 Ziyan Zhu
Ziyan Zhu Malin Ly1
Malin Ly1 Nazhen Liu
Nazhen Liu James J. Noël
James J. Noël
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mater. , 18 October 2022
Sec. Environmental Degradation of Materials
Volume 9 - 2022 | https://doi.org/10.3389/fmats.2022.1038310
This article is part of the Research Topic Nuclear Materials Degradation View all 6 articles
The electrochemical reduction of hydrogen peroxide has been studied in sodium chloride solutions containing various anions (bicarbonate/carbonate, sulphate) on Gd-UO2, Dy-UO2 and a SIMFUEL (UO2 doped to simulate spent nuclear fuel). The reaction was observed to proceed via the chemical oxidation of the surface to produce UV followed by its subsequent electrochemical reduction. The reaction was faster on the SIMFUEL surface due to the availability of the oxygen vacancies required to incorporate the OII ions necessary to maintain charge balance. Bicarbonate/carbonate, but not sulphate, was found to suppress peroxide reduction. This could be caused either by peroxide decomposition in solution or by the catalysis of peroxide reformation via the reaction of surface hydroxyl radicals with bicarbonate/carbonate to form carbonate radicals which subsequently decompose by reaction with water. The noble metal particles present in the SIMFUEL appear to play only a minor role in the reduction process.
The prospects for the long-term containment of spent nuclear fuel in a deep geologic repository are very good (Kwong, 2011; Hall and Keech, 2017; Hall et al., 2021). However, it is judicious to assume some containers may fail before radiation fields have decayed to innocuous levels. It is also reasonable to assume that container failure leading to contact of the fuel with groundwater will not occur until β/γ radiation fields are negligible (a few hundred years) and only α-radiation fields remain significant (Badley and Shoesmith, 2022). Hence, a clear understanding of the influence of H2O2, the key oxidant produced by the α-radiolysis of H2O, on fuel corrosion and radionuclide release is important to elucidate the full mechanism of UO2 corrosion. The influence of H2O2 on UO2 corrosion has been extensively studied and reviewed, with H2O2 shown to both cause corrosion and undergo decomposition (Sunder et al., 2004; Nilsson and Jonsson, 2011; Eriksen et al., 2012; Lousada et al., 2013; Wu and Shoesmith, 2014; Liu et al., 2018; Zhu et al., 2020). This means it will be both anodically oxidized and cathodically reduced on the UO2 surface with its cathodic reduction being the key reaction supporting both dissolution and decomposition by disproportionation.
The kinetics of reduction processes on UO2 surfaces have been shown to be dependent on the chemical state of the surface, with electrochemical and X-ray photoelectron spectroscopy (XPS) studies demonstrating that reduction is blocked when an insulating UVI oxide layer is present but proceeds rapidly on UIV/UV surfaces (Goldik et al., 2004; Goldik et al., 2005). Goldik et al. (2004), Goldik et al. (2005), Goldik et al. (2006a). Goldik et al. (2006b) interpreted the kinetics for O2 reduction using the mechanism proposed by Trunov and Presnov (1975a), Trunov and Presnov (1975b) on transition metal oxides and adopted by Hocking et al. (1991); Hocking et al. (1994) to explain the kinetics of O2 reduction on UO2. According to this mechanism, electron transfer to adsorbed O2 molecules occurs at donor-acceptor relay (DAR) sites comprising UIV and UV in adjacent locations. The cathodic reduction of O2 is slow on UO2 but accelerated by oxidation of the surface to U1–2xIV U2xV O2+x which provides the DAR sites to catalyze the electron relay process (Hocking et al., 1991; Hocking et al., 1994).
The kinetics of H2O2 reduction on UO2 are considerably faster than those of O2, since H2O2 can chemically create, rather than rely on existing, DAR sites, a process accompanied by the incorporation of an oxygen interstitial (OI) into an oxygen vacancy (OV),
These sites are subsequently electrochemically destroyed by electron transfer with the OI released (Hocking et al., 1991; Goldik et al., 2006b).
On SIMFUEL surfaces (UO2 doped with rare earths (REIII) and containing noble metal (ε) particles (Pd, Ru, Rh, Mo)), the kinetics of O2 reduction are catalyzed on the particle surfaces, leading to a notable increase in the overall reduction rate. However, electrochemical studies suggest only a minor influence of ε-particles on H2O2 reduction (Goldik et al., 2006a), since the rate on the DAR sites on the UO2 surface is already high. Goldik et al. (2005) demonstrated that the kinetics of H2O2 reduction varied with the [H2O2] and the degree of oxidation of the UO2 surface. The formation of an insulating UVI surface species suppressed the reduction by blocking DAR sites (Goldik et al., 2004).
The presence of REIII dopants in UO2 has been shown to stabilize the UO2 matrix with charge balance maintained by the creation of REIII-OV and UV sites (Razdan and Shoesmith, 2014; Liu et al., 2017a; Liu et al., 2017b). This study investigates the influence of REIII doping on the kinetics of H2O2 reduction using a combination of electrochemical techniques and Raman spectroscopy.
The materials used in this study were a 3 at% SIMFUEL (UO2 doped with Sr, Y, Ce, Nd, La, Zr, Ba, Pd, Ru, Rh, and Mo to simulate in-reactor irradiation), 6.0 wt% Gd2O3 in UO2 (Gd-UO2) and 12.9 wt% Dy2O3 in UO2 (Dy-UO2). The SIMFUEL and Dy-UO2 were fabricated by Canadian Nuclear Laboratories (Chalk River, Ontario, Canada) and the Gd-UO2 supplied by Cameco (Port Hope, Ontario, Canada). Rotating disc electrodes, approximately 2–3 mm thick and 12 mm in diameter, were cut from pellets and fabricated into electrodes using our previously described procedures (Santos et al., 2004). The resistivity values of the materials were: Gd-UO2 (50 Ω cm), Dy-UO2 (55 Ω cm) and SIMFUEL (82 Ω cm).
