 Shiyou Yang
Shiyou Yang Ming Jia
Ming Jia
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mech. Eng., 07 May 2019
Sec. Engine and Automotive Engineering
Volume 5 - 2019 | https://doi.org/10.3389/fmech.2019.00021
Biodiesel is a renewable, clean-burning diesel replacement, and may have superior brake thermal efficiency with certain blends compared to traditional diesel counterpart at higher compression ratios. The combustion chemistry process of biodiesel, which has not been well understood, is of great interests to some engine researchers. Researchers have developed some complicated chemical kinetic mechanisms for bio-diesel, which cannot be used in engine CFD (computational fluid dynamics) with current computational resources. The present work aims to construct a new chemical kinetic mechanism with a medium size for biodiesel combustion. Since 2016, H2/O2/CO/C1 and C2-C3 detailed sub-mechanisms (the C3 model contained in AramcoMech2.0) have been developed for accurately predicting laminar flame speeds, ignition delay times, and important species evolutions, and have been validated against a large array of experimental measurements over a wide range of conditions. In this paper, a 3-component biodiesel surrogate chemical kinetic mechanism constructed in 2015 based on decoupling methodology has been combined with the new “core” H2/O2/CO/C1~C3 detailed mechanism to generate a new bio-diesel chemical kinetic mechanism. In the surrogate mechanism construction, three skeletal sub-mechanisms are used for the three biodiesel components (MD (methyl decanoate), MD5D (methyl-5-decenoate), and n-decane). The final mechanism, which has 183 species and 1002 reactions, has been validated with available experiment data. It will be validated extensively with more experimental biodiesel data and applied to engine CFD for understanding biodiesel combustion.
Currently, fossil fuel is still the main source of energy for transportation. However, combustion of gasoline and diesel fuel emits carbon dioxide (CO2), particulate matter, oxides of nitrogen (a prime component of “smog”), carbon monoxide, and unburned hydrocarbons. When CO2 is released into the atmosphere, it functions as a heat-trapping greenhouse gas. Another problem for fossil fuels is that they are non-renewable. They are limited in supply and will be depleted one day. Due to the two problems (emissions and fuel supply limit) for internal combustion engines fueled with gasoline or diesel, while researchers continue to work on improving thermal efficiency and reducing emissions of I. C. engines, governments and some other researchers are putting more efforts on the development of new energy vehicles. New energy vehicles include HEV (hybrid electric vehicles), PHEV (plug-in hybrid electric vehicles), BEV (battery electric vehicles), HFCV (hydrogen fuel cell vehicles), and vehicles fueled with renewable energy sources.
There are five commonly used renewable energy sources: biomass, hydropower, geothermal, wind, and solar. Biodiesel is an organic biomass produced from vegetable oils and animal fats. Biodiesel has been chosen as an alternative fuel due to its similar physicochemical characteristics as fossil diesel fuel, which make it easy to be used in diesel engines with only minor or no modifications. Since biodiesel is a renewable, clean-burning diesel replacement, and may have superior brake thermal efficiency with certain blends compared to traditional diesel counterpart at higher compression ratios, the combustion chemistry process of biodiesel, which has not been well understood, is of great interests to some engine researchers in recent years. For a better understanding of the combustion process of biodiesel, an accurate chemical kinetic mechanism of biodiesel fuel is an indispensable prerequisite. Since it is still impossible to construct a detailed chemical kinetic mechanism consisting of all the components of real biodiesel fuel, it has been accepted that a surrogate biodiesel fuel with several components can capture certain combustion behaviors of a target fuel containing many components so that a mechanism with a suitable size is possible for engine CFD application.
