 Mirae Kim
Mirae Kim Jaeho Song2†
Jaeho Song2† Ilnam Kang
Ilnam Kang Jang-Cheon Cho
Jang-Cheon Cho- 1Department of Biological Sciences and Bioengineering, Inha University, Incheon, Republic of Korea
- 2Division of Microbiology, Honam National Institute of Biological Resources, Mokpo, Republic of Korea
- 3University of The Ryukyus, Okinawa, Japan
Despite previous culture-independent studies highlighting the prevalence of the order Burkholderiales in deep-sea environments, the cultivation and characterization of deep-sea Burkholderiales have been infrequent. A total of 243 deep-sea bacterial strains were isolated from various depths in the Northwest Pacific Ocean, with 33 isolates (13.6%) from a depth of 4000 m classified into Burkholderiales. Herein, we report the isolation and genome characteristics of strain SAORIC-580T, from a depth of 4000 m in the Northwest Pacific Ocean. The strain showed a close phylogenetic relationship with Limnobacter thiooxidans CS-K2T, sharing 99.9% 16S rRNA gene sequence identity. The complete whole-genome sequence of strain SAORIC-580T comprised 3.3 Mbp with a DNA G+C content of 52.5%. Comparative genomic analysis revealed average nucleotide identities between 79.4–85.7% and digital DNA-DNA hybridization values of 19.9–29.5% when compared to other Limnobacter genomes, indicating that the strain represents a novel species within the genus. Genomic analysis revealed unique adaptations to deep-sea conditions, including genes associated with phenol degradation, stress responses, cold adaptation, heavy metal resistance, signal transduction, and carbohydrate metabolism. The SAORIC-580T genome was found to be more abundant in the deep sea than at the surface in the trenches of the Northwest Pacific Ocean, suggesting adaptations to the deep-sea environment. Phenotypic characterization highlighted distinct differences from other Limnobacter species, including variations in growth conditions, enzyme activities, and phenol degradation capabilities. Chemotaxonomic markers of the strain included ubiquinone-10, major fatty acids such as C16:0, C16:1, and C18:1, and major polar lipids including phosphatidylethanolamine, phosphatidylglycerol, and diphosphatidylglycerol. Based on the polyphasic taxonomic data, it is concluded that strain SAORIC-580T (= KACC 21440T = NBRC 114111T) represents a novel species, for which the name Limnobacter profundi sp. nov. is proposed.
1 Introduction
The order Burkholderiales within the class Betaproteobacteria encompasses approximately 130 genera distributed across five families, according to validly published names as recorded on the List of Prokaryotic names with Standing in Nomenclature (LPSN) as of May 2024 (https://lpsn.dsmz.de). Members of this order display considerable phenotypic diversity, exemplified by variations in oxygen relationships (ranging from strict aerobes to facultative anaerobes), morphologies (including rods and cocci), temperature preferences (from psychrophiles to mesophiles), flagellar arrangements, and types of respiratory quinones (ubiquinone and methylmenaquinone) (De Ley et al., 1986; Garrity et al., 2005; Morotomi et al., 2011). Moreover, these bacteria exhibit a broad metabolic spectrum, capable of processes such as aerobic anoxygenic phototrophy, nitrogen fixation, oxidation of substances including hydrogen, iron, and thiosulfate, and degradation of plastic and aromatic compounds (Garrity et al., 2005; Imhoff et al., 2018; Syranidou et al., 2019). Burkholderiales bacteria are ubiquitously distributed across diverse ecological niches, including soil, freshwater, seawater, groundwater, activated sludge, coral reefs, and animal gastrointestinal tracts, with notably high abundance in certain deep-sea environments (Ginige et al., 2013; Moreira et al., 2015; Aguirre-Von-Wobeser et al., 2018; Meng et al., 2019; Li et al., 2019a).
Culture-independent analyses based on 16S rRNA gene sequences have revealed that Burkholderiales constitute up to 30–40% of bacterial communities in specific deep-sea samples, underscoring their significant presence and potential adaptations to the extreme conditions found below 200 m depth, characterized by high pressure, low temperature, and nutrient scarcity (Eloe et al., 2011a, b; Zhang et al., 2018; Sinha et al., 2019). Recent genomic studies have significantly advanced our understanding of the genetic features of deep-sea bacteria, including the enrichment of transposable elements, transcriptional regulators, toxin-antitoxin systems, and genes involved in the degradation of aromatic compounds (Martin-Cuadrado et al., 2007; Konstantinidis et al., 2009; Zhang et al., 2018, 2019). Despite these advances, the physiological and genetic diversity of deep-sea Burkholderiales remains largely unexplored due to the scarcity of isolated strains.
The genus Limnobacter, a member of the order Burkholderiales, was first proposed following the isolation and description of a thiosulfate-oxidizing bacterium from freshwater lake sediment (Spring et al., 2001). As of August 2024, the genus comprises 5 species with validly published names in LPSN. Members of the genus have been isolated from diverse habitats including freshwater lake sediment, volcanic deposits, humus soil, surface seawater, and marine algae (Lu et al., 2011; Vedler et al., 2013; Nguyen and Kim, 2017; Duan et al., 2020). This genus is characterized by the production of polyhydroxybutyrate (PHB) as a storage compound, being chemolithoheterotrophic bacteria that utilize thiosulfate as an energy source through biological oxidation, the presence of a single polar flagellum, and a rod-shaped cell morphology (Nguyen and Kim, 2017). Limnobacter genomes show a genome size ranging 3.3−3.4 Mbp, with an average G+C content of 52.5−53.2%, and include diverse metabolic pathways such as sulfur oxidation, citronellol degradation, and exopolysaccharide production (Chen et al., 2016; Duan et al., 2020; Naruki et al., 2024). Although species of the genus Limnobacter have been discovered in diverse environments, no deep-sea isolates of the genus have yet been validly published. However, in the literature survey conducted in this work, we found an abundance of 16S rRNA gene sequences associated with this genus in several deep-sea habitats, including the deep-sea water (6000 m) of the Puerto Rico Trench (Eloe et al., 2011b) and the sediment (3788 m) of the Indian Ocean (Khandeparker et al., 2014). Additionally, a significant number of metagenomic and metatranscriptomic reads retrieved from the deep waters of the Mariana Trench matched Limnobacter-related genomes (Dang et al., 2024). Therefore, the genus Limnobacter might be recognized as part of the deep-sea Burkholderiales.
In this study, we isolated a proteobacterial strain, designated SAORIC-580T, from seawater at a depth of 4000 m in the Northwest Pacific Ocean. The complete genome sequence of SAORIC-580T was obtained and comparatively analyzed against other Limnobacter species using Clusters of Orthologous Groups (COG) categories, Pfam domains, and Genomic Islands (GIs). SAORIC-580T features metabolic pathways, including phenol degradation, that suggest adaptation to the deep-sea environment. To the best of our knowledge, this genome represents the first genome in the order Burkholderiales cultivated from the deep sea. Phylogenetic and phenotypic analysis indicated that the strain belongs to the genus Limnobacter and is considered to represent a novel species within this genus. Based on these findings, we propose the name Limnobacter profundi for the new type strain, SAORIC-580T.
2 Materials and methods
2.1 Isolation of deep-sea bacteria and bacterial cultures
During the research cruise MR-11-05 aboard the RV ‘Mirai’ (Japan Agency for Marine-Earth Science and Technology [JAMSTEC]) as part of the K2S1 project (Honda et al., 2017), deep seawater samples were collected at depths of 1000, 2000, 3000, and 4000 m at S1 station (32°00’ N, 145°00’ E) in the Northwest Pacific Ocean in July 2011 and immediately transferred to the cleanroom of the RV. Total cells of each seawater sample were stained with 4’,6-diamidino-2-phenylindole (DAPI) and counted via epifluorescence microscopy (Nikon 80i, Nikon, Japan). An aliquot of each water sample (100 μl per plate) was spread on agar plates with two different compositions: one-fifth strength Marine Agar 2216E (1/5 MA; BD Difco, USA) and one-tenth strength R2A (1/10 R2A; BD Difco, USA), both dissolved in 1 L of seawater that had been aged in the dark for 1 year. After incubation at 10°C for 4 weeks in darkness, all colonies grown on the agar plates were transferred to new plates for pure cultivation and further analyses.
Among the colonies isolated from a depth of 4000 m, a beige-colored colony, designated SAORIC-580T, was maintained on R2A or in R2A broth at 30°C, with transfers every three days, and also preserved as glycerol suspensions (20%, v/v) at –80°C for long-term preservation. For phenotypic and chemotaxonomic comparisons, L. thiooxidans KACC 13837T (the type species of the genus Limnobacter), L. alexandrii KCTC 72281T, L. litoralis NBRC105857T, and L. humi KACC 18574T were obtained from the Korean Collection for Type Cultures (KCTC), the Korean Agricultural Culture Collection (KACC), and NITE Biological Resource Center (NBRC), respectively, and routinely cultured on R2A at 30°C.
2.2 Phylogenetic analyses of the deep-sea isolates
Bacterial cells were treated using InstaGene Matrix (Bio-Rad Laboratories, Hercules, CA, USA) according to the manufacturer’s instructions, yielding supernatants that can be used for PCR. The 16S rRNA genes of the isolates were amplified by PCR using bacterial universal primers 27F and 1492R (Weisburg et al., 1991). PCR amplifications were performed according to the following protocol: initial denaturation at 94°C for 5 min, followed by 30 cycles of denaturation at 94°C for 30 sec, annealing at 55°C for 60 sec, extension at 72°C for 90 sec, and a final extension at 72°C for 5 min. The resultant PCR products were digested with HindIII (Takara, Japan) at 37°C for 8 hours, visualized by electrophoresis on a 3% agarose gel, grouped using HindIII-based restriction fragment length polymorphism (RFLP) analyses, and representative PCR products were sequenced using a Sanger sequencer. The 16S rRNA gene sequences were analyzed using the BLASTn search in the GenBank database and the 16S-based ID application in the EzBioCloud (Yoon et al., 2017).
To determine the phylogenetic position of strain SAORIC-580T, the 16S rRNA gene sequences of the strain and its closely related species were aligned using the ClustalW program (Thompson et al., 1997). Phylogenetic trees were constructed from unambiguously aligned 1472 bp of the 16S rRNA gene sequences using the maximum-likelihood method (Felsenstein, 1981) with the GTR+G+I model, the neighbor-joining method (Saitou and Nei, 1987) with Jukes-Cantor correction, and the minimum evolution method (Rzhetsky and Nei, 1993) employing the Tree-Bisection-Reconnection (TBR) method, as implemented in MEGA X (Kumar et al., 2018). The topological integrity of the phylogenetic trees was confirmed through bootstrap analyses (Felsenstein, 1985) based on 1000 replications.
2.3 Whole genome sequencing and genome analyses
Genomic DNA of strain SAORIC-580T was extracted using the DNeasy Blood & Tissue kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Whole-genome sequencing of the strain was conducted at Macrogen (Seoul, Republic of Korea) using the Illumina MiSeq platform, employing a 2×300 paired-end run. Additionally, nanopore sequencing was performed. Genomic DNA was extracted with the MagAttract HMW DNA Kit (Qiagen, Hilden, Germany), and a sequencing library was prepared using the Ligation Sequencing Kit (SQK-LSK109, Oxford Nanopore Technologies, Oxford, UK) and Native Barcoding Expansion pack (EXP-NBD104, Oxford Nanopore Technologies, Oxford, UK), following the protocols provided by the manufacturer. Sequencing was then performed using an R9.4.1 flow cell on a MinION Mk1B device (Oxford Nanopore Technologies, Oxford, UK) and the resultant data were basecalled using Guppy (v3.4.5) in high accuracy mode. The quality of the long reads was checked using SeqKit (v2.8.0). After quality checking (Q20: 59.7%; Q30: 27.6%; N50: 11355 bp), all long-read sequences were used for assembly. A hybrid assembly combining both Illumina short reads and nanopore long reads was performed using Unicycler version 0.4.8 (Wick et al., 2017). The quality of the assembled genome was evaluated using CheckM2 version 1.0.2 (Chklovski et al., 2023). The CGView program was used to generate a circular genome map (Stothard and Wishart, 2005). The protein sequences were also used for comparative pan- and core-genome analyses using GET_HOMOLOGUES software with the orthoMCL algorithm (Contreras-Moreira and Vinuesa, 2013).
Genome annotation was performed using the Integrated Microbial Genomes Expert Review (IMG-ER) system (Markowitz et al., 2009) and the NCBI Prokaryotic Genome Annotation Pipeline (Tatusova et al., 2016). Comparative genomics involved genomes of L. thiooxidans DSM 13612T (IMG genome ID; 2795386119), L. alexandrii LZ-4T (2890930897), and Limnobacter sp. MED105 (640963026) against the SAORIC-580T genome (2887797443). Genomic relatedness between the strains was estimated using average nucleotide identity (ANIb) and digital DNA-DNA hybridization (dDDH) values, calculated by the OrthoANI algorithm on the EzBioCloud web service (Yoon et al., 2017) and the genome-to-genome distance calculator (GGDC 3.0) (Meier-Kolthoff et al., 2022), respectively. To infer genome-based phylogenetic trees, 92 universal bacterial core genes were extracted using the up-to-date bacterial core gene set and pipeline (UBCG2) (Kim et al., 2021) and analyzed using RAxML (Stamatakis, 2014) based on amino acid alignment.
