 Francois-Marie Allioux
Francois-Marie Allioux Brendan J. Holland2
Brendan J. Holland2 Ludovic F. Dumée
Ludovic F. Dumée- 1Institute for Frontier Materials, Deakin University, Geelong, VIC, Australia
- 2Centre for Chemistry and Biotechnology, School of Life and Environmental Sciences, Deakin University, Geelong, VIC, Australia
Biodiesel is a growing alternative to petroleum fuels and is produced by the catalyzed transesterification of fats in presence of an alcohol base. Transesterification processes using homogeneous catalysts are considered to be among the most efficient methods but rely on the feedstock quality and low water content in order to avoid undesirable saponification reactions. In this work, the electro-catalytic conversion of canola oil to biodiesel in a 1% aqueous methanolic and ethanolic reaction mixture was performed without the addition of external catalyst or cosolvent. An inexpensive stainless steel (SS) electrode and a hybrid SS electrode coated with an ion-exchange resin catalyst were used as cathode materials while the anode was composed of a plain carbon paper. The cell voltages were varied from 10 to 40 V and the reaction temperature maintained at 20 or 40°C. The canola oil conversion rates were found to be superior at 40°C without saponification reactions for cell voltages below 30 V. The conversion rates were as high as 87% for the hybrid electrode and 81% for the plain SS electrode. This work could inspire new process development for the conversion of high water content feedstock for the production of second-generation biodiesel.
Introduction
Biodiesels have become over the past decade a viable energy and renewable alternative to fossil fuel sources (Mardhiah et al., 2017). The culture of non-edible feedstocks or the use of vegetal or animal oils is particularly of interest as a route to value potential waste-products for direct energy generation (Ma and Hanna, 1999). Canola oil was extensively investigated as a first-generation feedstock, particularly, in Australia due to being the second largest exporter of canola seed in the world, with an annual production between 2 to 3 million tons of oilseeds1 (O’Connell et al., 2007). Biodiesels are produced through the transesterification of glycerides including tri, di and monoglycerides present in oils and fats, with alcohol molecules, such as methanol or ethanol (Mardhiah et al., 2017). Biodiesels produced using methanol consist of methyl esters (MEs) with variable carbon chain length and unsaturated bonds related to the oil and fat feedstock used (Yoon et al., 2014). Conventional transesterification of the triglycerides (TGs) with an alcohol (A) is a typical two-phase reaction due to the poor solubility of the alcohol into the TG phase (Tang et al., 2006). In order to increase the yield and rate of the reaction, a homogeneous acid or alkaline catalyst is usually introduced with excess alcohol molecules to shift the reaction equilibrium toward the product side (Ma and Hanna, 1999; Clements and Gerpen, 2004). The catalyst is dissolved in the alcohol phase and the transesterification reaction (Eq. 1) can be expressed as follows (Ma and Hanna, 1999; Dias et al., 2008):
The transesterification reaction of canola oil, for instance, occurs in the methanol phase where sodium methoxides, the reactive species, are formed by reaction of the methanol molecules with the sodium hydroxide catalyst (Sridhar, 1996). However, during the methoxide formation process, water molecules are formed and parasitic saponification reactions may occur, reducing the yield and quality of the final product (Mardhiah et al., 2017). In addition, for such systems, the removal of unreacted catalysts requires extra purification steps which may hamper the competitiveness of the process and increase the production cost of the biodiesels (Huang et al., 2008). Purification of the product esters phase is usually carried out by using warm distilled water baths (50°C) followed by drying with brines such as anhydrous sodium or magnesium sulfate (Sharma et al., 2008). Depending on the feedstock quality and until fulfilment of the standard (EN 14214 or ASTM D 6751), it can take up to 0.4 to 1 l of water to produce 1 l of purified biodiesel (Atadashi et al., 2011).
Various batch and continuous processes were developed for the production of biodiesel including both catalyzed and non-catalyzed systems (Ma and Hanna, 1999). Additions of a cosolvent, such as tetrahydrofuran or methyl tert-butyl ether, were investigated in order to accelerate the reaction rate. However, cosolvent-assisted processes require specific equipment to recover and recycle the solvents and to deal with such toxic volatile compounds in relatively large quantities at industrial levels (Demirbas, 2009). Non-catalytic supercritical processes were also developed at high temperature (350–400°C) and high pressure (>8 MPa). The limitations of supercritical processes include the cost of the equipment and the overall energy consumption making the biodiesel production relatively energy inefficient (Van Kasteren and Nisworo, 2007). Other catalytic processes include the use of enzymes, such as lipase, immobilized onto a material support (Bajaj et al., 2010). While the enzymatic reaction was found to be highly selective and non-water sensitive, the high cost and stability of the enzymes remain challenges to achieve cost competitive and efficient biodiesel production processes (Bajaj et al., 2010). Finally, the use of low temperature or cold plasma technologies were investigated for the conversion of vegetable oil to biodiesel (Abdul-Majeed et al., 2016; Cubas et al., 2016). Experimental corona plasma or plasma jet processes allowed for the injection of partially ionized gas into the vegetable oil and alcohol reaction mixture. Radical species, such as methoxides, were then directly produced in the reactor without the addition of catalysts (Cubas et al., 2016). High conversion rate (>95%) were successfully achieved but required expensive argon gas carrier as well as high voltages for plasma generation (Abdul-Majeed et al., 2016).
Recently, ion-exchange resins have been studied as a reusable heterogeneous catalyst and were shown to be promising means for the production of biodiesel since they offer high esters yield (>90%) and almost stoichiometric TGs to alcohols ratio (1:3.5, TG:A molar ratio). The availability of polymeric ion-exchange resin materials and their relative low cost also make them attractive for such large-scale operations (Shibasaki-Kitakawa et al., 2007, 2011; Atadashi et al., 2011). Both cation- and anion-exchange resins were found to be effective catalysts of the transesterification reaction (Mazzotti et al., 1997; Shibasaki-Kitakawa et al., 2007, 2011). However, continuous production is limited by the need to regenerate the ion-exchange resin bed with concentrated base or acid solutions (Shibasaki-Kitakawa et al., 2007, 2011). In order to tackle these issues, electro-assisted biodiesel production methods have been investigated combining the addition of a limited amount of water (<2 wt% of the reaction mixture) and an electrolyte support [sodium chloride (NaCl)] in the methanolic reaction mixture in presence of either a cosolvent and/or a heterogeneous catalysts (Guan and Kusakabe, 2009; Putra et al., 2016). During the electro-chemical process, the proton (H+) and hydroxide ions (OH−) formed at the surface of the electrodes, from water splitting, were found to act as acid and base catalysts enhancing the reaction rate (Guan and Kusakabe, 2009). Although these methods allow for a high yield of MEs (>95%), both were using a cosolvent to achieve a one-phase solution. In addition, the use of platinum electrodes (Guan and Kusakabe, 2009) or the addition of heterogeneous catalysts in the reaction mixture (Putra et al., 2016, 2017) rendered the methods less competitive compared to conventional homogeneous catalyzed transesterification methods.
