 Dulshani Kankanige
Dulshani Kankanige Liwan Liyanage1
Liwan Liyanage1 Michael D. O'Connor
Michael D. O'Connor- 1School of Computer, Data and Mathematical Sciences, Western Sydney University, Campbelltown, NSW, Australia
- 2Translational Health Research Institute, Western Sydney University, Campbelltown, NSW, Australia
- 3School of Medicine, Western Sydney University, Campbelltown, NSW, Australia
Protein interaction pathways and networks are critically-required for a vast range of biological processes. Improved discovery of candidate druggable proteins within specific cell, tissue and disease contexts will aid development of new treatments. Predicting protein interaction networks from gene expression data can provide valuable insights into normal and disease biology. For example, the resulting protein networks can be used to identify potentially druggable targets and drug candidates for testing in cell and animal disease models. The advent of whole-transcriptome expression profiling techniques—that catalogue protein-coding genes expressed within cells and tissues—has enabled development of individual algorithms for particular tasks. For example,: (i) gene ontology algorithms that predict gene/protein subsets involved in related cell processes; (ii) algorithms that predict intracellular protein interaction pathways; and (iii) algorithms that correlate druggable protein targets with known drugs and/or drug candidates. This review examines approaches, advantages and disadvantages of existing gene expression, gene ontology, and protein network prediction algorithms. Using this framework, we examine current efforts to combine these algorithms into pipelines to enable identification of druggable targets, and associated known drugs, using gene expression datasets. In doing so, new opportunities are identified for development of powerful algorithm pipelines, suitable for wide use by non-bioinformaticians, that can predict protein interaction networks, druggable proteins, and related drugs from user gene expression datasets.
Introduction
One aim of transcriptomic analyses is to accurately predict candidate druggable targets for subsequent—and more laborious—testing of unapproved, approved or repurposed drug candidates within cell and/or animal disease models (1). Accordingly, over the past two decades a range of algorithms have been independently developed to classify and collate gene and proteins from transcriptomic and proteomic data. As outlined below, significant progress has been made in creating and refining algorithms for analysis of transcriptomic data—including microarray and RNA-sequencing data. By understanding the approaches that underpin these algorithms, as well as their individual advantages and disadvantages, new opportunities are identified for development of algorithm pipelines that make prediction of protein networks, and associated druggable proteins/ drugs, more widely accessible.
Broad algorithm categories include algorithms for: gene/protein classification via gene ontology (GO) terms; prediction of protein-protein interaction (PPI) networks; and establishment of existing drug/drug target databases.
These tools are often designed to analyse gene or protein lists obtained from high-resolution and/or high-throughput transcriptomic or proteomic data. For example, GO tools are routinely used to analyse transcriptomic data to infer biological functions performed by a cell population. Similarly, PPI tools aim to infer specific cellular functions through prediction and visualisation of protein interaction networks. GO, PPI, and other genomic association algorithms have also been developed to identify druggable targets and associated drugs that might be useful for altering the biology of a cell population (as opposed to algorithms designed for drug target prediction).
In a few instances, multiple algorithms have been organised into an analysis pipeline to provide multiple, correlated outputs from input a single input such as transcriptomics data. Nevertheless, there remains significant scope to develop simplified and broadly accessible algorithm pipelines to predict protein networks, druggable targets, and drugs for subsequent cell- and animal-based studies.
This review therefore begins by examining the key approaches developed for pre-processing and analysis of gene expression data—as relates to protein network and target identification. This includes a review of gene enrichment analysis algorithms, then GO, PPI, and drug/target algorithms. This creates a conceptual framework for understanding current—and future potential—algorithm pipelines for predicting protein networks and druggable targets from gene expression data.
Algorithms for Gene Enrichment Analysis
Initial steps in analysing gene expression data routinely involve identification of genes/proteins that share a common property, such as being differentially expressed (2). It is important to note that co-expression, and/or co-differential expression, within transcriptomic datasets does not necessarily imply the co-expressed genes are functionally connected. Moreover, the presence of a gene transcript does not always correlate with levels and/or activity of the corresponding protein product (due to multiple layers of post-transcriptional regulation) (3). Nonetheless, co-expression and differential expression have for decades provided useful conceptual paradigms for initial steps in gene expression analyses.
