 Zuocheng Wang
Zuocheng Wang Eirini-Maria Paschalidou
Eirini-Maria Paschalidou Vincent Maurice
Vincent Maurice- PSL Research University, CNRS-Chimie ParisTech, Institut de Recherche de Chimie Paris, Research Group Physical Chemistry of Surfaces, Paris, France
An approach preventing contact to ambient air during transfer from liquid environment for electrochemical treatment to UHV environment for surface analysis by X-Ray Photoelectron Spectroscopy and Time-of-Flight Secondary Ion Mass Spectrometry was applied to study the mechanisms of Cr and Mo enrichments in the passive oxide film formed on 316L austenitic stainless steel. Starting from the air-formed native oxide-covered surface, exposures were conducted in aqueous sulfuric acid solution first at open circuit potential and then under anodic polarization in the passive range. At open circuit potential the thickness of the bi-layered oxide film was observed to decrease and the enrichments of both Cr(III) and Mo, mostly Mo(VI), to markedly increase as well as the film hydroxylation. This is due to preferential dissolution of the Fe(III) oxide/hydroxide, not compensated by oxide growth in the absence of an electric field established by anodic polarization. Anodic polarization in the passive domain causes the bi-layered structure of the oxide film to re-grow by oxidation of iron, chromium and molybdenum, without impacting the Cr enrichment and only slightly mitigating the Mo enrichment. De-hydroxylation of the inner layer is also promoted upon anodic polarization. These results show that the treatment of the surface oxide film in acid solution at open circuit potential enhances Cr and Mo enrichments and promotes hydroxylation. Passivation by anodic polarization allows dehydroxylation, yielding more Cr oxide, without markedly affecting the Mo enrichment, also beneficial for the corrosion resistance.
Introduction
Stainless steels (SS) are important technological materials of widespread application that combine excellent mechanical and corrosion properties. The high corrosion resistance is provided by the passive film, a surface oxide/hydroxide layer that is continuous and protective and does not exceed a few nanometers in thickness when formed at ambient temperature, as observed on Fe-Cr ferritic (Hashimoto et al., 1979; Seo and Sato, 1979; Hultquist et al., 1987; Mitchell and Graham, 1987; Marcus and Olefjord, 1988; Mischler et al., 1988, 1991; Calinski and Strehblow, 1989; Castle and Qiu, 1989a,b; Kirchheim et al., 1989; Yang et al., 1994; Haupt and Strehblow, 1995; Tan et al., 1995; Maurice et al., 1996; Oblonsky et al., 1998; Hamm et al., 2002; Schmuki, 2002; Keller and Strehblow, 2004) and Fe-Cr-Ni austenitic (Sugimoto and Sawada, 1977; Ogawa et al., 1978; Olefjord, 1980; Olefjord and Elfström, 1982; Yang et al., 1984; Olefjord et al., 1985; Brooks et al., 1986; Clayton and Lu, 1986, 1989; Lu et al., 1989; Olefjord and Wegrelius, 1990; De Vito and Marcus, 1992; Elbiache and Marcus, 1992; Habazaki et al., 1992; Macdonald, 1992; Olsson and Hörnström, 1994; Hakiki et al., 1998; Maurice et al., 1998, 2015; Bojinov et al., 2001; Yamamoto et al., 2009) stainless steels. The markedly Cr(III)-enriched composition of the passive film is a key factor for the corrosion resistance. In aqueous environments, it results from the competitive oxidation of iron and chromium with faster dissolution of Fe(II)/Fe(III) compared to Cr(III) species, especially in acid solutions (Keller and Strehblow, 2004).
On Fe-Cr-Ni austenitic stainless steels, the passive films contain no or very little Ni(II) species and the metallic alloy region underneath the oxide is enriched in nickel (Olefjord and Elfström, 1982; De Vito and Marcus, 1992; Olsson and Hörnström, 1994; Hakiki et al., 1998; Maurice et al., 1998, 2015; Bojinov et al., 2001; Yamamoto et al., 2009). SS grades alloyed with a few at% Mo better resist localized corrosion by pitting in chloride-containing environments. Their passive films are slightly enriched in Mo(IV) or Mo(VI) species with no alterations of the thickness (Olefjord and Elfström, 1982; Castle and Qiu, 1989a; Lu et al., 1989; Olefjord and Wegrelius, 1990; De Vito and Marcus, 1992; Hakiki et al., 1998). It has been proposed that the presence of molybdenum would mitigate the breakdown of the passive film preceding the initiation of localized corrosion (Sugimoto and Sawada, 1977; Olefjord, 1980; Olefjord and Elfström, 1982; Brooks et al., 1986; Clayton and Lu, 1986, 1989; Castle and Qiu, 1989a,b; Lu et al., 1989; Macdonald, 1992; Tan et al., 1995; Bojinov et al., 2001) or promote the passive film repair after breakdown (Ogawa et al., 1978; Hashimoto et al., 1979; Olefjord, 1980; Olefjord and Elfström, 1982; Yang et al., 1984).
More recently, it has been reported that the Cr(III) enrichment in the passive film may not be homogeneous at the nanometer scale (Massoud et al., 2014; Maurice et al., 2015) and that the heterogeneities of the Cr local distribution may be responsible for the passive film breakdown leading to the local failure of the corrosion resistance (Maurice and Marcus, 2018). The mechanisms of formation of the passive film include a pre-passivation stage during which a native oxide film is formed, most often in ambient air, and a subsequent stage where the native oxide film is altered, once exposed to the aqueous environment, to become the passive film. Hence, a thorough investigation of the Cr(III) enrichment mechanisms is necessary to develop the comprehensive knowledge on how to improve the resistance to local failure of passivity. It must address the initial stages of oxidation leading to pre-passivation of the SS surface (Ma et al., 2018, 2019) as well as the alterations of the native oxide film brought by immersion in the aqueous environment and by electrochemical anodic polarization in the passive range (Wang et al., 2019).
In the present study, we addressed the mechanisms by which the Cr and Mo enrichments in the native surface oxide film are modified when exposing a polycrystalline austenitic 316L SS to an acid aqueous solution. Time-of-Flight Secondary Ion Mass Spectroscopy (ToF-SIMS) elemental depth profile analysis and X-ray Photoelectron Spectroscopy (XPS) compositional surface analysis were applied to interrogate the modifications induced by immersion without and with application of anodic polarization in the passive domain. An experimental protocol avoiding contact to ambient air of the samples during transfer from liquid environment for electrochemical treatment to the different UHV platforms for surface analysis was adopted, enabling us to highlight the key effect of immersion under open circuit conditions on the Cr and Mo enrichments.
Experimental
Polycrystalline 316L austenitic SS samples of bulk composition Fe−19Cr−13Ni−2.7Mo (wt%) (Fe−20Cr−12Ni−1.6Mo at%) were used. The surfaces were prepared by mechanical polishing first with emery paper of successive 1,200 and 2,400 grades and then with diamond suspensions of successive 6, 3, 1, and 0.25 μm grades. Ultrasonicated baths of acetone, ethanol and Millipore® water (resistivity > 18 MΩ cm) were successively used for cleaning and rinsing after each polishing step. Filtered compressed air was used for drying.
Similarly to approaches developed in the past for surface analytical studies of passivity (Marcus et al., 1980; Olefjord and Elfström, 1982; Haupt et al., 1985), the experimental protocol was designed to avoid ambient air exposure of the electrochemically-treated samples. The samples, as received from surface preparation, were introduced in an Ar-filled (ΔP = ~200 Pa) glove box (Jacomex, France) equipped with a 3-electrode electrochemical cell for electrochemical treatment. The 3-electrode electrochemical cell was controlled by a Gamry 600 potentiostat. It included a Pt grid as counter electrode and saturated calomel reference electrode. The area (0.5 cm2) of the working electrode was delimited by a Viton O-ring. The electrolyte was a 0.05 M H2SO4 aqueous solution prepared from ultrapure chemicals (VWR®) and Millipore® water and bubbled with argon prior to introduction in the glove box. Oxygen and water vapor concentrations in the glove box evolved from <250 and <1,000 ppm after introduction of the electrolyte to <55 and < 200 ppm after performing the electrochemical treatments, respectively.
