. doi: 10.1152/ajplegacy.1959.196.2.381
38. Chi JT, Chang HY, Haraldsen G, Jahnsen FL, Troyanskaya OG, Chang DS, et al. Endothelial cell diversity revealed by global expression profiling. Proc Natl Acad Sci U S A. (2003) 100(19):10623–8. doi: 10.1073/pnas.1434429100
39. de Haan W, Oie C, Benkheil M, Dheedene W, Vinckier S, Coppiello G, et al. Unraveling the transcriptional determinants of liver sinusoidal endothelial cell specialization. Am J Physiol Gastrointest Liver Physiol. (2020) 318(4):G803–15. doi: 10.1152/ajpgi.00215.2019
40. Geraud C, Koch PS, Zierow J, Klapproth K, Busch K, Olsavszky V, et al. GATA4-dependent organ-specific endothelial differentiation controls liver development and embryonic hematopoiesis. J Clin Invest. (2017) 127(3):1099–114. doi: 10.1172/JCI90086
41. Coppiello G, Collantes M, Sirerol-Piquer MS, Vandenwijngaert S, Schoors S, Swinnen M, et al. Meox2/Tcf15 heterodimers program the heart capillary endothelium for cardiac fatty acid uptake. Circulation. (2015) 131(9):815–26. doi: 10.1161/CIRCULATIONAHA.114.013721
42. DeStefano JG, Jamieson JJ, Linville RM, Searson PC. Benchmarking in vitro tissue-engineered blood-brain barrier models. Fluids Barriers CNS. (2018) 15(1):32. doi: 10.1186/s12987-018-0117-2
43. Gray KM, Stroka KM. Vascular endothelial cell mechanosensing: new insights gained from biomimetic microfluidic models. Semin Cell Dev Biol. (2017) 71:106–17. doi: 10.1016/j.semcdb.2017.06.002
44. Cucullo L, Marchi N, Hossain M, Janigro D. A dynamic in vitro BBB model for the study of immune cell trafficking into the central nervous system. J Cereb Blood Flow Metab. (2011) 31(2):767–77. doi: 10.1038/jcbfm.2010.162
45. Benson BL, Li L, Myers JT, Dorand RD, Gurkan UA, Huang AY, et al. Biomimetic post-capillary venule expansions for leukocyte adhesion studies. Sci Rep. (2018) 8(1):9328. doi: 10.1038/s41598-018-27566-z
46. Vera D, Garcia-Diaz M, Torras N, Alvarez M, Villa R, Martinez E. Engineering tissue barrier models on hydrogel microfluidic platforms. ACS Appl Mater Interfaces. (2021) 13(12):13920–33. doi: 10.1021/acsami.0c21573
47. Moya ML, Hsu YH, Lee AP, Hughes CC, George SC. In vitro perfused human capillary networks. Tissue Eng Part C Methods. (2013) 19(9):730–7. doi: 10.1089/ten.tec.2012.0430
48. Natividad-Diaz SL, Browne S, Jha AK, Ma Z, Hossainy S, Kurokawa YK, et al. A combined hiPSC-derived endothelial cell and in vitro microfluidic platform for assessing biomaterial-based angiogenesis. Biomaterials. (2019) 194:73–83. doi: 10.1016/j.biomaterials.2018.11.032
49. Mathur T, Flanagan JM, Jain A. Tripartite collaboration of blood-derived endothelial cells, next generation RNA sequencing and bioengineered vessel-chip may distinguish vasculopathy and thrombosis among sickle cell disease patients. Bioeng Transl Med. (2021) 6(3):e10211. doi: 10.1002/btm2.10211
50. Wang L, Xiang M, Liu Y, Sun N, Lu M, Shi Y, et al. Human induced pluripotent stem cells derived endothelial cells mimicking vascular inflammatory response under flow. Biomicrofluidics. (2016) 10(1):014106. doi: 10.1063/1.4940041
51. Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY, Ingber DE. Reconstituting organ-level lung functions on a chip. Science. (2010) 328(5986):1662–8. doi: 10.1126/science.1188302
52. Iampietro C, Bellucci L, Arcolino FO, Arigoni M, Alessandri L, Gomez Y, et al. Molecular and functional characterization of urine-derived podocytes from patients with alport syndrome. J Pathol. (2020) 252(1):88–100. doi: 10.1002/path.5496
53. Musah S, Mammoto A, Ferrante TC, Jeanty SSF, Hirano-Kobayashi M, Mammoto T, et al. Mature induced-pluripotent-stem-cell-derived human podocytes reconstitute kidney glomerular-capillary-wall function on a chip. Nat Biomed Eng. (2017) 1:0069. doi: 10.1038/s41551-017-0069
54. Roye Y, Bhattacharya R, Mou X, Zhou Y, Burt MA, Musah S. A personalized glomerulus chip engineered from stem cell-derived epithelium and vascular endothelium. Micromachines (Basel). (2021) 12(8):967. doi: 10.3390/mi12080967
55. Cucullo L, Hossain M, Puvenna V, Marchi N, Janigro D. The role of shear stress in blood-brain barrier endothelial physiology. BMC Neurosci. (2011) 12:40. doi: 10.1186/1471-2202-12-40
56. Perruzzi CA, de Fougerolles AR, Koteliansky VE, Whelan MC, Westlin WF, Senger DR. Functional overlap and cooperativity among alphav and beta1 integrin subfamilies during skin angiogenesis. J Invest Dermatol. (2003) 120(6):1100–9. doi: 10.1046/j.1523-1747.2003.12236.x
57. Whelan MC, Senger DR. Collagen I initiates endothelial cell morphogenesis by inducing actin polymerization through suppression of cyclic AMP and protein kinase A. J Biol Chem. (2003) 278(1):327–34. doi: 10.1074/jbc.M207554200
58. Wang KC, Yeh YT, Nguyen P, Limqueco E, Lopez J, Thorossian S, et al. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc Natl Acad Sci U S A. (2016) 113(41):11525–30. doi: 10.1073/pnas.1613121113
59. Blaurock-Moller N, Groger M, Siwczak F, Dinger J, Schmerler D, Mosig AS, et al. CAAP48, A new sepsis biomarker, induces hepatic dysfunction in an in vitro liver-on-chip model. Front Immunol. (2019) 10:273. doi: 10.3389/fimmu.2019.00273
60. Khire TS, Salminen AT, Swamy H, Lucas KS, McCloskey MC, Ajalik RE, et al. Microvascular mimetics for the study of leukocyte-endothelial interactions. Cell Mol Bioeng. (2020) 13(2):125–39. doi: 10.1007/s12195-020-00611-6
61. Salminen AT, Tithof J, Izhiman Y, Masters EA, McCloskey MC, Gaborski TR, et al. Endothelial cell apicobasal polarity coordinates distinct responses to luminally versus abluminally delivered TNF-alpha in a microvascular mimetic. Integr Biol (Camb). (2020) 12(11):275–89. doi: 10.1093/intbio/zyaa022
62. Han S, Yan JJ, Shin Y, Jeon JJ, Won J, Jeong HE, et al. A versatile assay for monitoring in vivo-like transendothelial migration of neutrophils. Lab Chip. (2012) 12(20):3861–5. doi: 10.1039/c2lc40445a
63. Mossu A, Rosito M, Khire T, Li Chung H, Nishihara H, Gruber I, et al. A silicon nanomembrane platform for the visualization of immune cell trafficking across the human blood-brain barrier under flow. J Cereb Blood Flow Metab. (2019) 39(3):395–410. doi: 10.1177/0271678X18820584
64. Atchison L, Abutaleb NO, Snyder-Mounts E, Gete Y, Ladha A, Ribar T, et al. iPSC-Derived endothelial cells affect vascular function in a tissue-engineered blood vessel model of hutchinson-gilford progeria syndrome. Stem Cell Rep. (2020) 14(2):325–37. doi: 10.1016/j.stemcr.2020.01.005
