
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mech. Eng. , 13 December 2018
Sec. Micro- and Nanoelectromechanical Systems
Volume 4 - 2018 | https://doi.org/10.3389/fmech.2018.00021
 Alex J. Bastiaens1*
Alex J. Bastiaens1* Jean-Philippe Frimat1
Jean-Philippe Frimat1 Teun van Nunen1
Teun van Nunen1 Bart Schurink2Erik F. G. A. Homburg1
Bart Schurink2Erik F. G. A. Homburg1 Regina Luttge1*
Regina Luttge1*In this work we present advances in three dimensional (3D) neuronal cell culture systems based on a reversible assembly of a microbioreactor with a microelectrode array (MEA) to create a MEMS-based 3D cell culture system for in vitro neuro-electrophysiological recordings. A batch of six molds were milled in poly (methyl methacrylate). The molds were used for soft lithography of polydimethylsiloxane (PDMS). In the center of the PDMS shape, a porous polyethersulfone (PES) cylindrical tube was press-fitted to form a growth barrier between the culture chamber inside the PES tube and the microfluidic channel surrounding the PES tube. A thin layer of partially cured PDMS was used to seal the bottom of the microbioreactor and provide reversible adhesion with the glass surface of a MEA. SH-SY5Y cells were successfully differentiated inside the microbioreactors in Matrigel and demonstrated extended neuronal networks over a height of at least 184 micrometers within the system. In previous microbioreactor designs visibility was limited due to the closed top with the dispensing holes. The new open top design allows for a better evaluation of the cell culture by optical detection methods during the experiment. Electrophysiological activity was recorded within the microbioreactor using human induced pluripotent stem cell-derived cortical neurons cultured in Matrigel, in 3D, up until 21 days in vitro. In summary, we present advances made in the design, the fabrication process and integration of microbioreactors with MEAs. Optical imaging capabilities improved significantly with an open top and the culture time was further extended from 7 to 21 DIV without leakage or degradation thanks to introducing PES as a barrier material and an enhanced assembly procedure. The latter facilitated a sufficient long-term culture for neurons to mature in an environment free from flow-induced stress and provided a proof of principle for the recording of electrophysiological activity of cortical neurons cultured in 3D.
The advance of cell culture models employing three-dimensional (3D) matrices for cells has proven to be of vital importance in studying cellular physiological and pathological responses. Compared to two-dimensional (2D) cell culture, 3D cell culture better represents the in vivo situation, with more appropriate cell-cell and cell-matrix interactions and biochemical, and biophysical, microenvironments (Haycock, 2011; Huh et al., 2011; Baker and Chen, 2012). Hydrogels are often employed to create these 3D microenvironments (Tibbitt and Anseth, 2009; Verhulsel et al., 2014). However, the diffusion of oxygen and nutrients is a limiting factor. Therefore, 3D cell cultures are performed in culture platforms with microelectromechanical system (MEMS) features and often microfluidic components to compensate for such issues and create small, sustainable culture volumes typically in the microliter range that are accessible for optical, chemical, or electrical analysis. In this paper, we contribute to this research field with the advances made on our own microbioreactor design (Schurink and Luttge, 2013).
In vitro neuronal cell models benefit from these 3D cell culture platforms as cells can better express the neuronal phenotype and form more realistic neuronal networks (Kunze et al., 2009, 2011; LaPlaca et al., 2010; Li et al., 2012; Koutsopoulos and Zhang, 2013; Frimat et al., 2015). In neuroscience research, the study of the electrophysiological response of neurons and neuronal networks is vital to understand brain function and brain pathology. Planar microelectrode arrays (MEA) are a state-of-the-art tool in in vitro studies, which consists of a glass surface with an array of microelectrodes for neuron coupling that connect to an external electronical amplifying system for input and output via spring-loaded contact pins to contact pads on the chip. The contact between cells and electrodes determines the quality of the captured signals, which requires the use of low-impedance electrodes (Chung et al., 2015). Although neurons would interact in 2D with the electrodes of a MEA, having a 3D neuronal cell culture on top of a MEA could aid in more realistic neuronal network activity as well as drug and toxicity screenings. Several others have shown the relevance of in vitro systems for neuronal cell cultures that include electrophysiological measurements. For example, Soscia et al. reported on the development of a removable device that allows for regional seeding of different neuronal cell types onto custom-made MEAs without barriers (Soscia et al., 2017) and Kanagasabapathi et al. demonstrated a dual-compartment microfluidic system for segregated neuronal cell culture with functional connectivity (Kanagasabapathi et al., 2011). These systems show different 2D brain regions, which interact on one chip. An in vitro bioreactor with thick film, low impedance, gold electrodes has been also developed for neuronal recordings in a 3D neuronal cell culture volume (Bartsch et al., 2015, 2018), but cell culture within this system was not yet performed in a 3D cellular microenvironment such as a hydrogel. In another concept, a layer-by-layer approach of electrode arrays in a polydimethylsiloxane (PDMS) bioreactor was shown as a prototype for 3D neuronal cell culture that is compatible with commercial amplifiers for planar MEAs (Musick et al., 2009). This bioreactor concept requires fluid flow through the cell culture volume to refresh nutrients and wash away waste products. Compared to the systems with custom fabricated electrodes from Bartsch et al. and Musick et al. the devices of Soscia et al. and Kanagasabapathi et al. are more readily available for additional biological assays outside of electrophysiological experiments as these are relatively easy and fast to fabricate. However, all these devices have limitations to the extent in which 3D cell culture can be performed. We therefore see a need for a new concept, which facilitates 3D soft-tissue culture on currently available commercial MEA technology and implements a barrier between fluids and neuronal cell culture, to eliminate shear stress on the cells due to fluid flows and sustain long-term culture.
