 Mengshi Zhao
Mengshi Zhao Dongxu Li1
Dongxu Li1 Changhong Liu
Changhong Liu- 1State Key Laboratory of Pharmaceutical Biotechnology, Nanjing University, Nanjing, China
- 2Shanghai Engineering Research Center of Hadal Science and Technology, College of Marine Sciences, Shanghai Ocean University, Shanghai, China
- 3Laboratory for Marine Biology and Biotechnology, Qingdao National Laboratory for Marine Science and Technology, Qingdao, China
In anaerobic high hydrostatic pressure (HHP) sedimentary environments below the seafloor, fungi are found to dominate the eukaryotic communities, playing crucial ecological roles. However, the specific mechanisms by which fungi adapt to anaerobic HHP environments remain unclear. In this study, we investigated Schizophyllum commune 20R-7-F01 isolated from coal-bearing sediments at a depth of 2 km below the seafloor. By assessing the cell viability, biomass, and cell wall thickness changes of strain 20-7-1 under different HHP conditions, we observed that, compared to 0.1 MPa, strain 20-7-1 exhibited slower growth rates and decreased cell viability at 15 MPa and 35 MPa, yet demonstrated significant pressure tolerance. Transcriptomic and metabolomic analyses revealed that this strain activated the carbohydrate metabolic process to simultaneously utilize ethanol and lactic acid fermentation pathway. Additionally, it activates the oxidoreductase activity and hydrolase activity pathways to detoxify intracellular reactive oxygen species (ROS). Activation of the metal ion binding pathway increases the proportion of unsaturated fatty acids in the cell membrane, while instigation of the integral component of membrane pathway maintains cell wall structural stability. Furthermore, activation of the DNA repair pathway repairs DNA damage, demonstrating its comprehensive adaptive mechanisms against the HHP stress. These research findings deepen our understanding of fungal survival strategies and adaptation mechanisms in extreme environments, laying the groundwork for further exploration of their roles in cycling of carbon, nitrogen, sulfur, and other elements in the deep biosphere.
1 Introduction
The subseafloor sediment is one of the world’s largest and highly diverse microbial habitats, serving as a significant reservoir for organic carbon. Its depth extends vertically to depths of up to 2.5 kilometers below the seafloor (kmbsf), harboring approximately 2.93×1029 microbial cells, constituting between 0.18% to 3.6% of Earth’s total biomass (Bradley et al., 2022; Liu et al., 2017). Subseafloor sediments are characterized by aerobic and anaerobic microecosystems, exhibiting notable distinctions in bacterial and archaeal communities. Within anaerobic sediments, both bacterial and archaeal abundances decrease with increasing depth. In contrast, aerobic sediments show a depth-related decrease in bacterial abundance while archaeal abundance remains relatively consistent (Hoshino et al., 2020). In addition to bacteria and archaea, fungi are also significant constituents of subseafloor sediments. These fungi predominantly belong to the phyla Ascomycota, Basidiomycota, and Chytridiomycota. Their abundance and distribution are significantly influenced by total organic carbon content, electron acceptors, temperature, and salinity, with less clear relationships to depth (Inagaki et al., 2015). Most seafloor sediment fungi are closely related to terrestrial fungi, yet they typically possess smaller genomes adapted to resource-limited environments. These fungi exhibit unique gene structures, such as increased numbers of genes involved in DNA repair and heat shock families, to cope with extreme conditions like high pressure, high temperature, and high salinity (Liu et al., 2022). However, it remains unclear which specific genes and metabolic pathways play an important role in the survival and growth of these fungi in the extreme environments of the subseafloor biosphere.
HHP is a distinctive feature of the deep subseafloor sediment environments. Current research highlights that the magnitude of HHP profoundly influences microbial physiology and metabolism. Microorganisms have developed adaptive mechanisms to flourish in deep-sea sediment environments characterized by HHP, thereby ensuring their enduring survival under high-pressure conditions (Xiao X. et al., 2021; Zheng et al., 2023; Malas et al., 2024). Xiao et al. found in their study of the piezophilic bacterium Sporosarcina psychrophila DSM 6497 that the organism enhanced its antioxidant defense systems, accumulated compatible solutes intracellularly, and promoted membrane lipid synthesis to mitigate the damage caused by 50 MPa pressure (Wang et al., 2021). Amrani et al. discovered that the piezotolerant bacterium Desulfovibrio hydrothermalis, under 26 MPa pressure, not only significantly increased intracellular glutamate content but also enhanced the activities of cytochrome c oxidase and catechol oxidase to bolster ATP supply (Amrani et al., 2014). Although research on the mechanisms underlying fungal response to HHP remains limited, evidence suggests that Saccharomyces cerevisiae can respond to high-pressure environments by increasing the proportion of unsaturated fatty acids in cell membranes, activating heat shock protein families, and enhancing cell antioxidant capacity (Fernandes et al., 2004; Marx et al., 2011; Bravim et al., 2010; Abe, 2021). Recently, Peng et al. conducted cultivation experiments at 40 MPa with fungi isolated from the Mariana Trench sediments, including Aspergillus sp., revealing that these fungi’s tolerance to HHP correlated with the secretion of abundant secondary metabolites (Peng et al., 2021). Xiao et al., through genomic analysis, discovered that Penicillium bialowiezense A30 and Cladosporium colombiae 601, isolated from Mariana Trench sediments, exhibited extensive potential in carbon, nitrogen, and sulfur metabolism when exposed to 20 MPa pressure (Li et al., 2022). Limited research results suggest that fungi may possess distinct high-pressure adaptation mechanisms compared to bacteria or archaea, enabling their persistence in deep-sea sediment environments over millions of years.
Schizophyllum commune, belonging to the phylum Basidiomycota, order Schizophyllales, and family Schizophyllaceae, is a widely distributed fungus on Earth. It plays a crucial role in natural ecosystems by decomposing organic matter, such as tree wood, plant residues, and animal and plant remains, thereby facilitating the cycling of organic substances (Ohm et al., 2010; Tovar-Herrera et al., 2018). Li Feng et al., using high-throughput sequencing technology, discovered diverse and abundant fungal communities in sediments from the South China Sea, reaching depths of 897–1,434 m (within a pressure range of 9-15 MPa), including the presence of Schizophyllum fungi (Feng et al., 2021). Additionally, our earlier studies on fungal communities in lignite-bearing sediment samples obtained during IODP Expedition 337 (dating to 20 million years ago) revealed the presence of Schizophyllum fungi in sediments as deep as ~2 km (35 MPa), showing rich diversity and accounting for 5.8% of the total isolates obtained. Moreover, this subseafloor sediment Schizophyllum fungi exhibited specific anaerobic adaptation mechanisms, including enhanced ethanol and amino acid metabolism, increased mitochondrial and autophagosome abundance, and specialized pathways for carbon and nitrogen transformation, to cope with anaerobic environments (Zain Ul Arifeen et al., 2021a; Zain Ul Arifeen et al., 2021b; Ma et al., 2023; Huang et al., 2022). However, the specific pressure adaptation mechanisms of Schizophyllum fungi in in situ subseafloor (35 MPa) sediment environments remain unclear.
In this study we studied Schizophyllum commune 20R-7-F01 isolated from coal-bearing sediment samples retrieved at a depth of 2 kilometers below seafloor, aiming to investigate its growth mechanisms under high-pressure and anaerobic conditions using transcriptomics, metabolomics, and activity profiling techniques. Results offer novel insights into the diversity, distribution, and survival strategies of fungi within the deep subseafloor biosphere.
2 Materials and methods
2.1 Fungal strain and cultivation
Strain 20R-7-F01 (CGMCC11604) of S. commune was isolated in this laboratory from sediment samples collected at a depth of 1966 meters below seafloor (mbsf) off the northern coast of Japan (Liu et al., 2017), where in situ pressure is 30-35 MPa and temperatures range from 45°C to 47°C.The strain has been maintained at 4°C. For culturing, strain 20R-7-F01 was inoculated onto mPDA medium (potato extract, 200 g/L; glucose, 20 g/L; agar, 20 g/L) and incubated in darkness at 30°C for 3 days. Fresh mycelia were harvested from the colony edges, homogenized in sterilized glass homogenizers, and subsequently transferred to mPD medium. Cultures were then incubated in darkness at 30°C and 180 rpm on a shaker incubator for 3 days. Mycelia were collected using a sterile gauze filtration method within a laminar flow hood, washed three times with sterile double-distilled water, and centrifuged at 4°C and 12,000 rpm to obtain purified mycelia. Subsequently, 7 g of mycelia were weighed and inoculated into 170 ml sterile anaerobic culture bottles containing 160 ml of artificial seawater medium (CaCl2, 2.99 g; MgCl2, 4.17 g; KBr, 0.10 g; NH4Cl, 0.16 g; KCl, 5.05 g; NaCl, 33.43 g; H3BO3, 0.02 g; Na2SO4, 0.21 g; C6H12O6, 15 g; supplemented with 1‰ resazurin as an anaerobic indicator; prepared per liter of medium). A total of 45 bottles were inoculated. The bottles were then purged with 99.99% N2 for 15 minutes to remove oxygen from the culture bottles (Wang et al., 2018). 45 anaerobic culture bottles (equipped with sterile syringes inserted into their necks for pressure equilibrium) were placed into nine 1-liter stainless steel pressure vessels. The culture temperature was maintained at 30 °C using heaters, and the vessels were manually pressurized with water to achieve different hydrostatic pressures: 0.1, 15, and 35 MPa in respective vessels (Williams et al., 2019; Chen et al., 2021). After cultivation for 1, 3, and 5 days, five anaerobic culture bottles from each pressure vessel were sampled. Three bottles were used for biomass measurement, and one vial was subjected to fungal hyphal vitality assessment using the 0.4% Trypan Blue staining technique, the selective permeability of trypan blue dye causes non-viable cells to appear distinctly blue under microscopic examination, while viable cells, due to their intact cell membranes, remain unstained (Crowley et al., 2016; Lebeau et al., 2019). Additionally, fungal hyphae were fixed in 2.5% glutaraldehyde following the method by Zilan Xiao et al., washed with PBS, dehydrated with an ethanol gradient, embedded in epoxy resin, sectioned, and observed using a HITACHI H-7650 transmission electron microscope (TEM) (Xiao Z. et al., 2021). The remaining mycelium was partially cryopreserved in liquid nitrogen for transcriptomics and metabolomics studies, while another portion of the mycelium was used to measure intracellular ATP using the firefly luciferase bioluminescence assay. This method catalyzes luciferin oxidation by luciferase in the presence of ATP and oxygen, emitting light proportional to ATP concentration. Fungal cells were lysed to release ATP, and lysates were combined with luciferase-luciferin reagent. Luminescence was quantified using a luminometer, and ATP levels were determined via a standard curve (Liu et al., 2021).
