 Nils E. R. Zimmermann
Nils E. R. Zimmermann Matthew K. Horton
Matthew K. Horton Anubhav Jain
Anubhav Jain Maciej Haranczyk
Maciej Haranczyk- 1Lawrence Berkeley National Laboratory, Computational Research Division, Berkeley, CA, United States
- 2Department of Materials Science and Engineering, University of California, Berkeley, Berkeley, CA, United States
- 3Lawrence Berkeley National Laboratory, Energy Technologies Area, Berkeley, CA, United States
Structure–property relationships form the basis of many design rules in materials science, including synthesizability and long-term stability of catalysts, control of electrical and optoelectronic behavior in semiconductors, as well as the capacity of and transport properties in cathode materials for rechargeable batteries. The immediate atomic environments (i.e., the first coordination shells) of a few atomic sites are often a key factor in achieving a desired property. Some of the most frequently encountered coordination patterns are tetrahedra, octahedra, body and face-centered cubic as well as hexagonal close packed-like environments. Here, we showcase the usefulness of local order parameters to identify these basic structural motifs in inorganic solid materials by developing classification criteria. We introduce a systematic testing framework, the Einstein crystal test rig, that probes the response of order parameters to distortions in perfect motifs to validate our approach. Subsequently, we highlight three important application cases. First, we map basic crystal structure information of a large materials database in an intuitive manner by screening the Materials Project (MP) database (61,422 compounds) for element-specific motif distributions. Second, we use the structure-motif recognition capabilities to automatically find interstitials in metals, semiconductor, and insulator materials. Our Interstitialcy Finding Tool (InFiT) facilitates high-throughput screenings of defect properties. Third, the order parameters are reliable and compact quantitative structure descriptors for characterizing diffusion hops of intercalants as our example of magnesium in MnO2-spinel indicates. Finally, the tools developed in our work are readily and freely available as software implementations in the pymatgen library, and we expect them to be further applied to machine-learning approaches for emerging applications in materials science.
1. Introduction
Crystals consist of atoms that are arranged in periodic patterns in three dimensions (Sands, 1993). This regular arrangement is called the crystal structure which, together with the chemical composition, dictates the properties of a material (Morris, 2007). Typically, the crystal structure is described with approximations and abstractions (Morris, 2007). One approach is to focus on the immediate surrounding of each atom (first coordination shell) and to use the number of surrounding atoms (coordination number) and the pattern (structure motif) for structure description, the discipline of which was coined by Werner and which is today known as coordination chemistry (Werner, 1912). Among frequently occurring structure motifs are tetrahedra, octahedra, body-center and face-centered cubic as well as hexagonal close-packed motifs (Figure 1).
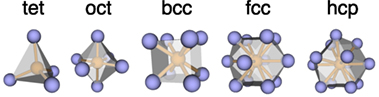
Figure 1. Basic structural motifs that frequently recur in various materials databases (from left to right): tetrahedron (tet), octahedron (oct) as well as body-centered cubic (bcc), face-centered cubic (fcc), and hexagonal close packed (hcp) motifs. The central atom and bonds are shown in orange, whereas the coordinating atoms are displayed in blue.
The occurrence of basic structural motifs in crystalline compounds has been shown to be important indictors for predicting materials properties in several scientific and technological contexts. Finding and quantitatively assessing primary building blocks of zeolite materials (SiO4 tetrahedra) can be used to predict the feasibility of synthesizing a (hypothetical) material (Li et al., 2013; Mazur et al., 2015) and to rate their likelihood for industrial deployment—for example, as a catalyst—(Zimmermann and Haranczyk, 2016). Design rules for novel battery materials are frequently developed employing information about the coordination pattern of the migrating ion (Rong et al., 2015) and the host structure (Li et al., 2009; Wang et al., 2015). Models based on structural fragments can be used to assess influencing factors to the superconductivity critical temperature (Isayev et al., 2015). Interstitials in dense inorganic materials are frequently found in positions where the interstitials assume tetrahedrally or octahedrally coordinated positions (Decoster et al., 2008, 2009a,b, 2010a,b, 2012; Pereira et al., 2011, 2012; Amorim et al., 2013; Silva et al., 2014).
Screening large databases for structure motif occurrence has hence the potential to find new candidate materials for various emerging applications. The inherent difficulty is to develop recognition tools that allow for reliable and rapid motif identification. There are two basic steps involved in the (automatic) identification of a coordination motif around a given atom: (i) neighbor finding and (ii) pattern matching. Neighbors can be found on the basis of interatomic distances—possibly in combination with typical bond lengths (Brunner, 1977; Hoppe, 1979; O’Keeffe and Brese, 1991)—or by a topology-based approach (Dirichlet, 1850; Voronoi, 1908; Mickel et al., 2013). For pattern matching, there exist two popular conceptual approaches: (i) using Monte Carlo (MC) moves (Shetty et al., 2002) and (ii) using order parameters (Steinhardt et al., 1983; Peters, 2009; Zimmermann et al., 2015). In the MC approach, an ideal structure motif is placed onto a central atom and its neighbors, and the ideal motif’s position and size are varied to yield a small root mean square deviation between the positions of the ideal motif and the neighbors of the central atom. In the systematically expandable (Santiso and Trout, 2011) order parameter approach, the bond angles of a given motif are used in mathematical functions to directly yield a measure of motif resemblance, thus, being a deterministic method. Note that the MC-based approach is expected to be much more time-consuming than the order parameter route.
We here develop an effective and computationally efficient approach for finding atomic neighbors and identifying motif types in inorganic materials using order parameters (Steinhardt et al., 1983; Peters, 2009; Zimmermann et al., 2015) for pattern matching. Furthermore, we introduce a testing framework (Einstein crystal test rig) for validation of any such motif-finding effort. We then apply our approach to the database provided by the Materials Project (Jain et al., 2013), where we use well-defined materials subsets for testing. Finally, the method is used to generate crystal structure representations of the Materials Project database, to determine potential interstitial sites in several materials, and to quantitatively characterize the coordination environment change along the jump-diffusion path of an intercalating ion.
2. Methods
We focus on local structural motifs that are based on a central atom and its first coordination shell. The two basic steps in identifying structural motifs are therefore:
1. finding bonded neighbors and
2. motif recognition.
More complex patterns such as those involving second-shell neighbors and cyclic motifs (rings) would require a more extensive analysis of the connectivity between atoms.
2.1. Bonding Identification
Bonds are determined on the basis of the distance, di,j, between two atoms i and j:
where pi is the position of atom i. We systematically investigate three different neighbor-finding methods, all of which work with a site-specific cutoff distance, rcut,i. In the first method (“min_dist”), we determine the (absolute) distance to the nearest neighbor, dmin,i, of a given site i and, subsequently, we consider all additional sites that are at maximum rcut,i = (1 + δ)dmin,i apart from site i (Figure 2), where δ denotes a (relative) neighbor-finding tolerance (distance). The other two approaches work similarly, except for the fact that we use dimensionless distances, , where l is a length being characteristic for the considered pair of atoms i and j. The following two approaches for the characteristic length are tested: the sum of atom (or, ion) radii (Shannon (1976); “min_VIRE”: ) and the typical bond length (O’Keeffe and Brese (1991); “min_OKeeffe”: ). The radii are calculated with a valence-ionic radius estimator (VIRE) implemented in pymatgen (Ong et al., 2013). The estimator uses a maximum a posteriori estimation method of the oxidation state of each site based on bond-valence sums (O’Keeffe and Brese, 1991). Furthermore, a first estimate of the coordination number is inferred from Voronoi decomposition (Dirichlet, 1850; Voronoi, 1908) as the number of faces making up the polyhedron, weighing each face’s contribution in proportion to its solid angle subtended by that face at the center (O’Keeffe, 1979). The oxidation state and coordination number estimates are subsequently used to calculate the atom radius on the basis of the ionic radius list provided by Shannon (1976). In the case when no oxidation states can be assigned, the estimator uses the atomic radii as provided by pymatgen (Ong et al., 2013).
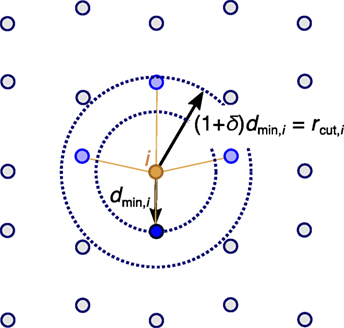
Figure 2. Atoms that are bonded (blue spheres) to a given central site i (orange) are identified on the basis of the nearest-neighbor distance, dmin,i (absolute or dimensionless) and an a priori chosen tolerance, δ, thus, defining a site-specific cutoff radius rcut,i = (1 + δ)dmin,i.