Metabolic pathways were reconstructed by additionally annotating each genome with Prokka (Seemann, 2014), which utilizes databases such as Prodigal, RNAmmer, Aragorn, SignalP, and Infernal with a default e-value cut-off of 1e-09. The resulting proteins were then queried in BlastKOALA (Kanehisa et al., 2016) and KofamKOALA (Aramaki et al., 2020). To analyze the Clusters of Orthologous Groups (COG) categories (Galperin et al., 2015) and Pfam domains (El-Gebali et al., 2019), protein sequences generated by Prokka were queried against the COG and Pfam databases using RPS-BLAST with an e-value cutoff of 0.01. Genomic islands within the SAORIC-580T genome were identified using IslandViewer4, employing the IslandPath-DIMOB method (Bertelli et al., 2017).
2.4 Metagenome fragment recruitment analyses
To explore the distribution of deep-sea bacteria, 12 bacterial genomes retrieved from the Northwest Pacific Ocean, including the SAORIC-580T genome, were matched against metagenomes obtained from the Japan Trench (DRA005791), Kuril Trench (DRP003768), Mariana Trench (DRA005792) and Ogasawara Trench (DRA005790) of the Northwest Pacific Ocean. The genomes utilized in these recruitment analyses include 10 genomes from Pseudomonadota, one from Bacteroidota, and one from Verrucomicrobiota. Prior to analysis, RNA-coding genes were masked within the isolate genomes. Subsequently, the metagenomes were quality-trimmed and randomly subsampled to yield 1 million reads each. Recruitment analysis was conducted using BlastN, with the isolate genomes serving as queries and the metagenomes as subject databases. Metagenomic reads that exhibited at least 95% similarity and an alignment length of 150 bp or more with the isolate genomes were considered successfully recruited (Hyatt et al., 2012).
2.5 Morphological, physiological, and biochemical characterization
Cell morphology, cell size, and the presence of flagella were observed using transmission electron microscopy (TEM, CM200, Philips, Bend, OR, USA). Cells were placed on a carbon-coated copper grid and stained with 2.0% uranyl acetate. Gram staining was performed using a Gram staining kit (bioMérieux, Marcy-l’Etoile, France). Catalase and oxidase activities were determined by spraying 3% (v/v) hydrogen peroxide solution and 1% (w/v) Kovac’s reagent (bioMérieux, Marcy-l’Etoile, France), respectively, to fresh colonies. Flagellum-based motility was evaluated in semi-solid marine agar containing 0.5% (w/v) agar, following incubation at 30°C for 3 days. Temperature range and optimum for growth were monitored at 4, 10, 15, 20, 25, 30, 37, 42, and 45°C, and pH range and optimum were assessed from pH 5.0 to 10.0 (at 1.0 pH intervals) using R2A broth. The pH was adjusted using the following buffering system: 1 M MES for pH 5.0−6.0, 1 M MOPS for pH 6.5−7.0, 1 M HEPES for pH 7.5−8.0, 1 M Tris for pH 8.5−9.0 and 0.5 M CHES for pH 9.5−10.0. NaCl requirement and tolerance tests were conducted with concentrations of 0−6% NaCl in 1% increments, using R2A broth as the basal medium. Growth under these conditions was monitored by measuring culture turbidity using a UV/Vis spectrophotometer (UV2600, Shimadzu, Kyoto, Japan). Anaerobic growth was monitored using an AnaeroPack system (Mitsubishi Gas Chemical, Tokyo, Japan) over a period of up to two weeks.
Phenol degradation was tested using a basal medium (8 g NaCl, 0.2 g KCl, 1.44 g Na2HPO4, 0.24 g KH2PO4 in 1 L deionized water) supplemented with NH4Cl at a final concentration of 10 µM. Phenol was added as the sole carbon source into the basal medium with final concentrations ranging from 0 to 4.5 mM in 0.5 mM increments. Cellular growth was monitored every two days using an Easy-Cyte flow cytometer (Millipore, MA, USA) following 1 h staining of a 100 µl culture aliquot with 1:2000 (v/v) dilution of SYBR-Green I (Invitrogen, Carlsbad, CA, USA). Additional biochemical tests were conducted using API 20 NE, API ZYM strips (bioMérieux, Marcy-l’Etoile, France) and GEN III MicroPlate (Biolog, Hayward, CA, USA), with results recorded according to the manufacturer’s guidelines after five days of incubation.
Analysis of cellular fatty acid profiles was carried out in accordance with the standard protocols provided by the MIDI/Hewlett Packard Microbial Identification system (Sasser, 1990). Fatty acid methyl esters (FAME) of strain SAORIC-580T and other Limnobacter strains were extracted from colonies cultured on identical plate sectors and were identified using the TSBA 6 database. Polar lipids were analyzed following methods outlined by Minnikin et al. (1984). Total polar lipids separated by two-dimensional thin-layer chromatography were identified by spraying with molybdophosphoric acid, and lipids containing specific functional groups were further identified by spraying with ninhydrin, molybdenum blue, and alpha-naphthol solutions. Determination of respiratory quinones was performed using thin-layer chromatography as previously described (Collins and Jones, 1981).
2.6 Nucleotide sequence accession numbers
The 16S rRNA gene sequence and the complete whole-genome sequence of strain SAORIC-580T has been deposited in the GenBank/EMBL/DDBJ database under the accession numbers OQ777717 and CP053084, respectively.
3 Results and discussion
3.1 Isolation of deep-sea bacteria in the Northwest Pacific Ocean
During a microbial isolation campaign from deep-sea environments, a total of 243 pure bacterial cultures (115 from 1/5MA and 127 from 1/10R2A) were successfully isolated from water samples collected in the Northwest Pacific Ocean in July 2011, as detailed in Supplementary Table S1. The quantity and taxonomic distribution of the isolates derived from each water sample are depicted in Figure 1 and Supplementary Tables S2–S5. Phylogenetic analysis based on 16S rRNA gene sequences revealed that these deep-sea bacterial isolates predominantly belonged to the phyla Pseudomonadota (formerly Proteobacteria, 223 strains, 91.8%), Bacteroidota (10 strains, 4.1%), and Actinomycetota (10 strains. 4.1%). The substantial prevalence of the phylum Pseudomonadota along with the frequent occurrence of the phyla Bacteroidota and Actinomycetota in the majority of deep-sea bacterial cultures, aligns with findings from previous studies (Kai et al., 2017; Sanz-Sáez et al., 2023). However, the distribution of cultured strains within the phylum Pseudomonadota, classified by class, and the prevalence of the most abundant species varied significantly with sampling depths. Isolates assigned to Alphaproteobacteria were dominant at depths of 1000 and 2000 m, Gammaproteobacteria at 3000 m, and Betaproteobacteria at 4000 m. Specifically, of the 82 and 64 isolates obtained from water samples at 1000 and 2000 m, respectively, the genus Sulfitobacter from the order Rhodobacterales of Alphaproteobacteria was the most abundant. From the 3000 m depth sample, the genus Alkanindiges within the order Pseudomonadales of Gammaproteobacteria was predominant. Notably, 42.9% of the isolates (33 out of 77 strains) cultured from the 4000 m sample were classified within the order Burkholderiales of the class Betaproteobacteria. The majority of these Burkholderiales isolates exhibited a close phylogenetic relationship to L. thiooxidans CS-K2T, showing more than 99% 16S rRNA gene sequence identity. The notable presence of Limnobacter spp. in several 16S rRNA gene studies from deep-sea ecosystems (Eloe et al., 2011b; Khandeparker et al., 2014), despite their scarcity in culture, coupled with the finding that the predominant bacterial isolates from a depth of 4000 m in this study were members of the genus Limnobacter, has led to further taxonomic and genomic investigations of a representative strain of the Limnobacter isolates.
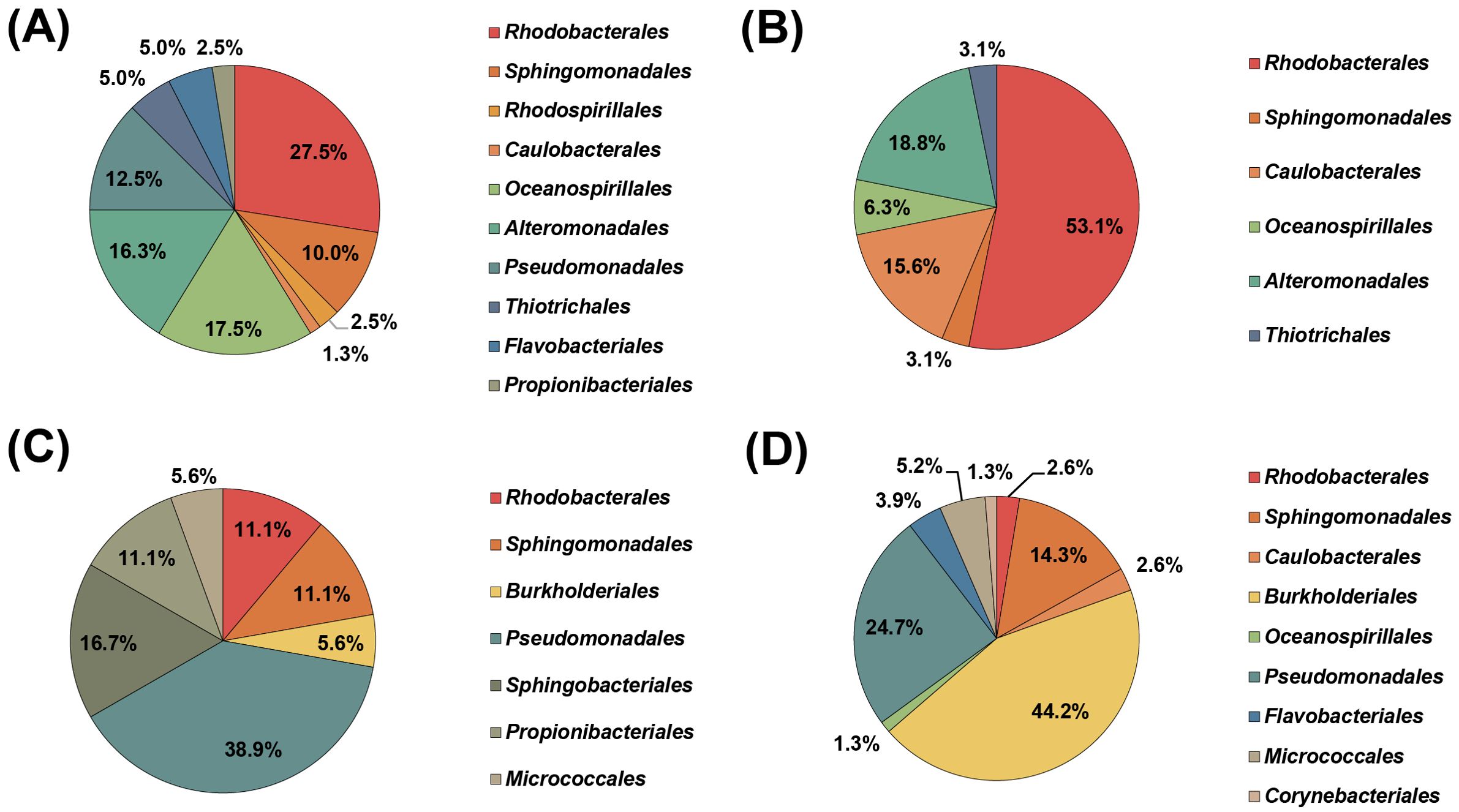
Figure 1. Order-level taxonomic distribution of bacterial isolates cultured from various depths at station S1 in the Northwest Pacific Ocean. Each pie chart represents results from different depths: 1000 m (A), 2000 m (B), 3000 m (C), and 4000 m (D).
3.2 Molecular phylogeny of SAORIC-580T, a representative of deep-sea Limnobacter
Among the 33 deep-sea Limnobacter strains that exhibited over 99% 16S rRNA gene sequence identity, a representative strain, designated SAORIC-580T, was selected for genomic and taxonomic analyses. The 16S rRNA gene sequence (1472 bp) analysis of strain SAORIC-580T showed that the strain is most closely related to L. thiooxidans CS-K2T (99.9% sequence identity), followed by L. alexandrii LZ-4T (99.2%), L. litoralis KP1-19T (97.7%), and L. humi UCM-39T (97.6%). In the phylogenetic tree constructed from the 16S rRNA gene sequences (Figure 2), strain SAORIC-580T formed a robust cluster with the type strains of the genus Limnobacter, supported by 99–100% bootstrap values, suggesting that the strain is a member of this genus.
