 Kuo-Jian Ma
Kuo-Jian Ma Yong-Lian Ye
Yong-Lian Ye Yu-Kang Li1,2
Yu-Kang Li1,2 Ge-Yi Fu
Ge-Yi Fu Yue-Hong Wu
Yue-Hong Wu Cong Sun
Cong Sun Xue-Wei Xu
Xue-Wei Xu- 1Ocean College, Zhejiang University, Zhoushan, China
- 2Key Laboratory of Marine Ecosystem Dynamics, Ministry of Natural Resources & Second Institute of Oceanography, Ministry of Natural Resources, Hangzhou, China
- 3National Deep Sea Center, Ministry of Natural Resources, Qingdao, China
- 4College of Life Sciences and Medicine, Zhejiang Sci-Tech University, Hangzhou, China
- 5Shaoxing Biomedical Research Institute of Zhejiang Sci-Tech University Co., Ltd, Zhejiang Engineering Research Center for the Development Technology of Medicinal and Edible Homologous Health Food, Shaoxing, China
The complete metabolism of carbohydrates, as the most abundant and structurally diverse organic matter on earth, requires the involvement of different carbohydrate-active enzymes (CAZymes). Flavobacteriales and Cytophagales are two groups whose members specialize in polysaccharide metabolism, but research on their polysaccharide metabolic patterns based on the overall CAZymes is scarce. In this study, we analyzed 702 filtered genomes of Flavobacteriales and Cytophagales and obtained 100,445 CAZymes. According to their taxonomic status and living environment, we explored the impact of taxonomic status, isolation source, and environmental condition on their potential polysaccharide metabolic patterns. The results indicated significant differences in the CAZyme composition among different taxonomic statuses or environments. Compared with the Flavobacteriales genomes, the genomes of Cytophagales possess more abundant and diverse CAZymes, but have fewer unique CAZyme families. Genomes from different families vary greatly in terms of CAZyme family diversity and composition, but relatively small divergences were found from families in the same order. Furthermore, our findings indicated that genomes from the marine and tidal flat environments share more similarities in CAZyme family composition and diversity compared with the terrestrial genomes. Extreme environments greatly constrain the types of CAZyme families present, and certain CAZyme families are significantly lower than those in normal environments. Although significant differences were found among genomes from both different taxonomic statuses and environments, the dimensionality reduction and the clustering analysis based on CAZyme composition indicated that evolutionary status is the main factor influencing the polysaccharide metabolic patterns of these strains. The correlations among CAZyme families indicated that the majority of these families are synergistically involved in polysaccharide metabolism. This study provides a comprehensive profile of the CAZymes in Flavobacteriales and Cytophagales, highlighting the role of evolutionary status in shaping the polysaccharide metabolic patterns and the prevalence of synergism among CAZyme families. These findings have implications for understanding microbial carbohydrate metabolism in different environments.
1 Introduction
Carbohydrates comprise the predominant organic matter on earth, constituting approximately 75% of the annually renewable biomass of approximately 200 billion tons (Lichtenthaler and Peters, 2004). Polysaccharides, which serve as the energy storage materials and structural components of organisms, are closely linked to life and can act as signal molecules to facilitate organism–environment interactions (Bar-On et al., 2018; Lapebie et al., 2019). Furthermore, polysaccharides play important roles in medicine, food, and chemical engineering (Li et al., 2016; Lovegrove et al., 2017; Shanmugam and Abirami, 2019; Meneguin et al., 2021). The terrestrial biomass is around two orders of magnitude higher than the marine biomass, and it is mainly composed of plant biomass (Bar-On et al., 2018). However, due to its huge area, half of the global net primary production is generated in the marine environment, which contains some unique polysaccharides, such as agar, alginate, carrageenan, fucoidan, laminarin, and ulvan (Field et al., 1998; Xu et al., 2022). Tidal flats are located in the intertidal zone between the high and low tide levels (Murray et al., 2019; Wang et al., 2020) and can receive carbohydrates from both marine and land. These distinct environments possess varying polysaccharide compositions and may harbor different enzymes required for their degradation.
According to the calculation by Laine, hexasaccharides could have more than 1.05 × 1012 possible isomers. As the most diverse macromolecules on earth, polysaccharides can be composed of different cyclic sugar monomers and replaced by different functional groups (Laine, 1994). Due to their great structural diversity, the complete metabolism of polysaccharides requires the involvement of different carbohydrate-active enzymes (CAZymes), including glycoside hydrolases (GHs), polysaccharide lyases (PLs), carbohydrate esterases (CEs), glycosyltransferases (GTs), auxiliary activities (AAs), and carbohydrate-binding modules (CBMs) (Drula et al., 2022). Among them, GHs, PLs, and CEs are responsible for the degradation of carbohydrates, GTs are mainly responsible for the synthesis of carbohydrates, AAs are involved in various redox transformations of carbohydrates, and CBMs can enormously enhance the catalytic activity of the active enzymes of corresponding carbohydrates (Wardman et al., 2022). To efficiently utilize diverse polysaccharides, these CAZymes act synergistically in the process of polysaccharide degradation (Sidar et al., 2020).
Bacteria employ numerous CAZymes and play an important role in the carbon cycle and human health. Heterotrophic bacteria, particularly the Bacteroidota species, are considered primary degraders of marine polysaccharides, playing a crucial role in the global carbon turnover (Falkowski et al., 1998; Reintjes et al., 2017). The utilization of polysaccharides by intestinal bacteria is critical for the stability of the gut microbiota and human health (Zafar and Saier, 2021; Cheng et al., 2022). Moreover, numerous CAZymes are employed by certain bacteria to facilitate the polysaccharide utilization process in biofuel production. Lignocellulolytic bacteria or their CAZymes can complete the pretreatment process of lignocellulosic biomass within a few hours to several days and without generating inhibitory compounds (Zabed et al., 2019; Bhujbal et al., 2022; Kumar et al., 2022). Bacteroidota (the Cytophaga–Flavobacterium–Bacteroides group) species specialize in the degradation of a wide range of complex carbohydrates, and metabolic flexibility makes them dominant in many carbohydrate-rich environments (Fernández-Gómez et al., 2013; Chen et al., 2021; McKee et al., 2021). Bacteroidota mainly relies on unclustered CAZyme genes and polysaccharide utilization loci (PULs) on genomes for polysaccharide degradation. The members Flavobacteriales and Cytophagales are widely distributed in marine and biofilm environments, typically forming the largest bacterial groups in some marine specimens (Anantapong and Kanjana-Opas, 2016). Furthermore, Flavobacteriales species are notable for their ability to degrade polysaccharides through CAZymes in PULs, and the PUL repertoires in different genera always correspond to their ecological niches (Kappelmann et al., 2019; Gavriilidou et al., 2020). On the other hand, Bacteroidales species are well-known intestinal bacteria that play a pivotal role in the degradation and utilization of complex carbohydrates within the human gut, contributing significantly to the maintenance of gut microbiota stability and overall human health.
PULs, as a gene cluster for the efficient utilization of polysaccharides, is increasingly becoming the focus of research on polysaccharide metabolism instead of the overall CAZymes in bacteria. Lapébie et al. estimated the diversity of glycan structures through the number of CAZyme combinations in PULs (Lapebie et al., 2019). While PUL-based polysaccharide metabolic mechanisms are common, many species of Bacteroidota heavily rely on a “PUL-free” approach, exploiting type 9 secretion system (T9SS) CAZymes to degrade polysaccharides such as cellulose and chitin (Lasica et al., 2017; Lauber et al., 2018; McKee et al., 2021). Therefore, considering only the CAZymes in PULs will significantly underestimate the bacterial polysaccharide metabolic ability. In order to gain deeper insights into the differences in the polysaccharide metabolic patterns among Flavobacteriales and Cytophagales species, as well as to explore the influence of the bacterial source on their polysaccharide metabolic patterns, we employed all the CAZymes in bacteria to assess their polysaccharide metabolic potential. Moreover, as the order of CAZymes in PULs does not appear to be significant (Larsbrink et al., 2014; Lapebie et al., 2019), it was feasible to use the CAZyme–species table to construct distance matrices. In summary, this study analyzes the differences in the polysaccharide metabolic patterns among Flavobacteriales and Cytophagales species and, according to their isolation source and environmental condition, reveals the impact of living environments on their polysaccharide metabolic patterns.
2 Materials and methods
2.1 Genome acquisition, screening, and classification
A total of 722 complete and scaffold genomes of Flavobacteriales and Cytophagales species were downloaded into a local server from the National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/). Notably, only RefSeq genomes were considered in this selection process. The quality of all 702 genomes was determined using Barrnap, tRNAscan-SE, and CheckM. Low-quality genomes were discarded. Briefly, the number of transfer RNA (tRNA) in each genome was determined using Barrnap (v0.9) with the parameter: –kingdom bac (https://github.com/tseemann/barrnap). tRNAscan-SE (v2.0.12) was used to predict the number of 16S ribosomal RNA (Chan et al., 2021). The completeness and the contamination of the manually downloaded genomes were assessed using CheckM (v1.2.2) with the parameter: lineage_wf (Parks et al., 2015). Genomes were retained only if their completeness was >95% and contamination was <5%. At the family level, the genomes of Cyclobacteriaceae, Cytophagaceae, Flavobacteriaceae, Hymenobacteraceae, Spirosomaceae, and Weeksellaceae accounted for more than 94% and were consequently selected for further analysis. In addition to the taxonomic status of these genomes, their isolation sources were also manually classified into three categories: land (soil, terrestrial organisms, freshwater, saline lakes, etc.), tidal flats (estuaries, mangroves, salt marshes, coastal sediments, etc.), and marine (marine sediments, marine organisms, seawater, etc.). Furthermore, the genomes were categorized into normal environments and extreme environments (e.g., hypersaline lake, desert soil, and glacier) based on the environmental conditions of their isolation. All of these pieces of information were manually collected from the corresponding literature. The genomes of strains without clear isolation information were also discarded. The number counts of the genomes in these groups are visualized in Figure 1A. A total of 702 high-quality RefSeq genomes (including 500 genomes belonging to Flavobacteriales and 202 genomes belonging to Cytophagales) and their related isolation information, taxonomic information, and environmental condition were obtained and are displayed in Supplementary Table S1.
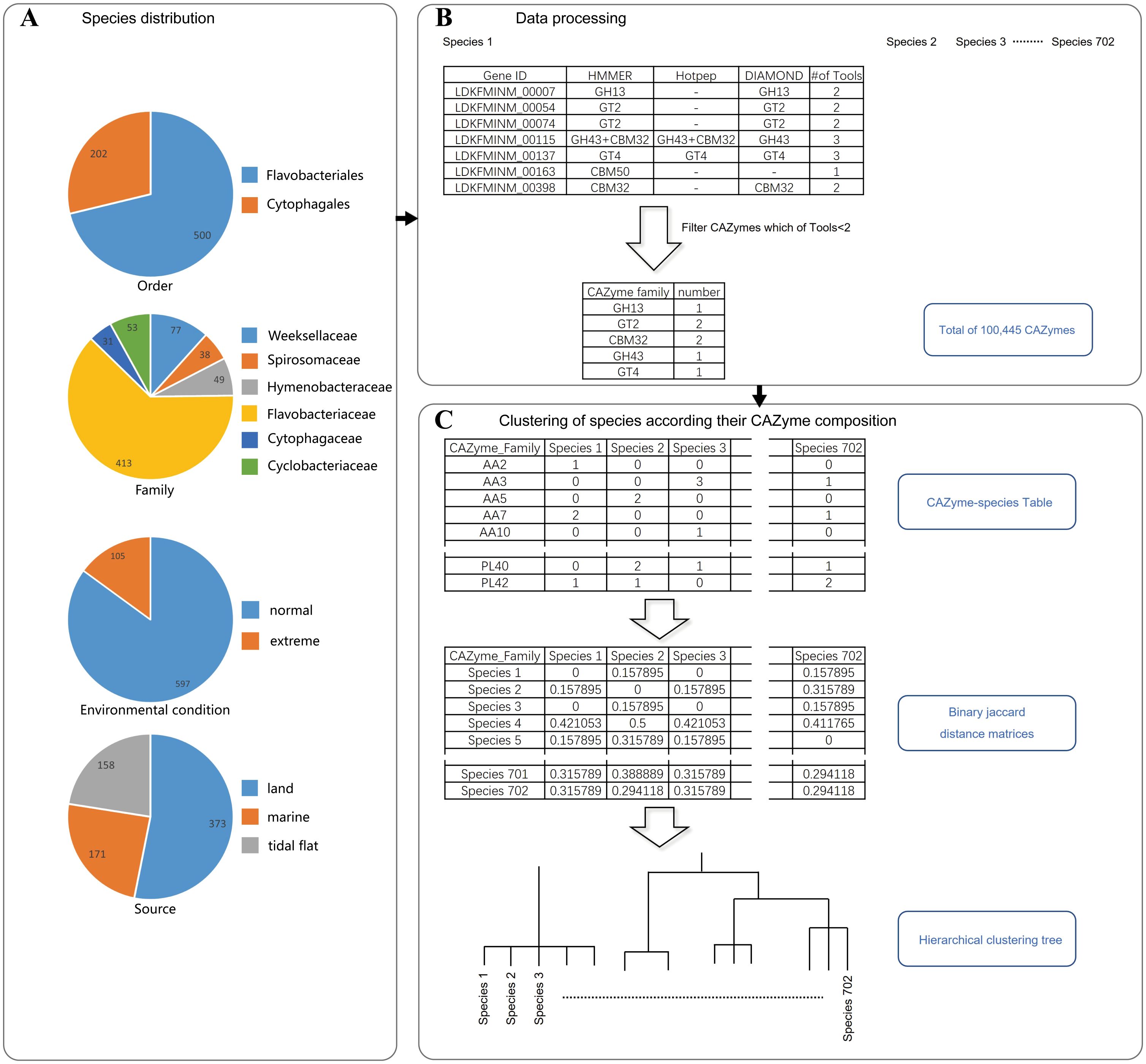
Figure 1. Species clustering pipeline. (A) Number of species in each group. A total of 702 filtered genomes were used for analysis. (B) Filtering of the carbohydrate-active enzymes (CAZymes) annotated using the HMMER, DIAMOND, and Hotpep tools within the dbCAN2 software. (C) Clustering of the species according to their CAZyme composition. Binary Jaccard distance matrices were calculated according to the CAZyme family composition.