Initial transcriptomic analysis steps—termed Gene Enrichment Analysis—make use of algorithms that access data from gene set libraries—in which individual genes are correlated with specific functional identifiers such as GO terms (4). Inputs required for Gene Enrichment Analysis include one or more lists of interesting genes, together with the species type. Outputs from Gene Enrichment Analysis include information such as GO terms and associated gene groupings, as well as related p-values derived by comparing the frequency of the GO term genes in the input list with their frequency in the genome.
Different approaches can be used to perform Gene Enrichment Analysis (Table 1), and these approaches can be categorised into one or more of the following algorithm classes.
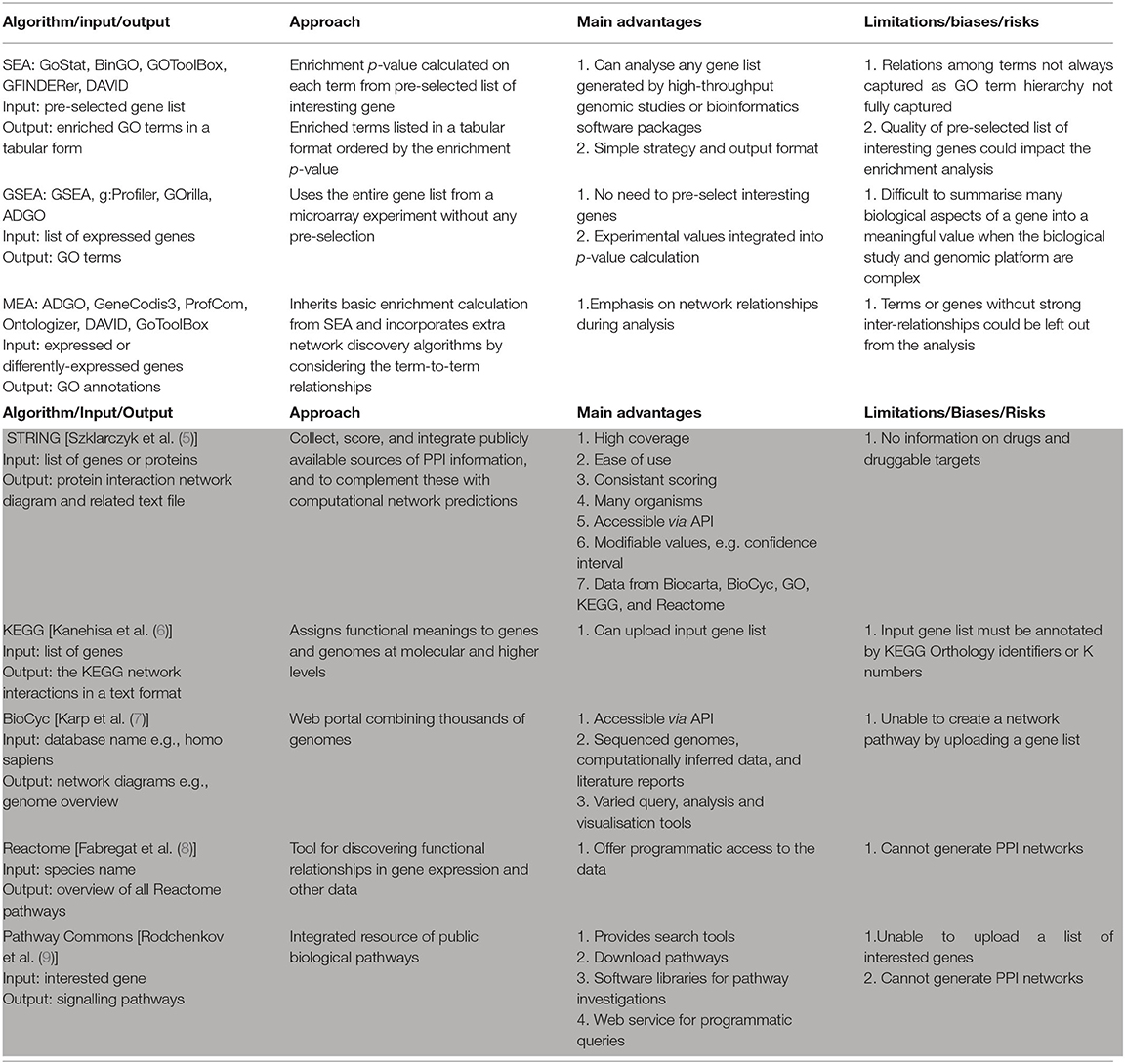
Table 1. Comparison of gene enrichment analysis tools (white background) and PPI pathway tools (grey background).