Biodiesel is mainly made up of five components: methyl palmitate (C17H34O2), methyl stearate (C19H38O2), methyl oleate (C19H36O2), methyl linoleate (C19H34O2), and methyl linolenate (C19H32O2). As seen in Figure 1, methyl palmitate and methyl stearate are saturated, while methyl oleate, methyl linoleate, and methyl linolenate contain one, two, and three double-bonds, respectively. Researchers at the Lawrence Livermore National Laboratory (LLNL) have proposed methyl decanoate (C11H22O2) as a biodiesel fuel surrogate based on the results showing that it does appear to be more representative of biodiesel fuel chemistry. Since biodiesel is typically composed of several unsaturated methyl esters, the methyl decanoate mechanism was expanded to include methyl-9-decenoate (C11H20O2), which has a double-bond at the ninth position on the carbon chain. C11H22O2 and C11H20O2 have detailed chemistry mechanisms available to-date at the LLNL. The LLNL mechanism also included n-heptane for use as an alkane surrogate for biodiesel blending. The combined methyl decanoate (MD) and methyl-9-decenoate (MD9D) mechanism from LLNL contains 3299 species and 10806 reactions. Westbrook et al. (Westbrook et al., 2011) built a detailed mechanism including all of the five components shown in Figure 1. The mechanism of Westbrook et al. is composed of about 4,800 species and 20,000 reactions, which is too huge to be applied in engine CFD modeling. It was reported in the literature that MD9D was not suitable for biodiesel surrogate due to its similar reaction activity with MD. Chang et al. (Chang et al., 2015) proposed a biodiesel surrogate model with the components of n-decane, MD, and MD5D (methyl-5-decenoate), since MD and MD5D were chosen to respectively represent the saturated methyl ester and unsaturated methyl ester, and n-decane was chosen to match the energy content and the C/H/O ratio of actual biodiesel fuel. Liu et al. (2016) developed a skeletal four-component biodiesel combustion mechanism (106 species and 263 reactions) comprising methyl decenoate, methyl-5-decenoate, n-decane and methyl linoleate using directed relation graph error propagation and sensitivity analysis (DRGEPSA). Wang et al. (Wang et al., 2015) developed a skeletal combustion mechanism (146 species and 652 reactions) of methyl decanoate (MD) as a surrogate for biodiesel fuels for compression ignition engine simulations by reducing a detailed mechanism using an integrated reduction method that contains directed relation graph method, sensitivity analysis, and reaction path analysis. Using the DRG (directed relation graph) based mechanism reduction methodology as used by Liu et al. (2016) and Wang et al. (2015), the H2/O2/CO/C1~C3 detailed mechanism may be affected. Li et al. (2019) proposed a four-part scheme to formulate a skeletal mechanism for heavy saturated methyl esters including low temperature oxidation, high temperature decomposition, ester group reactions and detailed C4-C0 chemistry. However, a mechanism including a C4 detailed mechanism will have a larger model size which is not practical to be used in engine CFD. In this work, the biodiesel skeletal mechanism of Chang et al. (2015) is selected as the baseline mechanism. However, a new “core” H2/O2/CO/C1~C3 detailed mechanism is used to replace the old “core” model. The reason is that the new “core” model has been validated against a large array of experimental measurements over a wide range of conditions, and compared to the old model the new model may accurately predict laminar flame speeds, ignition delay times, and important species evolutions.
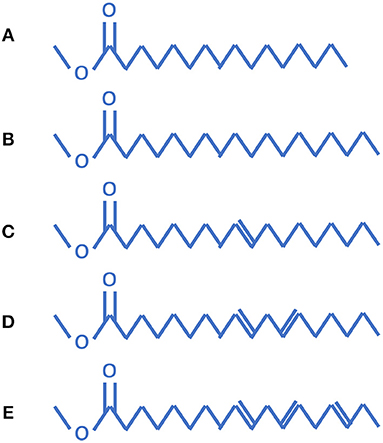
Figure 1. Five common components of biodiesel: (A) methyl palmitate, (B) methyl stearate, (C) methyl oleate, (D) methyl linoleate, and (E) methyl linolenate (Brakora et al., 2011).
The integration of detailed mechanisms of the surrogate components into multi-dimensional CFD modeling for engines still leads to extremely long computational times. Therefore, skeletal or reduced mechanisms that can shorten the computational time while maintaining accuracy are welcome for multi-dimensional CFD simulations of biodiesel engines. One method to develop a mechanism with a smaller size is to use the decoupling methodology proposed by Liu et al. (2013), which is of great interests by some researchers in recent years. The decoupling methodology considers the combustion process of a fuel as two parts: one part is ignition, which is largely dependent on the specific fuel, and the other part is flame propagating after ignition, which is mainly controlled by reactions involving small radicals and molecules of CO–C1 and is less dependent on the specific fuel. In Chang et al.'s work (Chang et al., 2015) the decoupling methodology was used to develop a skeletal chemical kinetic mechanism for biodiesel with 60 species and 172 reactions, showing a promising performance for engine applications incorporated with a multidimensional CFD modeling. In the mechanism of Chang et al. (2015), the “core” part CO–C1 sub-mechanism is mainly from Klippenstein et al. (2011) for the ignition of methanol at high pressures by using the ab initio transition state theory, and the transition part C2-C3 sub-mechanism including six species (C2H3, C2H4, C3H4, C3H5, C3H6, and C3H7) is from Patel et al. (2004) because of its small size. The range of the “core” part CO-C1 sub-mechanism used in Chang et al. (2015) is mainly for high pressures, and the C2-C3 sub-mechanism used by them may not be enough for a higher accuracy of laminar flame speed prediction over more operating conditions.