2.2 Genome annotation
PROKKA (v1.12) was used to predict the genes and corresponding amino acids within these genomes. The predicted SusC (an outer membrane TonB-dependent transporter) and SusD (a surface glycan-binding lipoprotein) genes within these genomes were extracted separately (Seemann, 2014). The amino acid sequences of the SusC and SusD genes were aligned using MAFFT v7.520 with default settings (Katoh and Standley, 2013). The CAZymes were all predicted using the HMMER, DIAMOND, and Hotpep tools within the dbCAN2 software based on the Carbohydrate-Active enZYmes Database v11 (CAZy database), and as recommended in the dbCAN2 documentation, the CAZyme hits were only considered if at least two of the three annotation tools were positive (Zhang et al., 2018; Drula et al., 2022). An in-house Python script was used to summarize the CAZymes annotated by at least two of the three annotation tools for each genome into the genomes–CAZymes abundance table (Figure 1B; Supplementary Table S2). The substrates of co-occurring CAZyme families were searched through the CAZy database (v11) and the Polysaccharide-Utilization Loci DataBase (PULDB) (Terrapon et al., 2018).
2.3 Statistical analysis
The species taxonomic information, the genomes–CAZymes abundance table, and the CAZyme classification information were imported into QIIME2 (v2022.11) for summary by different taxonomic statuses and sources using the analysis of composition of microbiomes (ANCOM) method (Mandal et al., 2015 988; Bolyen et al., 2019). The composition of CAZyme classes was visualized using Circos (Krzywinski et al., 2009). Alpha diversity was assessed by calculating the Shannon diversity and observed species (i.e., observed CAZymes) indices of the CAZyme families in different species. Differences in the CAZyme family diversity among bacteria from different taxonomic statuses, sources, and environmental conditions were compared using a t-test and/or the Wilcoxon test through pairwise comparison. On the other hand, differences in the relative abundance of the different CAZyme classes and families were compared using Welch’s t-test. Principal coordinate analysis (PCoA) was performed using binary Jaccard distance matrices to reveal the clustering patterns of the CAZymes from bacteria of different taxonomic ranks and sources. Permutational multivariate analysis of variance (PERMANOVA) was employed to assess the significance of the differences between different groups, and ggplot2 in R (v3.6.3) was used for visualization. The cluster tree for the species of Flavobacteriales and Cytophagales based on the composition of the CAZyme families was clustered using the unweighted pair group method with arithmetic mean (UPGMA, Figure 1C), and the clustering relationship of the CAZyme families in different species was visualized using ChiPlot (https://www.chiplot.online/). The “psych” package in R software was used to calculate the Spearman’s correlation coefficients between CAZyme families, and “igraph” was used to visualize these correlations. Other statistics were visualized using STAMP v2.1.3 and R software. A p-value less than 0.05 was considered significant, unless otherwise indicated (Parks et al., 2014).
3 Results
3.1 CAZyme diversity
To investigate the differences in the CAZyme families between different groups, the Shannon diversity indices were compared and the CAZyme indices (typically referring to the observed species index in microbial diversity) of the CAZyme families were observed. In terms of phylogenetic status, it was found that there were significantly more observed CAZyme families and higher Shannon diversity in Cytophagales species compared with those in Flavobacteriales species (padj < 2.22e−16 by t-test) (Figure 2), indicating greater polysaccharide metabolic potential in Cytophagales species. This difference was also reflected in the major families of Flavobacteriales (Flavobacteriaceae and Weeksellaceae) and Cytophagales (Cyclobacteriaceae, Cytophagaceae, Hymenobacteraceae, and Spirosomaceae). The species of Weeksellaceae and Flavobacteriaceae had the lowest Shannon diversity and observed CAZyme families on average, which were significantly lower than those in Cyclobacteriaceae, Cytophagaceae, Hymenobacteraceae, and Spirosomaceae species (Figure 2). In general, a pattern of Cytophagales > Flavobacteriales was found in both the Shannon diversity index (5.36 and 4.69 on average, respectively) and the observed CAZyme families (73.37 and 48.10 on average, respectively). Furthermore, the diversity of CAZymes across different environments was explored for each order (Supplementary Figure S1). For Flavobacteriales, the Shannon diversity of the CAZymes was significantly higher in land environments compared with marine environments (padj = 0.031 by t-test), while no significant difference was observed between the land and tidal flat environments. In contrast, for Cytophagales, no significant spatial differences in CAZyme diversity were detected among the land, marine, and tidal flat environments.
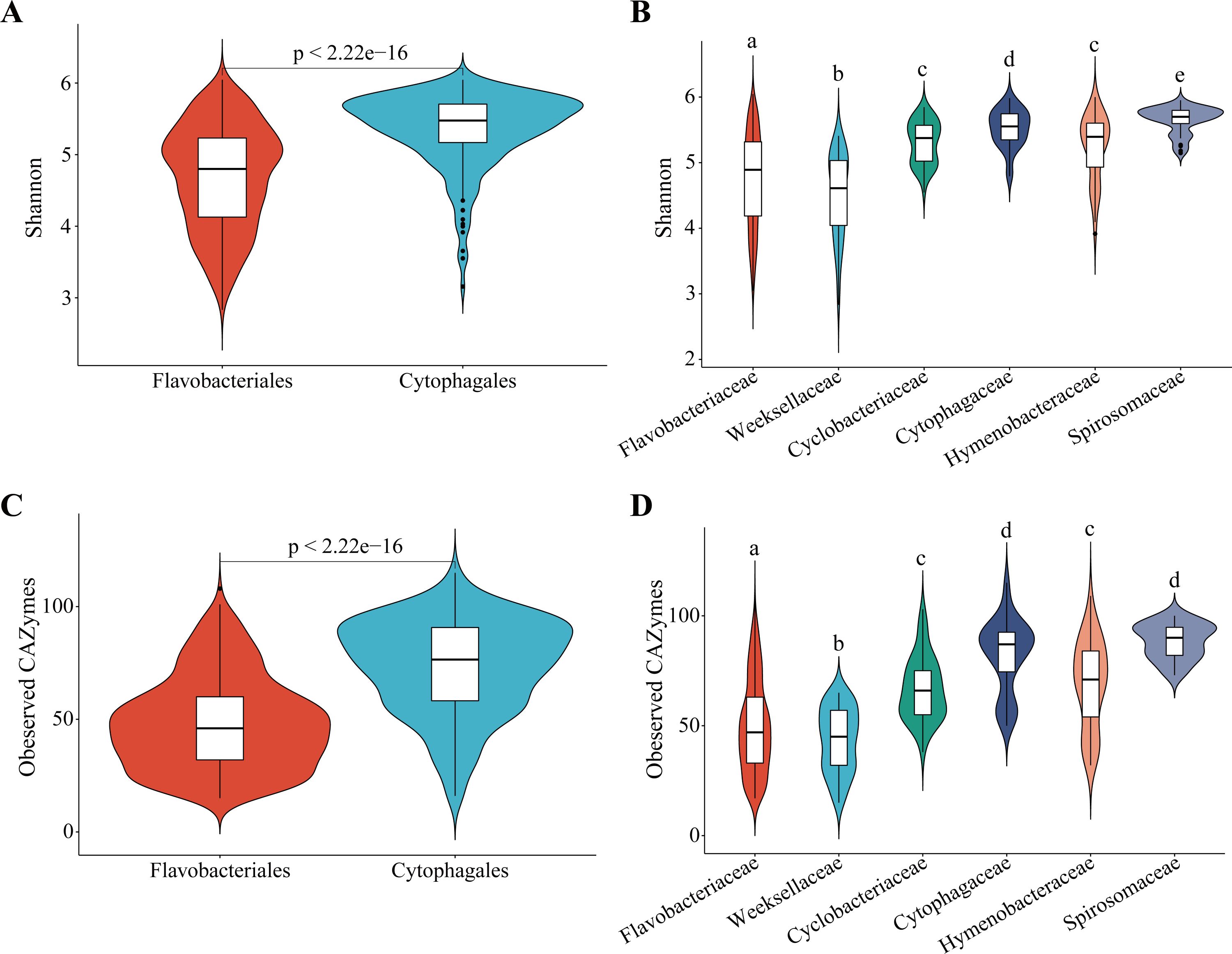
Figure 2. Alpha diversity characterized by the Shannon index and the observed carbohydrate-active enzyme (CAZyme) index. (A, B) Violin plots showing the distribution of the Shannon indices of the CAZymes among species from different orders (A) and major families (B). (C, D) Observed CAZyme indices of the CAZymes among species from different orders (C) and major families (D). A two-sided t-test was used to analyze the significance of the differences between different groups, groups with different lowercase letters are significantly different in the t-test, while groups with the same letter are not. Inner box plots show the median and quartiles.
When considering only the environmental origin, the observed CAZyme families in bacteria from land were significantly higher than those in bacteria from marine (padj = 0.03 by t-test), but no significant differences were observed in the CAZyme family diversity between tidal flats and marine/land, as well as between normal and extreme environments (Figure 3). Although not significant, a decreasing tendency of the Shannon diversity index (4.89, 4.88, and 4.86 on average, respectively) and observed CAZyme families (57.13, 54.16, and 52.64 on average, respectively) was observed from land to marine. Considering the diversity indices of the CAZymes, the disparity between distinct taxonomic groups was more pronounced than that between distinct environmental groups.

Figure 3. Alpha diversity characterized by the Shannon index and the observed carbohydrate-active enzyme (CAZyme) index. (A, B) Violin plots showing the distribution of the Shannon indices of the CAZymes among species from different sources (A) and environmental conditions (B). (C, D) Observed CAZyme indices of the CAZymes among species from different sources (C) and environmental conditions (D). A two-sided t-test was used to analyze the significance of the differences between different groups. Inner box plots show the median and quartiles.
3.2 CAZyme class composition
For the number of CAZymes in each genome, Cytophagales species were higher than Flavobacteriales species in all CAZyme classes (Figure 4A). The average numbers of AAs, CBMs, CEs, GHs, GTs, and PLs annotated in each genome of Cytophagales species were 247.4%, 130.6%, 125.5%, 70.6%, 32.3%, and 21.7% higher than those of Flavobacteriales, respectively. Moreover, in general, the average number of CAZymes in each genome of Cytophagales species was 62.3% higher than that in Flavobacteriales, showing a greater polysaccharide metabolic potential. Considering the great difference between the genome size of species from different orders (5.66 Mb on average for the species of Cytophagales and 3.86 Mb on average for the species of Flavobacteriales), the number of CAZymes per megabase genome was further analyzed (Figure 4B). The results showed that the average numbers of annotated AAs, CBMs, CEs, and GHs in each megabase genome of Cytophagales species were 136.7%, 57.1%, 53.6%, and 16.2% higher than those of Flavobacteriales, respectively, while the average numbers of annotated GTs and PLs in each megabase genome were 10.9% and 20.7% lower than those of Flavobacteriales, respectively. Furthermore, in general, the average number of CAZymes in each megabase genome of Cytophagales was 10.6% higher than that of Flavobacteriales. At the family level, for the species of Weeksellaceae, the numbers of CAZymes in all classes were lower than those of the species of other families, except for AAs and GTs, which were similar to those of the species of other families (Figures 4C, D). As members of Cytophagales, the species of Spirosomaceae, Hymenobacteraceae, and Cytophagaceae had relatively more CAZymes in the major CAZyme classes (e.g., GHs, CBMs, and CEs).
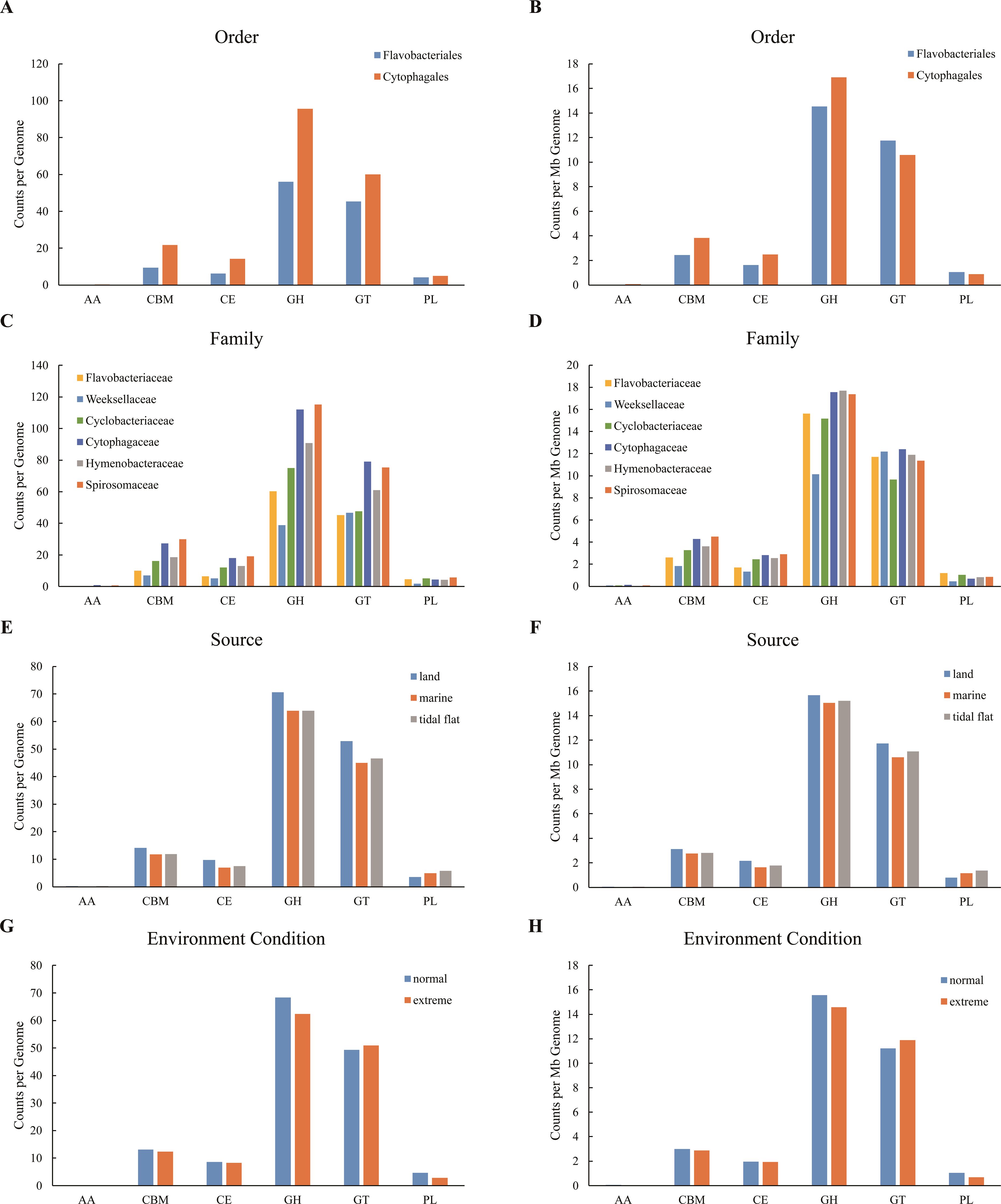
Figure 4. (A, C, E, G) Absolute number of each carbohydrate-active enzyme (CAZyme) class within each genome among different orders (A), major families (C), sources (E), and environmental conditions (G). (B, D, F, H) Absolute number of each CAZyme class per megabase (Mb) genome among different orders (B), major families (D), sources (F), and environmental conditions (H).