Gene Enrichment via Singular Enrichment Analysis (SEA)
For SEA, users typically first define a subset of interesting genes; this might include statistically-determining all the expressed genes or a list of genes differentially expressed across two treatment types. The SEA algorithm then analyses the user-defined gene list by (i) determining the frequency of genes in the list associated with a particular grouping (e.g., GO term), and (ii) statistically comparing that frequency with the expected frequency if the genes from that grouping were present by chance. The resulting groups of “enriched” genes, and their associated p-values, are then listed sequentially from smallest to largest p-value.
If the genome of the species being studied is sufficiently annotated, SEA algorithms can efficiently report gene groupings associated with key biological functions from large input gene lists. Widely-used SEA-based tools include GoStat (10), BinGO (11), GOToolBox (12), GFINDer (13), DAVID (14) etc. While these tools have proven useful, SEA algorithms have two inherent limitations. Firstly, the biological relevance of the output is dependent upon the quality of the user-defined input gene list. Secondly, SEA algorithms do not capture hierarchical relationship between the enriched GO terms identified through the SEA analysis—this can lead to hundreds of GO terms being reported for a single input gene list, which makes it difficult to interpret the biological relevance of the output.
Gene Enrichment via Gene Set Enrichment Analysis (GSEA)
As an alternative approach, GSEA algorithms do not require users to first partition gene expression data into lists of expressed/not-expressed or differentially-expressed genes (as occurs with SEA approaches). Instead, GSEA algorithms use the experimental data obtained for all genes. For example, by comparing the expression signals for all assayed genes on a microarray, GSEA algorithms can determine and then use a single expression fold-change parameter for each gene. This approach can avoid some issues or artefacts related to selection or exclusion of input genes (15). However, for more complex experimental data, the GSEA approach can be disadvantageous. For example, genes that have higher expression fold changes make larger contributions to the GSEA output, but this does not always reflect biological realities (e.g., small changes in transcription can have large effects on cell behaviour). Additionally, complexities in expression for individual genes can now be reliably assessed—such as differential promoter and/or exon usage, or multiple small nuclear polymorphisms. These complexities result in genes having multiple expression profiles, genomic locations, p-values, etc. Summarising these complexities into a single parameter value for a gene required by GSEA algorithms can be problematic or not possible. Examples commonly used GSEA approaches include GSEA (15), g:Profiler's g:GOSt tool (16), GOrilla (17), ADGO (18) etc.
Gene Enrichment via Modular Enrichment Analysis (MEA)
A third approach to gene enrichment involves MEA. These algorithms build upon the SEA approach by adding the ability to consider relationships between gene-associated terms such as GO terms (or in some cases, pathway information, protein domains, etc.). Examples include ADGO (18), GeneCodis3 (19), ProfCom (20), Ontologizer (21), DAVID (14) and GoToolBox (12). While the MEA approach can provide improved understanding of expression changes for well-characterised genes, it can inherently bias against genes that are more poorly characterised or that have fewer known relationships. Similar to SEA, the quality of the pre-selected gene list impacts the results of MEA algorithms.