A three compartment electrochemical cell was used in all experiments. A saturated calomel electrode (SCE) was used as the reference electrode, with a Pt mesh counter electrode spot welded to a Pt wire. The cell was placed inside a Faraday cage to prevent interference from external noise. Prior to each experiment, electrodes were wet-polished with 1200 SiC paper, rinsed with Type I H2O and cathodically cleaned at a potential (E) of −1.2 V (vs. SCE) for 2 min to reduce any air-formed oxide film.
A Solartron model 1287 potentiostat was used to apply potentials and record current responses. A potential scan rate of 10 mV s−1 was used in cyclic voltametric (CV) experiments with the current interrupt procedure adopted to compensate for electrode resistance (Britz and Brocke, 1975). Corrware (Scribner Associates) software was used to control instrumentation and analyze data. The rotation rate of the working electrode (WE) was controlled using an analytical model ASR rotator from Pine Instruments.
All solutions were prepared with Type I H2O with a resistivity of 18.2 MΩ cm, purified using a Millipore Milli-Q ion exchange column. Experiments were conducted at room temperature in solutions sparged with Ar [ultra high purity (Praxair)] for at least 30 min prior to each experiment, with sparging continued throughout each experiment. Experiments were performed in a 0.1 mol.L−1 NaCl solution, with the final pH adjusted to 9.7 using NaOH solution. When required, Na2CO3, Na2SO4 and H2O2 were added at the desired concentrations. All chemicals were reagent grade and purchased from Fisher Scientific.
X-ray photoelectron spectroscopy (XPS) was performed using a Kratos Axis NOVA spectrometer with a monochromatic Al Kα X-ray source (1486.6 eV). The instrument work function was calibrated to give a binding energy of 83.96 eV for the 4f7/2 line for metallic Au, and the spectrometer dispersion was adjusted to give a binding energy (BE) of 932.62 eV for the 2p3/2 line of metallic Cu. Survey scans were recorded over the energy range 0–1100 eV on an analysis area of 300 × 700 μm2 with a pass energy of 160 eV. High resolution spectra were recorded at a pass energy of 20 eV with a step size of 0.05 eV. The C 1s peak at 285.0 eV was used as a standard, when required, to correct for surface charging. Spectra were analyzed using CasaXPS software (version 2.3.14). The fractions of UIV/UV/UVI in the electrode surfaces were obtained by deconvolution of the high resolution XPS spectra using curve fitting procedures and BEs described elsewhere (Ilton et al., 2007; Schindler et al., 2009; Razdan et al., 2012).
Surface characterization of the electrodes using scanning electron microscopy, energy dispersive X-ray spectroscopy, X-ray diffraction and Raman spectroscopy have been published previously (He et al., 2007; He et al., 2012; Razdan et al., 2014; Razdan and Shoesmith, 2014). These analyses demonstrate that the REIII dopants were uniformly distributed in the UO2 matrix, not present as separate phases, with the noble metal (ε) particles predominantly, but not exclusively, located on grain boundaries (Zhu et al., 2019).
Figure 1 shows the fractions of UIV/UV/UVI, obtained from XPS analyses, in the surfaces of the three electrodes. As expected, the UV content increases with the extent of REIII doping from the lightly doped SIMFUEL to the heavily doped Dy-UO2. The small fractions of UVI present on all surfaces can be attributed to the presence of a thin air-formed layer since the electrodes were not subjected to cathodic reduction prior to recording the spectra. While XPS gives an indication of surface composition, Raman spectroscopy is a better indicator of the properties of the oxide matrix. Figure 2 shows the area ratio between the T2g Raman peak (445 cm−1) and the peak at 540 cm−1 calculated from the Raman spectroscopy measurements of Razdan and Shoesmith (2014). Since the T2g peak is attributed to the fundamental U-O stretch, it is characteristic of the undisturbed UO2 fluorite lattice, while the peak at 540 cm−1 is a measure of the REIII-OV clustering, a process which would be expected to limit the availability of the OV required for reaction 1 (He and Shoesmith, 2010).
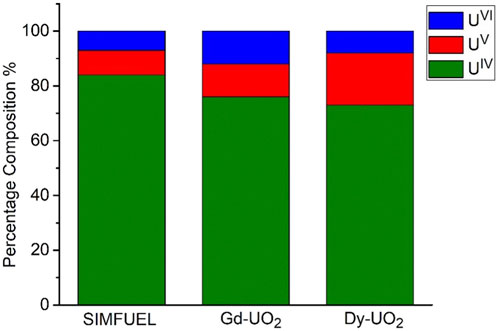
FIGURE 1. The fraction of U oxidation states on the surface of SIMFUEL, Gd-UO2 and Dy-UO2 after polishing and sonication.
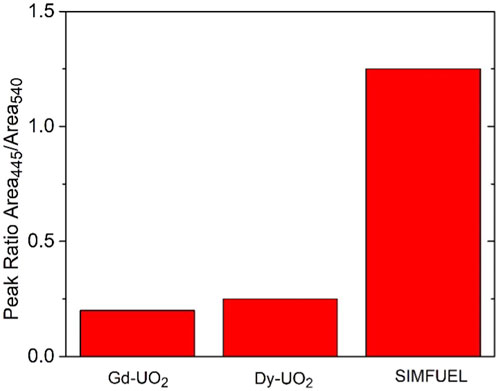
FIGURE 2. The area ratio of the Raman peaks recorded at 445 cm−1 and 540 cm−1 for the three materials (Nicol and Needes, 1973; Ofori, 2008).
