Abstract
Biological nitrogen fixation plays a crucial role in the marine nitrogen cycle, impacting global marine productivity and related carbon fluxes. The strains were analyzed by gene annotation, growth conditions and phylogenetic analysis of 16S rRNA gene sequences.These two strains were isolated from the coastal sediment at Xiaoshi Island in Weihai, China. The strains were analyzed by gene annotation, growth conditions and phylogenetic analysis of 16S rRNA gene sequences. It was revealed that strains D04T and AATT contain a set of nif gene clusters responsible for nitrogen fixation. Cell are yellow-colored, Gram-stain-negative, facultatively anaerobic, and rod-shaped bacteria. The optimal growth conditions for strain D04T were found to be at 33 °C, pH 7.0, and in 2% (w/v) NaCl, while strain AATT prefers growth conditions at 33 °C, pH 6.5, and in 3% (w/v) NaCl. The highest similarity of strains D04T and AATT was to Saccharicrinis fermentans NBRC 15936T, with a similarity of 94.1% and 94.8%, respectively. The 16S rRNA gene sequence similarity between the two strains was 96.6%. These novel strains were found to represent new taxa of the Marinilabiliaceae family, and we propose the names Plebeiobacterium marinum gen. nov., sp. nov. and Plebeiobacterium sediminum sp. nov. with type strains D04T (MCCC 1H00493T = KCTC 92026T) and AATT (MCCC 1H00485T = KCTC 92028T), respectively. In this study, nitrogen fixation genes were predicted for 53 strains from the whole order Marinilabiliales and it was found that nitrogen fixation gene clusters were present in 26 strains. These gene clusters were found in every family in the order, highlighting that the presence of nitrogen-fixing gene clusters in the order is common. Nitrogen-fixing bacteria in sediments play an important role in various biogeochemical cycles. Thus, understanding the oceanic nitrogen cycle can provide insights into the energy flow of marine systems.
1 Introduction
Nitrogen is an essential element for all living organisms. Molecular nitrogen (N2) comprises 78% of the Earth’s atmosphere and is also abundant in the ocean water column. However, N2 is highly stable and cannot be easily converted into a form that most organisms can utilize. The process of converting N2 into bioavailable NH3 is known as biological nitrogen fixation. (Weisburg et al., 1991; Chakraborty et al., 2021). In natural ecosystems, the supply of nitrogen to organisms plays a critical role in regulating primary productivity (Isobe and Ohte, 2014). It has been proved that microorganisms can mediate many N cycle processes, and microbial communities significantly contribute to N cycle processes (Isobe and Ohte, 2014). Biological nitrogen fixation is a critical aspect of the marine nitrogen cycle and plays a significant role in regulating global marine productivity and carbon flux (Sohm et al., 2011; Halm et al., 2012). It was long believed that the majority of nitrogen fixation in the ocean was carried out by cyanobacteria in surface waters. However, recent research has shown that nitrogen fixation also occurs in deeper waters, coastal regions, and even marine sediments, indicating that it is a widespread and significant phenomenon (Chakraborty et al., 2021; Luo et al., 2021). Similar to nitrogen-fixing microorganisms in terrestrial environments, a wide range of prokaryotes are capable of fixing nitrogen in the ocean. Recent studies have shown that nitrogen fixation can occur in marine sediments around seaweed communities, where heterotrophic sulfate-reducing bacteria play a significant role. These bacteria can fix nitrogen and provide bioavailable nitrogen directly to the seaweed, promoting its growth and productivity (Lehnen et al., 2016; Mohr et al., 2021). Nitrogen-fixing bacteria have been found to play a crucial role in nitrogen cycling in deep-sea sediments, with several groups identified, including Acidobacteria, Firmicutes, Nitrospirae, Gammaproteobacteria, and Deltaproteobacteria. These groups of bacteria are characterized by their ability to use a range of electron acceptors, including oxygen, nitrate, iron, sulfur, sulfate, and organic compounds, which allow them to couple nitrogen fixation with multiple biogeochemical cycles. This enables them to make important contributions to other elemental cycles, such as those of carbon, sulfur, and iron in deep-sea sediments (Kapili et al., 2020). Heterotrophic nitrogen-fixing bacteria have been found to be present in marine sediments, including aerobic, microaerobic, facultative, and specific anaerobic bacteria. The presence of these bacteria, such as Azotobacter, Clostridium, Enterobacter, Desulfobacter, Desulfovibrio, and Klebsiella, is critical for nitrogen cycling in marine ecosystems (Herbert, 1999).