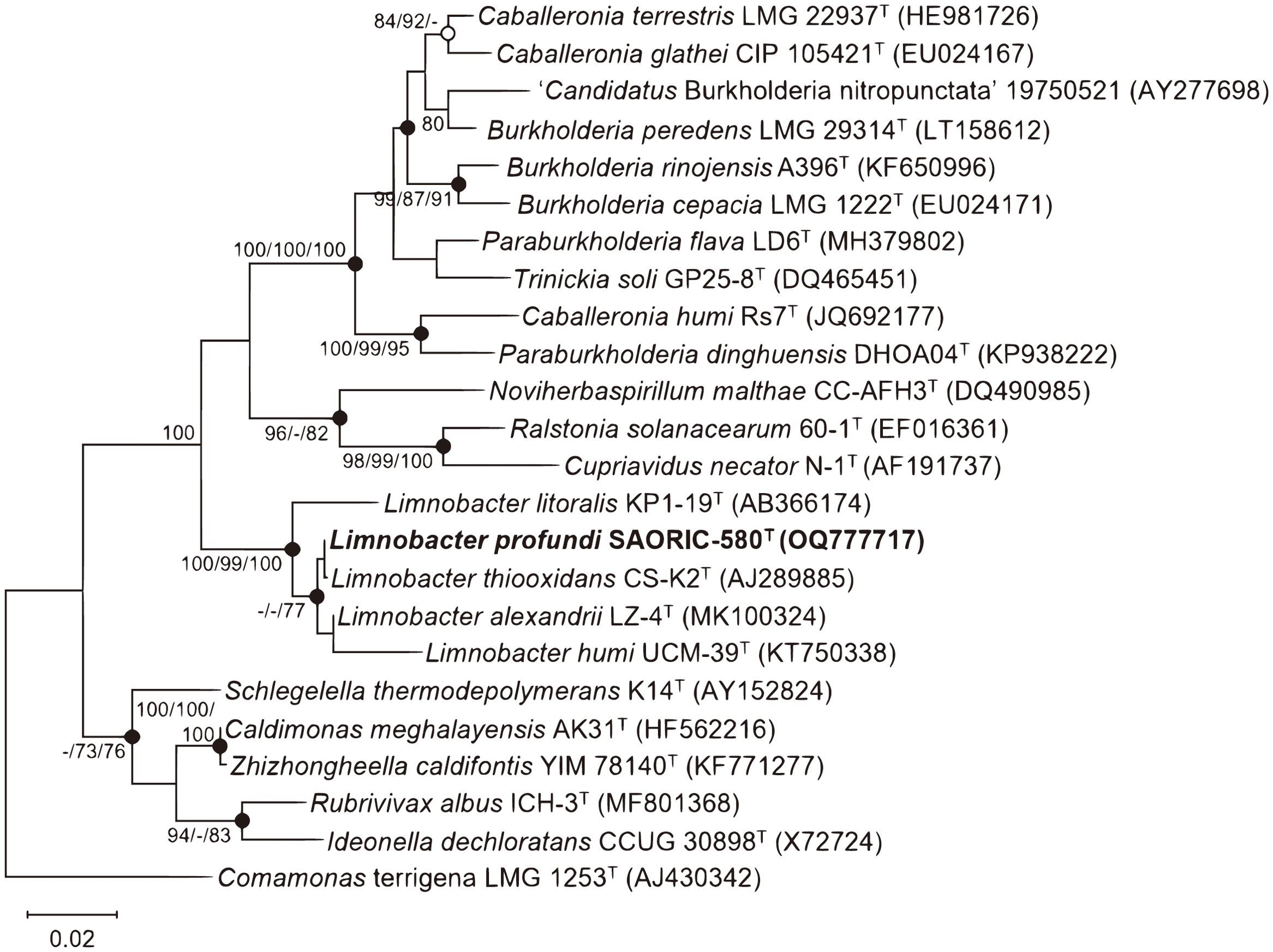
Figure 2. Maximum-likelihood phylogenetic tree based on 16S rRNA gene sequences showing the position of strain SAORIC-580T. Numbers at nodes represent bootstrap percentages (above 70%) obtained from maximum-likelihood (ML), neighbor-joining (NJ), and minimum evolution (ME) methods, respectively (ML/NJ/ME; −, less than 70%). Filled circles indicate nodes recovered by all treeing methods; open circles indicate nodes recovered by any two of the three methods. Comamonas terrigena LMG 1253T (AJ430342) was used as an outgroup. Bar, 0.02 substitutions per nucleotide position.
The whole genome sequence of strain SAORIC-580T was obtained to analyze genome-based molecular phylogenetic relationships. The SAORIC-580T genome shared 79.4% and 85.5% average nucleotide identity (ANI) with L. thiooxidans DSM 13612T and L. alexandrii LZ-4T genomes, respectively, and 21.8% and 29.5% digital DNA-DNA hybridization (dDDH) values with the two genomes, respectively (Supplementary Table S6). The ANI and dDDH values between SAORIC-580T and other Limnobacter genomes are well below the thresholds proposed for bacterial species demarcation, which are 95–96% for ANI and 70% for dDDH (Richter and Rossello-Mora, 2009; Chun et al., 2018). In the phylogenomic tree of Burkholderiales genomes (Figure 3), strain SAORIC-580T was clustered with the other Limnobacter genomes, suggesting that this novel strain represents a new species of the genus Limnobacter.
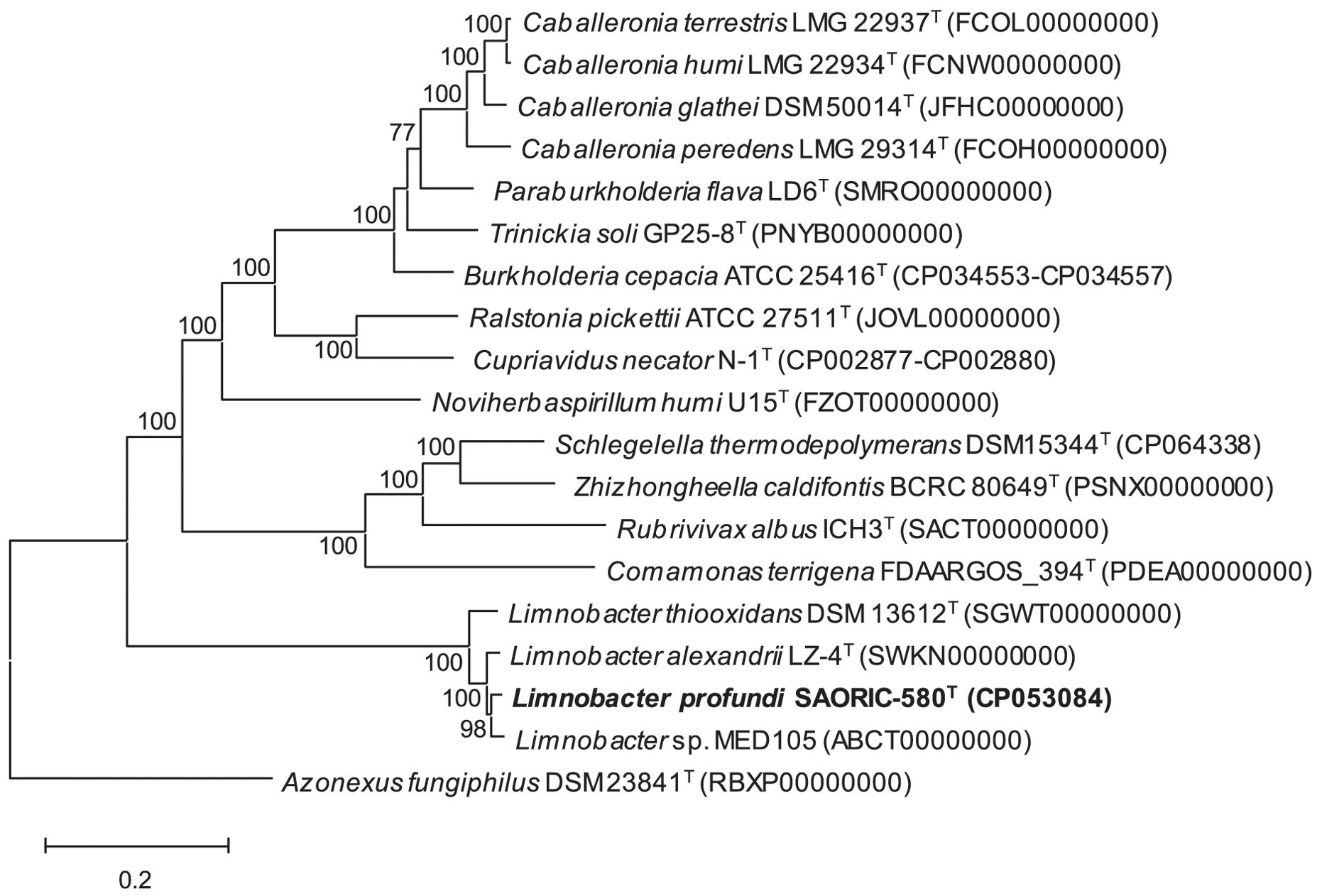
Figure 3. Phylogenomic tree generated using RAxML based on whole-genome sequences showing the position of strain SAORIC-580T. Values at nodes represent bootstrap support (above 70%) by RAxML. Azonexus fungiphilus DSM 23841T (RBXP00000000) was used as an outgroup. Bar, 0.2 substitutions per amino acid position.
3.3 Genome characteristics and comparative genome analysis of SAORIC-580T
The complete whole-genome sequence of strain SAORIC-580T, comprising a single circular chromosome of 3,297,222 bp, was obtained (Supplementary Figure S1). The genome completeness of the strain was estimated to be 99.8%, with 0.02% contamination. The SAORIC-580T genome encoded 3,077 protein-coding genes, 40 tRNA genes, and 6 rRNA genes (two rRNA operons) (Supplementary Table S6). Two copies of the 16S rRNA gene sequences (1,525 bp) on the genome exhibited 100% identity to each other and matched 100% with the Sanger sequencing counterpart amplified by PCR. The DNA G+C content of the SAORIC-580T genome was 52.5%, similar to those of other Limnobacter genomes (Supplementary Table S6). For genomic comparison within the genus Limnobacter, three strains were selected: L. thiooxidans DSM 13612T from freshwater lake sediment, L. alexandrii LZ-4T from the phycosphere of an alga, and Limnobacter sp. MED105 from surface seawater. The proportions of shared CDSs to total ORFs in each genome (0.72−0.76) and hypothetical proteins to total ORFs (0.43−0.45) were very similar across the four genomes (Supplementary Table S6). The major COG categories included general function prediction only (10.1%), signal transduction mechanisms (7.0%), cell wall/membrane/envelope biogenesis (6.9%), transcription (6.8%), energy production and conversion (6.7%), and translation (6.7%) (Supplementary Table S7). These results revealed no significant structural differences among the four genomes. Therefore, we examined the unique gene set and core gene set of the genomes using Get_HOMOLOGOUS, which resulted in a Venn diagram (Supplementary Figure S2). This analysis showed that SAORIC-580T shared 2329 common genes with the other Limnobacter genomes, but also possessed 199 unique genes. This suggests that SAORIC-580T not only has substantial gene content overlap with other Limnobacter strains but also harbors a specific set of genes not present in the other strains.
Given that genomic islands (GIs) can enhance bacterial adaptation to specific environments (Juhas et al., 2009), we investigated the presence of GIs in the SAORIC-580T genome. Three GIs were identified in the SAORIC-580T genome using IslandViewer4, with lengths of 5−36, 1599−1632, and 1836−1853 kb. Genetic maps of these GIs, designated GI1, GI2, and GI3, are illustrated in Supplementary Figure S3. Transposases or integrases, hallmark genes of mobile genetic elements (MGEs), were found in the GIs. MGEs play a role in enhancing genetic plasticity of microbes in stressful or extreme environments (Leduc and Ferroni, 1994; Nelson et al., 2011). The enrichment of transposases and integrases in deep-sea bacterial genomes and metagenomes is well documented (DeLong et al., 2006; Martin-Cuadrado et al., 2007; Lauro et al., 2008; Konstantinidis et al., 2009). Several transcriptional regulators, including the XRE family transcriptional regulators, terR, merR, araC, and tetR, were also found in the GIs. These regulators are known to be involved in stress response, oxidative stress resistance, multidrug resistance, and degradation of aromatics (Molina-Henares et al., 2006; Issa et al., 2018). Additionally, TonB-dependent receptors, DEAD/DEAH box helicase, and cytochrome P450 were found in GI1, GI2, and GI3, respectively. These proteins are known to be up-regulated under low temperature in marine bacteria (Ting et al., 2010) and play crucial roles in substrate transport and stress adaptation (Moeck and Coulton, 1998; Redder et al., 2015). Cytochromes P450 in several deep-sea bacteria exhibit features associated with adaption to high hydrostatic pressure (Davydov et al., 2013). Methyl-accepting chemotaxis sensory transducers and sel1 repeat proteins in GI2 may respond to environmental signals such as nutrient influxes and temperature or pressure fluctuations (Mittl and Schneider-Brachert, 2007). Genes related to heavy metal resistance, such as Co/Zn/Cd efflux systems, mercury transporters and reductases, copper resistance-related lipoproteins, and heavy metal response regulators, were also found in the GIs, consistent with the enrichment of heavy metal-resistant bacteria in the deep sea (Sayed et al., 2014; Gillard et al., 2019).