Recently, Kéromnès et al. (2013) and Metcalfe et al. (2013) have updated H2/O2/CO/C1 detailed sub-mechanism to characterize the kinetic and thermochemical properties of a large number of CO–C1 based hydrocarbon and oxygenated fuels over a wide range of experimental conditions. Metcalfe et al. (2013) and Burke et al. (2014) have developed a detailed C2-C3 sub-mechanism which is constructed in a hierarchical way for larger C2-C3 hydrocarbon species such as ethane, ethylene, acetylene, allene, propyne, and propene, as well as oxygenated species. The new C2-C3 detailed sub-mechanism has been validated against a large array of experimental measurements including data from shock tubes, rapid compression machines, flames, jet-stirred and plug-flow reactors. The new H2/O2/CO/C1 detailed sub-mechanism and the new C2-C3 detailed sub-mechanism are included in AramcoMech2.01.
In this paper, the biodiesel surrogate model proposed by Chang et al. (2015) is adopted. The surrogate model includes methyl decanoate (MD), methyl-5-decenoate (MD5D), and n-decane. The skeletal oxidation mechanism constructed by Chang et al. (2015) is used for each of the 3 biodiesel surrogate components. The three skeletal sub-mechanisms are next coupled with the above-mentioned latest C2-C3 and H2/O2/CO/C1 detailed sub-mechanisms to generate a new biodiesel mechanism with a medium size, which is practical under present computational conditions for biodiesel engine 3D (3 dimensional) CFD combustion modeling, while maintaining a higher accuracy.
The reaction paths of the new biodiesel surrogate chemical kinetic mechanism developed in the present work are summarized in Figure 2. The mechanism is constructed by integrating the skeletal sub-mechanisms for n-decane (C10H22), MD, and MD5D with the detailed C2-C3 sub-mechanism and the newly developed H2/O2/CO/C1 detailed sub-mechanism. A brief description of each sub-mechanism will be given below.
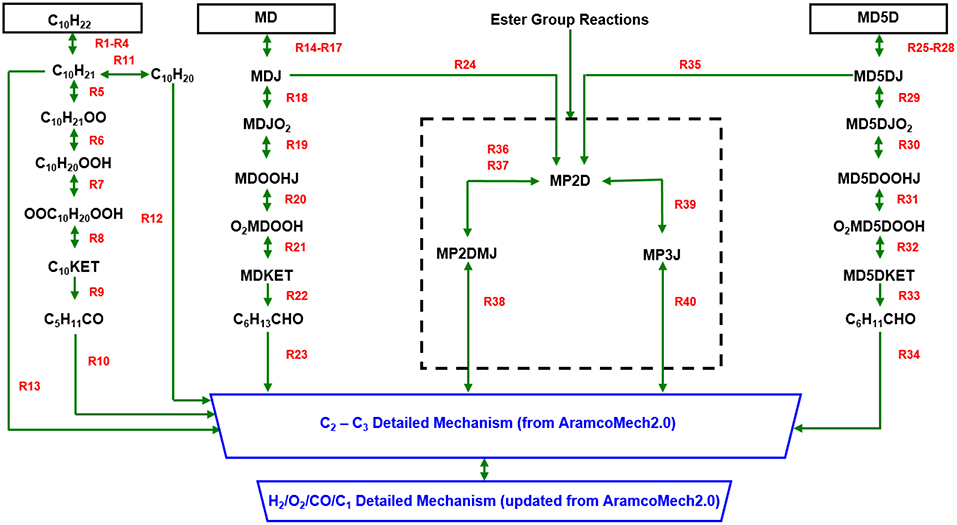
Figure 2. Overall reaction path diagram of updated 3-component biodiesel surrogate chemical kinetic mechanism.
