 Matthias Redies
1,2,3
Matthias Redies
1,2,3
 Gregor Michalicek
1
Gregor Michalicek
1
 Juba Bouaziz
1
Juba Bouaziz
1 Christian Terboven4Matthias S. Müller2,4
Christian Terboven4Matthias S. Müller2,4
 Stefan Blügel
1,2
Stefan Blügel
1,2
 Daniel Wortmann
1*
Daniel Wortmann
1*- 1 Peter Grünberg Institut and Institute for Advanced Simulation, Forschungszentrum Jülich, Jülich, Germany
- 2 JARA-CSD, Jülich, Germany
- 3 Department of Physics, RWTH Aachen University, Aachen, Germany
- 4 IT Center, RWTH Aachen University, Aachen, Germany
Virtual materials design requires not only the simulation of a huge number of systems, but also of systems with ever larger sizes and through increasingly accurate models of the electronic structure. These can be provided by density functional theory (DFT) using not only simple local approximations to the unknown exchange and correlation functional, but also more complex approaches such as hybrid functionals, which include some part of Hartree–Fock exact exchange. While hybrid functionals allow many properties such as lattice constants, bond lengths, magnetic moments and band gaps, to be calculated with improved accuracy, they require the calculation of a nonlocal potential, resulting in high computational costs, that scale rapidly with the system size. This limits their wide application. Here, we present a new highly-scalable implementation of the nonlocal Hartree-Fock-type potential into FLEUR—an all-electron electronic structure code that implements the full-potential linearized augmented plane-wave (FLAPW) method. This implementation enables the use of hybrid functionals for systems with several hundred atoms. By porting this algorithm to GPU accelerators, we can leverage future exascale supercomputers which we demonstrate by reporting scaling results for up to 64 GPUs and up to 12,000 CPU cores for a single k-point. As proof of principle, we apply the algorithm to large and complex iron garnet materials (YIG, GdIG, TmIG) that are used in several spintronic applications.
1 Introduction
Materials science aims to understand and predict material properties ever more accurately, so that new sophisticated materials can be discovered to drive innovation in domains that rely on them. While materials science has been around for millennia, it was only at the beginning of the last century that the arrival of quantum mechanics enabled the exact description of the microscopic properties in materials. However, the cost of calculating the exact solution to the Schrödinger equation grows exponentially with the size of the system and is therefore limited to very small systems. Density functional theory (DFT) replaces the 3N-dimensional wave function as the central quantity with the 3-dimensional ground-state density and thereby reduces the exponential computational cost to a polynomial one. While DFT is in principle exact, a key ingredient, the so-called exchange-correlation energy, has no known analytical expression. The approximations used for this term determine the accuracy with which material properties can be predicted. While the most commonly used approximations, the local density approximation (LDA) and the generalized gradient approximation (GGA), can predict certain properties with a high precision at a very low computational cost, they fail to predict some essential electronic properties in particular of electronically complex materials (Alberi et al., 2018).
DFT is increasingly being used in the context of high-throughput calculations, where hundreds of thousands of material candidates are screened using automated workflows (Yan et al., 2017; Mounet et al., 2018; Rosen et al., 2019). However, all of these calculations are limited to material classes and properties for which the underlying exchange-correlation functionals have a good predictive power. In order to enhance these calculations with material classes and properties for which LDA and GGA fail, it is necessary to rely on more accurate methods producing high-quality results. One class of accurate methods are the hybrid exchange-correlation functionals which are particularly suited to predicting electronic properties such as the band gap, the degree of charge localization and the polarization in materials with a stronger electron correlation (Cramer and Truhlar, 2009; Zhang et al., 2011; Burke, 2012; Becke, 2014; Garza and Scuseria, 2016).
Hybrid exchange-correlation functionals, such as PBE0 (Perdew et al., 1996) or HSE06 (Krukau et al., 2006) functionals, mix a portion of an orbital dependent exact exchange with the electron correlation described by other approximations, such as LDA or GGA (Becke, 1993). Their reliance on the orbital dependent exact exchange makes them computationally considerably more expensive than LDA or GGA. While an LDA or a GGA calculation grows with the 3rd power of the number of atoms, a hybrid exchange-correlation functional calculation typically grows with the 4th power of the number of atoms. Additionally, the computational cost of a hybrid calculation grows quadratically with the number of k-points used to sample the Brillouin zone, whereas for an LDA or a GGA calculation it only grows linearly. This large computational cost has prohibited precise predictions for systems with large unit cells, including a number of interesting material classes such as garnets (Rodic et al., 1999; Nakamoto et al., 2017) or materials of interest for solid-state batteries (Yu et al., 2016).
This article focuses on the implementation of hybrid functionals in the full-potential linearized augmented-plane-wave (FLAPW) (Wimmer et al., 1981) method as it is implemented in the open-source code FLEUR (Fleur, 2021). Unlike approaches employing the pseudopotential approximation, the FLAPW method treats all electrons explicitly and does not employ any approximations to represent the potential or density. It is therefore well suited for a wide range of systems, including systems containing heavy atoms that have d- and f-electrons. It is considered to be one of the most accurate DFT methods and has been used as a benchmark for other methods and codes (Lejaeghere et al., 2016). More specifically we focus here on the efficient implementation of the Hartree-Fock type exact exchange based on the bare Coulomb kernel as relevant for the PBE0 functional. Functionals based on the screened Coulomb kernel as HSE06 can be always expressed in terms of the matrix elements of the bare Coulomb kernel subtracted by matrix elements of a smooth function (Schlipf et al., 2011), whose numerical evaluation is not time critical and is not further discussed here.
There have been significant advances in bringing hybrid functionals to systems with hundreds or even thousands of atoms in other approaches, such as the projector augmented wave method (PAW) (Barnes et al., 2017; Carnimeo et al., 2019), the s-MTACE METHOD (Mandal et al., 2021), gaussian basis functions (Guidon et al., 2008) and atomic-orbitals method (Hakala and Foster, 2013; Lin et al., 2021). Even some all-electron methods have demonstrated their capability to calculate large systems with hybrid functionals (Ihrig et al., 2015; Levchenko et al., 2015). However, hybrid functionals within FLAPW have been constrained to very small systems (Betzinger et al., 2010; Schlipf et al., 2011; Blaha et al., 2020). The work presented here enables FLEUR’s hybrid functional implementation to run on the world’s most advanced supercomputers and use their immense computational power to investigate these large and interesting systems containing hundreds of atoms. Building on the pioneering work previously done on hybrid functionals in the FLAPW basis and in FLEUR specifically (Betzinger et al., 2010; Schlipf et al., 2011), we analyzed the performance and bottlenecks of this legacy implementation, and explored algorithmic improvements needed to calculate hundreds of atoms with the accuracy that FLAPW and hybrid functionals offer.