Figure 3 shows a CV recorded on the Dy-UO2 electrode over the potential range from –1.2 V to 0.4 V (vs. SCE) in a NaCl solution containing both H2O2 and HCO3−/CO32−. On the positive-going scan, a minor oxidation process is observed for E ≥ 0.1 V (vs. SCE), leading to a slight enhancement of the cathodic current on the negative-going scan in the E range from 0 V to −0.5 V (vs. SCE). In the absence of H2O2, a voltametric scan over the same potential range yielded a similar minor oxidation on the positive-going scan, leading to a shallow reduction peak between −0.6 V and −0.9 V (the potential range indicated by the horizontal arrow in Figure 3). Similar differences (not shown) were observed between the positive- and negative-going scans on the Gd-UO2 and SIMFUEL electrodes, with the currents on both the positive- and negative-going scans being significantly higher for the SIMFUEL, as shown for E = –0.4 V (vs. SCE) in Figure 4. Since the SIMFUEL contains ε-particles, there is a possibility they could act as catalytic locations for H2O2 reduction. However, a previous study on a series of SIMFUELs suggested that, while this was the case, the effect was small (Goldik et al., 2006a).
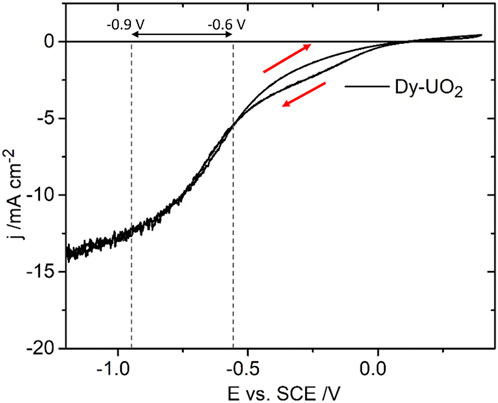
FIGURE 3. The current density (j) as a function of applied potential (E) recorded in a CV on Dy-UO2 in a 0.1 mol.L−1 NaCl solution containing 0.02 mol.L−1 of H2O2, and 0.05 mol.L−1 of NaHCO3 (pH = 9.7), electrode rotation rate (ω) = 8.33 Hz, and scan rate = 10 mV s−1. The red arrows indicate the scan direction; the dashed lines and horizontal arrow show the region −0.6 V to −0.9 V, within which oxide films are reduced.
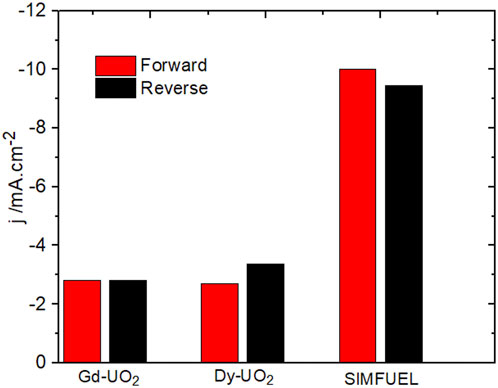
FIGURE 4. Cathodic currents recorded at −0.4 V in CVs performed on the three electrodes on the positive (forward)- and negative (reverse)-going scans.
Slightly enhanced currents on the negative-going scan recorded in H2O2-containing solutions have tentatively been assigned to the reduction of anodically-formed peroxycarbonate species (UVO2(HCO3) (H2O2)), which appear to catalyze H2O2 reduction (Goldik et al., 2006b), and possibly also to the reduction of UVI peroxycarbonate species (UVIO2(O2)x(CO3)y2−2x−2y) which would be expected at the [H2O2] and [CO3]tot ([CO3]tot = [HCO3−] + [CO32−]) involved in this study (Zhu et al., 2019). On occasion, currents on the negative-going scan were lower than those recorded on the positive-going scan, a feature attributable to the need to reduce anodically formed insulating UVI layers (UVIO3.2H2O/UO2CO3/UVIO4.4H2O) (Zhu et al., 2019) in the potential region indicated by the horizontal arrow in Figure 3. Figure 5 shows CVs recorded on the Gd-UO2 electrode in voltametric scans from E = –1.2 V (vs. SCE) to various more positive potential limits, showing that these anodic complications can be minimized if this limit is restricted to < 0 V (vs. SCE). Similar results (not shown) were observed on the other two materials.
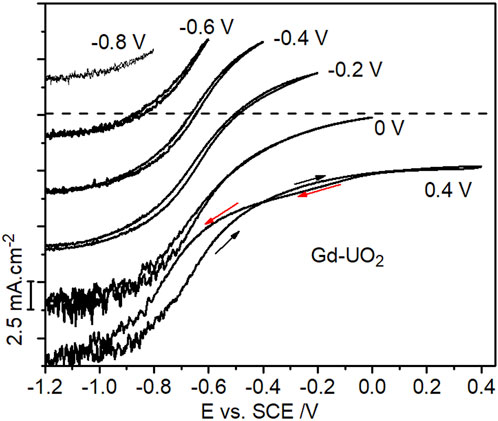
FIGURE 5. CVs recorded on the Gd-UO2 electrode from −1.2 V to different positive potential limits in a solution containing 0.1 mol.L−1 of NaCl, 0.05 mol.L−1 NaHCO3 and 0.02 mol.L−1 of H2O2 (pH = 9.7), electrode rotation rate = 8.33 Hz. The curves are offset by 5 mA cm−2 for clarity with the zero current point indicated for the scan to 0 V.
Figure 6 shows H2O2 reduction current densities, obtained from CVs, recorded as a function of electrode rotation rate (ω) at E = −1.1V, a potential in the apparently transport-controlled region (E ≤ −1.0 V (vs. SCE), Figure 5). Also shown is the diffusion-limited current calculated using the Levich equation (Santos et al., 2006) with a diffusion coefficient (D) for H2O2 = 1.32 × 10−5 cm2 s−1 Trunov and Presnov (1975a) and a kinematic viscosity for the electrolyte of 1.013 × 10−2 cm2 s−1 (Lide, 2005). While the currents are all linearly dependent on ω1/2, as expected according to the Levich relationship, they are lower than the calculated diffusion-limited current, demonstrating that total diffusion control was unattainable on all three materials. Also noteworthy are the relative values of the currents, which decrease with the level of REIII-doping (SIMFUEL > Gd-UO2 > Dy-UO2).
