 Daniele Rigotti
Daniele Rigotti Andrea Dorigato
Andrea Dorigato Alessandro Pegoretti
Alessandro Pegoretti- Department of Industrial Engineering and INSTM Research Unit, University of Trento, Trento, Italy
In this work, a commercial linear low density polyethylene (LLDPE) utilized for packaging applications was melt compounded with different amounts (from 10 up to 50 wt. %) of poly(3-hydroxybutyrate) [P(3HB)], with the aim to evaluate the possibility to partially replace LLDPE with a biodegradable matrix obtained from renewable resources. The processability, microstructural, and thermo-mechanical behavior of the resulting blends was investigated. Melt flow index (MFI) values of the LLDPE matrix were not much affected until a P(3HB) content of 20 wt.%, while for higher P(3HB) concentrations an evident decrease of the viscosity was detected. Scanning electron microscope (SEM) observations on the blends highlighted that at limited P(3HB) concentrations the secondary phase was homogeneously dispersed in form of isolated domains, while at a P(3HB) content of 50 wt.% a continuous layered morphology could be detected. Thermogravimetric analysis (TGA), differential scanning calorimetry (DSC) and Fourier transform infrared spectroscopy (FT-IR) did not evidence any chemical or physical interaction between the two polymer phases. Quasi-static tensile tests and dynamical mechanical analysis showed that the introduction of P(3HB) led to a pronounced stiffening effect, while the progressive drop of the yield and ultimate mechanical properties could be attributed to the weak interfacial adhesion and poor compatibility between the two matrices. The resistance to hydrolytic degradation of the LLDPE/P(3HB) blends was evaluated over a period of 100 days of immersion in water at 50°C. It was observed that the weight variation and the decrease of the tensile properties due to the hydrolytic process on the biodegradable phase were evident only for a P3HB content of 50 wt.%. In conclusion, this work showed that the partial replacement of LLDPE with a biobased P(3HB) could lead to the development of an innovative blend with good processability and mechanical properties, until a P(3HB) amount of 20 wt.%.
Introduction
In the last few decades global climate change and ecosystems deterioration have been the main driving forces for a progressive switch from a take-make-dispose economy based on continuous growth and increasing resource throughput to a circular economy in which resource input, waste, emission, and energy leakage are minimized by slowing, closing, and narrowing material and energy loops (Geissdoerfer et al., 2017). Circular Economy is receiving increasing attention as an effective way to prevent and/or reduce industrial waste and increase the efficiency of the production processes, reaching thus a better equilibrium and harmony between economy, environment, and society. This can be achieved through a better manufacturing strategy that focuses on long-lasting design, maintenance, repair, reuse, remanufacturing, refurbishing, and recycling (Ghisellini et al., 2016; Kalmykova et al., 2018). Accumulation of non-degradable plastics is one of the main problems for the environment and human health, because of the generation of secondary microplastics and nanoplastics, release of hazardous chemicals during manufacturing, use and the following landfilling, incineration, or improper disposal (Soroudi and Jakubowicz, 2013; Laycock et al., 2017). Thanks to their limited weight, flexibility and durability, plastics production has reached 380 million tons in 2015, and about 40% of this amount is applied in packaging field (Groh et al., 2019). For as concerns the management of the packaging waste, its reuse and recycling could reduce their environmental impact, delivering thus both economic and environmental benefits (Dilkes-Hoffman et al., 2018; Geueke et al., 2018). However, recycling would be neither practical nor economical for certain applications such as bags, agricultural mulch films, and food packaging (Pedroso and Rosa, 2005). Due to safety issues, it is particularly challenging to fully recycle food packaging waste to produce new food package, because of the presence of contaminants and/or chemicals that can directly migrate from the packaging into the food or beverage (Groh et al., 2019).
Considering the difficulties in recycling plastics for packaging applications, it is thus clear that biopolymers could represent a valuable solution to reduce the environmental burden associated to the life cycle of industrial packages. In fact, packaging is the largest market for biopolymers, accounting for about 58% of the total volume share. Despite this, biopolymers market represents today only a limited percentage of the total plastics production (Niaounakis, 2019). Processing biodegradable plastics under composting conditions at the end of their life could be preferred over recycling for these kinds of applications (Geueke et al., 2018). However, the end of life of biopolymers is a debatable issue. Even if recycling of biodegradable polymers does not exploit their biodegradability, the disposal of biopolymer articles through biodegradation has the disadvantage of discarding valuable raw materials and could lead to environmental problems such as the emission of methane. To reduce the consumption of renewable resources used for the synthesis of biopolymers is important to be able to recycle them. In this way, biopolymers waste may become a valuable alternative feedstock for monomers and intermediates (Hahladakis and Iacovidou, 2018).
Most of the traditional thermoplastic used as packaging materials are polyolefins, such as high-density polyethylene (HDPE), low-density polyethylene (LDPE), and polypropylene (PP). Polyethylene is a general-purpose thermoplastic polymer with good processing and mechanical properties as well as relative low price. In particular, linear low-density polyethylene (LLDPE), thanks to its elevated tear and impact strength, is generally applied in film production for the packaging. LLDPE is constituted by a linear hydrocarbon backbone with short chain branching, formed by a copolymer of ethylene and an α-olefin or diene (i.e., butene, hexane, or octene) (Dorigato et al., 2010a,b, 2011, 2013; Dorigato and Pegoretti, 2012, 2013). It finds large application in grocery bags, heavy duty shipping sacks, agricultural films, pipes, liners for consumers, landfills, and waste ponds (Hancox, 1992). An important issue for economic and environmental reasons is the problem of post-consumer recycling of this material, since the use of these plastics is continuously increasing (Hole and Hole, 2019).
Polyhydroxyalkanoates (PHAs) are generally considered a promising group of biopolymers from renewable resources and bacteria and they possess a high potential as bio-based and biodegradable plastic packaging materials in the transition toward a circular economy (Ragaert et al., 2019). Among all the PHAs, poly(3-hydroxybutyrate) [P(3HB)] is one of the most innovative thermoplastic. P(3HB) is synthesized by microorganism as an intracellular storage product under suitable growth conditions (abundant carbon source, limited sources of oxygen, phosphorous, or nitrogen). The carbon is assimilated and converted into hydroxyalkanoate monomers, polymerized, and stored in the cell cytoplasm. The resulting polymer is a biocompatible high molecular weight crystalline polyester that can be used to produce films and foils for the packaging industry and tissue engineering (Chen and Hajnal, 2015; Valentini et al., 2019). Moreover, P(3HB) can be degraded by microorganisms (i.e., bacteria, fungi, and algae) under different environmental conditions (Wang et al., 2014; Emadian et al., 2017; Pakalapati et al., 2018).