In terms of environment, compared with the tidal flat and marine species, the terrestrial species had more AAs, CBMs, CEs, GHs, and GTs, but fewer PLs (Figure 4E). Overall, the average number of CAZymes in the genomes of terrestrial species was 13.94% higher than that in marine species and 11.35% higher than that in intertidal species. With regard to the CAZymes per megabase genome, the terrestrial species also had more AAs, CBMs, CEs, GHs, GTs, but fewer PLs compared with the tidal flat and marine species, and the average number of CAZymes in each megabase genome of the terrestrial species was 7.46% higher than that in marine species and 3.80% higher than that in intertidal species (Figure 4F). For the different environmental conditions, species from normal environments had more AAs, CBMs, CEs, GHs, and PLs, but fewer GTs compared with species from extreme environments (Figure 4G). In general, the average number of CAZymes in the genomes of species in normal environments was 5.28% higher than that in extreme environments. With regard to the CAZymes per megabase genome, species from normal environments had more AAs, CBMs, CEs, GHs, and PLs, but fewer GTs compared with species from extreme environments (Figure 4H), and the average number of CAZymes in each megabase genome of species in normal environments was 2.54% higher than that in extreme environments.
To compare the polysaccharide metabolic potential among bacteria from different taxonomic statuses and sources, further analyses were conducted on the relative abundance of each CAZyme class in the different groups. For relative abundance, GHs and GTs were the major CAZyme classes in Flavobacteriales and Cytophagales, followed by CBMs, CEs, PLs, and AAs. A detailed composition of the CAZyme classes is shown in Figure 5A. The CAZyme class composition for bacteria from different families, sources, and environmental conditions showed the same pattern (Figures 5B-D). In addition, it was found that the CAZyme classes were significantly different among the species of Flavobacteriales and Cytophagales due to their different phylogenetic statuses. Compared with Flavobacteriales, Cytophagales tended to have higher relative abundance of CBMs, CEs, GHs, and AAs, while Flavobacteriales had relatively more GTs. No significant difference was observed between the relative abundance of PLs. At the family level, similar to Flavobacteriales and Cytophagales, the same differences were found between Flavobacteriaceae and Cytophagaceae, as shown in the extended error bar plot in Figure 5B, except that the relative abundance of PLs in Flavobacteriaceae was also significantly higher than that in Cytophagaceae. In general, the CAZyme composition exhibited a comparatively lower degree of divergence within the major families of Cytophagales (Cyclobacteriaceae, Cytophagaceae, Hymenobacteraceae, and Spirosomaceae), indicating a more conserved polysaccharide metabolic potential in Cytophagales species during their evolutionary history. In contrast, more significant variations were observed within the major families of Flavobacteriales (Flavobacteriaceae and Weeksellaceae) and between the major families of Cytophagales and Flavobacteriales. The detailed differences among other families are summarized in Supplementary Table S3. As members of Flavobacteriales, the species of Flavobacteriaceae and Weeksellaceae had higher numbers of GTs, particularly the Weeksellaceae species, and the number of GTs exceeded that of GHs, becoming the most abundant CAZyme class. In terms of environment, the bacteria isolated from marine and tidal flats had a similar CAZyme class composition and showed no significant difference (Supplementary Table S3). However, compared with the species isolated from marine and tidal flats, terrestrial species appeared to have significantly more CEs but fewer PLs, as shown in Figure 5C. Moreover, compared with the species in extreme environments, the species in normal environments had significantly more PLs, as there were more available polysaccharides (Figure 5D).
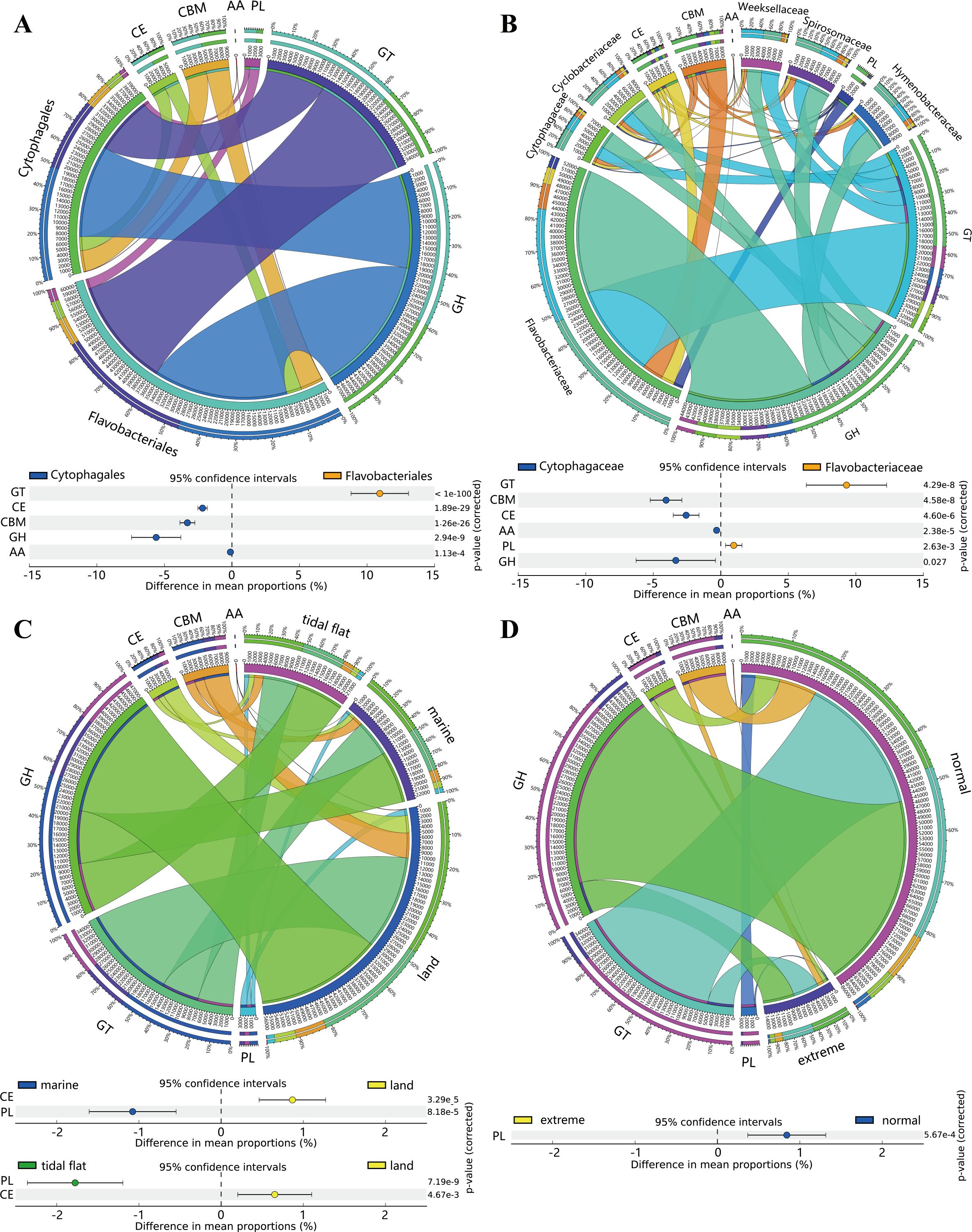
Figure 5. Proportions and differences of the carbohydrate-active enzyme (CAZyme) classes among species in different categories. (A–D) Proportions and differences of the CAZyme classes among species from different orders (A), major families (B), sources (C), and environmental conditions (D). padj < 0.05 are presented in the post-hoc plot. For Flavobacteriales and Cytophagales, 500 and 202 filtered genomes were used for the annotation of CAZymes, respectively.
3.3 CAZyme family composition
Due to the number of significant differences that were found in the composition and diversity of the CAZyme families among the different bacterial groups, detailed analyses of the differences in these CAZyme families were carried out. The CAZyme families were divided into two categories: i) CAZyme families that appeared in only one group (unique CAZyme families, i.e., present only in land or tidal flat or marine) and 2) CAZyme families that appeared in all groups (shared CAZyme families, i.e., CAZymes present in land, tidal flats, and marine).
i) We first analyzed the distribution of the unique CAZyme families in different groups. As presented in Figure 6A; Supplementary Table S4, there were 212 shared CAZyme families between Flavobacteriales and Cytophagales and 37 Flavobacteriales-specific CAZymes, which was higher than the 24 Cytophagales-specific CAZymes. This could be due to the higher number of Flavobacteriales species (500 species from Flavobacteriales and 202 species from Cytophagales). Flavobacteriaceae species showed the most unique CAZyme families due to the great quantity advantage of type species (413 species). The CAZyme family types in Weeksellaceae species differed greatly from the other five bacterial families, with only 28 CAZyme families present (Figure 6B).
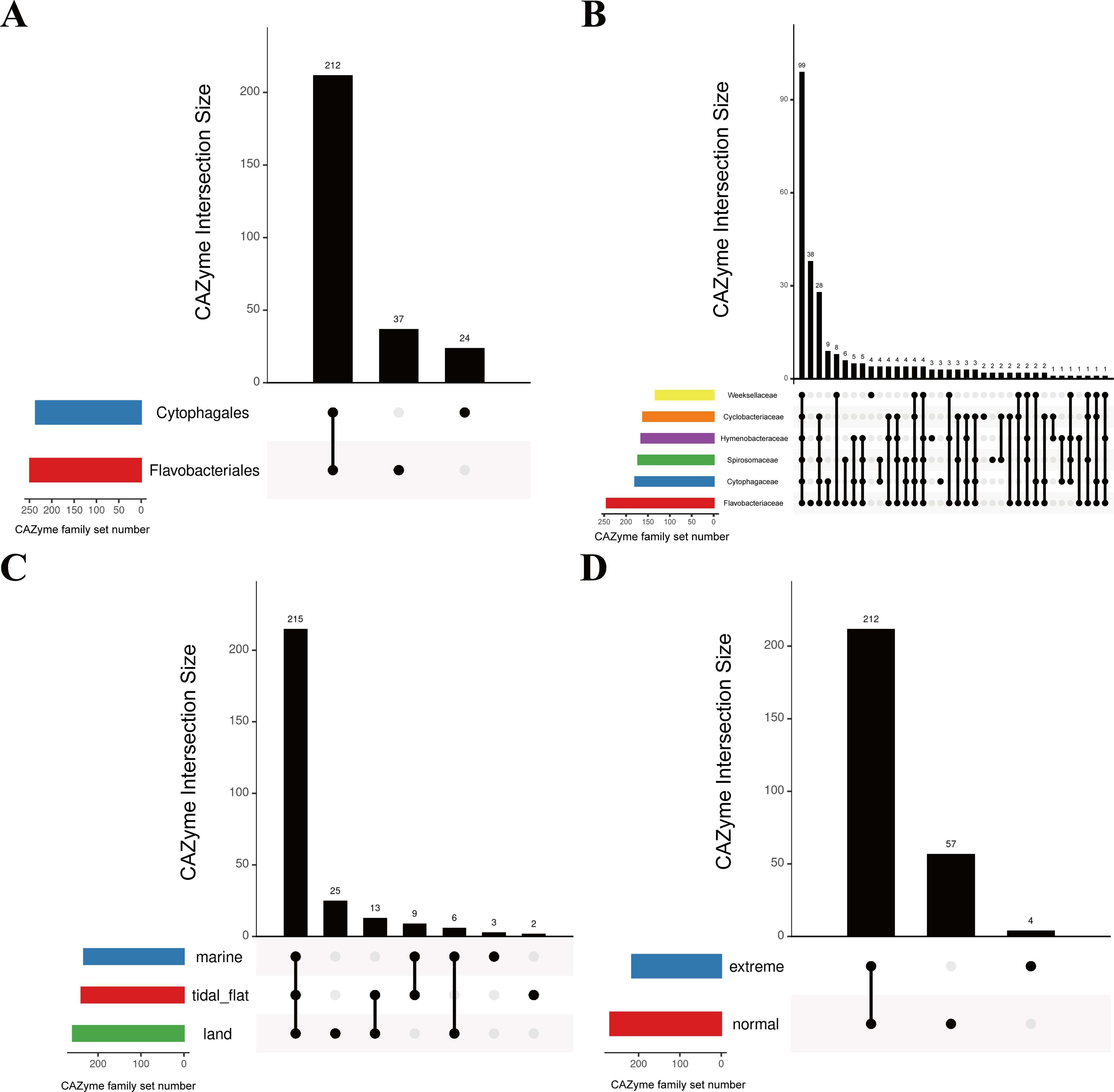
Figure 6. Upset plots showing the distribution of the carbohydrate-active enzyme (CAZyme) families in different orders (A), major families (B), sources (C), and environmental conditions (D). A total of 100,445 CAZymes belonging to 273 CAZyme families were used for statistics. The histograms show the counts of CAZyme families in each subset of overlap. The dots and lines at the bottom right represent the subsets of overlap. The strip at the bottom left show the counts of CAZyme families within each category.