As mentioned above, in our previous work, a microbioreactor was designed that can be placed onto glass surfaces, such as that of a MEA, to create a MEMS-based 3D neuronal cell culture system. In the microbioreactor, 3D cell culture could be maintained within a central chamber surrounded by an agarose membrane to mechanically separate the microfluidic channel from the culture medium. Although, this separation membrane between microfluidic channel and culture chamber effectively ensured compound exchange with the cell culture based on diffusion as preliminary demonstrated by primary rat cortical cell culture for 6 days in vitro (DIV) (Schurink and Luttge, 2013). For the practical purpose of scaling up this concept an additional iteration on the design was made. Instead of agarose, a polyethersulfone (PES) membrane, which is a biocompatible, low protein-binding material that does not degenerate over long periods of cell culture, was selected. This version was used so the microbioreactor could be successfully integrated with a capillary electrophoresis chip to perform biochemical analysis of metabolites of 3D cell cultures (Schurink, 2016). However, these previous studies with the PES membrane implemented in the microbioreactor were not described for their outcomes in cell culture yet. Also, the reusability of MEAs after the addition of a microbioreactor is important to reduce cost and waste in such experiments. Last, electrodes in MEAs block the imaging path and thereby limit the information that can be gained from neuronal cells behind these electrodes and the open-top microbioreactor introduced here may provide a means to improve visualization of the cell culture besides implementing the capability of electrical recordings.
In this study, we use two neuronal cell types. First SH-SY5Y cells, a human neuroblastoma cell line that can be differentiated toward the neuronal phenotype, was used in a pilot experiment to evaluate culture conditions for long-term culture in the microbioreactor with the PES barrier. Second, to proof the feasibility of relevant 3D neuronal cell cultures on MEAs, preliminary electrophysiological recordings from human induced pluripotent stem cell (iPSC)-derived cortical neurons were demonstrated.
The microbioreactor design is an iteration based on previous works in which the microbioreactor was created using brass and steel molds (Schurink and Luttge, 2013; Schurink, 2016). Here, the microbioreactor was fabricated by means of soft lithography of PDMS in poly (methyl methacrylate) (PMMA) molds and the use of porous polyethersulfone (PES) cylindrical membranes. The PES tube, which is generally used in water filtration applications (X-Flow Pentair, Enschede, the Netherlands), was chosen as a membrane due to its small pore size, biocompatibility and low-protein binding properties(Schurink, 2016). PMMA molds were made using the Roland MDX-40A (Roland DG, Geel, Belgium), a milling machine with micrometer precision range. This way a multitude of mold copies could be made, here, specifically a range of six identical molds. First, a PMMA plate with 10 mm thickness was placed in the machine and a 4 mm flat-bottomed roughing end mill (WNT, Roosendaal, Nederland) was used to mill coarse features, after which a 2 mm milling flat-bottomed end mill (WNT) was used to mill the detailed features. A 1 mm milling flat-bottomed end mill (WNT) was used to drill two 1.2 mm diameter holes into the PMMA mold based plate for flat-tipped 18-gauge needles (VWR, Amsterdam, the Netherlands) to be inserted. To ensure that PDMS could be removed from the mold after curing, the side walls of the mold were made from separate PMMA rings, which were laser cut from a PMMA plate of 8 mm thickness.
After assembly of the mold, PDMS (Sylgard® 184, Dow Corning, Wiesbaden, Germany) could be poured into the mold (Figure 1A). For this, the PDMS elastomer and curing agent were mixed in a 10:1 weight ratio and degassed for 10 min using a vacuum chamber and gently poured into a 10 ml syringe (VWR). Due to the viscous nature of the PDMS mixture and the small volume of each mold at ~1.1 ml, a syringe was used to inject the PDMS mixture into each unit of the mold. Next, the molds with PDMS were put in an oven at 65°C for 4 h to fully cure the PDMS. Using this new mold platform, six microbioreactors can be manufactured in one replica-molding batch.
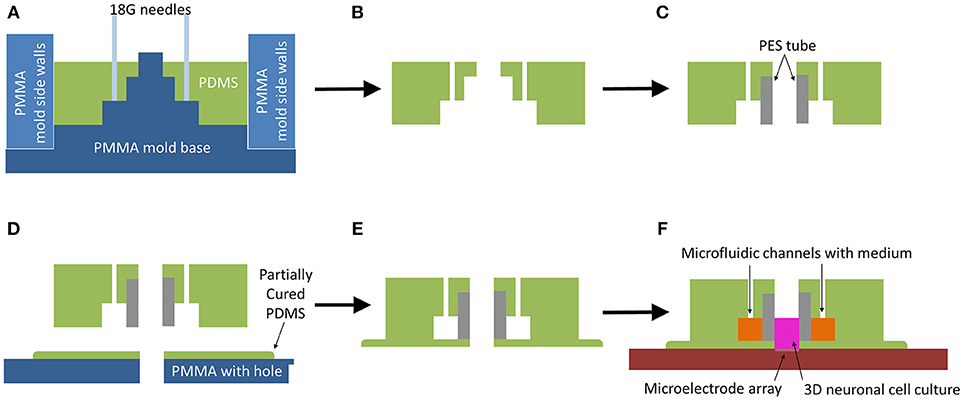
Figure 1. Microbioreactor mold fabrication process. (A) Schematic view of PDMS soft lithography in assembled mold. (B) After curing the PDMS in an oven at 65°C for 4 h, the PDMS is removed from the mold. (C) A porous PES cylinder is press-fitted into the opening of the PDMS, creating a membrane between the outer microfluidic channels and the central chamber for cell culture. (D) The PDMS-PES assembly is placed into a partially cured PDMS layer, obtained by curing PDMS on a PMMA plate for 17 min on a hotplate at 65°C, with the hole of the central chamber and the hole in the partially cured PDMS aligned by means of a pipette tip placed through the hole in the PMMA to guide the central chamber onto the partially cured PDMS. Afterwards, it is placed back onto the hot plate to fully cure at 65°C for 2 h. (E) The sealed and fully assembled microbioreactor is gently peeled off the PMMA plate. (F) The microbioreactor is placed onto a glass surface or MEA, allowing for 3D neuronal cell culture on the substrate.
The outer ring was used as side walls in the mold and the flat-tipped needles were removed from the mold so the PDMS part of the microbioreactor could also be removed from the mold plate (Figure 1B). The Porous PES tubes, providing a membrane between the cells in the culture and the fluidic channel, were cut to a length of 4 mm and press-fitted into the PDMS (Figure 1C).
A 2 mm thick plate of PMMA with six 3 mm diameter holes was warmed up on a hotplate at 65°C for 5 min, after which a thin layer of uncured PDMS was smeared over the whole plate. A 200 μL pipette tip was used to remove excess PDMS from the holes. The PMMA plate with PDMS was left on a hotplate at 65°C for 17 min for the PDMS to partially cure, after which microbioreactors were gently placed over the holes (Figure 1D). To make sure the hole in the PMMA aligned properly with the center of a microbioreactor, a 200 μL pipette tip was placed through the PMMA hole to center the microbioreactor. After centering, the pipette tip was removed and the PMMA plate with the microbioreactors was placed back onto the hotplate for another 2 h at 65°C to fully cure.