The samples, covered by the native oxide film formed in air, were first stored under Ar atmosphere, then immersed at open circuit potential (−0.26 ≤ UOCP ≤ −0.22 V/SCE) for 30 min and then passivated by a potential step to UPass = 0.5 V/SCE for 30 min. The selected UPass value corresponded to the minimum of passive current measured by linear sweep voltammetry (Figure 1). No cathodic pre-treatment was performed in order to avoid any reduction-induced alteration of the initial native oxide film exposed at UOCP. After electrochemical treatment, the samples were emerged from the electrolyte at free potential, rinsed with Millipore® water and dried with argon. They were then installed under argon in containers that were air tight sealed for transfer to surface analysis by XPS and ToF-SIMS.
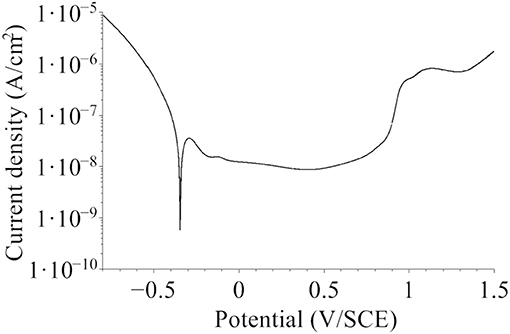
Figure 1. Polarization curve for the native oxide-covered 316L SS sample recorded in 0.05 M H2SO4 in the Ar-filled glove box after initial stabilization at open circuit potential for 30 min (dU/dt = 5 mV/s).
The air tight sealed containers with the samples under Ar atmosphere were transferred from the glove box used for electrochemical treatment to two separate Ar-filled glove boxes, one attached to the XPS spectrometer and the other attached to the ToF-SIMS spectrometer. In these glove boxes, the air tight sealed containers were opened under argon (ΔP = ~30 Pa, [O2] <5 ppm, [H2O] <1 ppm) and the samples mounted on the UHV holders for direct transfer to UHV. The three samples selected for comparative analysis were: (i) the initial native oxide-covered sample, as received from surface preparation and exposed to Ar atmosphere of the glove boxes, (ii) the sample treated at UOCP and (iii) the sample treated at UPass. For each sample, XPS analysis was performed prior to ToF-SIMS depth profile analysis because of the locally destructive nature of SIMS. The air tight sealed containers were used for transfer between the XPS and ToF-SIMS platforms.
XPS analysis was performed with a Thermo Electron ESCALAB 250 spectrometer (VG Scientific, United Kingdom) operating at about 10−9 mbar. The X-ray source was an AlKα monochromatized radiation (hν = 1486.6 eV). Survey spectra were recorded with a pass energy of 100 eV at a step size of 1 eV. High resolution spectra of the Fe 2p, Cr 2p, Ni 2p, Mo 3d, O 1s, S 2p, and C 1s core level regions were recorded with a pass energy of 20 eV at a step size of 0.1 eV. The take-off angle of the analyzed photoelectrons was 90°. The binding energies (BE) were calibrated by setting the C 1s signal corresponding to olefinic bonds (–CH2-CH2-) at 285.0 eV. Spectral reconstruction was performed by curve fitting with the CasaXPS software. Shirley type background subtraction and Lorentzian/Gaussian (70%/30%) peak shapes were used. Asymmetry of the peaks was taken into account for the metallic components (Cr0, Fe0, Mo0, Ni0). Symmetric but broader peak envelopes were used to account for the multiplet splitting of the oxide components (Cr3+, Fe3+, Mo4+/6+).
A ToF-SIMS 5 spectrometer (Ion ToF—Munster, Germany) operated at about 10−9 mbar was used for depth profile elemental analysis. The analysis interlaced topmost surface analysis in static SIMS conditions using a pulsed 25 keV Bi+ primary ion source delivering 1.2 pA current over a 100 × 100 μm2 area with sputtering using a 1 keV Cs+ sputter beam giving a 32 nA target current over a 600 × 600 μm2 area (300 × 300 μm2 for the sample treated at Upass). Analysis was centered inside the eroded crater to avoid edge effects. The profiles were recorded with negative secondary ions which have higher yield for oxide matrices than for metallic matrices. The Ion-Spec software was used for data acquisition and processing.
Results and Discussion
Bi-layered Chemical Structure of the Surface Oxide Films
Figure 2 presents the ToF-SIMS analysis of the elemental in-depth distribution for the native oxide-covered sample. In Figure 2a, the intensities of selected secondary ions characteristic of the oxide film (18O−, , , , and ) and substrate (, , and ) are plotted in logarithmic scale vs. sputtering time. The position of the “modified alloy” region between the oxide film and metallic bulk substrate regions, where Ni is found enriched in agreement with previous studies on austenitic stainless steels (Olefjord and Elfström, 1982; De Vito and Marcus, 1992; Olsson and Hörnström, 1994; Hakiki et al., 1998; Maurice et al., 1998, 2015; Bojinov et al., 2001; Yamamoto et al., 2009; Wang et al., 2019), was defined using the ions intensity profile and placed between 70 and 130 s of sputtering. This region is in principle metallic and does not contain any oxide species in a stratified model of the surface composition. The residual oxide species observed in this region in the ToF-SIMS profile may, at least partially, result from a profiling artifact caused by surface roughness due to the fact that the primary ions and sputtering ions impinge the surface at different angles. In the oxide film region, the most intense profile of the alloying elements are those of the and ions followed by the ions. The ions are the least intense.
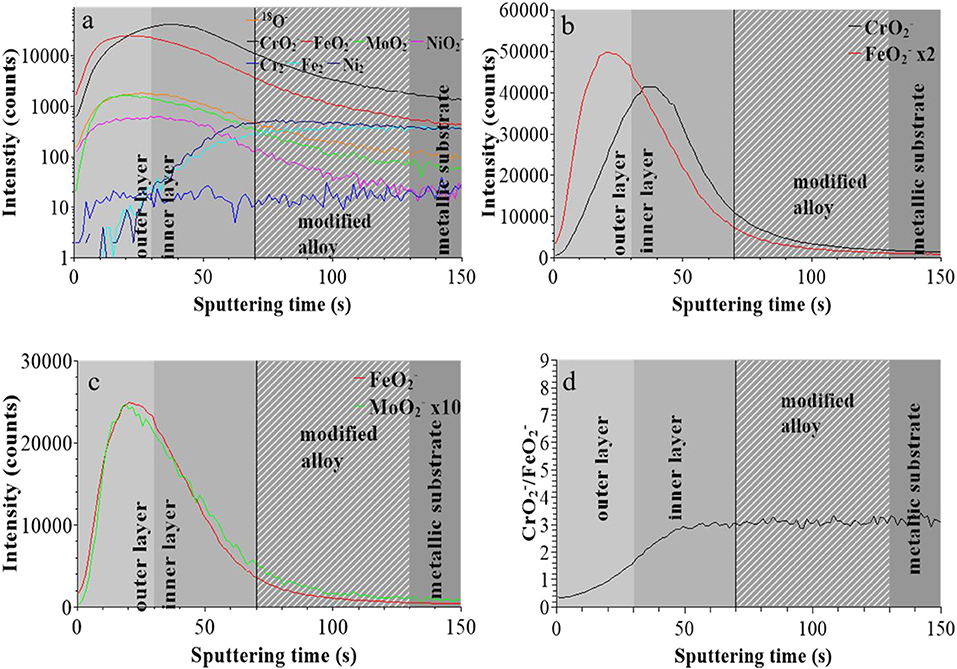
Figure 2. ToF-SIMS depth profiles for the native oxide film on 316L SS: (a) 18O−, , , , , , , and secondary ions, (b) and secondary ions, (c) and secondary ions, (d) / intensity ratio.