65. Tang Y, Soroush F, Sun S, Liverani E, Langston JC, Yang Q, et al. Protein kinase C-delta inhibition protects blood-brain barrier from sepsis-induced vascular damage. J Neuroinflammation. (2018) 15(1):309. doi: 10.1186/s12974-018-1342-y
66. McCloskey MC, Kasap P, Ahmad SD, Su SH, Chen KH, Mansouri M, et al. The modular mu SiM: a mass produced, rapidly assembled, and reconfigurable platform for the study of barrier tissue models in vitro. Adv Healthcare Mater. (2022) 11:2200804–00. doi: 10.1002/adhm.202200804. [Epub ahead of print]
67. Barkal LJ, Procknow CL, Alvarez-Garcia YR, Niu M, Jimenez-Torres JA, Brockman-Schneider RA, et al. Microbial volatile communication in human organotypic lung models. Nat Commun. (2017) 8(1):1770. doi: 10.1038/s41467-017-01985-4
68. Salminen AT, McCloskey MC, Ahmad SD, Romanick SS, Chen K, Houlihan W, et al. Molecular mechanisms underlying the heterogeneous barrier responses of two primary endothelial cell types to sphingosine-1-phosphate. Eur J Cell Biol. (2022) 101(3):151233. doi: 10.1016/j.ejcb.2022.151233
69. Lin Y, Weisdorf DJ, Solovey A, Hebbel RP. Origins of circulating endothelial cells and endothelial outgrowth from blood. J Clin Invest. (2000) 105(1):71–7. doi: 10.1172/JCI8071
70. Martin-Ramirez J, Hofman M, van den Biggelaar M, Hebbel RP, Voorberg J. Establishment of outgrowth endothelial cells from peripheral blood. Nat Protoc. (2012) 7(9):1709–15. doi: 10.1038/nprot.2012.093
71. Feitz WJC, van de Kar N, Cheong I, van der Velden T, Ortiz-Sandoval CG, Orth-Holler D, et al. Primary human derived blood outgrowth endothelial cells: an appropriate in vitro model to study shiga toxin mediated damage of endothelial cells. Toxins (Basel). (2020) 12(8):483. doi: 10.3390/toxins12080483
72. Ecklu-Mensah G, Olsen RW, Bengtsson A, Ofori MF, Hviid L, Jensen ATR, et al. Blood outgrowth endothelial cells (BOECs) as a novel tool for studying adhesion of plasmodium falciparum-infected erythrocytes. PLoS One. (2018) 13(10):e0204177. doi: 10.1371/journal.pone.0204177
73. Mathur T, Tronolone JJ, Jain A. Comparative analysis of blood-derived endothelial cells for designing next-generation personalized organ-on-chips. J Am Heart Assoc. (2021) 10(22):e022795. doi: 10.1161/JAHA.121.022795
74. Mathur T, Singh KA, R Pandian NK, Tsai SH, Hein TW, Gaharwar AK, et al. Organ-on-chips made of blood: endothelial progenitor cells from blood reconstitute vascular thromboinflammation in vessel-chips. Lab Chip. (2019) 19(15):2500–11. doi: 10.1039/C9LC00469F
75. Smadja DM, Melero-Martin JM, Eikenboom J, Bowman M, Sabatier F, Randi AM. Standardization of methods to quantify and culture endothelial colony-forming cells derived from peripheral blood: position paper from the international society on thrombosis and haemostasis SSC. J Thromb Haemost. (2019) 17(7):1190–4. doi: 10.1111/jth.14462
76. Jang S, Collin de l'Hortet A, Soto-Gutierrez A. Induced pluripotent stem cell-derived endothelial cells: overview, current advances, applications, and future directions. Am J Pathol. (2019) 189(3):502–12. doi: 10.1016/j.ajpath.2018.12.004
77. Xu M, He J, Zhang C, Xu J, Wang Y. Strategies for derivation of endothelial lineages from human stem cells. Stem Cell Res Ther. (2019) 10(1):200. doi: 10.1186/s13287-019-1274-1
78. Williams IM, Wu JC. Generation of endothelial cells from human pluripotent stem cells. Arterioscler Thromb Vasc Biol. (2019) 39(7):1317–29. doi: 10.1161/ATVBAHA.119.312265
79. Wang K, Lin RZ, Hong X, Ng AH, Lee CN, Neumeyer J, et al. Robust differentiation of human pluripotent stem cells into endothelial cells via temporal modulation of ETV2 with modified mRNA. Sci Adv. (2020) 6(30):eaba7606. doi: 10.1126/sciadv.aba7606
80. Adams WJ, Zhang Y, Cloutier J, Kuchimanchi P, Newton G, Sehrawat S, et al. Functional vascular endothelium derived from human induced pluripotent stem cells. Stem Cell Rep. (2013) 1(2):105–13. doi: 10.1016/j.stemcr.2013.06.007
81. Halaidych OV, Freund C, van den Hil F, Salvatori DCF, Riminucci M, Mummery CL, et al. Inflammatory responses and barrier function of endothelial cells derived from human induced pluripotent stem cells. Stem Cell Rep. (2018) 10(5):1642–56. doi: 10.1016/j.stemcr.2018.03.012
82. Orlova VV, van den Hil FE, Petrus-Reurer S, Drabsch Y, Ten Dijke P, Mummery CL. Generation, expansion and functional analysis of endothelial cells and pericytes derived from human pluripotent stem cells. Nat Protoc. (2014) 9(6):1514–31. doi: 10.1038/nprot.2014.102
83. Nishihara H, Gastfriend BD, Soldati S, Perriot S, Mathias A, Sano Y, et al. Advancing human induced pluripotent stem cell-derived blood-brain barrier models for studying immune cell interactions. FASEB J. (2020) 34(12):16693–715. doi: 10.1096/fj.202001507RR
84. Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. (2010) 37(1):13–25. doi: 10.1016/j.nbd.2009.07.030
85. Korczyn AD. Vascular parkinsonism–characteristics, pathogenesis and treatment. Nat Rev Neurol. (2015) 11(6):319–26. doi: 10.1038/nrneurol.2015.61
86. Mooradian AD, Chung HC, Shah GN. GLUT-1 expression in the cerebra of patients with Alzheimer's disease. Neurobiol Aging. (1997) 18(5):469–74. doi: 10.1016/S0197-4580(97)00111-5
87. Pan Y, Nicolazzo JA. Impact of aging, Alzheimer's disease and Parkinson's disease on the blood-brain barrier transport of therapeutics. Adv Drug Deliv Rev. (2018) 135:62–74. doi: 10.1016/j.addr.2018.04.009
88. Werring DJ, Brassat D, Droogan AG, Clark CA, Symms MR, Barker GJ, et al. The pathogenesis of lesions and normal-appearing white matter changes in multiple sclerosis: a serial diffusion MRI study. Brain. (2000) 123(Pt 8):1667–76. doi: 10.1093/brain/123.8.1667
89. Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol. (2017) 18(2):123–31. doi: 10.1038/ni.3666
90. Owens T, Bechmann I, Engelhardt B. Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol. (2008) 67(12):1113–21. doi: 10.1097/NEN.0b013e31818f9ca8
91. Galea I, Bernardes-Silva M, Forse PA, van Rooijen N, Liblau RS, Perry VH. An antigen-specific pathway for CD8 T cells across the blood-brain barrier. J Exp Med. (2007) 204(9):2023–30. doi: 10.1084/jem.20070064
92. Profaci CP, Munji RN, Pulido RS, Daneman R. The blood-brain barrier in health and disease: important unanswered questions. J Exp Med. (2020) 217(4):e20190062. doi: 10.1084/jem.20190062
93. Mastorakos P, McGavern D. The anatomy and immunology of vasculature in the central nervous system. Sci Immunol. (2019) 4(37):eaav0492. doi: 10.1126/sciimmunol.aav0492