2 Methods
The FLAPW method, implemented by FLEUR, partitions the unit cell of volume Ω into two kinds of domains. In spherical regions MT a centered around each atomic nucleus, a muffin-tin orbital basis (Andersen and Woolley, 1973) is used, relying on the products of spherical harmonics and radial functions. In between these spheres, in the so-called interstitial region (IS), a plane-wave basis is used. The resulting LAPW basis functions used to represent the wave functions are
Here k and G are the Bloch- and reciprocal lattice vectors, while σ indicates the spin.
In order to calculate the Hartree-Fock exact exchange, the Coulomb integral
2.1 Exact Exchange
Using this basis set the nonlocal exact exchange can be expressed as
where n, n′ and n′′ are band indices of the states
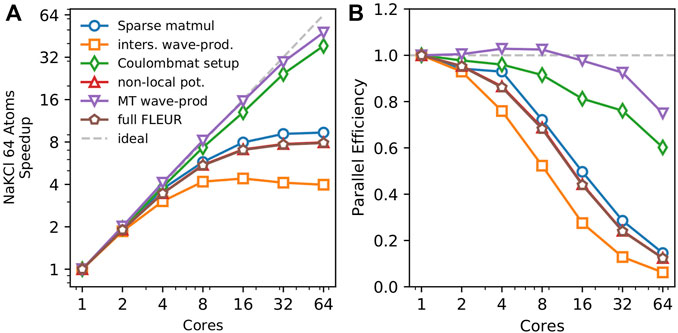
FIGURE 1. Strong scaling behavior with OpenMP on a single AMD EPYC 7742 64 core processor. The overall FLEUR iteration is shown with brown pentagons, while the calculation of the nonlocal potential is shown in red triangles. The four remaining lines show the major parts of the nonlocal potential. The red triangles indicating the nonlocal potential largely coincide with the brown pentagons indicating the full runtime, making it difficult to see them. The parallel efficiency of slightly above 100% in the routine for the setup mixed-product basis for 4 and 8 nodes is explained by cache effects. Depending on the number of cores used the stride of the parallelized loops executed on each core is changed, which can reduce the number of cache misses if this stride coincides with certain array dimensions.
The numerical evaluation of Eq. 2 represents the majority of the computational effort in a FLEUR calculation using hybrid functionals. In particular, the projection of the products of wave functions given in the LAPW basis set (Eq. 1) onto the mixed-product basis and the multiplication of these projections with the Coulomb matrix provide significant computational challenges. The implementation developed for this work relies on collecting data processed in the same way. It allows to exploit data parallelism on multiple levels, be it the use of a SIMD instruction set or an efficient and balanced use of multiple threads. Two significant changes have been made to the basic algorithm introduced in (Betzinger et al., 2010). First, contrary to the previous implementation, the projection onto the mixed-product basis in the interstitial region is now calculated by Fourier transforming the wave-functions into real-space and multiplying pairs of wave-functions there, before transforming them back into G-space. By employing fast Fourier transformations, this reduces the complexity of this calculation from
2.2 Shared Memory Parallelization
To enable the utilization of supercomputers with complex memory hierarchies, we rely on two classes of parallelization. We use shared memory parallelization to make full use of many-core CPUs or GPUs. While distributed memory parallelization is employed to distribute the calculation over hundreds of compute nodes. Shared memory parallelization is realized by utilizing libraries for standard math problems such as matrix-multiplications or Fourier transformations whenever possible. Code parts that do not fall within the mold of any standard math problem were parallelized using OpenMP on CPUs and OpenACC on GPUs. In Figure 1 the strong scaling behavior on a single node is shown. For strong scaling a fixed-size problem is calculated with an increasing amount of resources and the resulting speedup is measured. Here, the speedup is defined as
While some parts, such as the projection onto the mixed-product basis in the muffin-tins or the setup of the Coulomb matrix show excellent scaling, the speedup of the projection on the mixed-product basis in the interstitial exhibits a plateau around a speedup of 4 (see orange line). As mentioned previously, this algorithm relies on fast Fourier transformations, which have a low algorithmic intensity, meaning that only few calculations are performed compared to the number of load and store operations, making the algorithm more likely to be limited by the memory bandwidth rather than the available computational power, explaining the plateau in the speedup seen in Figure 1. Up until 8 cores, the FFT still benefits from the additional compute resources, beyond that the FFTs are not limited anymore by the computational power, but rather by the memory bandwidth, which does not increase with the number of assigned cores.
2.3 Distributed Memory Parallelization
The parallelism shown so far is limited to a single shared memory node and thus limited by the number of cores on a given node. Therefore, in order to scale the computational challenge posed by the hybrid exchange-correlation functionals to hundreds of nodes, we implemented three additional levels of distributed memory parallelism using MPI. The first two levels distribute the computations for different k- and q-points, while the third level parallelizes that of different occupied bands n′′. The distributed memory parallelization scheme is shown in Figure 2.
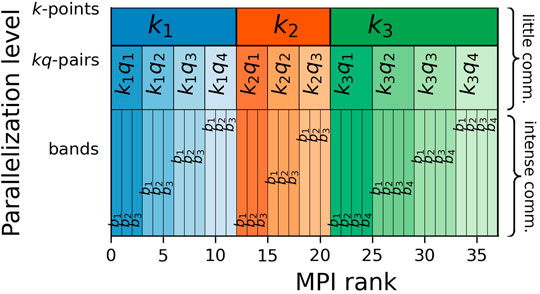
FIGURE 2. The distributed-memory parallelization of Eq. 2 is divided into three levels. For each k-point the exact exchange is calculated as an independent problem. At a k-point k i , the exact exchange is calculated as a sum over all q-points associated with k i , building the kq-pairs. These first two levels require very little communication, i.e., copying the final results to their destination for the k-points and a reduction within the sub-communicator of each k-point for the q-point sum. The third level of distributed-memory parallelization calculates groups of occupied bands n′′ in parallel. Here, a lot of inter-dependencies create a much larger communication demand compared to the first two levels. In a typical calculation of the non-local potential the workload is not uniformly distributed: Some k-points have more q-points than others and some kq-pairs might have more associated bands than others. The algorithm attempts to compensate this by assigning more nodes to heavier calculations.