The various and simplified metabolic pathways predicted from BlastKOALA and KofamKOALA are summarized in Figure 4. The SAORIC-580T and other Limnobacter genomes were all predicted to possess complete pathways for central carbon metabolism, including glycolysis, the tricarboxylic acid cycle (TCA), and the non-oxidative branch of the pentose phosphate pathway. Genes related to the SOX system for thiosulfate oxidation (soxABCDXYZ) and polyhydroxybutyrate (PHB) biosynthesis (phaABC) were identified in all four Limnobacter genomes (Supplementary Table S8). Although phenotypic tests were not conducted, the presence of these complete pathways suggests that SAORIC-580T has the potential to utilize thiosulfate and produce PHB, both of which are characteristic features of the genus Limnobacter (Nguyen and Kim, 2017). Furthermore, genes for assimilatory sulfate reduction, flagella assembly, and ABC transporters for tungstate, nitrate/nitrite/cyanate, molybdate, iron (III), phosphate, phosphonate, glutamate/aspartate, and branched-chain amino acids were encoded in all four Limnobacter genomes. However, the presence or absence of certain genes differed among the Limnobacter genomes. Notably, the genomes of SAORIC-580T, LZ-4T, and MED105, derived from marine environments, exhibited differences from the DSM 13612T genome, derived from lake sediment, in terms of the presence of the phenol degradation pathway (Figure 4). Phenol degradation by marine Limnobacter strains has been previously reported (Vedler et al., 2013). Additionally, the metabolic potential for the degradation of aromatic compounds has been well documented in deep-sea metagenomes (Zhang et al., 2018) and deep-sea bacterial genomes (Song et al., 2019a; Zhou et al., 2020). Conversely, only the DSM 13612T genome contained cbb3-type cytochrome oxidase, which plays an important role in various cellular processes under low oxygen conditions (Arai, 2011; Hamada et al., 2014). The presence of this enzyme in strain DSM 13612T may be related to the oxygen-deficient conditions of the lake sediment where the strain was isolated (Spring et al., 2001).
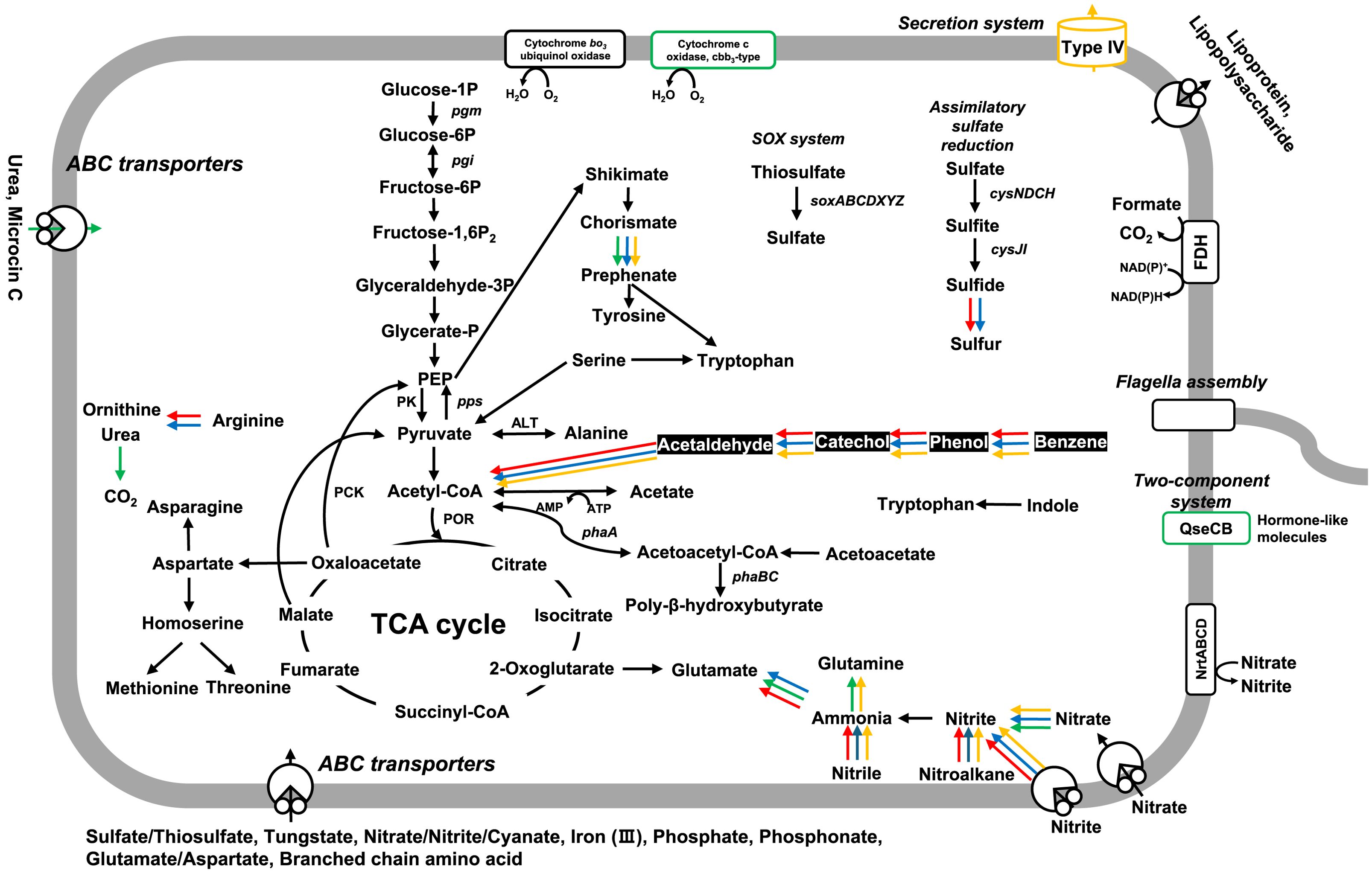
Figure 4. Schematic overview of predicted metabolic pathways in deep-sea Limnobacter genomes. The presence and absence of genes were predicted based on KEGG annotations. The color of the arrows indicates the presence of the corresponding gene in each Limnobacter genome as follows: Black arrow, all genomes; Red, SAORIC-580T; Green, DSM 13612T; Blue, LZ-4T; Yellow, MED105. The complete pathway for phenol degradation in marine Limnobacter genomes is highlighted with white font on a black background.
We further compared the SAORIC-580T genome to other genomes to gain insights into the genomic features of deep-sea bacteria. Annotation comparisons based on the Pfam database revealed that numerous protein families (Pfam entries; hereafter referred to as proteins) are either unique to or more abundantly represented in the SAORIC-580T genome. Specifically, the unique proteins and those with at least twice the number of copies in the SAORIC-580T genome compared to other genomes are listed in Supplementary Table S9. To investigate the unique genes of Limnobacter genomes from different environments, we categorized the four analyzed genomes into two groups based on their habitats: freshwater and marine (deep-sea, algae, surface seawater). The unique genes for each habitat are presented in Table 1. In the deep-sea isolate strain SAORIC-580T, five genes encoding haemolysin-type calcium binding protein-related domains and two genes encoding the C-terminal domain of ChrB were uniquely identified. Additionally, SAORIC-580T and other marine isolates displayed a distinctive set of genes, including glutathione S-transferase, sodium neurotransmitter symporter family proteins, and methane/phenol/alkene hydroxylase related to phenol degradation pathway. The SAORIC-580T genome showed an enrichment of PIN (PilT N-terminus) domain and tetratricopeptide repeat-like domain proteins (Supplementary Table S9), similar to another deep-sea bacterium (Song et al., 2019b). Tetratricopeptide repeat proteins are known to be up-regulated at low temperatures (Ting et al., 2010), suggesting their role in adapting to cold deep-sea conditions. The genome also contained an abundance of chromate transporter, chromate resistance protein, and Fe/Mn-superoxide dismutases (Fe/Mn-SOD), which aligns with the higher prevalence of heavy metal resistance genes in deep-sea bacteria (Simonato et al., 2006; Ivars-Martinez et al., 2008; Lauro et al., 2008). Chromium is commonly found in deep-sea environments (Goring-Harford et al., 2018), which may explain the enrichment of chromate-related genes. Heavy metals induce oxidative stress, and Fe/Mn-SOD helps mitigate this by converting superoxide to less harmful molecules (Li et al., 2019b). The genome also exhibited an enrichment of DnaB-like helicase, crucial for DNA replication, which could aid in coping with the pressures and low temperatures of deep-sea habitats where DNA replication is challenged (Campanaro et al., 2005; Goring-Harford et al., 2018). Additionally, polyphosphate kinase 2 (PPK2), essential for polyphosphate synthesis, was abundant, suggesting a role in adaptation to extreme environments (Seufferheld et al., 2008). Collectively, the SAORIC-580T genome harbors unique or enriched genes, suggesting its adaptation to the deep-sea environment and its potential involvement in deep-sea biogeochemical processes. Additionally, it is noteworthy that the adaptation of strain SAORIC-580T to the deep-sea may also be related to transcription, post translational modifications, and possibly proteins of unknown function.
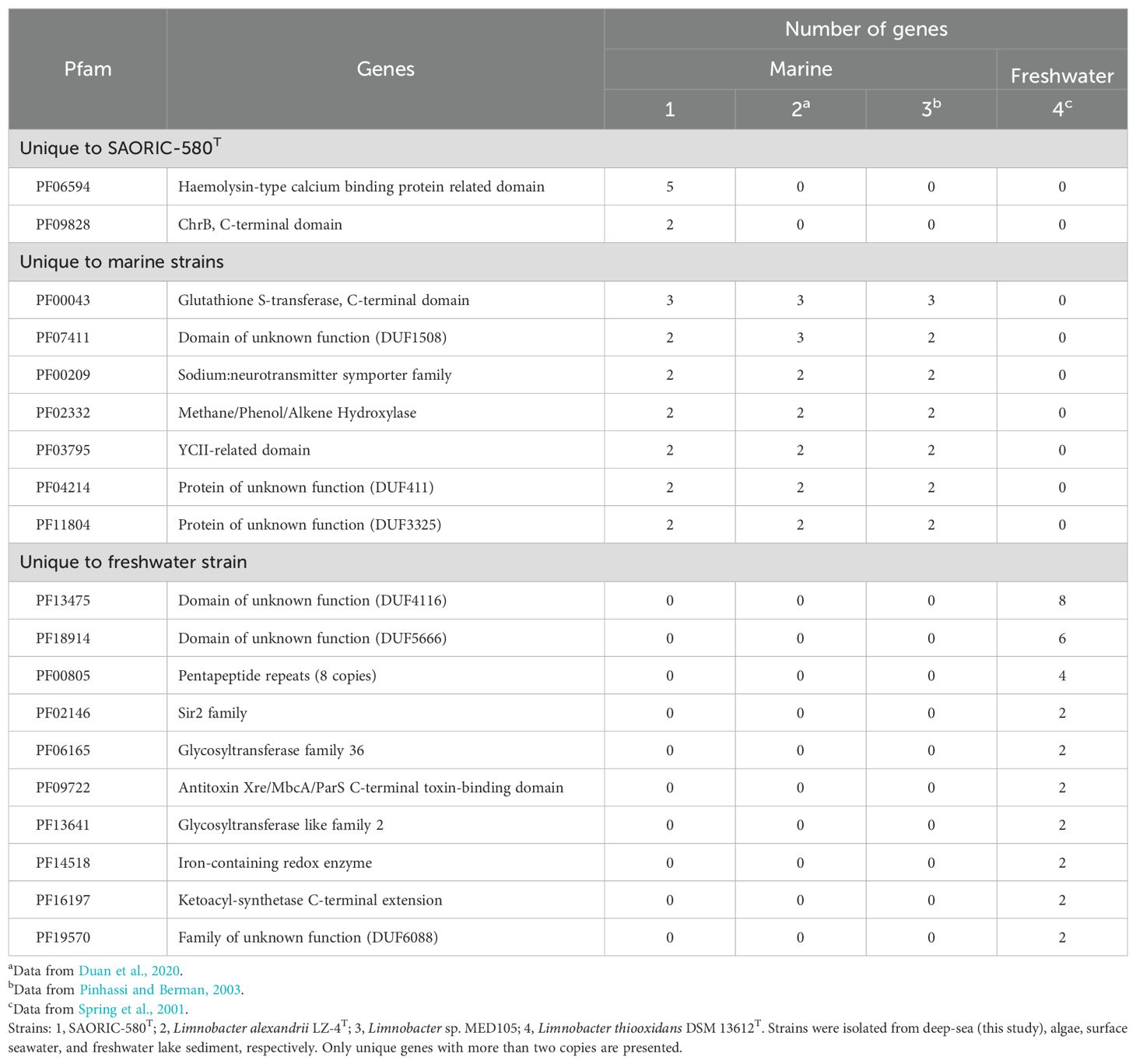
Table 1. Unique proteins in the genomes of bacteria isolated from marine or freshwater environments.