Electro-catalytic methods are, however, particularly attractive for the conversion of used cooking oils containing a high weight percentage of water (>5%), which provides a necessary acid and base catalyst source without the parasitic saponification reactions (Guan and Kusakabe, 2009; Putra et al., 2016).
In this work, a novel electro-catalytic method for the production of biodiesel from canola oil using a simple electro-catalytic process in an aqueous methanolic and ethanolic reaction mixture is presented. The transesterification reaction took place in a two-phase reaction mixture where an emulsion of canola oil in the alcohol phase is formed under strong stirring at either 40°C or room temperature. The electro-catalytic reactor was constituted of two electrodes. A 316L stainless steel (SS) mesh and hybrid SS/ion-exchange resin electrode, recently developed by our group, were used as the cathode materials as a plain carbon paper was used for the anode material (Dumée et al., 2014; Allioux et al., 2015). The water-splitting reactions at the surface of the electrodes were used to regenerate the ion-exchange resin coating and provide catalyst to the transesterification reaction. The effects of the applied voltage, electrode materials, reaction time, and temperature on the methyl and ethyl esters yield were studied and the reaction products were analyzed by high-performance liquid chromatography (HPLC).
Materials and Methods
Chemicals
Commercial grade canola oil (Coles brand) was obtained from a local supermarket. Reagent grade methanol and ethanol (ACS, >95% purity), NaCl, and sodium hydroxide were purchased from ChemSupply (SA, Australia) and used for the transesterification reactions. HPLC grade methanol, 2-propanol, and hexane were obtained from Sigma-Aldrich (NSW, Australia) and used for the HPLC analysis.
Electrodes Materials
Stainless steel woven wire mesh (316L grade, 70 × 70 mesh size, 180 ± 10 µm thick) and plain carbon paper (AvCarb® MGL 190, 190 µm thickness) were obtained from FuelCellStore (TX, USA). The hybrid electrodes consisted of the SS woven wire cloth coated with an anion- or cation-exchange resin paste, hybrid AEX, and CEX, respectively. The hybrid electrodes were prepared using a pore filling method across the porous SS wire mesh. The full fabrication details of the hybrid electrodes can be found elsewhere (Dumée et al., 2014; Allioux et al., 2015).
Electro-Catalytic Reactor
The electro-catalytic reactor consisted of an electrolysis cell containing two electrode plates (20 mm × 20 mm) separated by a distance of 30 mm (Figure 1). The working electrode (cathode) was made of the SS wire mesh or the hybrid electrode materials, and the carbon paper was used for the counter-electrode (anode). The electrolysis reaction was carried out using a constant potential varying from 10 to 40 V and was applied between the two electrodes at room temperature or at 40°C and under agitation.
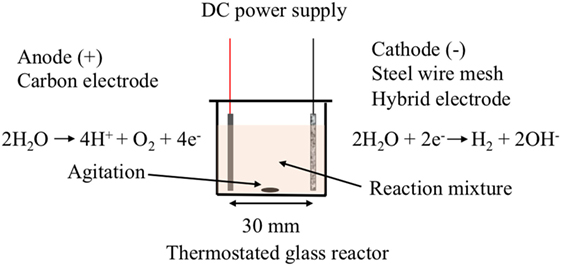
Figure 1. Schematic of the electro-catalysis cell and reactions occurring at the surface of the electrodes.
Reaction Mixtures
The electro-catalytic reactor was filled with 30 ml of a methanolic or ethanolic reaction mixture comprising of canola oil, methanol or ethanol, deionized water (DI water), and NaCl as the supporting electrolyte. The molar ratio of alcohol to canola oil was fixed at 10:1. In addition, 0.3 wt% of NaCl and 1 wt% of DI water was added based on the oil weight and on the total reaction mixture weight, respectively. In order to promote the dissociation of the ionic species, NaCl was first dissolved into the alcohol and water mixture before adding the oil into the electro-catalytic reactor. Conductivity and pH values of the reaction mixtures were recorded prior and post reaction using a conductivity meter (CondTPS, WP-81).
Conventional transesterification of canola oil using a homogeneous catalyst (sodium hydroxide, NaOH) was performed in a closed reactor at 40°C under agitation for 30 min. The canola oil was transesterified using a 10:1 M mixture of alcohol to canola oil ratio with a catalyst concentration of 1.5 wt% of the total oil weight.
HPLC Analysis
Samples Preparation
Small-volume samples of the reaction mixture (1 ml) were collected at the end of the experiment or at regular time intervals. The collected samples were allowed to settle for 24 h in order to separate the ester and glycerol phase by decantation and to allow the excess alcohol to evaporate. Both ester and oil phase were analyzed due to the high water content of the electro-catalytic method which may prevent the separation of MEs from the glycerol and unreacted TGs (Ma and Hanna, 1999). The sample solutions were then dissolved in 2-propanol/hexane (80:20, v/v%) to form 1% (v/v) solutions and filtered through a 0.45-µm pore size nylon syringe filter (Advantec MFS) prior to HPLC analysis.
HPLC Instrument and Methods
The HPLC system (Agilent Technologies 1200 Series liquid chromatography system, Agilent Technologies, VIC, Australia) was equipped with an ultraviolet detector, a solvent degasser, and quaternary pump. Agilent Chemstation software was used for system control and data acquisition. All separations were carried out using an Agilent Poroshell column (2.7 µm; EC-C18; 4.6 mm × 100 mm), with an injection volume of 10 µl. The temperature of the column oven and the wavelength of the detector were 40°C and 205 nm, respectively. A mobile phase gradient consisting of methanol and a 2-propanol/hexane (80:20, v/v%) mixture was pumped at a flow rate of 1.0 cm3 min−1 through the column. Samples were injected into 100% methanol, which was then linearly decreased to 50% methanol and 50% 2-propanol/hexane (80:20, v/v%) over 15 min. Each sample analysis was performed in at least duplicate and the relative error between consecutive measurements was evaluated at 5%. This method has previously been developed and successfully applied for the rapid quantitative determination of the yield of the transesterification reaction (Holčapek et al., 1999). The HPLC chromatograms of the raw canola oil, the methyl, and ethyl esters obtained from the transesterification of canola oil using a homogeneous catalyst are shown in Figure S1A in Supplementary Material. Deviations due to the methanolic and ethanolic reaction mixtures corresponded to approximately 60 AU as shown in Figure S1B in Supplementary Material.