Nowadays, the major challenge in the biopolymers research is to partially/totally replace conventional petroleum-based polymers with the biodegradable ones, in view of a more sustainable development. Therefore, blending biodegradable polymers with traditional plastics, such as polyethylene, has recently received considerable attention, in order to simultaneously improve/tailor the physical properties of the resulting materials and lower their environmental burden. A key point to be investigated in these systems is whether the biodegradable component could be effectively biodegraded by the microorganisms under certain conditions, and whether the remaining polyolefinic phase could be even degraded (Chandra and Rustgi, 1998). Several research efforts were thus made in this direction, through the development of different LDPE/biopolymer blends, such as LDPE/wax (Krupa and Luyt, 2000, 2001), LDPE/starch (Nguyen et al., 2016; Datta and Halder, 2019) and LDPE/PLA (Bhasney et al., 2019) systems. It was demonstrated that the degradation of polyethylene can be accelerated by environmental factors such as temperature, UV irradiation or the action of microorganisms (Restrepo-Flórez et al., 2014). For instance, Veethahavya et al. (2016) investigated the possibility to accelerate the biodegradation through microorganisms digestion of LDPE upon blending with natural polymers, finding that in liquid cultures the degradation rate of LDPE increased with the starch concentrations. Only a limited number of papers addressing the thermo-mechanical properties and the degradative behavior of LLDPE/P(3HB) blends are available in the open scientific literature. In these work, the attention was mainly focused on the morphology, highlighting that the blend components are thermodynamically incompatible and a clearly distinguishable interface is formed between the disperse phase (PHB) and the continuous matrix LDPE (Ol'khov et al., 2000; Pankova et al., 2010). With packaging application in mind, this immiscibility was exploited to regulate the resistance to hydrolysis and biodegradation through the control of water permeability studying the interaction between the two polymer phases against water according to Flory–Huggins theory (Pankova et al., 2010). Biodegradability of LDPE/PHB blends was extensively studied and also improved when natural additives such as castor oil or guar gum were inserted (Burlein and Rocha, 2014; Rocha and Moraes, 2015). Pro-oxidant additives, such as oxidized polyethylene wax, represent a promising solution to the problem of the environment contamination and could reduce the phase separation of LDPE with PHB and increased the biodegradation during aging in soil (Rosa et al., 2007).
Therefore, the present work aims to investigate the effect of blending P(3HB) to LLDPE at different concentrations (from 10 to 50 wt.%). A systematic investigation of the processability, of the microstructural and of the thermo-mechanical properties of the resulting blends was performed. Particular attention was devoted to the hydrolytic degradation of these systems, analyzing the water absorption, and the change of the thermal and mechanical performances of the samples as a function of the hydrolysis time.
Experimental Part
Materials
Poly(3-hydroxybutyrate) was provided by Biomer (Schwalbach, Germany) in form of white powder (Mw = 3.7 ± 0.2 · 105 Da, density = 1.18 g/cm3). Preliminary NMR studies on the obtained powder (not reported for sake of brevity) showed the characteristic signals of poly-3-hydroxybutyrate [P(3HB)] (i.e., C=O, -OCH-, -CH2-, and –CH3), excluding thus the presence of poly-hydroxyvalerate (PHV) or of poly-4-hydroxybutyrate [P(4HB)] in the purchased material. Linear low-density polyethylene used in this work was Flexirene® CL10 (density of 0.918 g/cm3, melting temperature of 120°C, melt flow index (MFI) at 190°C and 2.16 kg of 2.6 g/10 min), provided by Versalis Spa (San Donato Milanese, Italy) in form of granules.
Sample Preparation
Prior compounding, both LLDPE and P(3HB) were dried in an oven at 80°C for 24 h. The two polymers were mixed at different relative concentrations with a Thermo Haake Rheomix® 600 melt compounder at 190°C. After some preliminary trials, a rotor speed of 60 rpm and a compounding time of 10 min were selected, in order to provide a good homogenization of the blends. The resulting materials were then hot pressed at 190°C for 10 min in a Carver hot plate press, applying a pressure of 2.5 MPa, to obtain square sheets with a length of 20 cm and a thickness of about 1 mm. In this way, neat LLDPE, neat P(3HB) and LLDPE/P(3HB) blends with different P(3HB) concentrations, ranging from 10 up to 50 wt.%, were prepared. Samples were designated indicating the relative content of LLDPE and P(3HB) phases, i.e., LLDPE_x_PHB_y, where x and y are the weight percentage of LLDPE and P(3HB), respectively.
Experimental Techniques
In order to evaluate the processability of the resulting blends, melt flow index (MFI) measurements were performed according to ASTM D1238–04 standard, by using a Kyeness 4003DE plastomer at a temperature of 180°C under a load of 2.16 kg investigating about 10 g of material for each sample. This testing parameters were selected in order to evaluate the MFI under the same conditions for all the samples.
Morphological properties of the cryo-fractured surface of the LLDPE/PHB blends were determined by using a Zeiss Supra 40 high resolution field emission scanning electron microscope (FESEM), operating at an acceleration voltage of 4 kV. Prior to be observed, samples were covered with a conductive platinum/palladium coating deposited through a sputter coater.
FT-IR spectroscopy was conducted with a Perkin Elmer Spectrum One machine, in order to analyze the vibrational transitions inside molecules of the different samples. This analysis was performed in a wavenumber range between 650 and 4,000 cm−1.
DSC measurements were performed on one specimen for each sample in order to get information about the glass transition, crystallization, and melting temperature of the material. DSC tests were carried out with a Mettler DSC30 calorimeter under a nitrogen flow of 10 ml/min. A heating run from −100 to 200°C was followed by a cooling stage to −100°C and by a second heating run up to 200°C. All the scans were performed at a heating or cooling rate of 10°C/min. In this way, it was possible to determine the melting (Tm) and crystallization temperature (Tc) of LLDPE and P(3HB). The crystallinity degree (Xc) was computed as the ratio between the melting enthalpy of the samples and the reference value of the fully crystalline polymers, i.e., 293 J/g for LLDPE and 146 J/g for P(3HB) (Barham et al., 1984).
Knowing the critical issues of P(3HB) regarding its thermal degradation, it is extremely important to determine the thermal degradation of the blends. For this purpose, thermogravimetric analysis (TGA) was performed on one specimen for each sample through a TA Instruments TGAQ500 thermobalance, operating under a nitrogen flow of 10 ml/min in a temperature interval between 30 and 700°C, setting a heating rate of 10°C/min.
Viscoelastic behavior of the blends as a function of the temperature was investigated through dynamical mechanical analysis (DMA) with a TA Instrument DMA Q800. The tests were carried out in tensile mode on a rectangular specimen for each sample with dimension of 30 × 5 × 1 mm3 in a temperature range between −100 and 100°C, at a heating rate of 3°C/min and a testing frequency of 1 Hz.
Quasi-static tensile properties of the different polymer blends were evaluated at ambient temperature with an Instron 5969 electromechanical testing machine, testing ISO 527 1BA dogbone specimens, having a gage length of 30 mm. Elastic modulus (E) was evaluated at 0.25 mm/min with an extensometer with a gage length of 12.5 mm, setting a maximum deformation of 1%. E was computed as secant modulus between deformation levels of 0.05% and 0.25%. Tensile stress at yield (σy), stress at break (σb), and strain at break (εb) were determined without the use of an extensometer, setting a crosshead speed of 1 mm/min. At least five specimens were tested for each sample.