Although present in minority, fragment recruitment analysis suggested that bacterial genomes retrieved from the Northwestern Pacific Ocean (Supplementary Table S10) matched metagenome fragments obtained from various depths of the four trenches in the Northwest Pacific Ocean (Figure 5). Most deep-sea genomes corresponded more closely with deep-sea metagenomes. The abundance of Colwellia and Rubritalea genomes in the Kuril Trench was comparable to other metagenomic data derived from the Kuril Trench (Gorrasi et al., 2023a, b). Additionally, the presence of Sulfitobacter genomes in the Mariana Trench corresponded to findings from a culture-based experiment (Zhao et al., 2020). Notably, the SAORIC-580T genome was relatively enriched at depths of 10,899 m and 3,507 m in the Mariana Trench and Ogasawara Trench, respectively, compared to surface counterparts and other deep-sea bacterial genomes. The Mariana Trench is well known as the deepest of the world’s deep-sea trenches. The abundance of Limnobacter members in the Mariana Trench has also been found in other metagenomic data (Dang et al., 2024). These results suggest that strain SAORIC-580T thrives in the deep-sea environment of the Northwest Pacific Ocean and represents a member of deep-sea Burkholderiales.
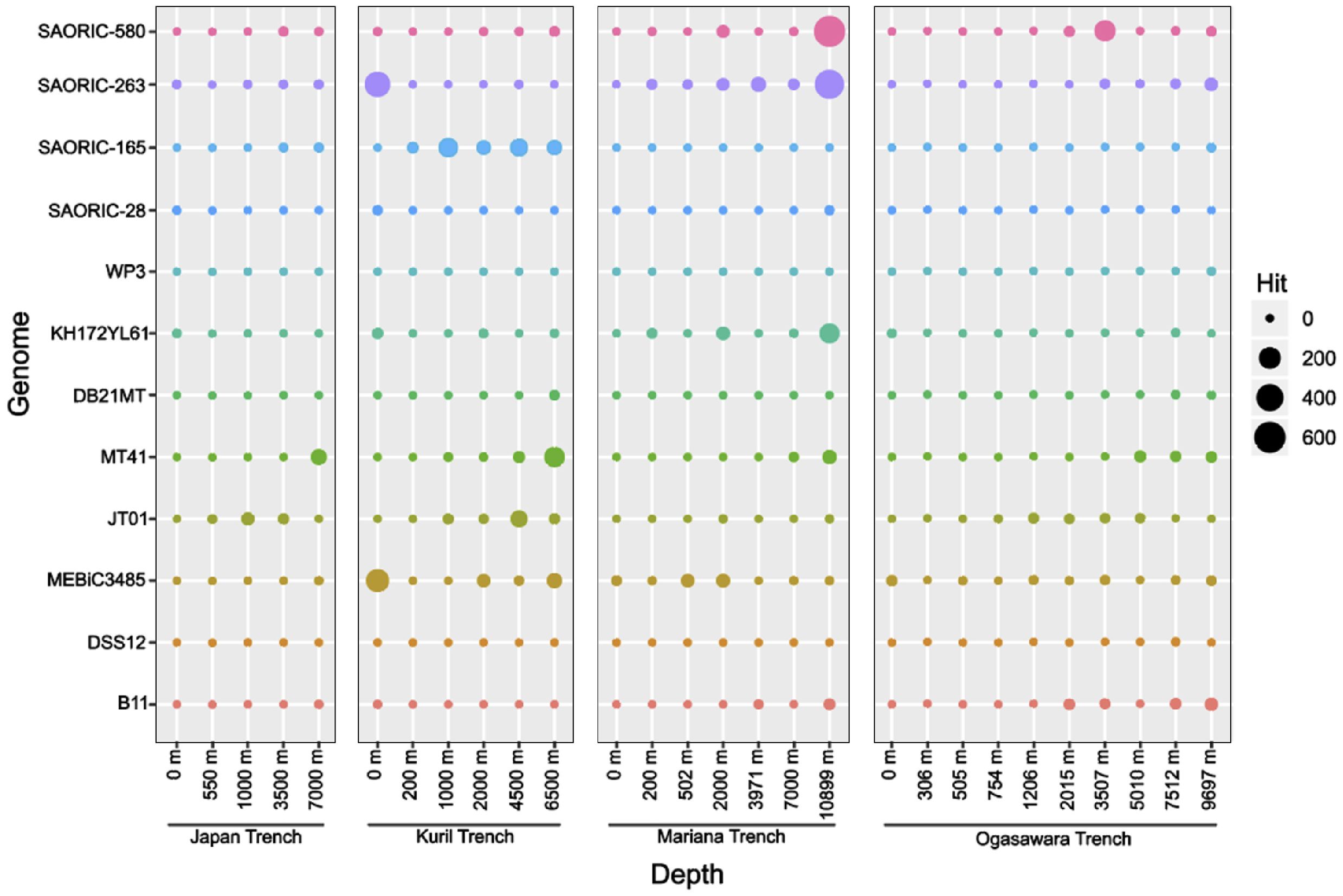
Figure 5. Relative abundance of deep-sea bacterial genomes analyzed by recruitment of metagenomic reads from four deep ocean trenches in the Pacific Ocean. Bubble plots show the hits of metagenomic reads from four trenches that matched genomic sequences of the deep-sea bacterial genomes. Full name of the bacterial strains: Limnobacter profundi sp. nov. SAORIC-580T, Sulfitobacter sp. SAORIC-263, Rubritalea sp. SAORIC-165, Rubrivirga sp. SAORIC-28, Shewanella sp. WP3, Psychrobacter sp. KH172YL61, Shewanella sp. DB21MT, Colwellia sp. MT41, Moritella sp. JT01, Pseudoalteromonas sp. MEBiC 03485, Shewanella sp. DSS12, and Altererythrobacter sp. B11. Bubble sizes indicate the number of metagenomic reads recruited to the genomes (y-axis).
Conclusively, the comparative genomic analysis of strain SAORIC-580T revealed its adaptation to the deep-sea environment, highlighted by the presence of diverse metabolic pathways and specific protein families associated with cold adaptation, and heavy metal resistance, distinguishing it from other Limnobacter strains. The genome contained unique or differentially abundant genes involved in various functions including signal transduction and mobile genetic elements, when compared to other Limnobacter genomes. Many of these genes were relevant to survival strategies that may enhance the ecological fitness in the deep-sea, suggesting that strain SAORIC-580T is well adapted to the deep-sea environment. To the best of our knowledge, the SAORIC-580T genome represents the first deep-sea genome to be reported for the order Burkholderiales and provides insight into the adaptation strategies used by its members.
3.4 Phenotypic characterization and taxonomic conclusion
Transmission electron microscopy (TEM) images showed that cells of strain SAORIC-580T are rod-shaped, measuring 0.4–0.5 × 1.0–2.1 µm, and have a polar flagellum (Supplementary Figure S4). The morphological, physiological, and biochemical characteristics of strain SAORIC-580T are listed in Table 2, Supplementary Table S11, and in the species description. Strain SAORIC-580T exhibited phenotypic differences from other Limnobacter species, such as growth range for temperature and NaCl concentration, catalase activity, several enzyme activities, and utilization of various carbon compounds. Phenol degradation by strain SAORIC-580T was tested using phenol added in concentrations ranging from 0 to 4.5 mM in 0.5 mM intervals (Figure 6). Strain SAORIC-580T could utilize phenol as a sole carbon source at concentrations up to 3.0–3.5 mM, which aligns with the phenol degradation pathway found in its genome sequence. Phenol degradation by marine Limnobacter strains and other marine bacteria has also been reported in other studies (Vedler et al., 2013; Li et al., 2019). Marine pollution from aromatic compounds poses serious threats to aquatic life and water quality, making it crucial to understand biodegradation processes by microorganisms (Anku et al., 2017). The ability of strain SAORIC-580T to degrade phenol suggests that the strain could play an important role in phenol degradation in marine environments.
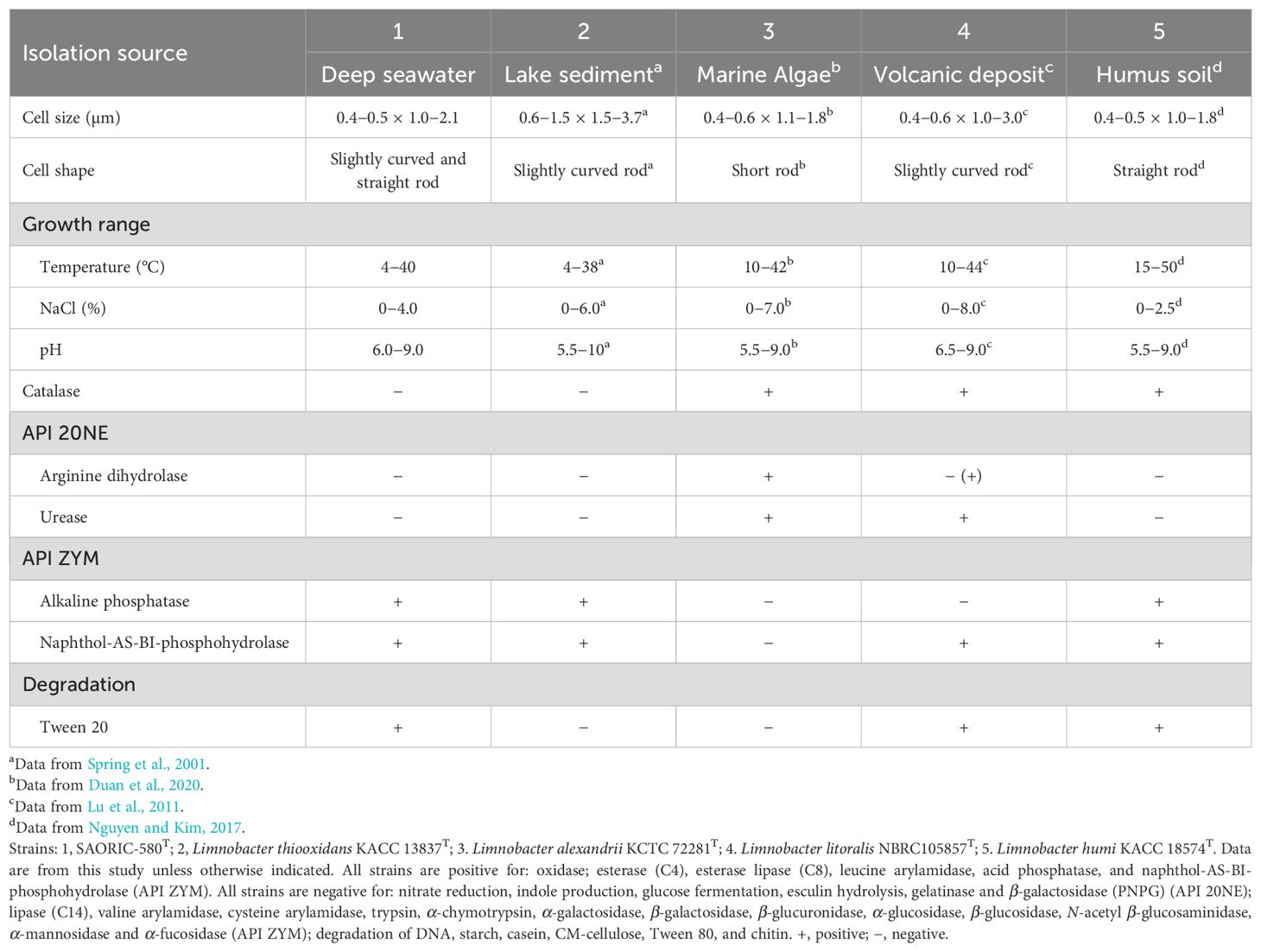
Table 2. Phenotypic characteristics of strain SAORIC-580T and four Limnobacter species.