Among bacteria from different sources, the terrestrial bacteria had the most unique CAZyme families (25), with only three and two unique CAZyme families being identified in marine and tidal flats, respectively (Figure 6C). Tidal flats shared more CAZyme families with terrestrial and marine bacteria, which may be attributed to the dual carbohydrate source in tidal flats. According to the analysis of the unique CAZyme families in different environmental conditions (Figure 6D), extreme environments greatly constrain the CAZyme family types. In contrast to the 57 unique CAZyme families present in strains from normal environments, only four unique CAZyme families were found in extreme environments.
ii) Secondly, differences in the shared CAZyme families among different groups were analyzed. The majority of the top 15 CAZyme families demonstrated significant variations between the Flavobacteriales and Cytophagales species (Figure 7A), particularly the most abundant GT2 and GT4 families (Supplementary Figure S2), both of which displayed considerable divergence. Among the 212 shared CAZyme families in Flavobacteriales and Cytophagales species, there were 25 and 59 significantly enriched CAZyme families, respectively (Supplementary Figure S3). The significantly enriched GT5 family in Flavobacteriales species is mainly glycogen glucosyltransferase and starch glucosyltransferase (Coutinho et al., 2003), while the GH73 family is mainly responsible for the degradation of β-1,4-glycosidic linkage between N-acetylglucosaminyl (NAG) and N-acetylmuramyl (NAM) (Grieb et al., 2020). Among the 59 significantly enriched CAZyme families found in Cytophagales species, the CE15 family showed the most noticeably significant enrichment trend. This family functions to cleave the covalent ester bonds between lignin and glucuronoxylan, playing an important role in hydrolyzing the recalcitrant lignin–carbohydrate–complex (LCC), which are major obstacles in the industrial enzyme biomass hydrolysis process (De Santi et al., 2017; Arnling Bååth et al., 2018; Kmezik et al., 2021). Furthermore, a substantial presence of unclassified GH (GH0) in Flavobacteriales and Cytophagales species was observed (Supplementary Figure S4). Specifically, the GH0 numbers both per genome and per megabase genome in Cytophagales species were significantly higher than those in Flavobacteriales species. In addition to the CAZyme families previously mentioned, significant numbers of susC/susD genes were also identified in Flavobacteriales and Cytophagales species, demonstrating significant taxonomic divergences (Supplementary Figure S5). Compared with Flavobacteriales species, Cytophagales species exhibited significantly higher abundance of susC/susD genes in each genome and per megabase genome.
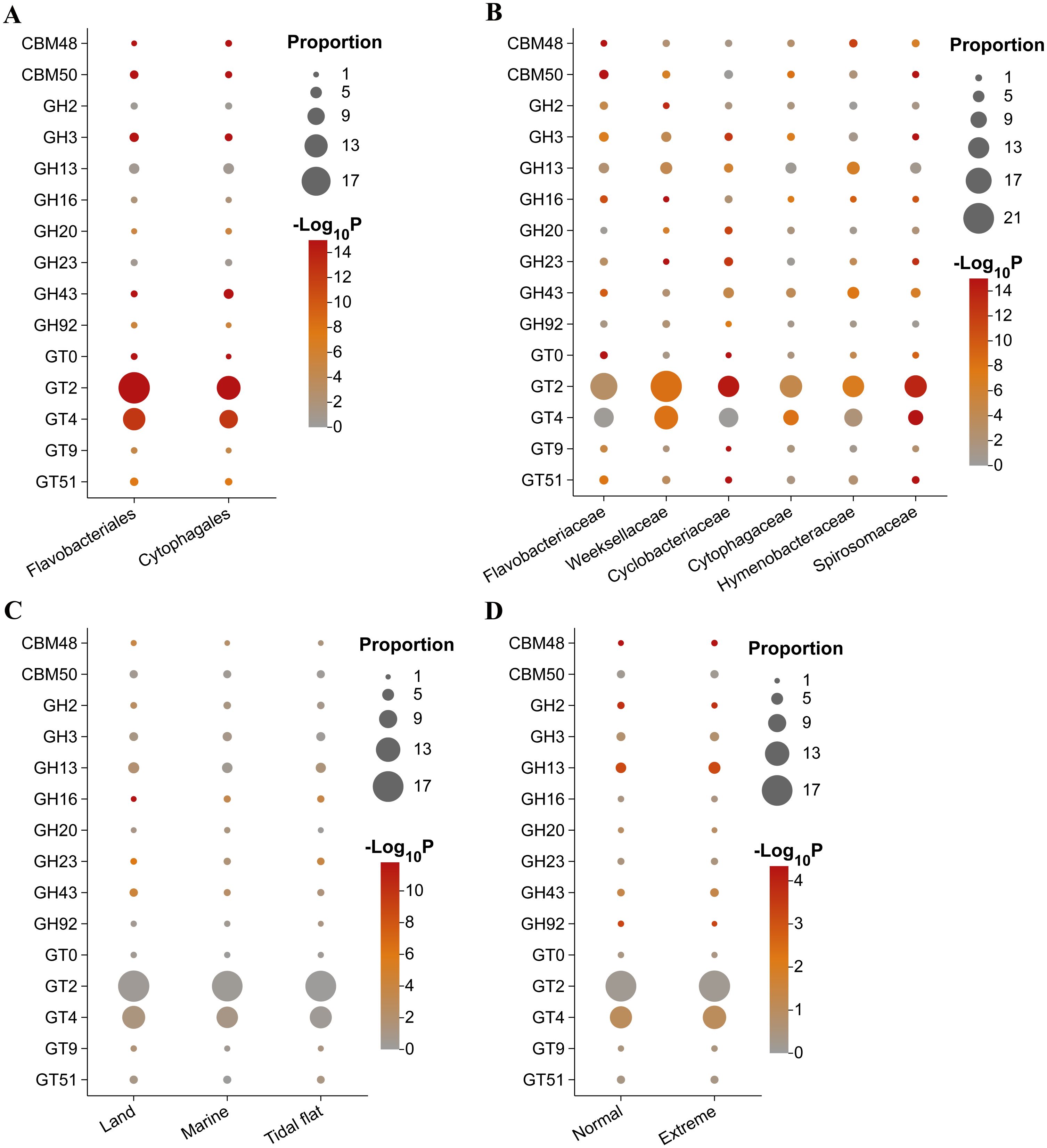
Figure 7. Differences in the proportions of carbohydrate-active enzyme (CAZyme) families among different orders (A), major families (B), sources (C), and environmental conditions (D). The significance of the differences for each group was determined by comparing a group to all other groups. The dot size was based on the proportion (in percentage) of CAZyme family in each group. The dot color indicates the significance of the enrichment. Only the top 15 abundant CAZyme families are displayed.
At the family level, as members of Flavobacteriales, the species of Weeksellaceae and Flavobacteriaceae had a significantly enriched GT2 family (Figure 7B), which was the most abundant CAZyme family, as described above. As the second most abundant CAZyme family, the GT4 family was significantly enriched in Weeksellaceae species. The species of Spirosomaceae, Cytophagaceae, and Cyclobacteriaceae exhibited a significant enrichment of the GH20 and GH43 families, which play a pivotal role in the degradation of chitin and hemicellulose (Morais et al., 2021; Mészáros et al., 2022), particularly the GH43 family, which was enriched in all four major families of Cytophagales.
In terms of environment, the composition of the top 15 CAZyme families in terrestrial bacteria exhibited slight variations compared with those in marine and tidal flat environments (Figure 7C). Nevertheless, considering all of the shared CAZyme families among the terrestrial, marine, and tidal flat bacteria, significant different polysaccharide metabolic patterns were observed (Supplementary Figures S5A, B). Compared with the marine and tidal flat bacteria, the terrestrial bacteria possessed significantly more GH25, GH77, and CBM20 families, indicating the degradation potential of terrestrial polysaccharides. The GH25 family comprises well-known bacterial lysozymes that play essential roles in oomycete pathogen antagonism for the terrestrial plant Arabidopsis thaliana and in the intestinal bacterial cell wall breakdown of dairy cows (Vollmer et al., 2008; Eitzen et al., 2021; Lin et al., 2023). The GH77 and CBM20 families are involved in intracellular maltose–maltodextrin metabolism and bind starch to promote starch hydrolysis (Son Tung et al., 2019; Marecek et al., 2021; Christensen et al., 2023). In addition, the CAZymes that were significantly enriched in bacteria from marine and tidal flats had stronger degradation potential for marine polysaccharides, such as the GH113, PL17, PL7, PL6, GH15 families. Of these, GH113 is a family with specificity toward mannans, which is distributed in both marine and terrestrial plants (del-Carmen-Rodriguez-Gacio et al., 2012; Terrett et al., 2019; Couturier et al., 2022). The GH15 family is generally known as glucoamylases (Trochine et al., 2022). However, a recent study has revealed that some enzymes in the GH15 family can also participate in the degradation of trehalose (Yuasa et al., 2018; Zhang et al., 2022). The PL6, PL7, and PL17 families all correspond to degrade alginate (Mathieu et al., 2016; Chernysheva et al., 2021). Different from the other groups, the bacteria from marine and tidal flats nearly had no significant difference in the composition of CAZyme families, except for the GH19 family enriched in marine and the PL28 family enriched in tidal flats (Supplementary Figure S5C). Of these, GH19 is a family of chitinases, while the PL28 family is associated with ulvan lyases. As shown in the ternary phase diagram, the top 100 CAZyme families in the three sources were distributed near the central axis (dashed blue line) between the marine and tidal flat environments (Supplementary Figure S5D), indicating that the bacteria from marine and tidal flats had a similar CAZyme family composition and are noticeably different from terrestrial bacteria.
As shown in Figure 7D, our analysis revealed a prominent enrichment of certain CAZyme families, specifically CBM48, GH13, and GH43, within the bacteria isolated from extreme environments. These enriched families are notably associated with the degradation of glycogen and hemicellulose (Ping et al., 2020; Morais et al., 2021).
Based on the CAZyme family composition (i.e., both unique and shared CAZyme families), beta diversity analyses (PCoA) were further conducted using binary Jaccard distance matrices to discover novel insights into the factors that influence their polysaccharide metabolic patterns (Figure 8). Species in the different orders could be well separated, and the clustering of the Cytophagales species was more compact than that of the Flavobacteriales species, indicating that the difference in the CAZyme family composition for the species of Cytophagales was smaller than that in Flavobacteriales. The main families of Cytophagales (Cyclobacteriaceae, Cytophagaceae, Hymenobacteraceae, and Spirosomaceae) clustered tightly together compared with the major families of Flavobacteriales. In Flavobacteriales, the CAZyme family composition between the Flavobacteriaceae and Weeksellaceae species significantly differed, and their clusters were far away from each other. Notably, consistent with the analysis above, the isolation sources greatly influenced the polysaccharide utilization patterns, with species from the tidal flat and marine environments clustering tightly together and were distinguished from terrestrial species, indicating higher similarity of the beta diversity of the CAZyme families between species from marine and tidal flats. Although the sources affected the polysaccharide metabolism patterns of these species, no clear distinction was found for the species from different environmental conditions. In order to reveal the main factor influencing the CAZyme family composition, a cluster tree was constructed using UPGMA (Figure 9). The species were mainly divided into two main groups (Flavobacteriales and Cytophagales). However, the species from different sources did not show a clear distinction, indicating that the evolutionary status plays a more pivotal role than the habitat in determining the CAZyme family composition of these bacteria.