In order to evaluate the resistance to hydrolysis, the blends were subjected to hydrolytic degradation tests, simulating a low run-off condition. The analysis was conducted in a thermostatically controlled bath at a constant temperature of 50°C. ISO 527 1BA dumbbell specimens were immersed in 80 ml of distilled water and the run-off condition was simulated by performing a periodically change of water every week, for a total duration of the tests of 100 days. Some samples were periodically extracted and dried in a vacuum oven at a temperature of 60° C for 90 h, in order to eliminate the absorbed water. The evaluation of hydrolytic degradation on the dried samples was performed by calculating the relative variation in weight of the specimens. Moreover, DSC tests were carried out on the same samples to monitor the trend of the crystallinity degree of LLDPE with the hydrolysis time, and also quasi-static tensile tests were performed to evaluate their elastic modulus and yield strength. For these tests, the same testing conditions applied for non-hydrolyzed samples were adopted.
Results and Discussions
One of the main issues in the production of polyethylene-based film for packaging applications is the processability of the resulting materials. Therefore, the evaluation of the viscosity of the produced blends in the molten state is of utmost importance. Figure 1 summarizes the MFI values of neat LLDPE and relative blends at different P(3HB) concentrations.
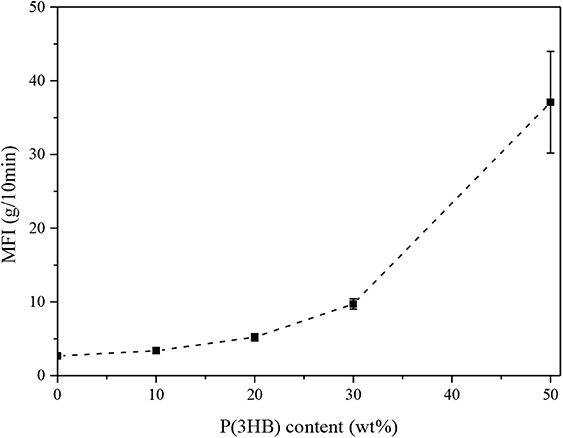
Figure 1. MFI values of neat LLDPE and LLDPE/P3HB blends (T = 180°C, load = 2.16 kg). Error bars represent the standard deviation.
It is interesting to observe how the MFI increases with the P(3HB) amount, thus indicating a progressive lowering of the viscosity of the blends in the molten state. For instance, at a P(3HB) loading of 50 wt.%, a MFI value of 37 g/10 min was reached. While the observed MFI increase is rather limited up to a biopolymer concentration of 20 wt.%, a more rapid increase can be detected at higher P(3HB) amounts. Considering that the selected LLDPE grade is generally used for the production of extruded film for food packaging, it is clear that the retention of the original MFI is a fundamental processability requirement for this process (Van Krevelen and Te Nijenhuis, 2009). MFI suggested for polyethylene in this manufacturing process is between 1 and 20 g/10 min (Patel, 2016) so it can be concluded that a P(3HB) content of 50 wt.% is probably not suitable for the production of extruded packaging films.
SEM analysis was then carried out, in order to appreciate the most important morphological features of the blends. In Figures 2A–D some representative micrographs of the cryofracture surfaces of LLDPE and of the relative blends are reported. As expected, the fracture profile of neat LLDPE is rather smooth (Figure 2A), while in the blends at low P(3HB) content the secondary phase is present in round domains homogenously distributed within the LLDPE matrix, with a mean size ranging from 2 to 4 μm (see Figures 2B,C). In these micrographs, the presence of the P(3HB) phase can be often detected by the holes produced during the cryofracturing operations. In any case, a rather limited interfacial adhesion between the two polymer phases, with an evident debonding, can be clearly seen. This is a first indication of the limited miscibility between the two constituents. At a P(3HB) amount of 50 wt.% a co-continuous morphology with a clear separation of the two phases can be seen (Figure 2D). The obtained microstructure depends on polymer molecular structure, composition, the method of blend preparation and also by the surface free energy (Venugopal and Krause, 1992; Walheim et al., 1997). LLDPE could have an average molecular weight higher than that of P(3HB) according to MFI, it is reasonable to consider that higher molecular weight polymers experiences a larger entropy penalty at the surface in respect of the lower molecular weight ones so the blend surface is expected to be depleted in LLDPE and enriched in P(3HB) as results of reduced conformational entropy (Wattenbarger et al., 1990; Bower, 2002). Moreover, the amplitude of entropically driven surface segregation is found to be proportional to the backbone density of substitute and small differences in this value could lead to strong surface segregation (Wu and Fredrickson, 1996). Therefore, the linear polymer, i.e., LDPE, concentration is expected to increase moving from the surface to the bulk respect to the concentration of the thicker “microbranched” polymer, i.e., P(3HB).

Figure 2. SEM micrographs of the cryofractured surface of (A) neat LLDPE, (B) LLDPE _90_PHB_10, (C) LLDPE _80_PHB_20, (D) LLDPE _50_PHB_50.
FT-IR spectra of neat matrices [i.e., LLDPE and P(3HB)] and of their relative blends are reported in Figure 3. Neat LLDPE sample shows characteristic peaks related to the antisymmetric (2,916 cm−1) and symmetric (2,848 cm−1) stretching vibrations of CH of saturated hydrocarbons and to the bending of the CH at 1,463 cm−1 (Gulmine et al., 2002). Increasing the amount of P(3HB) it is possible to observe the characteristic peak of this polyester, i.e., the stretching vibration of the C=O at 1,720 cm−1 (Padermshoke et al., 2005). From the FT-IR spectra of the blended samples it is not possible to detect any reflection different from those typical of the neat constituents, meaning that the two polymers are not miscible and that no physical and/or chemical interaction is present between the two polymer phases. Quite interestingly, the spectra of the LLDPE_50_PHB_50 is very similar to that of the neat PHB. According to the SEM images (see Figure 2D) and the previous considerations, it can be assumed that at elevated P(3HB) concentrations a superficial film of P(3HB) is formed on the samples, thus hiding the IR signal of the polyolefin phase.