Nitrogen plays a central role in marine biogeochemistry as a limiting element in biological production (Kuypers et al., 2018). Biological nitrogen fixation is the main process controlling the nitrogen supply of marine organisms (Zehr, 2011). Microbes use nitrogenase to catalyze biological nitrogen fixation, and nitrogenase is highly conserved during evolution. Nitrogen-fixing bacteria are divided into five clusters by constructing a gene phylogenetic tree (Zehr et al., 2003; Raymond et al., 2004). Three clusters (I, II, and III) contain genes encoding nitrogenase, while clusters IV and V are mainly homologous genes of nitrogenase genes and do not participate in nitrogen fixation (Gaby and Buckley, 2011). Cluster IV and V genes have various functions, including some genes involved in the biosynthesis of photopigments and some electron transfer reactions. (Gaby and Buckley, 2011). Most known nifH sequences belong to cluster I. Cluster I is composed entirely of nifH genes from the bacteria’s regular FeMo nitrogen-fixing enzyme (nifH). The cluster contains genes from all Cyanobacteria, most Proteobacteria, and certain Firmicutes and Actinobacteria. Cluster II contains sequences of certain methanogenic archaea that belong mainly to alternative FeV (vnfH) and FeFe (anfH) nitrogen-fixing enzymes, with relatively few sequences. Cluster III consists mainly of sequences of anaerobic bacteria and archaea, including spirochetes, methanogens, acetogens, sulfate-reducing bacteria, green sulfur bacteria, and clostridia (Gaby and Buckley, 2011). These genes include the key gene nifH, encoding the dinitrogenase reductase; nifD and nifK, encoding the MoFe dinitrogenase; as well as nifE, nifN, and nifB, encoding the FeMo cofactor biosynthesis machinery. The nifN and nifB genes were fused into a single gene (nifN-B) and two genes encoding PII-like nitrogen regulatory proteins (Sayavedra et al., 2021). nifA is an essential transcriptional activator for nif genes (Sarkar and Reinhold-Hurek, 2014). nifO seems to be necessary for nitrogen fixation in the presence of nitrate (Gutierrez et al., 1997).
Nitrogen fixation enzymes are mainly composed of two metalloproteins, including diazotrope in the catalytic part and diazoreductase in the electron transport part. Molybdenum-dependent nitrogen fixation enzymes are mainly composed of nifH (diazoreductase subunit), nifD, and nifK (diazotrope subunit) (López-Torrejón et al., 2016). The nifD and nifK genes encode the α- and β-subunits of diazoxide synthase, respectively, forming an α2β2-tetramer, the nifE and nifN genes encode another α2β2-tetramer necessary for metal cofactors, and nifB plays a major role in the biosynthesis of Fe and S donors for metal cofactors. The above six nif genes are used to define nif (nifHDKENB) (Pi et al., 2022).
