 Eric Edsinger1*†
Eric Edsinger1*† Leonid L. Moroz1,2†
Leonid L. Moroz1,2†- 1Whitney Laboratory for Marine Bioscience, University of Florida, St. Augustine, FL, United States
- 2Department of Neuroscience, College of Medicine, University of Florida, Gainesville, FL, United States
Functional and biodiversity genomics is essential for assessment and monitoring of planetary health and species-specific management in changing ecosystems. However, experimental knowledge of gene functions is limited to a few species, and dependencies on distantly related models. Combined with unrecognized degrees of lineage-specific gene family expansion, this means that traditional comparative methods are insufficient. Here, we introduce the concept of a hotspot, defined as innovations underlying the evolution of lineage-specific biology. We illustrate hotspots using molluscs having chromosome-scale genome assemblies and focus on heat-sensing TRPM channels and species living in environments of extreme heat stress (e.g., high intertidal and hydrothermal vent gastropods and bivalves). Integrating gene family, orthogroup, and domain-based methods with genomic hotspots (local homolog expansions on chromosomes), we show that conventional approaches overlook substantial amounts of species-specific gene family diversity due to limitations of distant homology detection. In contrast, local segmental duplications are often recent, lineage-specific genetic innovations reflecting emerging adaptions and can be identified for any genome. Revealed TRPM gene family diversification highlights unique neural and behavioral mechanisms that could be beneficial in predicting species’ resilience to heat stress. In summary, the identification of hotspots and their integration with other types of analyses illuminate evolutionary (neuro)genomic strategies that do not depend on knowledge from model organisms and unbiasedly reveal evolutionarily recent lineage-specific adaptations. This strategy enables discoveries of biological innovations across species as prospective targets for modeling, management, and biodiversity conservation.
1 Introduction
Environmental impacts, including record-setting marine heat waves (Li et al., 2023; Minière et al., 2023), are affecting global biodiversity and planetary health (Claudet et al., 2020; Armstrong McKay et al., 2022; Hansen et al., 2023; Lamboll et al., 2023; Minière et al., 2023). For marine ecosystems, recovery may be slow due to the massive heat-buffer capacity of oceans (Lubchenco et al., 2015; Hoegh-Guldberg et al., 2019; Claudet et al., 2020; Erskine et al., 2021; Jacquemont et al., 2022) but understanding how local species respond to accelerating environmental extremes is critical to biodiversity management. For example, Marine Protected Areas are isolated refugia with connectivity for benthic marine invertebrates provided by recruitment of pelagic swimming larvae (Christie et al., 2010; Moksnes and Jonsson, 2020; Lu et al., 2023; Muenzel et al., 2023). However, a larva’s binary decision to undergo settlement or not can be temperature sensitive (Da-Anoy et al., 2020; Viladrich et al., 2022; Weeriyanun et al., 2022), with implications for species survivorship and distribution in management. Powerful, accessible approaches to predict the adaptive potential of local species are needed for long-term modeling and mitigation of environmental impacts.
Biodiversity Genomics and the umbrella Earth BioGenome Project aim to produce reference genomes with chromosome assemblies for every eukaryotic species (Zoonomia Consortium, 2020; Blaxter et al., 2022; Cartney et al., 2022; Hogg et al., 2022; Lewin et al., 2022; of Life Project Consortium TDT et al., 2022; Sherkow et al., 2022; Stephan et al., 2022), including diverse spiralians (Lawniczak et al., 2022; Hawkins et al., 2023), with opportunities to address environmental stresses (Zoonomia Consortium, 2020; Blaxter et al., 2022; Cartney et al., 2022; Hogg et al., 2022; of Life Project Consortium TDT et al., 2022; Sherkow et al., 2022). Yet, genomic data are generally not accompanied by molecular-functional knowledge. Furthermore, there are limited tools to evaluate adaptive potential from diverse lineages (Lopez et al., 2018; Zoonomia Consortium, 2020; Hogg et al., 2022; Marx, 2022; of Life Project Consortium TDT et al., 2022; Stephan et al., 2022), and integrative approaches across fields, like neuroscience and conservation biology (Zoonomia Consortium, 2020; Marx, 2022; Michaiel and Bernard, 2022; Stephan et al., 2022; Anttonen et al., 2023; Doell et al., 2023).
Homology-based annotation of gene function is commonly used in the absence of direct molecular knowledge, wherein sequence and increasingly structural similarities enable mapping of gene function in genetic models, like humans, Drosophila melanogaster, and Caenorhabditis elegans, to a target species (Blaby-Haas and Merchant, 2019; de Crécy-Lagard et al., 2022; Bordin et al., 2023; Kim et al., 2023; Kirilenko et al., 2023; van Kempen et al., 2023; Svedberg et al., 2024). Still, gene families present in a target species but absent in a popular reference species can go mis-annotated or unannotated, or their presence in the genome can go unrecognized due to failures in assembly and/or structural annotation of gene models. Similarly, detection of the phylogenetic signal of remote homologs in sequence alignment becomes difficult at 20-35% sequence similarity (twilight zone) and goes beyond the theoretical limits at less than 20% (midnight zone) (Chang et al., [[NoYear]]; Chung and Subbiah, 1996; Rost, 1999; Koehl and Levitt, 2002), though, for example, deep-learning structural approaches are pushing these limits (Bordin et al., 2023; Kim et al., 2023; van Kempen et al., 2023; Pantolini et al., 2024; Svedberg et al., 2024). For instance, it is common for 25% or more of genes in a spiralian genome to go unannotated.
Genetic innovations underlying speciation adaptations can be most relevant to functional biodiversity assessments (Hahn et al., 2007; Toll-Riera et al., 2016; Villanueva-Cañas et al., 2017; Richter et al., 2018; Kim et al., 2022; Wu et al., 2022; Wu and Lambert, 2023; Mantica et al., 2024) but are the most likely to go undetected in current bioinformatic pipelines (detailed below; Figure 1A) (Blaby-Haas and Merchant, 2019; Peng and Zhao, 2024). Even the well-studied Drosophila has over 500 unannotated genes that arose during the recent evolution of its genus (Peng and Zhao, 2024). Overall, annotation methods that reduce reliance on distantly related species and highlight genetic innovations underlying lineage-specific biology are desirable.