Calculation of the Biodiesel Percentage Yield
The qualitative and semi-quantitative determination of the conversion of canola oil was carried out by comparing the sums of the areas of the peaks corresponding to the MEs and unreacted glycerides (mono-, di-, and triglycerides). The percentage yield was calculated as follows (Holčapek et al., 1999):
The conversion of canola oil to MEs was also assessed based on the volume difference between the initial quantity of oil in the electro-catalytic reactor (t0) and the remaining amounts of oil in the reactor at the end of the experiment (tf). The conversion yield was calculated from the following equation (Dubé et al., 2007):
Materials Characterization
The morphology of the plain carbon, SS wire mesh, and hybrid electrodes was characterized by scanning electron microscopy (JEOL Neoscope, JCM-5000) at 10 kV and 10 mm working distance. The samples were gold coated (model SCD 050 sputter coater, Bal-Tec) prior to imaging with a <5 nm thick layer of gold.
Thermogravimetry (TGA) experiments were performed under air with a Q50 TGA (TA instrument, USA). During the TGA experiments, the gas flow was maintained at 60 cm3 min−1 and the heating rate was fixed at 10°C min−1.
Attenuated total reflectance Fourier transform infrared spectra of the hybrid electrodes before and after electro-catalysis experiments were acquired on a Bruker Vetex-70 FTIR spectrometer and recorded in the range of 600–4,000 cm−1 at a resolution of 4 cm−1. A total of 64 spectra were averaged for each measurement. The hybrid electrodes were dried using Kimwipe® tissues to remove excess solutions prior TGA or FTIR analysis.
Results and Discussion
Electrode Materials Characterization
The SS wire mesh electrode (Figure 2A) consisted of large open mesh made of 100 µm diameter woven fibers with an open porosity of 55%. The hybrid electrode (Figures 2B,C) showed a dense, textured surface with visible ion-exchange resin catalyst embedded within the polymer binder (Allioux et al., 2015). The TGA analysis (Figure 2D) revealed an uptake of reactants and products of 16.45 and 16.75 wt% across the anion- and cation-exchange catalyst, respectively, which was associated to the weight loss up to 210°C for the anion-exchange catalyst and up to 250°C for the cation-exchange catalyst. This first weight loss was followed by the thermal degradation of the functional ion-exchange groups and polymer backbone at higher temperatures (Vinodh et al., 2011). The ion-exchange catalyst coatings were, therefore, stable above 40°C, which corresponded to the maximum temperature used for the electro-catalytic biodiesel production process. The TGA and DTG curves are showed in Figure S2 in Supplementary Material. The FTIR spectra of the anion-exchange catalyst before and after experiments are presented (Figure 2E). The biodiesel characteristic bands at 1,744, 1,450, and 1,170 cm−1 corresponding to the C=O ester bonds stretching, to the asymmetric stretching of –CH3 and O–CH3 respectively were detected at the surface of the hybrid materials (Rabelo et al., 2015). The coating of ion-exchange resin catalyst was, therefore, able to swell upon absorption of the reaction mixture and catalyze some of the TGs into MEs.
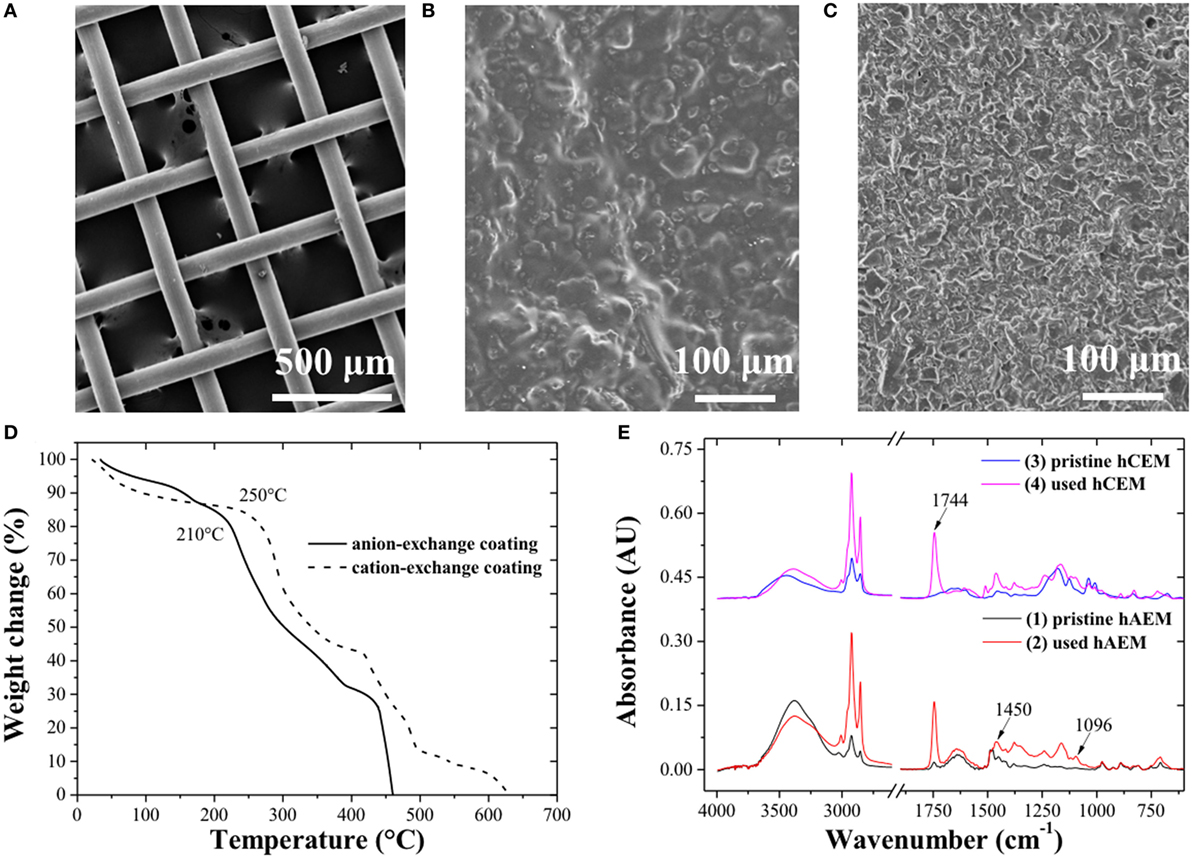
Figure 2. Scanning electron microscopy images of (A) the stainless steel wire mesh, (B) the hybrid anion-exchange, and (C) hybrid cation-exchange electrode. (D) Thermogravimetry and (E) FTIR analysis of the ion-exchange catalyst coating. FTIR spectra 3 and 4 were shifted from spectra 1 and 2 (by 0.40 AU).