Figure 2b shows that the and ions profiles peak at different positions, at 20 and 40 s, respectively. This is consistent with the native oxide film having a bilayer structure with iron and chromium oxides more concentrated in the outer and inner layers, respectively, as commonly reported for oxide films on stainless steels (Marcus and Olefjord, 1988; Mischler et al., 1991; Maurice et al., 2015; Wang et al., 2019). The interface between outer and inner layer was positioned at 30 s which is the median sputtering position between the two intensity maxima (Wang et al., 2019). Figure 2c shows that the ions profile peaks at the same position as the profile, meaning that Fe oxide and Mo oxide are concentrated in the outer layer and confirming previous data for native oxide film on 316L SS (Wang et al., 2019). Figure 2d shows that, starting from the extreme surface, the / intensity ratio continuously increases in the outer and inner parts of the oxide film before reaching saturation in the inner layer.
For the sample treated at UOCP (Figure 3), the “modified alloy” region was positioned between 45 to 110 s using the same method, suggesting a significant thickness decrease of the oxide film. The and intensity maxima are at 12 and 20 s of sputtering, respectively, with the median position at 16 s for the interface between outer and inner layers of the oxide film (Figure 3b). Compared to the native oxide film, the outer layer is much thinner (16 instead of 30 s) and the inner layer less altered (33 instead of 40 s) after treatment at UOCP. The maxima of the and ions profiles are still observed at the same sputtering time in the outer layer, however with a marked decrease in intensity for the ions compared to the ions suggesting the marked preferential loss of iron oxide (Figure 3c). Consistently, the profiles of the / intensity ratio, still increasing from extreme surface to inner part in the oxide film region, is overall higher (~6 vs. ~3 at saturation in the inner layer), suggesting an increase of the Cr enrichment of the oxide film due to iron oxide dissolution induced by immersion in sulfuric acid at open circuit potential.
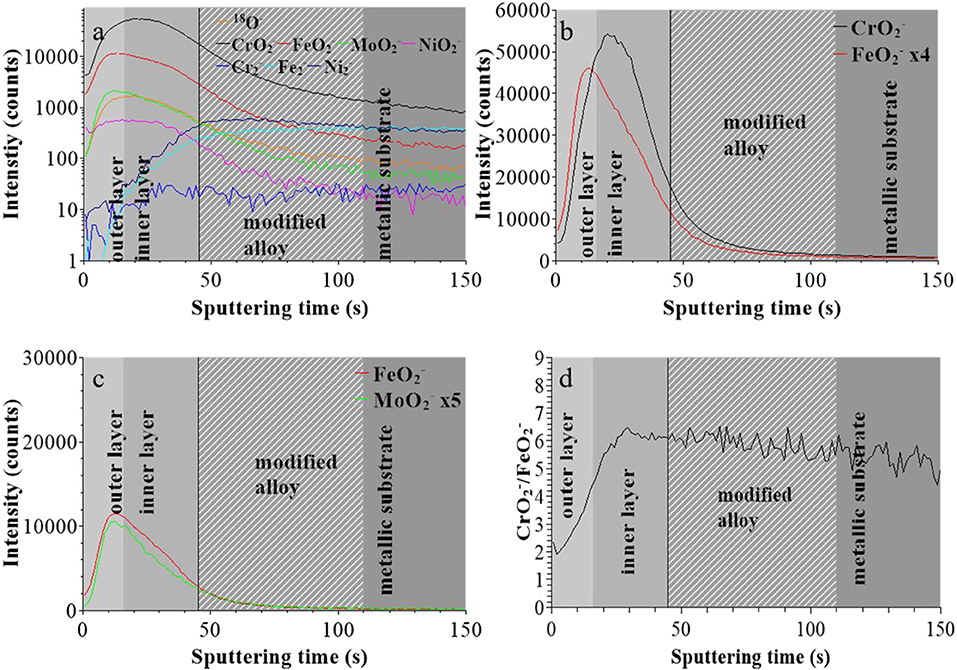
Figure 3. ToF-SIMS depth profiles for 316L SS immersed in 0.05 M H2SO4 at UOCP for 30 min: (a) 18O−, , , , , , , and secondary ions, (b) and secondary ions, (c) and secondary ions, (d) / intensity ratio.
For the sample treated at UPASS (Figure 4), the “modified alloy” region is observed between 18 and 36 s using the same positioning method. However, due to the smaller sputtered area during analysis (300 × 300 μm2 instead of 600 × 600 μm2), the sputtering rate was ~4 times higher in this experiment. After correction, this “modified alloy” region is between 72 and 144 s, which is similar to that of the native oxide-covered sample (70–130 s). Figures 4b,c shows that the bilayer structure persists after passivation with molybdenum concentrated in the outer layer. The interface between outer and inner layer was positioned at 28 s (after correction of the sputtering rate), meaning that the thickness of passive film is quite similar to that of the native oxide film and larger than after treatment at UOCP. The / intensity ratio shows a similar increasing profile than for the other two samples and reaches the value of ~6 at saturation in the inner layer, like for the film after treatment at UOCP and suggesting similar enrichment in Cr in the inner layer of the passive film.
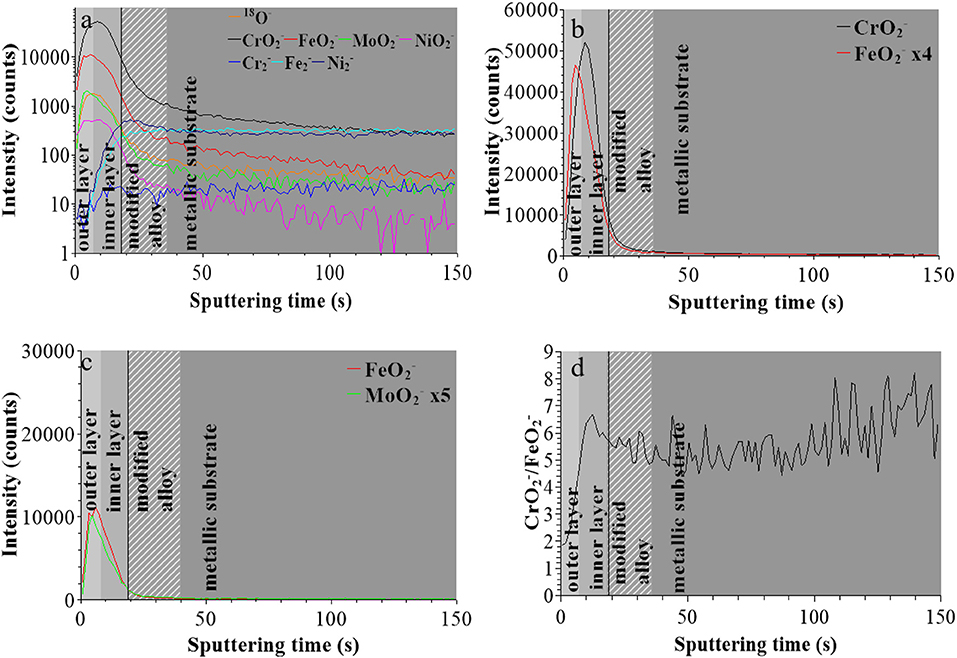
Figure 4. ToF-SIMS depth profiles for 316L SS passivated in 0.05 M H2SO4 at UPASS = 0.5 V/SCE for 30 min: (a) 18O−, , , , , , , and secondary ions, (b) and secondary ions, (c) and secondary ions, (d) / intensity ratio.
These data confirm that the in-depth chemical structure of the surface oxide films formed on 316L SS is bi-layered. This duplex structure is already formed in the initial native oxide film formed in air and persists after treatment at UOCP and UPass. Chromium oxide accumulates in the inner part to form the barrier layer of the oxide film whereas iron and molybdenum oxide accumulates in the outer part of the film that can be described as the exchange layer in contact with the environment. In sulfuric acid, the observed changes are the positions of the interfaces, reflecting the thickness variations of the oxide films, and the intensity ratios of the secondary ions characteristic of the oxidized alloying elements present in the films and reflecting the variations of the Cr and Mo enrichments induced by immersion and anodic passivation. Iron oxide preferentially dissolves but this is partially counter-balanced by anodic oxidation. The XPS data presented hereafter allow us to quantitatively discuss these electrochemically induced thickness and compositional changes.