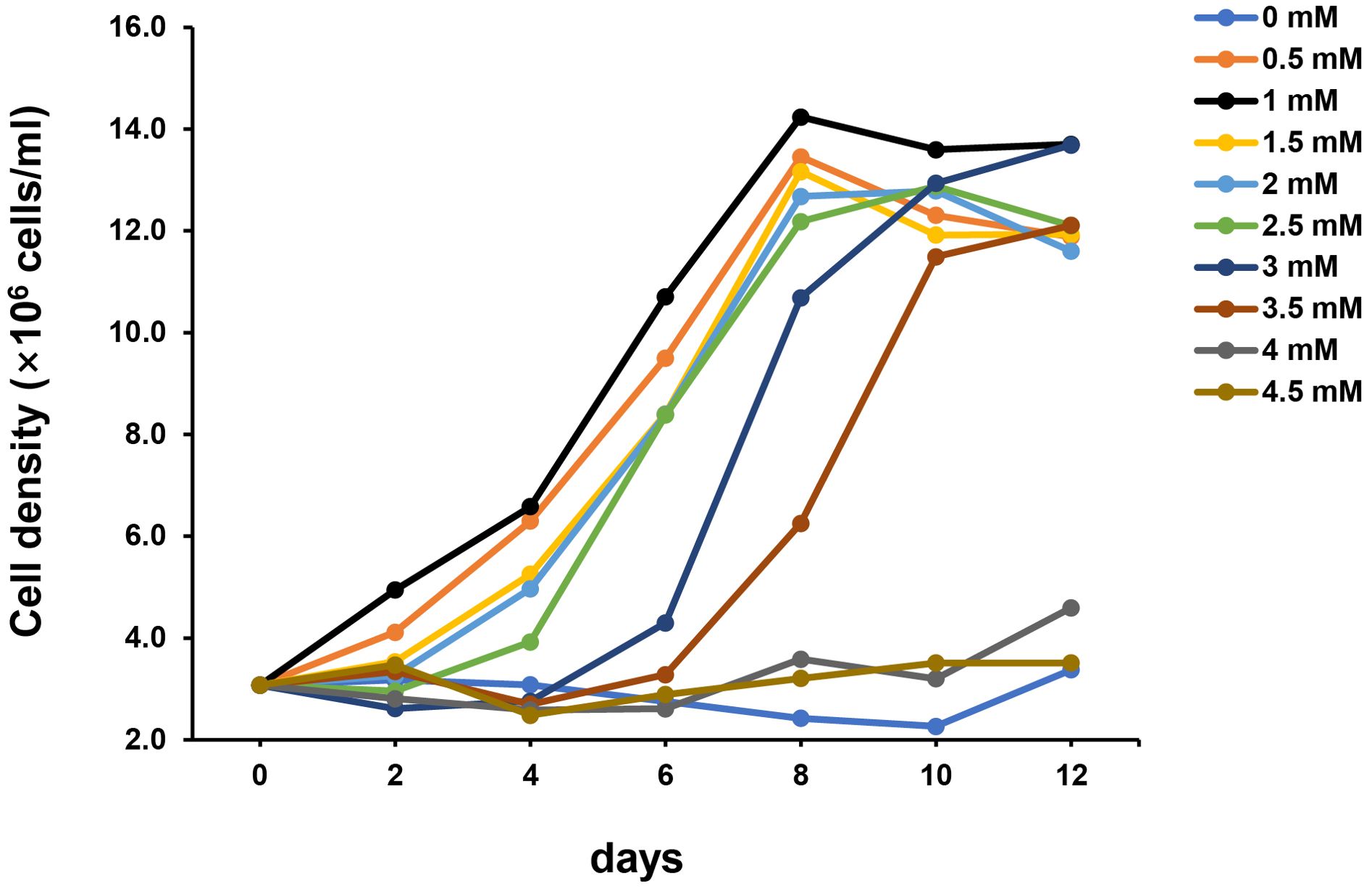
Figure 6. Growth of strain SAORIC-580T using phenol as a sole carbon source at different concentrations. Phenol as a sole carbon source was added in concentrations ranging from 0 to 4.5 mM in 0.5 mM increments to basal medium supplemented with 10 µM NH4Cl. The growth was measured at 30°C.
Fatty acid profiles of SAORIC-580T and closely related species are listed in Table 3. The major fatty acids (>10%) of strain SAORIC-580T were C16:0 (30.6%), summed feature 3 (C16:1 ω6c and/or C16:1 ω7c; 28.5%), and summed feature 8 (C18:1 ω7c and/or C18:1 ω6c; 17.3%). C16:0 and summed feature 3 were also major fatty acids of other Limnobacter species. However, strain SAORIC-580T differed from other Limnobacter species in the proportion of several fatty acids such as C14: 0, C17: 0 cyclo, and summed feature 8. The respiratory quinones detected in strain SAORIC-580T was Q-8. The major polar lipids were phosphatidylethanolamine (PE), phosphatidylglycerol (PG), and diphosphatidylglycerol (DPG), similar to the type species of the genus Limnobacter, L. thiooxidans KACC 13837T (Supplementary Figure S4).
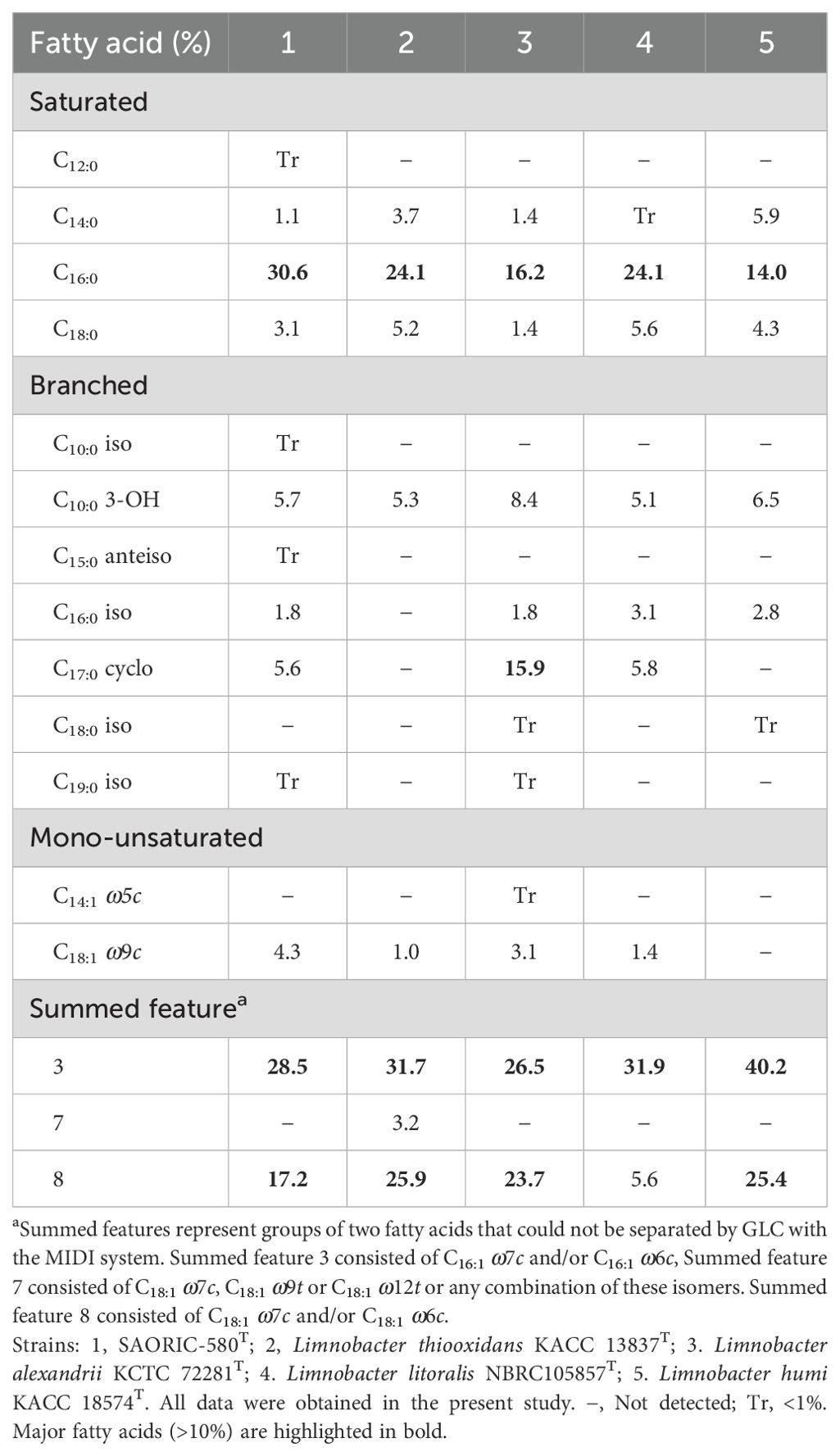
Table 3. Cellular fatty acids composition of strain SAORIC-580T and four Limnobacter species.
Chemotaxonomic characteristics and phylogenetic inference based on 16S rRNA gene and whole-genome sequences indicated that strain SAORIC-580T belongs to the genus Limnobacter. The low ANI and dDDH values between strain SAORIC-580T and the type strains of Limnobacter species suggest that strain SAORIC-580T should be classified as a novel species within the genus. Phenotypic analyses also showed that strain SAORIC-580T differs from other Limnobacter species in various characteristics. Therefore, the name Limnobacter profundi is proposed for this novel species, with SAORIC-580T as the type strain.
3.5 Description of Limnobacter profundi sp. nov.
Limnobacter profundi (pro.fun’di L. gen. n. profundi of/from the depths of the sea, referring to the environment where the type strain was isolated.)
Cells are Gram-stain-negative, motile, strictly aerobic, rod-shaped (0.4–0.5 µm in diameter and 1.0–2.1 µm in length). Colonies on R2A agar are circular, smooth with entire margins, and beige in color after 3 days of incubation at 30°C. Growth occurs in the presence of 0–4% (w/v) NaCl (optimum 0% NaCl), at pH 6.0–9.0 (optimum pH 7.0–8.0), and at 4–40°C (optimum 30°C). Oxidase-positive and catalase-negative. In the API 20NE strip, negative for nitrate reduction, indole production, glucose fermentation, arginine dihydrolase, urease, aesculin hydrolysis, gelatinase, and β-galactosidase (PNPG). In the API ZYM strip, positive for alkaline phosphatase, esterase (C4), esterase lipase (C8), leucine arylamidase, acid phosphatase, and naphthol-AS-BI-phosphohydrolase but negative for lipase (C14), valine arylamidase, cystine arylamidase, trypsin, α-chymotrypsin, α-galactosidase, β-galactosidase, β-glucuronidase, α-glucosidase, β-glucosidase, N-acetyl-β-glucosaminidase, α-mannosidase, and α-fucosidase. All macromolecules (casein, chitin, DNA, starch, CM-cellulose, and Tween 80) are not hydrolyzed except for Tween 20. In a carbon source oxidation test (GEN III microplate; Biolog), positive for l-alanine, l-aspartic acid, l-glutamic acid, glucuronamide, methyl pyruvate, l-lactic acid, α-ketoglutaric acid, d-malic acid, l-malic acid, bromosuccinic acid, Tween 40, β-hydroxy- d, l-butyric acid, propionic acid, and acetic acid but negative for dextrin, d-maltose, d-trehalose, d-cellobiose, gentiobiose, sucrose, d-turanose, stachyose, d-raffinose, α- d-lactose, d-melibiose, β-methyl- d-glucoside, d-salicin, N-acetyl- d-glucosamine, N-acetyl-β- d-mannosamine, N-acetyl- d-galactosamine, N-acetylneuraminic acid, α- d-glucose, d-mannose, d-fructose, d-galactose, 3-methyl glucose, d-fucose, l-fucose, l-rhamnose, inosine, d-sorbitol, d-arabitol, myo-inositol, glycerol, d-glucose-6-phosphate, d-fructose-6-phosphate, d-aspartic acid, d-serine, gelatin, glycyl- l-proline, l-arginine, l-histidine, l-pyroglutamic acid, l-serine, pectin, d-galacturonic acid, l-galactonic acid lactone, d-gluconic acid, d-glucuronic acid, mucic acid, quinic acid, d-saccharic acid, p-hydroxyphenlyacetic acid, d-lactic acid methyl ester, citric acid, γ-amino-butyric acid, α-hydroxybutyric acid, α-ketobutyric acid, acetoacetic acid, and formic acid. The major fatty acids are C16:0, summed feature 3 (C16:1 ω7c and/or C16:1 ω6c), and summed feature 8 (C18:1 ω7c and/or C18:1 ω6c). The polar lipids are phosphatidylethanolamine, phosphatidylglycerol, diphosphatidylglycerol, three unidentified phospholipids, two unidentified aminophospholipids, and four unidentified lipids. The major respiratory quinone is Q-8.
The type strain, SAORIC-580T (= KACC 21400T = NBRC 114111T), was isolated from deep-seawater (4000 m) in the northwestern Pacific Ocean. The length of the complete genome sequence of the type strain is 3.3 Mbp with a DNA G+C content of 52.5%. GenBank accession numbers of the 16S rRNA gene sequence and the complete whole-genome sequence of the type strain are OQ777717 and CP053084, respectively.