Conversion of Canola Oil in Methanolic and Ethanolic Reaction Mixture
The electro-catalytic conversion of canola oil in a methanolic reaction mixture was studied across a series of cell voltages at either 20 or 40°C. The percentage of canola oil converted into MEs after 1 h of reaction is shown in Figures 3A,B. The canola oil conversion rate to MEs was found to be lower at 20°C as compared to 40°C using the SS mesh electrode. However, when the electro-catalytic reaction was performed using the hybrid electrode coated with the anion-exchange resin (hAEM), the temperature of the reaction had very limited impact on the conversion rate of canola oil to MEs. As shown in Figures 3A,B, the conversion rate at 10 V was 19.9 ± 0.1% at 20°C and 21.2 ± 0.9% at 40°C with the hAEM electrode. Similarly, for a cell voltage of 20 V, the conversion rates at 20 or 40°C were comparable.
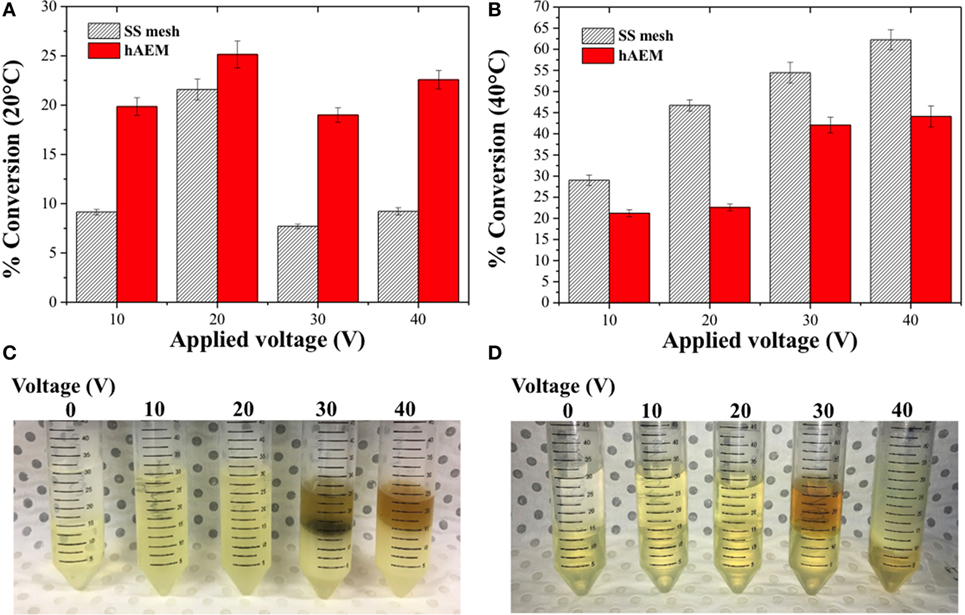
Figure 3. Effect of the cell voltage and temperature on the conversion of canola oil in methanolic reaction mixture at (A) 20°C and (B) at 40°C. Panels (C,D) are the photographs of the reaction products with the stainless steel (SS) mesh and hAEM electrodes at 40°C, respectively.
The conversion rates at 20°C increased with the applied voltage from 10 to 20 V, which was related to an increase formation of reactive species at the cathode (Guan and Kusakabe, 2009). However, the conversion rates were found to subsequently decrease when the experiments were performed at higher voltages, above 20 V, likely due to side reactions occurring in the reaction mixture. On the other hand, at 40°C, the conversion rates increased steadily by 15 and 22% between 20 and 40 V for both the SS mesh and hAEM electrodes. Side parasitic reactions, however, also occurred above 20 V. The apparition of a brown and cloudy phase, as seen on the photographs in Figures 3C,D, indicated that side reactions, such as hydrogenation and saponification, occurred at higher voltages (Ma and Hanna, 1999; Jang et al., 2005). The electro-catalytic conversion of canola oil was, therefore, greater at 40°C when using the plain SS mesh electrode. However, at 20°C, the hAEM electrode was found to perform better than the SS mesh electrode.
The canola oil-conversion rate was then studied as a function of the reaction time at 20 V and 40°C for the SS mesh and hAEM electrodes (Figure 4A). The HPLC chromatograms corresponding to the ester and canola oil phase sampled every hour are displayed Figures S1C–F in Supplementary Material. The conversion rate increased rapidly during the first hour of experiment to 45% for the SS mesh electrode and 34% for the hAEM electrode and further increased at a slow and steady pace to 58 and 39% at the end of the experiments for the SS mesh and hAEM electrodes, respectively. Figure 4B shows the electrode reaction rates in milliampere per second as a function of the reaction time. The electrode reaction rates correspond to the slopes of the intensity plotted as a function of time as displayed in Figures S3A,B in Supplementary Material. The slope of the curve reflects the rate of the electro-chemical reactions occurring at the surface of the electrodes. The values of the electrode reaction rate were shown to decrease over time while the cell voltage was kept constant. This trend indicated an increase of the reaction mixture resistance due to consumption of water and methanol molecules at the electrodes and during the transesterification reactions, respectively. The decrease in electrode reaction rate Figure 4B followed a similar trend as the canola oil conversion rate (Figure 4A). As seen in Figures 4A,B, and from the variation of the respective slopes, the majority of the electrolysis reactions, therefore, occurred during the first hour of the experiment.