Previous studies have primarily focused on nitrogen-fixing bacteria within the Proteobacteria phylum, with little attention paid to the potential role of Bacteroidetes. However, recent research has shown that several families of Bacteroidetes have the potential for nitrogen fixation. Gene sequencing analysis has predicted that Bacteroidetes families such as Marinilabiliaceae (Geofilum, Saccharicrinis, and Alkiflexus), Porphyromonadaceae (Paludibacter and Dysgonomonas), and Bacteroidaceae have nitrogen fixation potential (Inoue et al., 2015). Additionally, another study has also identified members of the Prolixibactaceae family, including Maribellus, Mangrovibacterium, and Draconibacillus, as Bacteroidetes with the potential for nitrogen fixation (Huang et al., 2020). Marinilabiliaceae, Prolixibacteraceae, and Marinifilaceae were transferred from the Bacteroides order to Marinilabiliales. (Wu et al., 2016). The current study of nitrogen-fixing enzymes is performed through the nifH gene (Zehr et al., 2003). This study annotates the obtained genome and analyzes the nitrogen fixation–related gene clusters of this order. The results of this study help to fill the gap in our knowledge of the nitrogen-fixing potential of this microbial group, and provide a theoretical basis for further research on nitrogen-fixing microbial communities in marine sediments.
As of the time of writing, the family Marinilabiliaceae is composed of twelve genera with a validly published and correct name1. These genera include Alkaliflexus, Alkalitalea, Anaerophaga, Breznakibacter, Carboxylicivirga, Geofilum, Labilibacter, Mangroviflexus, Marinilabilia, Natronoflexus, Saccharicrinis, and Thermophagus. The family encompasses a total of twenty-two species. Apart from the strains Alkaliflexus imshenetskii (Zhilina et al., 2004), Alkalitalea saponilacus (Zhao and Chen, 2012), and Natronoflexus pectinivorans (Sorokin et al., 2011), isolated from lakes and Anaerophaga thermohalophila (Denger et al., 2002) isolated from oil, others come from marine environments. Bacterial cells of the family Marinilabiliaceae are facultatively anaerobic and Gram-stain-negative, with menaquinone-7 (MK-7) as the major respiratory quinone. In this study, two yellow-colored, Gram-stain-negative, facultatively aerobic, and rod-shaped bacterium; strains D04T and AATT, were proposed as representing novel species belonging to the family Marinilabiliaceae.
2 Materials and methods
2.1 Sample collection, bacterial isolation, and preservation
A marine sediment sample was collected off the coast of Xiaoshi Island, Weihai, PR China (122°00′58′′ E, 37°31′36′′ N) and subjected to an enrichment culture technique as described by Mu (Mu et al., 2018). The sample was collected at a depth of approximately 10 cm and was kept cold and in the dark during transportation to the laboratory. Incubation was performed at 25°C for 8 months using a 350 ml sealed glass bottle. The bottle was shaken twice a day and kept sealed during the incubation. The sediment sample was serially diluted to 10−3 in sterilized seawater and 0.1 ml aliquots of each dilution were spread on to the surface of modified marine agar (MA) 2216. The modified MA medium used for the characterization of strains contained the following components (g L−1, pH 7.0): 5.0 tryptone, 1.0 yeast extract, 1.5 pyruvic acid sodium, and 2 ml vitamin mixture. The vitamin mixture contained the following components (mg L−1): 5.0 thiamine, 5.0 riboflavin, 5.0 nicotinic acid, 5.0 D-Ca-pantothenate, 10.0 pyridoxine-HCl, 2.0 biotin, 2.0 folic acid, 0.1 cobalamin, 5.0 lipoic acid and 5.0 p-aminobenzoic acid. After incubation for 7 days at 33°C, colonies of the two strains were selected from the plate and re-streaked to obtain pure cultures. These strains were then cultivated on modified MA 2216 at 33°C for further physiological, biochemical, and chemical analyses. Pure cultures of the strains were preserved at −80°C in a sterile 1% (w/v) saline solution supplemented with 15% (v/v) glycerol.