2.2 Transcriptomic analysis
The samples utilized for transcriptome sequencing and subsequent analysis were denoted as follows: “d1_01M”, “d3_01M”, “d5_01M”, “d1_15M”, “d3_15M”, “d5_15M”, “d1_35M”, “d3_35M”, and “d5_35M”. Herein, “dn” denotes the sampling time points (1, 3, and 5 days), while “nM” signifies the hydrostatic pressure applied to the cultured strain (0.1, 15, and 35 MPa). Total RNA was extracted using TRIzol reagent (TIANGEN, Beijing, China) following the manufacturer’s instruction. Subsequently, RNA samples were used for the construction of cDNA libraries. The cDNA libraries were sequenced on the Illumina HiSeq platform according to the default Illumina strand RNA protocol (Meiji, Shanghai, China). After obtaining clean reads by removing adapter sequences and reads with >10% N and low-quality sequences (>50% of each read with a Phred score Q ≤5) (Supplementary Table S1), the clean reads were then mapped to the assembled genome of strain 20R-7-F01 (BioProject ID: PRJNA544166) using TopHat2 (Kim et al., 2013). The expression level for each gene was calculated by quantifying the cDNA fragments Transcripts Per Million (TPM) by using Cufflinks software (Trapnell et al., 2012). All RNA-seq data generated for this study were deposited in the National Center for Biotechnology Information Sequence Read Archive under BioProject ID PRJNA1101667.
Differences in gene expression between samples were identified using the DESeq method. The count table was analyzed by DESeq2 (Love et al., 2014). In the process of analyzing differential expressed genes (DEGs), a threshold of p-value < 0.05 and | Log2 (Fold-change) | ≥ 1 was used to judge and select genes with significant differences (Anders and Huber, 2010). To further reveal the functions of the DEGs, Gene Ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis were conducted using the clusterProfiler package in R (v4.3.0). Annotated pathways were integrated and visualized using the Pathview package. The screening threshold for enriched gene annotation analysis was set at p-value < 0.05. DEGs related to 8 key pathways were subjected to hierarchical clustering, and the correlation between these DEGs was analyzed using the psych and reshape2 packages in R (v4.3.0). Subsequently, a correlation network of pathway genes was constructed using Cytoscape software version 3.9.1 (https://cytoscape.org/releasenotes.html).
2.3 Metabolomics analysis by untargeted LC-MS/MS
The mycelial samples utilized for untargeted metabolomic analysis were derived from the same source as those employed in transcriptomic analysis. Thus, identical labeling methods were applied for metabolomic profiling (Shah et al., 2012). Samples were extracted using methanol, followed by centrifugation to collect the supernatant into respective vials for subsequent analysis. LC-MS/MS analysis was conducted using a UHPLC system coupled with a TripleTOF 6600+ mass spectrometer (SCIEX). Samples were injected onto a Waters ACQUITY UPLC BEH C18 column, and the raw mass spectrometric data were converted to mzXML format using ProteoWizard. Peak extraction, alignment, and retention time correction were performed using the XCMS program. Peak areas were calibrated using the “SVR” method, with peaks exhibiting a missing value rate exceeding 50% across sample groups filtered out. Following calibration and filtration, peaks were annotated for qualitative and relative quantitative analysis using the mzCloud (https://www.mzcloud.org/) and ChemSpider (https://www.chemspider.com/) databases (Masuo et al., 2010). All raw data generated in this study are archived in the MetaboLights database under project number MTBLS10088.
Metabolites displaying significant differences, designated as Differential Metabolites (DEMs), were identified and screened using criteria of p-value < 0.05, | Log2(Fold-change) | ≥ 1, and Variable Importance in Projection (VIP) > 1.0. Hierarchical clustering analysis of these DEMs was subsequently performed using the heatmap package in R (v4.3.0).
2.4 Quantitative real-time PCR validation
The qRT-PCR experiment was conducted according to the method described by Zain Ul Arifeen et al. The fungus was cultured under identical conditions and durations as the RNA-seq samples. The qRT-PCR analysis was carried out utilizing the SYBR qPCR Master Mix reagent kit (Vazyme, Nanjing, China), along with specific primer pairs for each gene (Supplementary Table S2). The thermal cycle for qRT-PCR amplification was as follows:95°C for 30 s, followed by 43 cycles of 95°C for 10 s, 58.5°C for 30 s, and 72°C for 30 s. The relative expression of genes was calculated using the 2 −ΔΔCT method (Livak and Schmittgen, 2001).
2.5 Statistical analysis
The data were presented as mean ± standard deviation. One-way analysis of variance (ANOVA) or student’s t-test, performed using GraphPad Prism version 8.0.2to analyze significant differences between treatments (p < 0.05).
3 Results and discussion
3.1 Growth response of strain 20R-7-F01 to HHP
Strain 20R-7-F01 was cultivated under different pressure conditions for 1, 3, and 5 days, and biomass measurements revealed that the strain exhibited growth capability under varying pressure conditions, albeit with diminishing growth rate as pressure increased (Figure 1A). One day after inoculation, the biomass of the strain increased from the initial 330. mg to 351.8 mg, 336.8 mg, and 334.3 mg under pressures of 0.1 MPa, 15 MPa, and 35 MPa, respectively. Compared to biomass under atmospheric pressure, the biomass increase was 30.2% and 16.2% under 15 MPa and 35 MPa, respectively. With prolonged pressure stress, the biomass increase significantly rose under both conditions: after 3 days of cultivation, the biomass increase was 52.9% at 15 MPa and 24.7% at 35 MPa, and after 5 days, it further increased to 80.1% and 55.9%, respectively.This may indicate that the strain gradually adapts to pressure stresses of 15 MPa and 35 MPa, with the magnitude of biomass reduction showing a trend of gradual attenuation as the cultivation period lengthens. Additionally, Trypan blue staining results (Figure 1B) showed that compared to atmospheric pressure, hyphae at both 15 MPa and 35 MPa were stained blue, with deeper staining observed at 35 MPa. This indicates that pressure inhibited the mycelial activity of strain 20R-7-F01, with higher pressure exerting a more pronounced inhibitory effect. However, the strain remained viable, consistent with previous reports (Damare et al., 2006).These results may suggest that strain 20R-7-F01 possesses a certain level of stress tolerance that allows it to grow under high hydrostatic pressure. Therefore, to elucidate the pressure tolerance mechanisms of strain 20R-7-F01, we conducted transcriptomic and metabolomic analyses on strain 20R-7-F01exposed to different pressure conditions.
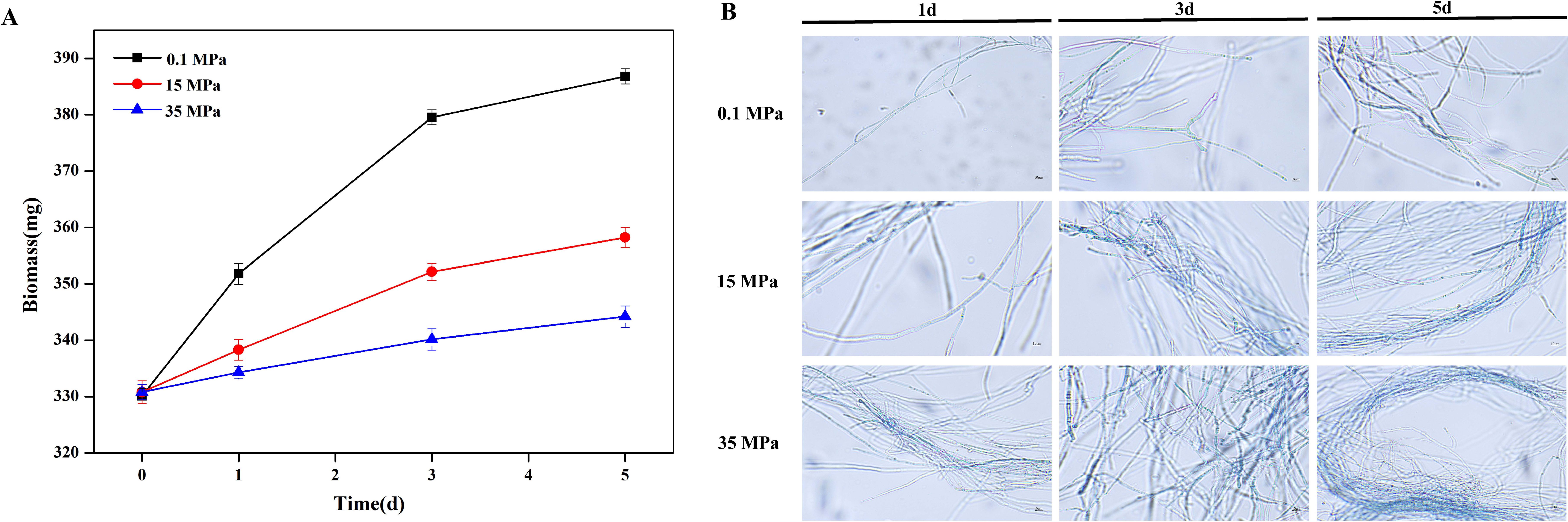
Figure 1. Effects of different pressure conditions on the biomass and activity of strain 20-7-1. (A) Variation in biomass of the strain under different pressures; (B) Variation in cellular activity of the strain under different pressures.