The low temperature oxidation processes of n-decane, MD, and MD5D are similar: a sequence of reaction pathways ṘH → Ṙ→ṘO2 → QOOH → O2QOOH → ketohydroperoxide → small species (i.e., reactions R1-R10 for n-decane, reactions R14-R23 for MD, and reactions R25-R34 for MD5D). These small species are then fed into the detailed C2-C3 sub-mechanism. Because of the different molecular structure between alkanes and methyl esters, at high temperatures a skeletal sub-mechanism containing the ester group is introduced. Since C2H3CO and CH3OCO are already included in the C2-C3 detailed sub-mechanism, the original reactions 39, 42, and 43 of Chang et al.'s model are removed in present work. In the detailed H2/O2 sub-mechanism, the reaction Ḣ+O2↔Ö+ȮH dominates the oxidation of all fuels undergoing oxidation at high temperatures (T ≥ 1,000 K). Based on two experimental datasets, a rate constant is adopted for this reaction over a temperature range of 1,100–3,370 K with a reduced uncertainty of < 10%. A low-pressure limit rate constant over a temperature range of 1,020–1,260 K and over a pressure range of 10–150 bar with a high-pressure limit rate constant over a lower temperature range of 300–900 K and over a pressure range of 1.5–950 bar are combined for the chain propagation reaction Ḣ+O2(+M)↔HȮ2(+M) which controls the low-temperature reactivity. Flame speed calculations are very sensitive to the recombination reaction Ḣ+ȮH(+M)↔H2O(+M) to form water. Increasing this reaction rate decreases reactivity. Kéromnès et al. (2013) have optimized the rate constant of this reaction to get the best agreement with experimental flame speed data. Flame speed predictions are also very sensitive to the reaction H2+ȮH↔Ḣ+H2O under fuel-lean conditions. For this reaction, a very recent rate constant measured using UV (Ultraviolet) laser absorption of ȮH radicals behind reflected shock waves is adopted over a temperature range of 902–1,518 K and a pressure range of 1.15–1.52 atm. In the detailed CO/C1/C2-C3 sub-mechanism, recent measurements are used for the reaction HĊO(+M)↔Ḣ+CO(+M). The reaction ĊH3+ȮH↔Products is very important in hydrocarbon flames. The recommended rate constant values from the literature have been refitted in AramcoMech2.0 to a single Arrhenius expression with a minimal increase in errors. The rate constant adopted for the reaction ĊH3+Ḣ(+M)↔CH4(+M), with a high-pressure limit of 1.27 × 1016T−0.630exp(-193/T) cm3mol−1s−1, is taken from GRI-Mechanism as it results in the best agreement with pressure-dependent methane/air flame speeds. A theoretical work has been adopted for the reaction C2H4+ȮH↔Products, which is a very important promoting reaction in ethane oxidation. There has been considerably more uncertainty in the assignment of the rate constant for reactions C2H2+Ö↔HĊCO+Ḣ/ĊH2+CO. The rate constants for the reaction of propene with hydroxyl radical C3H6+ȮH↔Products are determined both theoretically and experimentally. The theoretical method employed quantum chemical calculations, while the measurement method was based on a shock tube using laser absorption. The total rate constants from both theoretical and measurement methods are in very good agreement and thus are used in this work.
The reactions of the skeletal sub-mechanisms for n-decane (C10H22), MD, and MD5D are listed as below:
The new bio-diesel mechanism has been validated using Chemkin-PRO package for various reactant mixtures to investigate the effectiveness of each sub-mechanism: H2/O2/CO sub-mechanism, C1-C2 sub-mechanism, C3 sub-mechanism, MD skeletal mechanism, MD5D skeletal mechanism, and n-decane skeletal mechanism. The same entire mechanism was used in simulation for the validation of each sub-mechanism. To validate each sub-mechanism, various experimental data from the literature were used, including ignition delay times from shock tube or rapid compression machine, laminar flame speeds, species concentration profiles in jet-stirred reactor or flow reactor. Figures 3, 4 show the validation results of H2/O2/CO sub-mechanism.
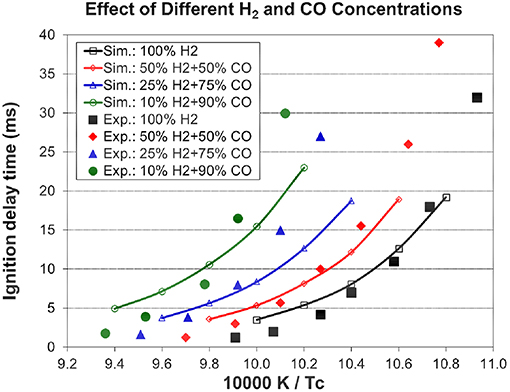
Figure 3. Effect of different H2 and CO concentrations on ignition delay times (Pc=70 bar, 12.5% [αH2+(1-α)CO)+6.25% O2+81.25% N2, experimental data are taken from Kéromnès et al. (2013)].
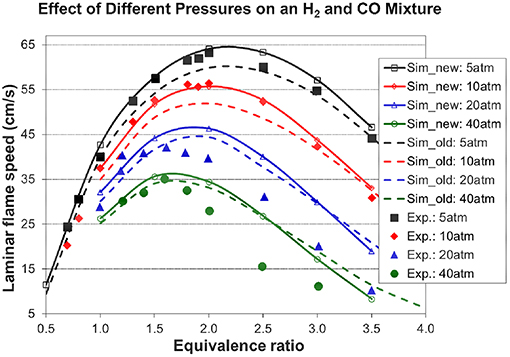
Figure 4. Comparison between measured flame speeds and calculated flame speeds from this work and (Chang et al., 2015) [95% CO + 5% H2 in O2 + 7 He, Ti = 298 K, experimental data are taken from Kéromnès et al. (2013)].