2.2 Morphological, physiological, and biochemical analyses
The morphological and physiological features of strains D04T and AATT were tested with cells grown on the modified MA at optimum growth conditions for 5 days. The experiment was repeated three times. Cell morphology and size were examined by light microscopy (E600; Nikon), transmission electron microscopy (JEM-1200, JEOL) and scanning electron microscopy (model Nova NanoSEM450; FEI). Gram reactions were carried out as described previously (CLSI, 2021). Gliding motility was examined in modified marine broth 2216 (MB; BD) supplemented with 0.3% agar as described previously (Bernardet et al., 2002). The growth temperature range was tested on the modified MA at various temperatures (4, 20, 25, 28, 30, 33, 37, 40, 45, and 50°C). To test for the pH range suitable for growth, bacterial strains were grown in modified marine broth 2216 with different buffers at a concentration of 20 mM. MES buffer was used for pH 5.5 and 6.0, PIPES buffer for pH 6.5 and 7.0, HEPES buffer for pH 7.5 and 8.0, Tricine buffer for pH 8.5, and CAPSO buffer for pH 9.0 and 9.5. For investigating growth at different NaCl concentrations, a medium containing 1 g/L−1 yeast extract, 5 g/L−1 peptone, and 20 g/L−1 agar prepared with artificial seawater [0.22% MgCl2, 0.15% CaCl2, 0.32% MgSO4, 0.02% NaHCO3, and 0.07% KCl, with 3% NaCl (w/v)] was used. The bacterial strains were grown in this medium at NaCl concentrations ranging from 0% to 9% in increments of 0.5%.
The susceptibility of the bacterial strains D04T and AATT to various antibiotics was tested using the disc diffusion method, as previously described (Du et al., 2014). A cell suspension of 0.5 McFarland standard was swabbed over the modified marine agar to create a uniform lawn before the aseptic placement of antibiotic discs onto the surface. After incubation for 5 days, the plates were examined for a clear zone of growth inhibition around the antibiotic discs. A total of 20 different antibiotic discs (with varying concentrations measured in micrograms per disc) were used to test the susceptibility of the bacterial strains D04T and AATT: lincomycin (2), streptomycin (10), erythromycin (15), clarithromycin (15), kanamycin (30), ampicillin (10), rifampin (5), tetracycline (30), penicillin (10), chloramphenicol (30), cefotaxime sodium (30), gentamycin (10), ofloxacin (5), norfloxacin (30), neomycin (30), carbenicillin (100), ceftriaxone (30) tobramycin (10), vancomycin (30), and polymyxin B (300). CLSI standards were strictly followed for cultivation and inhibition zone diameter reading (Gcrhardt et al., 1994).
Bacterial growth was monitored at 600 nm using a spectrophotometer. Catalase and oxidase activity were tested by pouring 3% H2O2 solution onto the cells and by using an oxidase test reagent (bioMérieux), respectively. Anaerobic growth was tested for 15 days at 30°C on the modified MA with or without 0.1% (w/v) NaNO3 in an anaerobic bag (hopebio). Hydrolyses of agar, starch, alginate, casein, CM-cellulose, DNA, and lipase (Tweens 20, 40, 60, and 80) were examined according to the method of CLSI (2021). API 20E and API 50CH tests (bioMérieux) were carried out according to the manufacturer’s instructions (except for salinity, which was adjusted to 3%). Enzyme activities were examined using the API ZYM test (bioMérieux). Oxidization of different compounds was tested in Biolog GEN III microplates according to the manufacturer’s instructions. All the API and Biolog tests were performed with three biological replicates each time and with two reference strains.
2.3 16S rRNA gene sequence analysis
The 16S rRNA gene sequences were amplified using PCR with the primer pair 27F (5’-AGAGTTTGATCMTGGCTCAG-3’) and 1492R (5’-TACGGYTACCTTGTTACGACTT-3’) (Jordan et al., 2007; Du et al., 2014). Purified PCR products were ligated into the pMD18-T vector (Takara) and cloned by following the manufacturer’s instructions. Sequencing was performed by BGI Co. Ltd (Qingdao, PR China). The nearly complete 16S rRNA gene sequences of strains D04T and AATT were obtained and compared with those available from the EzBioCloud2 (Yoon et al., 2017a) database for further phylogenetic analysis. Phylogenetic trees were constructed using MEGA version 11 employing the neighbor-joining (Saitou and Nei, 1987), maximum-likelihood (Felsenstein, 1981), and maximum-parsimony methods (Kumar, 1996). Bootstrap analyses were performed based on 1,000 replicates to estimate the confidence of branches in the generated phylogenetic trees (Felsenstein, 1985).