Algorithms for Construction of Protein Interaction Pathways
While gene enrichment analyses provide useful initial steps for identification of co-expressed genes -and associated grouping of functionally-related genes (e.g., via GO terms)—the outputs from gene enrichment algorithms do not include detailed information on protein interactions that might occur within the cell or tissue from which the gene expression data was generated. As a consequence, a range of algorithms have been developed to predict PPI networks using lists of expressed, co-expressed, or differentially-expressed genes. In predicting these PPI networks, it is important to note that gene expression and protein activity are not always correlated, and that protein interactions are not limited to direct physical binding. For example, proteins may also interact: indirectly, by sharing a substance in a metabolic pathway; by regulating each other transcriptionally; or by participating in larger multi-protein assemblies (5).
To provide information on potential PPI in a gene enrichment output, a range of tools have been developed to map interactions based on different levels of evidence—from experimental data to text associations in published literature. Examples of this evidence include experimental evidence (e.g., immunoprecipitation), genomic locations, and cooccurrence reports. The PPI algorithms typically ascribe different levels of confidence for each of these different evidence types to provide ranked output for predicted PPIs. In some instances, prediction of protein interaction networks can be progressed iteratively. For example, by first predicting networks from an input gene list, additional database searching can be performed to suggest additional proteins (not in the original input gene list) for incorporation into the predicted networks—if there is evidence for indicating the new proteins may interact with members of the first predicted network. Key PPI algorithms that have gained wide recognition within the field are briefly reviewed below.
KEGG Pathways
The Kyoto Encyclopaedia of Genes and Genomes (KEGG), is a widely used tool that integrates both manually curated and computationally generated databases into a single resource. KEGG provides information on genomes, PPI pathways, disease molecular pathophysiology, and drugs with known mechanisms of action. KEGG enables a gene list to be uploaded through an application programming interface (API), however, this function is somewhat limited as it requires the gene list to be annotated with KEGG Orthology identifiers (6).
BioCyC
The BioCyc database contains genome and predicted metabolic networks for >3,000 organisms. BioCyc enables programmable online access to information including reactions, metabolic pathways, and enzymes (22). BioCyC provides tools to query, analyse, and visualise genome and metabolism data, but does not generate PPI networks from a list of input genes (22).
Reactome
The Reactome pathway knowledgebase contains non-redundant curated information from other pathway databases such as CheEBI and UniProt (23). It supports API access to investigate unexpected functional relationships in gene expression profiles, however, similar to BioCyc it does not generate PPI networks.
Pathway Commons
Pathway Commons collates biological pathway information from sources such as BioGRID, HumanCyc, Reactome, NCI/Nature PID, etc. (24). To analyse pathway information, user datasets can be programmatically queried via a web service, software libraries and keyword searches. However, it cannot generate protein interaction pathways or upload a list of input genes.
String
STRING is a pathway database that collects, scores, and integrates public information relating to PPIs from various sources. The basic interaction units in STRING are links between proteins that are known to contribute to specific biologic processes—also known as functional associations. STRING extracts curated data from Biocarta, BioCyc, GO, KEGG, and Reactome (5). STRING has information for >5,000 organisms and >24 million proteins. It provides access through an API and can generate PPI network diagrams that can be user-customised by changing parameters such as the PPI confidence level. While gene lists can be uploaded, there still remain computational challenges including: (i) outputting PPI networks for each individual genes within a gene list (rather than interactions between members of an input gene list); (ii) comparing predicted PPI networks against the set of expressed genes for a tissue; and (iii) iterating these processes for all genes in an input gene list.
Algorithms for Drug/Target Discovery
A natural progression from having PPI networks predicted from gene expression data is the development of algorithms to correlate proteins in the networks with known small-molecule modulators of protein activity. Toward this end, improvements in genome sequencing have enabled development of pharmacogenetic databases that associate clinical, disease and other annotations (e.g., GO terms) with variations in sequences for particular genes (25). These databases can also link genes as drug targets with related drugs. Key algorithms for mapping proteins to known drugs are briefly reviewed below.