The simulation from the present mechanism has been performed and compared to experimental data under stoichiometric conditions with 100, 50, 25, and 10% H2 in the H2/CO fuel mixtures with nitrogen dilution at an end-of-compression pressure of 70 bar and an end-of-compression temperature range of 914–1,068 K. The experimental data in Figure 3 show the inhibiting effect of carbon monoxide on the syngas ignition delay times, which increase with increasing amounts of CO in the syngas mixture. The model captures this inhibiting effect accurately and its predictions are in very good agreement with the experimental results. The simulated laminar flame speeds from the present mechanism have been compared to the experimental data for the H2/CO fuel mixtures with helium dilution at an initial temperature of 298 K, an equivalence ratio range of 0.5–3.5, and various initial pressures. As shown in Figure 4, the present model (Sim_new) shows a good prediction compared to the experimental data, also the inhibiting effect of pressure on the syngas laminar flame speeds can be well captured by the present model. The results from the H2/O2/CO sub-mechanism used in Klippenstein et al. (2011), Liu et al. (2013), Chang et al. (2015) are also shown in Figure 4 for comparison (represented by Sim_old). Overall, the present mechanism is closer to experimental data than the old mechanism.
Figures 5, 6 show the validation results of C1-C2 sub-mechanism. Figure 5 shows the comparison of laminar flame speed of methane/air mixture versus equivalence ratio between simulation and experiment at 1, 5, and 10 atm with a constant initial temperature of 298 K. The simulation exhibits excellent agreement at both 5 and 10 atm. The good performance of the present mechanism is shown by the solid line with open symbol in this Figure, with it accurately predicting the effect of changing equivalence ratio and initial pressure. Figure 6 shows the ignition delay comparison between simulation and experimental shock tube data for C2H2/O2/Ar lean mixtures. Again, the ignition delay times from the experimental shock tube can be well simulated using the present mechanism.
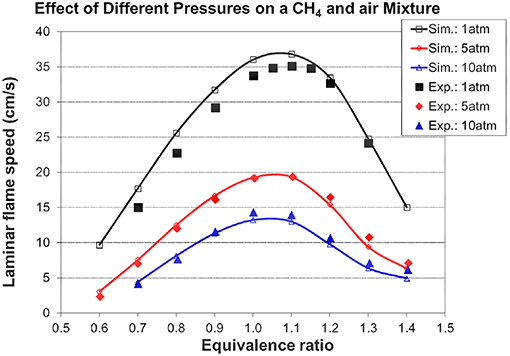
Figure 5. CH4/air laminar flame speed as a function of equivalence ratio at various pressures and an initial temperature of 298 K [experimental data are taken from Metcalfe et al. (2013)].
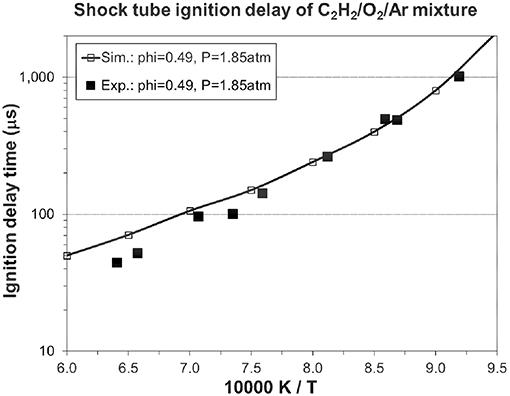
Figure 6. Ignition delay time in a shock tube: 0.5% C2H2, in Ar, φ = 0.49, p = 1.85 atm [experimental data are taken from (Metcalfe et al., 2013)].
Figures 7, 8A, 9 show the validation results of C3 sub-mechanism. Figure 7 shows the influence of equivalence ratio on ignition delay time of diluted propene/O2/Ar mixture at an initial pressure of 2 atm. The present mechanism can accurately capture the influence of equivalence ratio across the range of temperatures, showing faster ignition delay times for fuel-lean mixtures than fuel-rich mixtures. The effect of different dilution ratios (D = O2/(O2+N2) on the laminar flame speed of a propene, O2, and N2 mixture at p = 1 atm and T = 298 K was simulated and compared to experimental data. The dilution levels were: 0.18, 0.19, 0.20, 0.209 (air). Even minor modification of the dilution ratio will lead to significant change in the laminar flame speed as shown in Figure 8A, and this effect can be well captured by the present mechanism. Using the old mechanism of Klippenstein et al. (2011), Liu et al. (2013), Chang et al. (2015), the effect of different dilution levels on the laminar flame speed of the same mixture of Figure 8A at same condition is shown in Figure 8B. Compared to the results shown in Figure 8A from using the present new mechanism, the prediction from using the old mechanism shown in Figure 8B is much higher than the experiment data for equivalence ratio greater than 0.9. The reason is that the C2~C3 sub-mechanism of the old model is just a simply reduced mechanism. Figure 9 shows the simulation of concentration profiles of some species in a jet-stirred reactor with a C3H6/O2/He mixture and comparison with experimental data. The equivalence ratio is 2.19, the temperature ranges 800–1,100 K, and the pressure is near atmospheric. Overall the present mechanism can accurately predict the oxidation of propene in a JSR (jet-stirred reactor) under fuel-rich conditions, predicting the consumption of the fuel and oxygen and the formation of major intermediate and product species correctly.