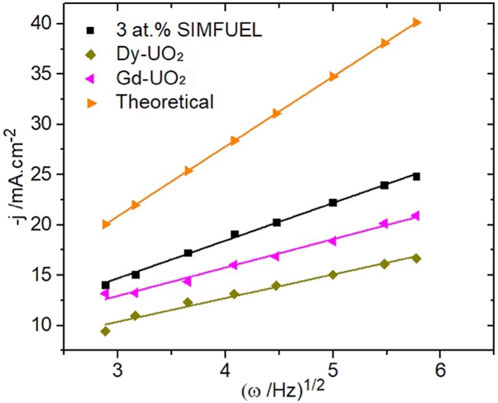
FIGURE 6. Levich plots of the current densities measured at −1.1 V for the three different UO2 electrodes in a 0.1 mol.L−1 NaCl solution containing 0.02 mol.L−1 of H2O2 and 0.05 mol.L−1 of NaHCO3 (pH = 9.7). The theoretical curve was calculated for this [H2O2] using the Levich equation Santos et al., 2006.
Currents for H2O2 reduction were measured as a function of potential and electrode rotation rate (ω). Using the Koutecky-Levich approach (Bard and Faulkner, 2000) and assuming a reaction order of 1 (the efficacy of choosing such a value is discussed in reference 13), currents were corrected for the influence of transport and the change in pH due to OH−production at the electrode surface to yield the kinetic currents plotted in the Tafel format in Figure 7. Two distinct regions of linear behaviour were observed, suggesting a change in the rate-determining reaction from the electrochemical reduction of the UV intermediate in region 1 (Figure 7) (reaction 2) to the chemical formation of UV in potential region 2 (Figure 7) (reaction 1). In region 1 the currents observed for Dy-UO2 and Gd-UO2 are effectively the same, while the currents for the SIMFUEL are significantly higher. The order of reactivity of the electrodes in region 1 (SIMFUEL > Gd-UO2 ∼ Dy-UO2) is the same as observed for their susceptibility to anodic dissolution at more positive potentials (Liu et al., 2017a). The similar Tafel slopes indicate that the mechanism of reduction is the same on all three materials. The transition between regions 1 and 2 was observed for all three materials and occurred over the potential range within which surface oxides become unstable and electrochemically reduced (as indicated by the horizontal arrow in Figure 3).
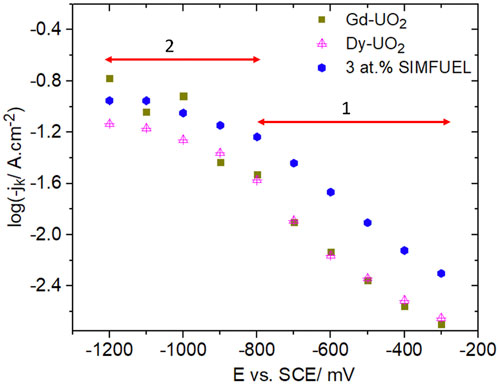
FIGURE 7. Plots of the kinetic current (jk) recorded on all three UO2 electrodes in a 0.1 mol.L−1 NaCl solution containing 0.02 mol.L−1 of H2O2 and 0.05 mol.L−1 of NaHCO3 (pH = 9.7).
Reaction orders in region 1 (Figure 7), measured over the range 0.01 ≤ [H2O2] ≤ 0.2 mol/L−1 in the absence of HCO3−/CO32− to avoid the complications discussed below, yielded values in the range 0.43–0.7 for the three materials. Attempts to measure reaction orders in region 2 were complicated by erratic currents attributable to the use of the current interrupt procedure, but yielded values in the range 1–1.5.
Figure 8 shows kinetic currents recorded for all three electrodes for two [CO3]tot (three for Dy-UO2) showing that an increase in [CO3]tot caused both a decrease in current and an increase in Tafel slope (as indicated in the figures) for all three materials. For Gd-UO2 and Dy-UO2, the Tafel slopes were effectively the same and increased slightly as [CO3]tot was increased. For both electrodes, but especially the Dy-UO2, the influence of [CO3]tot is minor at less negative potentials and increases as the potential is made more negative, indicating that when the electrochemical reaction 2 is slow compared to the chemical reaction 1, HCO3−/CO32− exerts only a minor influence on the overall reaction kinetics. For the SIMFUEL, the Tafel slopes were significantly higher, especially at the lower concentration. The transition from potential region 1 (E ≥ −0.9 V) to potential region 2 (E ≤ −0.9 V) was clear for the SIMFUEL and Dy-UO2 electrodes but only poorly defined for the Gd-UO2 electrode. The inclusion of data for a third higher [CO3]tot on Dy-UO2 confirmed that the influence of HCO3−/CO32− was most marked in region 2.
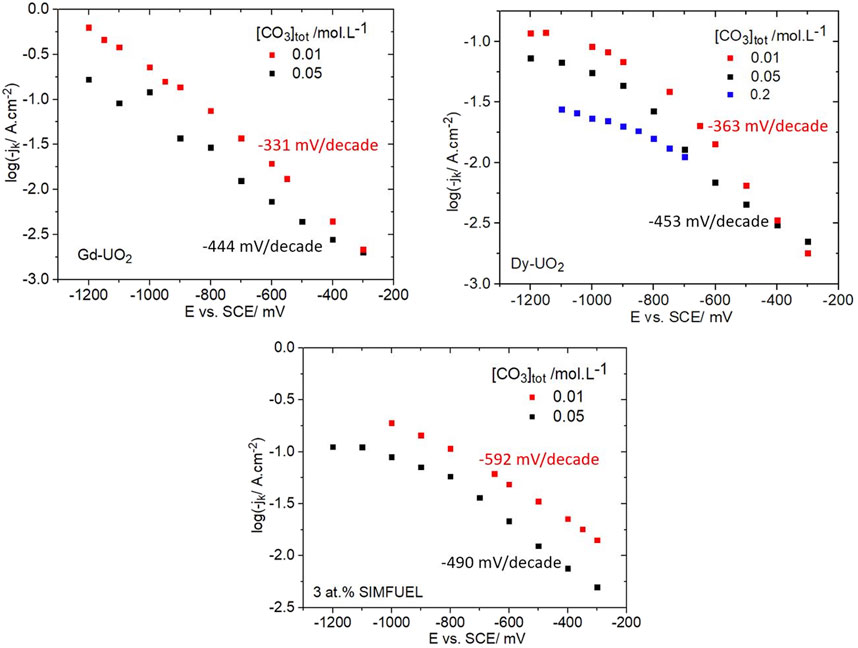
FIGURE 8. Plots of kinetic currents (jk) as a function of potential recorded on all three electrodes in a 0.1 mol.L−1 NaCl solution containing 0.02 mol.L−1 of H2O2 (pH = 9.7) and two different [CO3]tot (three for Dy-UO2). The Tafel slopes indicated on the plots were calculated over the potential region from −0.3 V to −0.9 V.