The sealed and cured microbioreactor was peeled off the PMMA plate (Figure 1E). A thin layer of PDMS surrounding the microbioreactor was left intact as part of the microbioreactor. This additional layer improves adhesion of the microbioreactor to the glass surface of either a MEA (Multi Channel Systems GmbH, Reutlingen, Germany) with configurations of either 60 or 120 electrodes, or a cover slip. Prior to cell culture, the microbioreactor was placed on either a cover slip or MEA (Figure 1F) and the top and inside of the microbioreactor were sterilized using 70% ethanol (VWR) for 30 min and rinsed thoroughly with demineralized water.
SH-SY5Y cells (Sigma Aldrich, Zwijndrecht, the Netherlands), a neuroblastoma cell line, were cultured in T75 flasks in culture medium containing Dulbecco's Modified Eagle's Medium (DMEM) and Ham's F12 medium in a 1:1 ratio (VWR), supplemented with 10% fetal bovine serum (FBS; lot no. 11113, Bovogen, East Keilor [VIC], Australia) and 1% penicillin/streptomycin (PS; Westburg, Leusden, the Netherlands) in an incubator at 37°C and 5% CO2. Cells were detached from the T75 flask using trypsin (Westburg) when reaching 70–80% confluency. The NucleoCounter NC-200 (ChemoMetec GmbH, Kaiserslautern, Germany) was used to determine the cell density of the cell suspension after which the number of cells required for cell seeding into the microbioreactors was calculated. All experiments were performed with cell cultures at passage 19.
To culture cells in the microbioreactor in 3D, Matrigel (VWR) was selected as a hydrogel for the 3D microenvironment (Koutsopoulos and Zhang, 2013; Benton et al., 2014). Cell suspension of SH-SY5Y cells in Matrigel was prepared by adding a 10X concentrated cell suspension in culture medium to the liquefied Matrigel, which is kept in an Eppendorf tube in ice water to ensure the Matrigel stays liquid-like. Pipettes used for handling Matrigel were stored in the freezer and taken out shortly before these handling steps to minimize temperature increase and thereby the gelling of the Matrigel, which starts at around ~10°C. Subsequently, a cell suspension at 10X concentration was added to the Matrigel at a 1:9 ratio to ensure that the final cell suspension in the Matrigel would be at 1X concentration.
Cell suspensions in the medium were prepared in the Matrigel at 4 × 105 cells ml−1 and 4 × 106 cells ml−1, referred to as low- and high-cell seeding density, respectively. For each cell seeding density, 15 μL of cells in the Matrigel were pipetted into the bottom of the PES tube of a microbioreactor at 0 DIV. To allow the Matrigel to gel, the microbioreactors were placed in the incubator at 37°C and 5% CO2 for 15 min. Next, 120 μL of medium was injected into the microfluidic channel and 50 μL of medium was pipetted on top of the cell culture in the culture chamber. For differentiation, at 1 DIV, the culture medium was replaced with the culture medium supplemented with 10 μM retinoic acid (RA; Sigma Aldrich) for 72 h to initiate differentiation of the cells (Dwane et al., 2013). Subsequently, at 4 DIV, medium was replaced with culture medium supplemented with 50 ng ml−1 brain-derived neurotrophic factor (BDNF; Sigma Aldrich) for 24 h to enhance the differentiation of the cells (Encinas et al., 2000; Teppola et al., 2016). After that, cells were kept in the culture medium until 21 DIV, and the medium was refreshed every 48 h. Cultures were fixed on 21 DIV by washing the samples twice in PBS and subsequently treating them with 3.7% paraformaldehyde (Merck Millipore, Amsterdam, Netherlands) for 1 h. The microbioreactors on the MEAs were removed prior to fixing the cells, as fixing is detrimental to the MEA. This was done by carefully lifting one side of the microbioreactor until the microbioreactor, including the 3D cell culture, came off the MEA, then transferring the microbioreactor, and its contents, to a cover slip.
For each of the cell densities used, experiments on microbioreactors on glass cover slips were performed in duplicate. SH-SY5Y neuronal cell cultures in 2D were performed as controls. The microbioreactors were placed onto two MEAs for 21 DIV at a low-cell seeding density.
Human iPSC-derived cortical neurons (Axol Bioscience, Cambridge, UK) were plated, differentiated and maintained according to manufacturer guidelines1 to generate a pure population of cortical neuronal without prior cell culture expansion in the microbioreactors on MEAs with 120 electrodes (Multi Channel Systems GmbH). In brief, the culture chamber and glass surface of the MEA were pre-coated with SureBond and ReadySet1 to ensure adhesion of cells to the surfaces 1 day prior to cell seeding. Cells were thawed and drop-wise added to an Eppendorf tube with 10 ml of pre-warmed, 37°C, Neural Expansion-XF Medium1, and centrifuged at 200 × g for 5 min, after which the medium was removed and cells were resuspended in Neural Plating-XF Medium1. Cells were then counted using the NucleoCounter NC-200. A 2D layer of cells at 1.5 × 105 cells cm−2 was added to the microbioreactors and left to adhere for 30 min prior to adding cells that were mixed with Matrigel to achieve a cell density of 8 × 106 cells ml−1 in the 3D volume. Then, 50 μL of the cell suspension in Matrigel was pipetted into each microbioreactor onto the 2D layer of cells. The microbioreactors were placed into an incubator for 24 h at 37°C and 5% CO2, after which half of the medium volume was replaced with Neural Maintenance-XF Medium1 to allow the cells to recover from thawing and seeding. After another 24 h, the medium was fully refreshed with Neural Differentiation-XF Medium1. Neural Differentiation-XF Medium was refreshed every 48 h until 10 DIV, after which half the volume of medium was refreshed every 48 h using Neural Maintenance-XF Medium.
Electrophysiological measurements were performed every day from 6 DIV onwards until 21 DIV using the 2100MEA-System (Multi Channel Systems GmbH) with 120 electrode head stage and associated software Multi Channel Experimenter (Multi Channel Systems GmbH). Each day, recordings of spontaneous activity of the neuronal cell culture were made for each sample.