For screening the equilibrium structures found in the Materials Project database, we will later use a single global tolerance, δ, that will be optimized on the basis of a diverse structure test set. By contrast, both the interstitial finding and the ionic diffusion path characterization proceed with an increasing tolerance (from δ = 0.1 in steps of 0.1 until a motif is found or δ = 0.8 reached) to find neighbors in these (non-equilibrium) configurations.
Finally, note that the “min_VIRE” and “min_OKeeffe” methods may seem more reliable because they introduce well-established chemical properties (atomic/ionic radii and bond lengths, respectively) that are directly connected to the bonded atoms. But our optimization data suggest that the more ad hoc “min_dist” method performs in fact best.
2.2. Motif Assessment
Once all neighbors of a central atom i are identified (Figure 3), the coordination pattern is evaluated. We use analytic order parameters (Steinhardt et al., 1983; Peters, 2009; Zimmermann et al., 2015) to perform the pattern recognition. The order parameters are typically designed in such a way to give a numerical value of 1 if the coordination pattern perfectly resembles the target motif and 0 if it is very different from the target motif.
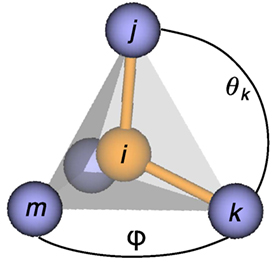
Figure 3. Definition of atom indices, i, j, k, and m, as well as angles, θ (polar) and φ (azimuth), which are used in the computation of the order parameters introduced by Peters (2009) and Zimmermann et al. (2015).
2.2.1. Order Parameters
Order parameters (OPs), q, are mathematical constructs which aim to provide a numerical measure of the immediate local environment around an atom. The simplest OP, qCN, is a coordination number (CN (Sprik, 1998)),
obtained from counting neighbors within a cutoff radius rcut,i around a central atom i. S is a weight that is 1 if an atom j is within a distance di,j < rcut,i of atom i and S = 0 otherwise. The next level of sophistication is a distance-weighted approach such as using the Fermi function (Sprik, 1998):
where κ−1 defines the transition width in which the contribution of an atom to the OP changes fastest to go from around 1 toward 0 as di,j increases (Sprik, 1998). Note that neither of these approaches provides information about how closely an environment resembles a given structural motif.
Bond-orientational order parameters, introduced by Steinhardt et al. (1983), can, to some extent, be used to discern different structural motifs (Mickel et al., 2013). Thermal motion and other small distortions (e.g., caused by relaxation to the ground state of a compound from an ideal initial prototype structure) can, however, yield overlap between the OP distributions so that identification of the motif type becomes difficult. Hence, there is a need to more reliably identify structural motifs with order parameters.
Peters (2009) and Zimmermann et al. (2015) have introduced order parameters based on pattern-matching ideas put forward by Shetty et al. (2002). The OPs specifically recognize body-centered cubic-like (Peters, 2009) as well as tetrahedral and octahedral environments (Zimmermann et al., 2015). The pattern-matching ansatz places a reward on local environments that are similar to the target structure, thus, resulting in a value, qi, of (close to) 1 for perfect resemblance. When the surrounding atoms are not in a configuration resembling the perfect prototype motif, penalties force the order parameter to attain values around zero. The pattern matching is achieved by setting up a spherical coordinate system around a central atom i with a subset of neighboring atoms j and k (Figure 3). This allows the determination of the remaining neighbors’ (m, …) polar angles, θ, and azimuth angles, φ. If a neighbor is not located at angles that are commensurate with the expected positions in the underlying structure motif, the decline in reward for being away from the perfect position follows a Gaussian function. Conversely, a full reward is given if any expected remaining position is exactly assumed. The procedure provides rotationally invariant order parameters because it is applied to each neighbor j being used as the North pole position and any remaining neighbor k for defining the prime meridian (cf., Figure 3); all possible combinations are then averaged. Below, we provide the definitions of the OPs that we use in this work.
The tetrahedral order parameter, qtet, is given by (Zimmermann et al., 2015):
where Nngh denotes the number of neighbors bonded to the central atom i in a motif, θk is the polar angle formed between the bonds of neighboring atoms j and k with their mutually bonded atom i, φ is the azimuth angle between bond i − m with the plane spanned by i, j, and k, and Δθ = 12° a parameter controlling the reward loss for increasingly non-ideal positions, which was optimized to distinguish tetrahedral and octahedral environments in NaCl wurtzite and conventional rocksalt (Zimmermann et al., 2015). Note that we use a slightly different variant of qtet as in the original formulation [cos2(1.5 φ) instead of cos(3 φ)] to avoid negative values.
The octahedral order parameter, qoct, is given by (Zimmermann et al., 2015):
where H(x) denotes the Heaviside function which is 1 if the argument x > 0 and 0 otherwise, Δθ1 = 12°, Δθ2 = 10°, and θthr is a threshold angle to distinguish second neighbors that are considered to be either in a “South pole” configuration or in a “prime meridian” position; we set this threshold to 160°. Note that we change the definition of qoct in a similar manner as we have done for qtet to avoid negative values.
The body-centered cubic order parameter, qbcc, is given by (Peters, 2009):
where Δθ1 = 12°, Δθ2 = 19.47°, and sgn(θ) is the signum function which is −1 for θ < 0, 0 if θ = 0, and 1 if θ > 0.
The mathematical definitions of the motif-specific order parameters qtet, qoct, and qbcc in equations (4)–(6) follow a mutual recipe:
1. The innermost sum gives the contribution of how closely neighbor m is located at its expected position with respect to polar angle, θm, and azimuth angle, φ. For the azimuth angle, squared cosine functions are preferably used to pinpoint locations around a circle to ensure that the OP strictly gives positive values.
2. The outermost sum accounts for neighbor k’s match with the expected polar angle, θk. The Gaussian functions are used with the polar angles to penalize deviations from expected positions.
3. The preceding factor normalizes the sums to give values between 0 and 1 based on combinatoric considerations.
For qoct and qbcc, there is furthermore the need to distinguish whether or not neighbor k is in (approximate) South pole position via the Heaviside function H(θk − θthr). And, qbcc also requires incorporating the alternating pattern of subsequent neighbors m being above and below the equator in bcc via the term 1.6 × sgn(θk − 90°) × (θm − 90°)/(Δθ2).
Finally, we use the bond-orientational order parameters (Steinhardt et al., 1983) q4 and q6 for identifying close-packed motifs—fcc and hcp—(Ackland and Jones, 2006):
where Yim are the spherical harmonics of degree i. Note that (i) the angles θ and φ are here with respect to a fixed frame of reference (Jungblut et al., 2013) and that (ii), while between 0 and 1, the values of the bond-orientational order parameters are typically not close to one for any motif, which is different to the behavior of qtet, qoct, and qbcc. Despite the fact that the bond-orientational order parameters q4 and q6 are frequently used in nucleation studies involving bcc, fcc, and hcp environments (ten Wolde et al., 1996; Peters, 2009; Jungblut et al., 2013; Limmer and Chandler, 2013), they are not highly reliable indicators to distinguish between all of these motifs when distortions (thermal noise) are introduced (Gasser et al., 2003). For this reason, we extensively use here those order parameters that were specifically designed for a given motif type (e.g., qtet for tetrahedra).
2.2.2. Motif-Recognition Criteria
Motif recognition is typically achieved on the basis of a threshold approach (Peters, 2009; Zimmermann et al., 2015): if a given order parameter, qi, is larger than an appropriate threshold, qi,thr, the coordination pattern is confirmed. Because of the design of these OPs, a threshold of 0.5 is often a reasonable a priori choice. The motif-specific order parameters qtet, qoct, and qbcc should then be ideally usable as stand-alone identifiers for tetrahedral, octahedral, and bcc-like coordination environments, respectively. However, Table 1 indicates that such an approach to defining criteria for motif recognition is not effective. For example, a site in a diamond structure would be identified as having both tetrahedral and bcc-like coordination; this is not surprising because the bcc motif can be viewed as two point-symmetric tetrahedra. Therefore, slightly more complex criteria must be developed to accurately distinguish structure motifs on the basis of order parameter values. We start with following set of criteria that allows us to identify all motifs separately and unambiguously for perfect prototype structures:
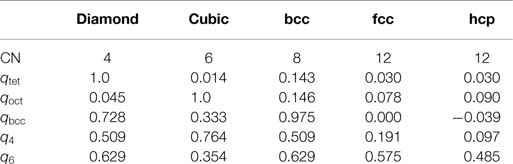
Table 1. Coordination numbers and order parameter values for different prototype structures.