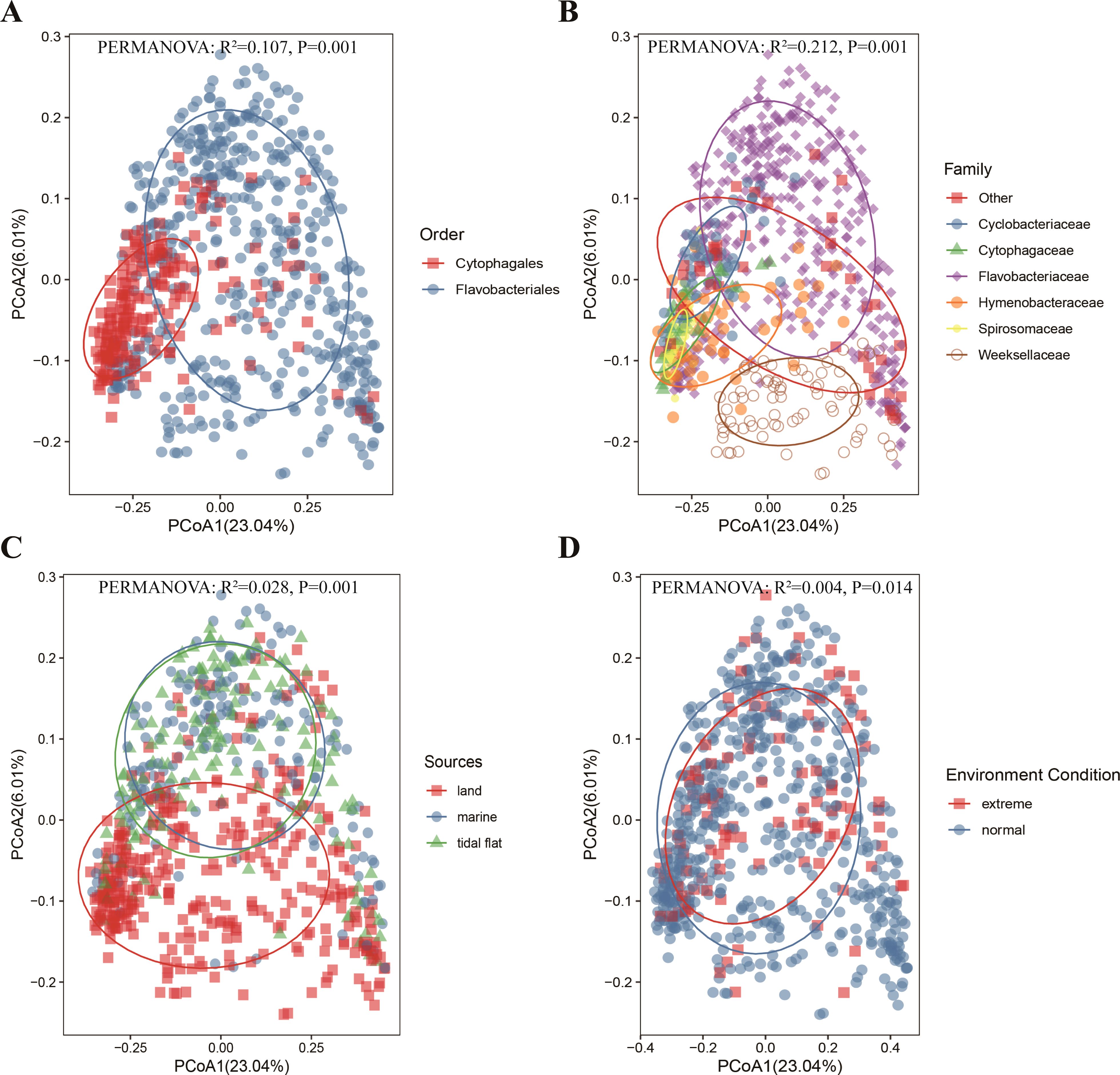
Figure 8. Principal coordinate analysis (PCoA) based on the binary Jaccard distance among different orders (A), major families (B), sources (C), and environmental conditions (D).
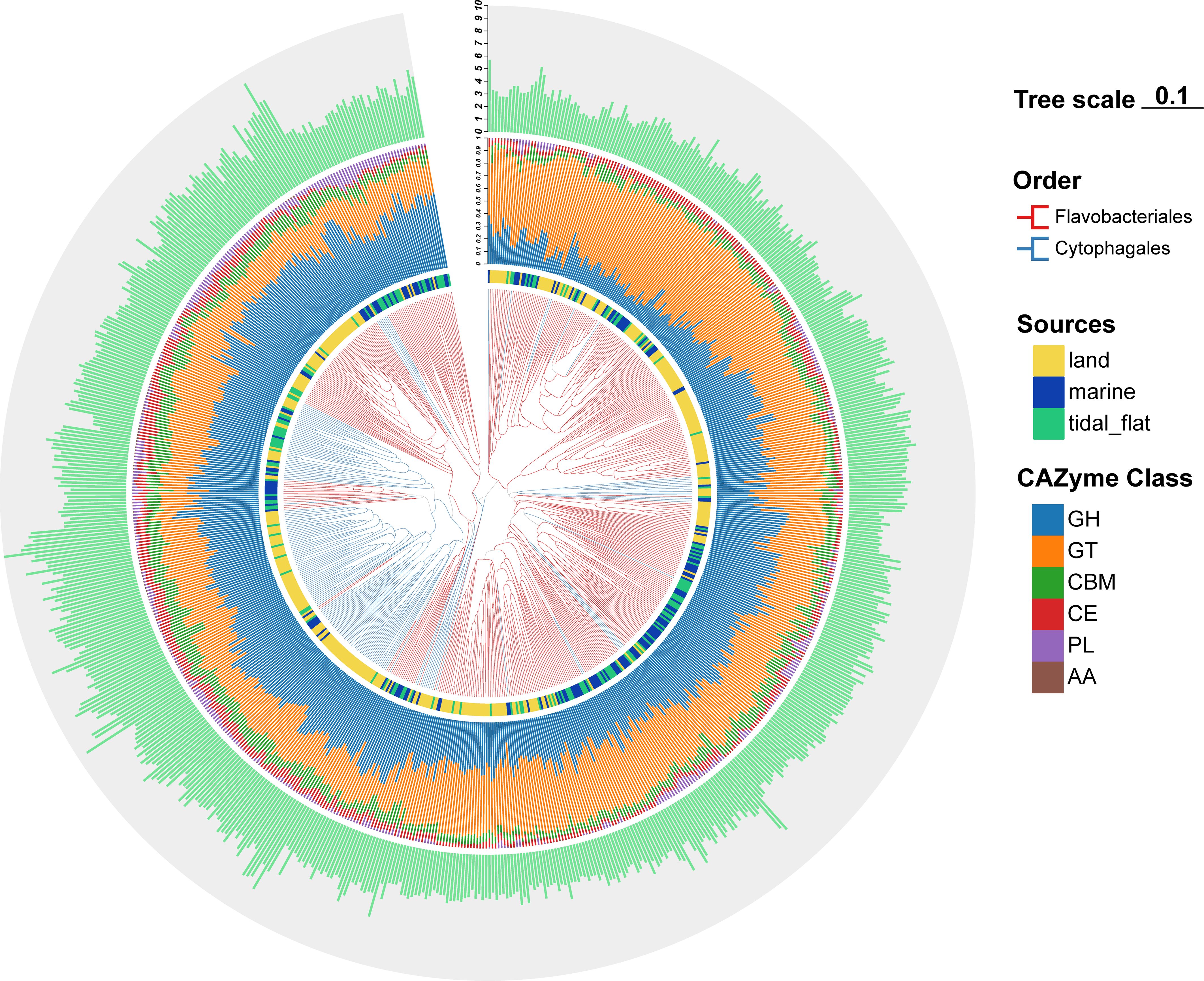
Figure 9. Unweighted pair group method with arithmetic mean (UPGMA) cluster tree based on the binary Jaccard distance. Branch colors represent the taxonomic status of the strains. The color on the inner ring refers to the sources of strains. The middle ring refers to the percentage of each type of carbohydrate-active enzyme (CAZyme) class, while the outer ring refers to the genomic sizes of the strains.
3.4 Co-occurrence networks of CAZymes
The co-occurrence patterns of the bacteria from Flavobacteriales and Cytophagales were further analyzed. As shown in Figure 10, the analysis revealed intricate patterns of correlation among the various families of CAZymes within the bacterial orders of Flavobacteriales and Cytophagales. In both co-occurrence networks, the vast majority of CAZyme families exhibited positive correlations, particularly within the Cytophagales species, where all of the significant relationships (correlation coefficients >0.6 and p < 0.05) were positive correlations, indicating the synergistic effects among these CAZyme families. Of the positively correlated CAZymes, the GH2, GH28, GH43, GH95, GH105, and CE20 families exhibited the highest connection degree in both co-occurrence networks, indicating the pivotal role of these CAZyme families in the bacterial metabolism of polysaccharides. Moreover, the CAZyme families of CBMs, CEs, GHs, GTs, and PLs demonstrated certain correlations with each other within the Flavobacteriales and Cytophagales groups in both co-occurrence networks, while the CAZyme families of AAs did not. Among them, a marked prevalence of GH class enzymes was observed, with most of the co-occurring CAZyme families belonging to them. Several CAZyme families exhibited significant positive correlations in both network graphs (e.g., between PL6 and PL7, and CE20 and GH43), which might suggest their synergistic roles in polysaccharide metabolism.
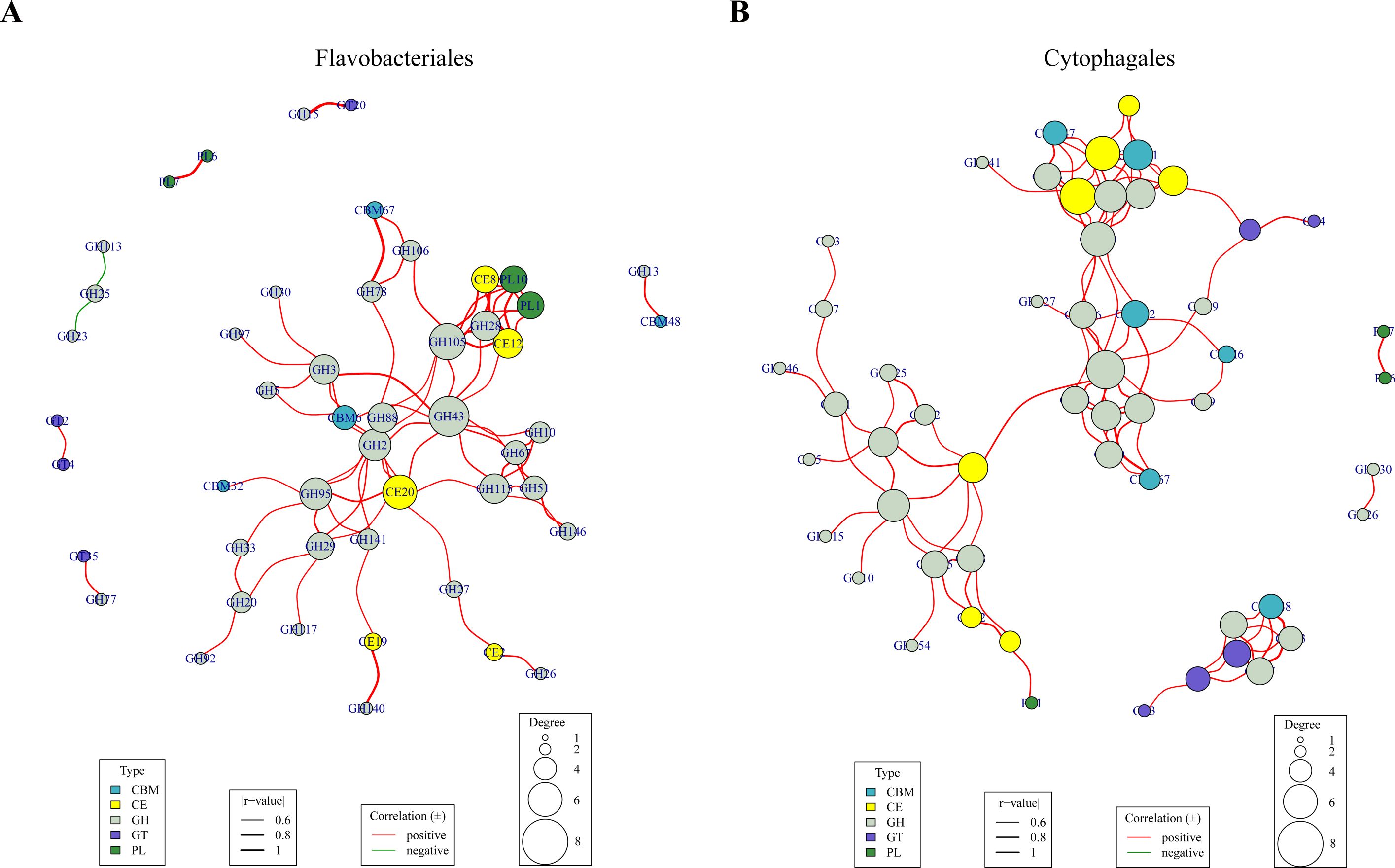
Figure 10. Co-occurrence networks of the carbohydrate-active enzyme (CAZyme) families for the Flavobacteriales (A) and Cytophagales (B) bacteria. Nodes represent the CAZyme families, while the size represents their connection degree. The thickness and the color of the lines were determined using Spearman’s correlation coefficients. Only correlation coefficients >0.6 and p < 0.05 are shown.
Despite these similarities, distinct differences were also evident between the two networks. The CAZyme families in Flavobacteriales and Cytophagales showed notable differences in the architecture of the co-occurrence networks. Flavobacteriales exhibited a higher number of modules, and the connections between various CAZyme families within the main module displayed greater intricacy and compactness. The co-occurrence network of Cytophagales exhibited a higher number of CBMs, CEs, and GTs compared with Flavobacteriales, along with greater connection degree, suggesting that they could play a more crucial synergistic role within Cytophagales species. Conversely, PLs were more prevalent in the co-occurrence network of Flavobacteriales.
4 Discussion
4.1 Differences in the polysaccharide metabolic patterns among different phylogenetic groups
Comprising the largest taxon in the phylum Bacteroidota, Flavobacteriales species are known for their ability to degrade polysaccharides (Gavriilidou et al., 2020). However, in this study, the overlooked polysaccharide metabolic potential of Cytophagales species was revealed. Notably, in terms of the absolute abundance of CAZymes per genome and per megabase genome, the CAZyme diversity, and the number of enriched CAZyme families, the Cytophagales species exhibited higher values compared with the Flavobacteriales species, indicating their greater polysaccharide metabolic potential.
As the differences in the CAZyme classes between Cytophagales and Flavobacteriales were substantial, a comprehensive comparison of their polysaccharide metabolic potential was further performed. The Flavobacteriales sourced from marine habitats accounted for 143/500, while the Cytophagales isolated from the marine environment only accounted for 28/202. The high counts of PLs per megabase genome in Flavobacteriales species could be attributed to their extensive distribution across tidal flat and marine environments (Gavriilidou et al., 2020). These environments are rich in marine polysaccharides containing abundant uronic acids (Furusawa et al., 2021; Yang et al., 2022), which necessitate various phospholipases for degradation (Michaud et al., 2003). In addition, a large number of GH0 was also found, with average numbers of 0.4 and 0.24 per megabase genome for the species of Cytophagales and Flavobacteriales, respectively (Supplementary Figure S4B). The greater number of unidentified GHs in Cytophagales species indicates that research on their CAZymes is still insufficient or incomplete. As a result, the average number of CAZymes belonging to each class per megabase genome of Cytophagales species was not noticeably higher than that of Flavobacteriales species.