Thickness and Composition of the Surface Oxide Films
The XPS Cr 2p3/2, Fe 2p3/2, Mo 3d, and O 1s core level spectra recorded for the native oxide-covered sample are shown in Figure 5 and those measured after treatment at UOCP and UPass are shown in Figures 6, 7, respectively. Table 1 compiles the values of the binding energies (BE), full widths at half maximum (FWHM) and relative intensities of the component peaks used for reconstruction of the experimental curves by curve fitting. It also includes the measured Ni 2p3/2, S 2s, and S 2p peak components.
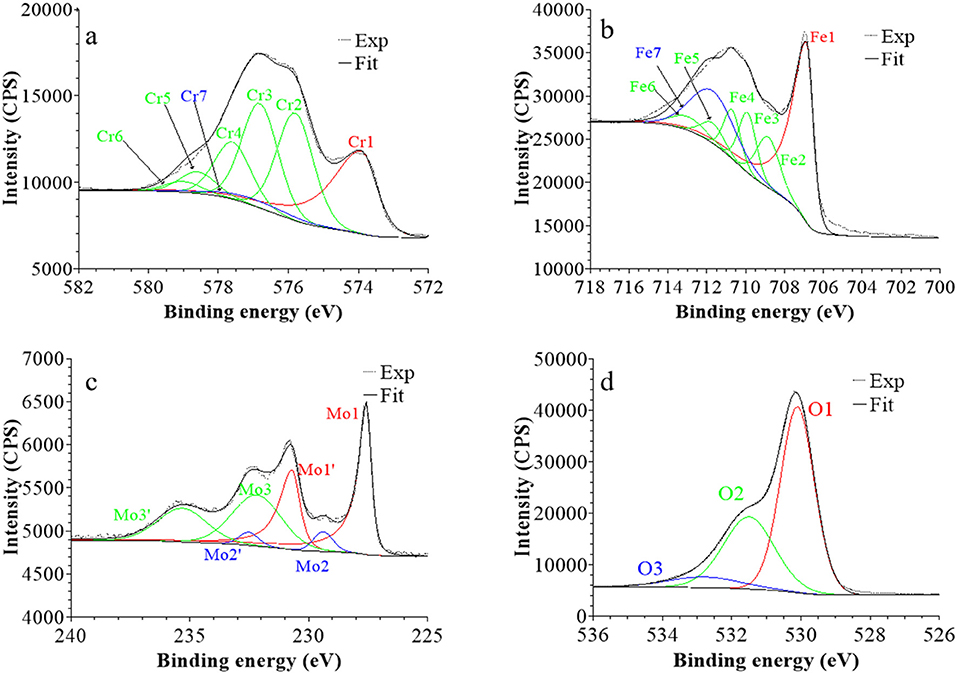
Figure 5. XPS core level spectra and their reconstruction for the native oxide film on 316L SS: (a) Cr 2p3/2, (b) Fe 2p3/2, (c) Mo 3d5/2−3/2, and (d) O 1s regions (take-off angle: 90°).
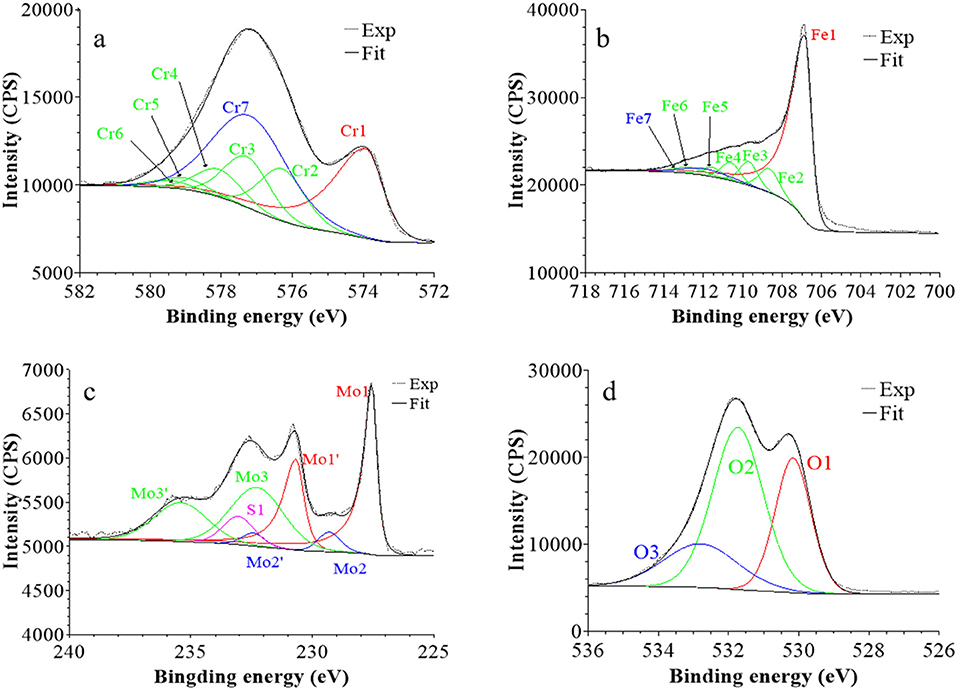
Figure 6. XPS core level spectra and their reconstruction for 316L SS immersed in 0.05 M H2SO4 at UOCP for 30 min: (a) Cr 2p3/2, (b) Fe 2p3/2, (c) Mo 3d5/2−3/2, and (d) O 1s regions (take-off angle: 90°).
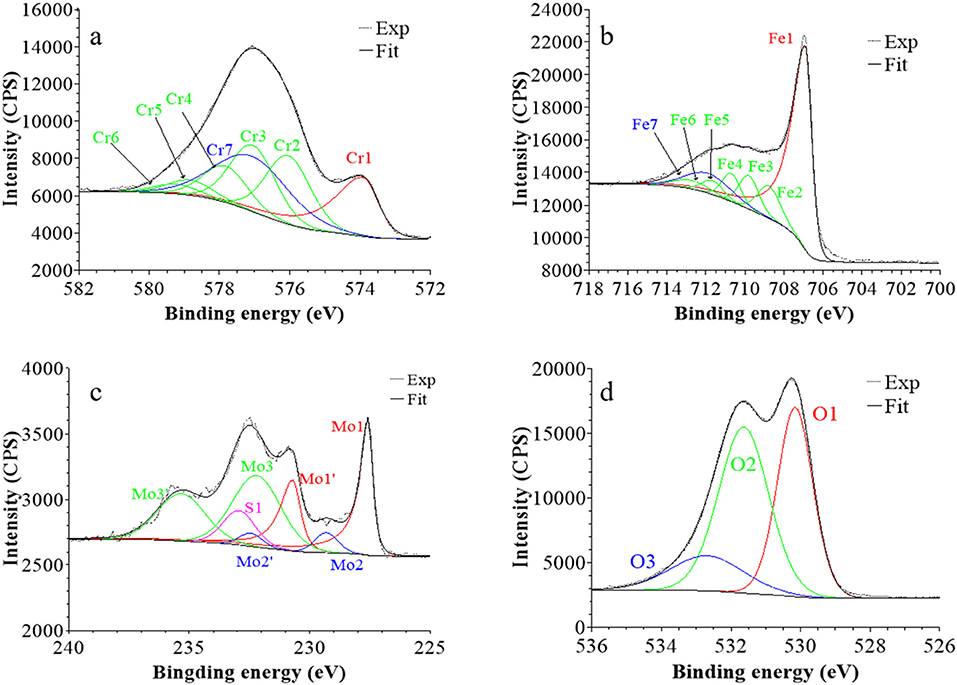
Figure 7. XPS core level spectra and their reconstruction for 316L SS passivated in 0.05 M H2SO4 at UPASS = 0.5 V/SCE for 30 min: (a) Cr 2p3/2, (b) Fe 2p3/2, (c) Mo 3d5/2−3/2, and (d) O 1s regions (take-off angle: 90°).