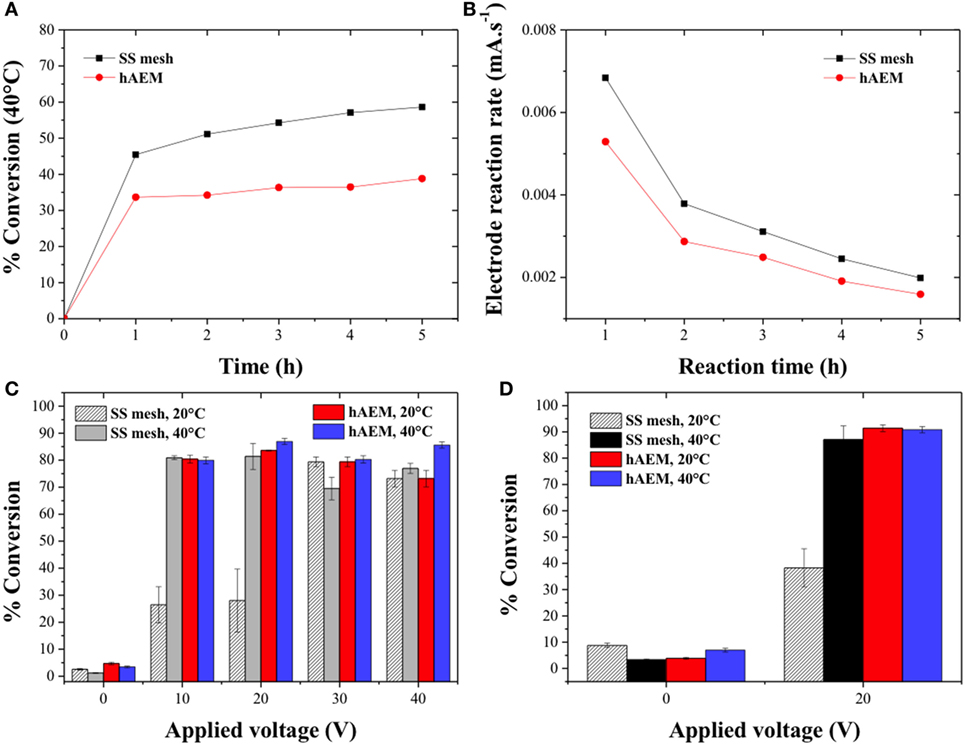
Figure 4. (A) Effect of the reaction time on the conversion of canola oil in methanolic solutions at 40°C for an applied voltage of 20 V and (B) electrode reaction rate across the electro-catalysis cell as a function of the reaction time. Quantitative determination of the conversion of canola oil from (C) a methanolic reaction mixture and (D) from an ethanolic reaction mixture as a function of the cell voltage.
The electro-assisted transesterification of canola oil may be rationalized as a two-step process (Guan and Kusakabe, 2009). First, the OH− ions formed at the cathode by water splitting (Figure 1) reacted with the methanol molecules to form reactive methoxide ions (CH3O−), with sodium ions (Na+) as counter-ions (Sridhar, 1996). Then, the methoxide ions attacked the TG molecules to form esters in a process similar to the reaction (1) (Guan and Kusakabe, 2009). No cosolvents were used to enhance the miscibility of the alcohol and canola oil system in the electro-catalytic process presented in this work (Tang et al., 2006). Therefore, the reaction was performed in a two-phase reaction mixture. The poor miscibility of the canola oil into the methanol phase was due to the high polarity of the methanol molecules (Takeuchi et al., 2009). However, the miscibility of the alcohol and canola oil system could be further increased using higher alcohols with longer hydrocarbons chain such as ethanol (Oliveira et al., 2011). The miscibility of the constituents of the reaction mixture (canola oil + alcohol + water + NaCl) was, therefore, only governed by the temperature and agitation conditions inside the electro-catalytic reactor, which may be tuned in order to increase the conversion rate or to favor phase and products separation. As the stirring was kept constant in the reactor, only the increase of temperature from 20 to 40°C promoted the miscibility of the system and, therefore, greater canola oil conversion rates (Tang et al., 2006; Oliveira et al., 2011).
The canola oil conversion is typically determined by HPLC analysis based on a method which was developed for the quantification of MEs in biodiesel produced from transesterification reactions with a homogeneous catalyst in a water-free environment (Holčapek et al., 1999). The electro-catalytic process without the addition of a cosolvent occurred in a two-phase reaction mixture and in the presence of large quantity of water and salts. Therefore, the HPLC analysis was primarily used as a qualitative and semi-quantitative tool for the conversion of canola oil to methyl and ethyl esters. In order to quantitatively measure the canola oil conversion rate, the volume difference between the initial and final volume of canola oil in the electro-catalytic reactor was calculated based on Eq. 3 (Dubé et al., 2007). The quantitative conversion rates of canola oil as a function of the applied voltage and reaction temperature in a methanolic and ethanolic reaction mixture are shown in Figures 4C,D, respectively. The conversion rate using the hAEM electrode was found to be comparable at both 20 and 40°C and on the order of 80% across all the cell voltages. The conversion rates of canola oil with the SS mesh electrode at 10 and 20 V were found to be three times greater at 40°C than at 20°C confirming the previous HPLC analysis and observations. However, the measured canola oil conversion rates at 30 and 40 V also accounted for the products of the side reactions and were, therefore, included into the overall conversion rates. The electro-catalytic conversion of canola oil to ethyl esters was also performed in an ethanolic reaction mixture at 20 V and at either 20 or 40°C for 1 h. The conversion rates are shown in Figure 4D and were found to follow a similar trend that the conversion rates of canola oil in methanol. The conversion rate observed at 20°C with the SS mesh electrode was 63% lower as compared to 40°C. However, the conversion of canola oil in an ethanolic reaction mixture at 20 V and 20°C was greater than in a methanolic reaction mixture due to the higher miscibility of the ethanol/canola oil system. At 40°C, the conversion rate in ethanol was similar to that of methanol obtained with the hAEM and SS mesh electrode systems. The conversion rates were only 7–14% lower when compared to other electro-catalytic conversion method using similar condition but with a cosolvent and more expensive platinum electrodes (Guan and Kusakabe, 2009) or 10–12% lower as compared to mixed electrolytic and heterogeneous catalyzed biodiesel production method (Putra et al., 2016). Despite the lower conversion rate due to the absence of cosolvent resulting in a two-phase reaction mixture, the separation of products and potentially the recycling of reactants were facilitated.