However, SusC and SusD, the outer membrane TonB-dependent transporter and surface glycan-binding protein, respectively, recognized as indicators of Bacteroidota PULs (Kappelmann et al., 2019; Wardman et al., 2022), provide additional evidence for the higher polysaccharide metabolic potential of Cytophagales species (Supplementary Figure S5). Cytophagales species exhibited higher average counts of SusC (63.97 per genome and 11.78 per megabase genome) and SusD (29.31 per genome and 5.39 per megabase genome) compared with Flavobacteriales species (33.57 per genome and 8.93 per megabase genome for SusC; 12.45 per genome and 3.31 per megabase genome for SusD). These results further indicated that Cytophagales species may possess more PULs associated with polysaccharide metabolism and could represent a new group with higher polysaccharide-degrading potential.
Among the taxon-specific CAZyme families, the GT113 and PL36 families comprised relatively large proportions in Flavobacteriales species, accounting for 0.048% and 0.035% of the total CAZymes, respectively (29 and 21 CAZymes, respectively), but were not found in Cytophagales species (Supplementary Table S4). Specifically, the GT113 domain adopts a GT-B fold and is the most similar GT to GT47. Wilson et al. suggested that the GT113 family in bacteria may be closely related to the eukaryotic GT47 family (Wilson et al., 2022). Moreover, 20 of the 21 PL36 family members were present in Flavobacterium species, as β-d-mannuronosyl linkage-specific lyases (EC 4.2.2.3, M-specific). The PL36 family can participate in the degradation of alginate, play an important role in alginate oligomer preparation, and fuel ethanol production from brown algal alginate (Enquist-Newman et al., 2014; Ghadam et al., 2017; Dong et al., 2019).
In contrast, the Cytophagales-specific CBM8 and GH93 families showed metabolic potential of terrestrial polysaccharides, accounting for 0.038% and 0.025% of the total CAZymes, respectively. CBM8 has been experimentally shown to bind cellulose (Liberato et al., 2022), while GH93 is an exo-α-l-1,5-arabinanase that plays an important role in the exo-mode degradation of arabinose, which is the major component in pectin (McKie et al., 1997; Seiboth and Metz, 2011; An et al., 2022).
More taxon-specific CAZyme families were found among different families. For example, the GH78 and GH9 families accounted for more than 1% of the total CAZymes in Cytophagaceae species, but were not detected in the Weeksellaceae species, showing their metabolic ability to degrade cellulose and polysaccharides containing l-rhamnose. Notably, most of the GH9 family endoglucanases have catalytic domains linked to CBM3 (Konar et al., 2022), which is well consistent with our study wherein the CBM3 family accounted for 0.16% of the total CAZymes in Cytophagaceae species, but was not found in Weeksellaceae species. Detailed differences of the CAZyme families in different taxonomic families are presented in Supplementary Table S4.
The GT2 and GT4 families were notably prevalent and emerged as the most abundant CAZymes in both orders (Supplementary Figure S2). They play important roles in critical glycan synthesis processes. The GT2 family is primarily associated with the synthesis of cell walls and extracellular β-glucans (Oehme et al., 2019), contributing significantly to the formation and modification of long polysaccharide structures, such as celluloses, chitin oligosaccharides, mannans, and glucans (Coutinho et al., 2003; Gómez-Silva et al., 2019). The GT4 family encompasses α-glucosyltransferase, α-mannosyltransferase, and glucosyltransferase, and even enzymes harness nucleotide-sugar and phospho-sugar donors. The GT2 and GT4 families have been speculated to be the original GTs, which evolved into other GTs in archaea (Coutinho et al., 2003). In addition, the GT2 and GT4 families in Flavobacteriaceae and Weeksellaceae species play a crucial role in the formation of biofilms, and they account for approximately 20% and 5% of the total biomass in the biofilm on household plastic surfaces within estuarine ecosystems, respectively (Lear et al., 2022). Moreover, the abundant presence of the GT2 and GT4 families has been proposed to exert a pivotal influence on the construction of biofilms on copper surfaces (Zhang et al., 2019c). Our analysis revealed noticeably high proportions of the GT2 and GT4 families in both Flavobacteriaceae and Weeksellaceae species (Figure 7B; Supplementary Figure S2), indicating that the species of Flavobacteriales (mainly composed of Flavobacteriaceae and Weeksellaceae species) could play an important role in the formation of coastal microbial biofilms.
4.2 Differences in the polysaccharide metabolic patterns among different environments
Compared with those in the tidal flat and marine environments, terrestrial bacteria exhibited distinct polysaccharide metabolism patterns. In general, the marine lineages of Bacteroidota demonstrate a higher peptidase/GH ratio than non-marine lineages (Fernández-Gómez et al., 2013; Kolton et al., 2013; Zhang et al., 2019a). Comparative genome analysis of six Vibrionaceae species also revealed that non-marine and free-living members of the Rumoiensis clade possess a greater abundance of GHs (Tanaka et al., 2020). Our comprehensive study of the CAZymes across all type strains of Cytophagales and Flavobacteriales indicated that there are not only more GHs but also more AAs, CBMs, CEs, and GTs (Figures 4E, F), as well as higher CAZyme diversity and more unique CAZymes, in terrestrial lineages (Figures 3C, 6C). This phenomenon might be attributed to the higher diversity and complexity of the terrestrial ecological environments, where vascular plants also possess a higher proportion of carbohydrates (15%–30%) compared with marine phytoplanktons (5%–25%) (Zhang et al., 2019a), further proving the importance of the terrestrial bacteria participating in terrestrial carbon source turnover (Delgado-Baquerizo et al., 2016; Žifčáková et al., 2017).
In addition, species in the tidal flat and marine environments showed high similarity in their CAZyme profiles, with most of their CAZyme classes exhibiting similar absolute and relative abundances (Figures 4E, F, 5C). A few CAZyme families were observed to be significantly different between the marine and tidal flat species (Supplementary Figure S6C). The GH19 family was enriched in the marine environment, and the PL28 family was enriched in tidal flats. As chitinases, the GH19 family plays an important role in the degradation of chitin, which has been estimated to be second to that of cellulose and widely distributed in shells of crustaceans (Zewude et al., 2022). The PL28 family was the most recently discovered ulvan lyase, although ulvan is present in marine algae such as the genus Ulva (Chlorophyta). However, with the influence of seasonal monsoons and ocean currents, they usually accumulate near tidal flats (Zhang et al., 2019b; Chen et al., 2020; Sun et al., 2022), hence the enrichment of the PL28 family in tidal flats. The similarity between the marine and tidal flat environments could be attributed to the frequent material exchange between them facilitated by the strong fluidity of seawater and tidal periodicity (Wang et al., 2020). Despite tidal flats being always considered to be the recipient of both marine and terrestrial organic carbon (Kubo and Kanda, 2017; Sasmito et al., 2020; Li et al., 2022), our analysis only presented very limited CAZyme similarity between the tidal flat and terrestrial species. This could be due to most of the terrestrial organic matter being consumed by freshwater microorganisms during transportation, resulting in only a minor fraction of the organic matter flux reaching the tidal flats, although they could reach tidal flats through rivers and underground rivers (Regnier et al., 2022).
Compared with the bacteria isolated from different sources, finite divergences of the CAZyme composition were found between species in extreme and normal environments. Extreme environments greatly limited the size of the CAZyme family categories (Figure 6D), with the average numbers of GHs and PLs in extreme environments also lower than those in normal environments (Figures 4G, H), illustrating the impact of environmental conditions on microbial ecological functions. In addition, species in extreme environments require encoding a large amount of GTs for survival. As critical enzymes in extracellular polysaccharide production, the GTs enriched in extreme environments may be related to the secretion of polysaccharides that help cells resist external environmental stress (Soumya and Nampoothiri, 2021). Moreover, some of the flagellar glycans synthesized by GTs may be associated with bacterial motility, biofilm formation, and colonization (Kint et al., 2022; Tomás et al., 2022), thereby supporting the survival of species that inhabit extreme environments. However, as shown in Supplementary Table S4, in contrast to that in extreme environments, the enriched GH17, GH128, and CBM85 in normal environments were associated with the degradation of β-1,3-glucans, which are widely present in animals, plants, and microbes. These polysaccharides, which are found in various normal environments such as soil, the gut, aquatic ecosystems, and plants, participate in numerous biological processes ranging from energy metabolism to cellular construction and are among the most abundant and commercially important polysaccharides (Santos et al., 2020; Walton, 2020). These observations underscore the intricate interplay between microbial communities and their surroundings, with the microbial CAZyme profiles being indicative of specific ecological niches. Such insights into the composition of CAZymes contribute to our understanding of the functional potential of microbial communities in different environments and shed light on their roles in nutrient transformation and ecosystem stability.
4.3 Phylogeny: the major factor shaping the polysaccharide metabolic pattern
In this study, the comparison between Flavobacteriales and Cytophagales revealed a substantial divergence in the CAZyme family diversity and composition, which are closely tied to their taxonomic positions. While environmental factors such as habitat (land or marine) undoubtedly influence microbial communities, they appear to play a secondary role in shaping the CAZyme diversity and composition compared with taxonomic lineage. Compared with different sources, strains from different taxa exhibited more significant divergence in CAZyme family diversity (Figures 2, 3). Similarly, strains belonging to different taxa displayed greater disparities in CAZyme class and family composition than those from different habitats (Figures 4-7).
Recent studies have indicated that CAZymes are phylogenetically conserved and vary significantly among microbial phyla (López-Mondéjar et al., 2022). At finer levels of taxonomic resolution, some researchers have found that the functional traits within Flavobacteriaceae are consistent with both single-copy marker gene-based phylogeny and 16S rRNA gene-based phylogeny rather than the habitat type (Liu et al., 2019; Silva et al., 2019; Gavriilidou et al., 2020). However, other studies have suggested that the PUL repertoire of Flavobacteriia species is more dependent on their distinct ecological niches, with phylogeny playing a secondary role (Kappelmann et al., 2019). In this study, the clusters for strains in different taxa and environments can be well separated, indicating that habitat type and phylogeny both influence the polysaccharide metabolic patterns of Cytophagales and Flavobacteriales species (Figure 8). However, the PERMANOVA results indicated that the explained variance grouping by taxon (10.7% and 21.2%, respectively) was greater than that of grouping by environment (2.8% and 0.4%, respectively), revealing that the CAZyme family composition was mainly affected by the taxonomic status. Furthermore, a cluster tree generated using UPGMA divided these strains into two major groups (Flavobacteriales and Cytophagales) (Figure 9), indicating that the composition of the CAZyme family was more affected by the phylogenetic status. Recent studies have also shown intra-clade diversity in encoding the potential and phylogenetic conservation of specific CAZymes at the genus level, reinforcing the idea that the phylogenetic status mainly shapes the CAZyme distribution in marine bacteria (Sun et al., 2021).
4.4 Synergism: the primary mechanism of Flavobacteriales and Cytophagales species in polysaccharide metabolism
In recent years, there has been a growing number of research works on the polysaccharide metabolism of the members of Bacteroidota, being the primary degraders of polysaccharides. However, unraveling the intricate interplay among CAZymes requires extensive and meticulous experimental investigations. In this study, co-occurrence analysis was employed to reveal the widespread positive associations among Flavobacteriales and Cytophagales species. A large number of positive correlations were found in both co-occurrence networks, indicating the reliability of the co-occurrence analysis. These results could suggest potential synergistic interactions among these CAZymes (Figure 10).
Many positively correlated CAZymes in the co-occurrence networks exhibited synergistic effects in polysaccharide metabolism. The GH15 and GT20 families in Flavobacteriales species exhibited an extremely high positive correlation. It was also found that their positions in PULs were always adjacent to each other, as shown in PULDB, and GH15 was related to trehalose metabolism. Furthermore, the co-occurrence of the PL6 and PL7 families indicated their synergism in cleaving alginate, while the co-occurring GH78, GH106, and CBM67 families showed the same ability in the degradation of polysaccharides containing α-l-rhamnose. Moreover, the co-occurring CBM48 family showed glycogen-binding function, which appended to GH13 modules (Machovic and Janecek, 2008; Ping et al., 2020). In contrast to some of the positively correlated CAZyme families showing synergistic effects, a number of CAZyme families with analogous functions were also observed to have negative correlations. For example, the negatively correlated CAZyme families GH23 and GH25 are both lysozymes, suggesting a potential functional substitution between them. Some other co-occurring CAZyme families might possess unknown collaborative or alternative mechanisms awaiting further exploration.
5 Conclusions
The results of this study revealed that Cytophagales possessed greater diversity and abundance of CAZymes compared with Flavobacteriales, suggesting a higher polysaccharide metabolic potential. Moreover, the findings demonstrated that taxonomic status has a more significant impact on the CAZyme composition and diversity than environmental conditions. While the strains in different environments, such as terrestrial, marine, and tidal flats, displayed distinct CAZyme compositions, the strains in the tidal flat and marine environments showed notable similarities, likely due to the frequent hydrodynamic disturbances that facilitate the biotic and abiotic exchanges between these two environments. This study highlighted the presence of synergistic interactions among various CAZyme families, indicating a cooperative mechanism in polysaccharide degradation. These findings provide valuable insights into the polysaccharide metabolic patterns across different habitats and taxa, contributing to our understanding of microbial carbohydrate metabolism and its ecological roles.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
KJM: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Visualization, Writing – original draft. YLY: Data curation, Writing – review & editing. YKL: Data curation, Writing – review & editing. GYF: Supervision, Writing – review & editing. YHW: Supervision, Validation, Writing – review & editing. CS: Conceptualization, Funding acquisition, Supervision, Validation, Writing – review & editing. XWX: Conceptualization, Funding acquisition, Project administration, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by National Natural Science Foundation of China (No. U23A2034 and 32370006), Zhejiang Provincial Natural Science Foundation of China under Grant No. LY24C010002 and Zhejiang Provincial High-level Talent Special Support Plan (No. 2021R51008).