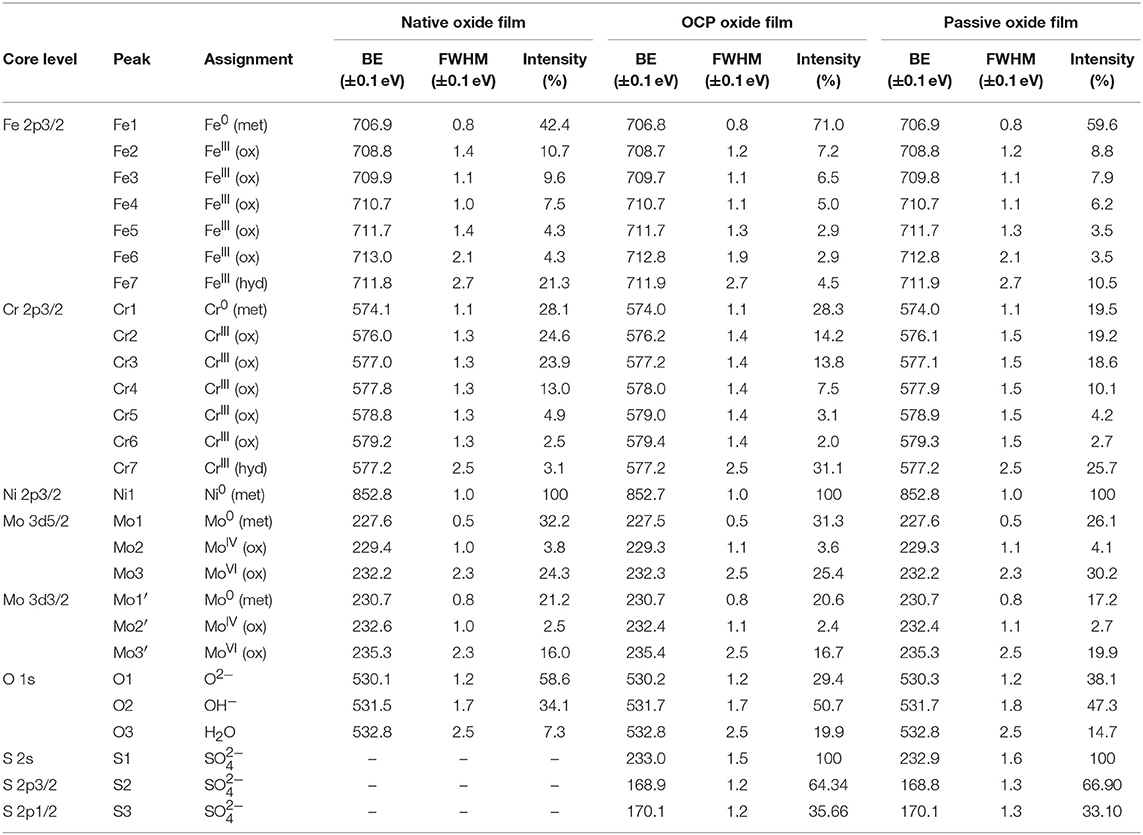
Table 1. BE, FWHM, and relative intensity values of the components measured by XPS on polycrystalline 316L SS after formation in ambient air of the native oxide film, after immersion for 30 min at OCP in 0.05 M H2SO4, and after passivation for 30 min at 0.5 V/SCE in 0.05 M H2SO4.
Following a previously proposed procedure (Yamashita and Hayes, 2008; Biesinger et al., 2011; Wang et al., 2019), the Fe 2p3/2 spectra were fitted with seven peaks, as exemplified in Figure 5 and detailed in Table 1. The first peak (Fe1) corresponds to metallic Fe0 in the substrate. The next five peaks (Fe2-Fe6) form a series at fixed BE intervals, FWHMs, and relative intensities and correspond to FeIII oxide in the surface oxide layers as previously determined from reference samples (Marcus et al., 1980; Haupt et al., 1985; Yamashita and Hayes, 2008; Biesinger et al., 2011). The additional peak (Fe7), needed at higher BE to optimize the fit, corresponds to FeIII hydroxide also in the oxide films. No series of five peaks, at lower BEs and associated to FeII oxide (Biesinger et al., 2011), was needed for reconstruction. After treatment at UOCP (Figure 6, Table 1), the FeIII to Fe0 intensity ratio markedly decreases, reflecting the loss of oxidized iron in the surface oxide film in agreement with the ToF-SIMS data. Within the range of ±0.1 eV allowed for fitting, one observes no significant changes of the BE and FWHM values of the metallic and oxidized iron components. After treatment at UPass (Figure 7, Table 1), the FeIII to Fe0 intensity ratio increases, indicating re-oxidation of iron. The BE and FWHM values remain unchanged.
Consistently, and also as proposed before (Biesinger et al., 2011; Payne et al., 2011; Wang et al., 2019), seven peaks were also used for reconstructing the Cr 2p spectra (see e.g., Figure 5, Table 1). The first one (Cr1) is associated to metallic Cr0 in the substrate, the next five peaks (Cr2-Cr6), also forming a well-defined series as previously determined from reference samples (Yamashita and Hayes, 2008; Biesinger et al., 2011; Payne et al., 2011), to CrIII oxide in the surface oxide layers, and the additional peak (Cr7) to CrIII hydroxide in the oxide film. No CrVI peak expected at a BE of ~579.5 eV (Biesinger et al., 2011) was needed for curve fitting. After treatment at UOCP (Figure 6, Table 1), the CrIII(ox) to Cr0 intensity ratio slightly decreases but the CrIII(hyd) to Cr0 intensity ratio markedly increases, indicating significant hydroxylation of chromium in the surface oxide. After treatment at UPass (Figure 7, Table 1), the CrIII(ox) to Cr0 intensity ratio increases markedly but the CrIII(hyd) to Cr0 intensity ratio only slightly, indicating re-oxidation and dehydroxylation of chromium. BE and FWHM values show no significant variations after treatment at UOCP and UPass.
The Mo 3d spectra were fitted with three 5/2–3/2 spin-orbit doublets (see e.g., Figure 5, Table 1; Yang et al., 1984; Brooks et al., 1986; Clayton and Lu, 1989; De Vito and Marcus, 1992; Di Castro and Ciampi, 1995; Biesinger et al., 2011; Maurice et al., 2015; Wang et al., 2019). One (Mo1/Mo1') corresponds to metallic Mo0 in the substrate and the other two, (Mo2/Mo2′) and (Mo3/Mo3′), to MoIV and MoVI in the surface oxide layers, respectively. The MoVI to MoIV intensity ratio is 6.4 in the native oxide film, indicating that Mo(VI) species are mostly present. After treatment at UOCP (Figure 6, Table 1), the MoIV+VI to Mo0 intensity ratio slightly increases in agreement with the Mo enrichment in the oxide outer layer observed by ToF-SIMS. It further increases after treatment at UPass (Figure 7, Table 1). The MoVI to MoIV ratio also increases after treatment at UOCP (7.1) and further after treatment at UPass (7.4), indicating further enrichment in Mo(VI) species after electrochemical treatment. An additional peak S1, assigned to the S 2s core level, was necessary for reconstruction of the Mo 3d spectra measured after the electrochemical treatments (Figures 6, 7, Table 1). The presence of sulfur originating from the sulfuric acid electrolyte was confirmed by the presence of a S 2p doublet assigned to sulfate species (Table 1), most likely adsorbed at the surface of the oxide films (Table 1).
The Ni 2p3/2 spectra were fitted using a single peak corresponding to metallic Ni0 in the substrate (Table 1; Haupt and Strehblow, 1995; Maurice et al., 2015; Wang et al., 2019), evidencing that the Ni oxide species measured by ToF-SIMS were at trace level below the detection limit of XPS in all three cases (< 0.5 at%).
The O 1s spectra were fitted with three components O1, O2, and O3 assigned to oxide (O2−), hydroxide (OH−) and water (H2O) ligands in the surface oxide films, respectively (see e.g., Figure 5, Table 1; Clayton and Lu, 1986; Castle and Qiu, 1989b; Biesinger et al., 2011; Maurice et al., 2015; Wang et al., 2019). The OH−/O2− intensity ratio increases from 0.58 for the native oxide film to 1.72 after treatment at UOCP and then decreases to 1.24 after treatment at UPass. Similarly, the H2O/O2− intensity ratio increases from 0.12 to 0.68 and then decreases to 0.39. Clearly, these variations reflect the markedly increased hydroxylation of the initial air-formed oxide film after immersion in the aqueous solution at OCP, showing that the oxide film not only decreases in thickness and gets enriched in Cr and Mo due to preferential iron oxide dissolution but also incorporates much more hydroxyl and water ligands. Anodic passivation causes competitive re-oxidation of the metallic elements leading to the oxide film growth in thickness, with the preferential formation of oxide ligands since the fractions of hydroxyl and water ligands decrease.