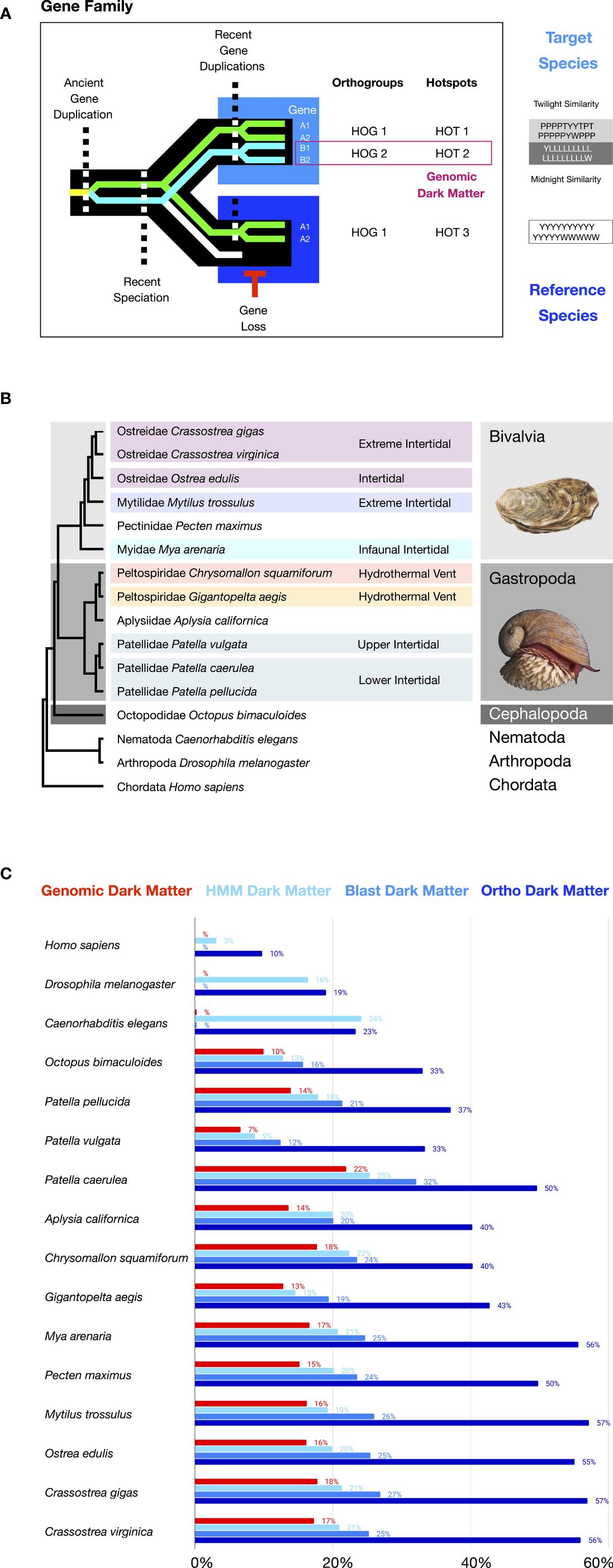
Figure 1 Genomic dark matter, Species16 species, and their proteomes. (A) A schematic highlighting one possible scenario where genomic dark matter arises due to patterns of gene and species evolution. (B) The phylogenetic tree for species and their habitats highlighting thermal stress. Molluscan classes are indicated in color blocks with cephalopods dark gray, gastropods medium gray, and bivalves light gray. Independent origins of heat-stress habitats (intertidal or hydrothermal vent) are indicated in color blocks. Four independent origins of intertidal habitats or upper-intertidal regions include 1) oyster - purple, 2) mussel - light purple, 3) Mya - light blue, and 4) snail - teal. Two independent origins of hydrothermal vent habitats are 1) Chrysomallon light red and 2) Gigantopelta light orange. (C) Percentage of genomic dark matter in each species, where genomic dark matter is defined as genes that lacked functional annotation based on sequence homology to reference species and their functionally assessed genes. Assessments for functional annotations were based on 1) HMM-based GO-Pfam domain and PANTHER gene family annotations, 2) top hit in reference genomes based on one-direct Blast annotations, and 3) Diamond-based genome clustering of all Species16 species annotations (see Methods for details). Supplementary material contains BUSCO values for the complete proteins percentages in respected reference species with sequenced genomes.
In deciphering those mechanisms, it was established that gene families commonly expand through segmental duplication of chromosome regions during DNA replication, generating new gene copies (paralogs within the initial species but becoming orthologs or homologs in subsequent derived species) physically adjacent to existing gene copies on a chromosome (Ohno, 1970; Zhang, 2003; Bergthorsson et al., 2007; Innan and Kondrashov, 2010). Evolutionary recent gene copies can diverge in function (division of labor), enabling biological novelties (Force et al., 1999; Bergthorsson et al., 2007; Hittinger and Carroll, 2007). Parallel processes, including deletions, inversions, and translocations, result in the spatial mixing of genes, with chromosomes forming “bags of genes” over time (Putnam et al., 2007; Putnam et al., 2008; Innan and Kondrashov, 2010; Session et al., 2016; Hart et al., 2018; Robert et al., 2022; Simakov et al., 2022; Yu et al., 2024). Thus, initial clusters of newly formed gene copies will eventually disperse in the evolution of genomes, meaning localized gene copies on a chromosome likely reflect recent evolutionary events and underlie lineage-specific biology. These genomic regions of evolutionary history could also act as catalytic sources of gene network innovations, taking advantage of the proximity of genes and their regulatory sites.
Here, we introduce the concept of hotspots with an emphasis on genome architectures. Genomic hotspots can be identified using simple bioinformatic methods for reference-free identification of gene copies locally clustered in a genome and integrated with genome-scale homology-based methods of molecular function gene annotation, orthogroups, and gene trees. We focus on genome-sequenced molluscs living in typical vs. extreme heat-stress environments. The selected molluscs include bivalves and gastropods species found in subtidal vs. intertidal or hydrothermal vent habitats (Supplementary Materials). We also emphasize evolutionary dynamics of the Transient Receptor Potential (TRP) ion channel superfamily, including the thermo-sensitive TRPM family (Lamas et al., 2019; Himmel and Cox, 2020; Szollosi, 2021; Kashio and Tominaga, 2022), which are diverse and expressed under heat stress in species used here (Zhang et al., 2012; Sun et al., 2020; Zeng et al., 2020; Fu et al., 2021; Lan et al., 2021; Moreira et al., 2021; Peng et al., 2021; Chi et al., 2023; Zhang et al., 2023). Our findings illustrate that approaches leveraging hotspots could enable predictions of adaptation and resilience in response to environmental change.
2 Materials and methods
Genome sources and computational methods are provided as Supplementary Materials. Python-Unix pipelines are provided as GitHub repository Hotspots_Paper_2024 v0.1.0-alpha.1 (GitHub Hotspots_Paper_2024 v0.1.0-alpha.1: https://github.com/000generic/Hotspots_Paper_2024/tree/v0.1.0-alpha.1). The repository is archived with a permanent DOI at Zenodo (Zenodo DOI: 10.5281/zenodo.11069191: https://zenodo.org/records/11069191).
3 Results
3.1 Hotspots highlight innovations underlying the origins and evolution of lineage-specific biology
Homology and the origins of novelties are at the core of evolutionary paradigms. Like others, we define homology as a state where biological features within or between individuals or species arise from the same ancestral feature in evolution. Use of the term ‘genomic dark matter’ has varied, including definitions based on regions of the genome resistant to assembly vs. regions of the genome resistant to functional annotation (Wilusz and Sharp, 2013; Bornberg-Bauer et al., 2015; Chi, 2016; Sedlazeck et al., 2018; Ebbert et al., 2019; Girardini et al., 2023). In the case of functional annotation, genomic dark matter was highlighted first and is commonly used in contexts of unannotated non-coding sequences, classically known as “junk DNA” and later ‘upgraded’ with recognition that the “junk” sequences included unannotated regulatory elements, transposons, and non-coding RNAs that are operational (ENCODE Project Consortium et al., 2007; Rosenbloom et al., 2010; Derrien et al., 2012; ENCODE Project Consortium, 2012; Harrow et al., 2012; Yip et al., 2012; ENCODE Project Consortium et al., 2020; Pang and Snyder, 2020; Sisu et al., 2020; Fagundes et al., 2022; Horton et al., 2023). Here, we define genomic dark matter simply as genomic structures resistant to functional annotation and highlight inclusion of both coding genes and non-coding regions. We also highlight the use of sequence homology approaches across species in performing structural and functional annotation in this definition, (Figure 1A). Finally, we introduce the concept of a hotspot and focus on its application in evolutionary genomics (Figure 1A). The rationale behind these terms and definitions is provided in Supplementary Materials.
We define a hotspot as the set of innovations underlying the evolution of lineage-specific biology (Figure 1A). Hotspots can be composed of structural components within and across hierarchical levels, from base elements to ecosystems. The term is scaleless. It can have diverse complex contexts, from molecular (e.g., genomic hotspots below) to cellular (e.g., neural circuit hotspots) to organismal (population hotspots; see also Supplementary Materials 3.2.4) and can include their cross-level integration.