DrugBank
DrugBank is a highly cited database that combines drug, drug-target, drug action and drug interaction information—but not disease-related or prescribing details. The database is updated daily and is accessible via an API, but does not allow users to upload gene list of interest (26).
PharmGKB
The Pharmacogenomics Knowledgebase (PharmGKB) is a widely used resource containing information curated from other databases including PubMed, DrugBank and the human small nucleotide polymorphism database, dbSNP. The main advantages of PharmGKB are it collates drug targets, drugs, and corresponding disease information in one platform, while providing access via an API (25).
ChEMBL
ChEMBL is an open access online database that extracts data from medicinal chemistry journals and integrates them with information on approved drugs. ChEMBL also exchanges data with other databases such as PubChem and the pharmacokinetics database, BindingDB. The data can be accessed using web services and in downloadable formats, although it does not provide integrated PPI/drug/target outputs (27).
TTD
The Therapeutic Target Database (TTD) provides information about drug targets, drugs, and related disease conditions. It extracts data from pathway databases including KEGG, MetaCyc, Reactome, and Wikipathways. TTD integrates drug/target/disease outputs, however, users can not upload gene lists or generate PPI networks. TTD also does not provide access via an API (28).
DGIdb
The drug–gene interaction database (DGIdb) integrates drug–gene interactions and potential druggable genes. It consolidates data from other databases such as GO, DrugBank, PharmGKB, the TTD and the National Cancer Institute (NCI). Users can upload gene lists to identify any associated drugs. However, similar to DrugBank, it does not capture related disease data (29).
DisGeNET
The disease-gene network database, DisGeNet, contains information on genes and their variants related only to human diseases. DisGeNET extracts data from different scientific literature and consolidates them using text mining tools. Data on DisGeNet database can be downloaded in many file formats, and accessed through an API (30).
PHAROS
PHAROS aims to provide insight on the druggable genome. It contains information on all human protein targets (both well- and poorly-characterised targets) including protein expression data, GO terms, disease associations and drug target interactions. While API access is possible, it is highly time-consuming as only one gene can be submitted at a time (31).
GPSnet
The genome-wide positioning systems network (GPSnet) is a recent algorithm focused on identifying cancers for which approved or investigational drugs might be repurposed to provide candidate new treatment options. GPSnet functions in two steps: (i) it analyses sequencing information from 15 cancer types (from ~5,000 patients); and then (ii) implements a GSEA algorithm and network proximity methods to identify candidate drugs for the targets identified in the first step (32). While this provides a powerful approach to identifying candidate new drug treatments, the information is not accessible through an API and the database currently focuses only on cancers.
Algorithm Pipelines for Predicting Druggable Targets/Drugs From Expression Data
As outlined above, a large range of algorithms have been developed for analysis of gene expression data. To date, only a small number of algorithm pipelines have been developed to progress from gene enrichment analysis, through GO analysis and protein network prediction, to drug target identification (Table 2). Of these pipelines, PHAROS, DGIdb and GPSnet are the most well-developed. PHAROS can generate PPI networks, identify drug targets and associated drugs when a list of genes is provided. However, PHAROS is human-specific and the API does not allow multiple genes as an input. Also, the data output is not grouped by GO terms, which would facilitate rapid assessment of PPI networks and associated drugs based on key biological processes or molecular functions. DGIdb integrates data from many other drug databases, and an input gene list can be uploaded. However, it does not capture disease information which is a significant disadvantage. GPSnet, a more recently developed algorithm, can perform GSEA and generate protein interaction networks. Unfortunately, it currently focuses only on cancer specific diseases and does not provide a free online too to access its data via an API.