94. Barkalow FJ, Goodman MJ, Gerritsen ME, Mayadas TN. Brain endothelium lack one of two pathways of P-selectin-mediated neutrophil adhesion. Blood. (1996) 88(12):4585–93. doi: 10.1182/blood.V88.12.4585.bloodjournal88124585
95. Cucullo L, Hossain M, Rapp E, Manders T, Marchi N, Janigro D. Development of a humanized in vitro blood-brain barrier model to screen for brain penetration of antiepileptic drugs. Epilepsia. (2007) 48(3):505–16. doi: 10.1111/j.1528-1167.2006.00960.x
96. Park TE, Mustafaoglu N, Herland A, Hasselkus R, Mannix R, FitzGerald EA, et al. Hypoxia-enhanced blood-brain barrier chip recapitulates human barrier function and shuttling of drugs and antibodies. Nat Commun. (2019) 10(1):2621. doi: 10.1038/s41467-019-10588-0
97. Sellgren KL, Hawkins BT, Grego S. An optically transparent membrane supports shear stress studies in a three-dimensional microfluidic neurovascular unit model. Biomicrofluidics. (2015) 9(6):061102. doi: 10.1063/1.4935594
98. Wang YI, Abaci HE, Shuler ML. Microfluidic blood-brain barrier model provides in vivo-like barrier properties for drug permeability screening. Biotechnol Bioeng. (2017) 114(1):184–94. doi: 10.1002/bit.26045
99. Wevers NR, Kasi DG, Gray T, Wilschut KJ, Smith B, van Vught R, et al. A perfused human blood-brain barrier on-a-chip for high-throughput assessment of barrier function and antibody transport. Fluids Barriers CNS. (2018) 15(1):23. doi: 10.1186/s12987-018-0108-3
100. Eigenmann DE, Xue G, Kim KS, Moses AV, Hamburger M, Oufir M. Comparative study of four immortalized human brain capillary endothelial cell lines, hCMEC/D3, hBMEC, TY10, and BB19, and optimization of culture conditions, for an in vitro blood-brain barrier model for drug permeability studies. Fluids Barriers CNS. (2013) 10(1):33. doi: 10.1186/2045-8118-10-33
101. Helms HC, Abbott NJ, Burek M, Cecchelli R, Couraud PO, Deli MA, et al. In vitro models of the blood-brain barrier: an overview of commonly used brain endothelial cell culture models and guidelines for their use. J Cereb Blood Flow Metab. (2016) 36(5):862–90. doi: 10.1177/0271678X16630991
102. Lu TM, Barcia Duran JG, Houghton S, Rafii S, Redmond D, Lis R. Human induced pluripotent stem cell-derived brain endothelial cells: current controversies. Front Physiol. (2021) 12:642812. doi: 10.3389/fphys.2021.642812
103. Santaguida S, Janigro D, Hossain M, Oby E, Rapp E, Cucullo L. Side by side comparison between dynamic versus static models of blood-brain barrier in vitro: a permeability study. Brain Res. (2006) 1109(1):1–13. doi: 10.1016/j.brainres.2006.06.027
104. Cecchelli R, Aday S, Sevin E, Almeida C, Culot M, Dehouck L, et al. A stable and reproducible human blood-brain barrier model derived from hematopoietic stem cells. PLoS One. (2014) 9(6):e99733. doi: 10.1371/journal.pone.0099733
105. Pedroso DC, Tellechea A, Moura L, Fidalgo-Carvalho I, Duarte J, Carvalho E, et al. Improved survival, vascular differentiation and wound healing potential of stem cells co-cultured with endothelial cells. PLoS One. (2011) 6(1):e16114. doi: 10.1371/journal.pone.0016114
106. Lippmann ES, Azarin SM, Palecek SP, Shusta EV. Commentary on human pluripotent stem cell-based blood-brain barrier models. Fluids Barriers CNS. (2020) 17(1):64. doi: 10.1186/s12987-020-00222-3
107. Qian T, Maguire SE, Canfield SG, Bao X, Olson WR, Shusta EV, et al. Directed differentiation of human pluripotent stem cells to blood-brain barrier endothelial cells. Sci Adv. (2017) 3(11):e1701679. doi: 10.1126/sciadv.1701679
108. Workman MJ, Svendsen CN. Recent advances in human iPSC-derived models of the blood-brain barrier. Fluids Barriers CNS. (2020) 17(1):30. doi: 10.1186/s12987-020-00191-7
109. Lippmann ES, Azarin SM, Kay JE, Nessler RA, Wilson HK, Al-Ahmad A, et al. Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat Biotechnol. (2012) 30(8):783–91. doi: 10.1038/nbt.2247
110. Lippmann ES, Al-Ahmad A, Azarin SM, Palecek SP, Shusta EV. A retinoic acid-enhanced, multicellular human blood-brain barrier model derived from stem cell sources. Sci Rep. (2014) 4:4160. doi: 10.1038/srep04160
111. Lu TM, Houghton S, Magdeldin T, Duran JGB, Minotti AP, Snead A, et al. Pluripotent stem cell-derived epithelium misidentified as brain microvascular endothelium requires ETS factors to acquire vascular fate. Proc Natl Acad Sci U S A. (2021) 118(8):e2016950118. doi: 10.1073/pnas.2016950118
112. Nishihara H, Perriot S, Gastfriend BD, Steinfort M, Cibien C, Soldati S, et al. Intrinsic blood-brain barrier dysfunction contributes to multiple sclerosis pathogenesis. Brain. (2022):awac019. doi: 10.1093/brain/awac019. [Epub ahead of print]35085379
113. Gillich A, Zhang F, Farmer CG, Travaglini KJ, Tan SY, Gu M, et al. Capillary cell-type specialization in the alveolus. Nature. (2020) 586(7831):785–9. doi: 10.1038/s41586-020-2822-7
114. Fuchs A, Weibel ER. Morphometrische untersuchung der verteilung einer spezifischen cytoplasmatischen organelle in endothelzellen der ratte. Zeitschrift für Zellforschung und Mikroskopische Anatomie. (1966) 73(1):1–9. doi: 10.1007/BF00348463
115. Wu S, Zhou C, King JA, Stevens T. A unique pulmonary microvascular endothelial cell niche revealed by weibel-palade bodies and Griffonia simplicifolia. Pulm Circ. (2014) 4(1):110–5. doi: 10.1086/674879
116. Doerschuk CM. Mechanisms of leukocyte sequestration in inflamed lungs. Microcirculation. (2001) 8(2):71–88. doi: 10.1111/j.1549-8719.2001.tb00159.x
117. Doerschuk CM. Leukocyte trafficking in alveoli and airway passages. Respir Res. (2000) 1(3):136–40. doi: 10.1186/rr24
118. Huertas A, Guignabert C, Barbera JA, Bartsch P, Bhattacharya J, Bhattacharya S, et al. Pulmonary vascular endothelium: the orchestra conductor in respiratory diseases: highlights from basic research to therapy. Eur Respir J. (2018) 51(4):1700745. doi: 10.1183/13993003.00745-2017
119. Niethamer TK, Stabler CT, Leach JP, Zepp JA, Morley MP, Babu A, et al. Defining the role of pulmonary endothelial cell heterogeneity in the response to acute lung injury. Elife. (2020) 9:e53072. doi: 10.7554/eLife.53072
120. Feuerhake F, Fuchsl G, Bals R, Welsch U. Expression of inducible cell adhesion molecules in the normal human lung: immunohistochemical study of their distribution in pulmonary blood vessels. Histochem Cell Biol. (1998) 110(4):387–94. doi: 10.1007/s004180050299