Here we focus on genomic hotspots formed as regions of chromosomes delineated by spatial clusters of gene paralogs. This is similar to synteny, in that the identity of genes in genomic proximity on a chromosome is evaluated, but is distinct, as syntenic methods are defined by identifying patterns across species while hotspots are defined internal to the target without outside reference. Methodologically, genomic hotspots are free of external requirements of high-quality genome assemblies and annotations outside a given target species or lineage, in contrast to syntenic approaches. Thus, although additional reference genomes can be useful in evaluating hotspots, they are not required in the identification and initial use of hotspots to guide deciphering of novelties and adaptions underlying lineage or species-specific biology and evolution.
To illustrate the ‘hotspot’ approach, we selected 16 genomes with chromosome-level assemblies. Initial assessments of assembly completeness were based on BUSCO Metazoa evaluation of processed proteomes having one representative single longest sequence per gene, with most species found to be 95% BUSCO Complete or better, but with exceptions of highly derived Caenorhabditis (75%), Patella vulgata (89%), Patella pellucida (86%) and Chrysomallon squamiformis (83%). These results are indicative of high-quality genome assemblies and structural gene model annotations (see the summary figure in Supplementary Materials). Additional details are provided in Supplementary Materials.
3.2 Genomic dark matter is prevalent in functional biodiversity annotations
We found substantial amounts of genomic dark matter in species after running commonly used functional annotation methods, highlighting limitations of these methods. To illustrate this, we performed three independent types of annotation for the proteomes, specifically: 1) by blasting against best-annotated reference genomes of model organisms (human, Drosophila, and Caenorhabditis) for one-direction top hit annotations, 2) by blast-based genome clustering for orthogroup annotations, and 3) by HMM-based identification of gene features for protein domain and gene family annotations. We then determined what percentage of genes were annotated or not for each method and across all methods, with genes going undetected in all three declared genomic dark matter.
For Blast annotations, unannotated genes ranged from 12-32% of the genome, with a 23% average (SD 5%) (Figure 1C). The method was intermediate in its ability to annotate but common e-value cut-offs of 1e-3 to 1e-10 mean there can be promiscuous domains and low-level false positives complicating the annotations in unknown ways.
For orthogroup annotations, we found unannotated genes ranged from 33-57% of the genome with a 47% average (SD 9) (Figure 1C). This method is the most powerful for inference of gene function, as its scope of comparison is restricted to orthogroup orthologs across species, thereby avoiding most false-positive issues. However, it is also the most conservative approach, producing exceedingly high levels of unannotated genes, more than double the other two methods, and lacking identification of deeper levels of homology commonly of interest.
For HMM-based domain annotation using Pfam and GO and HMM-based gene family assessment using PANTHER, unannotated genes ranged from 9-25% of the genome with a 19% average (SD 4) (Figure 1C). Although some degree of misannotation due to false positives is likely, it is thought that the highly sensitive information-rich aspects of how HMMs are built can reduce this issue in comparison to Blast and other tools (Girardini et al., 2023). Thus, the HMM method is the most effective for functional gene annotation in Species16 biodiversity genomes but still leaves significant numbers of genes unannotated.
Finally, we find that 7-22% of genes in a genome remained unannotated across all three methods, forming conservatively defined genomic dark matter (Figure 1C). These results highlight the degree to which reference-based methods for the functional annotation of genomes can fail in biodiversity assessments and illustrate the extent to which genetic novelty arises in evolution. We also found that for some species within a genus, their genomes exhibited quite different degrees of unannotated genomic dark matter, for example, 7% vs. 14% vs. 22% in the intertidal snails P.vulgata, P.pellucida, and Patella caerulea, respectively, suggesting dynamic patterns of gene innovations in recent speciation.
3.3 Genomic hotspots are common in genomes
We developed a simple stand-alone/reference-free method to identify genomic hotspots and found they are common in 16 bilaterian genomes, suggesting their identification can enable targeting of genes underlying species or lineage-specific biology. Focusing on molluscs, we blasted each predicted proteome against itself. We opted for an e-value cutoff of 1e-60 and identified all hits of a query gene located within a window of 20 genes centered on the query gene location in a chromosome or scaffold. These initial sets of genes were then merged based on overlapping membership between sets to form final genomic hotspot gene sets per genome.
We found that the number of hotspots and their genes ranged from 483 with 1,982 genes (average 31 genes per hotspot) in the shallow-water octopus Octopus bimaculoides to almost 8x as many in the intertidal infaunal clam Mya arenaria, with 3,747 hotspots and 11,982 genes (average 104 genes per hotspot). For initial test cases using nineteen species and focusing on hotspot identification in the sea hare Aplysia californica, small numbers of false positives arose at an e-value of 1e-40, most likely due to promiscuous domains or motifs. However, a substantially less restrictive e-value of 1e-10 was required to recover the Hox gene complex, an ancient chromosomal gene copy cluster and the most deeply studied and widely recognized (Wilusz and Sharp, 2013; Bornberg-Bauer et al., 2015; Chi, 2016; Sedlazeck et al., 2018; Ebbert et al., 2019; Fagundes et al., 2022). Also, while larger initial windows of 200 genes, rather than 20, sometimes detected additional hotspot members, the hits often appeared to be distantly related or due to a domain shared between unrelated gene families, greatly increasing false positives. At the same time windows smaller than 20 genes often lost likely hotspot members. Thus, we optimized for a window of 20 genes, as it best provided a stable core number of hotspot true positives, no obvious false positives, and reduced over-aggregation of distantly related hotspots.
3.4 Genomic hotspots are enriched in the TRP superfamily and TRPM family
To explore genomic hotspots in the context of gene family evolution, we focused on the TRP superfamily of ion channels and TRPM family within it. We identified all TRP superfamily members for 13 target mollusc species and 3 reference species (Species16). We identified in each species by reciprocal blast, using as queries a reference gene set of all human, Drosophila, and Caenorhabditis TRP proteins and then blasting back all target hit sequences against the reference proteomes. All target genes having a top hit back to a TRP family member in at least one reference proteome were accepted as candidate homologs. While TRP family size is 17, 22, and 32 genes respectively in Drosophila, Caenorhabditis, and human, it varied from 31 in Octopus to 167 in the upper intertidal mussel Mytilus trossulus, with an average of 81 genes per species (SD 35).
Next, we tested if the TRP superfamily is enriched for hotspots relative to the genome in general for each Species16 species. We found that while the average background density of genome hotspots per 100 genes varied from 3 in Octopus, the lowest of all, up to 10 in Mya, the average TRP hotspot density varied from 4 in the lower intertidal limpet Patella caerulea to 19 in Mya. Overall, the TRP superfamily was enriched for hotspots relative to the genome in nearly all selected species, suggesting that TRPs play important roles lineage-specific adaptations across molluscs.
3.5 Recent diversification of TRPs in molluscs
To further explore the role of TRPs in molluscan evolution, we assessed patterns of TRP superfamily evolution in Species16 species. Specifically, we constructed phylogenetic trees for the entire TRP superfamily (Figures 2A, B), and TRPM in particular (Figures 2C, D). We found that Mytilus exhibited the greatest diversification with 167 TRP genes (Figures 2E, F), including TRPM (Figures 2G, H). We find that the majority of TRP gene diversity across species lies outside reference species subbranches, indicating more recent lineage-specific expansions. We also find that hotspot sequences commonly form their own orthogroups, lacking functional annotation (genomic dark matter). Only rarely do hotspots belong to orthogroups that can be functionally annotated based on inclusion of reference genes (Figures 2B, D–H).