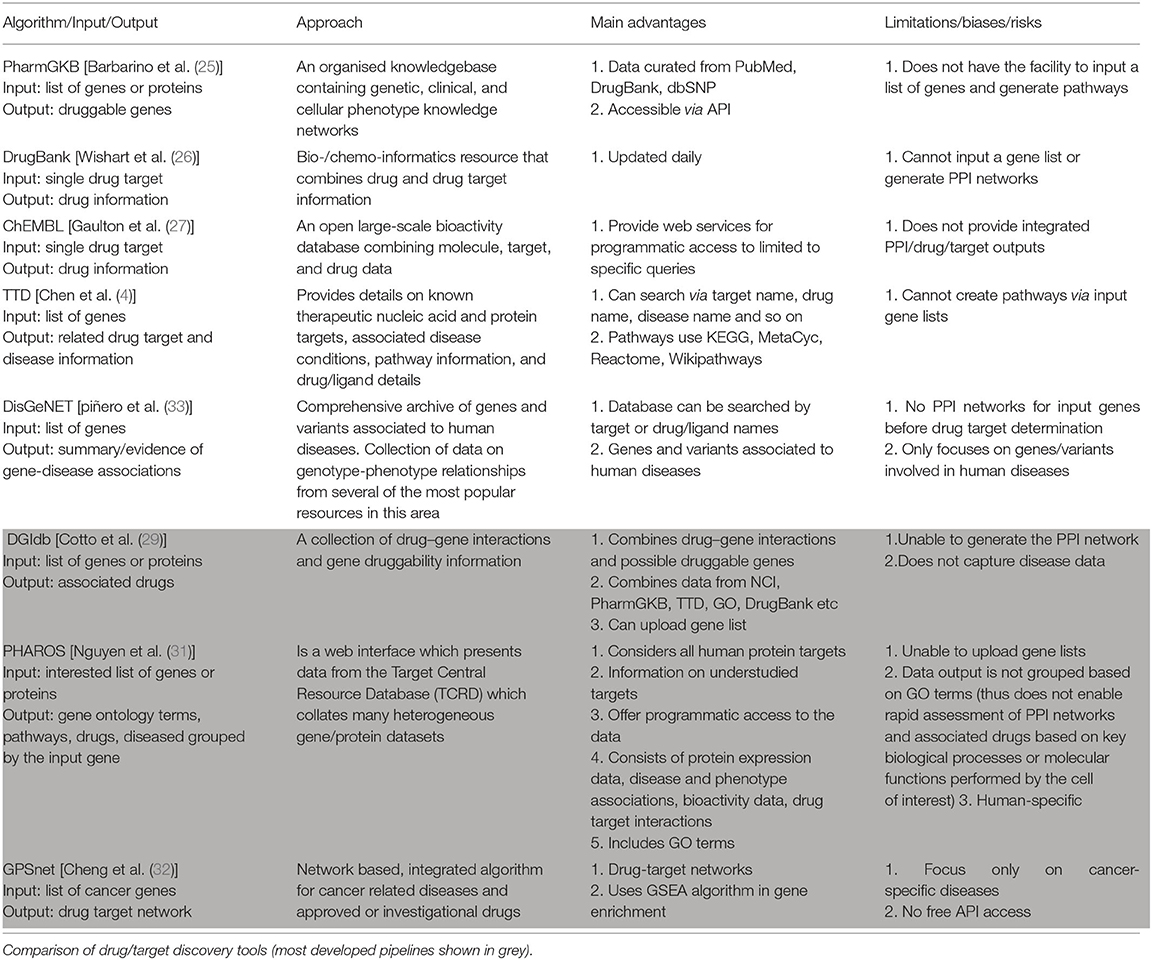
Table 2. Comparison of drug/target discovery tools.
Given the capabilities and limitations with the above-mentioned algorithms, there is a need and opportunity to develop an algorithm pipeline that: (i) enables users to upload gene expression data as an input (either expressed or differentially-expressed gene lists); (ii) groups genes based on their GO terms; (iii) creates additional outputs for each gene consisting of visualised PPI networks that include identified druggable targets and associated drugs; and (iv) has the potential to identify new pathways, and thus druggable target discovery, through modification of PPI network prediction parameters (for example, by modifying related parameters used by STRINGdb).