3.2 Transcriptome profiling of strain 20R-7-F01 under different pressure conditions
The analysis of significantly differentially expressed genes (DEGs), as shown in Supplementary Figure S1, indicates that in response to pressure stress, strain 20R-7-F01 requires regulation of a greater number of DEGs at the transcriptional level. At the first day of incubation, compared to atmospheric pressure conditions, the strain exhibited 2,367 and 2,910 DEGs at 15 MPa and 35 MPa, respectively, with 78.7% of the DEGs upregulated at 15 MPa and 77.1% upregulated at 35 MPa. This suggests that strain 20R-7-F01 predominately upregulates DEGs to cope with pressure stress, activating a series of gene clusters. However, with prolonged duration of pressure stress, DEGs showed varying trends at 15 MPa and 35 MPa. The number of upregulated DEGs significantly increased at 15 MPa over time, whereas the trend reversed at 35 MPa (Supplementary Figure S1). For instance, by the fifth day, DEGs increased to 3622 at 15 MPa, with 86.5% being upregulated, whereas at 35 MPa, despite an increase to 4704 in DEGs, only 47.1% were upregulated. The reasons behind the different patterns of DEGs changes in strain 20-7-1 in response to varying pressures might be that 15 MPa hydrostatic pressure represents a lower stress level for strain 20R-7-F01, allowing the strain to adapt gradually by continuously activating gene clusters to resist pressure stress. In contrast, 35 MPa imposes a higher stress level, resulting in extensive cellular damage that prevents the strain from adequately repairing pressure-induced injuries, potentially leading to cell death and thus a reduction in the number of upregulated DEGs in later stages. Similarly, numerous studies have demonstrated trends in microbial transcriptomic responses to pressure stress. For instance, Ishii et al. found that Escherichia coli required a greater number of DEGs with prolonged cultivation under 30 MPa and 50 MPa, with predominantly upregulated DEGs at 30 MPa and predominantly downregulated DEGs at 50 MPa (Ishii et al., 2005). Moreover, the transition from 0.1 MPa to 15 MPa or 35 MPa resulted in more pronounced changes in the number of DEGs compared to variations between 15 MPa and 35 MPa. This suggests that transitioning directly from 0.1 MPa to 15 MPa or 35 MPa necessitates broader gene regulation by the strain to adapt to pressure stress, whereas that from 15 MPa to 35 MPa is relatively gradual transition. Amrani et al. have substantiated this through transcriptomic analyses (Amrani et al., 2014).
Through GO enrichment analysis, we found that strain 20R-7-F01 exhibited similarity in key enriched pathways under different HHP conditions, encompassing carbohydrate metabolic process, oxidoreductase activity, DNA repair, ATP binding, hydrolase activity, metal ion binding, and integral component of membrane (Figure 2, Supplementary Figure S2). Surprisingly, the impact of pressure stress duration on these enriched pathways was minimal. This result may suggest that strain 20R-7-F01 likely has evolved adaptability to high-pressure environments over prolonged evolutionary periods. Even under different high-pressure conditions, the strain may have evolved common adaptive strategies, utilizing these shared pathways for adaptation. Notably, these pathways did not exhibit significant changes over short periods, underscoring their stability in biological responses. The KEGG enrichment analysis is consistent with the GO enrichment results, further validating the importance of these enriched pathways. (Supplementary Figure S3).
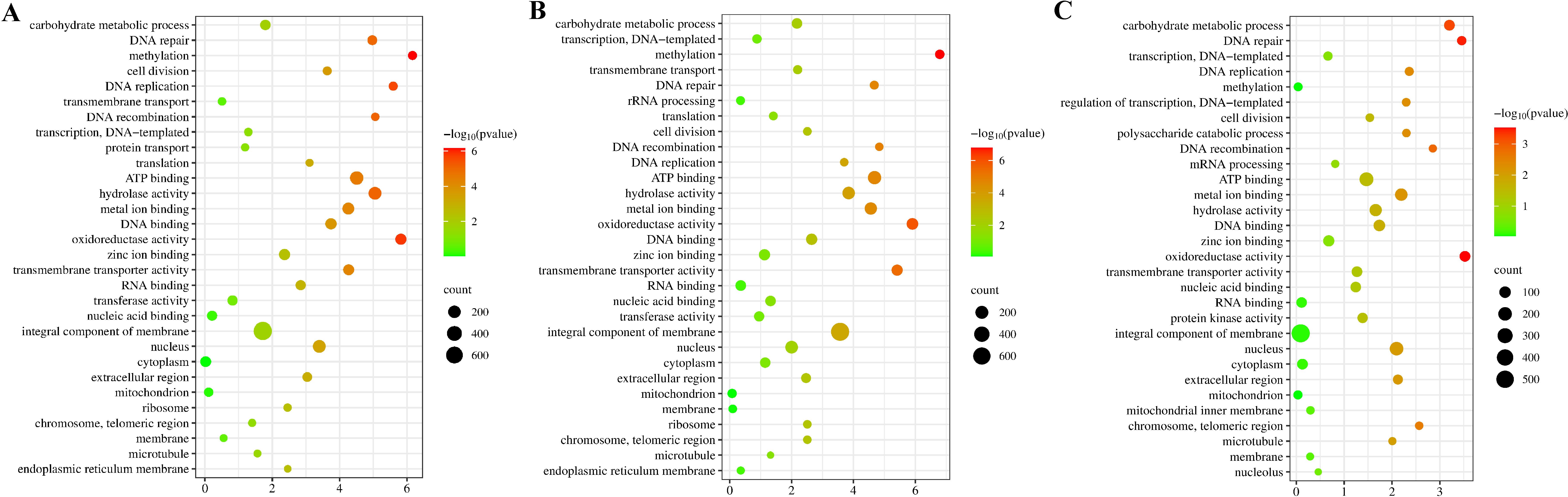
Figure 2. GO enrichment analysis of the DEGs on the third day. (A–C) GO enrichment analysis of DEGs between 15 MPa and 0.1 MPa, 35 MPa and 0.1 MPa, 35 MPa and 15 MPa. Description of the Top 30 Enriched GO.