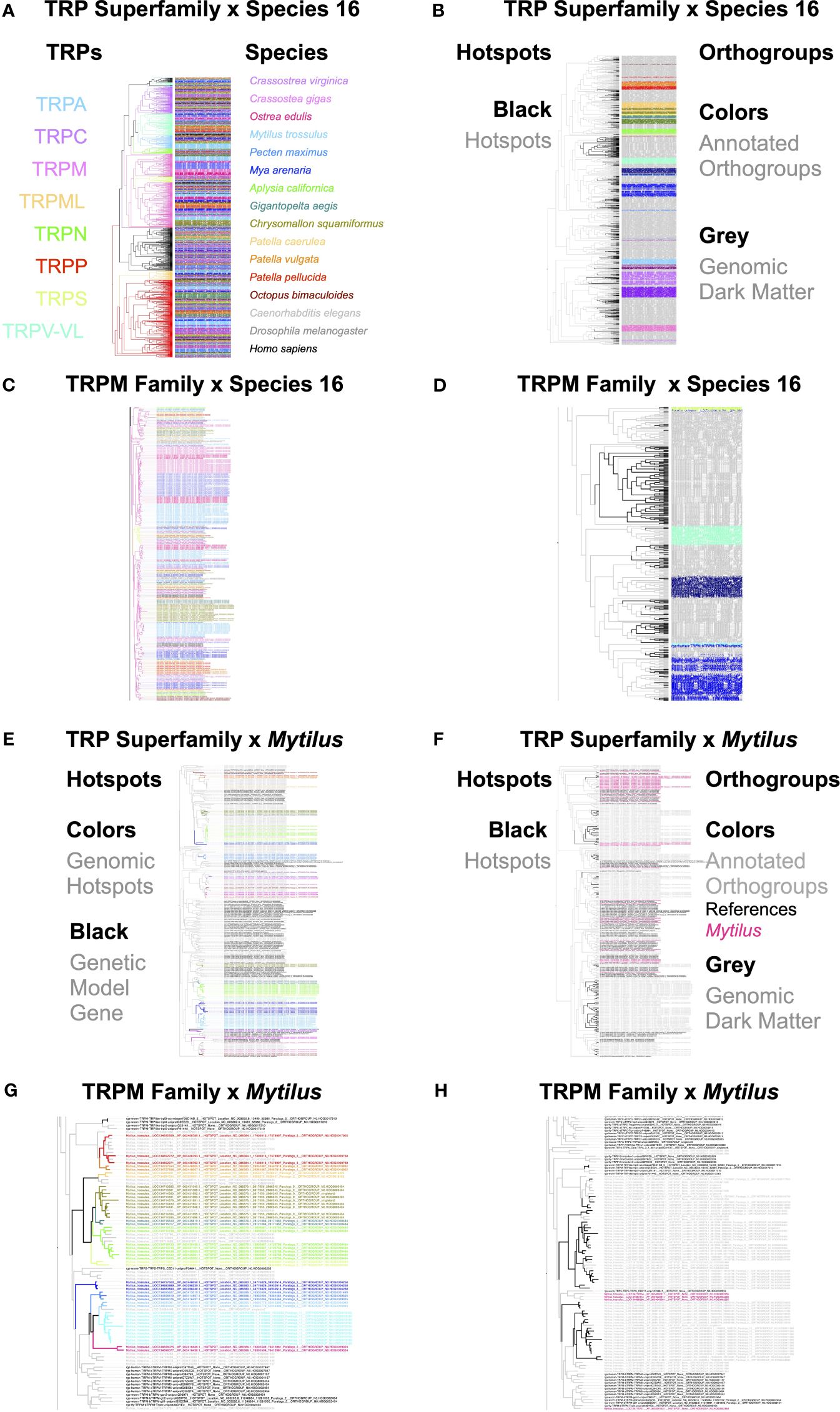
Figure 2 Integration of gene trees, orthogroups, and hotspots for the TRP superfamily in Species 16 species. TRP superfamily and TRPM family (A–D). These evolutionary expansions are visually evident in the gene trees as blocks of species-specific color (A, B). The color blocks are indicative of multiple paralogous gene copies in a species that arose within its immediate lineage on the Species16 species tree (Figure 1B). For a limited number of subbranches, we observed formation of many fine resolution rainbows of color, indicative of deeply conserved sequences with little lineage-specific evolution since the common ancestor (A–C). (A–H) Integration of gene families, hotspots, and/or orthogroups for TRPs or TRPM and either all Species16 species or Mytilus. (A) Species16 TRP gene tree with blocks of color on the species side indicative of lineage-specific gene family expansions. (B) Species16 integration of gene families, hotspots, and orthogroups on the TRP gene tree. Annotated orthogroups are orthogroups that include membership of at least one reference species gene. Genomic dark matter is all sequences not part of an annotated orthogroup. The tree is the same as in (A). (C) A Species16 TRPM gene tree with blocks of color on the species side indicative of lineage-specific gene family expansions. (D) Species16 TRPM integration of gene families, hotspots, and orthogroups. The tree is the same as in (C). (E) Integration of gene families, hotspots, and orthogroups on the TRP gene tree for Mytilus, highlighting individual hotspots in color. (F) Integration of gene families, hotspots, and orthogroups on the TRP gene tree for Mytilus, highlighting general patterns of hotspots vs orthogroups. The tree is the same as in (E). (G) Integration of gene families, hotspots, and orthogroups on the TRPM gene tree for Mytilus, highlighting individual hotspots in color. (H) Integration of gene families, hotspots, and orthogroups on the TRPM gene tree for Mytilus, highlighting general patterns of hotspots vs orthogroups. The tree is the same as in (G). Higher resolution trees can be found in the Supplementary Material.
Finally, we find that species that have independently evolved to live in extreme heat stress environments, such as those found in the intertidal or at hydrothermal vents (Figure 1B), have independently expanded the thermosensitive TRPM gene family, often extensively and uniquely so within the TRP superfamily (Figures 2C, D, G, H). In bivalves, this includes TRPM gene family expansions within each heat-stress tolerant lineage. In the lineage of oysters, Ostrea is lower intertidal and exhibits fewer expansions than the Crassostrea species C. gigas and C. virginica, which are found in the more extreme upper intertidal. Similarly, the mussel Mytilus and the clam Mya live in heat stress environments of the upper intertidal and infaunal intertidal mudflats, respectively, and exhibit a number of extensive lineage-specific TRPM gene family expansions. In contrast, the scallop Pecten maximus is closely related to oysters and mussels but is a subtidal species and has few TRPM genes and no substantial expansions in TRPM diversity (Figures 2C, D). In gastropods, the three Patella species are intertidal vs. Aplysia, which is a primarily shallow-water subtidal species. Although patterns of gene expansion are less striking, the Patella species have more TRPM genes than Aplysia and with more small-scale expansions of 1 or 2 genes (Figures 2C, D). The two hydrothermal vent gastropods, Chrysomallon squamiferum and Gigantopelta aegis, belong to the same family but they have adapted to the extreme heat stress independently (Sun et al., 2020; Zeng et al., 2020; Lan et al., 2021). Their genomes show striking patterns of parallel expansion in TRPM genes, less so than expansions found in bivalves but much greater than expansions seen in the other gastropods and Octopus (Figures 2C, D). Interestingly, the two main expansions in each species occur on the same branch within the greater TRPM gene tree (Figures 2C, D).
4 Discussion
The presented discussions of hotspots, homology and genomic dark matter agree with previous work (Wagner, 1989; Striedter and Northcutt, 1991; Hall, 1994; Abouheif, 1997; Chi, 2016; Strausfeld and Hirth, 2016; Ebbert et al., 2019; DiFrisco et al., 2023; Girardini et al., 2023; Rusin, 2023) but can help frame comparative genome–scale studies across functional biodiversity.