In addition, control experiments performed without any applied cell voltages showed extremely limited transesterification reactions. The open circuit potential values are shown in Figure S4 in Supplementary Material. The cell voltage using the SS mesh electrodes was found to be 30 and 70 mV at 20 and 40°C, respectively, which was substantially greater than with the hAEM (<10 mV at 20 or 40°C). However, the canola oil conversion rates were almost twice as high with the hAEM (4.7 ± 0.5% and 3.4 ± 0.3% at 20 and 40°C, respectively) than with the SS mesh electrode (2.5 ± 0.5% at 20°C and 1.2 ± 0.1% at 40°C). These results suggest that the conversion of canola oil when using the SS mesh electrode is exclusively due to the formation of reactive species by electrolysis as opposed to with the hAEM electrode, where the formation of reactive species is due to both the electro-chemical reactions and the catalytic activity of the anion-exchange resin.
The use of hybrid electrode coated with the cation-exchange resin (hCEM) (Figure 2C) was proven to be not as efficient as an electrode material with conversion rates below 15% when used as cathode material and below 22% when used as anode materials (Figure 5A). This may be due to reverse bias conditions occurring in the electro-catalytic cell, which are similar to the operation mode of bipolar membranes for electro-dialysis process (Xu, 2005; Xu and Huang, 2008). In this configuration, the negatively charged cation-exchange material facing the cathode resulted in the accumulation of cations near the surface of the cathode with an increased of parasitic side reactions. The photographs (Figure 5B) show a brown and cloudy solution at the end of the experiment with few detectable MEs (Figures 5C,D).
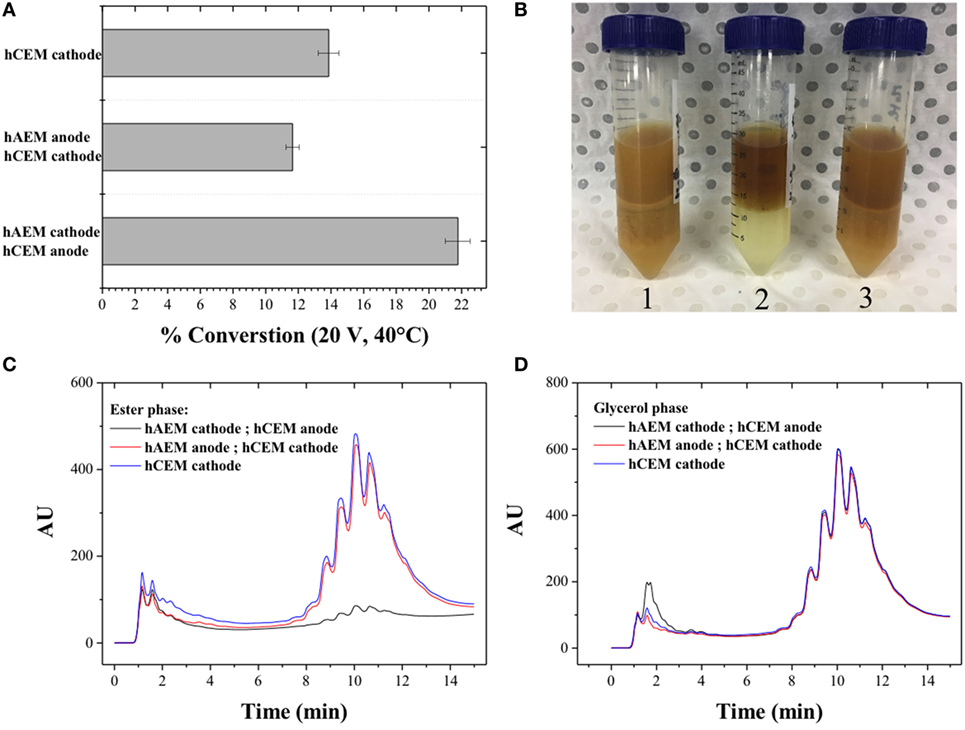
Figure 5. (A) Quantitative determination of the conversion of canola oil from a methanolic reaction mixture using three electrode configurations; (B) photographs of the canola oil conversion products; high-performance liquid chromatography signal of the reaction products in (C) the ester and (D) glycerol phase.
The electrical conductivity and pH values inside the electro-catalytic cell were monitored and reported in Figures 6A,B. The NaCl concentration (0.3 wt% of canola oil content) was below the solubility limit of the reaction mixture and the NaCl crystals were considered to be fully dissociated in the mixed water/alcohol phase at the start of the reaction. The pH values were found to increase from an initial value of 5 to above 9 within the duration of the experiment and were similar across the different experiments for an electrode material and at a given reaction temperature. The increase in pH can be explained by the formation of basic reactive species OH− and CH3O− during the electro-catalytic process. However, the pH values were found to be stable above an applied voltage of 10 V, which indicated that the electro-chemical reactions participated not only to the formation of reactive species but also to the formation of side products. The initial electrical conductivity value of the reaction mixture was found to be very low (355 μS) due to low water content (1 wt% of canola oil content) and high resistivity of the system methanol/canola oil. The decrease in electrical conductivity at the end of the experiments for an applied voltage of 10 and 20 V was attributed to the formation of MEs and consumption of water and methanol molecules. However, for the experiments performed at 30 and 40 V, the electrical conductivity was found to increase, which may be due to the products of the saponification reactions associated with formation of ionic and charged soap species.