Based on the ToF-SIMS results, the 2-layer model, previously proposed to calculate the thickness and composition of the outer and inner layers of the oxide films as well as the composition of the modified alloy underneath the oxide films (Wang et al., 2019), was used to process the XPS intensity data. This model assumes a mixed iron-chromium hydroxide outer layer and a mixed iron-chromium oxide inner layer. Molybdenum oxide is included in the outer layer and neglected in the inner layer. Assignment of the intensities of the different components was as follows: Cr2-Cr6 and Fe2-Fe4 components to the oxide film inner layer, Cr7, Fe5-Fe7, and Mo2-Mo3 components to the oxide film outer layer and Cr1, Fe1, Mo1, and Ni1 components to the modified alloy region underneath the oxide film. The results are presented in Table 2. The overall compositions of the oxide films were obtained by weighting the cation concentration value of each element by the fractional thickness of the inner and outer layers.
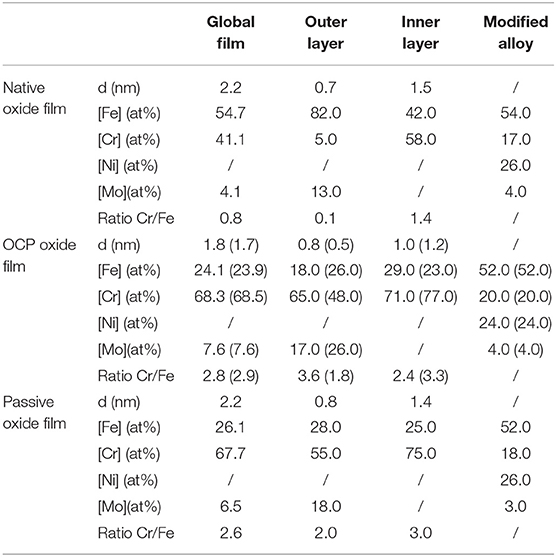
Table 2. Thickness and composition of the air-formed native oxide film on polycrystalline 316L SS and variations after immersion for 30 min at OCP in 0.05 M H2SO4, and after passivation for 30 min at 0.5 V/SCE in 0.05 M H2SO4 as calculated from the XPS data.
Let us first discuss the initial native oxide film. The total thickness is found to be 2.2 nm with outer and inner layers of 0.7 and 1.5 nm thickness, respectively. This total value is very close to that (2 nm) found following the same procedure on the same as-prepared surface of polycrystalline 316L samples (Wang et al., 2019). The overall composition of the film shows that chromium is already enriched in the air-formed native oxide film with CrIII ions representing 41% of the metal cations in the film. Consistently, Cr0 is found depleted (17 at% instead of 20 at% in the bulk alloy) in the modified alloy underneath the oxide film. Nickel, not in measurable amount in the oxide film, is confirmed to be enriched in the alloy underneath the oxide film (26 at% instead of 12 at% in the bulk alloy) in agreement with the ToF-SIMS profile. Concerning molybdenum, the calculated composition shows that it is enriched in the native oxide film (4 at% globally and 13 at% in the oxide outer layer instead of 1.6 at% in the bulk alloy). It is also found enriched in the alloy underneath the oxide film (4 at% instead of 1.6 at% in the bulk alloy). Compared to the more hydroxylated native oxide film formed on the same as-prepared surface of polycrystalline 316L samples (OH−/O2− intensity ratio of 0.93 instead of 0.58 in the present case; Wang et al., 2019), the present data show lower CrIII (41 vs. 55% of the metal cations) and MoIV/VI (4 vs. 6%) enrichments in the oxide film. This might be due to an aging effect, some studies having reported a decreasing Cr/Fe balance in the native oxide with aging in air (Yang et al., 1994; Maurice et al., 1996, 1998).
After treatment in sulfuric acid at UOCP, the results reported in Table 2 confirm the overall thickness decrease of the oxide film observed by ToF-SIMS. However, this decrease affects the inner layer only, in contrast with the ToF-SIMS data. The overall composition of the film shows the loss of Fe and the related increases of the Cr and Mo enrichments suggested by ToF-SIMS but the hierarchy of the Cr/Fe ratio between outer and inner layers is not respected. These discrepancies originate from the assignment of the Cr7 component, markedly increased in intensity. Since immersion at OCP causes iron oxide dissolution and pronounced hydroxylation of the film, it can be reasonably assumed that not only the outer layer but also the inner layer is hydroxylated, and thus that a fraction of the intensity of the Cr7 component can be assigned to the inner layer. The values reported in brackets in Table 2 have been calculated assigning 50% of the Cr7 intensity to the oxide inner layer. The hierarchy of the Cr/Fe ratio between outer and inner layers is now reproduced as well as the decrease in thickness of both the outer and inner layers. Based on these assumptions, the composition in the outer and inner layers are calculated as 26FeIII-48CrIII-26MoIV/VI and 23FeIII-77CrIII, respectively, vs. 82FeIII-5CrIII-13MoIV/VI and 42FeIII-58CrIII for the native oxide. The overall composition of the film also reflects the compositional changes, independently of the balance in the assignment of the Cr7 peak. The CrIII and MoIV/VI concentrations increase up to 68.5 and 7.6% in the oxide film, respectively, after treatment at UOCP, as a result of the preferential loss of iron oxide by dissolution. In the modified alloy underneath the oxide film, the calculated values confirm the Ni enrichment and suggest no preferential consumption at UOCP of one of the alloying elements if one considers a ±1 at% accuracy.
Clearly, these data show that the loss of oxide is caused by the preferential dissolution of iron oxide at open circuit potential. It is also shown that oxide growth is too slow to compensate the loss of oxide due to dissolution and, if occurring, does not preferentially consumes one of the alloying elements. To our knowledge, this is observed for the first time on an oxide pre-covered SS surface thanks to the transfer from liquid to UHV environments avoiding the contact with ambient air. Such a procedure was previously applied to ferritic Fe-17Cr samples but starting from an oxide-free surface, not an oxide pre-covered surface, and showed the preferential growth of Cr(III) oxide at anodic potential (Keller and Strehblow, 2004). The present data highlight that the alterations of the surface oxide film caused by immersion at open circuit potential results from the preferential dissolution of iron oxide.
After passivation at UPASS and compared to the treatment at UOCP, the overall thickness of the oxide film increases with both partitions growing (Table 2), which confirms the ToF-SIMS observation. The steady state thickness of the passive film increases due to faster growth of the surface oxide film. There are no significant variations of the overall composition of the oxide film and of the composition of the modified alloy region if one considers a ±1 at% accuracy of the calculated values, indicating that all alloying element are consumed at UPASS but with iron oxide preferentially dissolving in order to maintain the Cr enrichment. Cr remains enriched in both layers of the passive film and more predominantly in the inner layer. In the hydroxide outer layer, Mo would be less enriched than before re-growth of the oxide film under anodic polarization.
Compared to the native oxide film, there is no significant thickness variation after passivation at UPASS. However, the composition of the oxide film is markedly modified with further Cr(III) enrichment in both layers and Mo(IV-VI) enrichment in the outer layer, in agreement with the previous studies on the same polycrystalline 316L surface (Wang et al., 2019) and on a single-crystalline model Fe-Cr-Ni-Mo surface (Maurice et al., 2015). Thanks to the transfer procedure adopted here and avoiding the contact with ambient air, it is shown here that the preferential dissolution of Fe(III) is at the origin of these enrichments and essentially takes place at open circuit potential in the absence of any applied anodic polarization. Anodic passivation causes competitive re-growth of the oxide film and dehydroxylation of Fe(III), Cr(III) and Mo(IV-VI) oxide species It increases the steady state thickness of the passive film despite dissolution still occurring and counteracting the formation of the oxide species. The preferential dissolution of Fe(III) oxide species persists to maintain the Cr and Mo enrichments.