First, identification of genomic hotspots provides a reference-free means to identify candidate genes underlying the origins of lineage-specific biology, as their localization represents more recent evolutionary events due to the eventual dispersal of localized genes in eukaryotic genomes, with some notable exceptions, like Hox genes (Putnam et al., 2007; Putnam et al., 2008; Simakov et al., 2022; Schultz et al., 2023; Yu et al., 2024). It is striking that Species16 hotspots are predominately genomic dark matter and only more rarely associated with orthogroups and gene tree branches that include reference sequences of three model organisms used here. Future genome-scale statistical analyses and modeling could elucidate potential protection of older gene copies from hotspot formation and/or preferential utilization of new copies in more recent lineage-specific biology, perhaps due to associated gene regulatory elements that might be fully intact in older copies but variable in younger ones.
Second, the observed patterns of TRP family evolution are similar to previous studies in molluscs, including oysters (Himmel and Cox, 2020; Fu et al., 2021; Peng et al., 2021; Kashio and Tominaga, 2022). TRP gene family members form hotspots at substantially greater levels than observed background levels per genome. The TRP hotspots are predominately composed of unannotated genomic dark matter, which highlight the potential roles of TRP ion channels in lineage-specific biology. The number of TRP ion channels in bivalves and gastropods, with relatively simple nervous systems and behaviors, is much greater than that of humans and Octopus, which have independently evolved large brains (Moroz, 2009) and sophisticated behaviors suggesting functional pressures that limit gene diversification in complex nervous systems and/or lead to molecular expansion in simpler nervous systems (Moroz and Romanova, 2021; Moroz et al., 2021; Moroz and Romanova, 2022).
The TRPM gene family is recognized as a primary molecular sensor of temperature, including their elevated expression in response to heat stress in oysters and other molluscs (Himmel and Cox, 2020; Fu et al., 2021; Peng et al., 2021; Kashio and Tominaga, 2022; Chi et al., 2023; Zhang et al., 2023). Upper intertidal and hydrothermal vents are both environments featuring heat extremes, and bivalve and gastropod lineages have independently entered these environments with greatly expanded TRPM gene family diversity through lineage-specific hotspots.
Future studies that provide genome-scale evaluations of hotspot sets as clearly functional, and not degraded or established pseudogenes, and as having evidence of positive selection, will be important to support the idea that hotspots function in recently diverged species and critical for understanding lineage-specific adaptations. Similarly, at gene family levels, evidence of full-length transcripts that include signature domains or domain combinations will strengthen broader inclusion of genomic regions. It will also clarify that local expansions of genes by duplication are real and not a result of broken gene models or unrelated sequences sharing a similar domain in the region.
In summary, identification of the environmental molecular sensors of direct interest as part of newly emerging mechanistic work in functional biodiversity, enable new tools and resources to predict resilience and adaptability of a species facing rapid environmental change.
5 Conclusion
Overall, our findings highlight the idea that genomic hotspots represent relatively recent genetic innovations and that their unbiased reference-free identification can provide a novel and potentially powerful means to elucidate genomic mechanisms of evolution and the origins of genes underlying lineage-specific biology without any greater knowledge beyond the genome itself. TRP ion channels are important targets for understanding lineage-specific adaptations under regimes of environmental change and predicting outcomes for populations in response to such impacts.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
EE: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing. LM: Funding acquisition, Conceptualization, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by National Science Foundation (IOS - 2341882), and by the National Institutes of Health National Institute of Neurological Disorders and Stroke of under Award Number R01NS114491 and National Institute of Mental Health under Award Number 1R21MH119646-01. The content is solely the author’s responsibility and does not necessarily represent the official views of the National Institutes of Health or National Science Foundation.
Acknowledgments
We warmly thank J. Hsiao for assistance in initial design and coding to characterize and automate homolog neighborhoods (now genomic hotspots), and gene family phylogenetic trees, working with EE in the Chalasani Lab at the Salk Institute as a part of the GIGANTIC Project. We wish to thank S. Chalasani and lab for their encouragement and support. We also thank to NIH and NSF for financial support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2024.1434130/full#supplementary-material
References
Abouheif E. (1997). Developmental genetics and homology: a hierarchical approach. Trends Ecol. Evol. 12, 405–408. doi: 10.1016/S0169-5347(97)01125-7
Anttonen T., Burghi T., Duvall L., Fernandez M. P., Gutierrez G., Kermen F., et al. (2023). Neurobiology and changing ecosystems: mechanisms underlying responses to human-generated environmental impacts. J. Neurosci. 43, 7530–7537. doi: 10.1523/JNEUROSCI.1431-23.2023
Armstrong McKay D. I., Staal A., Abrams J. F., Winkelmann R., Sakschewski B., Loriani S., et al. (2022). Exceeding 1.5°C global warming could trigger multiple climate tipping points. Science 377, eabn7950. doi: 10.1126/science.abn7950
Bergthorsson U., Andersson D. I., Roth J. R. (2007). Ohno’s dilemma: evolution of new genes under continuous selection. Proc. Natl. Acad. Sci. U S A. 104, 17004–17009. doi: 10.1073/pnas.0707158104
Blaby-Haas C. E., Merchant S. S. (2019). Comparative and functional algal genomics. Annu. Rev. Plant Biol. 70(4), 605–638. doi: 10.1146/annurev-arplant-050718-095841
Blaxter M., Archibald J. M., Childers A. K., Coddington J. A., Crandall K. A., Di Palma F., et al. (2022). Why sequence all eukaryotes? Proc. Natl. Acad. Sci. U.S.A. 119 (4), e2115636118. doi: 10.1073/pnas.2115636118
Bordin N., Sillitoe I., Nallapareddy V., Rauer C., Lam S. D., Waman V. P., et al. (2023). AlphaFold2 reveals commonalities and novelties in protein structure space for 21 model organisms. Commun. Biol. 6, 160. doi: 10.1038/s42003-023-04488-9
Bornberg-Bauer E., Schmitz J., Heberlein M. (2015). Emergence of de novo proteins from “dark genomic matter” by “grow slow and moult. Biochem. Soc. Trans. 43, 867–873. doi: 10.1042/BST20150089
Cartney A. M. M., Anderson J., Liggins L., Hudson M. L., Anderson M. Z., TeAika B., et al. (2022). Balancing openness with Indigenous data sovereignty: An opportunity to leave no one behind in the journey to sequence all of life. Proc. Natl. Acad. Sci. 119, e2115860119. doi: 10.1073/pnas.2115860119
Chang G. S., Hong Y., Ko K. D., Bhardwaj G., Holmes E. C., Patterson R. L., et al. Phylogenetic profiles reveal evolutionary relationships within the twilight zone’’ of sequence similarity. Proc Natl Acad Sci U S A 105, 13474–9. doi: 10.1073/pnas.0803860105
Chi Y., Yang H., Shi C., Yang B., Bai X., Li Q. (2023). Comparative transcriptome and gene co-expression network analysis identifies key candidate genes associated with resistance to summer mortality in the Pacific oyster (Crassostrea gigas). Aquaculture 577, 739922. doi: 10.1016/j.aquaculture.2023.739922
Christie M. R., Tissot B. N., Albins M. A., Beets J. P., Jia Y., Ortiz D. M., et al. (2010). Larval connectivity in an effective network of marine protected areas. PloS One 5, e15715. doi: 10.1371/journal.pone.0015715
Chung S. Y., Subbiah S. (1996). A structural explanation for the twilight zone of protein sequence homology. Structure 4, 1123–1127. doi: 10.1016/S0969-2126(96)00119-0
Claudet J., Bopp L., Cheung W. W. L., Devillers R., Escobar-Briones E., Haugan P., et al. (2020). A roadmap for using the UN decade of ocean science for sustainable development in support of science, policy, and action. One Earth. 2, 34–42. doi: 10.1016/j.oneear.2019.10.012
Da-Anoy J. P., Cabaitan P. C., Conaco C. (2020). Warm temperature alters the chemical cue preference of Acropora tenuis and Heliopora coerulea larvae. Mar. pollut. Bull. 161, 111755. doi: 10.1016/j.marpolbul.2020.111755
de Crécy-Lagard V., Amorin de Hegedus R., Arighi C., Babor J., Bateman A., Blaby I., et al. (2022). A roadmap for the functional annotation of protein families: a community perspective. Database 2022, baac062. doi: 10.1093/database/baac062
Derrien T., Johnson R., Bussotti G., Tanzer A., Djebali S., Tilgner H., et al. (2012). The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789. doi: 10.1101/gr.132159.111
DiFrisco J., Love A. C., Wagner G. P. (2023). The hierarchical basis of serial homology and evolutionary novelty. J. Morphol. 284, e21531. doi: 10.1002/jmor.21531
Doell K. C., Berman M. G., Bratman G. N., Knutson B., Kühn S., Lamm C., et al. (2023). Leveraging neuroscience for climate change research. Nat. Clim Change 13, 1288–1297. doi: 10.1038/s41558-023-01857-4
Ebbert M. T. W., Jensen T. D., Jansen-West K., Sens J. P., Reddy J. S., Ridge P. G., et al. (2019). Systematic analysis of dark and camouflaged genes reveals disease-relevant genes hiding in plain sight. Genome Biol. 20, 97. doi: 10.1186/s13059-019-1707-2
ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. doi: 10.1038/nature11247
ENCODE Project Consortium, Birney E., Stamatoyannopoulos J. A., Dutta A., Guigó R., Gingeras T. R., et al. (2007). Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447, 799–816.