121. Li F, Li J, Wang PH, Yang N, Huang J, Ou J, et al. SARS-CoV-2 spike promotes inflammation and apoptosis through autophagy by ROS-suppressed PI3K/AKT/mTOR signaling. Biochim Biophys Acta Mol Basis Dis. (2021) 1867(12):166260. doi: 10.1016/j.bbadis.2021.166260
122. Miller AJ, Spence JR. In vitro models to study human lung development, disease and homeostasis. Physiology (Bethesda). (2017) 32(3):246–60. doi: 10.1152/physiol.00041.2016
123. Nichols JE, Niles JA, Vega SP, Argueta LB, Eastaway A, Cortiella J. Modeling the lung: design and development of tissue engineered macro- and micro-physiologic lung models for research use. Exp Biol Med (Maywood). (2014) 239(9):1135–69. doi: 10.1177/1535370214536679
124. Hermanns MI, Fuchs S, Bock M, Wenzel K, Mayer E, Kehe K, et al. Primary human coculture model of alveolo-capillary unit to study mechanisms of injury to peripheral lung. Cell Tissue Res. (2009) 336(1):91–105. doi: 10.1007/s00441-008-0750-1
125. Comhair SA, Xu W, Mavrakis L, Aldred MA, Asosingh K, Erzurum SC. Human primary lung endothelial cells in culture. Am J Respir Cell Mol Biol. (2012) 46(6):723–30. doi: 10.1165/rcmb.2011-0416TE
126. Taniguchi D, Matsumoto K, Machino R, Takeoka Y, Elgalad A, Taura Y, et al. Human lung microvascular endothelial cells as potential alternatives to human umbilical vein endothelial cells in bio-3D-printed trachea-like structures. Tissue Cell. (2020) 63:101321. doi: 10.1016/j.tice.2019.101321
127. Molema G, Aird WC. Vascular heterogeneity in the kidney. Semin Nephrol. (2012) 32(2):145–55. doi: 10.1016/j.semnephrol.2012.02.001
128. Kitagawa A, Tsuboi N, Yokoe Y, Katsuno T, Ikeuchi H, Kajiyama H, et al. Urinary levels of the leukocyte surface molecule CD11b associate with glomerular inflammation in lupus nephritis. Kidney Int. (2019) 95(3):680–92. doi: 10.1016/j.kint.2018.10.025
129. Molema G, Zijlstra JG, van Meurs M, Kamps J. Renal microvascular endothelial cell responses in sepsis-induced acute kidney injury. Nat Rev Nephrol. (2022) 18(2):95–112. doi: 10.1038/s41581-021-00489-1
130. Torres A, Munoz K, Nahuelpan Y, R Saez AP, Mendoza P, Jara C, et al. Intraglomerular monocyte/macrophage infiltration and macrophage-myofibroblast transition during diabetic nephropathy is regulated by the A2B adenosine receptor. Cells. (2020) 9(4):1051. doi: 10.3390/cells9041051
131. Zheng X, Soroush F, Long J, Hall ET, Adishesha PK, Bhattacharya S, et al. Murine glomerular transcriptome links endothelial cell-specific molecule-1 deficiency with susceptibility to diabetic nephropathy. PLoS One. (2017) 12(9):e0185250. doi: 10.1371/journal.pone.0185250
132. Devi S, Li A, Westhorpe CL, Lo CY, Abeynaike LD, Snelgrove SL, et al. Multiphoton imaging reveals a new leukocyte recruitment paradigm in the glomerulus. Nat Med. (2013) 19(1):107–12. doi: 10.1038/nm.3024
133. Finsterbusch M, Hall P, Li A, Devi S, Westhorpe CL, Kitching AR, et al. Patrolling monocytes promote intravascular neutrophil activation and glomerular injury in the acutely inflamed glomerulus. Proc Natl Acad Sci U S A. (2016) 113(35):E5172–81. doi: 10.1073/pnas.1606253113
134. Kuligowski MP, Kitching AR, Hickey MJ. Leukocyte recruitment to the inflamed glomerulus: a critical role for platelet-derived P-selectin in the absence of rolling. J Immunol. (2006) 176(11):6991–9. doi: 10.4049/jimmunol.176.11.6991
135. Zhu D, Tang Q, Yu B, Meng M, Liu W, Li J, et al. Major histocompatibility complexes are up-regulated in glomerular endothelial cells via activation of c-Jun N-terminal kinase in 5/6 nephrectomy mice. Br J Pharmacol. (2020) 177(22):5131–47. doi: 10.1111/bph.15237
136. Kitching AR, Hutton HL. The players: cells involved in glomerular disease. Clin J Am Soc Nephrol. (2016) 11(9):1664–74. doi: 10.2215/CJN.13791215
137. Sol M, Kamps J, van den Born J, van den Heuvel MC, van der Vlag J, Krenning G, et al. Glomerular endothelial cells as instigators of glomerular sclerotic diseases. Front Pharmacol. (2020) 11:573557. doi: 10.3389/fphar.2020.573557
138. Lee J, Kim K, Kim S. Kidney on chips. Methods Cell Biol. (2018) 146:85–104. doi: 10.1016/bs.mcb.2018.06.001
139. Lin NYC, Homan KA, Robinson SS, Kolesky DB, Duarte N, Moisan A, et al. Renal reabsorption in 3D vascularized proximal tubule models. Proc Natl Acad Sci U S A. (2019) 116(12):5399–404. doi: 10.1073/pnas.1815208116
140. CellSystems. Primary Human Glomerular Microvascular Endothelial Cells (ACBRI 128). Available at: https://cell-systems.com/products/primary-human-glomerular-microvascular-endothelial-cells-acbri-128?gclid=Cj0KCQjw1ZeUBhDyARIsAOzAqQJ8YLkmZuVzWgq3UdgA0P_dTYWCy5AdV6CdyUAFp2l-3Tminwg-uBsaAvVyEALw_wcB (updated 2022).
141. Petrosyan A, Cravedi P, Villani V, Angeletti A, Manrique J, Renieri A, et al. Author correction: a glomerulus-on-a-chip to recapitulate the human glomerular filtration barrier. Nat Commun. (2019) 10(1):4791. doi: 10.1038/s41467-019-12177-7
142. Strauss O, Phillips A, Ruggiero K, Bartlett A, Dunbar PR. Immunofluorescence identifies distinct subsets of endothelial cells in the human liver. Sci Rep. (2017) 7:44356. doi: 10.1038/srep44356
143. Knolle PA, Limmer A. Neighborhood politics: the immunoregulatory function of organ-resident liver endothelial cells. Trends Immunol. (2001) 22(8):432–7. doi: 10.1016/S1471-4906(01)01957-3
144. Racanelli V, Rehermann B. The liver as an immunological organ. Hepatology. (2006) 43(2 Suppl 1):S54–62. doi: 10.1002/hep.21060
145. Guidotti LG, Inverso D, Sironi L, Di Lucia P, Fioravanti J, Ganzer L, et al. Immunosurveillance of the liver by intravascular effector CD8(+) T cells. Cell. (2015) 161(3):486–500. doi: 10.1016/j.cell.2015.03.005
146. Chaudhry S, Emond J, Griesemer A. Immune cell trafficking to the liver. Transplantation. (2019) 103(7):1323–37. doi: 10.1097/TP.0000000000002690
147. Bonnardel J, T'Jonck W, Gaublomme D, Browaeys R, Scott CL, Martens L, et al. Stellate cells, hepatocytes, and endothelial cells imprint the kupffer cell identity on monocytes colonizing the liver macrophage niche. Immunity. (2019) 51(4):638–54.e9. doi: 10.1016/j.immuni.2019.08.017
148. Chosay JG, Fisher MA, Farhood A, Ready KA, Dunn CJ, Jaeschke H. Role of PECAM-1 (CD31) in neutrophil transmigration in murine models of liver and peritoneal inflammation. Am J Physiol. (1998) 274(4):G776–82. doi: 10.1152/ajpgi.1998.274.4.G776
149. Khandoga A, Kessler JS, Meissner H, Hanschen M, Corada M, Motoike T, et al. Junctional adhesion molecule-A deficiency increases hepatic ischemia-reperfusion injury despite reduction of neutrophil transendothelial migration. Blood. (2005) 106(2):725–33. doi: 10.1182/blood-2004-11-4416