The parallelization over k- and q-points requires very little communication and thus is very efficient, while the band-parallelization requires more communication. However, it allows us to limit the size of the largest matrix stored on a single node to n
basis size × n
total bands, which then has a size on the order of
Figure 3 singles out the strong scaling behavior of this 3rd MPI level for a single k- and q-point. All code parts except for the setup of the Coulomb matrix show a good scaling behavior with a parallel efficiency of over 50% on either 64 CPU nodes or 64 GPUs. The scaling behavior of the Coulomb-matrix setup is not critical, since it does not dominate the run time of the calculation even on 256 nodes. Additionally, it only scales linearly with the number of k-points whereas the other parts of the nonlocal potential scale quadratically with the number of k-points. To investigate the performance of our algorithm with multiple k-points we show the strong scaling behavior for a NaCl supercell with 64 atoms, 10 k-points and 205 kq-pairs in Figure 4. Here the scaling behavior of the Coulomb-matrix setup is the weakest once again, but it only accounts for less than 10%, even with 410 nodes. All other code parts show nearly perfect scaling. This is due to the fact that each kq-pair represents a largely independent problem that only requires little communication and the 3rd MPI level is only used with 205 and 410 nodes, since the parallelization over the 205 kq-pairs is preferred. SuperMUC-NG has two CPUs per node and therefore we assigned two MPI ranks to each node, resulting in a better performance compared to a one rank-per-node setup.
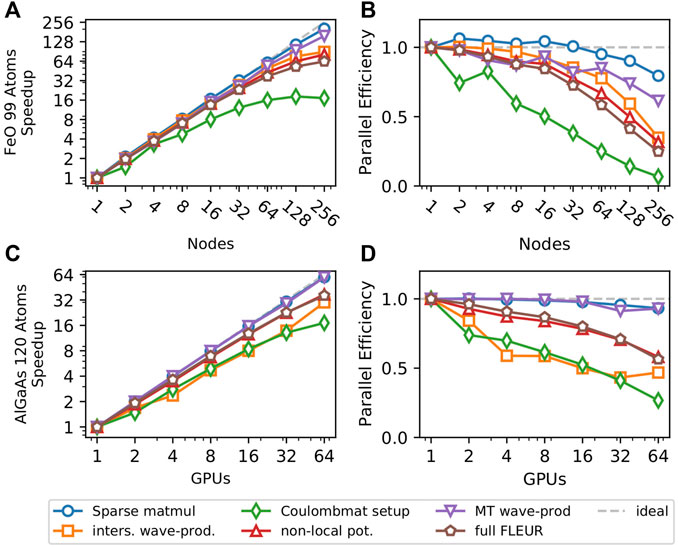
FIGURE 3. Scaling behavior of systems with a single k-point on two types of architectures. Panels (A) and (B) show scaling of a 99-atom FeO supercell with a vacancy defect on the CPU-based SuperMUC-NG supercomputer, while (C) and (D) show the scaling behavior of a 120-atom GaAs supercell with an Al defect on JURECA’s GPU partition. Panels (A) and (C) show the speedup, while (B) and (D) show the corresponding parallel efficiency.
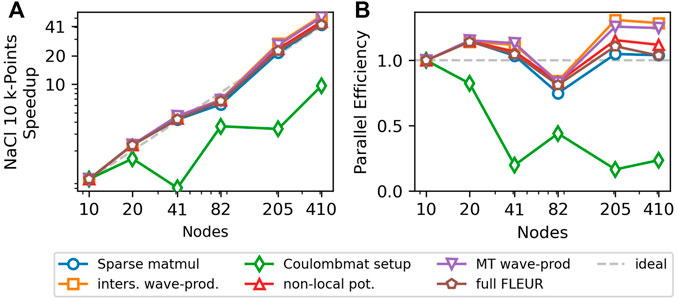
FIGURE 4. Strong scaling behavior of multiple k-points for a 64 atom NaCl supercell with a potassium (K) defect. The system has 10 k-points and 205 kq-pairs. The super-scalar behavior is caused by the fact, that the 205 kq-pairs are not evenly distributed on 10 nodes (20 MPI). Some nodes are assigned more kq-pairs and therefore need longer while the others sit idle. This effect disappears for 205 or 410 nodes, which allow for a perfectly even and therefore more efficient distribution. For 41 nodes, the speedup and parallel efficiency of the Coulomb-matrix setup drop drastically. This is due to the fact, that the Coulomb-matrix setup does not have a q-dependence, while the number of nodes is chosen to be optimal for the evaluation of Eq. 2. For 10 nodes (20 MPI), all k-points can be calculated in parallel on 2 processes each, while for 41 nodes (82 MPI), it is only possible to calculate 2 k-points in parallel, so that each is distributed over 41 processes, leading to an inefficient parallelization. In practical calculations this is mitigated by including more nodes (e.g., 45), so that both k- and kq-parallelization are efficient. However, even with 41 nodes, the Coulomb-matrix setup only accounts for 6% of the total runtime.
2.4 Weak Scaling
While the meaning of strong scaling is very intuitive, it does not necessarily reflect real life applications. Being able to calculate a system with twenty atoms in a minute or less may not advance science significantly. Certainly, science is advanced by being able to calculate increasingly bigger, more inhomogeneous and more complex systems in a reasonable time frame. Weak scaling deals with the latter. As discussed in the introduction, the computational demand of a hybrid functional calculation scales with
For simplicity and to focus on the ultimately limiting parallelization level, we use a single kq-pair and neglect the very efficient parallelization over different k- and q-vectors. In Figure 5 a gallium arsenide (GaAs) setup was scaled into supercells with a single nitrogen defect. Then, they were calculated with the parallelization chosen such that
where min(n atoms) is the number of atoms in the smallest supercell.
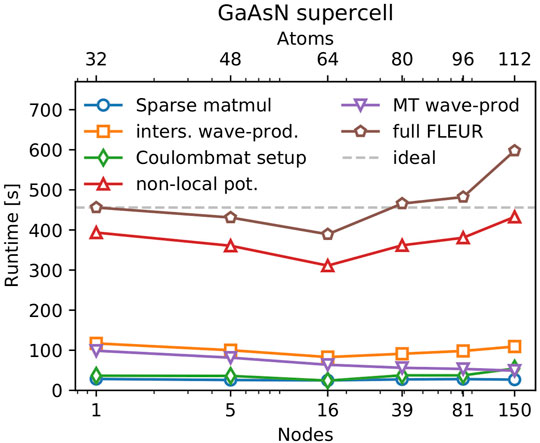
FIGURE 5. Weak scaling behavior of FLEUR’s hybrid functional calculations for a GaAs system which is scaled into a supercell with one Arsenic atom being substituted with Nitrogen. The y-axis shows the runtime for different code parts, while the bottom x-axis shows the number of nodes used. The top x-axis shows the number of atoms in that particular system.