2.4 Genome sequencing analysis
Genomic DNA of strains D04T and AATT was extracted and purified using a bacteria genomic DNA kit (Takara). The draft genome sequences of strains D04T and AATT were sequenced by Majorbio (MajorBio Co., Shanghai, China) using an Illumina MiSeq (Illumina, USA). The final genome was assembled with SOPA de novo version 2.04 (Luo et al., 2012). The resulting genomes were annotated using RAST (rapid annotation using subsystem technology online server)3 (Aziz et al., 2008). The molecular functions of genes and proteins are associated with ortholog groups and stored in the KEGG orthology (KO) database (Kanehisa et al., 2016). The genes involved in metabolic pathways and ortholog groups of proteins were analyzed using the4 and dbCAN2 meta servers5 (Zhang et al., 2018). The presence of gene clusters encoding secondary metabolites was predicted using antiSMASH 6.1.16 (Blin et al., 2021). The DNA G+C content was calculated based on the whole genome sequence. The AAI calculator estimates the average amino acid identity using both best hits (one-way AAI) and reciprocal best hits (two-way AAI) between two genomic datasets of proteins7. Average nucleotide identity (ANI) values were calculated using the ChunLab’s online ANI Calculator8 (Lee et al., 2016). The genome-to-genome distance calculator (GGDC 3.0)9 (Meier-Kolthoff et al., 2013) was used to calculate the digital DNA-DNA hybridization (dDDH).
2.5 Chemotaxonomic analyses
The reference strain Saccharicrinis fermentans DSM 9555T was obtained from the German Collection of Microorganisms and Cell Cultures GmbH and Labilibacter aurantiacus HQYD1T was obtained from our laboratory. Both strains were cultured on modified MA at 33°C and studied in parallel with strains D04T and AATT for physiological and chemotaxonomic comparisons. Cellular fatty acids and polar lipids were identified from a freeze-dried sample (40 mg and 50 mg) of cells grown to the exponential growth phase under optimal culture conditions on the modified MA. Cellular fatty acid methyl esters (FAMEs) were obtained from cells by saponification, methylation, and extraction following the MIDI protocol (Sasser, 1990). Cellular FAMEs were separated using gas chromatography and were identified and quantified using the Sherlock Microbial Identification System (version 6.1, MIDI 6890 with database TSBA6). Polar lipids were separated via two-dimensional silica gel TLC. Total lipid material was detected using molybdatophosphoric acid and the functional groups were determined using spray reagents specific for each group according to Fang et al. (2017). All polar lipid images were further analyzed as described by Minnikin et al. (1984). For respiratory quinone analyses, respiratory quinones were extracted from 200 mg of freeze-dried cell material using the two-stage method described by Minnikin et al. (1984). A silica-gel TLC plate (Merck Kieselgel 60 F254) was used to analyze the quinone type and the content of each quinone type was subsequently analyzed by HPLC according to the process described previously (Kroppenstedt, 1982).