ENCODE Project Consortium, Moore J. E., Purcaro M. J., Pratt H. E., Epstein C. B., Shoresh N., et al. (2020). Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710. doi: 10.1038/s41586-020-2493-4
Erskine E., Baillie R., Lusseau D. (2021). Marine Protected Areas provide more cultural ecosystem services than other adjacent coastal areas. One Earth. 4, 1175–1185. doi: 10.1016/j.oneear.2021.07.014
Fagundes N. J. R., Bisso-MaChado R., Figueiredo P. I. C. C., Varal M., Zani A. L. S. (2022). What we talk about when we talk about “Junk DNA”. Genome Biol. Evol. 14(5), evac055. doi: 10.1093/gbe/evac055
Force A., Lynch M., Pickett F. B., Amores A., Yan Y. L., Postlethwait J. (1999). Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151, 1531–1545. doi: 10.1093/genetics/151.4.1531
Fu H., Jiao Z., Li Y., Tian J., Ren L., Zhang F., et al. (2021). Transient Receptor Potential (TRP) Channels in the Pacific Oyster (Crassostrea gigas): Genome-Wide Identification and Expression Profiling after Heat Stress between C. gigas and C. angulata. Int. J. Mol. Sci. 22(6), 3222. doi: 10.3390/ijms22063222
Girardini K. N., Olthof A. M., Kanadia R. N. (2023). Introns: the “dark matter” of the eukaryotic genome. Front. Genet. 14, 1150212. doi: 10.3389/fgene.2023.1150212
Hahn M. W., Han M. V., Han S. G. (2007). Gene family evolution across 12 Drosophila genomes. PloS Genet. 3, e197. doi: 10.1371/journal.pgen.0030197
Hall B. K. (Ed.) (1994). Homology. The Hierarchical Basis of Comparative Biology (San Diego, California: Academic Press).
Hansen J. E., Sato M., Simons L., Nazarenko L. S., Sangha I., Kharecha P., et al. (2023). Global warming in the pipeline. Oxf Open Clim Chang 3(1), kgad008. doi: 10.1093/oxfclm/kgad008
Harrow J., Frankish A., Gonzalez J. M., Tapanari E., Diekhans M., Kokocinski F., et al. (2012). GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774. doi: 10.1101/gr.135350.111
Hart M. L. I., Vu B. L., Bolden Q., Chen K. T., Oakes C. L., Zoronjic L., et al. (2018). Genes relocated between Drosophila chromosome arms evolve under relaxed selective constraints relative to non-relocated genes. J. Mol. Evol. 86, 340–352. doi: 10.1007/s00239-018-9849-5
Hawkins S. J., Mieszkowska N., Mrowicki R., et al. (2023). The genome sequence of the common limpet, Patella vulgata (Linnaeus, 1758) [version 1; peer review: 3 approved]. Wellcome Open Res. 8, 418. doi: 10.12688/wellcomeopenres
Himmel N. J., Cox D. N. (2020). Transient receptor potential channels: current perspectives on evolution, structure, function and nomenclature. Proc. Biol. Sci. 287, 20201309. doi: 10.1098/rspb.2020.1309
Hittinger C. T., Carroll S. B. (2007). Gene duplication and the adaptive evolution of a classic genetic switch. Nature 449, 677–681. doi: 10.1038/nature06151
Hoegh-Guldberg O., Northrop E., Lubchenco J. (2019). The ocean is key to achieving climate and societal goals. Science 365, 1372–1374. doi: 10.1126/science.aaz4390
Hogg C. J., Ottewell K., Latch P., Rossetto M., Biggs J., Gilbert A., et al. (2022). Threatened Species Initiative: Empowering conservation action using genomic resources. Proc. Natl. Acad. Sci. U.S.A. 119, e2115643118. doi: 10.1073/pnas.2115643118
Horton C. A., Alexandari A. M., Hayes M. G. B., Marklund E., Schaepe J. M., Aditham A. K., et al. (2023). Short tandem repeats bind transcription factors to tune eukaryotic gene expression. Science 381, eadd1250. doi: 10.1126/science.add1250
Innan H., Kondrashov F. (2010). The evolution of gene duplications: classifying and distinguishing between models. Nat. Rev. Genet. 11, 97–108. doi: 10.1038/nrg2689
Jacquemont J., Blasiak R., Le Cam C., Le Gouellec M., Claudet J. (2022). Ocean conservation boosts climate change mitigation and adaptation. One Earth. 5, 1126–1138. doi: 10.1016/j.oneear.2022.09.002
Kashio M., Tominaga M. (2022). TRP channels in thermosensation. Curr. Opin. Neurobiol. 75, 102591. doi: 10.1016/j.conb.2022.102591
Kim G. B., Kim J. Y., Lee J. A., Norsigian C. J., Palsson B. O., Lee S. Y. (2023). Functional annotation of enzyme-encoding genes using deep learning with transformer layers. Nat. Commun. 14, 7370. doi: 10.1038/s41467-023-43216-z
Kim H., Kim H. W., Lee J. H., Park J., Lee H., Kim S., et al. (2022). Gene family expansions in Antarctic winged midge as a strategy for adaptation to cold environments. Sci. Rep. 12, 18263. doi: 10.1038/s41598-022-23268-9
Kirilenko B. M., Munegowda C., Osipova E., Jebb D., Sharma V., Blumer M., et al. (2023). Integrating gene annotation with orthology inference at scale. Science 380, eabn3107. doi: 10.1126/science.abn3107
Koehl P., Levitt M. (2002). Sequence variations within protein families are linearly related to structural variations. J. Mol. Biol. 323, 551–562. doi: 10.1016/S0022-2836(02)00971-3
Lamas J. A., Rueda-Ruzafa L., Herrera-Pérez S. (2019). Ion channels and thermosensitivity: TRP, TREK, or both? Int. J. Mol. Sci. 20(10), 2371. doi: 10.3390/ijms20102371
Lamboll R. D., Nicholls Z. R. J., Smith C. J., Kikstra J. S., Byers E., Rogelj J. (2023). Assessing the size and uncertainty of remaining carbon budgets. Nat. Clim Change 13, 1360–1367. doi: 10.1038/s41558-023-01848-5
Lan Y., Sun J., Chen C., Sun Y., Zhou Y., Yang Y., et al. (2021). Hologenome analysis reveals dual symbiosis in the deep-sea hydrothermal vent snail Gigantopelta aegis. Nat. Commun. 12, 1165. doi: 10.1038/s41467-021-21450-7
Lawniczak M. K. N., Darwin Tree of Life Barcoding collective, Wellcome Sanger Institute Tree of Life programme, Wellcome Sanger Institute Scientific Operations: DNA Pipelines collective, Tree of Life Core Informatics collective, Darwin Tree of Life Consortium (2022). The genome sequence of the blue-rayed limpet, Patella pellucida Linnaeus, 1758. Wellcome Open Res. 7, 126. doi: 10.12688/wellcomeopenres
Lewin H. A., Richards S., Lieberman Aiden E., Allende M. L., Archibald J. M., Bálint M., et al. (2022). The earth bioGenome project 2020: starting the clock. Proc. Natl. Acad. Sci. U.S.A. 119, 473–497. doi: 10.1073/pnas.2115635118