With ideal weak scaling behavior the runtime should be constant regardless of the size of the unit cell, since the computational cost in Eq. 3 is canceled out by the additional compute resources chosen in Eq. 4. Figure 5 shows that the hybrid functionals in FLEUR can be applied efficiently to a wide variety of system sizes. The time needed for the calculation of the nonlocal potential of the largest GaAs supercell is 9% larger compared to that of the smallest supercell and the full iteration runtime is 30% larger. The runtime does not monotonously increase as one would expect for the weak scaling of a simple algorithm performing a single task. In FLEUR, the situation is more complex, some parts of the code scale with
3 Application to Rare-Earth Iron Garnets
Yttrium iron garnet (Y3Fe5O12 or short YIG) is a complex ferrimagnetic insulator with a number of remarkable properties and applications, in the fields of magnonics (Serga et al., 2010), ultra-low temperature physics (Demokritov et al., 2006) and quantum computing (Tabuchi et al., 2015). This success has sparked interest in a related class of materials, the so-called rare-earth-iron garnets (RIGs), where the yttrium atom in the YIG structure is replaced with an element of the lanthanide series. Here applications range from materials with giant magnetostriction (Sayetat, 1986) to spin Seebeck insulators (Uchida et al., 2010). Despite great interest in these materials there is only a limited number of theoretical studies of their electronic structure. This is most likely due to the large unit cells with 160 atoms in the conventional and 80 atoms in the primitive unit cell.
The typical unit cell of a garnet is shown in Figure 6. The iron atoms in this structure have two types of environments. They are either in the centre of an octahedron or a tetrahedron spanned by neighbouring oxygen atoms. These different iron environments have a strong effect on the electronic structure, which is discussed in detail later in this paper. YIG and most RIGs are ferrimagnets, such that the magnetic moments of the 8 octahedral iron atoms point in the opposite direction with respect to the 12 tetrahedral iron atoms, which, for the RIGs discussed here, are aligned in parallel with the rare-earth elements. Only a very minor magnetic moment is induced in the yttrium and oxygen atoms.
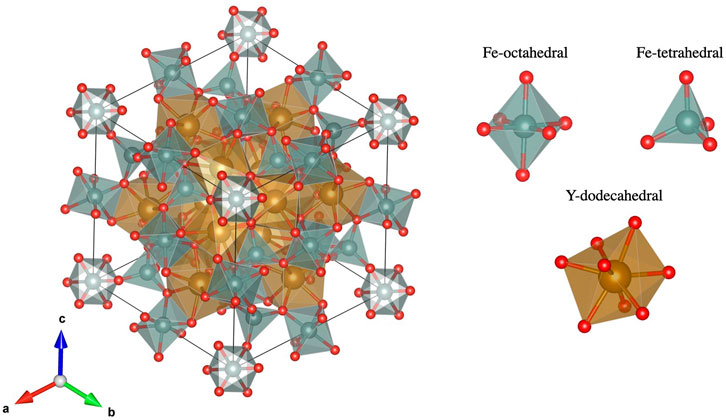
FIGURE 6. Unit cell of a garnet. Oxygen atoms are shown in red, while iron atoms are shown inside the grey polyhedra. The rare-earth or yttrium atoms are shown inside the golden dodecahedra. While the yttrium or rare-earth atoms are all symmetry equivalent the iron is present in two different environments. Structure from (Y3Fe5O12 crystal structure, 2012) and plotted with (Momma and Izumi, 2011).
3.1 Electronic Structure
In order to understand how the choice of the exchange-correlation functional affects the electronic structure of YIG we calculated the density-of-states (DOS) with PBE and with PBE0. All calculations shown in the paper were performed on a 2 × 2 × 2 k-point grid. We confirmed that the DOS is converged with this grid by comparing the PBE results to results on a denser k-point grid. We use a smearing of σ = 0.136 eV for all DOS calculations shown. The muffin-tin radii of Y, Gd, Tm, Fe and O are chosen to be r
Y/Gd/Tm = 2.8 a
0, r
Fe = 2.14 a
0 and r
O = 1.21 a
0. The structural information, e.g. the unit cell and the atomic positions used in this chapter are based on the experimental ones, exhibiting a
As expected, with a value of 0.44 eV, PBE massively underestimates the experimental band gap of 2.8 eV (Larsen and Metselaar, 1975), while PBE0 predicts an improved band gap of 1.83 eV. However, the experimental value relies on optical measurements, which are not sensitive to all transitions, potentially missing certain states and thus overestimating the real band gap.
In Figure 7A) the DOS of YIG is calculated using PBE as an exchange-correlation functional. In this figure, the antiferrimagnetic alignment of the iron atoms is visible: the occupied states associated with the tetrahedral iron atoms are mainly in the spin-up channel and the unoccupied ones are in the spin-down channel, while for the octahedral iron atoms the situation is reversed: below the Fermi level the octahedral iron states are mostly in the spin-down channel and above it in the spin-up one. Most states associated with the oxygen atoms are occupied, while the yttrium states are largely unoccupied. Below the Fermi level, the DOS in the interstitial region closely follows the oxygen DOS. Additionally, the DOS associated with both iron types also coincide with the oxygen and interstitial DOS. This indicates that the 2p-states of the oxygen and the 3d-states of iron hybridize for both iron environments. This analysis is supported by the number of valence electrons found in the different muffin-tin spheres, which are 6.5 and 6.2 electrons for iron atoms in the tetrahedral and octhedral environments, respectively, 1.1 valence electrons in the sphere of yttrium, and an average of 3.7 valence electrons in the spheres of oxygen. The large number of 164.1 electrons in the interstitial region additionally indicates a high degree of de-localization of these states. For the unoccupied octahedral iron states in contrast, we can see a clear signature of simple crystal field splitting of localized d-states: the three t 2g -states shift down and the two eg-states shift up leading to two distinct peaks with the lower one containing three and the higher one containing two states. Similarly, for the unoccupied tetrahedral Fe d-levels the e-states are shifted down, while the t 2-states are shifted up. This separation however, is not so clear as the shifts are smaller, the peaks still overlap and another splitting due to next-nearest neighbors can be seen.