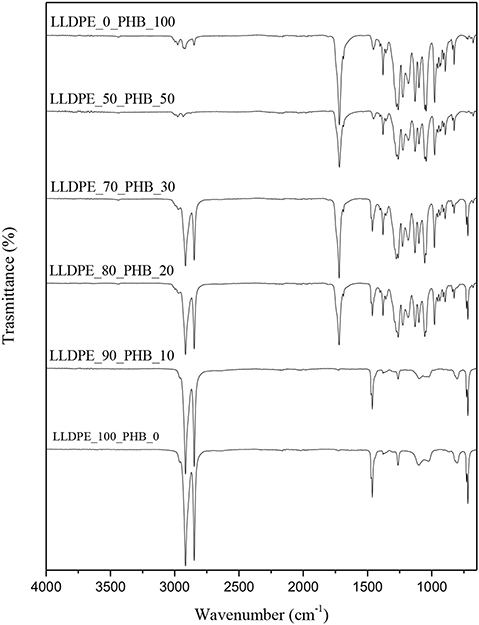
Figure 3. FT-IR spectra of neat LLDPE, neat P(3HB), and relative LLDPE/P(3HB) blends.
Thermal properties of the neat matrices and of the blended samples were investigated by using differential scanning calorimetry (DSC). In Figures 4A,B, representative DSC thermograms for the first heating and the cooling cycle are shown, while in Table 1 the most important results in terms melting temperature (Tm), crystallization temperature (Tc), and the degree of crystallinity (Xc) are summarized.
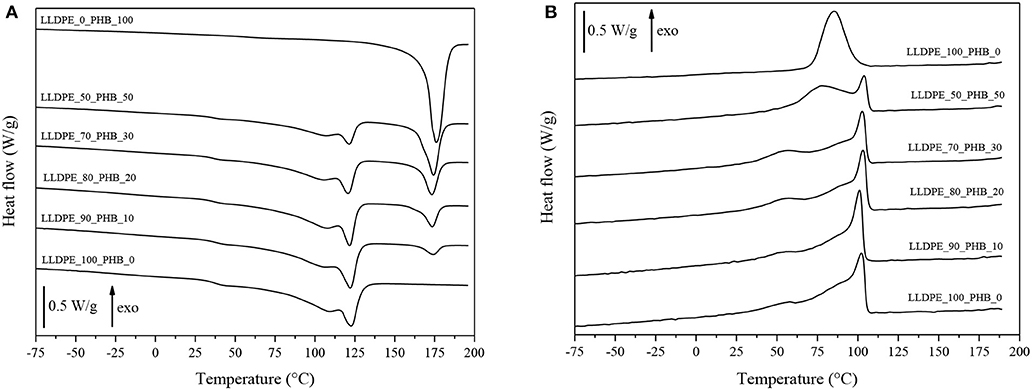
Figure 4. DSC thermograms of neat LLDPE, neat P(3HB), and relative LLDPE/P(3HB) blends. (A) First heating and (B) cooling scan.
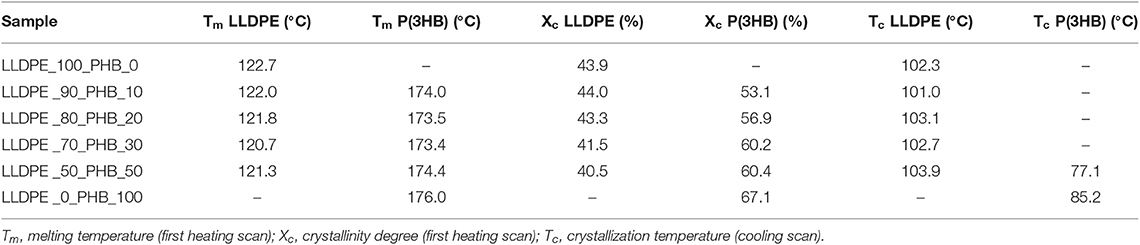
Table 1. Results of DSC tests neat LLDPE and relative LLDPE/P(3HB) blends (first heating scan).
Melting peaks of LLDPE and P(3HB) are clearly distinguishable in DSC thermograms collected during the first heating scan, and the intensity of these peaks is proportional to the relative amounts of the constituents within the blends. In the cooling scan, the crystallization peak of P(3HB) can be detected only for PHB contents higher than 50%, while for lower P(3HB) amounts it is hidden by the crystallization peak of the LLDPE constituent. From the results reported in Table 1 it can be concluded that the melting and the crystallization temperature of both the LLDPE and the PHB phases seem to be substantially unaffected by the relative composition of the blends. The same can be said for the crystallinity content of the LLDPE. Quite interestingly, the presence of the LLDPE in the blends seems to hinder the crystallization process of the PHB phase, lowering thus the Xc values, especially at elevated LLDPE contents. Further studies will be required to have a better comprehension of this aspect. However, also from DSC thermograms it can be concluded that the interaction between the two polymer constituents in the blends is rather limited, and immiscible blends are formed all the tested compositions.
Thermogravimetric analysis (TGA) was then performed, in order to evaluate the thermal stability of the LLDPE/P(3HB) blends. This is a very important point, due to the well-known processability limits of P(3HB) (Yeo et al., 2017). The curves of the weight loss as a function of the temperature and their corresponding derivative curves are presented in Figures 5A,B.
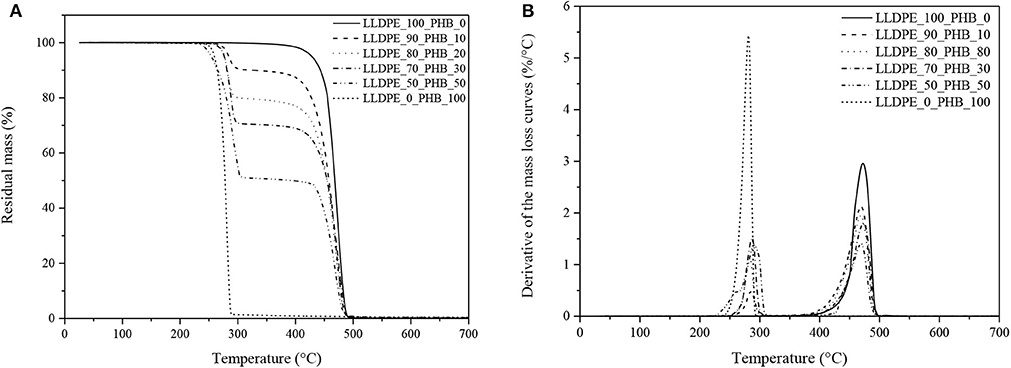
Figure 5. Thermogravimetric analysis on neat LLDPE, neat P(3HB) and relative LLDPE/P(3HB) blends. (A) Residual mass and (B) derivative of the mass loss curves.
The curves show two distinct degradation steps, relative to P(3HB) and LLDPE phases, respectively. LLDPE starts to degrade at temperatures higher than 400°C, while P(3HB) starts to thermally decompose at temperatures higher than 240°C. Considering that the melting temperature of P(3HB) from DSC tests resulted to be at 176°C (see Table 1), it can be concluded that the processability window of these blends, even if rather restricted, is still suitable for the production of extruded films for packaging applications. The degradation temperature of the LLDPE and P(3HB) phases within the blends, associated to the maximum mass loss rate, is respectively located at around 440 and 270°C, and is not substantially influenced by the relative concentration of the constituents in the blends. This confirms again the limited interaction between the two polymer phases. It is also interesting to notice that the residual mass at 700°C is very near to zero for all the tested compositions, meaning that both the neat samples and the blended specimens are completely decomposed into gaseous products even under an inert atmosphere. Due to the narrow processability window of P(3HB), the compounding temperature was set at 190°C, in order to guarantee the complete melting of the P(3HB) and minimize the risk of its thermal degradation.