150. McNab G, Reeves JL, Salmi M, Hubscher S, Jalkanen S, Adams DH. Vascular adhesion protein 1 mediates binding of T cells to human hepatic endothelium. Gastroenterology. (1996) 110(2):522–8. doi: 10.1053/gast.1996.v110.pm8566600
151. Shetty S, Lalor PF, Adams DH. Liver sinusoidal endothelial cells - gatekeepers of hepatic immunity. Nat Rev Gastroenterol Hepatol. (2018) 15(9):555–67. doi: 10.1038/s41575-018-0020-y
152. Ozkan A, Stolley D, Cressman ENK, McMillin M, DeMorrow S, Yankeelov TE, et al. The influence of chronic liver diseases on hepatic vasculature: a liver-on-a-chip review. Micromachines (Basel). (2020) 11(5):487. doi: 10.3390/mi11050487
153. Poisson J, Lemoinne S, Boulanger C, Durand F, Moreau R, Valla D, et al. Liver sinusoidal endothelial cells: physiology and role in liver diseases. J Hepatol. (2017) 66(1):212–27. doi: 10.1016/j.jhep.2016.07.009
154. Ehrlich A, Duche D, Ouedraogo G, Nahmias Y. Challenges and opportunities in the design of liver-on-chip microdevices. Annu Rev Biomed Eng. (2019) 21:219–39. doi: 10.1146/annurev-bioeng-060418-052305
155. Beckwitt CH, Clark AM, Wheeler S, Taylor DL, Stolz DB, Griffith L, et al. Liver ‘organ on a chip’. Exp Cell Res. (2018) 363(1):15–25. doi: 10.1016/j.yexcr.2017.12.023
156. Koui Y, Kido T, Ito T, Oyama H, Chen SW, Katou Y, et al. An in vitro human liver model by iPSC-derived parenchymal and non-parenchymal cells. Stem Cell Rep. (2017) 9(2):490–8. doi: 10.1016/j.stemcr.2017.06.010
157. Gage BK, Liu JC, Innes BT, MacParland SA, McGilvray ID, Bader GD, et al. Generation of functional liver sinusoidal endothelial cells from human pluripotent stem-cell-derived venous angioblasts. Cell Stem Cell. (2020) 27(2):254–69.e9. doi: 10.1016/j.stem.2020.06.007
158. De Smedt J, van Os EA, Talon I, Ghosh S, Toprakhisar B, Furtado Madeiro Da Costa R, et al. PU.1 drives specification of pluripotent stem cell-derived endothelial cells to LSEC-like cells. Cell Death Dis. (2021) 12(1):84. doi: 10.1038/s41419-020-03356-2
159. Smith AR, Gouon-Evans V. c-Maf: the magic wand that turns on LSEC fate. Cell Stem Cell. (2022) 29(4):491–3. doi: 10.1016/j.stem.2022.03.011
160. Ajalik RE, Alenchery RG, Cognetti JS, Zhang VZ, McGrath JL, Miller BL, et al. Human organ-on-a-chip microphysiological systems to model musculoskeletal pathologies and accelerate therapeutic discovery. Front Bioeng Biotechnol. (2022) 10. doi: 10.3389/fbioe.2022.846230
161. Latroche C, Gitiaux C, Chrétien F, Desguerre I, Mounier R, Chazaud B. Skeletal muscle microvasculature: a highly dynamic lifeline. Physiology. (2015) 30(6):417–27. doi: 10.1152/physiol.00026.2015
162. Filipowska J, Tomaszewski KA, Niedźwiedzki Ł, Walocha JA, Niedźwiedzki T. The role of vasculature in bone development, regeneration and proper systemic functioning. Angiogenesis. (2017) 20(3):291–302. doi: 10.1007/s10456-017-9541-1
163. Haywood L, Walsh DA. Vasculature of the normal and arthritic synovial joint. Histol Histopathol. (2001) 16(1):277–84. doi: 10.14670/HH-16.277
164. Fox AJS, Bedi A, Rodeo SA. The basic science of human knee menisci: structure, composition, and function. Sports Health. (2012) 4(4):340–51. doi: 10.1177/1941738111429419
165. Tempfer H, Traweger A. Tendon vasculature in health and disease. Front Physiol. (2015) 6. doi: 10.3389/fphys.2015.00330
166. Fenwick SA, Hazleman BL, Riley GP. The vasculature and its role in the damaged and healing tendon. Arthritis Res. (2002) 4(4):252–60. doi: 10.1186/ar416
167. Fournier DE, Kiser PK, Shoemaker JK, Battié MC, Séguin CA. Vascularization of the human intervertebral disc: a scoping review. JOR SPINE. (2020) 3(4):e1123. doi: 10.1002/jsp2.1123
168. Fenwick SA, Gregg PJ, Rooney P. Osteoarthritic cartilage loses its ability to remain avascular. Osteoarthritis Cartilage. (1999) 7(5):441–52. doi: 10.1053/joca.1998.0238
169. Osaki T, Sivathanu V, Kamm RD. Crosstalk between developing vasculature and optogenetically engineered skeletal muscle improves muscle contraction and angiogenesis. Biomaterials. (2018) 156:65–76. doi: 10.1016/j.biomaterials.2017.11.041
170. Langen UH, Pitulescu ME, Kim JM, Enriquez-Gasca R, Sivaraj KK, Kusumbe AP, et al. Cell–matrix signals specify bone endothelial cells during developmental osteogenesis. Nat Cell Biol. (2017) 19(3):189–201. doi: 10.1038/ncb3476
171. Ramasamy SK, Kusumbe AP, Schiller M, Zeuschner D, Bixel MG, Milia C, et al. Blood flow controls bone vascular function and osteogenesis. Nat Commun. (2016) 7(1):13601. doi: 10.1038/ncomms13601
172. Itkin T, Gur-Cohen S, Spencer JA, Schajnovitz A, Ramasamy SK, Kusumbe AP, et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature. (2016) 532(7599):323–8. doi: 10.1038/nature17624
173. Kopp H-G, Avecilla ST, Hooper AT, Rafii S. The bone marrow vascular niche: home of HSC differentiation and mobilization. Physiology. (2005) 20(5):349–56. doi: 10.1152/physiol.00025.2005
174. Chou DB, Frismantas V, Milton Y, David R, Pop-Damkov P, Ferguson D, et al. On-chip recapitulation of clinical bone marrow toxicities and patient-specific pathophysiology. Nature Biomedical Engineering. (2020) 4(4):394–406. doi: 10.1038/s41551-019-0495-z
175. Millar NL, Silbernagel KG, Thorborg K, Kirwan PD, Galatz LM, Abrams GD, et al. Tendinopathy. Nature Reviews Disease Primers. (2021) 7(1):1. doi: 10.1038/s41572-020-00234-1
176. Lehner C, Gehwolf R, Ek J, Korntner S, Bauer H, Bauer HC, et al. The blood-tendon barrier: identification and characterisation of a novel tissue barrier in tendon blood vessels. European Cells and Materials. (2016) 31:296–311. doi: 10.22203/eCM.v031a19
177. Maffioletti SM, Sarcar S, Henderson ABH, Mannhardt I, Pinton L, Moyle LA, et al. Three-dimensional human iPSC-derived artificial skeletal muscles model muscular dystrophies and enable multilineage tissue engineering. Cell Rep. (2018) 23(3):899–908. doi: 10.1016/j.celrep.2018.03.091
178. Avolio E, Alvino VV, Ghorbel MT, Campagnolo P. Perivascular cells and tissue engineering: current applications and untapped potential. Pharmacol Ther. (2017) 171:83–92. doi: 10.1016/j.pharmthera.2016.11.002
179. Dorrier CE, Jones HE, Pintaric L, Siegenthaler JA, Daneman R. Emerging roles for CNS fibroblasts in health, injury and disease. Nat Rev Neurosci. (2022) 23(1):23–34. doi: 10.1038/s41583-021-00525-w
180. Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. (2011) 21(2):193–215. doi: 10.1016/j.devcel.2011.07.001