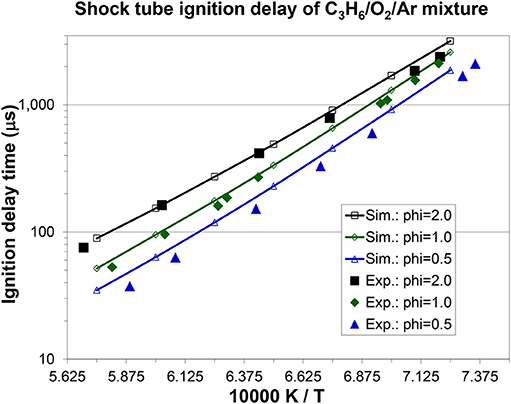
Figure 7. Effect of equivalence ratio on propene ignition delay time under dilute conditions: C3H6/4% + O2, in Ar, p = 2 atm [experimental data are taken from Burke et al. (2015)].
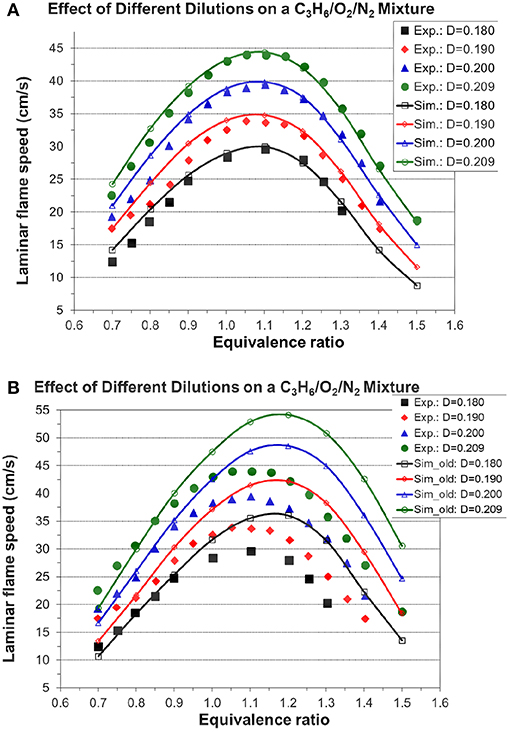
Figure 8. Effect of different dilution levels on the laminar flame speed of a propene, O2, and N2 mixture at p = 1 atm and T = 298 K, dilution level D = O2/(O2+N2) [experimental data are taken from Burke et al. (2015)]. (A) Results from the present new mechanism. (B) Results from the old mechanism of Liu et al. (Klippenstein et al., 2011; Liu et al., 2013; Chang et al., 2015).
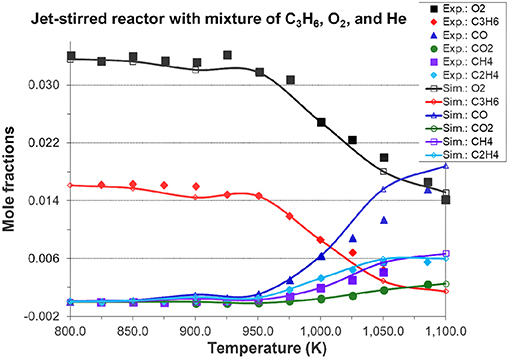
Figure 9. Simulated species mole fraction in a jet-stirred reactor with a mixture of 1.64% propene, 3.38% O2 in He, φ = 2.19, p = 1.05 atm, residence time=2.0 s, and compared to jet-stirred reactor experimental data [experimental data are taken from Burke et al. (2014)].
The predicted ignition delay times of MD/air and MD5D/air at φ = 1 and different pressures are compared with the experimental data in Figure 10A. The predicted ignition delay times for MD/air mixture match very well with the experimental data over most of the temperature range. The NTC (negative temperature coefficient) behavior of the experimental ignition for MD/air can also be captured using the new mechanism. The agreement between the predicted and experimental ignition delay times for MD5D/air is good over the whole range of temperatures. For the ignition delay times of MD5D, the magnitude of the NTC behavior becomes less obvious than MD. The predicted ignition delay times of MD/air and MD5D/air (MD: green dot curve; MD5D: red dashed curve) using the old model of Chang et al. (2015) are also shown in Figure 10A for comparison with the new mechanism. Since the simulation of ignition delay times is largely determined by the skeletal mechanisms while we didn't change the skeletal mechanisms of n-decane and MD/MD5D, overall the results from the new model are very close and similar to the old model. The slight difference is that the MD/air mixture using the old model has a larger NTC. The reason is that the ester group reactions in the new mechanism had minor changes while the reaction rate coefficients have not yet been reoptimized. The re-optimization of the reaction rate coefficients will be finished in our next stage of work. Figure 10B shows the predicted ignition delay times of MD/air and MD5D/air at φ = 1.5 and different pressures as well as the comparison to the experimental data. The results from the new model are still very close and similar to the old model except the slightly larger NTC behavior for the MD/air mixture using the old model.