Conflict of interest
Author CS was employed by the company Shaoxing Biomedical Research Institute of Zhejiang Sci-Tech University Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2025.1551618/full#supplementary-material
References
An J., Xu W., Meng X., Chen G., Zhang W., Liu W. (2022). Biochemical characterization of a thermophilic exo-arabinanase from the filamentous fungus Rasamsonia emersonii. J. Biosci. Bioeng. 133, 316–322. doi: 10.1016/j.jbiosc.2021.12.010
Anantapong T., Kanjana-Opas A. (2016). Microbial community analysis of aquatic gliding bacteria (Cytophaga-flavobacterium-bacteroides) by fluorescence in situ hybridization (FISH). Walailak J. Sci. Technol. 14, 969–980. Available at: https://wjst.wu.ac.th/index.php/wjst/article/view/2417.
Arnling Bååth J., Mazurkewich S., Knudsen R. M., Poulsen J.-C. N., Olsson L., Lo Leggio L., et al. (2018). Biochemical and structural features of diverse bacterial glucuronoyl esterases facilitating recalcitrant biomass conversion. Biotechnol. Biofuels 11, 213. doi: 10.1186/s13068-018-1213-x
Bar-On Y. M., Phillips R., Milo R. (2018). The biomass distribution on Earth. Proc. Natl. Acad. Sci. United States America 115, 6506–6511. doi: 10.1073/pnas.1711842115
Bhujbal S. K., Ghosh P., Vijay V. K., Rathour R., Kumar M., Singh L., et al. (2022). Biotechnological potential of rumen microbiota for sustainable bioconversion of lignocellulosic waste to biofuels and value-added products. Sci. Total Environ. 814, 152773. doi: 10.1016/j.scitotenv.2021.152773
Bolyen E., Rideout J. R., Dillon M. R., Bokulich N. A., Abnet C. C., Al-Ghalith G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Chan P. P., Brian Y., Allysia J., Todd M. (2021). tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096. doi: 10.1093/nar/gkab688
Chen Y.-J., Leung P. M., Wood J. L., Bay S. K., Hugenholtz P., Kessler A. J., et al. (2021). Metabolic flexibility allows bacterial habitat generalists to become dominant in a frequently disturbed ecosystem. ISME J. 15, 2986–3004. doi: 10.1038/s41396-021-00988-w
Chen Y., Song D., Li K., Gu L., Wei A., Wang X. (2020). Hydro-biogeochemical modeling of the early-stage outbreak of green tide (Ulva prolifera) driven by land-based nutrient loads in the Jiangsu coast. Mar. pollut. Bull. 153, 111028. doi: 10.1016/j.marpolbul.2020.111028
Cheng J., Hu J., Geng F., Nie S. (2022). Bacteroides utilization for dietary polysaccharides and their beneficial effects on gut health. Food Sci. Hum. Wellness 11, 1101–1110. doi: 10.1016/j.fshw.2022.04.002
Chernysheva N., Bystritskaya E., Likhatskaya G., Nedashkovskaya O., Isaeva M. (2021). Genome-wide analysis of PL7 alginate lyases in the genus zobellia. Molecules 26, 2387. doi: 10.3390/molecules26082387
Christensen S. J., Madsen M. S., Zinck S. S., Hedberg C., Sorensen O. B., Svensson B., et al. (2023). Enzymatic potato starch modification and structure-function analysis of six diverse GH77 4-alpha-glucanotransferases. Int. J. Biol. Macromol. 224, 105–114. doi: 10.1016/j.ijbiomac.2022.10.107
Coutinho P. M., Deleury E., Davies G. J., Henrissat B. (2003). An evolving hierarchical family classification for glycosyltransferases. J. Mol. Biol. 328, 307–317. doi: 10.1016/S0022-2836(03)00307-3
Couturier M., Touvrey-Loiodice M., Terrapon N., Drula E., Buon L., Chirat C., et al. (2022). Functional exploration of the glycoside hydrolase family GH113. PloS One 17, e0267509. doi: 10.1371/journal.pone.0267509
del-Carmen-Rodriguez-Gacio M., Iglesias-Fernandez R., Carbonero P., Matilla A. J. (2012). Softening-up mannan-rich cell walls. J. Exp. Bot. 63, 3975–3988. doi: 10.1093/jxb/ers096
Delgado-Baquerizo M., Maestre F. T., Reich P. B., Jeffries T. C., Gaitan J. J., Encinar D., et al. (2016). Microbial diversity drives multifunctionality in terrestrial ecosystems. Nat. Commun. 7, 10541. doi: 10.1038/ncomms10541
De Santi C., Gani O. A., Helland R., Williamson A. (2017). Structural insight into a CE15 esterase from the marine bacterial metagenome. Sci. Rep. 7, 17278. doi: 10.1038/s41598-017-17677-4
Dong F., Xu F., Chen X.-L., Li P.-Y., Li C.-Y., Li F.-c., et al. (2019). Alginate lyase aly36B is a new bacterial member of the polysaccharide lyase family 36 and catalyzes by a novel mechanism with lysine as both the catalytic base and catalytic acid. J. Mol. Biol. 431, 4897–4909. doi: 10.1016/j.jmb.2019.10.023
Drula E., Garron M.-L., Dogan S., Lombard V., Henrissat B., Terrapon N. (2022). The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res. 50, D571–D577. doi: 10.1093/nar/gkab1045
Eitzen K., Sengupta P., Kroll S., Kemen E., Doehlemann G. (2021). A fungal member of the Arabidopsis thaliana phyllosphere antagonizes Albugo laibachii via a GH25 lysozyme. Elife 10, e65306. doi: 10.7554/eLife.65306
Enquist-Newman M., Faust A. M. E., Bravo D. D., Santos C. N. S., Raisner R. M., Hanel A., et al. (2014). Efficient ethanol production from brown macroalgae sugars by a synthetic yeast platform. Nature 505, 239–243. doi: 10.1038/nature12771
Falkowski P. G., Barber R. T., Smetacek V. (1998). Biogeochemical controls and feedbacks on ocean primary production. Science 281, 200–206. doi: 10.1126/science.281.5374.200
Fernández-Gómez B., Richter M., Schüler M., Pinhassi J., Acinas S. G., González J. M., et al. (2013). Ecology of marine Bacteroidetes: a comparative genomics approach. ISME J. 7, 1026–1037. doi: 10.1038/ismej.2012.169
Field C. B., Behrenfeld M. J., Randerson J. T., Falkowski P. (1998). Primary production of the biosphere: Integrating terrestrial and oceanic components. Science 281, 237–240. doi: 10.1126/science.281.5374.237
Furusawa G., Azami N. A., Teh A.-H. (2021). Genes for degradation and utilization of uronic acid-containing polysaccharides of a marine bacterium Catenovulum sp. CCB-QB4. PeerJ 9, e10929. doi: 10.7717/peerj.10929
Gavriilidou A., Gutleben J., Versluis D., Forgiarini F., van Passel M. W., Ingham C. J., et al. (2020). Comparative genomic analysis of Flavobacteriaceae: insights into carbohydrate metabolism, gliding motility and secondary metabolite biosynthesis. BMC Genomics 21, 569. doi: 10.1186/s12864-020-06971-7
Ghadam P., Akhlaghi F., Ali A. A. (2017). One-step purification and characterization of alginate lyase from a clinical Pseudomonas aeruginosa with destructive activity on bacterial biofilm. Iranian J. Basic Med. Sci. 20, 467–473. doi: 10.22038/IJBMS.2017.8668
Gómez-Silva B., Vilo-Muñoz C., Galetović A., Dong Q., Castelán-Sánchez H. G., Pérez-Llano Y., et al. (2019). Metagenomics of Atacama lithobiontic extremophile life unveils highlights on fungal communities, biogeochemical cycles and carbohydrate-active enzymes. Microorganisms 7, 619. doi: 10.3390/microorganisms7120619
Grieb A., Francis T. B., Krüger K., Orellana L. H., Amann R., Fuchs B. M. (2020). Candidatus Abditibacter, a novel genus within the Cryomorphaceae, thriving in the North Sea. System. Appl. Microbiol. 43, 126088. doi: 10.1016/j.syapm.2020.126088
Kappelmann L., Krüger K., Hehemann J.-H., Harder J., Markert S., Unfried F., et al. (2019). Polysaccharide utilization loci of North Sea Flavobacteriia as basis for using SusC/D-protein expression for predicting major phytoplankton glycans. ISME J. 13, 76–91. doi: 10.1038/s41396-018-0242-6
Katoh K., Standley D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kint N., Unay J., Viollier P. H. (2022). Specificity and modularity of flagellin nonulosonic acid glycosyltransferases. Trends Microbiol. 30, 109–111. doi: 10.1016/j.tim.2021.10.005
Kmezik C., Krska D., Mazurkewich S., Larsbrink J. (2021). Characterization of a novel multidomain CE15-GH8 enzyme encoded by a polysaccharide utilization locus in the human gut bacterium Bacteroides eggerthii. Sci. Rep. 11, 17662. doi: 10.1038/s41598-021-96659-z
Kolton M., Sela N., Elad Y., Cytryn E. (2013). Comparative genomic analysis indicates that niche adaptation of terrestrial flavobacteria is strongly linked to plant glycan metabolism. PloS One 8, e76704. doi: 10.1371/journal.pone.0076704
Konar A., Aich S., Katakojwala R., Datta S., Mohan S. V. (2022). A processive GH9 family endoglucanase of Bacillus licheniformis and the role of its carbohydrate-binding domain. Appl. Microbiol. Biotechnol. 106, 6059–6075. doi: 10.1007/s00253-022-12117-4
Krzywinski M., Schein J., Birol İ., Connors J., Gascoyne R., Horsman D., et al. (2009). Circos: An information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kubo A., Kanda J. (2017). Seasonal variations and sources of sedimentary organic carbon in Tokyo Bay. Mar. pollut. Bull. 114, 637–643. doi: 10.1016/j.marpolbul.2016.10.030
Kumar R., Kim T. H., Basak B., Patil S. M., Kim H. H., Ahn Y., et al. (2022). Emerging approaches in lignocellulosic biomass pretreatment and anaerobic bioprocesses for sustainable biofuels production. J. Clean. Product. 333, 130180. doi: 10.1016/j.jclepro.2021.130180
Laine R. A. (1994). A calculation of all possible oligosaccharide isomers both branched and linear yields 1.05 × 10(12) structures for a reducing hexasaccharide: the Isomer Barrier to development of single-method saccharide sequencing or synthesis systems. Glycobiology 4, 759–767. doi: 10.1093/glycob/4.6.759
Lapebie P., Lombard V., Drula E., Terrapon N., Henrissat B. (2019). Bacteroidetes use thousands of enzyme combinations to break down glycans. Nat. Commun. 10, 2043. doi: 10.1038/s41467-019-10068-5
Larsbrink J., Rogers T. E., Hemsworth G. R., McKee L. S., Tauzin A. S., Spadiut O., et al. (2014). A discrete genetic locus confers xyloglucan metabolism in select human gut Bacteroidetes. Nature 506, 498–502. doi: 10.1038/nature12907
Lasica A. M., Ksiazek M., Madej M., Potempa J. (2017). The type IX secretion system (T9SS): highlights and recent insights into its structure and function. Front. Cell. Infect. Microbiol. 7. doi: 10.3389/fcimb.2017.00215
Lauber F., Deme J. C., Lea S. M., Berks B. C. (2018). Type 9 secretion system structures reveal a new protein transport mechanism. Nature 564, 77–82. doi: 10.1038/s41586-018-0693-y
Lear L., Padfield D., Dowsett T., Jones M., Kay S., Hayward A., et al. (2022). Bacterial colonisation dynamics of household plastics in a coastal environment. Sci. Total Environ. 838, 156199. doi: 10.1016/j.scitotenv.2022.156199
Li S., Xiong Q., Lai X., Li X., Wan M., Zhang J., et al. (2016). Molecular modification of polysaccharides and resulting bioactivities. Compr. Rev. Food Sci. Food Saf. 15, 237–250. doi: 10.1111/1541-4337.12161
Li T., Zhang G., Wang S., Mao C., Tang Z., Rao W. (2022). The isotopic composition of organic carbon, nitrogen and provenance of organic matter in surface sediment from the Jiangsu tidal flat, southwestern Yellow Sea. Mar. pollut. Bull. 182, 114010. doi: 10.1016/j.marpolbul.2022.114010
Liberato M. V., Campos B. M., Tomazetto G., Crouch L. I., Garcia W., de Mattos Zeri A. C., et al. (2022). Unique properties of a Dictyostelium discoideum carbohydrate-binding module expand our understanding of CBM–ligand interactions. J. Biol. Chem. 298, 101891. doi: 10.1016/j.jbc.2022.101891
Lichtenthaler F. W., Peters S. (2004). Carbohydrates as green raw materials for the chemical industry. Comptes Rendus Chimie 7, 65–90. doi: 10.1016/j.crci.2004.02.002
Lin L., Lai Z., Yang H., Zhang J., Qi W., Xie F., et al. (2023). Genome-centric investigation of bile acid metabolizing microbiota of dairy cows and associated diet-induced functional implications. ISME J. 17, 172–184. doi: 10.1038/s41396-022-01333-5
Liu J., Xue C.-X., Sun H., Zheng Y., Meng Z., Zhang X.-H. (2019). Carbohydrate catabolic capability of a Flavobacteriia bacterium isolated from hadal water. System. Appl. Microbiol. 42, 263–274. doi: 10.1016/j.syapm.2019.01.002
López-Mondéjar R., Tláskal V., da Rocha Ulisses N., Baldrian P. (2022). Global distribution of carbohydrate utilization potential in the prokaryotic tree of life. mSystems 7, e00829-00822. doi: 10.1128/msystems.00829-22