181. Dessalles CA, Babataheri A, Barakat AI. Pericyte mechanics and mechanobiology. J Cell Sci. (2021) 134(6):jcs240226. doi: 10.1242/jcs.240226
182. Zhang ZS, Zhou HN, He SS, Xue MY, Li T, Liu LM. Research advances in pericyte function and their roles in diseases. Chin J Traumatol. (2020) 23(2):89–95. doi: 10.1016/j.cjtee.2020.02.006
183. Rudziak P, Ellis CG, Kowalewska PM. Role and molecular mechanisms of pericytes in regulation of leukocyte diapedesis in inflamed tissues. Mediators Inflamm. (2019) 2019:4123605. doi: 10.1155/2019/4123605
184. Caporarello N, D'Angeli F, Cambria MT, Candido S, Giallongo C, Salmeri M, et al. Pericytes in microvessels: from “mural” function to brain and retina regeneration. Int J Mol Sci. (2019) 20(24):6351. doi: 10.3390/ijms20246351
185. Gronloh MLB, Arts JJG, van Buul JD. Neutrophil transendothelial migration hotspots - mechanisms and implications. J Cell Sci. (2021) 134(7):jcs255653. doi: 10.1242/jcs.255653
186. Voisin MB, Nourshargh S. Neutrophil transmigration: emergence of an adhesive cascade within venular walls. J Innate Immun. (2013) 5(4):336–47. doi: 10.1159/000346659
187. Pieper C, Galla HJ. Ultra structure analysis of cell-cell interactions between pericytes and neutrophils in vitro. Biochem Biophys Res Commun. (2014) 445(1):180–3. doi: 10.1016/j.bbrc.2014.01.159
188. Pieper C, Pieloch P, Galla HJ. Pericytes support neutrophil transmigration via interleukin-8 across a porcine co-culture model of the blood-brain barrier. Brain Res. (2013) 1524:1–11. doi: 10.1016/j.brainres.2013.05.047
189. Proebstl D, Voisin MB, Woodfin A, Whiteford J, D'Acquisto F, Jones GE, et al. Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J Exp Med. (2012) 209(6):1219–34. doi: 10.1084/jem.20111622
190. Sava P, Cook IO, Mahal RS, Gonzalez AL. Human microvascular pericyte basement membrane remodeling regulates neutrophil recruitment. Microcirculation. (2015) 22(1):54–67. doi: 10.1111/micc.12173
191. Kumar A, D'Souza SS, Moskvin OV, Toh H, Wang B, Zhang J, et al. Specification and diversification of pericytes and smooth muscle cells from mesenchymoangioblasts. Cell Rep. (2017) 19(9):1902–16. doi: 10.1016/j.celrep.2017.05.019
192. Lauridsen HM, Pober JS, Gonzalez AL. A composite model of the human postcapillary venule for investigation of microvascular leukocyte recruitment. FASEB J. (2014) 28(3):1166–80. doi: 10.1096/fj.13-240986
193. Maier CL, Pober JS. Human placental pericytes poorly stimulate and actively regulate allogeneic CD4 T cell responses. Arterioscler Thromb Vasc Biol. (2011) 31(1):183–9. doi: 10.1161/ATVBAHA.110.217117
194. Torok O, Schreiner B, Schaffenrath J, Tsai HC, Maheshwari U, Stifter SA, et al. Pericytes regulate vascular immune homeostasis in the CNS. Proc Natl Acad Sci U S A. (2021) 118(10):e2016587118. doi: 10.1073/pnas.2016587118
195. Li P, Wu Y, Goodwin AJ, Halushka PV, Wilson CL, Schnapp LM, et al. Generation of a new immortalized human lung pericyte cell line: a promising tool for human lung pericyte studies. Lab Invest. (2021) 101(5):625–35. doi: 10.1038/s41374-020-00524-y
196. Smyth LCD, Rustenhoven J, Park TI, Schweder P, Jansson D, Heppner PA, et al. Unique and shared inflammatory profiles of human brain endothelia and pericytes. J Neuroinflammation. (2018) 15(1):138. doi: 10.1186/s12974-018-1167-8
197. Gauer S, Yao J, Schoecklmann HO, Sterzel RB. Adhesion molecules in the glomerular mesangium. Kidney Int. (1997) 51(5):1447–53. doi: 10.1038/ki.1997.198
198. Hsu J, Rappaport J, Muro S. Specific binding, uptake, and transport of ICAM-1-targeted nanocarriers across endothelial and subendothelial cell components of the blood-brain barrier. Pharm Res. (2014) 31(7):1855–66. doi: 10.1007/s11095-013-1289-8
199. Knittel T, Dinter C, Kobold D, Neubauer K, Mehde M, Eichhorst S, et al. Expression and regulation of cell adhesion molecules by hepatic stellate cells (HSC) of rat liver: involvement of HSC in recruitment of inflammatory cells during hepatic tissue repair. Am J Pathol. (1999) 154(1):153–67. doi: 10.1016/S0002-9440(10)65262-5
200. Stebbins MJ, Gastfriend BD, Canfield SG, Lee MS, Richards D, Faubion MG, et al. Human pluripotent stem cell-derived brain pericyte-like cells induce blood-brain barrier properties. Sci Adv. (2019) 5(3):eaau7375. doi: 10.1126/sciadv.aau7375
201. Wanjare M, Kusuma S, Gerecht S. Defining differences among perivascular cells derived from human pluripotent stem cells. Stem Cell Rep. (2014) 2(5):561–75. doi: 10.1016/j.stemcr.2014.03.004
202. Xu J, Gong T, Heng BC, Zhang CF. A systematic review: differentiation of stem cells into functional pericytes. FASEB J. (2017) 31(5):1775–86. doi: 10.1096/fj.201600951RRR
203. Faal T, Phan DTT, Davtyan H, Scarfone VM, Varady E, Blurton-Jones M, et al. Induction of mesoderm and neural crest-derived pericytes from human pluripotent stem cells to study blood-brain barrier interactions. Stem Cell Rep. (2019) 12(3):451–60. doi: 10.1016/j.stemcr.2019.01.005
204. Blanchard JW, Bula M, Davila-Velderrain J, Akay LA, Zhu L, Frank A, et al. Reconstruction of the human blood-brain barrier in vitro reveals a pathogenic mechanism of APOE4 in pericytes. Nat Med. (2020) 26(6):952–63. doi: 10.1038/s41591-020-0886-4
205. Lyu Z, Park J, Kim KM, Jin HJ, Wu H, Rajadas J, et al. A neurovascular-unit-on-a-chip for the evaluation of the restorative potential of stem cell therapies for ischaemic stroke. Nat Biomed Eng. (2021) 5(8):847–63. doi: 10.1038/s41551-021-00744-7
206. Pediaditakis I, Kodella KR, Manatakis DV, Le CY, Hinojosa CD, Tien-Street W, et al. Modeling alpha-synuclein pathology in a human brain-chip to assess blood-brain barrier disruption. Nat Commun. (2021) 12(1):5907. doi: 10.1038/s41467-021-26066-5
207. Badolato R, Sozzani S. Lymphocyte trafficking in health and disease. Basel, Boston: Birkhauser (2006). x, 247.
208. Irimia D, Wang X. Inflammation-on-a-Chip: probing the immune system ex vivo. Trends Biotechnol. (2018) 36(9):923–37. doi: 10.1016/j.tibtech.2018.03.011
209. Janeway C. Immunobiology: the immune system in health and disease. 6th ed. New York: Garland Science (2005). xxiii, 823.
210. Hong C-W. Current understanding in neutrophil differentiation and heterogeneity. Immune Netw. (2017) 17(5):298–306. doi: 10.4110/in.2017.17.5.298
211. Blumenreich MS. The white blood cell and differential count. In: Walker HK, Hall DW, Hurst JW, editors. Clinical methods: The history, physical, and laboratory examinations. 3rd ed. Boston, MA: Butterworths (1990) Chapter 153.