4 Conclusion
In this study, a significant proportion of cultured bacteria from a depth of 4000 m in the Northwest Pacific Ocean were classified into the genus Limnobacter of the order Burkholderiales, with the representative strain SAORIC-580T further characterized. This strain, identified as a novel species member of the genus Limnobacter, demonstrates a range of genomic adaptations to the extreme conditions of the deep sea, including unique or differentially abundant genes associated with cold adaptation, heavy metal resistance, and signal transduction. These genomic features suggest specialized survival strategies that enhance ecological fitness in the deep-sea environment. The SAORIC-580T genome is the first deep-sea genome reported for the order Burkholderiales, providing an insight into the adaptation mechanisms of these bacteria in the deep-sea biosphere.
Additionally, taxonomic analyses confirmed the distinctiveness of strain SAORIC-580T from other Limnobacter species, revealing differences in temperature and NaCl growth ranges, enzyme activities, and carbon compound utilization. The ability of SAORIC-580T to degrade phenol, along with chemotaxonomic properties, further supports its classification as a novel species within the genus Limnobacter. Based on these findings, we propose the name Limnobacter profundi for this new species, with SAORIC-580T as the type strain. This study not only enriches our understanding of the genetic and physiological diversity of deep-sea Burkholderiales but also underscores the specific adaptability of the Limnobacter isolates to extreme marine environments.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, OQ777717 https://www.ncbi.nlm.nih.gov/genbank/, CP053084.
Author contributions
MK: Data curation, Formal analysis, Methodology, Writing – original draft. JS: Data curation, Formal analysis, Investigation, Methodology, Writing – original draft. SS: Investigation, Methodology, Writing – review & editing. KK: Conceptualization, Formal analysis, Investigation, Project administration, Writing – review & editing. IK: Conceptualization, Investigation, Resources, Software, Supervision, Writing – review & editing. J-CC: Conceptualization, Funding acquisition, Investigation, Project administration, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by the High Seas Bioresources Program of the Korea Institute of Marine Science & Technology Promotion (KIMST), funded by the Ministry of Oceans and Fisheries (KIMST-20210646), the Mid-Career Research Program (NRF-2022R1A2C3008502), and Science Research Center Program (NRF-2018R1A5A1025077) through the National Research Foundation (NRF) funded by the Ministry of Sciences and ICT.
Acknowledgments
We are grateful to the officer and crew of R/V Mirai (Japan Agency for Marine-Earth Science and Technology [JAMSTEC]) for their assistance and support in sample collection.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2024.1449548/full#supplementary-material
References
Aguirre-Von-Wobeser E., Rocha-Estrada J., Shapiro L. R., de la Torre M. (2018). Enrichment of Verrucomicrobia, Actinobacteria and Burkholderiales drives selection of bacterial community from soil by maize roots in a traditional milpa agroecosystem. PloS One 13, e0208852. doi: 10.1371/journal.pone.0208852
Anku W. W., Mamo M. A., Govender P. P. (2017). “Phenolic compounds in water: sources, reactivity, toxicity and treatment methods,” in Phenolic compounds-natural sources, importance and applications. Eds. Soto-Hernández M., Palma-Tenango M., García-Mateos R. (London, United Kingdom: IntecOpen), 419–443.
Arai H. (2011). Regulation and function of versatile aerobic and anaerobic respiratory Metabolism in Pseudomonas aeruginosa. Front. Microbiol. 2, 103. doi: 10.3389/fmicb.2011.00103
Aramaki T., Blanc-Mathieu R., Endo H., Ohkubo K., Kanehisa M., Goto S., et al. (2020). KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinform 36, 2251–2252. doi: 10.1093/bioinformatics/btz859
Bertelli C., Laird M. R., Williams K. P., Group S.F.U.R.C., Lau B. Y., Hoad G., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Campanaro S., Vezzi A., Vitulo N., Lauro F. M., D’angelo M., Simonato F., et al. (2005). Laterally transferred elements and high pressure adaptation in Photobacterium profundum strains. BMC Genom. 6, 1–15. doi: 10.1186/1471-2164-6-122
Chen Y., Feng X., He Y., Wang F. (2016). Genome analysis of a Limnobacter sp. identified in an anaerobic methane-consuming cell consortium. Front. Mar. Sci. 3, 257. doi: 10.3389/fmars.2016.00257
Chklovski A., Parks D. H., Woodcroft B. J., Tyson G. W. (2023). CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat. Methods 20, 1203–1212. doi: 10.1038/s41592-023-01940-w
Chun J., Oren A., Ventosa A., Christensen H., Arahal D. R., Da Costa M. S., et al. (2018). Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 68, 461–466. doi: 10.1099/ijsem.0.002516
Collins M. D., Jones D. (1981). A note on the separation of natural mixtures of bacterial ubiquinones using reverse-phase partition thin-layer chromatography and high performance liquid chromatography. J. Appl. Bacteriol. 51, 129–134. doi: 10.1111/j.1365-2672.1981.tb00916.x
Contreras-Moreira B., Vinuesa P. (2013). GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 79, 7696–7701. doi: 10.1128/AEM.02411-13
Dang Y. R., Cha Q. Q., Liu S. S., Wang S. Y., Li P. Y., Li C. Y., et al. (2024). Phytoplankton-derived polysaccharides and microbial peptidoglycans are key nutrients for deep-sea microbes in the Mariana Trench. Microbiome 12, 77. doi: 10.1186/s40168-024-01789-x
Davydov D. R., Sineva E. V., Davydova N. Y., Bartlett D. H., Halpert J. R. (2013). CYP261 enzymes from deep sea bacteria: a clue to conformational heterogeneity in cytochromes P450. Biotechnol. Appl. Biochem. 60, 30–40. doi: 10.1002/bab.2013.60.issue-1
De Ley J., Segers P., Kersters K., Mannheim W., Lievens A. (1986). Intra-and intergeneric similarities of the Bordetella ribosomal ribonucleic acid cistrons: proposal for a new family, Alcaligenaceae. Int. J. Syst. Evol. Microbiol. Int. J. Syst. Evol. Microbiol. 36, 405–414. doi: 10.1099/00207713-36-3-405
DeLong E. F., Preston C. M., Mincer T., Rich V., Hallam S. J., Frigaard N.-U., et al. (2006). Community genomics among stratified microbial assemblages in the ocean’s interior. Sci 311, 496–503. doi: 10.1126/science.1120250
Duan Y., Jiang Z., Wu Z., Sheng Z., Yang X., Sun J., et al. (2020). Limnobacter alexandrii sp. nov., a thiosulfate-oxidizing, heterotrophic and EPS-bearing Burkholderiaceae isolated from cultivable phycosphere microbiota of toxic Alexandrium catenella LZT09. Antonie Van Leeuwenhoek 113, 1689–1698. doi: 10.1007/s10482-020-01473-8
El-Gebali S., Mistry J., Bateman A., Eddy S. R., Luciani A., Potter S. C., et al. (2019). The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432. doi: 10.1093/nar/gky995
Eloe E. A., Malfatti F., Gutierrez J., Hardy K., Schmidt W. E., Pogliano K., et al. (2011a). Isolation and characterization of a psychropiezophilic alphaproteobacterium. Appl. Environ. Microbiol. 77, 8145–8153. doi: 10.1128/AEM.05204-11
Eloe E. A., Shulse C. N., Fadrosh D. W., Williamson S. J., Allen E. E., Bartlett D. H. (2011b). Compositional differences in particle-associated and free-living microbial assemblages from an extreme deep-ocean environment. Environ. Microbiol. Rep. 3, 449–458. doi: 10.1111/j.1758-2229.2010.00223.x
Felsenstein J. (1981). Evolutionary trees from gene frequencies and quantitative characters: finding maximum likelihood estimates. Evol 35, 1229–1242. doi: 10.2307/2408134
Felsenstein J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evol 39, 783–791. doi: 10.2307/2408678
Galperin M. Y., Makarova K. S., Wolf Y. I., Koonin E. V. (2015). Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–D269. doi: 10.1093/nar/gku1223
Garrity G., Bell J., Lilburn T. (2005). “Order I. Burkholderiales,” in Bergey’s Manual of Systematic Bacteriology. Eds. Brenner D. J., Krieg N. R., Staley J T., Garrity G. M. (Springer, New York), 575.
Gillard B., Chatzievangelou D., Thomsen L., Ullrich M. S. (2019). Heavy-metal-resistant microorganisms in deep-sea sediments disturbed by mining activity: An application toward the development of experimental in vitro systems. Front. Marr Sci. 6, 462. doi: 10.3389/fmars.2019.00462
Ginige M. P., Kaksonen A. H., Morris C., Shackelton M., Patterson B. M. (2013). Bacterial community and groundwater quality changes in an anaerobic aquifer during groundwater recharge with aerobic recycled water. FEMS Microbiol. Ecol. 85, 553–567. doi: 10.1111/1574-6941.12137
Goring-Harford H. J., Klar J., Pearce C. R., Connelly D. P., Achterberg E. P., James R. H. (2018). Behaviour of chromium isotopes in the eastern sub-tropical Atlantic Oxygen Minimum Zone. Geochim. Cosmochim. Acta 236, 41–59. doi: 10.1016/j.gca.2018.03.004
Gorrasi S., Brandt A., Pittino F., Franzetti A., Pasqualetti M., Muñoz-Palazon B., et al. (2023a). Uncovering the prokaryotic diversity of the bathyal waters above the kuril–kamchatka trench. J. Mar. Sci. Eng. 11, 2145. doi: 10.3390/jmse11112145
Gorrasi S., Franzetti A., Brandt A., Minzlaff U., Pasqualetti M., Fenice M. (2023b). Insights into the prokaryotic communities of the abyssal-hadal benthic-boundary layer of the Kuril Kamchatka Trench. Environ. Microbiome 18, 67. doi: 10.1186/s40793-023-00522-9
Hamada M., Toyofuku M., Miyano T., Nomura N. (2014). cbb3-type cytochrome c oxidases, aerobic respiratory enzymes, impact the anaerobic life of Pseudomonas aeruginosa PAO1. J. Bacteriol. 196, 3881–3889. doi: 10.1128/JB.01978-14
Honda M. C., Wakita M., Matsumoto K., Fujiki T., Siswanto E., Sasaoka K., et al. (2017). Comparison of carbon cycle between the western Pacific subarctic and subtropical time-series stations: highlights of the K2S1 project. J. Oceanogr. 73, 647–667. doi: 10.1007/s10872-017-0423-3
Hyatt D., LoCascio P. F., Hauser L. J., Uberbacher E. C. (2012). Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 28, 2223–2230. doi: 10.1093/bioinformatics/bts429
Imhoff J. F., Rahn T., Künzel S., Neulinger S. C. (2018). Photosynthesis is widely distributed among Proteobacteria as demonstrated by the phylogeny of PufLM reaction center proteins. Front. Microbiol. 8, 2679. doi: 10.3389/fmicb.2017.02679
Issa H. B., Phan G., Broutin I. (2018). Functional mechanism of the efflux pumps transcription regulators from Pseudomonas aeruginosa based on 3D structures. Front. Mol. Biosci. 5, 57. doi: 10.3389/fmolb.2018.00057
Ivars-Martinez E., Martin-Cuadrado A.-B., D’auria G., Mira A., Ferriera S., Johnson J., et al. (2008). Comparative genomics of two ecotypes of the marine planktonic copiotroph Alteromonas macleodii suggests alternative lifestyles associated with different kinds of particulate organic matter. ISME J. 2, 1194–1212. doi: 10.1038/ismej.2008.74
Juhas M., van der Meer J. R., Gaillard M., Harding R. M., Hood D. W., Crook D. W. (2009). Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 33, 376–393. doi: 10.1111/j.1574-6976.2008.00136.x
Kai W., Peisheng Y., Rui M., Wenwen J., Zongze S. (2017). Diversity of culturable bacteria in deep-sea water from the South Atlantic Ocean. Bioengineered 8, 572–584. doi: 10.1080/21655979.2017.1284711
Kanehisa M., Sato Y., Morishima K. (2016). BlastKOALA and ghostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Khandeparker R., Meena R. M., Deobagkar D. (2014). Bacterial diversity in deep-sea sediments from Afanasy Nikitin seamount, equatorial Indian Ocean. Geomicrobiol. J. 31, 942–949. doi: 10.1080/01490451.2014.918214