DMA tests were performed to evaluate the viscoelastic properties of the blends and their thermal transitions. DMA curves showing the trends of the storage modulus (E′) and of the loss tangent (tanδ) are shown in Figures 6A,B. At a general level, it can be seen that the progressive introduction of the P(3HB) within the blends leads to an important stiffening effect, with an increase of the storage modulus and a corresponding lowering of the tanδ values.
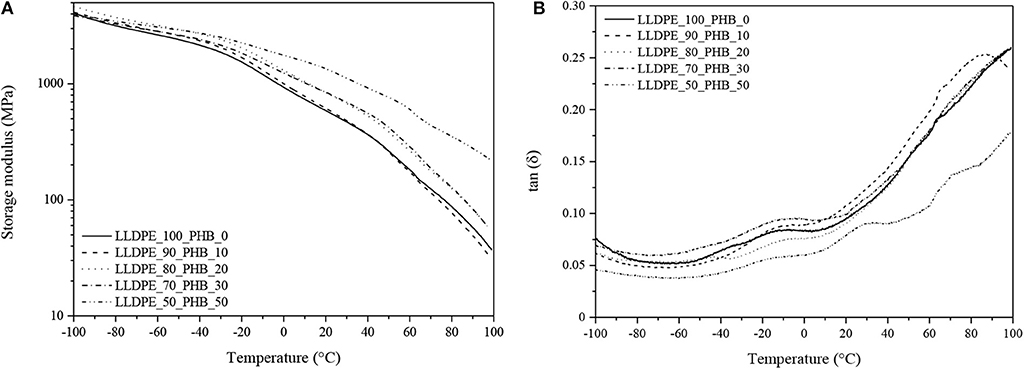
Figure 6. Dynamical mechanical analysis of neat LLDPE, neat P(3HB) and relative LLDPE/P(3HB) blends (frequency = 1 Hz). (A) Storage modulus and (B) loss tangent curves.
Differently from DSC thermograms, in DMA curves of the neat P(3HB) sample it is possible to detect the presence of the Tg, located at around 30°C (see tanδ peak in Figure 6B). This signal is still visible in the LLDPE_50_PHB_50 blends, while for lower PHB contents it practically disappears. Therefore, the introduction of a relatively stiff polymer like P(3HB) can increase the dimensional stability of the blends at ambient temperature, but for temperatures above 30°C (i.e., higher than the Tg of PHB) a strong decrease of the dimensional stability of the material can occur in the blends at elevated PHB amounts. This issue must be taken in serious consideration for the intended application of the blends as food packaging films.
Representative stress-strain curves of neat LLDPE and of the relative blends at different PHB amounts are reported in Figure 7, while in Table 2 the results of the most important tensile properties are summarized.
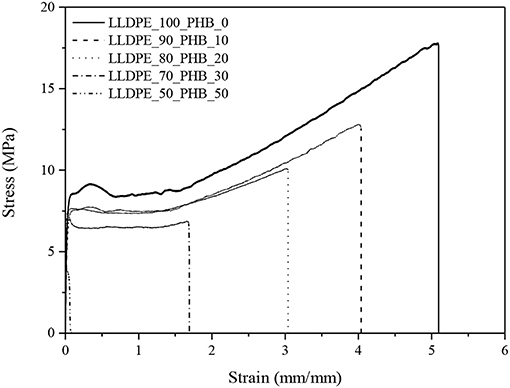
Figure 7. Representative stress-strain curves from quasi-static tensile tests of neat LLDPE and relative LLDPE/P(3HB) blends.
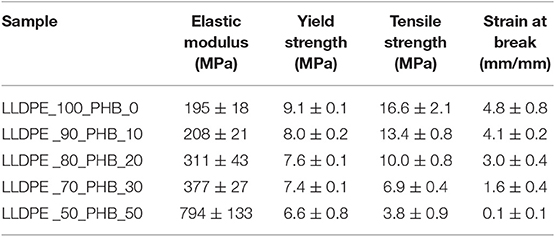
Table 2. Results of quasi-static tensile tests on neat LLDPE and relative LLDPE/P(3HB) blends.
In these tests, it was not possible to obtain the values of the tensile properties of neat P(3HB), because it was too brittle. According to DMA analysis, the progressive introduction of a stiff polymer like P(3HB) leads to a remarkable increase of the elastic modulus (E). For instance, with a P(3HB) amount of 30 wt.% it is possible to double the original stiffness of the LLDPE matrix, and raising the P(3HB) concentration to 50 wt.% the elastic modulus is even four times higher. As a drawback, ultimate elongation values (εb) are strongly reduced. Even if until a P(3HB) concentration of 20 wt.% the observed εb drop is not dramatic, a harsher decrease can be seen at elevated P(3HB) loadings. Similar results regarding the elastic modulus and the elongation at break were found by Burlein and Rocha, the stiffness of LDPE has been doubled with an addition of 30% of P(3HB) and a drop in the elongation at break from 100 to 16% have been reported increasing the concentration from 20 to 30% (Burlein and Rocha, 2014). The same considerations are valid also for the tensile strength (σb). It is also interesting to notice that also the yield strength values (σy) are progressively reduced upon the PHB addition. According to the indication reported in our previous works of polyolefin based nanocomposites (Dorigato et al., 2012), an enhancement of the yield strength is generally related to a rather strong filler-matrix interaction, otherwise σy would decrease. In fact, in polyolefins filled with traditional microfillers (talk, mica, calcium carbonate) the increase in the stiffness of the material is generally accompanied to an heavy drop of the yield stress, because these fillers do not bear the load in the direction of deformation (Ahmed and Jones, 1990; Nielsen and Landel, 1994; Galeski, 2003). Thus, the low degree of miscibility and the rather limited interfacial interaction between the two polymeric constituents of these blends detected in microstructural and thermal analysis seems to be supported by the observed drop of the yield (and also of the failure) tensile properties.
Hydrolytic degradation tests were finally performed, in order to evaluate the influence of the PHB introduction on the thermo-mechanical behavior of the blends at different hydrolysis treatment times. In these tests, the characterization activity was focused on the evaluation of the variation of the weight, of the crystallinity degree and of the quasi-static tensile performances (normalized with respect to the mechanical properties before the treatment). The trends of these properties for the different blends formulations as a function of the treatment time is graphically represented in Figures 8A–D. From weight variation curves (see Figure 8A) it is possible to notice that the weight gain is more intense in the first 10 days of hydrolytic treatment, while a stabilization occurs for longer times. It can be generally concluded that the weight gain is rather limited for all the tested compositions, and only with a P(3HB) content of 50 wt.% a maximum weight variation of 0.25% can be registered. For as concerns the thermal properties, it can be seen from Figure 8B that the variation of the LLDPE crystallinity degree is completely absent or rather limited at all the PHB contents, without any clear correlation with the treatment duration and/or the P(3HB) concentration.