Conclusion
As reviewed here, much progress has been made in developing a wide variety of algorithms for gene expression analyses. However, a gap in the field exists for algorithm pipelines that combine all these important analysis tools into unified and simplified packages suitable for broad use by non-bioinformaticians. These algorithm pipelines should allow users to upload gene expression datasets from human and non-human species. They should also provide integrated outputs including GO terms, protein network predictions, and drug target, drug and disease information. Astute combination and modification of existing algorithms could address this gap. Once realised, these new algorithm pipelines could accelerate identification and prioritisation of drug targets, and associated drugs—in order to minimise the time and labour costs associated with testing unapproved, approved or repurposed drug candidates in cell and animal models of disease.
Author Contributions
All authors were involved in outlining, writing, and reviewing the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Pushpakom S, Iorio F, Eyers PA, Escott KJ, Hopper S, Wells A, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov. (2019) 18:41–58. doi: 10.1038/nrd.2018.168
2. Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. (2009) 37:1–13. doi: 10.1093/nar/gkn923
3. Wang X, Liu Q, Zhang B. Leveraging the complementary nature of RNA-Seq and shotgun proteomics data. Proteomics. (2014) 14:2676–87. doi: 10.1002/pmic.201400184
4. Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. (2013) 14:128–128. doi: 10.1186/1471-2105-14-128
5. Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. (2019) 47:D607–13. doi: 10.1093/nar/gky1131
6. Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. (2017) 45:D353–61. doi: 10.1093/nar/gkw1092
7. Karp PD, Billington R, Caspi R, Fulcher CA, Latendresse M, Kothari A, et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief Bioinform. (2019) 20:1085–93. doi: 10.1093/bib/bbx085
8. Fabregat A, Korninger F, Viteri G, Sidiropoulos K, Marin-Garcia P, Ping P, et al. Reactome graph database: Efficient access to complex pathway data. PLoS Comput Biol. (2018) 14:e1005968. doi: 10.1371/journal.pcbi.1005968
9. Rodchenkov I, Babur O, Luna A, Aksoy BA, Wong JV, Fong D, et al. Pathway commons 2019 update: integration, analysis and exploration of pathway data. Nucleic Acids Res. (2020) 48:D489–97. doi: 10.1093/nar/gkz946
10. Beissbarth T, Speed TP. GOstat: find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics. (2004) 20:1464–5. doi: 10.1093/bioinformatics/bth088
11. Maere S, Heymans K, Kuiper M. BiNGO: a cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. (2005) 21:3448–9. doi: 10.1093/bioinformatics/bti551
12. Martin D, Brun C, Remy E, Mouren P, Thieffry D, Jacq B. GOToolBox: functional analysis of gene datasets based on Gene Ontology. Genome Biol. (2004) 5:R101–R101. doi: 10.1186/gb-2004-5-12-r101
13. Masseroli M, Martucci D, Pinciroli F. GFINDer: Genome Function INtegrated Discoverer through dynamic annotation, statistical analysis, and mining. Nucleic Acids Res. (2004) 32:W293–300. doi: 10.1093/nar/gkh432
14. Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, et al. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. (2007) 35:W169–75. doi: 10.1093/nar/gkm415
15. Aravind S, Pablo T, Vamsi KM, Sayan M, Benjamin LE, Michael AG, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceed Nat Acad Sci PNAS. (2005) 102:15545–50. doi: 10.1073/pnas.0506580102
16. Reimand J, Kull M, Peterson H, Hansen J, Vilo J. g:Profiler–a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. (2007) 35:W193–200. doi: 10.1093/nar/gkm226
17. Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. (2009) 10:48–48. doi: 10.1186/1471-2105-10-48