The fourth-order bond-orientational order parameter q4 is hence not necessary to identify all motifs. However, as we will explain later, small modifications to these criteria are needed to more accurately distinguish non-ideal motifs. In particular, we will merge the fcc and hcp criteria into a single close-packed rule (q6 > 0.4 and qtet, qoct, qbcc < 0.4), and we will decrease the threshold of the tetrahedral order parameter to 0.3.
2.2.3. Validation
Validation is essential for reliable quantitative structure–property relationships (QSPRs) (Tropsha et al., 2003). We follow a three-step hierarchical approach for our structure-motif assessment on the basis of order parameters using perfect prototype structures. First, the responses of the tetrahedral and octahedral order parameters are measured when a single neighbor in the corresponding prototype structure is subjected to defined perturbations. We focus here on qtet and qoct because those motifs are particularly important throughout materials science and in corresponding design rules. Second, we randomly, but systematically, perturb the locations of sites in all prototype structures to mimic the effect of small distortions in equilibrium structures on all order parameters. This provides a more detailed insight into the sensitivity of order parameters to motif distortion, and it provides the necessary data for the next validation level. Third, we calculate motif-recognition likelihoods based on the histograms of the second validation level, which provides a first reasonable value of the tolerance, δ, that is later used for neighbor finding in materials from a database.
In Figure 4, we display the response of the tetrahedral (Figure 4A) and octahedral (Figure 4B) order parameter to perturbations of a single tagged neighbor in the respective perfect structure motif along the azimuth (orange) and polar angle (blue) as well as along an intermediate path (purple). The order parameters decline from 1 almost linearly to approximately 0.37 and 0.8 for polar and azimuth angular perturbations up to 27° and 18° for qtet and qoct, respectively. The response along the intermediate perturbation path is similar. This indicates that the OPs can be used as (linear) measures of tetrahedrality and octahedrality for non-negligible perturbations of single neighbors, making the tetrahedral OP, for example, also attractive for structure analysis of liquid and solid water phases (Tao et al., 2017). Figure 4 indicates that configurations with two atoms nearly overlapping score relatively high, which, however, does not pose a problem because those arrangements are very unlikely (i.e., two atoms will not be present at such close distances).
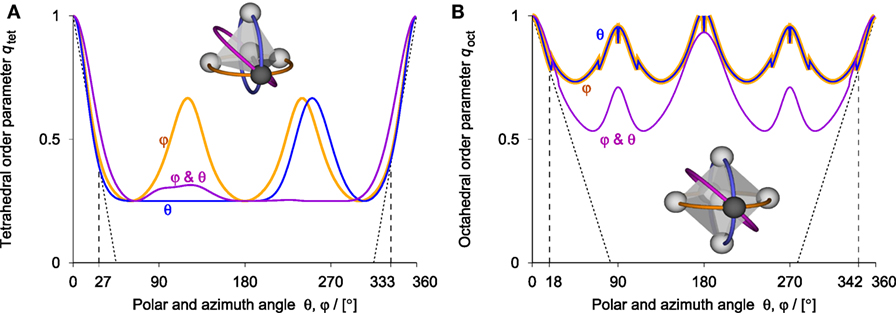
Figure 4. Responses of (A) tetrahedral, qtet, and (B) octahedral order parameter, qoct, to well-defined perturbations of a single neighbor in the corresponding perfect structure motif along the azimuth (orange) and polar (blue) angle as well as an intermediate path (purple). Peaks at intermediate angles indicate overlapping atoms.
Next, we apply Gaussian-distributed random perturbations to all sites in the prototype structures via the polar form of Box–Muller transforms (Box and Muller, 1958) as implemented in numpy (van der Walt et al., 2011). The perturbations resemble the spatial distribution behavior of atoms in Einstein crystals (Einstein, 1906; Frenkel and Ladd, 1984; Aragones et al., 2012), the procedure of which we therefore call Einstein crystal test rig. Each lattice atom oscillates independently around its equilibrium position with a distribution width of σES. We refer the reader to the Supplementary Material (Section 1.1 in Supplementary Material) for more details on the Einstein crystal calculations. Figure 5 indicates that the OPs respond to Einsteinian perturbations of magnitude σES/did = 0.02 and a relative cutoff radius of rcut/did = 1.1 with relative frequency distributions of finite width. The distributions confirm the validity of both: (i) our proposed structure motif-recognition criteria and (ii) that qtet and qoct are in particular well-behaved measures for the degree of a given motif. The data, however, also suggest that the ability to distinguish between fcc and hcp on the basis of q6 will be limited. Therefore, we redefine those structure motif criteria (equations (11) and (12)) to provide a single criterion for close-packed motifs (cp = fcc + hcp):
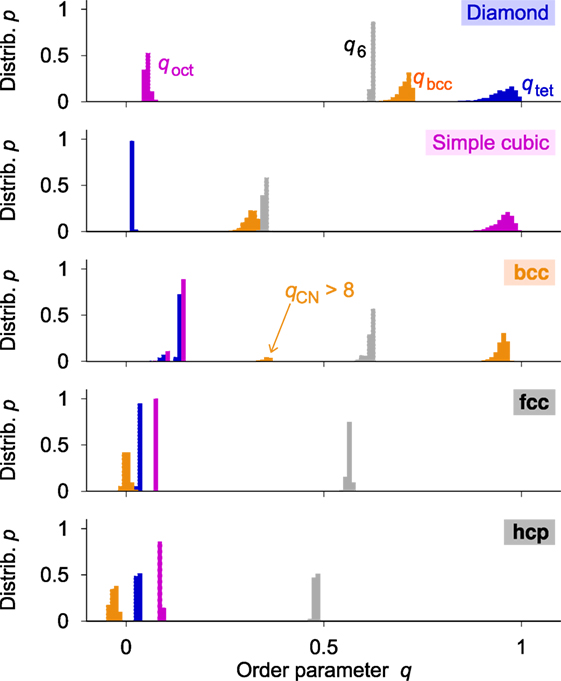
Figure 5. Relative frequency distribution, p, of all four order parameters qtet, qoct, qbcc, and q6 in all five prototype structures diamond, cubic, bcc, fcc, and hcp when the positions of atoms are subject to Einstein crystal-like perturbations away from their ideal location (here: Gaussian width of perturbations σES/did = 0.02, and cutoff radius for neighbor finding relative to the ideal neighbor distance in the perfect prototype structure rcut/did = 1.1). Note that the small intermediate peak at qbcc = 0.35 obtained with the bcc prototype structure (central panel) is caused by configurations that have a non-ideal number of neighbors (qCN > 8)—not by the order parameter definition itself.
In this context, the approach by Honeycutt and Andersen (1987) is worthwhile noting, which compresses information about local environments—specifically, the number of shared near neighbors of a pair of atoms and the connectivity among the shared neighbors—into a four-integer index. This index can, however, not easily be used as an automatic motif recognition tool for hcp–fcc distinction because of two reasons. First, it requires visual inspection of the index-underlying graphs because the fourth integer is an arbitrary enumeration (cf., indices 1,421 and 1,422 occurring in fcc and hcp). Second, the index is computed for pairs of atoms so that the index itself cannot be used to directly characterize the entire coordination environment of a single atom. We are currently working toward solving the hcp–fcc distinction problem via definition of additional order parameters, resulting in an order parameter feature vector that might enable distinction between hcp and fcc.
To quantitatively assess our structure-motif recognition capabilities, we systematically expand the Einstein crystal sensitivity test approach to various distortion degrees, σES, and relative cutoff radii, rcut/did. For this purpose, we use following basic likelihood function (Sivia, 2012), :
where Nstr and Nmot are the number of tested prototype structures and motifs, respectively (both are 4 here: tet, oct, bcc, and cp), Ni,j is the number of times that structure i is recognized as consisting of motif j of all Nsamp samples for i and j (here: Nsamp = 1,000), and is the Kronecker delta function, which is 1 if i = j and 0 otherwise. Since we have merged fcc and hcp to one cp motif, we average data from fcc and hcp evaluations so that each of the four regrouped motifs (tet, oct, bcc, and cp) has an equal weight on . The likelihood function thus represents the joint probability to do both: to correctly identify true motifs and to correctly reject false motifs. Furthermore, the particular form of as products of the separate motif and structure likelihoods more stringently requires that all motifs in all structures are correctly identified and rejected, respectively, to an acceptable degree. A mere arithmetical mean would favor compensation effects in which high recognition scores achieved with one motif and/or structure would balance entirely unsuccessful recognitions with low scores.