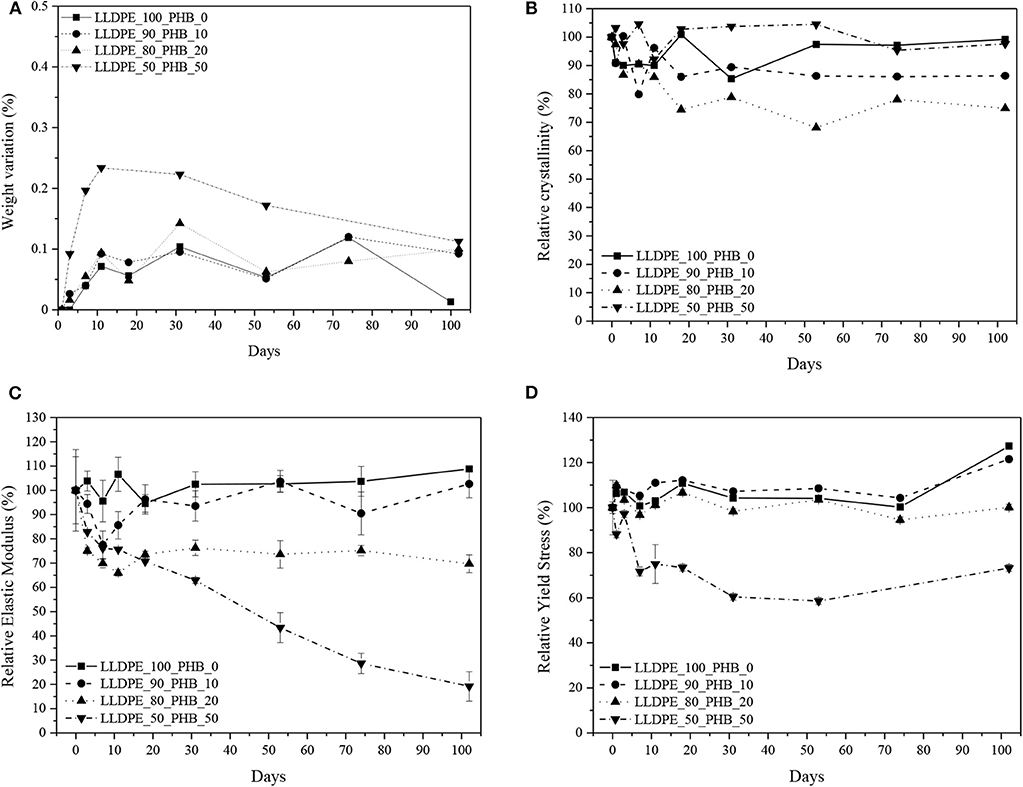
Figure 8. Results of hydrolytic degradation tests on neat LLDPE and relative LLDPE/P(3HB) blends. (A) Weight variation, (B) relative crystallinity degree of LLDPE, (C) relative elastic modulus, (D) relative yield stress. Error bars represent the standard deviation.
More interesting information can be obtained from the analysis of the mechanical properties. From the trends of the relative elastic modulus reported in Figure 8C, it can be seen that the introduction of P(3HB) determines a fast lowering of the stiffness of the material in the first 10 days of hydrolysis, while for longer times the observed decrease is less intense. The drop of the stiffness of the material seems to be proportional to the P(3HB) amount. For instance, the relative variation of the elastic modulus after 100 days with a P(3HB) content of 20 wt.% is about 30%, while increasing the P(3HB) concentration up to 50 wt.% the relative drop is as high as 80%. This means that after 100 days of hydrolytic treatment the pristine dimensional stability of this blend is completely compromised. A similar trend can be detected considering the stress at yield values (see Figure 8D), even if in this case the observed drop is more limited.
The relative variation of the σy values is practically negligible until a P(3HB) concentration of 20 wt.%, while increasing the P(3HB) amount up to 50 wt.% a σy drop of about 30% can be seen after 100 days. The observed decrease of the mechanical properties at a P(3HB) content of 50 wt.% could be directly correlated to the morphological features of this blend. In fact, SEM micrographs highlighted that at this blend composition a change in the morphology can be observed, with a formation of a continuous P(3HB) phase with layered structure (see Figure 2D). In these conditions, it can be hypothesized that the hydrolytic degradation process can freely take place in the P(3HB) phase, that is not entrapped anymore in the LLDPE matrix. It can be therefore concluded that the prepared blends demonstrate a good hydrolytic degradation resistance until a P(3HB) concentration of 20 wt.%, with a good retention of the original thermo-mechanical properties even after a prolonged treatment.
Conclusions
Different amounts of Poly(3-hydroxybutyrate) P(3HB) were melt compounded with a Linear Low Density Polyethylene (LLDPE) matrix, and the resulting materials were characterized from a microstructural and thermo-mechanical point of view, with the aim to evaluate the possibility to partially replace LLDPE with a novel biobased plastic.
MFI measurements evidenced that the pristine processability of the LLDPE matrix was not substantially affected until a P(3HB) content of 20 wt.%, while for higher P(3HB) amounts an evident drop of the viscosity was observed. SEM micrographs highlighted that the two polymer constituents have a limited miscibility and a scarce interfacial interaction degree, while at a P(3HB) content of 50 wt.% a co-continuous phase can be detected. Also DSC, TGA, and FT-IR measurements did not highlight any chemical or physical interaction between the two polymer phases. A pronounced increase of the stiffness of the blends with the P(3HB) amount was detected through DMA and quasi-static tensile tests, while the low compatibility between the constituents determined a heavy drop of both the yield and the failure tensile properties. Hydrolytic degradation tests, performed over a timeframe of 100 days, showed that the decrease of the mechanical properties in the treated blends was evident only for P(3HB) contents higher than 20 wt.%. It was therefore demonstrated that the substitution of LLDPE with a biobased plastic like P(3HB) at a relative amount of 20 wt.% leads to the development of a material with suitable processability requirements and good mechanical properties, even after a prolonged hydrolytic degradation treatment.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
DR performed most of the experimental activities. AD analyzed the data and wrote the manuscript. AP conceived the project and corrected the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Mr. Andrea Bettega is gratefully acknowledged for his support to the experimental work. The NMR analyses were performed by Dr. Emanuela Callone using the equipment of the Klaus Müller NMR Lab. of the Industrial Engineering Department at the University of Trento.