Conclusion
Starting from the air-formed native oxide-covered surface, ToF-SIMS and XPS were combined to study the mechanisms of Cr and Mo enrichments leading to passivation of 316L austenitic stainless steel.
The native oxide film formed in ambient air on the 316L surface prepared by mechanical polishing was found to have a thickness of 2.2 nm. It is a mixed Cr(III)-Fe(III) hydroxylated oxide with a bi-layered structure highly enriched in Cr(III) oxide in the inner layer. Iron is concentrated as Fe(III) hydroxide in the outer layer together with molybdenum, mostly present as Mo(VI). Nickel is below the XPS detection limit. Immersion at open circuit potential in aqueous acid solution causes hydroxylation and preferential dissolution of Fe(III) from both layers of the film, decreasing the thickness and promoting the enrichments in Cr and Mo. In the absence of an anodizing electric field, oxide growth and thickness increase do not happen. Upon anodic polarization in the passive domain, dehydroxylation is promoted in the inner layer and oxidation of iron, chromium and molybdenum takes place, leading to re-growth of the oxide film and increase of the steady-state thickness without affecting the overall Cr enrichment but slightly mitigating the Mo enrichment. Cr and Mo enrichments persist owing to preferential dissolution of Fe(III).
These results were obtained using an experimental approach preventing contact to ambient air of the samples during transfer from liquid environment for electrochemical treatment to UHV environment for surface analysis. They show that the treatment of the surface native oxide film in acid solution, in the absence of applied electric field, promotes the Cr and Mo enrichments known as beneficial to the resistance to passivity breakdown and initiation of localized corrosion.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
The study was conceived by VM and PM. ZW performed surface preparation, experiments and data acquisition, and analysis under the supervision of E-MP, AS, and SZ. ZW wrote the manuscript first draft revised by all. All authors approved the final version and discussed the data interpretation.
Funding
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (ERC Advanced Grant no. 741123). Région Île-de-France is acknowledged for partial funding of the ToF-SIMS equipment.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Biesinger, M. C., Payne, B. P., Grosvenor, A. P., Lau, L. W. M., Gerson, A. R., and Smart, R. S. C. (2011). Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni, Appl. Surf. Sci. 257, 2717–2730. doi: 10.1016/j.apsusc.2010.10.051
Bojinov, M., Fabricius, G., Laitinen, T., Mäkelä, K., Saario, T., and Sundholm, G. (2001). Influence of molybdenum on the conduction mechanism in passive films on iron–chromium alloys in sulphuric acid solution. Electro. Acta. 46, 1339–1358. doi: 10.1016/S0013-4686(00)00713-1
Brooks, A. R., Clayton, C. R., Doss, K., and Lu, Y. C. (1986). On the role of Cr in the passivity of stainless steel. J. Electrochem. Soc. 133, 2459–2464. doi: 10.1149/1.2108450
Calinski, C., and Strehblow, H. H. (1989). ISS depth profiles of the passive layer on Fe/Cr alloys. J. Electrochem. Soc. 136, 1328–1331. doi: 10.1149/1.2096915
Castle, J. E., and Qiu, J. H. (1989a). A co-ordinated study of the passivation of alloy steels by plasma source mass spectrometry and x-ray photoelectron spectroscopy−1. characterization of the passive film. Corrosion Sci. 29, 591–603. doi: 10.1016/0010-938X(89)90010-3
Castle, J. E., and Qiu, J. H. (1989b). A co-ordinated study of the passivation of alloy steels by plasma source mass spectrometry and x-ray photoelectron spectroscopy—II. growth kinetics of the passive film. Corrosion Sci. 29, 605–616. doi: 10.1016/0010-938X(89)90011-5
Clayton, C. R., and Lu, Y. C. (1986). A bipolar model of the passivity of stainless steel: the role of Mo addition. J. Electrochem. Soc. 133, 2465–2473. doi: 10.1149/1.2108451
Clayton, C. R., and Lu, Y. C. (1989). A bipolar model of the passivity of stainless steels—III. The mechanism of MoO42– formation and incorporation. Corrosion Sci. 29, 881–898. doi: 10.1016/0010-938X(89)90059-0
De Vito, E., and Marcus, P. (1992). XPS study of passive films formed on molybdenum-implanted austenitic stainless steels. Surf. Interf. Anal. 19, 403–408. doi: 10.1002/sia.740190175
Di Castro, V., and Ciampi, S. (1995). XPS study of the growth and reactivity of FeMnO thin films. Surf. Sci. 331–333, 294–299. doi: 10.1016/0039-6028(95)00190-5
Elbiache, A., and Marcus, P. (1992). The role of molybdenum in the dissolution and the passivation of stainless steels with adsorbed Sulphur. Corrosion Sci. 33, 261–269. doi: 10.1016/0010-938X(92)90150-2
Habazaki, H., Kawashima, A., Asami, K., and Hashimoto, K. (1992). The corrosion behavior of amorphous Fe-Cr-Mo-PC and Fe-Cr-WPC alloys in 6 M HCl solution. Corrosion Sci. 33, 225–236. doi: 10.1016/0010-938X(92)90147-U
Hakiki, N. E., Belo, M. D. C., Simões, A. M. P., and Ferreira, M. G. S. (1998). Semiconducting properties of passive films formed on stainless steels influence of the alloying elements. J. Electrochem. Soc. 145, 3821–3829. doi: 10.1149/1.1838880
Hamm, D., Ogle, K., Olsson, C. O. A., Weber, S., and Landolt, D. (2002). Passivation of Fe–Cr alloys studied with ICP-AES and EQCM. Corrosion Sci. 44, 1443–1456. doi: 10.1016/S0010-938X(01)00147-0
Hashimoto, K., Asami, K., and Teramoto, K. (1979). An X-ray photo-electron spectroscopic study on the role of molybdenum in increasing the corrosion resistance of ferritic stainless steels in HCl. Corrosion Sci. 19, 3–14. doi: 10.1016/0010-938X(79)90003-9
Haupt, S., Collisi, U., Speckmann, H. D., and Strehblow, H. H. (1985). Specimen transfer from the electrolyte to the UHV in a closed system and some examinations of the double layer on Cu. J. Electroanal. Chem. 194, 79–190. doi: 10.1016/0022-0728(85)85002-6
Haupt, S., and Strehblow, H. H. (1995). A combined surface analytical and electrochemical study of the formation of passive layers on FeCr alloys in 0.5 M H2SO4. Corrosion Sci. 37, 43–54. doi: 10.1016/0010-938X(94)00104-E
Hultquist, G., Seo, M., Leitner, T., Leygraf, C., and Sato, N. (1987). The dissolution behaviour of iron, chromium, molybdenum and copper from pure metals and from ferritic stainless steels. Corrosion Sci. 27, 937–946. doi: 10.1016/0010-938X(87)90060-6
Keller, P., and Strehblow, H. H. (2004). XPS investigations of electrochemically formed passive layers on Fe/Cr-alloys in 0.5 M H2SO4. Corrosion Sci. 46, 1939–1952. doi: 10.1016/j.corsci.2004.01.007
Kirchheim, R., Heine, B., Fischmeister, H., Hofmann, S., Knote, H., and Stolz, U. (1989). The passivity of iron-chromium alloys. Corrosion Sci. 29, 899–917. doi: 10.1016/0010-938X(89)90060-7
Lu, Y. C., Clayton, C. R., and Brooks, A. R. (1989). A bipolar model of the passivity of stainless steels—II. The influence of aqueous molybdate. Corrosion Sci. 29, 863–880. doi: 10.1016/0010-938X(89)90058-9