Immunofluorescence staining was performed on the cells in the Matrigel and on 2D samples for control. The SH-SY5Y cells were stained with anti-β-Tubulin III (Sigma Aldrich) and anti-mouse IgG Alexa 555 (Thermofisher Scientific, Bleiswijk, the Netherlands) as primary and secondary antibodies to selectively stain SH-SY5Y that differentiated into a neuron-like phenotype (Agholme et al., 2010). Cells were permeabilized for 1 h with 0.1% Triton X-100 (Merck Millipore), then washed twice for 30 min with PBS. Then, for 4 h a blocking buffer of 10% FBS in PBS was applied, followed by incubation overnight with 1:100 primary antibody and 10% FBS in PBS. Cells were washed 4 times for 2 h with 10% FBS in PBS, then incubated overnight with 1:100 secondary antibody and 10% FBS in PBS. Additionally, two drops ml-1 NucBlue®; (Thermofisher Scientific) were added to the secondary antibody solution to stain the cell nuclei. In the case of the cell cultures on the MEAs, two drops ml-1 Actingreen®; (Thermofisher Scientific) to stain the cytoskeletal protein F-actin in the cells was also added. Finally, samples were rinsed four times for 1 h with PBS prior to imaging. To ensure cell cultures would not dry out during confocal imaging, PBS was replaced with the embedding medium Mowiol (Sigma Aldrich), which is typically used to mount 2D samples on cover slips prior to imaging, but can also be used for 3D samples.
During the experiments, cell cultures were studied using phase-contrast imaging with the EVOS FL microscope (Thermofisher Scientific) to obtain qualitative observations on culture viability, cell density, neuronal outgrowths, and network formation on 1 and 14 DIV. Confocal laser scanning microscopy was performed as end-point analysis on stained cell cultures to generate z-stack images, stacked 2D images forming a 3D representation, using the Leica TCS SP5X (Leica, Amsterdam, the Netherlands).
After the pilot experiment using SH-SY5Y cell culture within the microbioreactor on the MEAs and prior to fixing the cell culture, the microbioreactor was carefully lifted from the MEA. To ensure no chemicals or cell remnants would dry out on the MEA, the surface was covered in PBS until cleaning was performed. The MEA manufacturer guidelines were followed for cleaning the MEAs after cell culture. A 1% Terg-A-Zyme (Sigma Aldrich) solution in distilled water was prepared and placed onto the MEA overnight at room temperature. Afterwards, the MEA was thoroughly rinsed with distilled water and left to dry prior to optical inspection.
A microbioreactor design was realized (Figures 2A,B) by means of PDMS soft lithography using a micromilled PMMA mold and assembled with an insert, a porous PES tube, which forms a growth barrier between the culture chamber and microfluidic channel (Figure 2C). The PDMS shapes resulting from the molds were reproducible over multiple production runs without damaging or deteriorating the molds.
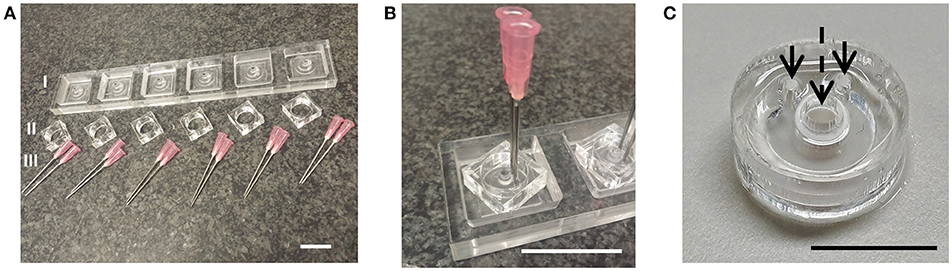
Figure 2. Microbioreactor mold and fabrication result. (A) Parts of the mold for the fabrication of the PDMS part of the microbioreactor. The primary mold I was made using micromilling of PMMA, which allowed for the production of multiple copies. To ensure the PDMS could be removed after soft lithography from the mold, the side walls II were made from separate pieces of PMMA. For the inlets of the microfluidics channels, flat-tipped needles III were inserted into the mold. (B) The assembled mold. Scale bars in (A,B) denote 3 cm. (C) Resulting PDMS microbioreactor with PES tube, with arrows denoting inlets to the microfluidic channel and the dashed arrow denoting the culure chamber inside the PES tube. Scale bar denotes 1 cm.
The PES tube locked into the PDMS firmly and the thin layer of partially cured PDMS underneath the microbioreactor ensured the PES was locked in place and when inserted into the microbioreactor, no fluids would circumvent the PES tube. The thin layer of PDMS also ensured that if a minimal amount of liquid seeped in between the PDMS and the glass surface, losing the adhesion and placement of the microbioreactor was avoided. Last, the thin layer of PDMS smoothened out any surface roughness from the micromilling on the bottom of the PDMS part of the microbioreactor, again aiding the adhesion between the PDMS and the glass. The microbioreactor was placed on the MEA after lifting the microbioreactor from the PMMA plate, using a microscope for overlay of the culture chamber with the electrodes during placement. Due to this manual handling, placement was not particularly well-overlaid with the area of the microelectrodes and the small edges of the thin layer of the PDMS could still be seen within the inner part of the PES tube (Figure 5A). Nevertheless, the area with microelectrodes was fully accessible for the 3D cell culture.
Long-term cell culture of 21 DIV was performed using both SH-SY5Y cells and human iPSC-derived cortical neurons in the microbioreactor. The SH-SY5Y cells were cultured at low- and high-cell seeding densities on glass cover slips and at low-cell seeding density on MEAs. Human iPSC-derived cortical neurons were cultured in microbioreactor on MEAs. These experiments were performed to demonstrate the long-term functionality of the MEMS-based 3D neuronal cell culture system that is provided by integrating the microbioreactor with a MEA.
SH-SY5Y cell cultures, both in 3D and 2D control samples, remained viable over the course of the experiment (Figures 3A–F). Due to the upgrade of the design having a culture chamber with an open top, cell cultures could be tracked well-during the experiments. Already at the end of one DIV, after adding the RA, cells could be seen with outgrowths starting to form toward creating a neuronal network of connected cells (Figures 3A–C). Cells continued to show differentiation through their morphological changes, in which outgrowths could be seen growing in the Matrigel (Figures 3D,E) and in the 2D control samples (Figure 3F). In 3D, the outgrowths between groups of cells within the 3D microenvironment formed neuronal networks.although as of 14 DIV, individual outgrowths for cells were poorly distinguishable within the cell culture, as these had grown into dense networks, particularly in the case of the high-cell seeding density.