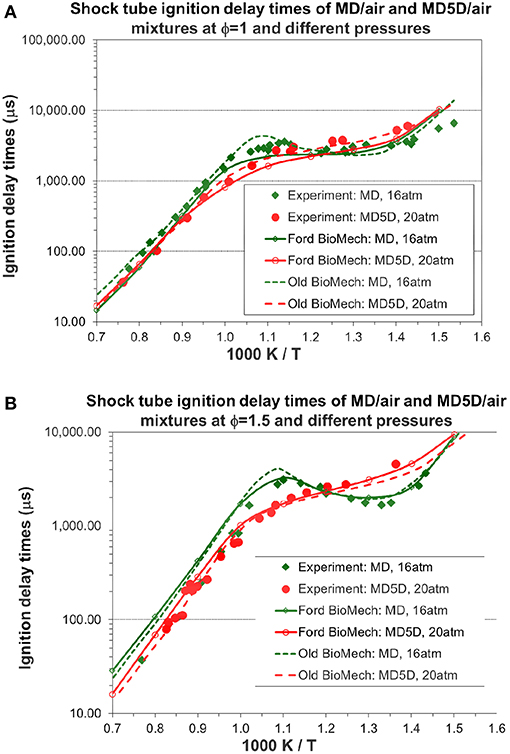
Figure 10. Simulated shock tube ignition delay times of MD/air and MD5D/air mixtures and comparison with shock tube experimental data [experimental data are taken from Chang et al. (2015)]. (A) Equivalence ratio = 1.0. (B) Equivalence ratio = 1.5.
The n-decane skeletal mechanism has been validated by Jia's team (Chang et al., 2015). So, the n-decane skeletal mechanism was not simulated in this work. However, considering other parts of the entire new mechanism may change the performance of the n-decane skeletal mechanism, it will be validated by us in the near future. Also, more experimental data for different biodiesel blends under various operating conditions will be collected to validate the present mechanism extensively in the near future. Finally, the present mechanism will be applied to biodiesel engine 3D CFD combustion modeling for understanding the combustion process. Figure 11 compares the experimental and simulated mole fractions of several major species (O2, CO, CO2) for the oxidation of RME (n-decane/MD/MD5D mixture) in a JSR at pressure = 10 atm and equivalence ratio = 1.0. Overall, the agreement between the experimental data and simulated results using the new mechanism is reasonable.
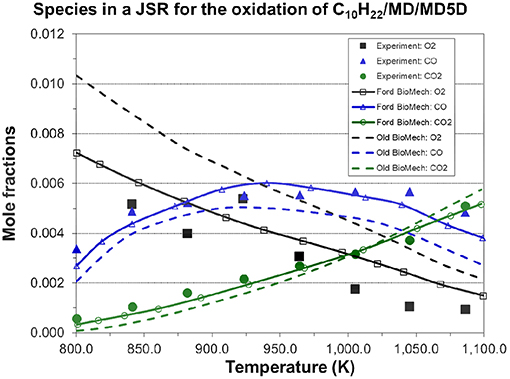
Figure 11. Simulated species concentrations in a jet-stirred reactor for the oxidation of RME (C10H22/MD/MD5D=0.035/0.0025/0.0475 mol.% for RME in the simulation) φ = 1.0, p = 10.0 atm, residence time = 1.0 s, and compared to jet-stirred reactor experimental data [experimental data are taken from Chang et al. (2015)].
A new chemical kinetic mechanism with 183 species and 1002 reactions for biodiesel based on decoupling methodology and latest C3 model (including latest H2/O2/CO/C1 and C2-C3 detailed sub-mechanisms) has been constructed. The mechanism aims to predict laminar flame speeds, ignition delay times, and important species evolutions accurately. In the construction of the new mechanism, the new “core” sub-mechanisms are coupled with the skeletal sub-mechanisms of MD, MD5D, and n-decane. The entire mechanism has been validated with available experiment data from small to large molecules of biodiesel. The existing validation results from this work show that the new mechanism has a very good performance. Especially the new mechanism has a better laminar flame speed prediction than published mechanisms. The n-decane skeletal mechanism and more experiment data for different biodiesel blends under various operating conditions will be validated extensively in the near future. Finally, the present mechanism will be tested in biodiesel engine 3D CFD combustion modeling.