References
Ahmed, S., and Jones, F. R. (1990). A review of particluate reinforcement theories for polymer composites. J. Mater. Sci. 25, 4933–4942. doi: 10.1007/BF00580110
Barham, P. J., Keller, A., Otun, E. L., and Holmes, P. A. (1984). Crystallization and morphology of a bacterial thermoplastic: poly-3-hydroxybutyrate. J. Mater. Sci. 19, 2781–2794. doi: 10.1007/BF01026954
Bhasney, S. M., Bhagabati, P., Kumar, A., and Katiyar, V. (2019). Morphology and crystalline characteristics of polylactic acid [PLA]/linear low density polyethylene [LLDPE]/microcrystalline cellulose [MCC] fiber composite. Compos. Sci. Technol. 171, 54–61. doi: 10.1016/j.compscitech.2018.11.028
Bower, D. (2002). An Introduction to Polymer Physics. Cambridge: Cambridge University Press. doi: 10.1017/CBO9780511801280
Burlein, G. A. D., and Rocha, M. C. G. (2014). Mechanical and morphological properties of LDPE/PHB blends filled with castor oil pressed cake. Mater. Res. 17, 97–105. doi: 10.1590/S1516-14392013005000196
Chandra, R., and Rustgi, R. (1998). Biodegradable polymers. Prog. Polym. Sci. 23, 1273–1335. doi: 10.1016/S0079-6700(97)00039-7
Chen, G. Q., and Hajnal, I. (2015). The ‘PHAome’. Trends Biotechnol. 33, 559–564. doi: 10.1016/j.tibtech.2015.07.006
Datta, D., and Halder, G. (2019). Effect of media on degradability, physico-mechanical and optical properties of synthesized polyolefinic and PLA film in comparison with casted potato/corn starch biofilm. Process Safety Environ. Protect. 124, 39–62. doi: 10.1016/j.psep.2019.02.002
Dilkes-Hoffman, L. S., Lane, J. L., Grant, T., Pratt, S., Lant, P. A., and Laycock, B. (2018). Environmental impact of biodegradable food packaging when considering food waste. J. Clean. Prod. 180, 325–334. doi: 10.1016/j.jclepro.2018.01.169
Dorigato, A., D'amato, M., and Pegoretti, A. (2012). Thermo-mechanical properties of high density polyethylene - fumed silica nanocomposites: effect of filler surface area and treatment. J. Polymer Res. 19, 9889–9899. doi: 10.1007/s10965-012-9889-2
Dorigato, A., Dzenis, Y., and Pegoretti, A. (2013). Filler aggregation as a reinforcement mechanism in polymer nanocomposites. Mech. Mater. 61, 79–90. doi: 10.1016/j.mechmat.2013.02.004
Dorigato, A., and Pegoretti, A. (2012). Fracture behaviour of linear low density polyethylene - fumed silica composites. Eng. Fract. Mech. 79, 213–224. doi: 10.1016/j.engfracmech.2011.10.014
Dorigato, A., and Pegoretti, A. (2013). (Re)processing effects on linear low-density polyethylene/silica nanocomposites. J. Polymer Res. 20, 92–101. doi: 10.1007/s10965-013-0092-x
Dorigato, A., Pegoretti, A., Fambri, L., Lonardi, C., Slouf, M., and Kolarik, J. (2011). Linear low density polyethylene - cycloolefin copolymer blends. Express Polymer Lett. 5, 23–37. doi: 10.3144/expresspolymlett.2011.4
Dorigato, A., Pegoretti, A., and Kolarik, J. (2010a). Nonlinear tensile creep of linear low density polyethylene/fumed silica nanocomposites: time-strain superposition and creep prediction. Polymer Compos. 31, 1947–1955. doi: 10.1002/pc.20993
Dorigato, A., Pegoretti, A., and Penati, A. (2010b). Linear low-density polyethylene/silica micro- and nanocomposites: dynamic rheological measurements and modelling. Express Polymer Lett. 4, 115–129. doi: 10.3144/expresspolymlett.2010.16
Emadian, S. M., Onay, T. T., and Demirel, B. (2017). Biodegradation of bioplastics in natural environments. Waste Manag. 59, 526–536. doi: 10.1016/j.wasman.2016.10.006
Galeski, A. (2003). Strength and toughness of crystalline polymer systems. Prog. Polym. Sci. 28, 1643–1699. doi: 10.1016/j.progpolymsci.2003.09.003
Geissdoerfer, M., Savaget, P., Bocken, N. M. P., and Hultink, E. J. (2017). The Circular Economy – A new sustainability paradigm? J. Clean. Prod. 143, 757–768. doi: 10.1016/j.jclepro.2016.12.048
Geueke, B., Groh, K., and Muncke, J. (2018). Food packaging in the circular economy: overview of chemical safety aspects for commonly used materials. J. Clean. Prod. 193, 491–505. doi: 10.1016/j.jclepro.2018.05.005
Ghisellini, P., Cialani, C., and Ulgiati, S. (2016). A review on circular economy: the expected transition to a balanced interplay of environmental and economic systems. J. Clean. Prod. 114, 11–32. doi: 10.1016/j.jclepro.2015.09.007
Groh, K. J., Backhaus, T., Carney-Almroth, B., Geueke, B., Inostroza, P. A., Lennquist, A., et al. (2019). Overview of known plastic packaging-associated chemicals and their hazards. Sci. Total Environ. 651, 3253–3268. doi: 10.1016/j.scitotenv.2018.10.015
Gulmine, J. V., Janissek, P. R., Heise, H. M., and Akcelrud, L. (2002). Polyethylene characterization by FTIR. Polym. Test. 21, 557–563. doi: 10.1016/S0142-9418(01)00124-6
Hahladakis, J. N., and Iacovidou, E. (2018). Closing the loop on plastic packaging materials: what is quality and how does it affect their circularity? Sci. Total Environ. 630, 1394–1400. doi: 10.1016/j.scitotenv.2018.02.330
Hancox, N. J. (1992). Introduction to Industrial Polymers, 2nd Edn. Munich: Hanser. doi: 10.1016/0261-3069(93)90097-F
Hole, G., and Hole, A. S. (2019). Recycling as the way to greener production: a mini review. J. Clean. Prod. 212, 910–915. doi: 10.1016/j.jclepro.2018.12.080
Kalmykova, Y., Sadagopan, M., and Rosado, L. (2018). Circular economy – from review of theories and practices to development of implementation tools. Resourc. Conserv. Recyc. 135, 190–201. doi: 10.1016/j.resconrec.2017.10.034
Krupa, I., and Luyt, A. S. (2000). Thermal properties of uncross-linked and cross-linked LLDPE/wax blends. Polym. Degrad. Stab. 70, 111–117. doi: 10.1016/S0141-3910(00)00097-5
Krupa, I., and Luyt, A. S. (2001). Thermal and mechanical properties of extruded LLDPE/wax blends. Polym. Degrad. Stab. 73, 157–161. doi: 10.1016/S0141-3910(01)00082-9
Laycock, B., Nikolić, M., Colwell, J. M., Gauthier, E., Halley, P., Bottle, S., et al. (2017). Lifetime prediction of biodegradable polymers. Prog. Polym. Sci. 71, 144–189. doi: 10.1016/j.progpolymsci.2017.02.004
Nguyen, D. M., Do, T. V. V., Grillet, A. C., Ha Thuc, H., and Ha Thuc, C. N. (2016). Biodegradability of polymer film based on low density polyethylene and cassava starch. Int. Biodeterior. Biodegrad. 115, 257–265. doi: 10.1016/j.ibiod.2016.09.004
Niaounakis, M. (2019). Recycling of biopolymers – the patent perspective. Eur. Polym. J. 114, 464–475. doi: 10.1016/j.eurpolymj.2019.02.027
Nielsen, L. E., and Landel, R. F. (1994). Mechanical Properties of Polymers and Composites. New York, NY: Dekker.