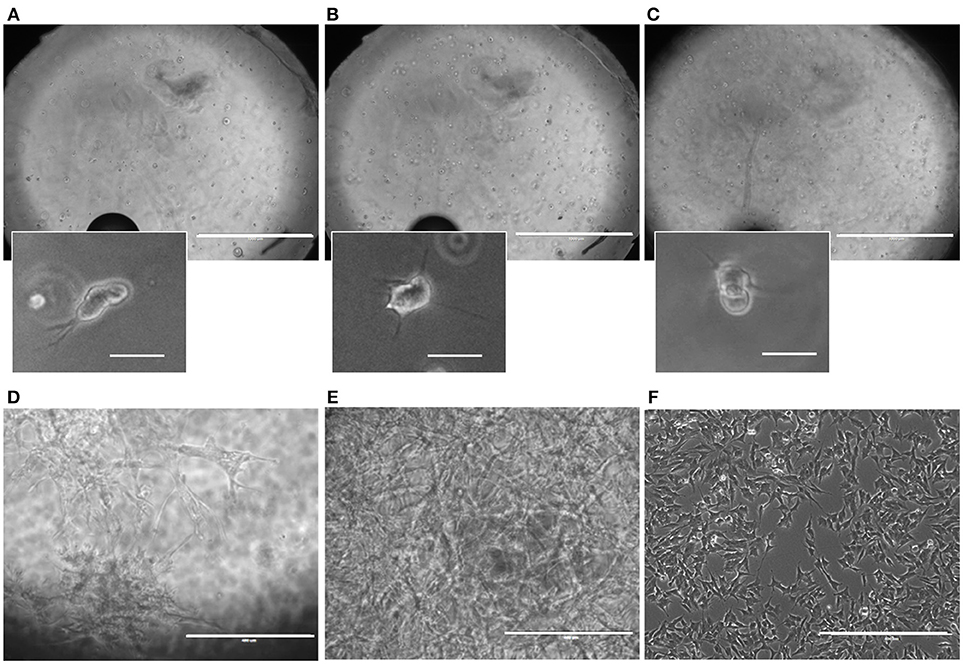
Figure 3. Microbioreactor on MEA before and during neuronal cell culture. (A–C) Representative overview images taken of the SH-SY5Y neuroblastoma culture in the Matrigel within the microbioreactor on a glass cover slip at 1 DIV at focal planes near the cover slip (A), the middle (B), and the top (C) of the 3D cell culture. Small images indicate enlarged sections of the large images, showing SH-SY5Y cells that have started to produce outgrowths. The scale bars denote 1 mm for the overview images and 100 μm for the enlarged sections. (D) A representative image of differentiated SH-SY5Y cells at 14 DIV for 4 × 105 cells ml−1 in a microbioreactor on top of a cover slip. The scale bar denotes 400 μm. (E) Representative image of differentiated SH-SY5Y cells at 14 DIV for 4 × 106 cells ml−1 in a microbioreactor on top of a cover slip. The scale bar denotes 400 μm. (F) A representative image of 2D control culture of differentiated SH-SY5Y cells at 14 DIV. The scale bar denotes 400 μm. For all images, the brightness was increased by 20% to increase visibility.
At the end of these pilot experiments, cells were fixed and stained on the glass cover slips. For the cells in the microbioreactor on the MEAs, we first removed the cell culture from the MEA, otherwise reuse of the MEA would be hampered due to the fixation chemicals, as fixed cell remnants do not wash off the MEA surface using Terg-A-Zyme. After lifting the microbioreactor off the MEA, it was carefully placed on a glass cover slip to reseal the system and allow for fixing and staining. Due to the microbioreactor being wet from the culture medium, a thin film of medium would form between the cover slip and the microbioreactor, making the microbioreactor slide on top of the cover slip. By placing the microbioreactor and cover slip on a hydrophobic surface, here a layer of Parafilm (Sigma Aldrich), during fixing and staining, no leaking of liquid from the microbioreactor would occur, ensuring that the cultures stayed wet. Confocal laser scanning resulted in z-stacks of the 3D cell culture volume (Figure 4). These z-stacks confirmed that a 3D volume of Matrigel with cells distributed throughout the gel was in place after 21 DIV, although comparatively more cells were in the bottom layer (Figure 4A) compared to the higher regions of the culture volume (Figures 4B,C). In particular it was observed that most cells in the upper layers of the 3D volume stained for β-Tubulin III, therefore confirming the expression of the neuronal phenotype and showing longer outgrowths. Relatively fewer cells near the bottom layer of the 3D volume showed this neuron-specific protein and also fewer and shorter outgrowths.