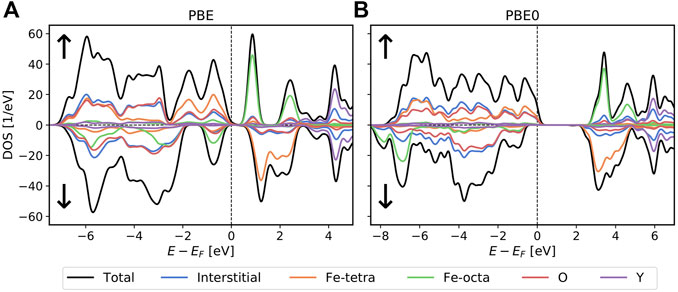
FIGURE 7. DOS of YIG calculated with PBE in (A) and with PBE0 in (B) on a 2 × 2 × 2 k-grid. In both calculations we use a
In Figure 7B) we show the DOS calculated using the hybrid exchange-correlation functional PBE0. The results are qualitatively different from the PBE results with the most significant change seen in the different behavior of the two types of Fe in the PBE results: While the occupied tetrahedral iron 3d-states still hybridize with the 2p-states of the surrounding oxygen atoms, most of the octahedral iron 3d-states are now strongly localized and form a double peak in the DOS at around −6.5 eV.
Such a localization effect of the d-states can also be reproduced in a PBE+U treatment (Chen et al., 2021). However, in these simulations the d-states of both Fe types show the same behavior. The different tendency to localize can also be seen in these simulation by the different values of U used for the different atoms to achieve the localization. Hence, the result strongly suggests that the local Coulomb interaction exhibits different strength in the two environments of Fe. This effect can be caused by the different initial localization of the d-states as well as by different interactions and screening effects of the surrounding. Such a difference can also be seen in the unoccupied spectrum which is again dominated by a crystal field splitting of the d-states. However, in the octahedral environment this effect is again much clearer while the tetrahedral d-states form a broad band with several peaks also indicating next-nearest neighbor effects.
Finally, we would like to point out that the octahedral Fe d-states show a rather complex sub-structure with a large peak at − 7 eV and a minor peak at − 8 eV. This is not a crystal field splitting but rather shows that different states with a different degree of localization are formed. While the lower peak is clearly separated from the O p-states, some remaining hybridization can be identified for the larger peak.
Further investigations of the consequences of these differences between the Fe atoms is beyond the scope of this paper, but we expect this electronic structure to have some influence, e.g. on magnetic interactions and the transition temperature.
3.2 Magnetic Moment
In the introduction we discussed that some key applications of YIG are related to its magnetic properties. Therefore, we want to investigate the precision of our predictions for magnetic properties with different exchange-correlation functionals. In Table 1 we compare the magnetic moments predicted for the different iron atom types. We use the magnetic moment inside the muffin-tin sphere to assign the moment to a specific atom. Therefore, the magnetic moment depends on the choice of the radius of the muffin-tin sphere and, strictly speaking, is not uniquely defined. The magnetization calculated for the oxygen and yttrium atoms is negligible regardless of the computational method used. The total magnetic moment per unit formula was 5 μB for every functional. This agreement is expected, since YIG is a magnetic insulator, which constrains the total magnetization per unit cell to integer values.
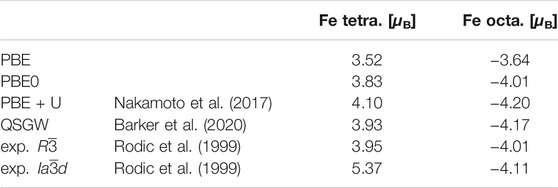
TABLE 1. The magnetic moments within the different muffin-tins of both Fe types in units of μ B.
While PBE predicts the magnetic moments of the two iron types within only ±0.5 μB of the experimental value for the
3.3 Rare-earth-iron Garnets
As two representatives of the rare-earth-iron garnet group, we chose to examine Gd3Fe5O12 (GdIG) and Tm3Fe5O12 (TmIG) more closely. We selected these materials, because a lot of interesting experimental (Fechine et al., 2008; Phan et al., 2009; Lassri et al., 2011; Lee et al., 2020; Vilela et al., 2020; Vu et al., 2020) and even some theoretical work using the FLAPW method (Lassri et al., 2011) has been published for these materials.
In Figure 8 we present the density of states for GdIG and TmIG calculated with the PBE0 exchange-correlation functional. Reaching numerical self-consistency for TmIG was challenging with PBE, which is the starting point for any PBE0 calculation. We achieved self-consistency by using a few hundred straight mixing iterations with a low mixing parameter, followed by a set of Anderson mixing iterations until convergence was reached. With a converged PBE as a starting density the convergence of PBE0 is straight forward. This difficult convergence is caused by the metallic behavior of TmIG with PBE as a functional. After the non-local potential is included, a gap opens up and all later density convergence cycles do not exhibit this problematic metallic behavior. GdIG converged without problems both for PBE and PBE0.
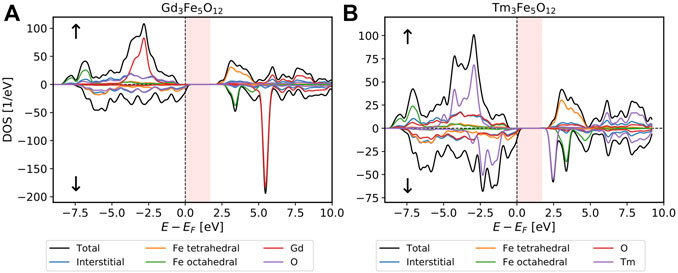
FIGURE 8. The density-of-states is calculated for GdIG in (A) and for TmIG in (B) using PBE0 on a 2 × 2 × 2 k-point grid. Both calculations were performed with a
For GdIG the band gap was calculated to be 1.7 eV with PBE0. Literature values obtained using PBE + U suggest a gap of 1.6 eV (Nakamoto et al., 2017). For TmIG we also predict a band gap of 1.7 eV using PBE0. To our knowledge, this is the first prediction for the band gap of TmIG. We are not aware of any experimental results regarding the band gap in either system.