Ol'khov, A. A., Iordanskii, A. L., Zaikov, G. E., Shibryaeva, L. S., Litvinov, I. A., and Vlasov, S. V. (2000). Morphologically special features of poly(3-hydroxybutyrate)/low-density polyethylene blends. Polym. Plast. Technol. Eng. 39, 783–792. doi: 10.1081/PPT-100101403
Padermshoke, A., Katsumoto, Y., Sato, H., Ekgasit, S., Noda, I., and Ozaki, Y. (2005). Melting behavior of poly(3-hydroxybutyrate) investigated by two-dimensional infrared correlation spectroscopy. Spectrochim. Acta Part A 61, 541–550. doi: 10.1016/j.saa.2004.05.004
Pakalapati, H., Chang, C. K., Show, P. L., Arumugasamy, S. K., and Lan, J. C. W. (2018). Development of polyhydroxyalkanoates production from waste feedstocks and applications. J. Biosci. Bioeng. 126, 282–292. doi: 10.1016/j.jbiosc.2018.03.016
Pankova, Y. N., Shchegolikhin, A. N., Iordanskii, A. L., Zhulkina, A. L., Ol'khov, A. A., and Zaikov, G. E. (2010). The characterization of novel biodegradable blends based on polyhydroxybutyrate: the role of water transport. J. Mol. Liq. 156, 65–69. doi: 10.1016/j.molliq.2010.04.018
Patel, R. M. (2016). “2 - Polyethylene,” in Multilayer Flexible Packaging, 2nd Edn, ed J. R. Wagner (William Andrew Publishing), 17–34. doi: 10.1016/B978-0-323-37100-1.00002-8
Pedroso, A. G., and Rosa, D. S. (2005). Mechanical, thermal and morphological characterization of recycled LDPE/corn starch blends. Carbohydr. Polym. 59, 1–9. doi: 10.1016/j.carbpol.2004.08.018
Ragaert, P., Buntinx, M., Maes, C., Vanheusden, C., Peeters, R., Wang, S., et al. (2019). “Polyhydroxyalkanoates for food packaging applications,” in Reference Module in Food Science (Elsevier). doi: 10.1016/B978-0-08-100596-5.22502-X
Restrepo-Flórez, J. M., Bassi, A., and Thompson, M. R. (2014). Microbial degradation and deterioration of polyethylene – A review. Int. Biodeterior. Biodegrad. 88, 83–90. doi: 10.1016/j.ibiod.2013.12.014
Rocha, M. C. G., and Moraes, L. R. D. C. (2015). Low density polyethylene (LDPE) blends based on Poly(3-Hydroxi-Butyrate) (PHB) and Guar Gum (GG) biodegradable polymers. Polímeros 25, 42–48. doi: 10.1590/0104-1428.1495
Rosa, D. D. S., Gaboardi, F., Guedes, C. D. G. F., and Calil, M. R. (2007). Influence of oxidized polyethylene wax (OPW) on the mechanical, thermal, morphological and biodegradation properties of PHB/LDPE blends. J. Mater. Sci. 42, 8093–8100. doi: 10.1007/s10853-007-1701-z
Soroudi, A., and Jakubowicz, I. (2013). Recycling of bioplastics, their blends and biocomposites: a review. Eur. Polym. J. 49, 2839–2858. doi: 10.1016/j.eurpolymj.2013.07.025
Valentini, F., Dorigato, A., Rigotti, D., and Pegoretti, A. (2019). Polyhydroxyalkanoates/fibrillated nanocellulose composites for additive manufacturing. J. Polym. Environ. 27, 1333–1341. doi: 10.1007/s10924-019-01429-8
Van Krevelen, D. W., and Te Nijenhuis, K. (eds.). (2009). “Chapter 24 - processing properties,” in Properties of Polymers, 4th Edn (Amsterdam: Elsevier), 799–818. doi: 10.1016/B978-0-08-054819-7.00024-8
Veethahavya, K. S., Rajath, B. S., Noobia, S., and Kumar, B. M. (2016). Biodegradation of low density polyethylene in aqueous media. Proc. Environ. Sci. 35, 709–713. doi: 10.1016/j.proenv.2016.07.072
Venugopal, G., and Krause, S. (1992). Development of phase morphologies of poly(methyl methacrylate)-polystyrene-toluene mixtures in electric fields. Macromolecules 25, 4626–4634. doi: 10.1021/ma00044a025
Walheim, S., Böltau, M., Mlynek, J., Krausch, G., and Steiner, U. (1997). Structure formation via polymer demixing in spin-cast films. Macromolecules 30, 4995–5003. doi: 10.1021/ma9619288
Wang, Y., Yin, J., and Chen, G. Q. (2014). Polyhydroxyalkanoates, challenges and opportunities. Curr. Opin. Biotechnol. 30, 59–65. doi: 10.1016/j.copbio.2014.06.001
Wattenbarger, M. R., Chan, H. S., Evans, D. F., and Dill, K. A. (1990). Surface-induced enhancement of internal structure in polymers and proteins. J. Chem. Phys. 93, 8343–8351. doi: 10.1063/1.459317
Wu, D. T., and Fredrickson, G. H. (1996). Effect of architecture in the surface segregation of polymer blends. Macromolecules 29, 7919–7930. doi: 10.1021/ma9602278
Keywords: polyethylene, polyhydroxyalkanoates, blends, mechanical properties, degradation
Citation: Rigotti D, Dorigato A and Pegoretti A (2020) Thermo-Mechanical Behavior and Hydrolytic Degradation of Linear Low Density Polyethylene/Poly(3-hydroxybutyrate) Blends. Front. Mater. 7:31. doi: 10.3389/fmats.2020.00031
Received: 24 October 2019; Accepted: 30 January 2020;
Published: 18 February 2020.
Edited by:
Alfonso Maffezzoli, University of Salento, ItalyReviewed by:
Debora Puglia, University of Perugia, ItalyMauro Zammarano, National Institute of Standards and Technology (NIST), United States
Copyright © 2020 Rigotti, Dorigato and Pegoretti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniele Rigotti, ZGFuaWVsZS5yaWdvdHRpLTFAdW5pdG4uaXQ=; Alessandro Pegoretti, YWxlc3NhbmRyby5wZWdvcmV0dGlAdW5pdG4uaXQ=