Experiments in solutions containing SO42− (not shown) rather than HCO3−/CO32− showed no significant effect of this anion on H2O2 reduction currents, confirming that the suppression of the H2O2 reduction current was specific to HCO3−/CO32−. This absence of an influence is not surprising since, unlike HCO3−/CO32−, SO42− is not a strong complexant for soluble U species (Grenthe et al., 1992) and its influence on the electrochemistry of UO2 has been shown to be limited to acidic solutions (Nicol and Needes, 1973; Ofori, 2008).
Previously, we adopted the kinetic analysis developed for the reduction of H2O2 on Cu (Vazquez et al., 1994; Ceré et al., 1999) to interpret the kinetics of its reduction on UO2 (Goldik et al., 2006a). According to this analysis, the transition from a potential-dependent current at low cathodic overpotentials to a chemically-controlled current at high overpotentials can be attributed to the reaction scheme (1) and (2), involving chemical oxidation of the UO2 surface to produce a UV intermediate, followed by its electrochemical reduction. The formation of UV is known to proceed via the initial formation of surface OH● radicals (Barreiro Fidalgo et al., 2018; Maier et al., 2020),
The formation of a UV surface state is also the first step in the anodic oxidation/dissolution of UO2, a reaction also involving the incorporation of OII into interstitial oxygen vacancy (OV) sites in the UO2 matrix to produce a thin U1–2xIV U2xV O2+x surface layer (Goldik et al., 2004).
The involvement of OVs can also explain the order of H2O2 reduction currents as demonstrated by Raman spectroscopy, Figure 2. Using the peak ratio as a measure of OV content, it is clear that their availability, and hence ability to support reaction 1, is in the order observed for H2O2 reduction currents,
The similarity in reduction currents for Gd-UO2 and Dy-UO2, despite the major differences in doping level, suggest that there may be a limit to the impact of REIII doping on the kinetics of H2O2 reduction. An identical similarity was observed in their resistance to anodic dissolution (Liu et al., 2017b). The ability of UO2 to function catalytically in this manner is similar to the well-recognized behaviour of CeO2, which also possesses a fluorite lattice containing OV and can be readily oxidized and reduced between CeIII and CeIV. Both oxides possess the ability to incorporate and release O as indicated in reactions 1 and 2.
According to our previous studies (Goldik et al., 2005; Goldik et al., 2006a; Goldik et al., 2006b), the kinetic balance between reactions 1 and 2 determines both the Tafel slope and the reaction order with respect to H2O2, both of which are defined by the rate constants for these reactions (Vazquez et al., 1994; Ceré et al., 1999). The Tafel slope for reaction 2 is given by
and the reaction order (m) by
with
and kc and ke are the rate constants for reactions 1 and 2, respectively.
Based on these relationships, the large Tafel slopes and fractional reaction orders can be explained in terms of the relative rates of these two reactions. If the rate of reaction 1, the formation of the intermediate UV state, was slow (kc[H2O2] << ke), the overall reaction would be chemically controlled: X would become small, the reaction order (m) approach 1, and the Tafel slope become very large. If the rate of reaction 2, the electrochemical reduction of the UV intermediate, was slow (kc[H2O2] >> ke), the overall reaction would be electrochemically controlled and X would tend to ∞, m to 0, and the Tafel slope would approach the value of 120 mV/decade, as expected for a one-electron-transfer-controlled process.
The plots in Figure 7 are consistent with such an analysis. In region 1, the large Tafel slopes and fractional reaction orders are consistent with an overall reaction under mixed chemical/electrochemical control. All three materials exhibit similar values, with the only significant difference being the higher currents measured on the SIMFUEL, attributed (above) to the higher availability of OV. In region 2, the switch to an almost potential-independent current and a reaction order in the region of 1 confirms that the overall reaction becomes chemically controlled at large negative potentials, when the electrochemical reaction would be rapid. The order of the current values changes, with that for the heavily doped Dy-UO2 being less than the approximately equal values observed for Gd-UO2 and SIMFUEL.
The absence of any influence of SO42–, but a clear influence of HCO3−/CO32−, on the H2O2 reduction current indicates a direct involvement of the latter anion in the surface reduction process. A possibility is that the suppressed current on all three materials can be explained by the well documented ability of HCO3−/CO32− to form peroxycarbonates which catalyze H2O2 decomposition in slightly alkaline solutions (Richardson et al., 2000)
HCO4− has been shown to be a kinetically more rapid oxidant than H2O2 for various species (Richardson et al., 2000; Regino and Richardson, 2007), and could be so for UO2, providing only minor consumption by decomposition on the time scale of our experiments. This would lead to an increase in the rate of reaction 1 and the overall reaction, a possible extension of region 1 to more negative potentials, and a decrease in the Tafel slope. The observation that Tafel slopes increase when the [CO3]tot is increased does not support this argument.