The electronic structure of these two systems has a few striking similarities with that of YIG. The 3d-states of both types of iron atoms hybridize with the oxygen 2p-states in PBE, while with PBE0 the octahedral iron states show localization and a strong shift to lower energies. This again highlights the difference of the tetrahedral and octahedral oxygen environment of the iron atoms causing different effective interactions at these atoms and casting doubt on simple PBE+U predictions for these garnet systems. For the unoccupied octahedral iron states we can see the typical signature of crystal field splitting and in the tetrahedral case this signature is weaker. The additional 4f-states of the rare-earth elements in the spin-up channel are strongly localized in PBE. In PBE0 they show a slightly larger bandwidth, indicating increased hybridization with the oxygen 2p-states which could be understood due to the decrease of hybridization of these states with the octahedral Fe d-states. As expected, Gd has no occupied 4f-states in the spin-down channel, while the 4f-states of Tm are partially occupied, causing a metallic behavior in PBE. In PBE0 the increased interaction provided by the exchange term opens a gap in the Tm 4f-band.
In Table 2 the magnetic moments of all atom types are given. For GdIG we predict a total magnetization per formula unit of 16.0 μB and for TmIG we predict 1.75 μB for PBE as well as PBE0. Notice, that the formula unit contains 20 atoms, while the primitive unit cell contains 80. This means, while the magnetic moment per formula unit is not integer, it is integer per unit cell.
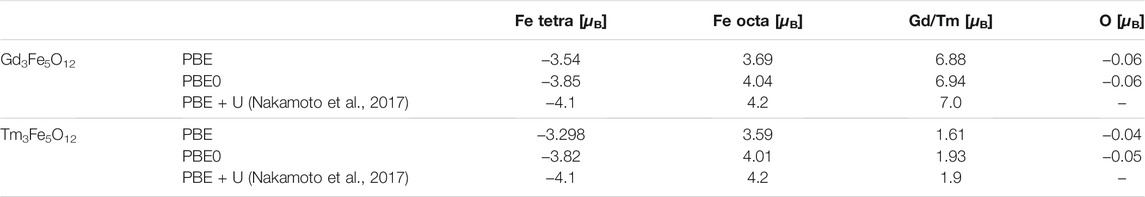
TABLE 2. Predicted magnetic moments of GdIG and TmIG for each atom type, given in units of [μ B].
The predicted total magnetic moments are in exact agreement with experimental results for GdIG (Geller et al., 1965), while they are in good agreement with the experimental value of 1.2 μB for the TmIG. This experimental value would correspond to a total magnetic moment of 4.8 μB for the primitive unit cell. PBE + U shows a tendency to predict larger magnetic moments for almost all atoms: 4.2 μB for the octahedral iron, − 4.1 μB for the tetrahedral iron, 7.0 μB for Gd and 1.9 μB for Tm (Nakamoto et al., 2017).
4 Conclusion
In this article we presented a highly scalable implementation of hybrid exchange-correlation functionals in the LAPW basis. In this work we focused on the scalable implementation of the Hartree-Fock exact exchange, which corresponds to the implementation of the PBE0 functional, but screened functionals like HSE06 are related by an additional fast computation of a smooth function. The combination of shared and distributed memory parallelization allows to calculate a broad range of systems with high efficiency. Combining all three MPI levels gives us an outlook on the scaling potential of this algorithm. If we were for example to calculate the GaAs system with 120 atoms and we would use 8 k-points we would get 125 kq-pairs. Figure 3 shows that for this system a single kq-pair has a good parallel performance even if distributed over more than 32 GPUs. Therefore, it is reasonable to assume that the calculation of the nonlocal potential for a system with 8 k-points would still have good scaling with
Using the new implementation of the hybrid functional code, we performed simulations of the electronic structure of iron based garnet materials. The significant improvement in the obtained band gap as well as the changes in the electronic structures discussed in detail demonstrate the significance and power of this treatment for these technological relevant material class. Our results suggests an experimental reevaluation of the structure-magnetism relation of the yttrium iron garnet (YIG), Y3Fe5O12.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
MR, DW, and GM performed and analyzed the initial performance measurements. MR, DW, and GM designed the 3-Level MPI parallelism with feedback form CT and MM. CT and MM suggested solutions for the performance bottlenecks in MPIs one-sided communication. MR, DW, GM implemented shared-memory parallelism using OpenMP, while CT contributed memory blocking for certain OpenMP kernels. MR performed the performance measurements and the electronic structure calculation for the garnet materials. JB and SB helped with the analysis and understanding of the electronic structure of the garnet materials. JB created Figure 5.
Funding
The authors gratefully acknowledge the computing time granted through JARA on the supercomputer JURECA at Forschungszentrum Jülich as well as the Gauss Centre for Supercomputing e.V. (www.gauss-centre.eu). for funding this project by providing computing time on the SuperMUC-NG Supercomputer. The work is funded by the JARA-CSD School for Simulation and Data Science (SSD) and by the MaX Center of Excellence funded by the EU through the H2020-INFRAEDI-2018 (project: GA 824143).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to acknowledge fruitful discussions with Christoph Friedrich.