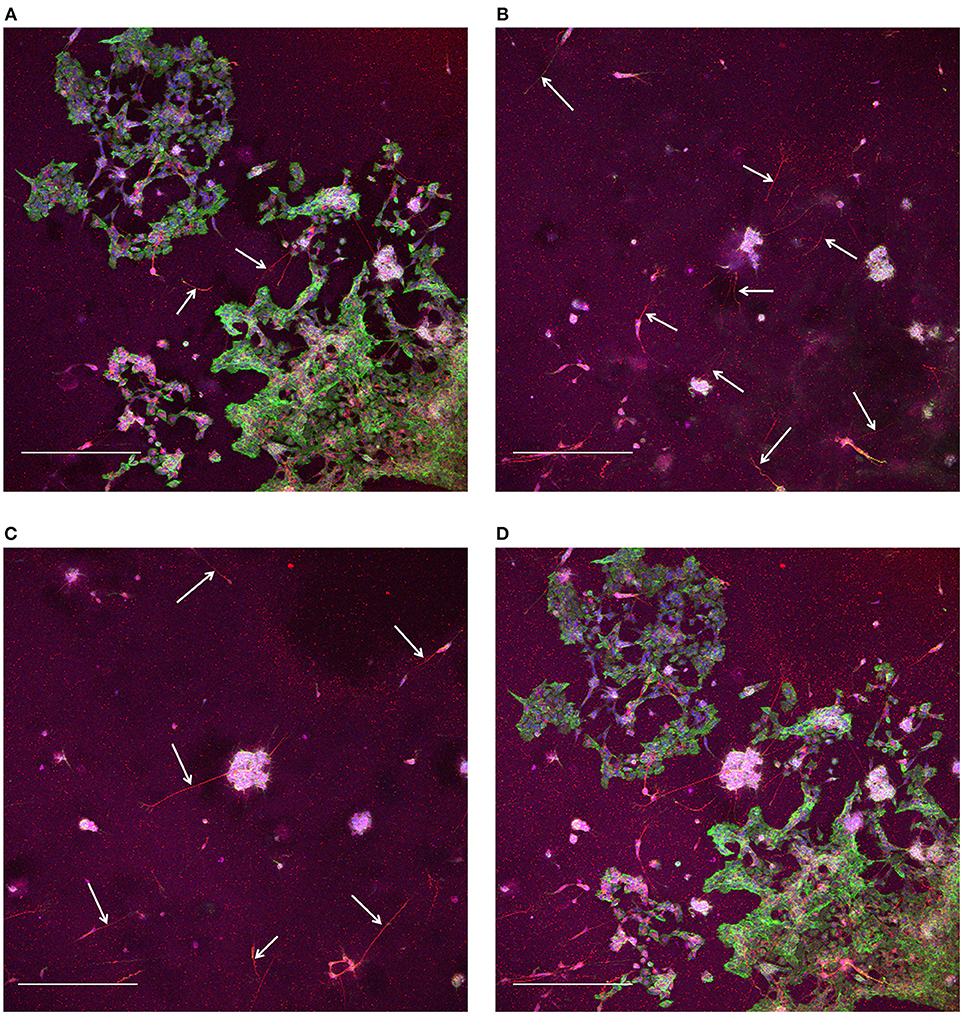
Figure 4. Confocal laser scanning microscopy of 3D SH-SY5Y cells in the microbioreactor. (A) Maximum intensity projection of slices for the range of 0–33.8 μm away from the bottom surface. (B) Maximum intensity projection of slices for the range of 35.1–106.6 μm away from the bottom surface. (C) Maximum intensity projection of slices for the range of 107.9–184.6 μm away from the bottom surface. (D) Maximum intensity projection the complete z-stack with a range of 184.6 μm and slices at a 1.3 μm interval. The staining shows red for neuron-specific protein β-Tubulin III, green for actin, and blue for cell nuclei for (A–D). The scale bars denote 400 μm. White arrows in (A–C) indicate neuronal outgrowths.
After the experiments were performed with the microbioreactors on the MEAs, it was demonstrated that the MEAs can be cleaned for future experiments. Following manufacturer guidelines on cleaning and drying the MEAs, little debris was left on the surface with no visible cell bodies or outgrowth remnants as compared to the MEA prior to the experiment upon optical inspection (Figure 5).

Figure 5. A MEA surface before and after a 3D SH-SY5Y cell experiment in a microbioreactor on top of a MEA. (A) Tile stitched image derived from 2 × 2 images at 4 × magnification of a microbioreactor on top of a MEA prior to the experiment. The dark circle is the porous PES tube that functions as a membrane between the microfluidic channel and the inner culture chamber that is centered on the microelectrodes. Scale bar denotes 1 mm. (B) After the long-term cell culture of SH-SY5Y cells inside a microbioreactor on a MEA, the cell culture was removed from the MEA for further analysis. The MEA was cleaned and imaged to assess the debris left after the cell culture. The scale bar denotes 1 mm. The image brightness and contrast were increased by 40% to increase visibility.
Recordings of electrophysiological activity were successfully performed using human iPSC-derived cortical neurons in 3D within the microbioreactor on top of a MEA. Over the course of time, from the first measurements at 6 DIV until measurements at 21 DIV, electrodes were able to record spontaneous spiking activity from the cortical neurons (Figure 6). The recordings show amplitudes in the 100 μV range at 7 DIV (Figure 6A), to amplitudes higher than 400 μV at 14 and 21 DIV (Figures 6B,C). The recordings also show that when comparing 7, 14, and 21 DIV, that over time the number of bursts of activity and the number of locations where activity is recorded increases (Figures 6D–F).
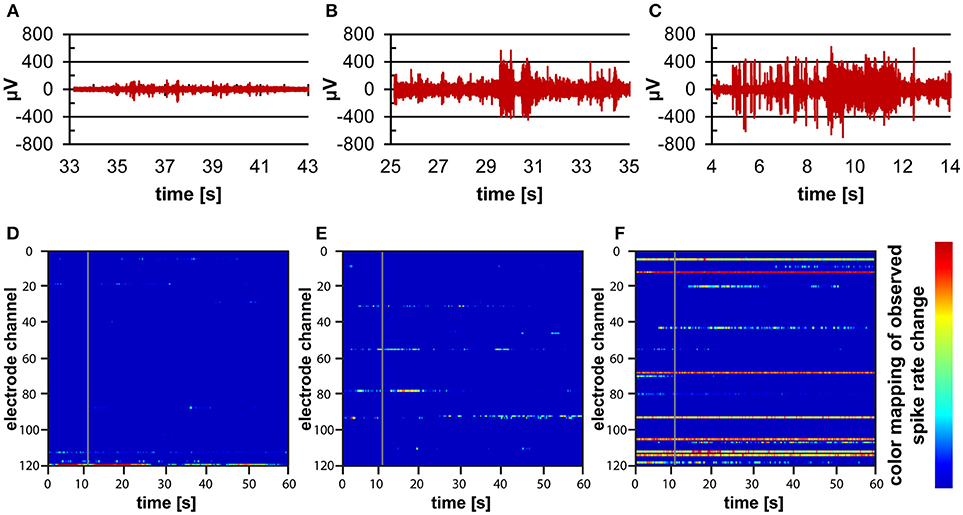
Figure 6. Electrophysiological measurements on human iPSC-derived cortical neurons within a microbioreactor on a MEA. (A–C) Filtered data recordings of spontaneous activity of one electrode for 10 s at 7 DIV (A), 14 DIV (B), and 21 DIV (C) of the 3D cell culture, showing the increase in spontaneous activity of cells. (D–F) Raster plots showing 60 s of spontaneous activity for all electrodes of the MEA at 7 DIV (D), 14 DIV (E), and 21 DIV (F) of the 3D neuronal cell culture. The color bar to the right displays the color associated with high (red) to low (blue) spiking activity rates, showing that over time more electrodes pick up more spontaneous activity from the cells.