18. Nam D, Kim S-B, Kim S-K, Yang S, Kim S-Y, Chu I-S. ADGO: analysis of differentially expressed gene sets using composite GO annotation. Bioinformatics. (2006) 22:2249–53. doi: 10.1093/bioinformatics/btl378
19. Tabas-Madrid D, Nogales-Cadenas R, Pascual-Montano A. GeneCodis3: a non-redundant and modular enrichment analysis tool for functional genomics. Nucleic Acids Res. (2012) 40:W478–83. doi: 10.1093/nar/gks402
20. Antonov AV, Schmidt T, Wang Y, Mewes HW. ProfCom: a web tool for profiling the complex functionality of gene groups identified from high-throughput data. Nucleic Acids Res. (2008) 36:W347–51. doi: 10.1093/nar/gkn239
21. Bauer S, Grossmann S, Vingron M, Robinson PN. Ontologizer 2.0-a multifunctional tool for GO term enrichment analysis and data exploration. Bioinformatics. (2008) 24:1650–1. doi: 10.1093/bioinformatics/btn250
22. Caspi R, Altman T, Billington R, Dreher K, Foerster H, Fulcher CA, et al. the metacyc database of metabolic pathways and enzymes and the biocyc collection of pathway/genome databases. Nucleic Acids Res. (2014) D459–71. doi: 10.1093/nar/gkt1103
23. Vastrik I, D'Eustachio P, Schmidt E, Joshi-Tope G, Gopinath G, Croft D, et al. Reactome: a knowledge base of biologic pathways and processes. Genome Biol. (2007) 8:R39. doi: 10.1186/gb-2007-8-3-r39
24. Cerami EG, Gross BE, Demir E, Rodchenkov I, Babur O, Anwar N, et al. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. (2011) 39:D685–90. doi: 10.1093/nar/gkq1039
25. Barbarino JM, Whirl-Carrillo M, Altman RB, Klein TE. PharmGKB: a worldwide resource for pharmacogenomic information. Wiley Interdisciplin Rev. (2018)0.10:e1417-n/a. doi: 10.1002/wsbm.1417
26. Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. (2018) 46:D1074-D1082. doi: 10.1093/nar/gkx1037
27. Mendez D, Gaulton A, Bento AP, Chambers J, De Veij M, Félix E, et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. (2019) 47:D930–40. doi: 10.1093/nar/gky1075
28. Zhu F, Han B, Kumar P, Liu X, Ma X, Wei X, et al. Update of TTD: therapeutic target database. Nucleic Acids Res. (2010) 38:D787–91. doi: 10.1093/nar/gkp1014
29. Cotto KC, Wagner AH, Feng YY, Kiwala S, Coffman AC, Spies G, et al. DGIdb 3.0: a redesign and expansion of the drug-gene interaction database. Nucleic Acids Res. (2018) 46:D1068–73. doi: 10.1093/nar/gkx1143
30. Piñero J, Bravo À, Queralt-Rosinach N, Gutiérrez-Sacristán A, Deu-Pons J, Centeno E, et al. DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. (2017) 45:D833–9. doi: 10.1093/nar/gkw943
31. Nguyen D-T, Mathias S, Bologa C, Brunak S, Fernandez N, Gaulton A, et al. Pharos: Collating protein information to shed light on the druggable genome. Nucleic Acids Res. (2017) 45:D995–D1002. doi: 10.1093/nar/gkw1072
32. Cheng F, Lu W, Liu C, Fang J, Hou Y, Handy DE, et al. A genome-wide positioning systems network algorithm for in silico drug repurposing. Nat Commun. (2019) 10:3476–3414. doi: 10.1038/s41467-019-10744-6
Keywords: gene expression analyses, bioinformatics, pipeline, gene ontology, protein interaction pathways, drug targets, drugs
Citation: Kankanige D, Liyanage L and O'Connor MD (2022) Application of Transcriptomics for Predicting Protein Interaction Networks, Drug Targets and Drug Candidates. Front. Med. Technol. 4:693148. doi: 10.3389/fmedt.2022.693148
Received: 09 April 2021; Accepted: 05 January 2022;
Published: 09 March 2022.
Edited by:
Indira Ghosh, Jawaharlal Nehru University, IndiaReviewed by:
Wenyan Gong, Hangzhou Normal University, ChinaChiquito J. Crasto, Texas Tech University, United States
Copyright © 2022 Kankanige, Liyanage and O'Connor. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael D. O'Connor, bS5vY29ubm9yQHdlc3Rlcm5zeWRuZXkuZWR1LmF1