Since HCO3−/CO32– is a strong complexant for oxidized states of U, both as surface species(UV/UVI) (Keech et al., 2011) and as soluble species (UVIO2(CO3)x(2−2x)+) (Mühr-Ebert et al., 2019; Grenthe and Gaona, 2020), it would be expected to accelerate the formation of UV via reaction 1 and to stabilize it against subsequent electrochemical reduction by reaction 2. Such a process could lead to a suppression of the overall current, as observed, but not to an increased Tafel slope indicative of a slower chemical reaction relative to the rate of the electrochemical reaction.
Since the reaction of H2O2 proceeds through radical surface intermediates (reactions 3 and 4) and HCO3−/CO32− has been shown to form radical species via reaction with radiolytically-produced ●OH (Haygarth et al., 2010),
it is likely that such carbonate radicals are formed on the UO2 surface and recombine to reform H2O2,
with CO2 rapidly hydrolyzing to HCO3− at the pH employed in our study. Alternative pathways for this radical decay process have been demonstrated in radiolytic studies (Richardson et al., 2000; Barreiro Fidalgo et al., 2018; Maier et al., 2020). While speculative, some support for such a reformation of H2O2 is provided in studies of the electrochemical reduction of O2 on UO2 (Nicol and Needes, 1973; Ofori, 2008), a reaction which can proceed via the formation of H2O2 as an intermediate:
In the absence of HCO3−/CO32−, this reaction proceeds efficiently with the release to solution of only minimal amounts of H2O2. In the presence of HCO3−/CO32− measurable amounts of H2O2 are released to solution, consistent with its partial stabilization in the presence of these anions.
The influence of [CO3]tot on H2O2 reduction on the SIMFUEL is somewhat different from that observed on the other two materials, especially at low negative potentials in region 1. According to the reaction scheme proposed, this would suggest a very marked influence of HCO3−/CO32− in reducing the rate of reaction 2 relative to reaction 1 by stabilizing the chemically formed UV state. However, an increased Tafel slope, as observed (Figure 7), suggests the opposite. Previous studies on a series of SIMFUELs (Santos et al., 2006) indicate a small influence of ε-particles on H2O2 reduction currents at less negative potentials (lower cathodic overpotentials). Thus, a possibility for the enhanced influence of HCO3−/CO32− in this potential range is that the formation of ●OH leading to ●CO3− formation is accelerated on these particles. This could then either lead to reformation of H2O2 (reaction 12) or to its decomposition (reactions 8 and 9). As chemical processes, these reactions would not generate electrochemical current but would reduce the fraction of H2O2 undergoing current-generating electrochemical reduction. The decomposition of H2O2 on noble metals is well characterized (McKee, 1969; Serra-Maia et al., 2018).
Presently this suite of reactions cannot be separated, leaving it possible that the overall process is a complex mix of them all.
The kinetics of H2O2 reduction have been studied on REIII-doped UO2 specimens and a SIMFUEL, more lightly REIII-doped but also containing noble metal particles. Reduction was shown to proceed via the chemical formation followed by electrochemical reduction of UV sites in the UO2 surface, with the rate dependent on the availability of OV in the UO2 matrix. This availability was significantly greater for the SIMFUEL, leading to more rapid H2O2 reduction. Also, on the SIMFUEL, the reduction may be slightly enhanced on the noble metal particles.
In the presence of HCO3−/CO32−, the process is complicated, possibly by the anions ability to cause H2O2 decomposition and to react with ●OH radicals, formed by H2O2 on the surface to yield ●CO3−. These radicals appear to catalyze the reformation of H2O2 thereby reducing the extent of its electrochemical reduction.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
ZZ: Methodology, Validation, Formal analysis, Investigation, Data curation, Writing—original draft, Visualization. ML: Methodology, data curation, Visualization. NL: Methodology, Supervision. JN: Methodology, Resources, Writing—review and editing, Supervision, Project administration, Funding acquisition. DS: Conceptualization, Methodology, Resources, Writing—review and editing, Supervision, Project administration, Funding acquisition.
This research was funded under the Industrial Research Chair and Collaborative Research and Development (CRDPJ 507465–16) agreements between the Natural Sciences and Engineering Research Council (NSERC, Ottawa) and the Nuclear Waste Management Organization (NWMO, Toronto).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Badley, M., and Shoesmith, D. (2022). The corrosion/dissolution of used nuclear fuel in a deep geological repository. Toronto: Nuclear Waste Management Organization. NWMO-TR-2022-09.
Bard, A. J., and Faulkner, L. R. (2000). Electrochemical methos fundamentals and applications. Second ed. New York: John Wiley & Sons, 819.
Barreiro Fidalgo, A., Kumagai, Y., and Jonsson, M. (2018). The role of surface-bound hydroxyl radicals in the reaction between H2O2 and UO2. J. Coord. Chem. 71 (11-13), 1799–1807. doi:10.1080/00958972.2018.1466287
Britz, D., and Brocke, W. A. (1975). Elimination of iR-drop in electrochemical cells by the use of a current-interruption potentiostat. J. Electroanal. Chem. Interfacial Electrochem. 58 (2), 301–311. doi:10.1016/s0022-0728(75)80088-x
Ceré, S., Vazquez, M., de Sánchez, S. R., and Schiffrin, D. J. (1999). Surface redox catalysis and reduction kinetics of hydrogen peroxide on copper–nickel alloys. J. Electroanal. Chem. 470 (1), 31–38. doi:10.1016/s0022-0728(99)00207-7
Eriksen, T. E., Shoesmith, D. W., and Jonsson, M. (2012). Radiation induced dissolution of UO2 based nuclear fuel – a critical review of predictive modelling approaches. J. Nucl. Mater. 420 (1-3), 409–423. doi:10.1016/j.jnucmat.2011.10.027
Goldik, J. S., Nesbitt, H. W., Noël, J. J., and Shoesmith, D. W. (2004). Surface electrochemistry of UO2 in dilute alkaline hydrogen peroxide solutions. Electrochimica Acta 49 (11), 1699–1709. doi:10.1016/j.electacta.2003.11.029
Goldik, J. S., Noël, J. J., and Shoesmith, D. W. (2006b). Surface electrochemistry of UO2 in dilute alkaline hydrogen peroxide solutions. Electrochimica Acta 51 (16), 3278–3286. doi:10.1016/j.electacta.2005.09.019
Goldik, J. S., Noel, J. J., and Shoesmith, D. W. (2006a). The effects of simulated fission products in the reduction of hydrogen peroxide on simulated nuclear fuel electrodes. J. Electrochem. Soc. 153 (9), E151–E159. doi:10.1149/1.2213419
Goldik, J. S., Noël, J. J., and Shoesmith, D. W. (2005). The electrochemical reduction of hydrogen peroxide on uranium dioxide electrodes in alkaline solution. J. Electroanal. Chem. 582 (1), 241–248. doi:10.1016/j.jelechem.2004.11.007
Grenthe, I., Fuger, J., Konings, R. J., Lemire, R. J., Muller, A. B., Nguyen-Trung, C., et al. (1992). Chemical thermodynamics of uranium, 1. Amsterdam: North-Holland.