After observing the growth of strain 20R-7-F01 under different pressures, we found minimal changes in its gene expression levels three days later (Supplementary Figure S4). Therefore, we proceeded with GO enrichment pathway analysis and hierarchical clustering analysis based on the third-day data (Figures 3, 4). Additionally, we validated the expression levels of relevant genes using qPCR (see Supplementary Table 1, Supplementary Figure S5). Comparing to 0.1 MPa, we observed significant upregulation of genes involved in carbohydrate metabolic processes, particularly for those related to glycolysis, ethanol fermentation, and lactic acid fermentation, in strain 20R-7-F01 under 15 MPa and 35 MPa conditions. For instance, under 15 MPa, ALDH gene encoding aldehyde dehydrogenase were upregulated 9.9-fold, and LDHD gene encoding Lactose dehydrogenase were upregulated 2.7-fold. Under 35 MPa, ALDH gene encoding aldehyde dehydrogenase were upregulated 46.7-fold, and LDHD gene encoding Lactose dehydrogenase were upregulated 4.9-fold. Additionally, compared to 0.1 MPa, genes involved in the TCA cycle were significantly suppressed in strain 20R-7-F01 under HHP conditions. These results suggest that strain 20R-7-F01 may possess a unique set of mechanisms to cope with HHP. Most deep-sea sediment microbes under high-pressure conditions either enhance energy production through upregulation of the TCA cycle and oxidative phosphorylation-related gene expression or rely on enhanced ethanol or lactic acid fermentation to support growth. For instance, S. psychrophila DSM 6497 responds to 50 MPa pressure by upregulating expression of Fe-S cluster assembly genes, which may improve intracellular electron transfer efficiency and ATP production (Wang et al., 2021). Alternatively, piezophilic bacteria like Photobacterium profundum SS9 under anaerobic 28 MPa conditions primarily depend on stickland fermentation for energy production to sustain growth (Bartlett et al., 2008). Pressure-tolerant bacteria such as Lb. bulgaricus and M. sediminis YLB-01 can rely on lactic acid fermentation at 30 MPa under anaerobic conditions for energy production (Qiu et al., 2022). Here, strain 20R-7-F01 under HHP conditions may employ both ethanol and lactic acid fermentation, which are always independent of the TCA cycle. Compared to relying on a single ethanol or lactic acid fermentation pathway, strain 20R-7-F01’s ability to utilize both fermentation processes could more effectively enhance energy production, thereby aiding its adaptation to high-pressure stress. Compared with 0.1 MPa, genes involved in oxidative stress (Yap1, MET, GLT, GSS, GST, SOD, katE, CAT, catB, srpA) in the oxidoreductase activity enrichment pathway under HHP were significantly upregulated. Among Yap1, a crucial transcription factor widely present in eukaryotes including yeast and mammals, is known to activate in response to oxidative damage, translocate to the nucleus, and bind to specific target genes, thereby enhancing the transcription of downstream antioxidant genes to bolster cellular resistance against oxidative stress (Li et al., 2022; Delaunay et al., 2000). Additionally, MET, GLT, GSS, GST genes are involved in the synthesis of antioxidants such as cysteine and glutathione in deep-sea sediment microbes. For instance, Saccharomyces cerevisiae BT0510 accumulates cysteine by upregulating the cysteine synthase MET gene under 50 MPa, which directly reduces superoxide radicals to stable compounds, thereby enhancing antioxidant capacity (Bravim et al., 2016). Similarly, in response to 50 MPa, the piezophilic bacterium S. psychrophila DSM6497 stimulates glutamate synthesis by upregulating the GLT gene, which not only helps maintain cellular osmotic balance but also neutralizes hydroxyl radicals to alleviate oxidative stress damage (Wang et al., 2021). Genes encoding glutathione synthase (GSS) and glutathione transferase (GST) are commonly reported, encoding enzymes that promote intracellular glutathione synthesis and transport it to oxidative damage sites to neutralize free radicals (El-Bahr, 2013; Ifeanyi, 2018). Antioxidant enzymes encoded by SOD, katE, CAT, catB, srpA genes also play significant roles in deep-sea bacterial antioxidant defenses (Alkadi, 2020). For example, S. psychrophila DSM 6497 significantly upregulates the katE gene under 50 MPa to catalyze the reduction of H2O2 to H2O and O2, thereby mitigating oxidative damage within cells (Wang et al., 2021). It is noteworthy that the reported involvement of these antioxidant-related genes in response to HHP oxidative stress in the filamentous fungus is unprecedented, indicating that strain 20R-7-F01 also possesses an antioxidant system as to piezophilic bacteria. Additionally, we found significant upregulation of genes involved in hydrolase activity pathways, particularly those related to peptide hydrolysis. For instance, the APE gene was upregulated 5.4-fold at 15 MPa and 6.9-fold at 35 MPa. It has been reported that fungi such as Saccharomyces cerevisiae activate APE peptidases to hydrolyze damaged proteins into peptides as an antioxidant mechanism (Li and Wilson, 2014; Rao et al., 1998). This suggests that strain 20R-7-F01 employed multiple antioxidant strategies to protect its cells from damage under HHP stress. In comparison to 0.1 MPa, strain 20R-7-F01 under HHP conditions exhibited significant upregulation of genes involved in metal ion binding pathways, particularly those related to unsaturated fatty acid synthesis enzymes (FAD2, SCD, desC). The gene encoding the polyunsaturated fatty acid FAD2 has previously been demonstrated in Saccharomyces cerevisiae to enhance membrane fluidity in response to freeze and salt stress (Rodríguez-Vargas et al., 2007). Similarly, it has been documented in piezophilic yeast Kluyveromyces lactis to enhance membrane fluidity under low temperature and low oxygen conditions (Santomartino et al., 2019). There are some reports on its role in enhancing membrane fluidity under HHP conditions. Likewise, genes encoding monounsaturated fatty acid synthesis enzymes SCD and desC, although established in mice for converting stearic acid (C18:0) to oleic acid (C18:1) to maintain membrane fluidity, have not been extensively documented for this purpose in filamentous fungi (Griffiths et al., 2013). This indicates that strain 20R-7-F01 adapts to HHP by modulating the composition and structure of cell membranes to enhance membrane fluidity, thereby increasing cellular adaptability to high pressure. In comparison to 0.1 MPa, under HHP conditions, strain 20R-7-F01 showed significant upregulation of genes involved in the integral component of membrane pathway, particularly those associated with maintaining stability of the inner cell wall polysaccharides (Mid, Rho1, Pkc1, FKS, CHS3, Bck1, Slt2). These genes, including Mid, Rho1, Pkc1, FKS, CHS3, Bck1, and Slt2, act as key components in the CWI (Cell Wall Integrity) signaling cascade in Saccharomyces cerevisiae, where they are activated to maintain the structural integrity of the inner cell wall (Quilis et al., 2021; Gow et al., 2017). Specifically, the highly glycosylated integral membrane sensor (encoded by Mid) is activated when the cell wall is compressed, subsequently activating Rho1 through the MAPK cascade. Rho1-GTPase not only activates FKS on the plasma membrane to promote β-(1,3)-glucan synthesis but also transmits the signal to protein kinase C (Pkc1), which activates CHS3 on the plasma membrane to enhance chitin synthesis. Concurrently, activated Pkc1 stimulates the mitogen-activated protein kinase Slt2, which upregulates genes involved not only in repairing damaged cell walls but also in enhancing the expression of the cell cycle regulator SWI6 to ensure normal cell cycle progression. However, this mechanism has not been reported in deep-sea sediment fungi. With regards to this, we conducted TEM electron microscopy observations of cell walls under different HHP conditions (Supplementary Figure S6), revealing that the cell wall thickness of strain 20-7-1 under HHP was greater than at 0.1 MPa. For instance, on the fifth day, the average cell wall thickness at 15 MPa was approximately 0.6 µm thicker than at 0.1 MPa, while at 35 MPa, it was around 0.8 µm thicker than at 0.1 MPa. This indicates that strain 20-7-1 responds to HHP conditions by modulating the expression of genes involved in the structural integrity of the inner cell wall skeleton. This adaptive response likely enhances the stability of polysaccharides in the cell wall, resulting in increased cell wall thickness. Compared to 0.1 MPa, under HHP conditions, the ATM/ATR-Chk1-CDC25 regulatory pathway has been identified as the primary DNA repair mechanism in strain 20-7-1. Studies indicate that ATM and ATR are activated in the hypoxic tumor microenvironment to suppress the cell cycle and upregulate DNA repair genes (Jing et al., 2019; Lama-Sherpa and Shevde, 2020). Additionally, ATM and ATR have been shown to activate DNA repair genes in Arabidopsis thaliana in response to radiation-induced DNA damage (Manian et al., 2021). Similarly, in yeast facing stressors such as radiation, chemical inducers, and ionizing radiation, ATM and ATR have been demonstrated to initiate DNA damage signaling cascades through Rad53 and Chk1, thereby regulating cell cycle progression and DNA damage repair (Smith et al., 2010). However, investigations into fungal DNA repair mechanisms regulated by ATM and ATR under pressure stress have not been reported. Compared to 0.1 MPa, when exposed to HHP, the strain exhibited significant upregulation of ATM/ATR genes. Activated ATM/ATR directly upregulate DNA repair genes such as RAD9A, RFC1, RFA1, and EXO1. However, the focus of DNA repair differed between pressures of 15 MPa and 35 MPa. Under 15 MPa stress, ATM/ATR predominantly upregulated genes involved in repairing localized base damage, such as NTH, MBD4, and UNG. Conversely, under 35 MPa stress, ATM/ATR predominantly upregulated genes involved in repairing breaks across entire stretches of DNA, including XPA and XRCC5. This indicates that the DNA repair mechanisms under HHP were influenced by the pressure level, exhibiting distinct adaptive strategies. Furthermore, to prevent the propagation of erroneous or damaged DNA to progeny, the strain activates Chk1 via ATM/ATR signaling cascades, leading to significant downregulation of the CDC25 cell cycle regulatory gene. Consequently, the strain temporarily arrested the cell cycle at the G2 phase. This mechanism of cell cycle arrest provides strain 20R-7-F01 with ample time to repair DNA damage or correct errors, thereby mitigating the risk of propagating flawed or damaged DNA to descendant cells. Transcriptomic analysis suggests that besides the ATM/ATR-Chk1-CDC25 regulatory pathway, mitochondrial damage-induced cytochrome c release also contributed to cell cycle arrest. Accumulation of intracellular H2O2 reportedly damages mitochondrial membranes, releasing cytochrome c, which activates the caspase-3 gene and triggers apoptosis, consequently inhibiting CDC25 and ultimately causing cell cycle arrest at the G2 phase (Shyur et al., 2010). Currently, it has been demonstrated that under HHP, strain 20-7-1 exhibited significant upregulation of the caspase-3 gene involved in mitochondrial membrane channels. Further validation of this pathway’s role is warranted.

Figure 3. Results of hierarchical cluster analysis of DEGs observed in strain 20R-7-F01 under various pressure conditions. (A–C) illustrate the differential expression trends of DEGs at 0.1MPa (control), 15MPa, and 35MPa on the third day.