References
Alberi, K., Nardelli, M. B., Zakutayev, A., Mitas, L., Curtarolo, S., Jain, A., et al. (2018). The 2019 Materials by Design Roadmap. J. Phys. D: Appl. Phys. 52, 013001. doi:10.1088/1361-6463/aad926
Andersen, O. K., and Woolley, R. G. (1973). Muffin-Tin Orbitals and Molecular Calculations: General Formalism. Mol. Phys. 26, 905–927. doi:10.1080/00268977300102171
Barker, J., Pashov, D., and Jackson, J. (2020). Electronic Structure and Finite Temperature Magnetism of Yttrium Iron Garnet. Electron. Struct. 2, 044002. doi:10.1088/2516-1075/abd097
Barnes, T. A., Kurth, T., Carrier, P., Wichmann, N., Prendergast, D., Kent, P. R. C., et al. (2017). Improved Treatment of Exact Exchange in Quantum Espresso. Computer Phys. Commun. 214, 52–58. doi:10.1016/j.cpc.2017.01.008
Becke, A. D. (1993). A New Mixing of Hartree-Fock and Local Density‐Functional Theories. J. Chem. Phys. 98, 1372–1377. doi:10.1063/1.464304
Becke, A. D. (2014). Perspective: Fifty Years of Density-Functional Theory in Chemical Physics. J. Chem. Phys. 140, 18A301. doi:10.1063/1.4869598
Betzinger, M., Friedrich, C., and Blügel, S. (2010). Hybrid Functionals within the All-Electron FLAPW Method: Implementation and Applications of PBE0. Phys. Rev. B 81, 195117. doi:10.1103/PhysRevB.81.195117
Blaha, P., Schwarz, K., Tran, F., Laskowski, R., Madsen, G. K. H., and Marks, L. D. (2020). Wien2k: An APW+lo Program for Calculating the Properties of Solids. J. Chem. Phys. 152, 074101. doi:10.1063/1.5143061
Burke, K. (2012). Perspective on Density Functional Theory. J. Chem. Phys. 136, 150901. doi:10.1063/1.4704546
Carnimeo, I., Baroni, S., and Giannozzi, P. (2019). Fast Hybrid Density-Functional Computations Using Plane-Wave Basis Sets. Electron. Struct. 1, 015009. doi:10.1088/2516-1075/aaf7d4
Chen, J., Hsu, H.-S., and Lo, F.-Y. (2021). Spin-Dependent Optical Transitions in Yttrium Iron Garnet. Mater. Res. Express 8, 026101. doi:10.1088/2053-1591/abe013
Cramer, C. J., and Truhlar, D. G. (2009). Density Functional Theory for Transition Metals and Transition Metal Chemistry. Phys. Chem. Chem. Phys. 11, 10757–10816. doi:10.1039/B907148B
Demokritov, S. O., Demidov, V. E., Dzyapko, O., Melkov, G. A., Serga, A. A., Hillebrands, B., et al. (2006). Bose-Einstein Condensation of Quasi-Equilibrium Magnons at Room Temperature under Pumping. Nature 443, 430–433. doi:10.1038/nature05117
Fechine, P. B. A., Moretzsohn, R. S. T., Costa, R. C. S., Derov, J., Stewart, J. W., Drehman, A. J., et al. (2008). Magnego-Dielectric Properties of the Y3Fe5O12 and Gd3Fe5O12 Dielectric Ferrite Resonator Antennas. Microw. Opt. Technol. Lett. 50, 2852–2857. doi:10.1002/mop.23824
Fleur (2021). Forschungszentrum Jülich - PGI-1 & IAS-1. Available at: https://www.flapw.de.
Friedrich, C., Schindlmayr, A., and Blügel, S. (2009). Efficient Calculation of the Coulomb Matrix and its Expansion Around k=0 within the FLAPW Method. Computer Phys. Commun. 180, 347–359. doi:10.1016/j.cpc.2008.10.009
Garza, A. J., and Scuseria, G. E. (2016). Predicting Band Gaps with Hybrid Density Functionals. J. Phys. Chem. Lett. 7, 4165–4170. doi:10.1021/acs.jpclett.6b01807
Gd3FeO12 crystal structure (2012). Datasheet from “Pauling File Multinaries Edition – 2012” in Springermaterials (Japan: Springer-Verlag Berlin Heidelberg & Material Phases Data System (MPDS), Switzerland & National Institute for Materials Science NIMS). Available at: https://materials.springer.com/isp/crystallographic/docs/sd_0308674. Copyright 2016 .
Geller, S., Remeika, J. P., Sherwood, R. C., Williams, H. J., and Espinosa, G. P. (1965). Magnetic Study of the Heavier Rare-Earth Iron Garnets. Phys. Rev. 137, A1034–A1038. doi:10.1103/PhysRev.137.A1034
Guidon, M., Schiffmann, F., Hutter, J., and VandeVondele, J. (2008). Ab Initio Molecular Dynamics Using Hybrid Density Functionals. J. Chem. Phys. 128, 214104. doi:10.1063/1.2931945
Hakala, M. H., and Foster, A. S. (2013). Computationally Efficient Implementation of Hybrid Functionals in SIESTA. Tech. Rep.
Ihrig, A. C., Wieferink, J., Zhang, I. Y., Ropo, M., Ren, X., Rinke, P., et al. (2015). Accurate Localized Resolution of Identity Approach for Linear-Scaling Hybrid Density Functionals and for Many-Body Perturbation Theory. New J. Phys. 17, 093020. doi:10.1088/1367-2630/17/9/093020
Krukau, A. V., Vydrov, O. A., Izmaylov, A. F., and Scuseria, G. E. (2006). Influence of the Exchange Screening Parameter on the Performance of Screened Hybrid Functionals. J. Chem. Phys. 125, 224106. doi:10.1063/1.2404663
Larsen, P. K., and Metselaar, R. (1975). Defects and the Electronic Properties of Y3Fe5O12. J. Solid State. Chem. 12, 253–258. doi:10.1016/0022-4596(75)90315-1
Lassri, H., Hlil, E. K., Prasad, S., and Krishnan, R. (2011). Magnetic and Electronic Properties of Nanocrystalline Gd3Fe5O12 Garnet. J. Solid State. Chem. 184, 3216–3220. doi:10.1016/j.jssc.2011.09.034
Lee, A. J., Ahmed, A. S., Flores, J., Guo, S., Wang, B., Bagués, N., et al. (2020). Probing the Source of the Interfacial Dzyaloshinskii-Moriya Interaction Responsible for the Topological Hall Effect in Metal/Tm3Fe5O12 Systems. Phys. Rev. Lett. 124, 107201. doi:10.1103/PhysRevLett.124.107201
Lejaeghere, K., Bihlmayer, G., Björkman, T., Blaha, P., Blügel, S., Blum, V., et al. (2016). Reproducibility in Density Functional Theory Calculations of Solids. Science 351, aad3000. doi:10.1126/science.aad3000
Levchenko, S. V., Ren, X., Wieferink, J., Johanni, R., Rinke, P., Blum, V., et al. (2015). Hybrid Functionals for Large Periodic Systems in an All-Electron, Numeric Atom-Centered Basis Framework. Computer Phys. Commun. 192, 60–69. doi:10.1016/j.cpc.2015.02.021