212. Mayadas TN, Cullere X, Lowell CA. The multifaceted functions of neutrophils. Annu Rev Pathol. (2014) 9:181–218. doi: 10.1146/annurev-pathol-020712-164023
213. Cowland JB, Borregaard N. Granulopoiesis and granules of human neutrophils. Immunol Rev. (2016) 273(1):11–28. doi: 10.1111/imr.12440
214. Lawrence SM, Corriden R, Nizet V. The ontogeny of a neutrophil: mechanisms of granulopoiesis and homeostasis. Microbiol Mol Biol Rev. (2018) 82(1):e00057–17. doi: 10.1128/MMBR.00057-17
215. Ohms M, Möller S, Laskay T. An attempt to polarize human neutrophils toward N1 and N2 phenotypes in vitro. Front Immunol. (2020) 11:532. doi: 10.3389/fimmu.2020.00532
216. Rosales C. Neutrophil: a cell with many roles in inflammation or several cell types? Front Physiol. (2018) 9:113. doi: 10.3389/fphys.2018.00113
217. Silvestre-Roig C, Fridlender ZG, Glogauer M, Scapini P. Neutrophil diversity in health and disease. Trends Immunol. (2019) 40(7):565–83. doi: 10.1016/j.it.2019.04.012
218. Ng LG, Ostuni R, Hidalgo A. Heterogeneity of neutrophils. Nat Rev Immunol. (2019) 19(4):255–65. doi: 10.1038/s41577-019-0141-8
219. Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil kinetics in health and disease. Trends Immunol. (2010) 31(8):318–24. doi: 10.1016/j.it.2010.05.006
220. Tak T, Tesselaar K, Pillay J, Borghans JA, Koenderman L. What's your age again? Determination of human neutrophil half-lives revisited. J Leukoc Biol. (2013) 94(4):595–601. doi: 10.1189/jlb.1112571
221. Hajishengallis G, Chavakis T, Hajishengallis E, Lambris JD. Neutrophil homeostasis and inflammation: novel paradigms from studying periodontitis. J Leukoc Biol. (2015) 98(4):539–48. doi: 10.1189/jlb.3VMR1014-468R
222. Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med. (2011) 17(11):1381–90. doi: 10.1038/nm.2514
223. Witko-Sarsat V, Rieu P, Descamps-Latscha B, Lesavre P, Halbwachs-Mecarelli L. Neutrophils: molecules, functions and pathophysiological aspects. Lab Invest. (2000) 80(5):617–53. doi: 10.1038/labinvest.3780067
224. Scapini P, Cassatella MA. Social networking of human neutrophils within the immune system. Blood. (2014) 124(5):710–9. doi: 10.1182/blood-2014-03-453217
225. Riddle RB, Jennbacken K, Hansson KM, Harper MT. Endothelial inflammation and neutrophil transmigration are modulated by extracellular matrix composition in an inflammation-on-a-chip model. Sci Rep. (2022) 12(1):6855. doi: 10.1038/s41598-022-10849-x
226. Gjorevski N, Avignon B, Gérard R, Cabon L, Roth AB, Bscheider M, et al. Neutrophilic infiltration in organ-on-a-chip model of tissue inflammation. Lab Chip. (2020) 20(18):3365–74. doi: 10.1039/D0LC00417K
227. Moarefian M, Davalos RV, Burton MD, Jones CN. Electrotaxis-on-chip to quantify neutrophil migration towards electrochemical gradients. Front Immunol. (2021) 12:674727. doi: 10.3389/fimmu.2021.674727
228. Gröger M, Rennert K, Giszas B, Weiß E, Dinger J, Funke H, et al. Monocyte-induced recovery of inflammation-associated hepatocellular dysfunction in a biochip-based human liver model. Sci Rep. (2016) 6(1):21868. doi: 10.1038/srep21868
229. Sharifi F, Htwe SS, Righi M, Liu H, Pietralunga A, Yesil-Celiktas O, et al. A foreign body response-on-a-chip platform. Adv Healthcare Mater. (2019) e1801425. doi: 10.1002/adhm.201801425
230. Ronaldson-Bouchard K, Teles D, Yeager K, Tavakol DN, Zhao Y, Chramiec A, et al. A multi-organ chip with matured tissue niches linked by vascular flow. Nature Biomedical Engineering. (2022) 6(4):351–71. doi: 10.1038/s41551-022-00882-6
231. de Haan L, Suijker J, van Roey R, Berges N, Petrova E, Queiroz K, et al. A microfluidic 3D endothelium-on-a-chip model to study transendothelial migration of T cells in health and disease. Int J Mol Sci. (2021) 22(15):8234. doi: 10.3390/ijms22158234
232. Park JY, Kim HO, Kim KD, Kim SK, Lee SK, Jung H. Monitoring the status of T-cell activation in a microfluidic system. Analyst. (2011) 136(13):2831–6. doi: 10.1039/c1an15038c
233. Avci E, Akkaya-Ulum YZ, Yoyen-Ermis D, Esendagli G, Balci-Peynircioglu B. A method for high-purity isolation of neutrophil granulocytes for functional cell migration assays. Turkish Journal of Biochemistry. (2019) 44(6):810–21. doi: 10.1515/tjb-2019-0089
234. Martin SJ, Bradley JG, Cotter TG. HL-60 cells induced to differentiate towards neutrophils subsequently die via apoptosis. Clin Exp Immunol. (1990) 79(3):448–53. doi: 10.1111/j.1365-2249.1990.tb08110.x
235. Majumder A, Suknuntha K, Bennin D, Klemm L, Brok-Volchanskaya VS, Huttenlocher A, et al. Generation of human neutrophils from induced pluripotent stem cells in chemically defined conditions using ETV2 modified mRNA. STAR Protoc. (2020) 1(2):100075. doi: 10.1016/j.xpro.2020.100075
236. Guo Y, Gao F, Wang Q, Wang K, Pan S, Pan Z, et al. Differentiation of HL-60 cells in serum-free hematopoietic cell media enhances the production of neutrophil extracellular traps. Exp Ther Med. (2021) 21(4):353. doi: 10.3892/etm.2021.9784
237. Yaseen R, Blodkamp S, Lüthje P, Reuner F, Völlger L, Naim HY, et al. Antimicrobial activity of HL-60 cells compared to primary blood-derived neutrophils against Staphylococcus aureus. J Negat Results Biomed. (2017) 16(1):2. doi: 10.1186/s12952-017-0067-2
238. Brok-Volchanskaya VS, Bennin DA, Suknuntha K, Klemm LC, Huttenlocher A, Slukvin I. Effective and rapid generation of functional neutrophils from induced pluripotent stem cells using ETV2-modified mRNA. Stem Cell Rep. (2019) 13(6):1099–110. doi: 10.1016/j.stemcr.2019.10.007
239. van Furth R, Beekhuizen H. Monocytes. In: Delves PJ, editors. Encyclopedia of immunology. 2nd ed. Oxford: Elsevier (1998). p. 1750–4.
240. Guilliams M, Mildner A, Yona S. Developmental and functional heterogeneity of monocytes. Immunity. (2018) 49(4):595–613. doi: 10.1016/j.immuni.2018.10.005
241. Hume DA, Irvine KM, Pridans C. The mononuclear phagocyte system: the relationship between monocytes and macrophages. Trends Immunol. (2019) 40(2):98–112. doi: 10.1016/j.it.2018.11.007
242. Riddy DM, Goy E, Delerive P, Summers RJ, Sexton PM, Langmead CJ. Comparative genotypic and phenotypic analysis of human peripheral blood monocytes and surrogate monocyte-like cell lines commonly used in metabolic disease research. PLoS One. (2018) 13(5):e0197177. doi: 10.1371/journal.pone.0197177
243. Spiller KL, Wrona EA, Romero-Torres S, Pallotta I, Graney PL, Witherel CE, et al. Differential gene expression in human, murine, and cell line-derived macrophages upon polarization. Exp Cell Res. (2016) 347(1):1–13. doi: 10.1016/j.yexcr.2015.10.017
244. Cao X, Yakala GK, Van Den Hil FE, Cochrane A, Mummery CL, Orlova VV. Differentiation and functional comparison of monocytes and macrophages from hiPSCs with peripheral blood derivatives. Stem Cell Rep. (2019) 12(6):1282–97. doi: 10.1016/j.stemcr.2019.05.003
245. Takata K, Kozaki T, Lee CZW, Thion MS, Otsuka M, Lim S, et al. Induced-Pluripotent-Stem-Cell-Derived primitive macrophages provide a platform for modeling tissue-resident macrophage differentiation and function. Immunity. (2017) 47(1):183–98.e6. doi: 10.1016/j.immuni.2017.06.017
246. Yanagimachi MD, Niwa A, Tanaka T, Honda-Ozaki F, Nishimoto S, Murata Y, et al. Robust and highly-efficient differentiation of functional monocytic cells from human pluripotent stem cells under Serum- and feeder cell-free conditions. PLoS One. (2013) 8(4):e59243. doi: 10.1371/journal.pone.0059243
247. Cui D, Franz A, Fillon SA, Jannetti L, Isambert T, Fundel-Clemens K, et al. High-yield human induced pluripotent stem cell-derived monocytes and macrophages are functionally comparable with primary cells. Front Cell Dev Biol. (2021) 9:656867. doi: 10.3389/fcell.2021.656867
248. Cano RLE, Lopera HDE. Introduction to T and B lymphocytes. In: Anaya JM, Shoenfeld Y, Rojas-Villarraga A, Levy RA, Cervera R, editors. Autoimmunity: From bench to bedside. Bogota, Colombia: El Rosario University Press © 2013 Universidad del Rosario. (2013) Chapter 5.
251. Constantin G, Laudanna C. Lymphocyte-endothelial cell interaction. In: Badolato R, Sozzani S, editors. Lymphocyte trafficking in health and disease. Birkhäuser Basel: Basel (2006). p. 39–54.