Lovegrove A., Edwards C. H., De Noni I., Patel H., El S. N., Grassby T., et al. (2017). Role of polysaccharides in food, digestion, and health. Crit. Rev. Food Sci. Nutr. 57, 237–253. doi: 10.1080/10408398.2014.939263
Machovic M., Janecek S. (2008). Domain evolution in the GH13 pullulanase subfamily with focus on the carbohydrate-binding module family 48. Biologia 63, 1057–1068. doi: 10.2478/s11756-008-0162-4
Mandal S., Van Treuren W., White R. A., Eggesbo M., Knight R., Peddada S. D. (2015). Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb. Ecol. Health Dis. 26, 27663. doi: 10.3402/mehd.v26.27663
Marecek F., Moller M. S., Svensson B., Janecek S. (2021). A putative novel starch-binding domain revealed by in silico analysis of the N-terminal domain in bacterial amylomaltases from the family GH77. 3 Biotech. 11, 229. doi: 10.1007/s13205-021-02787-8
Mathieu S., Henrissat B., Labre F., Skjak-Braek G., Helbert W. (2016). Functional exploration of the polysaccharide lyase family PL6. PloS One 11, e0159415. doi: 10.1371/journal.pone.0159415
McKee L. S., La Rosa S. L., Westereng B., Eijsink V. G., Pope P. B., Larsbrink J. (2021). Polysaccharide degradation by the Bacteroidetes: mechanisms and nomenclature. Environ. Microbiol. Rep. 13, 559–581. doi: 10.1111/1758-2229.12980
McKie V. A., Black G. W., Millward-Sadler S. J., Hazlewood G. P., Laurie J. I., Gilbert H. J. (1997). Arabinanase A from Pseudomonas fluorescens subsp. cellulosa exhibits both an endo- and an exo- mode of action. Biochem. J. 323, 547–555. doi: 10.1042/bj3230547
Meneguin A. B., Silvestre A. L. P., Sposito L., de Souza M. P. C., Sabio R. M., Araujo V. H. S., et al. (2021). The role of polysaccharides from natural resources to design oral insulin micro- and nanoparticles intended for the treatment of Diabetes mellitus: A review. Carbohydr. Polymers 256, 117504. doi: 10.1016/j.carbpol.2020.117504
Mészáros Z., Petrásková L., Kulik N., Pelantová H., Bojarová P., Křen V., et al. (2022). Hypertransglycosylating variants of the GH20 β-N-acetylhexosaminidase for the synthesis of chitooligomers. Adv. Synth. Catal. 364, 2009–2022. doi: 10.1002/adsc.202200046
Michaud P., Da Costa A., Courtois B., Courtois J. (2003). Polysaccharide lyases: recent developments as biotechnological tools. Crit. Rev. Biotechnol. 23, 233–266. doi: 10.1080/07388550390447043
Morais M. A. B., Coines J., Domingues M. N., Pirolla R. A. S., Tonoli C. C. C., Santos C. R., et al. (2021). Two distinct catalytic pathways for GH43 xylanolytic enzymes unveiled by X-ray and QM/MM simulations. Nat. Commun. 12, 367. doi: 10.1038/s41467-020-20620-3
Murray N. J., Phinn S. R., DeWitt M., Ferrari R., Johnston R., Lyons M. B., et al. (2019). The global distribution and trajectory of tidal flats. Nature 565, 222–225. doi: 10.1038/s41586-018-0805-8
Oehme D. P., Shafee T., Downton M. T., Bacic A., Doblin M. S. (2019). Differences in protein structural regions that impact functional specificity in GT2 family β-glucan synthases. PloS One 14, e0224442. doi: 10.1371/journal.pone.0224442
Parks D. H., Imelfort M., Skennerton C. T., Hugenholtz P., Tyson G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Parks D. H., Tyson G. W., Hugenholtz P., Beiko R. G. (2014). STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124. doi: 10.1093/bioinformatics/btu494
Ping Q., Zheng M., Dai X., Li Y. (2020). Metagenomic characterization of the enhanced performance of anaerobic fermentation of waste activated sludge with CaO2 addition at ambient temperature: Fatty acid biosynthesis metabolic pathway and CAZymes. Water Res. 170, 115309. doi: 10.1016/j.watres.2019.115309
Regnier P., Resplandy L., Najjar R. G., Ciais P. (2022). The land-to-ocean loops of the global carbon cycle. Nature 603, 401–410. doi: 10.1038/s41586-021-04339-9
Reintjes G., Arnosti C., Fuchs B. M., Amann R. (2017). An alternative polysaccharide uptake mechanism of marine bacteria. ISME J. 11, 1640–1650. doi: 10.1038/ismej.2017.26
Santos C. R., Costa P. A. C. R., Vieira P. S., Gonzalez S. E. T., Correa T. L. R., Lima E. A., et al. (2020). Structural insights into β-1,3-glucan cleavage by a glycoside hydrolase family. Nat. Chem. Biol. 16, 920–929. doi: 10.1038/s41589-020-0554-5
Sasmito S. D., Kuzyakov Y., Lubis A. A., Murdiyarso D., Hutley L. B., Bachri S., et al. (2020). Organic carbon burial and sources in soils of coastal mudflat and mangrove ecosystems. Catena 187, 104414. doi: 10.1016/j.catena.2019.104414
Seemann T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Seiboth B., Metz B. (2011). Fungal arabinan and L-arabinose metabolism. Appl. Microbiol. Biotechnol. 89, 1665–1673. doi: 10.1007/s00253-010-3071-8
Shanmugam M., Abirami R. G. (2019). Microbial polysaccharides - chemistry and applications. J. Biol. Active Prod. Nat. 9, 73–78. doi: 10.1080/22311866.2019.1571944
Sidar A., Albuquerque E. D., Voshol G. P., Ram A. F. J., Vijgenboom E., Punt P. J. (2020). Carbohydrate binding modules: diversity of domain architecture in amylases and cellulases from filamentous microorganisms. Front. Bioeng. Biotechnol. 8. doi: 10.3389/fbioe.2020.00871
Silva S. G., Blom J., Keller-Costa T., Costa R. (2019). Comparative genomics reveals complex natural product biosynthesis capacities and carbon metabolism across host-associated and free-living Aquimarina (Bacteroidetes, Flavobacteriaceae) species. Environ. Microbiol. 21, 4002–4019. doi: 10.1111/1462-2920.14747
Son Tung N., Phuong Duy T.-L., Ho G. T., Le L. Q., Le Minh B., Bao Khanh H., et al. (2019). Interaction of carbohydrate binding module 20 with starch substrates. Rsc Adv. 9, 24833–24842. doi: 10.1039/c9ra01981b
Soumya M. P., Nampoothiri K. M. (2021). An overview of functional genomics and relevance of glycosyltransferases in exopolysaccharide production by lactic acid bacteria. Int. J. Biol. Macromol. 184, 1014–1025. doi: 10.1016/j.ijbiomac.2021.06.131
Sun Z.-Z., Ji B.-W., Zheng N., Wang M., Cao Y., Wan L., et al. (2021). Phylogenetic distribution of polysaccharide-degrading enzymes in marine bacteria. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.658620
Sun Y., Liu J., Xia J., Tong Y., Li C., Zhao S., et al. (2022). Research development on resource utilization of green tide algae from the Southern Yellow Sea. Energy Rep. 8, 295–303. doi: 10.1016/j.egyr.2022.01.168
Tanaka M., Kumakura D., Mino S., Doi H., Ogura Y., Hayashi T., et al. (2020). Genomic characterization of closely related species in the Rumoiensis clade infers ecogenomic signatures to non-marine environments. Environ. Microbiol. 22, 3205–3217. doi: 10.1111/1462-2920.15062
Terrapon N., Lombard V., Drula E., Lapébie P., Al-Masaudi S., Gilbert H. J., et al. (2018). PULDB: the expanded database of Polysaccharide Utilization Loci. Nucleic Acids Res. 46, D677–D683. doi: 10.1093/nar/gkx1022
Terrett O. M., Lyczakowski J. J., Yu L., Iuga D., Franks W. T., Brown S. P., et al. (2019). Molecular architecture of softwood revealed by solid-state NMR. Nat. Commun. 10, 4978. doi: 10.1038/s41467-019-12979-9
Tomás J. M., Fulton K. M., Twine S. M., Merino S. (2022). Generation of null mutants to elucidate the role of bacterial glycosyltransferases in bacterial motility. J. Visual. Exper. 181), e63231. doi: 10.3791/63231
Trochine A., Bellora N., Nizovoy P., Duran R., Greif G., de Garcia V., et al. (2022). Genomic and proteomic analysis of Tausonia pullulans reveals a key role for a GH15 glucoamylase in starch hydrolysis. Appl. Microbiol. Biotechnol. 106, 4655–4667. doi: 10.1007/s00253-022-12025-7
Vollmer W., Joris B., Charlier P., Foster S. (2008). Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol. Rev. 32, 259–286. doi: 10.1111/j.1574-6976.2007.00099.x
Walton P. H. (2020). Enzymes knuckle down to the job. Nat. Chem. Biol. 16, 815–816. doi: 10.1038/s41589-020-0585-y
Wang X., Xiao X., Zou Z., Chen B., Ma J., Dong J., et al. (2020). Tracking annual changes of coastal tidal flats in China during 1986–2016 through analyses of Landsat images with Google Earth Engine. Remote Sens. Environ. 238, 110987. doi: 10.1016/j.rse.2018.11.030
Wardman J. F., Bains R. K., Rahfeld P., Withers S. G. (2022). Carbohydrate-active enzymes (CAZymes) in the gut microbiome. Nat. Rev. Microbiol. 20, 542–556. doi: 10.1038/s41579-022-00712-1
Wilson L. F. L., Dendooven T., Hardwick S. W., Echevarría-Poza A., Tryfona T., Krogh K. B. R. M., et al. (2022). The structure of EXTL3 helps to explain the different roles of bi-domain exostosins in heparan sulfate synthesis. Nat. Commun. 13, 3314. doi: 10.1038/s41467-022-31048-2
Xu J., Cai Z., Zhou J. (2022). Research progress of carbohydrate-active enzymes on marine bacteria. Acta Microbiol. Sin. 62, 1286–1307. doi: 10.13343/j.cnki.wsxb.20210495
Yang J., Li S., Liu Y., Li R., Long L. (2022). Structural and biochemical basis of a marine bacterial glycoside hydrolase family 2 β-glycosidase with broad substrate specificity. Appl. Environ. Microbiol. 88, e02226–e02221. doi: 10.1128/AEM.02226-21
Yuasa M., Okamura T., Kimura M., Honda S., Shin Y., Kawakita M., et al. (2018). Two trehalose-hydrolyzing enzymes from Crenarchaeon Sulfolobus acidocaldarius exhibit distinct activities and affinities toward trehalose. Appl. Microbiol. Biotechnol. 102, 4445–4455. doi: 10.1007/s00253-018-8915-7
Zabed H. M., Akter S., Yun J., Zhang G., Awad F. N., Qi X., et al. (2019). Recent advances in biological pretreatment of microalgae and lignocellulosic biomass for biofuel production. Renewable Sustain. Energy Rev. 105, 105–128. doi: 10.1016/j.rser.2019.01.048
Zafar H., Saier M. H. Jr. (2021). Gut Bacteroides species in health and disease. Gut Microbes 13, 1848158. doi: 10.1080/19490976.2020.1848158
Zewude D. A., Noguchi T., Sato K., Izawa H., Ifuku S. (2022). Production of chitin nanoparticles by bottom-up approach from alkaline chitin solution. Int. J. Biol. Macromol. 210, 123–127. doi: 10.1016/j.ijbiomac.2022.05.006
Zhang Y., He P., Li H., Li G., Liu J., Jiao F., et al. (2019b). Ulva prolifera green-tide outbreaks and their environmental impact in the Yellow Sea, China. Natl. Sci. Rev. 6, 825–838. doi: 10.1093/nsr/nwz026
Zhang Y., Ma Y., Zhang R., Zhang B., Zhai X., Li W., et al. (2019c). Metagenomic resolution of functional diversity in copper surface-associated marine biofilms. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.02863
Zhang H., Yohe T., Huang L., Entwistle S., Wu P., Yang Z., et al. (2018). dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46, W95–W101. doi: 10.1093/nar/gky418
Zhang H., Yoshizawa S., Sun Y., Huang Y., Chu X., González J. M., et al. (2019a). Repeated evolutionary transitions of flavobacteria from marine to non-marine habitats. Environ. Microbiol. 21, 648–666. doi: 10.1111/1462-2920.14509
Zhang J., Yu X., Guan B., Hu Y., Li X., Zeng J., et al. (2022). Identification and characterization of a novel cold-adapted GH15 family trehalase from the psychrotolerant microbacterium phyllosphaerae LW106. Fermentation 8, 471. doi: 10.3390/fermentation8100471
Keywords: Flavobacteriales, Cytophagales, carbohydrate-active enzymes, polysaccharide metabolism, comprehensive genomic analysis
Citation: Ma K-J, Ye Y-L, Li Y-K, Fu G-Y, Wu Y-H, Sun C and Xu X-W (2025) Polysaccharide metabolic pattern of Cytophagales and Flavobacteriales: a comprehensive genomics approach. Front. Mar. Sci. 12:1551618. doi: 10.3389/fmars.2025.1551618
Received: 03 January 2025; Accepted: 03 February 2025;
Published: 04 March 2025.
Edited by:
Michaela M. Salcher, Biology Centre of the Czech Academy of Sciences, CzechiaCopyright © 2025 Ma, Ye, Li, Fu, Wu, Sun and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xue-Wei Xu, eHV4d0BzaW8ub3JnLmNu; Cong Sun, bWljaGFlbF9zY0BzaW5hLmNvbQ==