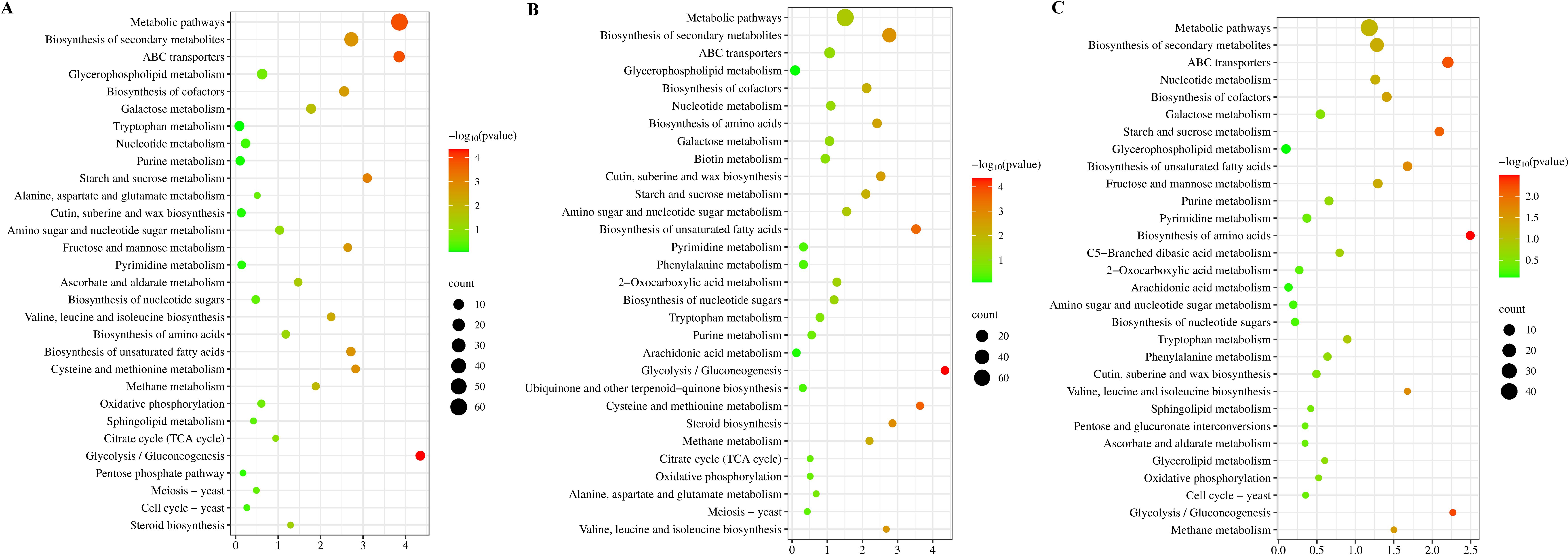
Figure 4. KEGG enrichment analysis of the DEMs. (A–C): KEGG enrichment analysis of DEMs between 15 MPa and 0.1 MPa, 35 MPa and 0.1 MPa, 35 MPa and 15 MPa on the third day. Description of the Top 30 Enriched GO.
3.3 Metabolome profiles of strain 20R-7-F01 under different pressures
The significant differential metabolite (DEMs) analysis results (Supplementary Figure S7) indicate an increasing trend in responsive metabolites under pressure stress. However, unlike the changes observed in DEGs, the number of DEMs in strain 20R-7-F01 decreases in the later stages when facing 15 MPa and 35 MPa pressures. This trend is similar to the response of the psychrophilic microbe Microbacterium sediminis YLB-01 to 30 MPa conditions, suggesting a gradual restoration of metabolic balance as cells adapt to pressure. Initially elevated concentrations of stress metabolites decline, leading to a reduction in the number of significantly differential metabolites. It is worth noting that, there is a notable increase in the proportion of upregulated DEGs in strain responses to HHP, potentially reflecting the organism’s strategic allocation of energy and resources to repair pressure-induced damage and maintain essential physiological functions. This selective induction may prioritize specific DEMs to support these processes effectively.
Following KEGG enrichment analysis of the DEMs, key enriched pathways include biosynthesis of amino acids, cysteine and methionine metabolism, biosynthesis of unsaturated fatty acids, glycolysis/gluconeogenesis, and starch and sucrose metabolism (Supplementary Figure S8). Similarly, we found minimal changes in its gene expression levels three days later (Supplementary Figure S9). Therefore, we proceeded with KEGG enrichment pathway analysis and hierarchical clustering analysis based on the third-day data (Figures 4, 5). Through hierarchical clustering analysis, we observed that under HHP compared to 0.1 MPa, metabolites such as glucose, glucose-6-phosphate, ethanol, and lactate in the glycolysis/gluconeogenesis enriched pathway were significantly upregulated. Conversely, metabolites like succinic acid and citrate in the citrate cycle enriched pathway were significantly downregulated. This is likely due to the significant upregulation of genes associated with glycolysis and anaerobic fermentation, and the downregulation of genes involved in the TCA cycle in strain 20R-7-F01 under HHP conditions. Similarly, Xia et al. found that the psychrophilic bacterium YLB-01 coped with 30 MPa, 4 °C stress by inhibiting the TCA cycle and increasing lactate content (Qiu et al., 2022). In contrast, strain 20-7-1 can simultaneously produce ethanol and lactate to provide additional energy sources, aiding in meeting the increased energy demands induced by HHP. Furthermore, compared to 0.1 MPa, biosynthesis of amino acids-enriched pathway under HHP showed significant upregulation of branched-chain amino acids (BCAAs: leucine, isoleucine, valine) and small peptides. Previous studies by Zain Ul Arifeen et al. suggest that the increased production of amino acids, particularly BCAAs, by strain 20R-7-F01 under anaerobic conditions is a crucial energy-generating mechanism supporting its growth (Zain Ul Arifeen et al., 2021). Here, we further demonstrate that biosynthesis of BCAAs under HHP conditions may continue to be a supportive growth mechanism. Moreover, the increased levels of branched-chain amino acids and small peptides in strain 20R-7-F01 under HHP likely exhibit antioxidant activity to some extent, aiding in mitigating oxidative stress caused by HHP. They may act by scavenging free radicals or enhancing the activity of antioxidant enzymes, thereby protecting cells from oxidative damage, like antioxidant strategies observed in some piezophilic bacteria under HHP (Dewapriya and Kim, 2014). Compared to 0.1 MPa, under HHP, the enriched biosynthesis of unsaturated fatty acids pathway in strain 20R-7-F01 showed significant upregulation of linoleic acid, eicosapentaenoic acid, and other unsaturated fatty acids. Previous studies have demonstrated that increasing unsaturated fatty acid levels under HHP conditions can enhance membrane fluidity, thereby contributing to the integrity and stability of cell membranes under pressure (Scoma et al., 2019; Tamby et al., 2023). Distinctively, strain 20R-7-F01 under HHP exhibits a predominance of polyunsaturated fatty acids among the increased unsaturated fatty acids, which is advantageous for enhancing membrane stability, improving antioxidant capacity, increasing membrane permeability, and modulating intracellular signaling pathways. Furthermore, compared to 0.1 MPa, the enriched pathway of cysteine and methionine metabolism under HHP showed significant upregulation of homocysteine, methionine, glutathione, and other metabolites. These metabolites are known for their efficient antioxidant functions, suggesting that strain 20R-7-F01 may increase the levels of these antioxidants to protect cells from oxidative damage. Additionally, under HHP conditions, the enriched pathway of starch and sucrose metabolism exhibited significant upregulation of biosynthesis of polysaccharides such as trehalose, trehalose, D-mannitol, D-galactose, etc. These polysaccharides are important constituents of the cell wall. Strain 20R-7-F01 cells may regulate polysaccharide synthesis pathways to respond to external pressure changes, including enhancing cell wall strength, stability, and defense capabilities.