Li Z., England M. H., Groeskamp S. (2023). Recent acceleration in global ocean heat accumulation by mode and intermediate waters. Nat. Commun. 14, 6888. doi: 10.1038/s41467-023-42468-z
Lopez J. V., Kamel B., Medina M., Collins T., Baums I. B. (2018). Multiple facets of marine invertebrate conservation genomics. Annu. Rev. Anim. Biosci. 7, 473–97. doi: 10.1146/annurev-animal-020518-115034
Lu J., Chen Y., Wang Z., Zhao F., Zhong Y., Zeng C., et al. (2023). Larval dispersal modeling reveals low connectivity among national marine protected areas in the yellow and east China seas. Biology 12(3), 396. doi: 10.3390/biology12030396
Lubchenco J., Barner A. K., Cerny-Chipman E. B., Reimer J. N. (2015). Sustainability rooted in science. Nat. Geosci. 8, 741–745. doi: 10.1038/ngeo2552
Mantica F., Iñiguez L. P., Marquez Y., Permanyer J., Torres-Mendez A., Cruz J., et al. (2024). Evolution of tissue-specific expression of ancestral genes across vertebrates and insects. Nat. Ecol. Evol. doi: 10.1038/s41559-024-02398-5
Marx V. (2022). Conservation genomics in practice. Nat. Methods 19, 522–525. doi: 10.1038/s41592-022-01477-4
Michaiel A. M., Bernard A. (2022). Neurobiology and changing ecosystems: Toward understanding the impact of anthropogenic influences on neurons and circuits. Front. Neural Circuits. 16, 995354. doi: 10.3389/fncir.2022.995354
Minière A., von Schuckmann K., Sallée J. B., Vogt L. (2023). Robust acceleration of Earth system heating observed over the past six decades. Sci. Rep. 13, 22975. doi: 10.1038/s41598-023-49353-1
Moksnes P. O., Jonsson P. R. (2020). Larval connectivity and marine protected area networks. PLoS One. 5, e15715. doi: 10.1093/oso/9780190648954.003.0015
Moreira C., Stillman J. H., Lima F. P., Xavier R., Seabra R., Gomes F., et al. (2021). Transcriptomic response of the intertidal limpet Patella vulgata to temperature extremes. J. Therm Biol. 101, 103096. doi: 10.1016/j.jtherbio.2021.103096
Moroz L. L. (2009). On the independent origins of complex brains and neurons. Brain Behav. Evol. 74, 177–190. doi: 10.1159/000258665
Moroz L. L., Romanova D. Y. (2021). Selective advantages of synapses in evolution. Front. Cell Dev. Biol. 9, 726563. doi: 10.3389/fcell.2021.726563
Moroz L. L., Romanova D. Y. (2022). Alternative neural systems: What is a neuron? (Ctenophores, sponges and placozoans). Front. Cell Dev. Biol. 10, 1071961. doi: 10.3389/fcell.2022.1071961
Moroz L. L., Romanova D. Y., Kohn A. B. (2021). Neural versus alternative integrative systems: molecular insights into origins of neurotransmitters. Philos. Trans. R Soc. Lond B Biol. Sci. 376, 20190762. doi: 10.1098/rstb.2019.0762
Muenzel D., Critchell K., Cox C., Campbell S. J., Jakub R., Suherfian W., et al. (2023). Integrating larval connectivity into the marine conservation decision-making process across spatial scales. Conserv. Biol. 37, e14038. doi: 10.1111/cobi.14038
of Life Project Consortium TDT, Blaxter M., Mieszkowska N., Palma F. D., Holland P., Durbin R., et al. (2022). Sequence locally, think globally: The Darwin Tree of Life Project. Proc. Natl. Acad. Sci. 119, e2115642118. doi: 10.1073/pnas.2115642118
Ohno S. (1970). Evolution by gene duplication. 1st ed. (Berlin, Heidelberg: Springer-Verlag), 160. doi: 10.1007/978-3-642-86659-3
Pang B., Snyder M. P. (2020). Systematic identification of silencers in human cells. Nat. Genet. 52, 254–263. doi: 10.1038/s41588-020-0578-5
Pantolini L., Studer G., Pereira J., Durairaj J., Tauriello G., Schwede T. (2024). Embedding-based alignment: combining protein language models with dynamic programming alignment to detect structural similarities in the twilight-zone. Bioinformatics 40, btad786. doi: 10.1093/bioinformatics/btad786
Peng C., Yang Z., Liu Z., Wang S., Yu H., Cui C., et al. (2021). A Systematical Survey on the TRP Channels Provides New Insight into Its Functional Diversity in Zhikong Scallop (Chlamys farreri). Int. J. Mol. Sci. 22, 11075. doi: 10.3390/ijms222011075
Peng J., Zhao L. (2024). The origin and structural evolution of de novo genes in Drosophila. Nat. Commun. 15, 810. doi: 10.1038/s41467-024-45028-1
Putnam N. H., Butts T., Ferrier D. E. K., Furlong R. F., Hellsten U., Kawashima T., et al. (2008). The amphioxus genome and the evolution of the chordate karyotype. Nature 453, 1064–1071. doi: 10.1038/nature06967
Putnam N. H., Srivastava M., Hellsten U., Dirks B., Chapman J., Salamov A., et al. (2007). Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science 317, 86–94. doi: 10.1126/science.1139158
Richter D. J., Fozouni P., Eisen M. B., King N. (2018). Gene family innovation, conservation and loss on the animal stem lineage. Elife 7, e2115859119. doi: 10.7554/eLife.34226
Robert N. S. M., Sarigol F., Zieger E., Simakov O. (2022). SYNPHONI: scale-free and phylogeny-aware reconstruction of synteny conservation and transformation across animal genomes. Bioinformatics 38, 5434–5436. doi: 10.1093/bioinformatics/btac695
Rosenbloom K. R., Dreszer T. R., Pheasant M., Barber G. P., Meyer L. R., Pohl A., et al. (2010). ENCODE whole-genome data in the UCSC Genome Browser. Nucleic Acids Res. 38, D620–D625. doi: 10.1093/nar/gkp961
Rost B. (1999). Twilight zone of protein sequence alignments. Protein Eng. 12, 85–94. doi: 10.1093/protein/12.2.85
Rusin L. Y. (2023). Evolution of homology: From archetype towards a holistic concept of cell type. J. Morphol. 284, e21569. doi: 10.1002/jmor.21569
Schultz D. T., Haddock S. H. D., Bredeson J. V., Green R. E., Simakov O., Rokhsar D. S. (2023). Ancient gene linkages support ctenophores as sister to other animals. Nature 618, 110–117. doi: 10.1038/s41586-023-05936-6
Sedlazeck F. J., Lee H., Darby C. A., Schatz M. C. (2018). Piercing the dark matter: bioinformatics of long-range sequencing and mapping. Nat. Rev. Genet 19, 329–346. doi: 10.1038/s41576-018-0003-4