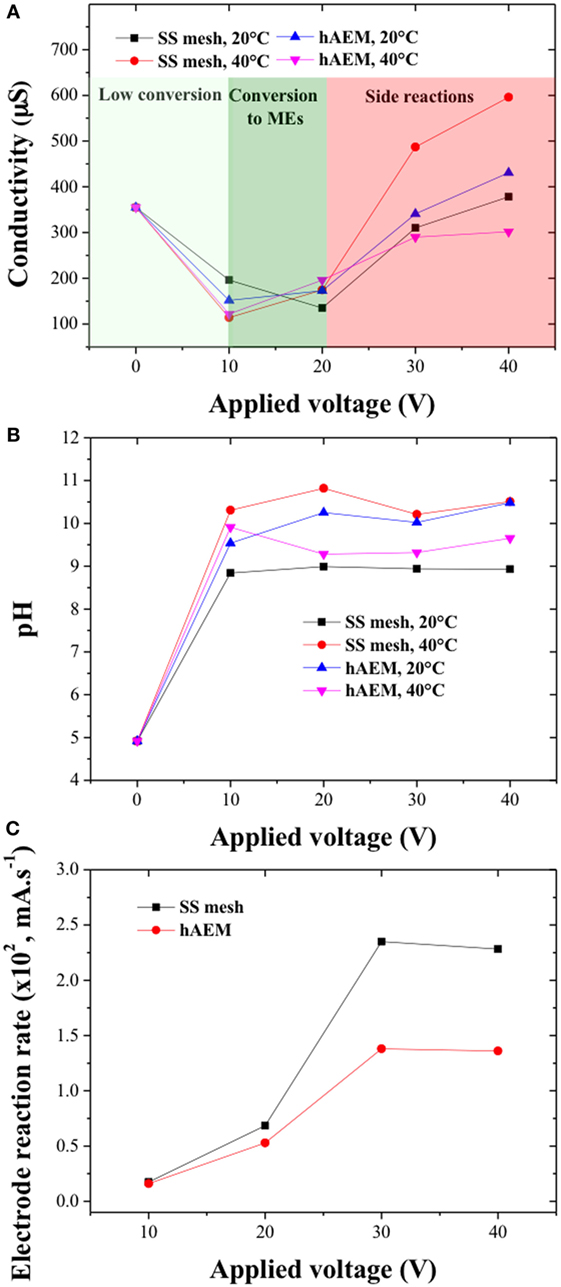
Figure 6. (A) pH and (B) conductivity values of the canola oil conversion products as a function of the cell voltage; (C) electrode reaction rates as a function of the cell voltage for the stainless steel (SS) mesh and hAEM electrodes at 40°C.
The change in electrical conductivity within the electro-catalytic cell showed similar trends for the SS mesh and hybrid AEM electrodes suggesting that the high cell voltages are the main factor affecting the rate of the side reactions. Figure 6C shows the electrode reaction rate values as a function of the cell voltages for the SS mesh and hAEM electrodes at 40°C. The electrode reaction rates increased with the cell voltage up to 30 V and became stable at higher cell voltage. These results indicate that above 30 V, the electro-catalytic process is limited by the rate of formation of ionic species near the surface of the electrodes.
Conclusion
The conversion of canola oil to methyl and ethyl esters was successfully performed without the addition of cosolvent or catalysts in a simple electro-catalytic reactor. Cell voltages above 20 V resulted in undesirable saponification side reactions, which were related to excessive water splitting occurring in such conditions. The plain SS electrode performed better at 40°C while the overall conversion rates were greater with the hybrid electrode at 20°C due to the catalytic activity of the anion-exchange resin. The use of hybrid materials combining the electrical conductivity of the metal support and the reactivity of the ion-exchange resin could inspire new process development for the conversion of high water content feedstock for the production of second-generation biodiesel.
Author Contributions
The manuscript was written through contributions of all authors.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
F-MA would like to thank the Institute for Frontier Materials, Deakin University, VIC, Australia for funding his Ph.D. scholarship and AINSE Ltd. for providing financial assistance (PGRA Award—30290). LD acknowledges Deakin University for his Alfred Deakin Post-doctoral Fellowship.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fmats.2017.00022/full#supplementary-material.
Footnote
References
Abdul-Majeed, W. S., AAl-Thani, G. S., and Al-Sabahi, J. N. (2016). Application of flying jet plasma for production of biodiesel fuel from wasted vegetable oil. Plasma Chem. Plasma Process. 36, 1517–1531. doi: 10.1007/s11090-016-9735-0
Allioux, F.-M., He, L., She, F., Hodgson, P. D., Kong, L., and Dumée, L. F. (2015). Investigation of hybrid ion-exchange membranes reinforced with non-woven metal meshes for electro-dialysis applications. Sep. Purif. Technol. 147, 353–363. doi:10.1016/j.seppur.2015.03.007
Atadashi, I. M., Aroua, M. K., Abdul Aziz, A. R., and Sulaiman, N. M. N. (2011). Membrane biodiesel production and refining technology: a critical review. Renew. Sustain. Energ. Rev. 15, 5051–5062. doi:10.1016/j.rser.2011.07.051
Bajaj, A., Lohan, P., Jha, P. N., and Mehrotra, R. (2010). Biodiesel production through lipase catalyzed transesterification: an overview. J. Mol. Catal. B Enzym. 62, 9–14. doi:10.1016/j.molcatb.2009.09.018
Clements, L. D., and Gerpen, J. (2004). Biodiesel Production Technology. Batelle, OH: NREL-National Renewable Energy Laboratory. NREL/SR-510-36244.
Cubas, A. L. V., Machado, M. M., Pinto, C. R. S. C., Moecke, E. H. S., and Dutra, A. R. A. (2016). Biodiesel production using fatty acids from food industry waste using corona discharge plasma technology. Waste Manag. 47, 149–154. doi:10.1016/j.wasman.2015.05.040
Demirbas, A. (2009). Progress and recent trends in biodiesel fuels. Energy Convers. Manag. 50, 14–34. doi:10.1016/j.enconman.2008.09.001
Dias, J. M., Alvim-Ferraz, M. C. M., and Almeida, M. F. (2008). Comparison of the performance of different homogeneous alkali catalysts during transesterification of waste and virgin oils and evaluation of biodiesel quality. Fuel 87, 3572–3578. doi:10.1016/j.fuel.2008.06.014
Dubé, M. A., Tremblay, A. Y., and Liu, J. (2007). Biodiesel production using a membrane reactor. Bioresour. Technol. 98, 639–647. doi:10.1016/j.biortech.2006.02.019
Dumée, L. F., Allioux, F.-M., Reis, R., Duke, M., Gray, S., Tobin, M., et al. (2014). Qualitative spectroscopic characterization of the matrix–silane coupling agent interface across metal fibre reinforced ion exchange resin composite membranes. Vib. Spectrosc. 75, 203–212. doi:10.1016/j.vibspec.2014.05.011
Guan, G., and Kusakabe, K. (2009). Synthesis of biodiesel fuel using an electrolysis method. Chem. Eng. J. 153, 159–163. doi:10.1016/j.cej.2009.06.005
Holčapek, M., Jandera, P., Fischer, J., and Prokeš, B. (1999). Analytical monitoring of the production of biodiesel by high-performance liquid chromatography with various detection methods. J. Chromatogr. A 858, 13–31. doi:10.1016/S0021-9673(99)00790-6
Huang, H.-J., Ramaswamy, S., Tschirner, U., and Ramarao, B. (2008). A review of separation technologies in current and future biorefineries. Sep. Purif. Technol. 62, 1–21. doi:10.1016/j.seppur.2007.12.011
Jang, E. S., Jung, M. Y., and Min, D. B. (2005). Hydrogenation for low trans and high conjugated fatty acids. Compr. Rev. Food Sci. Food Saf. 4, 22–30. doi:10.1111/j.1541-4337.2005.tb00069.x
Ma, F., and Hanna, M. A. (1999). Biodiesel production: a review. Bioresour. Technol. 70, 1–15. doi:10.1016/S0960-8524(99)00025-5
Mardhiah, H. H., Ong, H. C., Masjuki, H. H., Lim, S., and Lee, H. V. (2017).A review on latest developments and future prospects of heterogeneous catalyst in biodiesel production from non-edible oils. Renew. Sustain. Energ. Rev. 67, 1225–1236. doi:10.1016/j.rser.2016.09.036
Mazzotti, M., Neri, B., Gelosa, D., Kruglov, A., and Morbidelli, M. (1997). Kinetics of liquid-phase esterification catalyzed by acidic resins. Ind. Eng. Chem. Res. 36, 3–10. doi:10.1021/ie9606348
O’Connell, D., Batten, D., O’Connor, M., May, B., Raison, J., Keating, B., et al. (2007). Biofuels in Australia: An Overview of Issues and Prospects. Canberra: Rural Industries Research and Development Corporation.