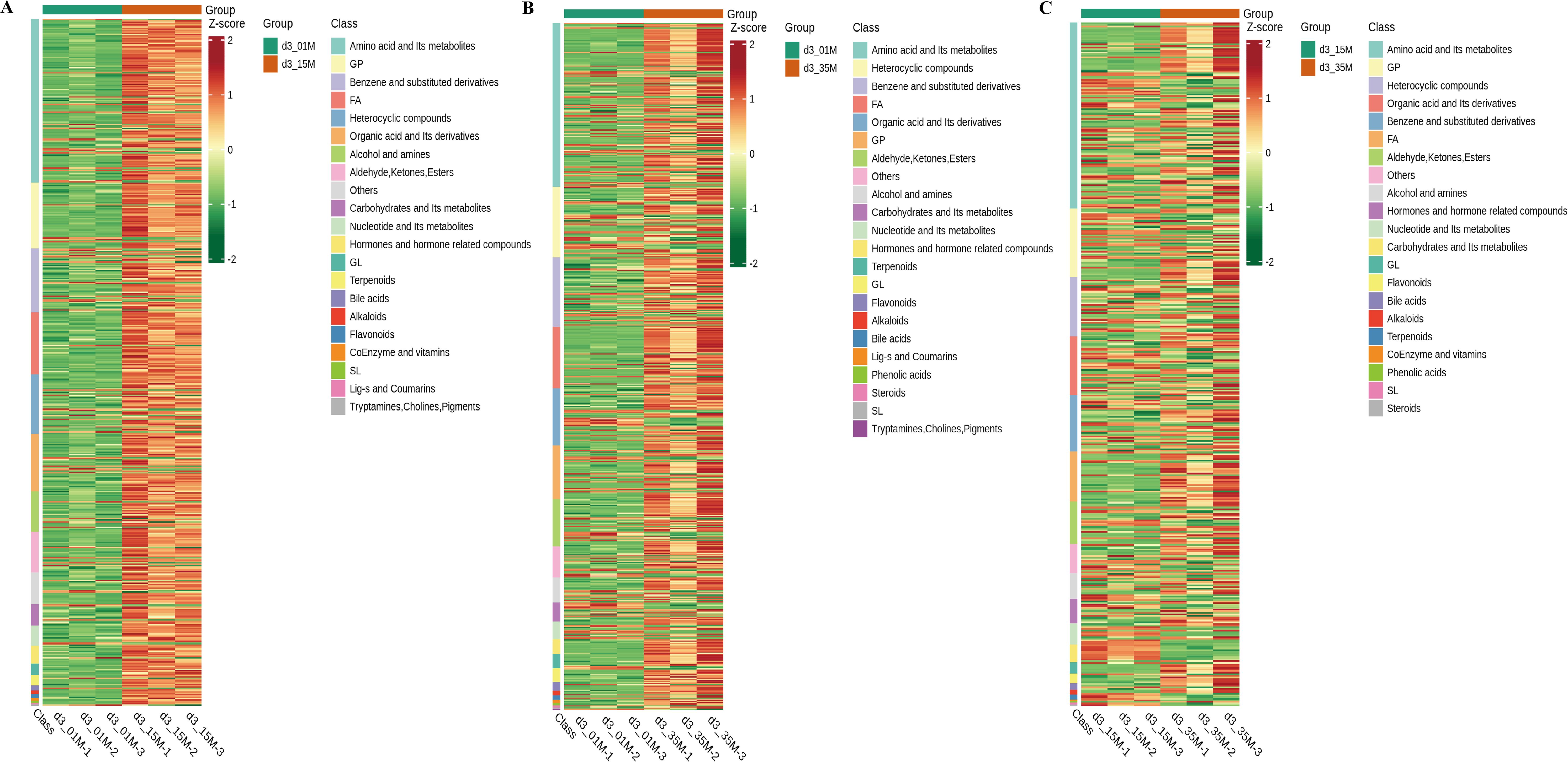
Figure 5. Results of hierarchical cluster analysis of DEMs observed in strain 20R-7-F01 under various pressure conditions. (A–C) illustrate the differential expression trends of DEMs at 0.1MPa (control), 15MPa, and 35MPa on the third day.
3.4 Response mechanism of strain 20R-7-F01 to HHP
Through transcriptomic and metabolomic analyses, we have delineated the adaptive mechanisms employed by strain 20R-7-F01 in response to HHP. However, the interrelationships among these mechanisms remain to be elucidated. To address this, we conducted a correlation network analysis integrating core DEGs from transcriptomic enriched pathways and core DEMs from metabolomic enriched pathways, as illustrated in Figure 6. The entire correlation network comprises 179 nodes and 926 edges. Specifically, the 100 DEGs nodes encompass genes associated with anaerobic fermentation, TCA cycle, DNA repair, cell wall polysaccharide synthesis, antioxidant defenses, unsaturated fatty acid biosynthesis, protein and peptide hydrolysis, and cell cycle regulation. Additionally, the 79 DEMs nodes include metabolites such as ethanol and lactate, unsaturated fatty acids, amino acids and derivatives, small peptides, and polysaccharides. Moreover, we applied a correlation coefficient threshold ≥ 0.9, where coefficients above 0.9 indicate strong positive correlations and coefficients below 0.9 indicate strong negative correlations. This analytical approach facilitated the identification of connections between DEGs and DEMs, offering insights into their interactions and potential coordination in strain 20R-7-F01’s response to HHP stress. The response mechanisms of strain 20-7-1 to HHP closely parallel evolutionary strategies observed in deep-sea organisms for coping with high-pressure environments, focusing specifically on optimizing energy allocation. Core to these strategies are anaerobic fermentation-related genes, which exhibited significant correlations among themselves and with other genes and metabolites, totaling 369 correlation edges. Among these, 306 edges showed strong positive correlations, while 63 exhibited strong negative correlations. Notably, within the 306 highly positively correlated edges, many involved associations between genes related to DNA repair, antioxidant defenses, and other pathways critical for resisting HHP stress. This supports the notion that strain 20R-7-F01 likely produced excess energy under high-pressure conditions compared to atmospheric pressure environments, with this surplus energy predominantly allocated to activating pathways such as DNA repair, antioxidant defenses, and unsaturated fatty acid synthesis, rather than for its own growth and development. Our measurements of intracellular ATP concentration under different pressures further confirmed that the ATP concentration per unit cell mass was indeed higher under high pressure compared to atmospheric pressure (Supplementary Figure S9). This may explain why its growth and development were somewhat constrained in high-pressure environments. Simultaneously, transcriptomic and metabolomic analyses reveal a close coordination in strain 20R-7-F01’s response to HHP stress, jointly contributing to the maintenance of cellular stability and adaptability. There are clear positive correlations between relevant DEGs and DEMs; for instance, genes associated with polyunsaturated fatty acid synthesis like FAD2 and their products such as linolenic acid, as well as genes involved in monounsaturated fatty acid synthesis like SCD and desC and their product octadecenoic acid, all exhibited significant positive correlations. In summary, strain 20R-7-F01 may effectively adapt to HHP stress through optimized energy allocation, prioritizing survival needs overgrowth and development, with close coordination between transcriptomic and metabolomic responses maintaining its adaptability and survival capabilities in high-pressure environments.
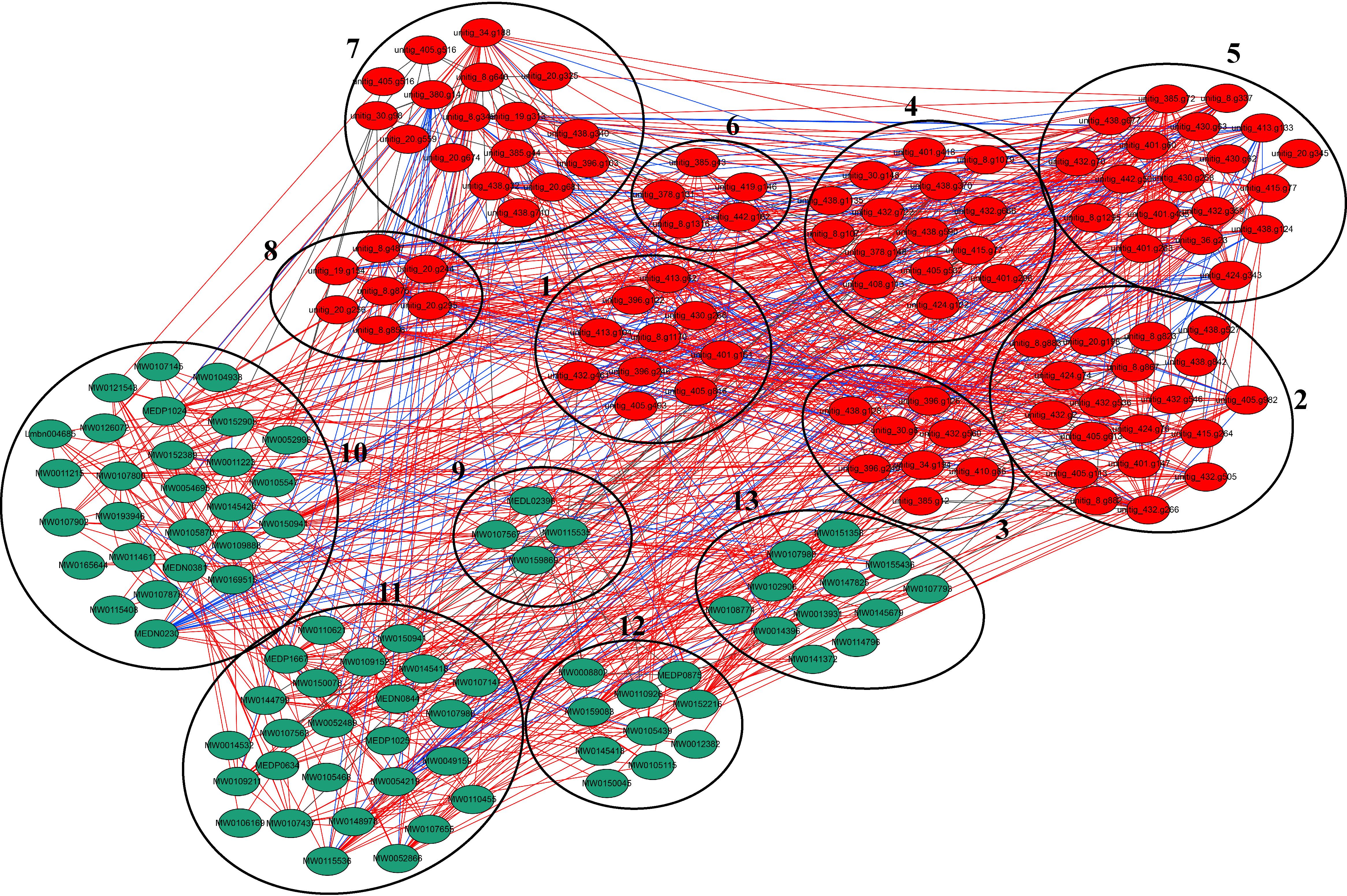
Figure 6. Correlation network of DEGs and DEMs in strain 20R-7-F01. 1: anaerobic fermentation-related genes; 2: DNA repair-related genes; 3: cell wall polysaccharide synthesis-related genes; 4: antioxidant-related genes; 5: unsaturated fatty acid synthesis-related genes; 6: protein and peptide hydrolysis-related genes; 7: TCA cycle-related genes; 8: cell cycle regulation-related genes; 9: ethanol and lactic acid; 10: unsaturated fatty acids; 11: amino acids and their derivatives; 12: small peptides; 13: polysaccharides. Red nodes represent DEGs, green nodes represent DEMs, and the lines between nodes represent correlations, where red represents p ≥ 0.9 and blue represents p ≤ -0.9.