Kim J., Na S.-I., Kim D., Chun J. (2021). UBCG2: Up-to-date bacterial core genes and pipeline for phylogenomic analysis. J. Microbiol. 59, 609–615. doi: 10.1007/s12275-021-1231-4
Konstantinidis K. T., Braff J., Karl D. M., Delong E. F. (2009). Comparative metagenomic analysis of a microbial community residing at a depth of 4000 meters at station ALOHA in the North Pacific subtropical gyre. Appl. Environ. Microbiol. 75, 5345–5355. doi: 10.1128/AEM.00473-09
Kumar S., Stecher G., Li M., Knyaz C., Tamura K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lauro F. M., Tran K., Vezzi A., Vitulo N., Valle G., Bartlett D. H. (2008). Large-scale transposon mutagenesis of Photobacterium profundum SS9 reveals new genetic loci important for growth at low temperature and high pressure. J.Bacteriol 190, 1699–1709. doi: 10.1128/JB.01176-07
Leduc L., Ferroni G. (1994). The chemolithotrophic bacterium Thiobacillus ferrooxidans. FEMS Microbiol. Rev. 14, 103–119. doi: 10.1111/j.1574-6976.1994.tb00082.x
Li H., Meng F., Duan W., Lin Y., Zheng Y. (2019). Biodegradation of phenol in saline or hypersaline environments by bacteria: A review. Ecotoxicol. Environ. Saf. 184, 109658. doi: 10.1016/j.ecoenv.2019.109658
Li S., Hu S., Shi S., Ren L., Yan W., Zhao H. (2019a). Microbial diversity and metaproteomic analysis of activated sludge responses to naphthalene and anthracene exposure. RSC Adv. 9, 22841–22852. doi: 10.1039/C9RA04674G
Li Y., Kong X., Zhang H. (2019b). Characteristics of a novel manganese superoxide dismutase of a hadal sea cucumber (Paelopatides Sp.) from the Mariana Trench. Mar. Drugs 17, 84. doi: 10.3390/md17020084
Lu H., Sato Y., Fujimura R., Nishizawa T., Kamijo T., Ohta H. (2011). Limnobacter litoralis sp. nov., a thiosulfate-oxidizing, heterotrophic bacterium isolated from a volcanic deposit, and emended description of the genus Limnobacter. Int. J. Syst. Evol. Microbiol. 61, 404–407. doi: 10.1099/ijs.0.020206-0
Markowitz V. M., Mavromatis K., Ivanova N. N., Chen I.-M. A., Chu K., Kyrpides N. C. (2009). IMG ER: a system for microbial genome annotation expert review and curation. Bioinform 25, 2271–2278. doi: 10.1093/bioinformatics/btp393
Martin-Cuadrado A. B., Lopez-Garcia P., Alba J. C., Moreira D., Monticelli L., Strittmatter A., et al. (2007). Metagenomics of the deep Mediterranean, a warm bathypelagic habitat. PloS One 2, e914. doi: 10.1371/journal.pone.0000914
Meier-Kolthoff J. P., Carbasse J. S., Peinado-Olarte R. L., Göker M. (2022). TYGS and LPSN: a database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 50, D801–D807. doi: 10.1093/nar/gkab902
Meng Q., Xu X., Zhang W., Cheng L., Men M., Xu B., et al. (2019). Diversity and abundance of denitrifiers during cow manure composting. Rev. Argent. Microbiol. 51, 191–200. doi: 10.1016/j.ram.2018.08.003
Minnikin D. E., O’donnell A. G., Goodfellow M., Alderson G., Athalye M., Schaal A., et al. (1984). An integrated procedure for the extraction of bacterial isoprenoid quinones and polar lipids. J. Microbiol. Methods 2, 233–241. doi: 10.1016/0167-7012(84)90018-6
Mittl P. R., Schneider-Brachert W. (2007). Sel1-like repeat proteins in signal transduction. Cell Signal. 19, 20–31. doi: 10.1016/j.cellsig.2006.05.034
Moeck G. S., Coulton J. W. (1998). TonB-dependent iron acquisition: mechanisms of siderophore-mediated active transport. Mol. Microbiol. 28, 675–681. doi: 10.1046/j.1365-2958.1998.00817.x
Molina-Henares A. J., Krell T., Eugenia Guazzaroni M., Segura A., Ramos J. L. (2006). Members of the IclR family of bacterial transcriptional regulators function as activators and/or repressors. FEMS Microbiol. Rev. 30, 157–186. doi: 10.1111/j.1574-6976.2005.00008.x
Moreira A. P. B., Meirelles P. M., Santos E. D. O., Amado-Filho G. M., Francini-Filho R. B., Thompson C. C., et al. (2015). Turbulence-driven shifts in holobionts and planktonic microbial assemblages in St. Peter and St. Paul Archipelago, Mid-Atlantic Ridge, Brazil. Front. Microbiol. 6, 1038. doi: 10.3389/fmicb.2015.01038
Morotomi M., Nagai F., Watanabe Y. (2011). Parasutterella secunda sp. nov., isolated from human faeces and proposal of Sutterellaceae fam. nov. in the order Burkholderiales. Int. J. Syst. Evol. Microbiol. 61, 637–643. doi: 10.1099/ijs.0.023556-0
Naruki M., Watanabe A., Warashina T., Morita T., Arakawa K. (2024). Complete genome sequence of Limnobacter thiooxidans CS-K2T, isolated from freshwater lake sediments in Bavaria, Germany. Microbiol. Resour. Announc. 13, e00992–e00923. doi: 10.1128/mra.00992-23
Nelson W. C., Wollerman L., Bhaya D., Heidelberg J. F. (2011). Analysis of insertion sequences in thermophilic cyanobacteria: exploring the mechanisms of establishing, maintaining, and withstanding high insertion sequence abundance. Appl. Environ. Microbiol. 77, 5458–5466. doi: 10.1128/AEM.05090-11
Nguyen T. M., Kim J. (2017). Limnobacter humi sp. nov., a thiosulfate-oxidizing, heterotrophic bacterium isolated from humus soil, and emended description of the genus Limnobacter Spring et al., 2001. J. Microbiol. 55, 508–513. doi: 10.1007/s12275-017-6645-7
Pinhassi J., Berman T. (2003). Differential growth response of colony-forming α-and γ-proteobacteria in dilution culture and nutrient addition experiments from Lake Kinneret (Israel), the eastern Mediterranean Sea, and the Gulf of Eilat. Appl. Environ. Microbiol. 69, 199–211. doi: 10.1128/AEM.69.1.199-211.2003
Redder P., Hausmann S., Khemici V., Yasrebi H., Linder P. (2015). Bacterial versatility requires DEAD-box RNA helicases. FEMS Microbiol. Rev. 39, 392–412. doi: 10.1093/femsre/fuv011
Richter M., Rossello-Mora R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Rzhetsky A., Nei M. (1993). Theoretical foundation of the minimum-evolution method of phylogenetic inference. Mol. Biol. Evol. 10, 1073–1095. doi: 10.1093/oxfordjournals.molbev.a040056
Saitou N., Nei M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Sanz-Sáez I., Sánchez P., Salazar G., Sunagawa S., De Vargas C., Bowler C., et al. (2023). Top abundant deep ocean heterotrophic bacteria can be retrieved by cultivation. ISME Commun. 3, 92. doi: 10.1038/s43705-023-00290-0
Sasser M. (1990). “Identification of bacteria by gas chromatography of cellular fatty acids,” in MIDI Technical Note 101 (MIDI Inc, Newark: DE).
Sayed A., Ghazy M. A., Ferreira A. J., Setubal J. C., Chambergo F. S., Ouf A., et al. (2014). A novel mercuric reductase from the unique deep brine environment of Atlantis II in the Red Sea. J. Biol. Chem. 289, 1675–1687. doi: 10.1074/jbc.M113.493429
Seemann T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinform 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Seufferheld M. J., Alvarez H. M., Farias M. E. (2008). Role of polyphosphates in microbial adaptation to extreme environments. Appl. Environ. Microbiol. 74, 5867–5874. doi: 10.1128/AEM.00501-08
Simonato F., Campanaro S., Lauro F. M., Vezzi A., D’angelo M., Vitulo N., et al. (2006). Piezophilic adaptation: a genomic point of view. J. Biotechnol. 126, 11–25. doi: 10.1016/j.jbiotec.2006.03.038
Sinha R. K., Krishnan K., Thomas F. A., Binish M., Mohan M., Kurian P. J. (2019). Polyphasic approach revealed complex bacterial community structure and function in deep sea sediment of ultra-slow spreading Southwest Indian Ridge. Ecol. Indic. 96, 40–51. doi: 10.1016/j.ecolind.2018.08.063
Song J., Jang H. J., Joung Y., Kang I., Cho J. C. (2019a). Sulfitobacter profundi sp. nov., isolated from deep seawater. J. Microbiol. 57, 661–667. doi: 10.1007/s12275-019-9150-3
Song J., Kang I., Joung Y., Yoshizawa S., Kaneko R., Oshima K., et al. (2019b). Genome analysis of Rubritalea profundi SAORIC-165T, the first deep-sea verrucomicrobial isolate, from the northwestern Pacific Ocean. J. Microbiol. 57, 413–422. doi: 10.1007/s12275-019-8712-8
Spring S., Kampfer P., Schleifer K. H. (2001). Limnobacter thiooxidans gen. nov., sp. nov., a novel thiosulfate-oxidizing bacterium isolated from freshwater lake sediment. Int. J. Syst. Evol. Microbiol. 51, 1463–1470. doi: 10.1099/00207713-51-4-1463
Stamatakis A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. J. Bioinform. 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stothard P., Wishart D. S. (2005). Circular genome visualization and exploration using CGView. Bioinform 21, 537–539. doi: 10.1093/bioinformatics/bti054
Syranidou E., Karkanorachaki K., Amorotti F., Avgeropoulos A., Kolvenbach B., Zhou N.-Y., et al. (2019). Biodegradation of mixture of plastic films by tailored marine consortia. J. Hazard. Mater. 375, 33–42. doi: 10.1016/j.jhazmat.2019.04.078
Tatusova T., Dicuccio M., Badretdin A., Chetvernin V., Nawrocki E. P., Zaslavsky L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Thompson J. D., Gibson T. J., Plewniak F., Jeanmougin F., Higgins D. G. (1997). The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882. doi: 10.1093/nar/25.24.4876
Ting L., Williams T. J., Cowley M. J., Lauro F. M., Guilhaus M., Raftery M. J., et al. (2010). Cold adaptation in the marine bacterium, Sphingopyxis alaskensis, assessed using quantitative proteomics. Environ. Microbiol. 12, 2658–2676. doi: 10.1111/j.1462-2920.2010.02235.x
Vedler E., Heinaru E., Jutkina J., Viggor S., Koressaar T., Remm M., et al. (2013). Limnobacter spp. as newly detected phenol-degraders among Baltic Sea surface water bacteria characterised by comparative analysis of catabolic genes. Syst. Appl. Microbiol. 36, 525–532. doi: 10.1016/j.syapm.2013.07.004
Weisburg W. G., Barns S. M., Pelletier D. A., Lane D. J. (1991). 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. Res. 173, 697–703. doi: 10.1128/jb.173.2.697-703.1991
Wick R. R., Judd L. M., Gorrie C. L., Holt K. E. (2017). Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PloS Comput. Biol. 13, e1005595. doi: 10.1371/journal.pcbi.1005595
Yoon S.-H., Ha S.-M., Kwon S., Lim J., Kim Y., Seo H., et al. (2017). Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 67, 1613–1617. doi: 10.1099/ijsem.0.001755
Zhang W.-J., Cui X.-H., Chen L.-H., Yang J., Li X.-G., Zhang C., et al. (2019). Complete genome sequence of Shewanella benthica DB21MT-2, an obligate piezophilic bacterium isolated from the deepest Mariana Trench sediment. Mar. Genom. 44, 52–56. doi: 10.1016/j.margen.2018.09.001
Zhang X., Xu W., Liu Y., Cai M., Luo Z., Li M. (2018). Metagenomics reveals microbial diversity and metabolic potentials of seawater and surface sediment from a hadal biosphere at the Yap Trench. Front. Microbiol. 9, 2402. doi: 10.3389/fmicb.2018.02402
Zhao X., Liu J., Zhou S., Zheng Y., Wu Y., Kogure K., et al. (2020). Diversity of culturable heterotrophic bacteria from the Mariana Trench and their ability to degrade macromolecules. Mar. Life Sci. Technol. 2, 181–193. doi: 10.1007/s42995-020-00027-1
Keywords: Limnobacter profundi, deep sea, Burkholderiales, genome, novel species, phenol degradation
Citation: Kim M, Song J, Shin SY, Kogure K, Kang I and Cho J-C (2024) Cultivation of deep-sea bacteria from the Northwest Pacific Ocean and characterization of Limnobacter profundi sp. nov., a phenol-degrading bacterium. Front. Mar. Sci. 11:1449548. doi: 10.3389/fmars.2024.1449548
Received: 15 June 2024; Accepted: 15 October 2024;
Published: 04 November 2024.
Edited by:
Taichi Yokokawa, Japan Agency for Marine-Earth Science and Technology (JAMSTEC), JapanReviewed by:
Ana Paula B. Moreira, Federal University of Rio de Janeiro, BrazilSujata Dabolkar, Government College of Arts, Science and Commerce, Quepem, India
Copyright © 2024 Kim, Song, Shin, Kogure, Kang and Cho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jang-Cheon Cho, Y2hvamNAaW5oYS5hYy5rcg==
†These authors have contributed equally to this work