Grenthe, I., and Gaona, X. (2020). Second update on the chemical thermodynamics of uranium, neptunium, plutonium, americium and technetium. Paris, France: OECD/NEA Publishing. Report No. NEA-7500.
Hall, D. S., Behazin, M., Jeffrey Binns, W., and Keech, P. G. (2021). An evaluation of corrosion processes affecting copper-coated nuclear waste containers in a deep geological repository. Prog. Mater. Sci. 118, 100766. doi:10.1016/j.pmatsci.2020.100766
Hall, D. S., and Keech, P. G. (2017). An overview of the Canadian corrosion program for the long-term management of nuclear waste. Corros. Eng. Sci. Technol. 52 (1), 2–5. doi:10.1080/1478422x.2016.1275419
Haygarth, K. S., Marin, T. W., Janik, I., Kanjana, K., Stanisky, C. M., and Bartels, D. M. (2010). Carbonate radical formation in radiolysis of sodium carbonate and bicarbonate solutions up to 250 °C and the mechanism of its second order decay. J. Phys. Chem. A 114 (5), 2142–2150. doi:10.1021/jp9105162
He, H., Broczkowski, M., O’Neil, K., Ofori, D., Semenikhin, O., and Shoesmith, D. (2012). Corrosion of nuclear fuel (UO2) inside a failed nuclear waste container. Toronto, Ontario: Nuclear Waste Management Organization.
He, H., Keech, P. G., Broczkowski, M. E., Noël, J. J., and Shoesmith, D. W. (2007). Characterization of the influence of fission product doping on the anodic reactivity of uranium dioxide. Can. J. Chem. 85 (10), 702–713. doi:10.1139/v07-056
He, H., and Shoesmith, D. (2010). Raman spectroscopic studies of defect structures and phase transition in hyper-stoichiometric UO(2+x). Phys. Chem. Chem. Phys. 12 (28), 8108–8117. doi:10.1039/b925495a
Hocking, W., Betteridge, J. S., and Shoesmith, D. W. (1991). The cathodic reduction of oxygen on uranium dioxide in dilute alkaline aqueous solution. Canada: Atomic Energy of Canada Limited Report, 1572–6657.
Hocking, W. H., Betteridge, J. S., and Shoesmith, D. W. (1994). The cathodic reduction of oxygen on uranium dioxide in dilute alkaline aqueous solution. J. Electroanal. Chem. 379 (1), 339–351. doi:10.1016/0022-0728(94)87156-6
Ilton, E. S., Boily, J.-F., and Bagus, P. S. (2007). Beam induced reduction of U(VI) during X-ray photoelectron spectroscopy: The utility of the U4f satellite structure for identifying uranium oxidation states in mixed valence uranium oxides. Surf. Sci. 601 (4), 908–916. doi:10.1016/j.susc.2006.11.067
Keech, P. G., Goldik, J. S., Qin, Z., and Shoesmith, D. W. (2011). The anodic dissolution of SIMFUEL (UO2) in slightly alkaline sodium carbonate/bicarbonate solutions. Electrochimica Acta 56 (23), 7923–7930. doi:10.1016/j.electacta.2010.12.024
Kwong, G. (2011). Status of corrosion studies for copper used fuel containers under low salinity conditions. Toronto, ON: Nuclear Waste Management Organization. Report NWMO TR-2011-14.
Liu, N., He, H., Noël, J. J., and Shoesmith, D. W. (2017a). The electrochemical study of Dy2O3 doped UO2 in slightly alkaline sodium carbonate/bicarbonate and phosphate solutions. Electrochimica Acta 235, 654–663. doi:10.1016/j.electacta.2017.03.075
Liu, N., Kim, J., Lee, J., Youn, Y.-S., Kim, J.-G., Kim, J.-Y., et al. (2017b). Influence of Gd doping on the structure and electrochemical behavior of UO2. Electrochimica Acta 247, 496–504. doi:10.1016/j.electacta.2017.07.006
Liu, N., Zhu, Z., Noël, J. J., and Shoesmith, D. W. (2018). “Corrosion of nuclear fuel inside a failed waste container.” In Encyclopedia of interfaical chemistry: Surface science and electrochemistry (Amsterdam: Elsevier), Vol. 6, 172–182.
Lousada, C. M., Trummer, M., and Jonsson, M. (2013). Reactivity of H2O2 towards different UO2-based materials: The relative impact of radiolysis products revisited. J. Nucl. Mater. 434 (1-3), 434–439. doi:10.1016/j.jnucmat.2011.06.003
Maier, A. C., Kegler, P., Klinkenberg, M., Baena, A., Finkeldei, S., Brandt, F., et al. (2020). On the change in UO 2 redox reactivity as a function of H 2 O 2 exposure. Dalton Trans. 49 (4), 1241–1248. doi:10.1039/c9dt04395k
McKee, D. (1969). Catalytic decomposition of hydrogen peroxide by metals and alloys of the platinum group. J. Catal. 14 (4), 355–364. doi:10.1016/0021-9517(69)90326-1
Mühr-Ebert, E. L., Wagner, F., and Walther, C. (2019). Speciation of uranium: Compilation of a thermodynamic database and its experimental evaluation using different analytical techniques. Appl. Geochem. 100, 213–222. doi:10.1016/j.apgeochem.2018.10.006
Nicol, M. J., and Needes, C. R. S. (1973). The mechanism of the anodic dissolution of uranium dioxide. Johannesburg, South Africa: National Institute for Metallurgy Report No. 7079.