Ma, L., Wiame, F., Maurice, V., and Marcus, P. (2018). New insight on early oxidation stages of austenitic stainless steel from in situ XPS analysis on single-crystalline Fe−18Cr−13Ni. Corrosion Sci. 140, 205–216. doi: 10.1016/j.corsci.2018.06.001
Ma, L., Wiame, F., Maurice, V., and Marcus, P. (2019). Origin of nanoscale heterogeneity in the surface oxide film protecting stainless steel against corrosion. npj Mater. Degradat. 3:29. doi: 10.1038/s41529-019-0091-4
Macdonald, D. D. (1992). The point defect model for the passive state. J. Electrochem. Soc. 139, 3434–3449. doi: 10.1149/1.2069096
Marcus, P., and Olefjord, I. (1988). Round Robin on combined electrochemical and AES/ESCA characterization of the passive films on Fe-Cr and Fe-Cr-Mo alloys. Corrosion Sci. 11, 569–576. doi: 10.1002/sia.740111106
Marcus, P., Oudar, J., and Olefjord, I. (1980). Studies of the influence of sulphur on the passivation of nickel by Auger electron spectroscopy and electron spectroscopy for chemical analysis. Mater. Sci. Eng. 42, 191–197. doi: 10.1016/0025-5416(80)90028-2
Massoud, T., Maurice, V., Wiame, F., Klein, L. H., Seyeux, A., and Marcus, P. (2014). Nanostructure and local properties of oxide layers grown on stainless steel in simulated pressurized water reactor environment. Corrosion Sci. 84, 198–203. doi: 10.1016/j.corsci.2014.03.030
Maurice, V., and Marcus, P. (2018). Current developments of nanoscale insight into corrosion protection by passive oxide films. Curr. Opin. Solid State Mater. Sci. 22, 156–167. doi: 10.1016/j.cossms.2018.05.004
Maurice, V., Peng, H., Klein, L. H., Seyeux, A., Zanna, S., and Marcus, P. (2015). Effects of molybdenum on the composition and nanoscale morphology of passivated austenitic stainless steel surfaces. Faraday Discussions 180, 151–170. doi: 10.1039/C4FD00231H
Maurice, V., Yang, W., and Marcus, P. (1996). XPS and STM study of passive films formed on Fe-22Cr (110) single-crystal surface. J. Electrochem. Soc. 143, 1182–1200. doi: 10.1149/1.1836616
Maurice, V., Yang, W., and Marcus, P. (1998). X-Ray photoelectron spectroscopy and scanning tunneling microscopy study of passive films formed on (100) Fe-18Cr-13Ni single-crystal surfaces. J. Electrochem. Soc. 145, 909–920. doi: 10.1149/1.1838366
Mischler, S., Mathieu, H. J., and Landolt, D. (1988). Investigation of a passive film on an iron-chromium alloy by AES and XPS. Surf. Interf. Anal. 11, 182–188. doi: 10.1002/sia.740110403
Mischler, S., Vogel, A., Mathieu, H. J., and Landolt, D. (1991). The chemical composition of the passive film on Fe-24Cr and Fe-24Cr-11Mo studied by AES, XPS and SIMS Corrosion Sci. 32, 925–944. doi: 10.1016/0010-938X(91)90013-F
Mitchell, D. F., and Graham, M. (1987). Comparison of Auger and SIMS analysis of a thin passive oxide film on iron−25% chromium. Surf. Interf. Anal. 10, 259–261. doi: 10.1002/sia.740100507
Oblonsky, L. J., Ryan, M. P., and Isaacs, H. S. (1998). In situ determination of the composition of surface films formed on Fe-Cr alloys. J. Electrochem. Soc. 145, 1922–1932. doi: 10.1149/1.1838577
Ogawa, H., Omata, H., Itoh, I., and Okada, H. (1978). Okada, Auger electron spectroscopic and electrochemical analysis of the effect of alloying elements on the passivation behavior of stainless steels. Corrosion 34, 52–60. doi: 10.5006/0010-9312-34.2.52
Olefjord, I. (1980). The passive state of stainless steels. Mater. Sci. Eng. 42, 161–171. doi: 10.1016/0025-5416(80)90025-7
Olefjord, I., Brox, B., and Jelvestam, U. (1985). Surface composition of stainless steels during anodic dissolution and passivation studied by ESCA. J. Electrochem. Soc. 132, 2854–2861. doi: 10.1149/1.2113683
Olefjord, I., and Elfström, B.-O. (1982). The composition of the surface during passivation of stainless steels. Corrosion 38, 46–52. doi: 10.5006/1.3577318
Olefjord, I., and Wegrelius, L. (1990). Surface analysis of passive state. Corrosion Sci. 31, 89–98. doi: 10.1016/0010-938X(90)90095-M
Olsson, C. O. A., and Hörnström, S. E. (1994). An AES and XPS study of the high alloy austenitic stainless steel 254 SMO® tested in a ferric chloride solution. Corrosion Sci. 36, 141–151. doi: 10.1016/0010-938X(94)90115-5
Payne, B. P., Biesinger, M. C., and McIntyre, N. S. (2011). X-ray photoelectron spectroscopy studies of reactions on chromium metal and chromium oxide surfaces, Journal of Electron Spectroscopy and Related Phenomena. 184, 29–37. doi: 10.1016/j.elspec.2010.12.001
Schmuki, P. (2002). From Bacon to barriers: a review on the passivity of metals and alloys. J. Solid State Electrochem. 6, 145–154. doi: 10.1007/s100080100219
Seo, M., and Sato, N. (1979). Differential composition profiles in depth of thin anodic oxide films on iron-chromium alloy. Surf. Sci. 86, 601–609. doi: 10.1016/0039-6028(79)90440-0
Sugimoto, K., and Sawada, Y. (1977). The role of molybdenum additions to austenitic stainless steels in the inhibition of pitting in acid chloride solutions. Corrosion Sci. 17, 425–445.
Tan, M.-W., Akiyama, E., Kawashima, A., Asami, K., and Hashimoto, K. (1995). The effect of air exposure on the corrosion behavior of amorphous Fe-8Cr-Mo-13P-7C alloys in 1 M HCl. Corrosion Sci. 37, 1289–1301. doi: 10.1016/0010-938X(95)00035-I
Wang, Z., Di Franco, F., Seyeux, A., Zanna, S., Maurice, V., and Marcus, P. (2019). Passivation-induced physicochemical alterations of the native surface oxide film on 316L austenitic stainless steel, J. Electrochem. Soc. 166:C3376–C3388. doi: 10.1149/2.0321911jes
Yamamoto, T., Fushimi, K., Seo, M., Tsuri, S., Adachi, T., and Habazaki, H. (2009). Depassivation–repassivation behavior of type-312L stainless steel in NaCl solution investigated by the micro-indentation. Corrosion Sci. 51, 1545–1553. doi: 10.1016/j.corsci.2008.11.020
Yamashita, T., and Hayes, P. (2008). Analysis of XPS spectra of Fe2+ and Fe3+ ions in oxide materials, Appl. Surf. Sci. 254, 2441–2449. doi: 10.1016/j.apsusc.2007.09.063
Yang, W., Ni, R. C., Hua, H.-Z., and Pourbaix, A. (1984). The behavior of chromium and molybdenum in the propagation process of localized corrosion of steels. Corrosion Sci. 24, 691–707. doi: 10.1016/0010-938X(84)90059-3
Keywords: stainless steel, passivation, oxide film, surface analysis, Cr enrichment
Citation: Wang Z, Paschalidou E-M, Seyeux A, Zanna S, Maurice V and Marcus P (2019) Mechanisms of Cr and Mo Enrichments in the Passive Oxide Film on 316L Austenitic Stainless Steel. Front. Mater. 6:232. doi: 10.3389/fmats.2019.00232
Received: 25 June 2019; Accepted: 05 September 2019;
Published: 24 September 2019.
Edited by:
Daniel John Blackwood, National University of Singapore, SingaporeReviewed by:
VS Raja, Indian Institute of Technology Bombay, IndiaWolfram Fürbeth, DECHEMA Forschungsinstitut (DFI), Germany
Copyright © 2019 Wang, Paschalidou, Seyeux, Zanna, Maurice and Marcus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vincent Maurice, dmluY2VudC5tYXVyaWNlQGNoaW1pZXBhcmlzdGVjaC5wc2wuZXU=; Philippe Marcus, cGhpbGlwcGUubWFyY3VzQGNoaW1pZXBhcmlzdGVjaC5wc2wuZXU=