SY worked on the mechanism integration and validation. MJ provided the skeletal mechanisms and decoupling methodology.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
BEV, battery electric vehicles; CFD, computational fluid dynamics; DRG, directed relation graph; DRGEPSA, directed relation graph error propagation and sensitivity analysis; HEV, hybrid electric vehicles; HFCV, hydrogen fuel cell vehicles; JSR, jet-stirred reactor; LLNL, Lawrence Livermore National Laboratory; MD, methyl decanoate; MD5D, methyl-5-decenoate; MD9D, methyl-9-decenoate; NTC, negative temperature coefficient; PHEV, plug-in hybrid electric vehicles; UV, Ultraviole.
Brakora, J., Ra, Y., and Reitz, R. D. (2011). Combustion Model for Biodiesel-Fueled Engine Simulations using Realistic Chemistry and Physical Properties. SAE Technical Paper 2011-01-0831.
Burke, S. M., Burke, U., Mathieu, O., Osorio, I., Keesee, C., Morones, A., et al. (2015). An experimental and modeling study of propene oxidation. part 2: ignition delay time and flame speed measurements. Combust. Flame 162, 296–314. doi: 10.1016/j.combustflame.2014.07.032
Burke, S. M., Metcalfe, W. K., Herbinet, O., Battin-Leclerc, F., Haas, F. M., Santner, J., et al. (2014). An experimental and modeling study of propene oxidation. part 1: speciation measurements in jet-stirred and flow reactors. Combust. Flame 161, 2765–2784. doi: 10.1016/j.combustflame.2014.05.010
Chang, Y., Jia, M., Li, Y., Zhang, Y., Xie, M., Wang, H., et al. (2015). Development of a skeletal oxidation mechanism for biodiesel surrogate. Proc. Combust. Inst. 35, 3037–3044. doi: 10.1016/j.proci.2014.09.009
Kéromnès, A., Metcalfe, W. K., Heufer, K. A., Donohoe, N., Das, A. K., Sung, C. J., et al. (2013). An experimental and detailed chemical kinetic modeling study of hydrogen and syngas mixture oxidation at elevated pressures. Combust. Flame 160, 995–1011. doi: 10.1016/j.combustflame.2013.01.001
Klippenstein, S. J., Harding, L. B., Davis, M. J., Tomlin, A. S., and Skodje, R. T. (2011). Uncertainty driven theoretical kinetics studies for CH3OH Ignition: HO2+CH3OH and O2+CH3OH. Proc. Combust. Inst. 33, 351–357. doi: 10.1016/j.proci.2010.05.066
Li, H., Yang, W., Zhou, D., and Yu, W. (2019). Skeletal mechanism construction for heavy saturated methyl esters in real biodiesel fuels. Fuel 239, 263–271. doi: 10.1016/j.fuel.2018.11.020
Liu, T., Jiaqiang, E., Yang, W., Hui, A., and Cai, H. (2016). Development of a skeletal mechanism for biodiesel blend surrogates with varying fatty acid methyl esters proportion. Appl. Energy 162, 278–288. doi: 10.1016/j.apenergy.2015.10.090
Liu, Y., Jia, M., Xie, M., and Pang, B. (2013). Improvement on a skeletal chemical kinetic model of iso-octane for internal combustion engine by using a practical methodology. Fuel 103, 884–891. doi: 10.1016/j.fuel.2012.07.046
Metcalfe, W. K., Burke, S. M., Ahmed, S. S., and Curran, H. J. (2013). A hierarchical and comparative kinetic modeling study of C1–C2 hydrocarbon and oxygenated fuels. Int. J. Chem. Kinet. 45, 638–675. doi: 10.1002/kin.20802
Patel, A., Kong, S. C., and Reitz, R. D. (2004). Development and Validation of Reduced Reaction Mechanism for HCCI Engine Simulation. SAE Technical Paper 2004-01-0558
Wang, X., Liu, H., Zheng, Z., and Yao, M. (2015). A skeletal mechanism of a biodiesel surrogate fuel for compression ignition engines. Energy Fuels 29, 1160–1171. doi: 10.1021/ef502142c
Keywords: bio-diesel, chemical kinetic mechanism, decoupling methodology, internal combusion engine, detailed chemical kinetic mechanism
Citation: Yang S and Jia M (2019) A Bio-Diesel Chemical Kinetic Mechanism Based on Decoupling Methodology and Detailed H2/O2/CO/C1~C3 Mechanism. Front. Mech. Eng. 5:21. doi: 10.3389/fmech.2019.00021
Received: 13 December 2018; Accepted: 08 April 2019;
Published: 07 May 2019.
Edited by:
Tiegang Fang, North Carolina State University, United StatesReviewed by:
Alpaslan Atmanli, National Defense University, TurkeyCopyright © 2019 Yang and Jia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shiyou Yang, c3lhbmczM0B1d2FsdW1uaS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.