Lin, P., Ren, X., and He, L. (2021). Efficient Hybrid Density Functional Calculations for Large Periodic Systems Using Numerical Atomic Orbitals. J. Chem. Theor. Comput. 17, 222–239. doi:10.1021/acs.jctc.0c00960
Mandal, S., Kar, R., Kloeffel, T., Meyer, B., and Nair, N. N. (2021). Improving the Scaling and Performance of Multiple Time Stepping Based Molecular Dynamics with Hybrid Density Functionals. ArXiv, 07670. doi:10.1002/jcc.26816
Momma, K., and Izumi, F. (2011). VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data. J. Appl. Cryst. 44, 1272–1276. doi:10.1107/S0021889811038970
Mounet, N., Gibertini, M., Schwaller, P., Campi, D., Merkys, A., Marrazzo, A., et al. (2018). Two-Dimensional Materials from High-Throughput Computational Exfoliation of Experimentally Known Compounds. Nat. Nanotech 13, 246–252. doi:10.1038/s41565-017-0035-5
Nakamoto, R., Xu, B., Xu, C., Xu, H., and Bellaiche, L. (2017). Properties of Rare-Earth Iron Garnets from First Principles. Phys. Rev. B 95, 024434. doi:10.1103/PhysRevB.95.024434
Perdew, J. P., Ernzerhof, M., and Burke, K. (1996). Rationale for Mixing Exact Exchange with Density Functional Approximations. J. Chem. Phys. 105, 9982–9985. doi:10.1063/1.472933
Phan, M. H., Morales, M. B., Chinnasamy, C. N., Latha, B., Harris, V. G., and Srikanth, H. (2009). Magnetocaloric Effect in Bulk and Nanostructured Gd3Fe5O12 Materials. J. Phys. D: Appl. Phys. 42, 115007. doi:10.1088/0022-3727/42/11/115007
Rodic, D., Mitric, M., Tellgren, R., Rundlof, H., and Kremenovic, A. (1999). True Magnetic Structure of the Ferrimagnetic Garnet Y3Fe5O12 and Magnetic Moments of Iron Ions. J. Magnetism Magn. Mater. 191, 137–145. doi:10.1016/S0304-8853(98)00317-5
Rosen, A. S., Notestein, J. M., and Snurr, R. Q. (2019). Identifying Promising Metal-Organic Frameworks for Heterogeneous Catalysis via High-Throughput Periodic Density Functional Theory. J. Comput. Chem. 40, 1305–1318. doi:10.1002/jcc.25787
Sayetat, F. (1986). Huge Magnetostriction in Tb3Fe5O12, Dy3Fe5O12, Ho3Fe5O12, Er3Fe5O12 Garnets. J. Magnetism Magn. Mater. 58, 334–346. doi:10.1016/0304-8853(86)90456-7
Schlipf, M., Betzinger, M., Friedrich, C., Ležaić, M., and Blügel, S. (2011). HSE Hybrid Functional within the FLPAW Method and its Application to GdN. Phys. Rev. B 84, 125142. doi:10.1103/PhysRevB.84.125142
Serga, A. A., Chumak, A. V., and Hillebrands, B. (2010). YIG Magnonics. J. Phys. D: Appl. Phys. 43, 264002. doi:10.1088/0022-3727/43/26/264002
Shavitt, I. (1959). A Calculation of the Rates of the Ortho‐Para Conversions and Isotope Exchanges in Hydrogen. J. Chem. Phys. 31, 1359–1367. doi:10.1063/1.1730599
Tabuchi, Y., Ishino, S., Noguchi, A., Ishikawa, T., Yamazaki, R., Usami, K., et al. (2015). Coherent Coupling between a Ferromagnetic Magnon and a Superconducting Qubit. Science 349, 405–408. doi:10.1126/science.aaa3693
Tm3Fe5O12 crystal structure (2012). Datasheet from “Pauling File Multinaries Edition – 2012” in Springermaterials (Japan: Springer-Verlag Berlin Heidelberg & Material Phases Data System (MPDS), Switzerland & National Institute for Materials Science NIMS). Available at: https://materials.springer.com/isp/crystallographic/docs/sd_0312594. Copyright 2016 .
Uchida, K., Xiao, J., Adachi, H., Ohe, J., Takahashi, S., Ieda, J., et al. (2010). Spin Seebeck Insulator. Nat. Mater 9, 894–897. doi:10.1038/nmat2856
Vilela, G. L. S., Abrao, J. E., Santos, E., Yao, Y., Mendes, J. B. S., Rodríguez-Suárez, R. L., et al. (2020). Magnon-Mediated Spin Currents in Tm3Fe5O12/Pt with Perpendicular Magnetic Anisotropy. Appl. Phys. Lett. 117, 122412. doi:10.1063/5.0023242
Vu, N. M., Meisenheimer, P. B., and Heron, J. T. (2020). Tunable Magnetoelastic Anisotropy in Epitaxial (111) Tm3Fe5O12 Thin Films. J. Appl. Phys. 127, 153905. doi:10.1063/1.5142856
Wimmer, E., Krakauer, H., Weinert, M., and Freeman, A. J. (1981). Full-Potential Self-Consistent Linearized-Augmented-Plane-Wave Method for Calculating the Electronic Structure of Molecules and surfaces: O2 molecule. Phys. Rev. B 24, 864–875. doi:10.1103/PhysRevB.24.864
Y3Fe5O12 crystal structure (2012). Datasheet from “Pauling File Multinaries Edition – 2012” in Springermaterials (Japan: Springer-Verlag Berlin Heidelberg & Material Phases Data System (MPDS), Switzerland & National Institute for Materials Science NIMS). Available at: https://materials.springer.com/isp/crystallographic/docs/sd_0310635. Copyright 2016.
Yan, Q., Yu, J., Suram, S. K., Zhou, L., Shinde, A., Newhouse, P. F., et al. (2017). Solar Fuels Photoanode Materials Discovery by Integrating High-Throughput Theory and Experiment. Proc. Natl. Acad. Sci. USA 114, 3040–3043. doi:10.1073/pnas.1619940114
Yu, S., Schmidt, R. D., Garcia-Mendez, R., Herbert, E., Dudney, N. J., Wolfenstine, J. B., et al. (2016). Elastic Properties of the Solid Electrolyte Li7La3Zr2O12 (LLZO). Chem. Mater. 28, 197–206. doi:10.1021/acs.chemmater.5b03854
Keywords: Density Functional Theory (DFT), Rare-Earth Iron Garnets, High-Performance Computing (HPC), PBE0, Hybrid Functionals, YIG, GdIG, TmIG
Citation: Redies M, Michalicek G, Bouaziz J, Terboven C, Müller MS, Blügel S and Wortmann D (2022) Fast All-Electron Hybrid Functionals and Their Application to Rare-Earth Iron Garnets. Front. Mater. 9:851458. doi: 10.3389/fmats.2022.851458
Received: 09 January 2022; Accepted: 22 February 2022;
Published: 21 March 2022.
Edited by:
Zhenyu Li, University of Science and Technology of China, ChinaReviewed by:
Honghui Shang, Institute of Computing Technology (CAS), ChinaMohan Chen, Peking University, China
Wei Hu, Lawrence Berkeley National Laboratory, United States
Copyright © 2022 Redies, Michalicek, Bouaziz, Terboven, Müller, Blügel and Wortmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel Wortmann, ZC53b3J0bWFubkBmei1qdWVsaWNoLmRl