There exists a very localized optimal region of parameters where is maximal (close to 1), which is found in the vicinity of small perturbations and small relative cutoff radii (Figure 6A). We expected a more broad distribution at vanishing degrees of distortion, which is, in fact, observable once the results from the bcc prototype structure are removed (Figure 6B). This is a reflection of the well-known issue that, in the bcc structure, second nearest neighbors are close to nearest neighbors (cf., Mickel et al. (2013) and Figure 5). As a result, the bcc issue sets certain limits to using a global tolerance for successful neighbor finding.
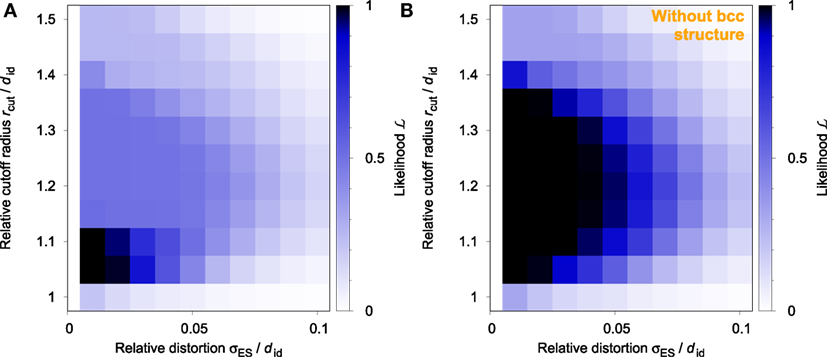
Figure 6. Likelihood, , to successfully identify the correct structural motif in any one of the five prototype structures (diamond, cubic, bcc, fcc, and hcp) with varying degree of relative distortion, σES/did, and varying relative cutoff radius, rcut/did, for finding bonded nearest neighbors: (A) all structures and (B) without bcc structure.
3. Results
We now apply the validated order parameters based structure motif recognition criteria and the order parameters themselves (as a degree of perfect-motif resemblance) to automatically find structure motifs in a large materials database (Jain et al., 2013), determine interstitials (Broberg et al., 2016), and analyze the coordination environment along solid-state jump-diffusion paths (Rong et al., 2015).
3.1. Motif Recognition in Materials Project Database
We aim to apply our structure-motif recognition approach on the entire Materials Project (Jain et al., 2013) database, which currently contains over 67,000 inorganic materials.1 The bulk of Materials Project’s (MP’s) database has originated (Jain et al., 2013) from the Inorganic Crystal Structure Database (Bergerhoff et al., 1983; Belsky et al., 2002). Prior to dissemination, structures are relaxed with electronic density functional theory (Hohenberg and Kohn, 1964; Kohn and Sham, 1965), typically using the Vienna ab initio Simulation Package (VASP (Kresse and Hafner, 1993)) in the generalized gradient approximation (GGA) with + U corrections for transition metal oxides.
3.1.1. Optimizing Neighbor Finding and Motif Criteria
Before turning to the results from the entire database, we determine the most suitable neighbor-finding method and the optimal tolerance parameter, δ, given a reasonable test (sub)set from the MP database. Our initial test set, for which the number of different motif sites, Ni, is well known, consists of materials that are members of the structure groups listed in Table 2. Materials belonging to a given structure group were found by scanning the MP database for structures that are similar to a reference structure. Similarity between structures is hereby determined with a structure matcher algorithm implemented in pymatgen (Ong et al., 2013) using default parameters, except for turning off species matching (i.e., we match the frameworks of the structures). The references were ideal prototype structures in the case of unary materials (diamond-like, simple cubic, bcc, fcc, and hcp) and the canonical structure in the MP database for all other materials (zinc blende: mp-10695; rocksalt: mp-22862; CsCl-like: mp-22851; MgAl2O4-spinel: mp-3536), respectively. For the resulting 1,025 test structures, we calculate the order parameter values qCN, qtet, qoct, qbcc, and q6 of all Nsites sites in each structure. Then, we determine the numbers of different structure motifs (Ntet, Noct, Nbcc, and Ncp) in each material on the basis of our recognition criteria (equations (8)–(10) and (13)) as well as the number of times that a site was assigned to more than a single structure motif (Nmulti).
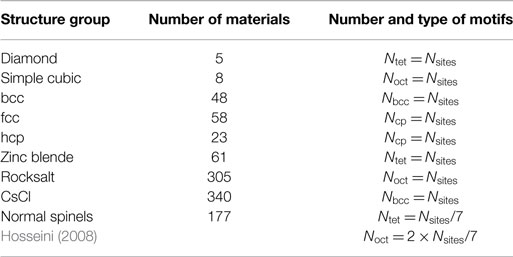
Table 2. Structure groups defining the initial test set.
The different neighbor-finding settings are compared by averaging the fraction, prec,i, of correctly recognized number of expected motifs j, in each structure i, Nj,i,
overall 1,025 test structures. Note that we put a very stringent criterion on multiple assignments: if a structure contains a single site that is assigned to different motifs, the structure has no positive contribution on the recognition fraction (i.e., prec,i = 0). Furthermore, we address the problem that, in spinels, the target number of tetrahedral and octahedral sites, respectively, can be overpredicted by using following functional form for :
The results in Figure 7 indicate that all three neighbor-finding approaches yield high recognition rates (>85%) for the chosen test set if the respective tolerance parameter is small enough (≪0.1). Furthermore, the prediction quality decreases precipitously when the neighbor-finding tolerance, δ, reaches a value of around 0.1 for any of the three methods. The best performing method is the minimum distance-based approach with 0.03 ≤ δ ≤ 0.08. For all following analyses, we use this method where we, however, employ a slightly larger tolerance (δ = 0.1) because this tolerance yields a very low average coordination number of 3 for all sites in the entire Materials Project database.
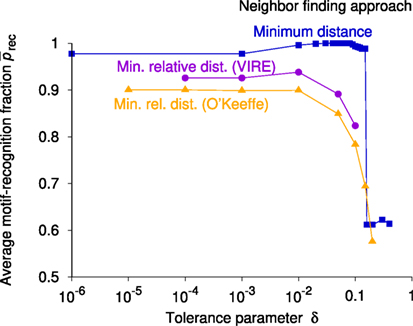
Figure 7. Average fractions of sites for which the expected motif types are correctly predicted, , as functions of the tolerance parameter, δ, involved in the respective neighbor-finding method. Three different neighbor-finding methods are tested: minimum distance (blue squares), minimum relative distance using a valence-ionic radius estimator (VIRE; purple circles) as implemented in pymatgen (Ong et al., 2013), and minimum relative distance using the bond-valence parameters according to O’Keeffe and Brese (1991), orange triangles.
Apart from being effective, the minimum distance-based neighbor-finding method is also computationally exceptionally efficient. Calculating all order parameters requires only 0.026 s per site on a compute node of NERSC’s2 Edison cluster (time was averaged over all sites in the MP database). That is, analyzing a structure with 100 sites should take 2–3 s, assuming the entire procedure roughly scales linearly with the number of sites. Note (i) that we decrease the tetrahedral OP threshold to 0.3 because tests on Jahn–Teller active structures suggest 0.5 being too strict and (ii) that we also require the coordination number, qCN, to match the ideal motif value. Thus, the final set of rules that we use for motif determination across the entire Materials Project database are:
In Algorithm 1, we also provide a pseudocode implementation of our motif recognition method.

Algorithm 1. Motif recognition.
3.1.2. Relative Motif Occurrence
The MP database screening results are presented in Figure 8A as relative motif occurrences for each element separately, and they agree with several previous observations (Cotton and Wilkinson, 1980; Brown, 1988). For example, Li occurs similarly often as a tetrahedral site as it occurs as an octahedral site, but bcc-like Li motifs are rare and close-packed ones are absent. These motif frequencies follow trends of Li for 4-, 6-, 8-, and 12-fold coordination that were already established by Brown (1988). Furthermore, we see comparably good agreement between motif occurrence and Brown’s coordination statistics for Na, Ca, Ba, Mn, Fe, Co, B, As, and La. Examples with some differences but still overall favorable agreement include K, Sr, Pb, Ni, Cd, Sb, and Bi, whereas we obtain very different dominant motif vs coordination number distributions for Tl, Hg, Al, and In. In light of these differences, we note that our material set is far larger (and more diverse) than the set that Brown used; there is a 100-fold difference between the number of sites that we have analyzed (>15,000,000) in comparison to the previous study by Brown (1988) (14,000).