Oliveira, M. B., Barbedo, S., Soletti, J. I., Carvalho, S. H. V., Queimada, A. J., and Coutinho, J. A. P. (2011). Liquid–liquid equilibria for the canola oil biodiesel + ethanol + glycerol system. Fuel 90, 2738–2745. doi:10.1016/j.fuel.2011.03.017
Putra, R. S., Liyanita, A., Arifah, N., Puspitasari, E., Sawaludin, and Hizam, M. N. (2017). Enhanced electro-catalytic process on the synthesis of FAME using CaO from eggshell. Energy Proc. 105, 289–296. doi:10.1016/j.egypro.2017.03.316
Putra, R. S., Pratama, K., Antono, Y., Idris, M., Rua, J., and Ramadhani, H. (2016). Enhanced electrocatalytic biodiesel production with chitosan gel (hydrogel and xerogel). Proc. Eng. 148, 609–614. doi:10.1016/j.proeng.2016.06.522
Rabelo, S. N., Ferraz, V. P., Oliveira, L. S., and Franca, A. S. (2015). FTIR analysis for quantification of fatty acid methyl esters in biodiesel produced by microwave-assisted transesterification. Int. J. Environ. Sci. Dev. 6, 964. doi:10.7763/IJESD.2015.V6.730
Sharma, Y., Singh, B., and Upadhyay, S. (2008). Advancements in development and characterization of biodiesel: a review. Fuel 87, 2355–2373. doi:10.1016/j.fuel.2008.01.014
Shibasaki-Kitakawa, N., Honda, H., Kuribayashi, H., Toda, T., Fukumura, T., and Yonemoto, T. (2007). Biodiesel production using anionic ion-exchange resin as heterogeneous catalyst. Bioresour. Technol. 98, 416–421. doi:10.1016/j.biortech.2005.12.010
Shibasaki-Kitakawa, N., Tsuji, T., Kubo, M., and Yonemoto, T. (2011). Biodiesel production from waste cooking oil using anion-exchange resin as both catalyst and adsorbent. Bioenerg. Res. 4, 287–293. doi:10.1007/s12155-011-9148-0
Sridhar, S. (1996). Electrodialysis in a non-aqueous medium: production of sodium methoxide. J. Memb. Sci. 113, 73–79. doi:10.1016/0376-7388(95)00217-0
Takeuchi, T., Pereira, C., Braga, M., Marostica, M., Leal, P., and Meireles, M. (2009). Extracting Bioactive Compounds for Food Products – Theory and Applications. Boca Raton: CRC Press.
Tang, Z., Du, Z., Min, E., Gao, L., Jiang, T., and Han, B. (2006). Phase equilibria of methanol–triolein system at elevated temperature and pressure. Fluid Phase Equilib. 239, 8–11. doi:10.1016/j.fluid.2005.10.010
Van Kasteren, J., and Nisworo, A. (2007). A process model to estimate the cost of industrial scale biodiesel production from waste cooking oil by supercritical transesterification. Resourc. Conservat. Recycl. 50, 442–458. doi:10.1016/j.resconrec.2006.07.005
Vinodh, R., Padmavathi, R., and Sangeetha, D. (2011). Separation of heavy metals from water samples using anion exchange polymers by adsorption process. Desalination 267, 267–276. doi:10.1016/j.desal.2010.09.039
Xu, T. (2005). Ion exchange membranes: state of their development and perspective. J. Memb. Sci. 263, 1–29. doi:10.1016/j.memsci.2005.05.002
Xu, T., and Huang, C. (2008). Electrodialysis-based separation technologies: a critical review. AIChE J. 54, 3147–3159. doi:10.1002/aic.11643
Keywords: biodiesel, electro-catalytic conversion, ion-exchange resin, porous stainless steel electrodes, hybrid electrodes
Citation: Allioux F-M, Holland BJ, Kong L and Dumée LF (2017) Electro-Catalytic Biodiesel Production from Canola Oil in Methanolic and Ethanolic Solutions with Low-Cost Stainless Steel and Hybrid Ion-Exchange Resin Grafted Electrodes. Front. Mater. 4:22. doi: 10.3389/fmats.2017.00022
Received: 25 May 2017; Accepted: 30 June 2017;
Published: 20 July 2017
Edited by:
Alessandro Pegoretti, University of Trento, ItalyReviewed by:
Rodrigo Cercená, UNESC, Coordenação de Aperfeicoamento de Pessoal de Nível Superior (CAPES), BrazilKenan Song, Massachusetts Institute of Technology, United States
Copyright: © 2017 Allioux, Holland, Kong and Dumée. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francois-Marie Allioux, Zi5hbGxpb3V4QHJlc2VhcmNoLmRlYWtpbi5lZHUuYXU=