252. Faley S, Seale K, Hughey J, Schaffer DK, VanCompernolle S, McKinney B, et al. Microfluidic platform for real-time signaling analysis of multiple single T cells in parallel. Lab Chip. (2008) 8(10):1700–12. doi: 10.1039/b719799c
253. Giese C, Lubitz A, Demmler CD, Reuschel J, Bergner K, Marx U. Immunological substance testing on human lymphatic micro-organoids in vitro. J Biotechnol. (2010) 148(1):38–45. doi: 10.1016/j.jbiotec.2010.03.001
254. Kirschbaum M, Jaeger MS, Schenkel T, Breinig T, Meyerhans A, Duschl C. T cell activation on a single-cell level in dielectrophoresis-based microfluidic devices. J Chromatogr A. (2008) 1202(1):83–9. doi: 10.1016/j.chroma.2008.06.036
255. Moura Rosa P, Gopalakrishnan N, Ibrahim H, Haug M, Halaas O. The intercell dynamics of T cells and dendritic cells in a lymph node-on-a-chip flow device. Lab Chip. (2016) 16(19):3728–40. doi: 10.1039/C6LC00702C
256. Kim SK, Moon WK, Park JY, Jung H. Inflammatory mimetic microfluidic chip by immobilization of cell adhesion molecules for T cell adhesion. Analyst. (2012) 137(17):4062–8. doi: 10.1039/c2an35424a
257. Ren X, Getschman AE, Hwang S, Volkman BF, Klonisch T, Levin D, et al. Investigations on T cell transmigration in a human skin-on-chip (SoC) model. Lab Chip. (2021) 21(8):1527–39. doi: 10.1039/D0LC01194K
258. Lin F, Butcher EC. T cell chemotaxis in a simple microfluidic device. Lab Chip. (2006) 6(11):1462–9. doi: 10.1039/B607071J
259. Deng J, Wang ES, Jenkins RW, Li S, Dries R, Yates K, et al. CDK4/6 Inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov. (2018) 8(2):216–33. doi: 10.1158/2159-8290.CD-17-0915
260. Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, et al. Ex vivo profiling of PD-1 blockade using organotypic tumor spheroids. Cancer Discov. (2018) 8(2):196–215. doi: 10.1158/2159-8290.CD-17-0833
261. Moore N, Doty D, Zielstorff M, Kariv I, Moy LY, Gimbel A, et al. A multiplexed microfluidic system for evaluation of dynamics of immune-tumor interactions. Lab Chip. (2018) 18(13):1844–58. doi: 10.1039/C8LC00256H
262. Pavesi A, Tan AT, Koh S, Chia A, Colombo M, Antonecchia E, et al. A 3D microfluidic model for preclinical evaluation of TCR-engineered T cells against solid tumors. JCI Insight. (2017) 2(12):e89762. doi: 10.1172/jci.insight.89762
263. Boussommier-Calleja A, Li R, Chen MB, Wong SC, Kamm RD. Microfluidics: a new tool for modeling cancer-immune interactions. Trends Cancer. (2016) 2(1):6–19. doi: 10.1016/j.trecan.2015.12.003
264. Hammel JH, Cook SR, Belanger MC, Munson JM, Pompano RR. Modeling immunity in vitro: slices, chips, and engineered tissues. Annu Rev Biomed Eng. (2021) 23:461–91. doi: 10.1146/annurev-bioeng-082420-124920
265. Salminen AT, Allahyari Z, Gholizadeh S, McCloskey MC, Ajalik R, Cottle RN, et al. In vitro studies of transendothelial migration for biological and drug discovery. Front Med Technol. (2020) 2:600616. doi: 10.3389/fmedt.2020.600616
266. Xie H, Appelt JW, Jenkins RW. Going with the flow: modeling the tumor microenvironment using microfluidic technology. Cancers (Basel). (2021) 13(23):6052. doi: 10.3390/cancers13236052
267. Kobayashi T, Kumagai S, Doi R, Afonina E, Koyama S, Nishikawa H. Isolation of tumor-infiltrating lymphocytes from preserved human tumor tissue specimens for downstream characterization. STAR Protoc. (2022) 3(3):101557. doi: 10.1016/j.xpro.2022.101557
268. Tan YS, Lei YL. Isolation of tumor-infiltrating lymphocytes by ficoll-paque density gradient centrifugation. Methods Mol Biol. (2019) 1960:93–9. doi: 10.1007/978-1-4939-9167-9_8
269. Ben-Avi R, Farhi R, Ben-Nun A, Gorodner M, Greenberg E, Markel G, et al. Establishment of adoptive cell therapy with tumor infiltrating lymphocytes for non-small cell lung cancer patients. Cancer Immunol Immunother. (2018) 67(8):1221–30. doi: 10.1007/s00262-018-2174-4
270. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don't see). Nat Rev Immunol. (2014) 14(6):377–91. doi: 10.1038/nri3667
271. Colin-York H, Kumari S, Barbieri L, Cords L, Fritzsche M. Distinct actin cytoskeleton behaviour in primary and immortalised T-cells. J Cell Sci. (2019) 133(5):jcs232322. doi: 10.1242/jcs.232322
272. Abraham RT, Weiss A. Jurkat T cells and development of the T-cell receptor signalling paradigm. Nat Rev Immunol. (2004) 4(4):301–8. doi: 10.1038/nri1330
273. Riley JL. PD-1 signaling in primary T cells. Immunol Rev. (2009) 229(1):114–25. doi: 10.1111/j.1600-065X.2009.00767.x
274. Lin Z, Fillmore GC, Um TH, Elenitoba-Johnson KS, Lim MS. Comparative microarray analysis of gene expression during activation of human peripheral blood T cells and leukemic jurkat T cells. Lab Invest. (2003) 83(6):765–76. doi: 10.1097/01.LAB.0000073130.58435.E5
275. Ando M, Nishimura T, Yamazaki S, Yamaguchi T, Kawana-Tachikawa A, Hayama T, et al. A safeguard system for induced pluripotent stem cell-derived rejuvenated T cell therapy. Stem Cell Rep. (2015) 5(4):597–608. doi: 10.1016/j.stemcr.2015.07.011
276. Maeda T, Nagano S, Ichise H, Kataoka K, Yamada D, Ogawa S, et al. Regeneration of CD8αβ T cells from T-cell-derived iPSC imparts potent tumor antigen-specific cytotoxicity. Cancer Res. (2016) 76(23):6839–50. doi: 10.1158/0008-5472.CAN-16-1149
277. Nishimura T, Kaneko S, Kawana-Tachikawa A, Tajima Y, Goto H, Zhu D, et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. (2013) 12(1):114–26. doi: 10.1016/j.stem.2012.11.002
278. Themeli M, Kloss CC, Ciriello G, Fedorov VD, Perna F, Gonen M, et al. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat Biotechnol. (2013) 31(10):928–33. doi: 10.1038/nbt.2678
279. Vizcardo R, Masuda K, Yamada D, Ikawa T, Shimizu K, Fujii S, et al. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell. (2013) 12(1):31–6. doi: 10.1016/j.stem.2012.12.006
Keywords: vascular heterogeneity, transmigration, endothelial cell, pericyte, inflammation, tissue chip, microphysiological system, organ-on-chip
Citation: McCloskey MC, Zhang VZ, Ahmad SD, Walker S, Romanick SS, Awad HA and McGrath JL (2022) Sourcing cells for in vitro models of human vascular barriers of inflammation. Front. Med. Technol. 4:979768. doi: 10.3389/fmedt.2022.979768
Received: 27 June 2022; Accepted: 29 September 2022;
Published: 21 October 2022.
Edited by:
Qasem Ramadan, Alfaisal University, Saudi ArabiaReviewed by:
Jennifer Frattolin, Imperial College London, United KingdomShane Browne, Royal College of Surgeons in Ireland, Ireland
© 2022 McCloskey, Zhang, Ahmad, Walker, Romanick, Awad and McGrath. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James McGrath am1jZ3JhdGhAYm1lLnJvY2hlc3Rlci5lZHU=
Specialty Section: This article was submitted to Pharmaceutical Innovation, a section of the journal Frontiers in Medical Technology