4 Conclusion
We studied Schizophyllum commune strain 20R-7-F01 isolated from coal-bearing sediments at depths exceeding 2,000 meters below the seafloor, investigating its adaptation mechanisms in anaerobic high-pressure environments through physiological, biochemical, transcriptomic, and metabolomic analyses. The findings seem to indicate that the fungus employed multiple strategies in response to high hydrostatic pressure by activating: the carbohydrate metabolic process to concurrently utilize ethanol and lactic acid in fermentation, the integral component of membrane pathway to uphold cell wall structural stability, the metal ion binding pathway to augment the ratio of unsaturated fatty acids within the cell membrane, the DNA repair pathway to rectify DNA damage, and the oxidoreductase activity pathway and hydrolase activity pathway to eliminate intracellular ROS. These insights deepen our understanding of fungal survival strategies and adaptation mechanisms in extreme subseafloor environments, laying a foundation for further research into their roles in Earth’s carbon, nitrogen, sulfur cycles, in the deep biosphere.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA1101667. Further inquiries can be directed to the corresponding authors.
Ethics statement
The manuscript presents research on animals that do not require ethical approval for their study.
Author contributions
MZ: Conceptualization, Data curation, Methodology, Software, Writing – original draft. DL: Resources, Validation, Writing – review & editing. JL: Investigation, Resources, Writing – review & editing. JF: Funding acquisition, Supervision, Writing – review & editing. CL: Funding acquisition, Resources, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (no. 41973073, 41773083, and 92251303).
Acknowledgments
We thank the editor and reviewer for their valuable comments and suggestions. We are also thankful to our lab fellow and all those who assisted during this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2024.1471465/full#supplementary-material
References
Abe F. (2021). Molecular responses to high hydrostatic pressure in eukaryotes: genetic insights from studies on saccharomyces cerevisiae. Biology 10, 1305. doi: 10.3390/biology10121305
Alkadi H. (2020). A review on free radicals and antioxidants. Infect. Disord. Drug Targets 20, 16–26. doi: 10.2174/1871526518666180628124323
Amrani A., Bergon A., Holota H., Tamburini C., Garel M., Ollivier B., et al. (2014). Transcriptomics reveal several gene expression patterns in the piezophile Desulfovibrio hydrothermalis in response to hydrostatic pressure. PloS One 9, e106831. doi: 10.1371/journal.pone.0106831
Anders S., Huber W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. doi: 10.1186/gb-2010-11-10-r106
Bartlett D. H., Ferguson G., Valle G. (2008). Adaptations of the psychrotolerant piezophile Photobacterium profundum strain SS9. High-Pressure Microbiol. 18, 319–337. doi: 10.1128/9781555815646
Bradley J. A., Arndt S., Amend J. P., Burwicz-Galerne E., LaRowe D. E. (2022). Sources and fluxes of organic carbon and energy to microorganisms in global marine sediments. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.910694
Bravim F., de Freitas J. M., Fernandes A. A., Fernandes P. M. (2010). High hydrostatic pressure and the cell membrane: stress response of Saccharomyces cerevisiae. Ann. New York Acad. Sci. 1189, 127–132. doi: 10.1111/j.1749-6632.2009.05182.x
Bravim F., Mota M. M., Fernandes A. A., Fernandes P. M. (2016). High hydrostatic pressure leads to free radicals accumulation in yeast cells triggering oxidative stress. FEMS Yeast Res. 16, fow052. doi: 10.1093/femsyr/fow052
Chen P., Zhou H., Huang Y., Xie Z., Zhang M., Wei Y., et al. (2021). Revealing the full biosphere structure and versatile metabolic functions in the deepest ocean sediment of the Challenger Deep. Genome Biol. 22, 207. doi: 10.1186/s13059-021-02408-w
Crowley L. C., Marfell B. J., Christensen M. E., Waterhouse N. J. (2016). Measuring cell death by trypan blue uptake and light microscopy. Cold Spring Harbor Protoc. 2016 (7). doi: 10.1101/pdb.prot087155
Damare S., Raghukumar C., Raghukumar S. (2006). Fungi in deep-sea sediments of the Central Indian Basin. Deep Sea Res. Part I: Oceanographic Res. Papers 53, 14–27. doi: 10.1016/J.DSR.2005.09.005
Delaunay A., Isnard A. D., Toledano M. B. (2000). H2O2 sensing through oxidation of the Yap1 transcription factor. EMBO J. 19, 5157–5166. doi: 10.1093/emboj/19.19.5157
Dewapriya P., Kim S. K. (2014). Marine microorganisms: An emerging avenue in modern nutraceuticals and functional foods. Food Res. Int. 56, 115–125. doi: 10.1016/j.foodres.2013.12.022
El-Bahr S. M. (2013). Biochemistry of free radicals and oxidative stress. Biochemistry 1, 11–11. doi: 10.5567/sciintl.2013.111.117
Feng L., Song Q., Jiang Q., Li Z. (2021). The horizontal and vertical distribution of deep-sea sediments fungal community in the South China Sea. Front. Mar. Sci. 8. doi: 10.3389/fmars.2021.592784
Fernandes P. M., Domitrovic T., Kao C. M., Kurtenbach E. (2004). Genomic expression pattern in Saccharomyces cerevisiae cells in response to high hydrostatic pressure. FEBS Lett. 556, 153–160. doi: 10.1016/s0014-5793(03)01396-6
Gow N. A. R., Latge J. P., Munro C. A. (2017). The fungal cell wall: structure, biosynthesis, and function. Microbiol. Spectr. 5 (3). doi: 10.1128/microbiolspec.FUNK-0035-2016
Griffiths B., Lewis C. A., Bensaad K., Ros S., Zhang Q., Ferber E. C., et al. (2013). Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab. 1, 3. doi: 10.1186/2049-3002-1-3
Hoshino T., Doi H., Uramoto G. I., Wörmer L., Adhikari R. R., Xiao N., et al. (2020). Global diversity of microbial communities in marine sediment. Proc. Natl. Acad. Sci. United States America 117, 27587–27597. doi: 10.1073/pnas.1919139117
Huang X., Liu X., Xue Y., Pan B., Xiao L., Wang S., et al. (2022). Methane production by facultative anaerobic wood-rot fungi via a new halomethane-dependent pathway. Microbiol. Spectr. 10, e0170022. doi: 10.1128/spectrum.01700-22
Inagaki F., Hinrichs K. U., Kubo Y., Bowles M. W., Heuer V. B., Hong W. L., et al. (2015). DEEP BIOSPHERE. Exploring deep microbial life in coal-bearing sediment down to ~2.5 km below the ocean floor. Sci. (New York N.Y.) 349, 420–424. doi: 10.1126/science.aaa6882
Ishii A., Oshima T., Sato T., Nakasone K., Mori H., Kato C. (2005). Analysis of hydrostatic pressure effects on transcription in Escherichia coli by DNA microarray procedure. Extremophiles: Life under Extreme Conditions 9, 65–73. doi: 10.1007/s00792-004-0414-3
Jing X., Yang F., Shao C., Wei K., Xie M., Shen H., et al. (2019). Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 18, 157. doi: 10.1186/s12943-019-1089-9
Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S. L. (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. doi: 10.1186/gb-2013-14-4-r36
Lama-Sherpa T. D., Shevde L. A. (2020). An emerging regulatory role for the tumor microenvironment in the DNA damage response to double-strand breaks. Mol. Cancer research: MCR 18, 185–193. doi: 10.1158/1541-7786
Lebeau P. F., Chen J., Byun J. H., Platko K., Austin R. C. (2019). The trypan blue cellular debris assay: a novel low-cost method for the rapid quantification of cell death. MethodsX 6, 1174–1180. doi: 10.1016/j.mex.2019.05.010
Li M., Wilson III, D. M. (2014). Human apurinic/apyrimidinic endonuclease 1. Antioxidants Redox Signaling 20, 678–707. doi: 10.1089/ars.2013.5492
Li J., Zhang Y., Lv Y., Xiao X. (2022). Isolation and characterization of piezotolerant fungi from mariana trench sediment. Deep Sea Res. Part I: Oceanographic Res. Papers. Deep Sea Res. Part I: Oceanographic Res. Papers 190, 103873. doi: 10.1016/j.dsr.2022.103873
Li S., Zhou X., Zeng R., Lin L., Zou X., Yan Y., et al. (2022). YAP1 silencing attenuated lung injury/fibrosis but worsened diaphragmatic function by regulating oxidative stress and inflammation response in mice. Free Radical Biol. Med. 193, 485–498. doi: 10.1016/j.freeradbiomed.2022.10.323