Nilsson, S., and Jonsson, M. (2011). H2O2 and radiation induced dissolution of UO2 and SIMFUEL pellets. J. Nucl. Mater. 410 (1–3), 89–93. doi:10.1016/j.jnucmat.2011.01.020
Ofori, D. (2008). The influence of corrosion product deposits on UO2. London, ON: University of Western Ontario.
Razdan, M., Hall, D. S., Keech, P. G., and Shoesmith, D. W. (2012). Electrochemical reduction of hydrogen peroxide on SIMFUEL (UO2) in acidic pH conditions. Electrochimica Acta 83, 410–419. doi:10.1016/j.electacta.2012.05.163
Razdan, M., and Shoesmith, D. W. (2014). Influence of trivalent-dopants on the structural and electrochemical properties of uranium dioxide (UO2). J. Electrochem. Soc. 161 (3), H105–H113. doi:10.1149/2.047403jes
Razdan, M., Trummer, M., Zagidulin, D., Jonsson, M., and Shoesmith, D. W. (2014). Electrochemical and surface characterization of uranium dioxide containing rare-earth oxide (Y2O3) and metal (Pd) particles. Electrochimica Acta 130, 29–39. doi:10.1016/j.electacta.2014.02.134
Regino, C. A. S., and Richardson, D. E. (2007). Bicarbonate-catalyzed hydrogen peroxide oxidation of cysteine and related thiols. Inorganica Chim. Acta 360 (14), 3971–3977. doi:10.1016/j.ica.2007.05.020
Richardson, D. E., Yao, H., Frank, K. M., and Bennett, D. A. (2000). Equilibria, kinetics, and mechanism in the bicarbonate activation of hydrogen peroxide: Oxidation of sulfides by peroxymonocarbonate. J. Am. Chem. Soc. 122 (8), 1729–1739. doi:10.1021/ja9927467
Santos, B. G., Nesbitt, H. W., Noël, J. J., and Shoesmith, D. W. (2004). X-ray photoelectron spectroscopy study of anodically oxidized SIMFUEL surfaces. Electrochimica Acta 49 (11), 1863–1873. doi:10.1016/j.electacta.2003.12.016
Santos, B. G., Noël, J. J., and Shoesmith, D. W. (2006). The effect of pH on the anodic dissolution of SIMFUEL (UO2). J. Electroanal. Chem. 586 (1), 1–11. doi:10.1016/j.jelechem.2005.09.021
Schindler, M., Hawthorne, F. C., Freund, M. S., and Burns, P. C. (2009). XPS spectra of uranyl minerals and synthetic uranyl compounds. I: The U 4f spectrum. Geochimica Cosmochimica Acta 73 (9), 2471–2487. doi:10.1016/j.gca.2008.10.042
Serra-Maia, R., Bellier, M., Chastka, S., Tranhuu, K., Subowo, A., Rimstidt, J. D., et al. (2018). Mechanism and kinetics of hydrogen peroxide decomposition on platinum nanocatalysts. ACS Appl. Mat. Interfaces 10 (25), 21224–21234. doi:10.1021/acsami.8b02345
Sunder, S., Miller, N. H., and Shoesmith, D. W. (2004). Corrosion of uranium dioxide in hydrogen peroxide solutions. Corros. Sci. 46 (5), 1095–1111. doi:10.1016/j.corsci.2003.09.005
Trunov, A., and Presnov, V. (1975a). Oxygen electroreduction on semiconductor catalysis II: Processes at oxide semiconductor-electrolyte interface. Elektrokhimiya 11 (1), 77–84.
Trunov, A., and Presnov, V. (1975b). Oxygen electroreduction on semiconductor catalysis III: The importance of 3d electrodes of oxide semiconductor. Elektrokhimiya 11 (1), 290–292.
Vazquez, M. V., de Sanchez, S. R., Calvo, E. J., and Schiffrin, D. J. (1994). The electrochemical reduction of hydrogen peroxide on polycrystalline copper in borax buffer. J. Electroanal. Chem. 374 (1), 179–187. doi:10.1016/0022-0728(94)03342-0
Wu, L., and Shoesmith, D. W. (2014). An electrochemical study of H2O2 oxidation and decomposition on simulated nuclear fuel (SIMFUEL). Electrochimica Acta 137, 83–90. doi:10.1016/j.electacta.2014.06.002
Zhu, Z., Noël, J. J., and Shoesmith, D. W. (2020). Hydrogen peroxide decomposition on simulated nuclear fuel bicarbonate/carbonate solutions. Electrochimica Acta 340, 135980. doi:10.1016/j.electacta.2020.135980
Keywords: electrochemistry, SIMFUEL, hydrogen peroxide, UO2, rare earth dopants
Citation: Zhu Z, Ly M, Liu N, Noël JJ and Shoesmith DW (2022) The kinetics of hydrogen peroxide reduction on rare earth doped UO2 and SIMFUEL. Front. Mater. 9:1038310. doi: 10.3389/fmats.2022.1038310
Received: 06 September 2022; Accepted: 30 September 2022;
Published: 18 October 2022.
Edited by:
Hongbo Cong, University of Akron, United StatesReviewed by:
Qiao Yanxin, Jiangsu University of Science and Technology, ChinaCopyright © 2022 Zhu, Ly, Liu, Noël and Shoesmith. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ziyan Zhu, enpodTQ2QHV3by5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.