Session A. M., Uno Y., Kwon T., Chapman J. A., Toyoda A., Takahashi S., et al. (2016). Genome evolution in the allotetraploid frog Xenopus laevis. Nature 538, 336–343. doi: 10.1038/nature19840
Sherkow J. S., Barker K. B., Braverman I., Cook-Deegan R., Durbin R., Easter C. L., et al. (2022). Ethical, legal, and social issues in the Earth BioGenome Project. Proc. Natl. Acad. Sci. U.S.A. 119, 397. doi: 10.1073/pnas.2115859119
Simakov O., Bredeson J., Berkoff K., Marletaz F., Mitros T., Schultz D. T., et al. (2022). Deeply conserved synteny and the evolution of metazoan chromosomes. Sci. Adv. 8, eabi5884. doi: 10.1126/sciadv.abi5884
Sisu C., Muir P., Frankish A., Fiddes I., Diekhans M., Thybert D., et al. (2020). Transcriptional activity and strain-specific history of mouse pseudogenes. Nat. Commun. 11, 3695. doi: 10.1038/s41467-020-17157-w
Stephan T., Burgess S. M., Cheng H., Danko C. G., Gill C. A., Jarvis E. D., et al. (2022). Darwinian genomics and diversity in the tree of life. Proc. Natl. Acad. Sci. U.S.A. 119. doi: 10.1073/pnas.2115644119
Strausfeld N. J., Hirth F. (2016). Introduction to “Homology and convergence in nervous system evolution. Philos. Trans. R Soc. Lond B Biol. Sci. 371, 20150034. doi: 10.1098/rstb.2015.0034
Striedter G. F., Northcutt R. G. (1991). Biological hierarchies and the concept of homology. Brain Behav. Evol. 38, 177–189. doi: 10.1159/000114387
Sun J., Chen C., Miyamoto N., Li R., Sigwart J. D., Xu T., et al. (2020). The Scaly-foot Snail genome and implications for the origins of biomineralised armour. Nat. Commun. 11, 1657. doi: 10.1038/s41467-020-15522-3
Svedberg D., Winiger R. R., Berg A., Sharma H., Tellgren-Roth C., Debrunner-Vossbrinck B. A., et al. (2024). Functional annotation of a divergent genome using sequence and structure-based similarity. BMC Genomics 25, 6. doi: 10.1186/s12864-023-09924-y
Szollosi A. (2021). Two decades of evolution of our understanding of the transient receptor potential melastatin 2 (TRPM2) cation channel. Life 11, 397. doi: 10.3390/life11050397
Toll-Riera M., San Millan A., Wagner A., MacLean R. C. (2016). The genomic basis of evolutionary innovation in pseudomonas aeruginosa. PloS Genet. 12, e1006005. doi: 10.1371/journal.pgen.1006005
van Kempen M., Kim S. S., Tumescheit C., Mirdita M., Lee J., Gilchrist C. L. M., et al. (2023). Fast and accurate protein structure search with Foldseek. Nat. Biotechnol 42, 243–246. doi: 10.1038/s41587-023-01773-0
Viladrich N., Linares C., Padilla-Gamiño J. L. (2022). Lethal and sublethal effects of thermal stress on octocorals early life-history stages. Glob Chang Biol. 28, 7049–7062. doi: 10.1111/gcb.16433
Villanueva-Cañas J. L., Ruiz-Orera J., Agea M. I., Gallo M., Andreu D., Albà M. M. (2017). New genes and functional innovation in mammals. Genome Biol. Evol. 9, 1886–1900. doi: 10.1093/gbe/evx136
Wagner G. P. (1989). The biological homology concept. Annu. Rev. Ecol. Syst. 20, 51–69. doi: 10.1146/annurev.es.20.110189.000411
Weeriyanun P., Collins R. B., Macadam A., Kiff H., Randle J. L., Quigley K. M. (2022). Predicting selection-response gradients of heat tolerance in a widespread reef-building coral. J. Exp. Biol. 225. doi: 10.1242/jeb.243344
Wilusz J. E., Sharp P. A. (2013). A circuitous route to noncoding RNA. Science 340, 440–441. doi: 10.1126/science.1238522
Wu B., Hao W., Cox M. P. (2022). Reconstruction of gene innovation associated with major evolutionary transitions in the kingdom Fungi. BMC Biol. 20, 144. doi: 10.1186/s12915-022-01346-8
Wu L., Lambert J. D. (2023). Clade-specific genes and the evolutionary origin of novelty; new tools in the toolkit. Semin. Cell Dev. Biol. 145, 52–59. doi: 10.1016/j.semcdb.2022.05.025
Yip K. Y., Cheng C., Bhardwaj N., Brown J. B., Leng J., Kundaje A., et al. (2012). Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcription-related factors. Genome Biol. 13, R48. doi: 10.1186/gb-2012-13-9-r48
Yu H., Li Y., Han W., Bao L., Liu F., Ma Y., et al. (2024). Pan-evolutionary and regulatory genome architecture delineated by an integrated macro- and microsynteny approach. Nat. Protoc. doi: 10.1038/s41596-024-00966-4
Zeng X., Zhang Y., Meng L., Fan G., Bai J., Chen J., et al. (2020). Genome sequencing of deep-sea hydrothermal vent snails reveals adaptions to extreme environments. Gigascience 9. doi: 10.1093/gigascience/giaa139
Zhang J. (2003). Evolution by gene duplication: an update. Trends Ecol. Evol. 18, 292–298. doi: 10.1016/S0169-5347(03)00033-8
Zhang G., Fang X., Guo X., Li L., Luo R., Xu F., et al. (2012). The oyster genome reveals stress adaptation and complexity of shell formation. Nature 490, 49–54. doi: 10.1038/nature11413
Zhang Y., Nie H., Yan X. (2023). Transient receptor potential (TRP) channels in the Manila clam (Ruditapes philippinarum): Characterization and expression patterns of the TRP gene family under heat stress in Manila clams based on genome-wide identification. Gene 854, 147112. doi: 10.1016/j.gene.2022.147112
Keywords: homology, genomic dark matter, hotspot, functional and biodiversity genomics, TRP ion channels, heat stress resilience, climate change, molluscs
Citation: Edsinger E and Moroz LL (2024) Genomic hotspots: localized chromosome gene expansions identify lineage-specific innovations as targets for functional biodiversity and predictions of stress resilience. Front. Mar. Sci. 11:1434130. doi: 10.3389/fmars.2024.1434130
Received: 17 May 2024; Accepted: 26 June 2024;
Published: 01 August 2024.
Edited by:
Nathan James Kenny, University of Otago, New ZealandReviewed by:
Leandro Aristide, National Scientific and Technical Research Council (CONICET), ArgentinaMarco Gerdol, University of Trieste, Italy
Copyright © 2024 Edsinger and Moroz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric Edsinger, MDAwZ2VuZXJpY0BnbWFpbC5jb20=
†ORCID: Eric Edsinger, orcid.org/0000-0002-1012-1506
Leonid L. Moroz, orcid.org/0000-0002-1333-3176