In this work we present advances in 3D neuronal cell culture systems based on a reversible assembly of a microbioreactor with a MEA to create a MEMS-based 3D cell culture system for in vitro neuro-electrophysiological recordings.
The fabrication of the microbioreactor was scalable thanks to the use of a micromilled mold platform to scale up the microbioreactor replica molding process from one to six per batch. This makes the platform useful to experiments requiring multiple repetitions, as it is often the case in biological studies. Is the option for a scalable fabrication process also facilitates higher throughput experiments. Any seeping of fluid in between the PDMS and glass would often result in the microbioreactor gradually detaching from the glass and drifting from its original position, showing the importance of starting with a completely dry MEA and microbioreactor and gentle addition of fluids, as the resulting pressure on the systems may cause deformation of the PDMS and thereby local detachment of PDMS from the surface. The thin layer of PDMS underneath aided in improved adhesion between the microbioreactor and the surface of the glass. In our experience, the use of the thin layer of PDMS and starting with completely dry components worked better with regard to adhesion to the glass surface compared to when the microbioreactor was directly placed onto the glass substrates after dipping the microbioreactor into the partially cured PDMS. Potentially, the partially cured PDMS would allow the microbioreactor to take the shape of a glass surface. However, this better fit did not result in better adhesion. Also, in the case that any of the partially cured PDMS would leech into the surrounding glass surface of the MEA, this would result in a deteriorated performance of the electrodes due to fouling. Considering the importance of a reversible bond between PDMS and the glass surface, plasma treatment was not used, as this creates a covalent bond between the PDMS and glass.
SH-SY5Y cell cultures were successfully differentiated and neuronal outgrowths formed neuronal networks throughout the 3D volume within the system for a height of at least 184 μm (Figure 4). Confocal microscopy also revealed that detachment of the microbioreactor with cell culture from the MEA was not detrimental for the imaging of the neuronal networks contained in the microbioreactor. Although part of the cell culture that had adhered to the MEA may not have been detached together with the microbioreactor, an overview of cells throughout the 3D volume of the cell culture were successfully imaged. In previous microbioreactor designs, due to the closed top with the dispensing holes, visibility was limited compared to this improved design, which allows for better evaluation of the cell culture by optical detection methods during the experiment.
After the experiments, the MEAs can be successfully cleaned as checked by optical assessment underneath a microscope, allowing for their reusability. Also, both the fully cured PDMS and the PES material are inert and should not leave chemical residues on the MEA surface that might not get removed using the cleaning agent. Nevertheless, electrochemical impedance testing before and after the experiment and after cleaning could further eliminate doubts to whether the microbioreactor addition to the MEA negatively affects the performance of the electrodes.
The culture of human iPSC-derived cortical neurons within our MEMS-based 3D cell culture system demonstrates the feasibility of performing electrophysiological recordings on 3D neuronal cell cultures for at least 21 days. Previous research using self-assembled glass beads as a 3D scaffold has already shown significant differences in network dynamics between 2D and 3D neuronal cell culture (Frega et al., 2014). Hence, as a next step, we will expand our experiments with the soft-scaffold microbioreactor using the human iPSC-derived cortical neurons to investigate 3D neuronal network activity.
In conclusion, we showed advances made in the design, the fabrication process and integration of microbioreactors with MEAs. Optical imaging capabilities improved significantly with an open top and the culture time was further extended from 7 to 21 DIV including the successful recording of electrophysiological activity of cortical neuronal networks cultured in 3D. Cell culture was possible without leakage or degradation, due to introducing PES as a barrier material and the enhanced assembly procedure. The latter allowed a sufficiently long-term culture for neurons to mature and to eliminate fluid flow induced stresses on the cell culture. With these advances in the design of our 3D neuronal cell culture system human-derived 3D neuronal cell networks can now be performed.
RL acquired funding; AB, J-PF, TvN, BS, EH, and RL conceptualization and design of the work; AB, J-PF, TvN, and EH data acquisition; AB, J-PF, TvN, and BS data analysis; AB, J-PF, TvN, and RL data interpretation; AB, J-PF, TvN, BS, EH, and RL preparation of the manuscript; AB, J-PF, TvN, BS, EH, and RL final approval of the version to be published.
This work was financially supported by the European Research Council (ERC), Grant No. 280281 and the Eindhoven University of Technology.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors are grateful for the experimental support of the members of the Microfab/lab at the Eindhoven University of Technology.
2D, Two-dimensional; Three-dimensional; BDNF, Brain-derived neurotrophic factor; DIV, Days in vitro; DMEM, Dulbecco's modified Eagle's medium; FBS, Fetal bovine serum; MEA, Microelectrode array; MEMS, Microelectromechanical systems; PBS, Phosphate buffered saline; PES,Polyethersulfone; PDMS, Polydimethylsiloxane; PMMA, Poly(methyl methacrylate); PS, Penicillin/streptomycin; RA, Retinoic acid.
1. ^Axol Bioscience. Human iPSC-Derived Neural Stem Cells, Protocol version 5.0 Available online at: https://docs.axolbio.com/wp-content/uploads/Protocol-Human-iPSC-Derived-Neural-Stem-Cells-Version-5.0.pdf (Accessed April 2, 2018).