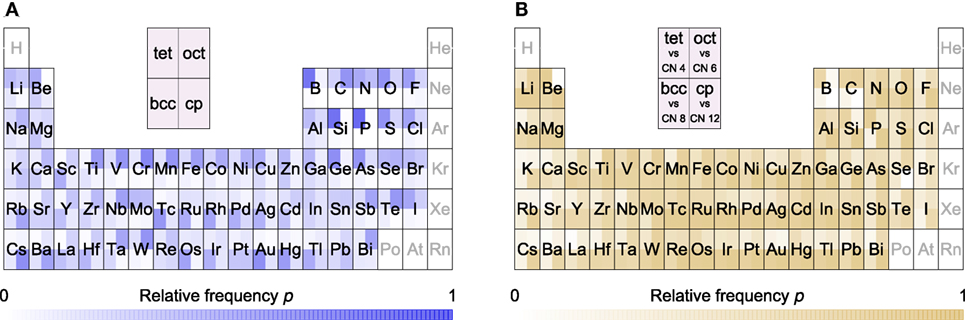
Figure 8. (A) Relative occurrences of motifs (tetrahedra, octahedra, bcc-like, and close packed) for each element in the Materials Project (MP) database; (B) relative occurrence of each recognizable motif (e.g., tet) to all local environments with the corresponding coordination number (e.g., 4). Note that each site contributes with a factor of 1/Nsites, where Nsites denotes the total number of sites (atoms) found in the primitive unit cell of the material in question (i.e., the frequencies are materials weighted, not site-weighted). Also, note that we filtered out 9% of the MP structures because we discarded noble gas-containing materials and materials that seem very unstable (energy above the convex hull of 0.2 eV per atom or more); in addition, we exclude the results for hydrogen sites. Section 1.2 in Supplementary Material lists how many sites of a given element were used for generating these figures.
To continue the discussion of how our structure motif data relate to results from literature and common wisdom we note that (i) S and Se occur in tetrahedral coordination, (ii) S, Se, and Te can be seen in octahedra, but (iii) 8-fold coordination only occurs for Te and (iv) close-packed motifs of S, Se, and Te are not known (Cotton and Wilkinson, 1980). Silicon is known as a strong tetrahedron-former (e.g., in zeolites (Baerlocher et al., 2007)). This is confirmed when comparing both panels in Figure 8; Figure 8B gives the frequency at which a motif (here: tet) occurs relative to all motifs—recognizable and unrecognizable—with the same coordination number (here: 4). Figure 8B indicates that 86% of all 4-fold coordinated Si motifs are tetrahedra. Surprisingly, the ratio at which lithium occurs in 4-fold coordination as a tetrahedron is low (47%). By contrast, the low tetrahedral coordination fraction of Ag and Cu motifs (both 41%) was expected because these elements are frequently found in square-planar environments.
An intriguing observation for us is that the known decline in tetrahedral site preference (Navrotsky and Kleppa, 1967; Burdett et al., 1982; Rong et al., 2015) as we go from Li over Mg to Ca in spinel is reflected in the MP database as a whole. The relative occurrence of Li as a tetrahedron among all 4 recognizable motifs is is 42%, Mg 20%, and Ca 13%. Zn typically takes a place between Li and Mg in spinels (Rong et al., 2015), which is, however, different in the MP database (46%).
The unexpected motif prevalences that are observed for some elements may hint at the fact that our neighbor finding method could benefit from further study and improvement. However, the overall approach represents a fast, fully automatic method to determine coordination environment motifs over large crystal databases that is relatively trustworthy and intuitive.
3.2. Interstitial Finding
Interstitials in dense inorganic materials are frequently found at sites that resemble basic structural motifs. Tetrahedral and octahedral coordination environments are particularly prevailing in isolated (i.e., non-complex) interstitials. This is evidenced by a series (Decoster et al., 2008, 2009a,b, 2010a,b, 2012; Pereira et al., 2011, 2012; Amorim et al., 2013; Silva et al., 2014) of β− emission channeling measurements (Hofsäss and Lindner, 1991; Silva et al., 2013) which were conducted at CERN’s ISOLDE beamline. Those measurements inspired us to develop our Interstitialcy Finding Tool (InFiT), which is already used by the Python Charged Defect Tools (PyCDT)—a Python package for automatic setup and analysis of isolated charged defect calculations (Broberg et al., 2016).
The key idea of InFiT is to perform a systematic structure-motif search on a regular grid (Δl ≈ 0.2 Å) that is spanned in the unit cell of a periodic material (Figure 9). For each point of the grid that is not closer than 1 Å to any crystal atom, the algorithm (Broberg et al., 2016) goes through following steps:
• Place an atom of the target interstitial type at this trial point.
• Perform a loop of increasing neighbor finding tolerance, δ, starting from 0.1 in steps of 0.1 up to δ = 0.8:
• Get all neighbors and determine motif type.
• If the motif type is recognized, consider the trial position for further evaluation, store the corresponding order parameter value, qi, and stop the δ-loop.
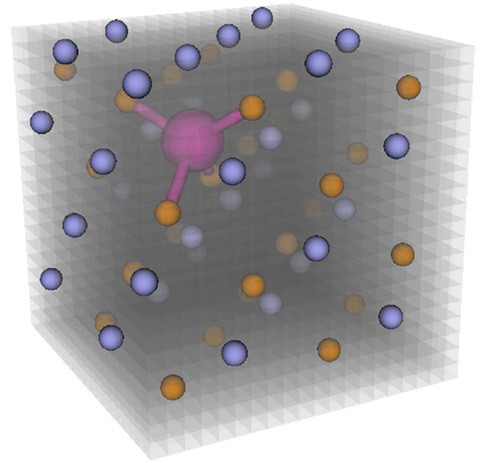
Figure 9. The Interstitialcy Finding Tool (InFiT) performs structure-motif recognition on a regular grid in the unit cell.
After a list of tentative interstitial sites is thus created, two pruning measures are taken. First, a distance-based clustering of the trial sites is performed. From each resulting motif-specific cluster, only one site is retained: the one with the highest order parameter value for the given motif type. Second, the (typically few) remaining sites are tested for (and pruned by) symmetrical equivalence.
There exists one symmetrically distinct tetrahedral interstitial site in diamond-like materials (Decoster et al., 2008), bcc (Seletskaia et al., 2005), fcc (Rosato et al., 1989), as well as hcp (Igarashi et al., 1991), where the latter three prototype structures also have each one symmetrically distinct octahedral site. Furthermore, 2 tetrahedral sites are (geometrically) possible in zinc blende-like materials, each one with different (host) atom coordination. Therefore, we use Er in diamond-like Ge (cf., Decoster et al. (2008)), Mn in sphalerite-like GaAs (cf., Pereira et al. (2011)), He in iron (bcc structure; cf., Seletskaia et al. (2005)), self-interstitials in Cu (fcc structure; cf., Rosato et al. (1989)), as well as O in α-Ti (hcp structure; cf., Yu et al. (2015)) to test our interstitial finding tool InFiT. The results in Table 3 and Figure 10 indicate that, apart from hcp-Ti, we always find the correct number and type of sites. For hcp-Ti, we find 2 similarly looking tetrahedral interstitials, which only get identified as symmetrically equivalent when the corresponding symmetry threshold is increased significantly (i.e., to three times the largest distance between any two face-connected grid points). As for the octahedral interstitials in Ti, we find the expected interstitial with a usual value of the neighbor-finding threshold parameter (δ = 0.2). However, we also find two very distorted octahedra with as large a δ as 0.8, the observation of which is used here to define a maximum reasonable value of δ (<0.8). A relative high neighbor-finding threshold (0.7) is desirable in some case, as the snapshots and complementary data for Fe in Figure 10 highlight (cf., octahedral site). That example also underlines that a low tetrahedral OP threshold (0.3) can be necessary to find interstitials.