Liu X., Huang X., Chu C., Xu H., Wang L., Xue Y., et al. (2022). Genome, genetic evolution, and environmental adaptation mechanisms of Schizophyllum commune in deep subseafloor coal-bearing sediments. iScience 25, 104417. doi: 10.1016/j.isci.2022.104417
Liu C. H., Huang X., Xie T. N., Duan N., Xue Y. R., Zhao T. X., et al. (2017). Exploration of cultivable fungal communities in deep coal-bearing sediments from ∼1.3 to 2.5 km below the ocean floor. Environ. Microbiol. 19, 803–818. doi: 10.1111/1462-2920.13653
Liu Y., Yang K., Jia Y., Shi J., Tong Z., Fang D., et al. (2021). Gut microbiome alterations in high-fat-diet-fed mice are associated with antibiotic tolerance. Nat. Microbiol. 6, 874–884. doi: 10.1038/s41564-021-00912-0
Livak K. J., Schmittgen T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Love M. I., Huber W., Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/s13059-014-0550-8
Ma Y., Zhao M., Zhou F., Liu X., Liu C. (2023). Anaerobic production and biosynthesis mechanism of exopolysaccharides in Schizophyllum commune 20R-7-F01. Int. J. Biol. Macromol 253, 127339. doi: 10.1016/j.ijbiomac.2023.127339
Malas J., Russo D. C., Bollengier O., Malaska M. J., Lopes R. M. C., Kenig F., et al. (2024). Biological functions at high pressure: transcriptome response of Shewanella oneidensis MR-1 to hydrostatic pressure relevant to Titan and other icy ocean worlds. Front. Microbiol. 15. doi: 10.3389/fmicb.2024.1293928
Manian V., Orozco-Sandoval J., Diaz-Martinez V. (2021). Detection of genes in arabidopsis thaliana L. Responding to DNA damage from radiation and other stressors in spaceflight. Genes 12, 938. doi: 10.3390/genes12060938
Marx G., Moody A., Bermúdez-Aguirre D. (2011). A comparative study on the structure of Saccharomyces cerevisiae under nonthermal technologies: high hydrostatic pressure, pulsed electric fields and thermo-sonication. Int. J. Food Microbiol. 151, 327–337. doi: 10.1016/j.ijfoodmicro.2011.09.027
Masuo S., Terabayashi Y., Shimizu M., Fujii T., Kitazume T., Takaya N. (2010). Global gene expression analysis of Aspergillus nidulans reveals metabolic shift and transcription suppression under hypoxia. Mol. Genet. Genomics 284(6), 415–424. doi: 10.1007/s00438-010-0576-x
Ifeanyi O. E. (2018). A Review on Free Radicals and Antioxidants. 4, 123–133. doi: 10.22192/ijcrms.2018.04.02.019
Ohm R. A., de Jong J. F., Lugones L. G., Aerts A., Kothe E., Stajich J. E., et al. (2010). Genome sequence of the model mushroom Schizophyllum commune. Nat. Biotechnol. 28, 957–963. doi: 10.1038/nbt.1643
Peng Q., Li Y., Deng L., Fang J., Yu X. (2021). High hydrostatic pressure shapes the development and production of secondary metabolites of Mariana Trench sediment fungi. Sci. Rep. 11, 11436. doi: 10.1038/s41598-021-90920-1
Qiu X., Cao X., Jian H., Wu H., Xu G., Tang X. (2022). Transcriptomic Analysis Reveals that Changes in Gene Expression Contribute to Microbacterium sediminis YLB-01 Adaptation at Low Temperature Under High Hydrostatic Pressure. Curr. Microbiol. 79, 95. doi: 10.1007/s00284-022-02786-9
Quilis I., Gomar-Alba M., Igual J. C. (2021). The CWI pathway: A versatile toolbox to arrest cell-cycle progression. J. fungi (Basel Switzerland) 7, 1041. doi: 10.3390/jof7121041
Rao M. B., Tanksale A. M., Ghatge M. S., Deshpande V. V. (1998). Molecular and biotechnological aspects of microbial proteases. Microbiol. Mol. Biol. Reviews: MMBR 62, 597–635. doi: 10.1128/MMBR.62.3.597-635.1998
Rodríguez-Vargas S., Sánchez-García A., Martínez-Rivas J. M., Prieto J. A., Randez-Gil F. (2007). Fluidization of membrane lipids enhances the tolerance of Saccharomyces cerevisiae to freezing and salt stress. Appl. Environ. Microbiol. 73, 110–116. doi: 10.1128/AEM.01360-06
Santomartino R., Camponeschi I., Polo G., Immesi A., Rinaldi T., Mazzoni C., et al. (2019). The hypoxic transcription factor KlMga2 mediates the response to oxidative stress and influences longevity in the yeast Kluyveromyces lactis. FEMS Yeast Res. 19, foz020. doi: 10.1093/femsyr/foz020
Scoma A., Garrido-Amador P., Nielsen S. D., Røy H., Kjeldsen K. U. (2019). The polyextremophilic bacterium clostridium paradoxum attains piezophilic traits by modulating its energy metabolism and cell membrane composition. Appl. Environ. Microbiol. 85, e00802–e00819. doi: 10.1128/AEM.00802-19
Shah S. H., Kraus W. E., Newgard C. B. (2012). Metabolomic profiling for the identification of novel biomarkers and mechanisms related to common cardiovascular diseases: form and function. Circulation 126, 1110–1120. doi: 10.1161/CIRCULATIONAHA.111.060368
Shyur L. F., Lee S. H., Chang S. T., Lo C. P., Kuo Y. H., Wang S. Y. (2010). Taiwanin A inhibits MCF-7 cancer cell activity through induction of oxidative stress, upregulation of DNA damage checkpoint kinases, and activation of p53 and FasL/Fas signaling pathways. Phytomedicine: Int. J. Phytotherapy Phytopharmacology 18, 16–24. doi: 10.1016/j.phymed.2010.06.005
Smith J., Tho L. M., Xu N., Gillespie D. A. (2010). The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 108, 73–112. doi: 10.1016/B978-0-12-380888-2.00003-0
Tamby A., Sinninghe Damsté J. S., Villanueva L. (2023). Microbial membrane lipid adaptations to high hydrostatic pressure in the marine environment. Front. Mol. Biosci. 9. doi: 10.3389/fmolb.2022.1058381
Tovar-Herrera O. E., Martha-Paz A. M., Pérez-LLano Y., Aranda E., Tacoronte-Morales J. E., Pedroso-Cabrera M. T., et al. (2018). Schizophyllum commune: An unexploited source for lignocellulose degrading enzymes. MicrobiologyOpen 7, e00637. doi: 10.1002/mbo3.637
Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Wang W., Wang A., Luo G., Ma F., Wei X., Bi Y. (2018). S1P1 receptor inhibits kidney epithelial mesenchymal transition triggered by ischemia/reperfusion injury via the PI3K/Akt pathway. Acta Biochim. Biophys. Sin. 50, 651–657. doi: 10.1093/abbs/gmy058
Wang H., Zhang Y., Bartlett D. H., Xiao X. (2021). Transcriptomic analysis reveals common adaptation mechanisms under different stresses for moderately piezophilic bacteria. Microbial Ecol. 81, 617–629. doi: 10.1007/s00248-020-01609-3
Williams B. T., Cowles K., Bermejo Martínez A., Curson A. R. J., Zheng Y., Liu J., et al. (2019). Bacteria are important dimethylsulfoniopropionate producers in coastal sediments. Nat. Microbiol. 4, 1815–1825. doi: 10.1038/s41564-019-0527-1
Xiao Z., Gong N., Zhou X., Zhu L., He X., Zheng J., et al. (2021). Developmental characteristics of sporogenous hyphae: a new observation between Brassica juncea var. tumida and Albugo candida. Eur. J. Plant Pathol. 162, 343–355. doi: 10.1007/s10658-021-02406-5
Xiao X., Zhang Y., Wang F. (2021). Hydrostatic pressure is the universal key driver of microbial evolution in the deep ocean and beyond. Environ. Microbiol. Rep. 13, 68–72. doi: 10.1111/1758-2229.12915
Zain Ul Arifeen M., Chu C., Yang X., Liu J., Huang X., Ma Y., et al. (2021a). The anaerobic survival mechanism of Schizophyllum commune 20R-7-F01, isolated from deep sediment 2 km below the seafloor. Environ. Microbiol. 23, 1174–1185. doi: 10.1111/1462-2920.15332
Zain Ul Arifeen M., Ma Z. J., Wu S., Liu J. Z., Xue Y. R., Liu C. H. (2021b). Effect of oxygen concentrations and branched-chain amino acids on the growth and development of sub-seafloor fungus, Schizophyllum commune 20R-7-F01. Environ. Microbiol. 23, 6940–6952. doi: 10.1111/1462-2920.15738
Keywords: anaerobic, high hydrostatic pressure, Schizophyllum commune 20R-7-F01, transcriptome, metabolome, correlation network analysis
Citation: Zhao M, Li D, Liu J, Fang J and Liu C (2024) Pressure-tolerant survival mechanism of Schizophyllum commune 20R-7-F01 isolated from deep sediments 2 kilometers below the seafloor. Front. Mar. Sci. 11:1471465. doi: 10.3389/fmars.2024.1471465
Received: 30 July 2024; Accepted: 08 October 2024;
Published: 11 November 2024.
Edited by:
Danny Ionescu, Leibniz-Institute of Freshwater Ecology and Inland Fisheries (IGB), GermanyReviewed by:
Doug Bartlett, University of California, San Diego, United StatesGong Linfeng, Ministry of Natural Resources, China
Copyright © 2024 Zhao, Li, Liu, Fang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiasong Fang, anNmYW5nQHNob3UuZWR1LmNu; Changhong Liu, Y2hsaXVAbmp1LmVkdS5jbg==