Agholme, L., Lindström, T., Kågedal, K., Marcusson, J., and Hallbeck, M. (2010). An in vitro model for neuroscience: differentiation of SH-SY5Y cells into cells with morphological and biochemical characteristics of mature neurons. J. Alzheimers Dis. 20, 1069–1082. doi: 10.3233/JAD-2010-091363
Baker, B. M., and Chen, C. S. (2012). Deconstructing the third dimension – how 3D culture microenvironments alter cellular cues. J. Cell Sci. 125, 3015–3024. doi: 10.1242/jcs.079509
Bartsch, H., Baca, M., Fernekorn, U., Müller, J., Schober, A., and Witte, H. (2018). Functionalized thick film impedance sensors for use in in vitro cell culture. Biosensors 8:37. doi: 10.3390/bios8020037
Bartsch, H., Himmerlich, M., Fischer, M., Demkó, L., Hyttinen, J., and Schober, A. (2015). LTCC-based multi-electrode arrays for 3D in vitro cell cultures. J. Ceram. Sci. Technol. 6, 315–324. doi: 10.4416/JCST2015-00056
Benton, G., Arnaoutova, I., George, J., Kleinman, H. K., and Koblinski, J. (2014). Matrigel: from discovery and ECM mimicry to assays and models for cancer research. Adv. Drug Deliv. Rev. 79, 3–18. doi: 10.1016/j.addr.2014.06.005
Chung, T., Wang, J. Q., Wang, J., Cao, B., Li, Y., and Pang, S. W. (2015). Electrode modifications to lower electrode impedance and improve neural signal recording sensitivity. J. Neural Eng. 12:056018. doi: 10.1088/1741-2560/12/5/056018
Dwane, S., Durack, E., and Kiely, P. A. (2013). Optimising parameters for the differentiation of SH-SY5Y cells to study cell adhesion and cell migration. BMC Res. Notes 6:366. doi: 10.1186/1756-0500-6-366
Encinas, M., Iglesias, M., Liu, Y., Wang, H., Muhaisen, A., Ceña, V., et al. (2000). Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated, neurotrophic factor-dependent, human neuron-like cells. J. Neurochem. 75, 991–1003. doi: 10.1046/j.1471-4159.2000.0750991.x
Frega, M., Tedesco, M., Massobrio, P., Pesce, M., and Martinoia, S. (2014). Network dynamics of 3D engineered neuronal cultures: a new experimental model for in vitro electrophysiology. Sci. Rep. 4:5489. doi: 10.1038/srep05489
Frimat, J.-P., Xie, S., Bastiaens, A., Schurink, B., Wolbers, F., den Toonder, J., et al. (2015). Advances in 3D neuronal cell culture. J. Vac. Sci. Technol. B, Nanotechnol. Microelectron. 33:06F902. doi: 10.1116/1.4931636
Haycock, J. W. (2011). 3D Cell culture: a review of current approaches and techniques. Methods Mol. Biol. 695, 1–15. doi: 10.1007/978-1-60761-984-0_1
Huh, D., Hamilton, G. A., and Ingber, D. E. (2011). From 3D cell culture to organs-on-chips. Trends Cell Biol. 21, 745–754. doi: 10.1016/j.tcb.2011.09.005
Kanagasabapathi, T. T., Ciliberti, D., Martinoia, S., Wadman, W. J., and Decré, M. M. J. (2011). Dual-compartment neurofluidic system for electrophysiological measurements in physically segregated and functionally connected neuronal cell culture. Front. Neuroeng. 4:13. doi: 10.3389/fneng.2011.00013
Koutsopoulos, S., and Zhang, S. (2013). Long-term three-dimensional neural tissue cultures in functionalized self-assembling peptide hydrogels, Matrigel and Collagen I. Acta Biomater. 9, 5162–5169. doi: 10.1016/j.actbio.2012.09.010
Kunze, A., Bertsch, A., Giugliano, M., and Renaud, P. (2009). Microfluidic hydrogel layers with multiple gradients to stimulate and perfuse three-dimensional neuronal cell cultures. Procedia Chem. 1, 369–372. doi: 10.1016/j.proche.2009.07.092
Kunze, A., Giugliano, M., Valero, A., and Renaud, P. (2011). Micropatterning neural cell cultures in 3D with a multi-layered scaffold. Biomaterials 32, 2088–2098. doi: 10.1016/j.biomaterials.2010.11.047
LaPlaca, M. C, Vernekar, V. N., Shoemaker, J. T., and Cullen, D. K. (2010). Three-dimensional Neuronal Cultures, Methods in Bioengineering: 3D Tissue Engineering (Norwood, MA: Artech House), 187–204.
Li, X(James)., Valadez, A. V., Zuo, P., and Nie, Z. (2012). Microfluidic 3D cell culture: potential application for tissue-based bioassays. Bioanalysis 4, 1509–1525. doi: 10.4155/bio.12.133
Musick, K., Khatami, D., and Wheeler, B. C. (2009). Three-dimensional micro-electrode array for recording dissociated neuronal cultures. Lab Chip 9:2036. doi: 10.1039/b820596e
Schurink, B. (2016). Microfabrication and Microfluidics for 3D Brain-on-Chip. Ph.D. thesis. University of Twente, Enschede.
Schurink, B., and Luttge, R. (2013). Hydrogel/poly-dimethylsiloxane hybrid bioreactor facilitating 3D cell culturing. J. Vac. Sci. Technol. B, Nanotechnol. Microelectron. Mater. Process. Meas. Phenom. 31, 06F903. doi: 10.1116/1.4831762
Soscia, D., Belle, A., Fischer, N., Enright, H., Sales, A., Osburn, J., et al. (2017). Controlled placement of multiple CNS cell populations to create complex neuronal cultures. PLoS ONE 12:e0188146. doi: 10.1371/journal.pone.0188146
Teppola, H., Sarkanen, J. R., Jalonen, T. O., and Linne, M. L. (2016). Morphological differentiation towards neuronal phenotype of SH-SY5Y neuroblastoma cells by estradiol, retinoic acid and cholesterol. Neurochem. Res. 41, 731–747. doi: 10.1007/s11064-015-1743-6
Tibbitt, M. W., and Anseth, K. S. (2009). Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng. 103, 655–663. doi: 10.1002/bit.22361
Keywords: SH-SY5Y cells, 3D cell culture, microelectrode array, microbioreactor, neuro-electrophysiological recording
Citation: Bastiaens AJ, Frimat J-P, van Nunen T, Schurink B, Homburg EFGA and Luttge R (2018) Advancing a MEMS-Based 3D Cell Culture System for in vitro Neuro-Electrophysiological Recordings. Front. Mech. Eng. 4:21. doi: 10.3389/fmech.2018.00021
Received: 31 July 2018; Accepted: 03 December 2018;
Published: 13 December 2018.
Edited by:
Massimo Mastrangeli, Delft University of Technology, NetherlandsReviewed by:
Rongyu Tang, Beijing Institute of Technology, ChinaCopyright © 2018 Bastiaens, Frimat, van Nunen, Schurink, Homburg and Luttge. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alex J. Bastiaens, YS5qLmJhc3RpYWVuc0B0dWUubmw=
Regina Luttge, ci5sdXR0Z2VAdHVlLm5s
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.