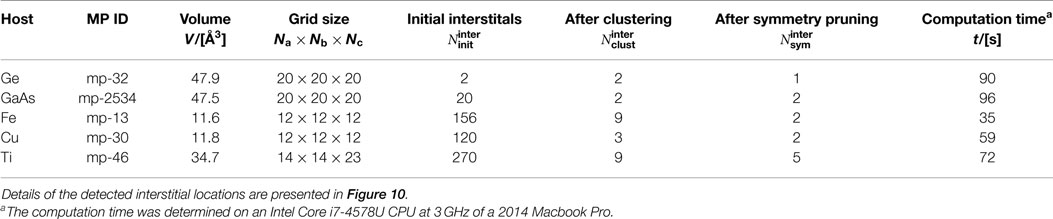
Table 3. Interstitials found in primitive unit cells.
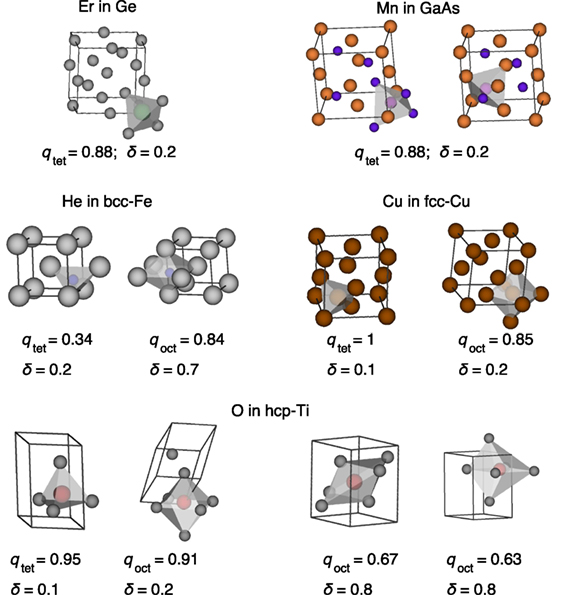
Figure 10. Interstitials found in five different materials with our tool InFiT (here: conventional unit cell for clarity).
Finally, we highlight three additional points with respect to InFiT. First, Table 3 shows the effectiveness of the clustering prune step, especially for the dense metals (from several 100 points to less than 20). Second, on an Intel Core i7-4578U CPU at 3 GHz, the interstitial finding took between 35 and 96 s for the five test systems (cf., Table 3), which translates into 0.011–0.034 s per grid point. And third, we successfully tested 19 additional diamond and sphalerite-like structures that are frequently investigated in the context of charged defects, and we always obtained the expected number of tetrahedral interstitials (1 and 2 for diamond and sphalerite-like materials, respectively; cf., Section 1.3 in Supplementary Material).
3.3. Diffusion Path Characterization
The ease of ion migration through an intercalant host can be often correlated with specific coordination environments—CN and pattern—(Rong et al., 2015). Since both tetrahedral and octahedral coordination play particularly important roles in predicting ion transport through promising new cathode materials for rechargeable batteries (Rong et al., 2015), we show the usefulness of the two order parameters qtet and qoct on the example of magnesium jump-diffusion hops in (empty) spinel manganese oxide (mvc-15009). The jump-diffusion path is obtained from a method that Zimmermann, Haranczyk, and co-workers are currently aiming to publish: the potential of electrostatics finite ion size (PfEFIS) method. It reliably estimates migration barriers deduced from nudged-elastic band (NEB; Mills et al. (1995)) calculations on the basis of electrostatic data (estimate SE around 50 meV). For our particular example, the estimate gives 711 meV (Figure 11; black circles and line), which agrees well with the NEB barrier (776 meV; cf., Rong et al. (2015)) while achieving a speed-up factor of 10,000. The diffusion path for Mg in spinel goes from a tetrahedral site over an intermediate octahedral site back to a tetrahedral site (Rong et al., 2015). The solid blue line in Figure 11 indicates the change in qtet along the path, whereas the dotted orange line depicts the change of the octahedral OP, qoct. Clearly, the order parameters help visualize the change in coordination environment along the diffusion path—in a quantitative and physically meaningful (Wu et al., 2017) way.
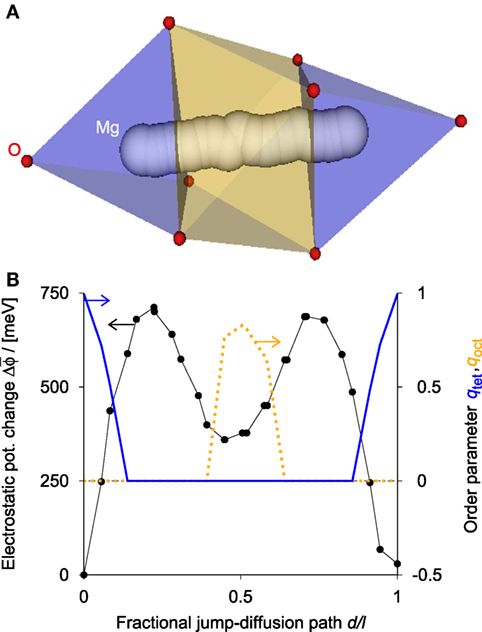
Figure 11. (A) A magnesium ion (white spheres) hops in Mn2O4-spinel from a site at which it is coordinated by four oxygen atoms (red spheres) in a tetrahedral manner (blue polyhedron) through an intermediate O-octahedron (orange) back into a tetrahedral site. (B) Change in electrostatic potential, (left abscissa, black circles and line) and tetrahedral (blue solid line), qtet, as well as octahedral order parameter (orange dotted line), qoct (both: right abscissa) as functions of the fractional jump-diffusion path, d/l. Note that the electrostatic potential is averaged over a spherical volume of radius 1.475 Å; Zimmermann and co-workers are currently aiming to publish the underlying method. Furthermore, our diffusion path characterization method is robust as the additional spinel examples in the Supplemental Material (Figure S2 in Supplementary Material) underscore.
4. Conclusion
We have shown here that order parameters (Steinhardt et al., 1983; Peters, 2009; Zimmermann et al., 2015), when paired with efficient and effective neighbor finding methods, can be reliably used as fast automatic structure-motif finding and coordination environment assessment tools, regardless of a material’s chemistry. We introduced an effective validation framework—the Einstein crystal test rig—which subjects all atoms in a (prototype) structure to well-defined (random) distortions, thus, systematically sounding out the robustness of any motif recognition approach. We then applied our approach successfully to three important applications in (computational) materials science: (i) mapping the structural character of a materials database via element-specific relative structure-motif occurrence plots, (ii) effective interstitial finding (InFiT tool developed here; cf., Broberg et al. (2016)), and ion jump-diffusion path characterization (Rong et al., 2015). Our effective and efficient motif-recognition and assessment capabilities are freely available through the Python package pymatgen (Ong et al., 2013).3 We ultimately emphasize that materials science is currently undergoing a “change of paradigm: from description to prediction” (Heine, 2014). Thus, we expect these tools to be useful in future machine-learning (Jain et al., 2016; Ward and Wolverton, 2017) applications as descriptors that capture much of the most basic—but essential (Wagner and Rondinelli, 2016)—information of a given material: the crystal structure.
Author Contributions
NZ, with continuous advice from both AJ and MH, developed the main concept of the study, conducted the calculations as well as the (majority of) tests and analyses, and prepared the manuscript. MKH performed additional tests of structure motif recognition. All authors discussed the results and implications and commented extensively on the manuscript at all stages.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer, JJ, and handling Editor declared their shared affiliation.
Acknowledgments
We thank Alireza Faghaninia for helpful comments on the symmetry pruning implementation of InFiT.
Funding
This work was intellectually led by the U.S. Department of Energy (DOE) Basic Energy Sciences (BES) program—the Materials Project—under Grant No. EDCBEE. MKH acknowledges support of BASF Corporation through the California Research Alliance. This research used resources of the National Energy Research Scientific Computing Center, which is supported by the Office of Science of the U.S. Department of Energy under Contract No. DEAC02-05CH11231. Lawrence Berkeley National Laboratory is funded by the DOE under award DE-AC02-05CH11231.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/article/10.3389/fmats.2017.00034/full#supplementary-material.
Footnotes
- ^Current numbers are available on the Materials Project website: http://materialsproject.org/.
- ^NERSC: National Energy Research Scientific Computing Center (http://www.nersc.gov/).
- ^The pymatgen library is, for example, available via GitHub: https://github.com/materialsproject/pymatgen/.
References
Ackland, G. J., and Jones, A. P. (2006). Applications of local crystal structure measures in experiment and simulation. Phys. Rev. B 73, 054104. doi:10.1103/PhysRevB.73.054104
Amorim, L. M., Wahl, U., Pereira, L. M. C., Decoster, S., Silva, D. J., da Silva, M. R., et al. (2013). Precise lattice location of substitutional and interstitial Mg in AlN. Appl. Phys. Lett. 103, 262102. doi:10.1063/1.4858389
Aragones, J. L., Sanz, E., and Vega, C. (2012). Solubility of NaCl in water by molecular simulation revisited. J. Chem. Phys. 136, 244508. doi:10.1063/1.4728163
Baerlocher, C., McCusker, L. B., and Olsen, D. H. (2007). Atlas of Zeolite Framework Types, 6th Edn. Amsterdam, The Netherlands: Elsevier.
Belsky, A., Hellenbrandt, M., Karen, V. L., and Luksch, P. (2002). New developments in the inorganic crystal structure database (ICSD): accessibility in support of materials research and design. Acta Cryst. B 58, 364–369. doi:10.1107/S0108768102006948
Bergerhoff, G., Hundt, R., Sievers, R., and Brown, I. D. (1983). The inorganic crystal structure data base. J. Chem. Inf. Comput. Sci. 23, 66–69. doi:10.1021/ci00038a003
Box, G. E. P., and Muller, M. E. (1958). A note on the generation of random normal deviates. Ann. Math. Stat. 29, 610–611. doi:10.1214/aoms/1177706645
Broberg, D., Medasani, B., Zimmermann, N. E. R., Canning, A., Haranczyk, M., Asta, M., et al. (2016). PyCDT: A python toolkit for modeling point defects in semiconductors and insulators. arXiv: 1611.07481.
Brown, I. D. (1988). What factors determine cation coordination numbers? Acta Cryst. B Struct. Sci. 44, 545–553. doi:10.1107/S0108768188007712
Brunner, G. O. (1977). Definition of coordination and its relevance in structure types AlB2 and NiAs. Acta Cryst. A33, 226–227. doi:10.1107/S0567739477000461
Burdett, J. K., Price, G. D., and Price, S. L. (1982). Role of the crystal-field theory in determining the structures of spinels. J. Am. Chem. Soc. 104, 92–95. doi:10.1021/ja00365a019
Cotton, F. A., and Wilkinson, G. (1980). Advance Inorganic Chemistry – A Comprehensive Text, 4 Edn. New York, USA: John Wiley & Sons.
Decoster, S., Cottenier, S., De Vries, B., Emmerich, H., Wahl, U., Correia, J. G., et al. (2009a). Transition metal impurities on the bond-centered site in germanium. Phys. Rev. Lett. 102, 065502. doi:10.1103/PhysRevLett.102.065502
Decoster, S., De Vries, B., Wahl, U., Correia, J. G., and Vantomme, A. (2009b). Lattice location study of implanted In in Ge. J. Appl. Phys. 105, 083522. doi:10.1063/1.3110104
Decoster, S., Cottenier, S., Wahl, U., Correia, J. G., Pereira, L. M. C., Lacasta, C., et al. (2010a). Diluted manganese on the bond-centered site in germanium. Appl. Phys. Lett. 97, 151914. doi:10.1063/1.3501123
Decoster, S., Cottenier, S., Wahl, U., Correia, J. G., and Vantomme, A. (2010b). Lattice location study of ion implanted Sn and Sn-related defects in Ge. Phys. Rev. B 81, 155204. doi:10.1103/PhysRevB.81.155204
Decoster, S., De Vries, B., Wahl, U., Correia, J. G., and Vantomme, A. (2008). Experimental evidence of tetrahedral interstitial and bond-centered Er in Ge. Appl. Phys. Lett. 93, 141907. doi:10.1063/1.2996280
Decoster, S., Wahl, U., Cottenier, S., Correia, J. G., Mendonça, T., Amorim, L. M., et al. (2012). Lattice position and thermal stability of diluted As in Ge. J. Appl. Phys. 111, 053528. doi:10.1063/1.3692761
Dirichlet, G. L. (1850). Über die Reduction der positiven quadratischen Formen mit drei unbestimmten ganzen Zahlen. J. Reine Angew. Math. 40, 209–227. doi:10.1515/crll.1850.40.209
Einstein, A. (1906). Die Plancksche Theorie der Strahlung und die Theorie der spezifischen Wärme. Ann. Phys. 22, 180–190. doi:10.1002/andp.19063270110
Frenkel, D., and Ladd, A. J. C. (1984). New Monte Carlo method to compute the free energy of arbitrary solids. Application to the fcc and hcp phases of hard spheres. J. Chem. Phys. 81, 3188–3193. doi:10.1063/1.448024
Gasser, U., Schofield, A., and Weitz, D. A. (2003). Local order in a supercooled colloidal fluid observed by confocal microscopy. J. Phys. Condens. Matter 15, S375–S380. doi:10.1088/0953-8984/15/1/351
Heine, T. (2014). Grand challenges in computational materials science: from description to prediction at all scales. Front. Mater. 1:7. doi:10.3389/fmats.2014.00007
Hofsäss, H., and Lindner, G. (1991). Emission channeling and blocking. Phys. Rep. 201, 121–183. doi:10.1016/0370-1573(91)90121-2
Hohenberg, P., and Kohn, W. (1964). Inhomogeneous electron gas. Phys. Rev. B 136, B864–B871. doi:10.1103/PhysRev.136.B864
Honeycutt, J. D., and Andersen, H. C. (1987). Molecular dynamics study of melting and freezing of small Lennard–Jones clusters. J. Phys. Chem. 91, 4950–4963. doi:10.1021/j100303a014
Hoppe, R. (1979). Effective coordination numbers (ECoN) and mean fictive ionic radii (MEFIR). Z. Kristallogr. 150, 23–52. doi:10.1524/zkri.1979.150.1-4.23
Hosseini, S. M. (2008). Structural, electronic and optical properties of spinel MgAl2O4 oxide. Phys. Stat. Sol. B 245, 2800–2807. doi:10.1002/pssb.200844142
Igarashi, M., Khantha, M., and Vitek, V. (1991). N-body interatomic potentials for hexagonal close-packed metals. Philos. Mag. B 63, 603–627. doi:10.1080/13642819108225975
Isayev, O., Fourches, D., Muratov, E. N., Oses, C., Rasch, K., Tropsha, A., et al. (2015). Materials cartography: representing and mining materials space using structural and electronic fingerprints. Chem. Mater. 27, 735–743. doi:10.1021/cm503507h
Jain, A., Hautier, G., Ong, S. P., and Persson, K. (2016). New opportunities for materials informatics: resources and data mining techniques for uncovering hidden relationships. J. Mater. Res. 31, 977–994. doi:10.1557/jmr.2016.80
Jain, A., Ong, S. P., Hautier, G., Chen, W., Richards, W. D., Dacek, S., et al. (2013). Commentary: The Materials Project: a materials genome approach to accelerating materials innovation. APL Mater. 1, 011002. doi:10.1063/1.4812323
Jungblut, S., Singraber, A., and Dellago, C. (2013). Optimising reaction coordinates for crystallisation by tuning the crystallinity definition. Mol. Phys. 111, 3527–3533. doi:10.1080/00268976.2013.832820
Kohn, W., and Sham, L. J. (1965). Self-consistent equations including exchange and correlation effects. Phys. Rev. A 140, A1133–A1138. doi:10.1103/PhysRev.140.A1133
Kresse, G., and Hafner, J. (1993). Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561. doi:10.1103/PhysRevB.47.558
Li, X., Xu, Y., and Wang, C. (2009). Suppression of Jahn–Teller distortion of spinel LiMn2O4 cathode. J. Alloys Compd. 479, 310–313. doi:10.1016/j.jallcom.2008.12.081
Li, Y., Yu, J., and Xu, R. (2013). Criteria for zeolite frameworks realizable for target synthesis. Angew. Chem. Int. Ed. 52, 1673–1677. doi:10.1002/anie.201206340
Limmer, D. T., and Chandler, D. (2013). The putative liquid-liquid transition is a liquid-solid transition in atomistic models of water. II. J. Chem. Phys. 138, 214504. doi:10.1063/1.4807479
Mazur, M., Wheatley, P. S., Navarro, M., Roth, W. J., Položij, M., Mayoral, A., et al. (2015). Synthesis of ‘unfeasible’ zeolites. Nat. Chem. 8, 58–62. doi:10.1038/NCHEM.2374
Mickel, W., Kapfer, S. C., Schröder-Turk, G. E., and Mecke, K. (2013). Shortcomings of the bond orientational order parameters for the analysis of disordered particulate matter. J. Chem. Phys. 138, 044501. doi:10.1063/1.4774084
Mills, G., Jónsson, H., and Schenter, G. K. (1995). Reversible work transition state theory: application to dissociative adsorption of hydrogen. Surf. Sci. 324, 305–337. doi:10.1016/0039-6028(94)00731-4
Morris, J. W. Jr. (2007). A Survey of Materials Science I. Structure. Berkeley, USA: University of California.
Navrotsky, A., and Kleppa, O. J. (1967). Thermodynamics of cation distributions in simple spinels. J. Inorg. Nucl. Chem. 29, 2701–2714. doi:10.1016/0022-1902(67)80008-3
O’Keeffe, M. (1979). Proposed rigorous definition of coordination number. Acta Cryst. A 35, 772–775. doi:10.1107/S0567739479001765
O’Keeffe, M., and Brese, N. E. (1991). Atom sizes and bond lengths in molecules and crystals. J. Am. Chem. Soc. 113, 3226–3229. doi:10.1021/ja00009a002
Ong, S. P., Richards, W. D., Jain, A., Hautier, G., Kocher, M., Cholia, S., et al. (2013). Python materials genomics (pymatgen): a robust, open-source python library for materials analysis. Comput. Mater. Sci. 68, 314–319. doi:10.1016/j.commatsci.2012.10.028
Pereira, L. M. C., Wahl, U., Decoster, S., Correia, J. G., Amorim, L. M., da Silva, M. R., et al. (2012). Stability and diffusion of interstitial and substitutional Mn in GaAs of different doping types. Phys. Rev. B 86, 125206. doi:10.1103/PhysRevB.86.125206
Pereira, L. M. C., Wahl, U., Decoster, S., Correia, J. G., da Silva, M. R., Vantomme, A., et al. (2011). Direct identification of interstitial Mn in heavily p-type doped GaAs and evidence of its high thermal stability. Appl. Phys. Lett. 98, 201905. doi:10.1063/1.3592568
Peters, B. (2009). Competing nucleation pathways in a mixture of oppositely charged colloids: out-of-equilibrium nucleation revisited. J. Chem. Phys. 131, 244103. doi:10.1063/1.3271024
Rong, Z., Malik, R., Canepa, P., Gautam, G. S., Liu, M., Jain, A., et al. (2015). Materials design rules for multivalent ion mobility in intercalation structures. Chem. Mater. 27, 6016–6021. doi:10.1021/acs.chemmater.5b02342
Rosato, V., Guillope, M., and Legrand, B. (1989). Thermodynamical and structural properties of f.c.c. transition metals using a simple tight-binding model. Philos. Mag. A 59, 321–336. doi:10.1080/01418618908205062
Santiso, E. E., and Trout, B. L. (2011). A general set of order parameters for molecular crystals. J. Chem. Phys. 134, 064109. doi:10.1063/1.3548889
Seletskaia, T., Osetsky, Y., Stoller, R. E., and Stocks, G. M. (2005). Magnetic interactions influence the properties of helium defects in iron. Phys. Rev. Lett. 94, 046403. doi:10.1103/PhysRevLett.94.046403
Shannon, R. D. (1976). Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. A32, 751–767. doi:10.1107/S0567739476001551
Shetty, R., Escobedo, F. A., Choudhary, D., and Clancy, P. (2002). A novel algorithm for characterization of order in materials. J. Chem. Phys. 117, 4000–4009. doi:10.1063/1.1494986
Silva, D. J., Wahl, U., Correia, J. G., Pereira, L. M. C., Amorim, L. M., da Silva, M. R., et al. (2014). Lattice location and thermal stability of implanted nickel in silicon studied by on-line emission channeling. J. Appl. Phys. 115, 023504. doi:10.1063/1.4861142
Silva, M. R., Wahl, U., Correia, J. G., Amorim, L. M., and Pereira, L. M. C. (2013). A versatile apparatus for on-line emission channeling experiments. Rev. Sci. Instrum. 84, 073506. doi:10.1063/1.4813266
Sivia, D. S. (2012). Data Anlaysis – A Bayesian Tutorial, 2 Edn. Oxford, UK: Oxford Science Publishing.
Sprik, M. (1998). Coordination numbers as reaction coordinates in constrained molecular dynamics. Faraday Discuss. 110, 437–445. doi:10.1039/a801517a
Steinhardt, P. J., Nelson, D. R., and Ronchetti, M. (1983). Bond-orientational order in liquids and glasses. Phys. Rev. B 28, 784–805. doi:10.1103/PhysRevB.28.784
Tao, Y., Zou, W., Jia, J., Li, W., and Cremer, D. (2017). Different ways of hydrogen bonding in water – why does warm water freeze faster than cold water? J. Chem. Theory Comput. 13, 55–76. doi:10.1021/acs.jctc.6b00735
ten Wolde, P. R., Ruiz-Montero, M. J., and Frenkel, D. (1996). Numerical calculation of the rate of crystal nucleation in a Lennard–Jones system at moderate undercooling. J. Chem. Phys. 104, 9932–9947. doi:10.1063/1.471721
Tropsha, A., Gramatica, P., and Gombar, V. K. (2003). The importance of being earnest: validation is the absolute essential for successful application and interpretation of QSPR models. QSAR Comb. Sci. 22, 69–77. doi:10.1002/qsar.200390007
van der Walt, S., Colbert, S. C., and Varoquaux, G. (2011). The NumPy array: a structure for efficient numerical computation. Comput. Sci. Eng. 13, 22–30. doi:10.1109/MCSE.2011.37
Voronoi, G. (1908). Nouvelles applications des paramètres continus à la théorie des formes quadratiques. Sur quelques propriétés des formes quadratiques positives parfaites. J. Reine Angew. Math. 133, 97–178.
Wagner, N., and Rondinelli, J. M. (2016). Theory-guided machine learning in materials science. Front. Mater. 3:28. doi:10.3389/fmats.2016.00028
Wang, Y., Richards, W. D., Ong, S. P., Miara, L. J., Kim, J. C., Mo, Y., et al. (2015). Design principles for solid-state lithium superionic conductors. Nat. Mater. 14, 1026–1031. doi:10.1038/NMAT4369
Ward, L., and Wolverton, C. (2017). Atomistic calculations and materials informatics: a review. Curr. Opin. Solid State Mater. Sci. 21, 167–176. doi:10.1016/j.cossms.2016.07.002
Werner, A. (1912). Zur Kenntnis des asymmetrischen Kobaltatoms. V. Ber. Deu. Chem. Gesell. 45, 121–130. doi:10.1002/cber.19120450116
Wu, H., Lorenson, A., Anderson, B., Witteman, L., Wu, H., Meredig, B., et al. (2017). Robust fcc solute diffusion predictions from ab-initio machine learning methods. Comput. Mater. Sci. 134, 160–165. doi:10.1016/j.commatsci.2017.03.052
Yu, Q., Qi, L., Tsuru, T., Traylor, R., Rugg, D., Morris, J. W. Jr., et al. (2015). Origin of dramatic oxygen solute strengthening effect in titanium. Science 347, 635–639. doi:10.1126/science.1260485
Zimmermann, N. E. R., and Haranczyk, M. (2016). History and utility of zeolite framework-type discovery from a data-science perspective. Cryst. Growth Des. 16, 3043–3048. doi:10.1021/acs.cgd.6b00272
Keywords: materials science, crystal structure, descriptors, databases, interstitials, intercalation, diffusion
Citation: Zimmermann NER, Horton MK, Jain A and Haranczyk M (2017) Assessing Local Structure Motifs Using Order Parameters for Motif Recognition, Interstitial Identification, and Diffusion Path Characterization. Front. Mater. 4:34. doi: 10.3389/fmats.2017.00034
Received: 13 July 2017; Accepted: 18 October 2017;
Published: 13 November 2017
Edited by:
Zhenyu Li, University of Science and Technology of China, ChinaReviewed by:
Yi Liu, Shanghai University, ChinaZhen Zhou, Nankai University, China
Jun Jiang, University of Science and Technology of China, China
Copyright: © 2017 Zimmermann, Horton, Jain and Haranczyk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nils E. R. Zimmermann, bmVyekBsYmwuZ292