3 Results and discussion
3.1 Morphology, physiology, and biochemical characteristics
The newly discovered strains D04T and AATT of bacteria belonging to the Marinilabiliaceae family were facultative anaerobes, Gram-negative, rod-shaped, oxidase-positive, and catalase-negative. Strain D04T had a width of 0.3 μm and a length of 3.4–4.1 μm, while strain AATT had a width of 0.4–0.5 μm and a length of 0.6–1.4 μm (Supplementary Figure 1). Strain D04T also demonstrated gliding motility, while strain AATT did not. Both strains grew as yellow-colored, smooth, and circular colonies on modified MA medium at 33°C. They were oxidase-positive, catalase-negative, and reduced nitrate to nitrite. The growth temperature range of the bacterial strains studied was between 20°C and 40°C and they were unable to grow at temperatures of 4°C and 45°C. Strains D04T and AATT showed optimal growth at a temperature of 33°C. The pH range for growth of strain D04T was 6.5–7.5, with optimal growth occurring at pH 7.0. Strain AATT was able to grow at pH values ranging from 6.0–7.5, with optimal growth occurring at a pH of 6.5. In terms of salt tolerance, strain D04T was able to grow at NaCl concentrations ranging from 0% to 8.0% w/v, with an optimum NaCl concentration of 2.0%. Strain AATT was able to grow at NaCl concentrations ranging from 0.5% to 8.0% w/v, with an optimum NaCl concentration of 3.0%. The addition of a vitamin mixture to the medium increased their growth. The strains were positive for the hydrolysis of starch but negative for casein, CM-cellulose, alginate, and Tweens 20, 40, 60, and 80. Strain AATT was positive for DNA, while strain D04T was negative. Both strains were resistant to antibiotics such as tobramycin, vancomycin, tetracycline, norfloxacin, neomycin, gentamycin, ofloxacin, streptomycin, kanamycin, and polymyxinB. However, strain D04T was susceptible to erythromycin, ampicillin, penicillin, chloramphenicol, rifampin, and lincomycin, while strain AATT was between resistant and sensitive to cefotaxime sodium and carbenicillin.
Strains D04T and AATT share similar morphological and biochemical characteristics with the other two species in the Marinilabiliaceae family, including being rod-shaped, Gram-stain-negative, and facultatively aerobic. They also showed positive results for various enzyme activities such as alkaline phosphatase, esterase (C4), esterase lipase (C8), acid phosphatase, and naphthol-AS-BI-phosphohydrolase, as well as for tryptophan deaminase, Voges–Proskauer reaction, and arabinose. However, there were some differential phenotypic characteristics between strains D04T and AATT and the two reference strains, as shown in Table 1.
Table 1
| Characteristic | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Colony color | Yellow | Yellow | Orange a* | Orange b* |
| Temperature (°C) (optimum temperature) | 20–40 (33) | 20–40 (33) | 19–37 (30–32) a* | 4–37 (28) b* |
| NaCl (%) (optimum NaCl%) | 0–8.0 (2.0) | 0.5–8.0 (3.0) | 1.0–6.0 (2.5) a* | 1.0–5.0 (2.0-3.0) b* |
| pH (optimum pH) | 6.5–7.5 (7.0) | 6.0–7.6 (6.5) | 7.0–9.0 (8.0) a* | 6.0–8.5 (7.0–7.5) b* |
| Nitrate reduction | + | + | −a* | −b* |
| Enzyme activity (API ZYM): | ||||
| leucine arylamidase | − | + | − | − |
| cystine arylamidase | − | + | − | − |
| trypsin | − | + | − | + |
| β-galactosidase | + | + | + | − |
| α-glucosidase | − | + | − | − |
| β-glucosidase | + | + | − | − |
| N-acetyl-β-glucosaminidase | − | + | − | − |
| Utilization of (API 20E) | ||||
| arginine dihydrolase | − | − | + | + |
| ornithine decarboxylase | − | − | − | + |
| urease | − | − | − | + |
| gelatinase | − | − | + | + |
| glucose | − | − | + | + |
| mannitol | − | − | + | − |
| inositol | − | − | + | − |
| sorbitol | − | − | + | − |
| sucrose | − | − | + | − |
| melibiose | − | − | + | + |
| amygdalin | − | − | + | − |
| Acid production from (API 50CH) | ||||
| mannose | − | − | + | − |
| erythritol | − | + | − | − |
| l-arabinose | + | + | + | − |
| l-xylose | – | – | + | − |
| d-adonitol | + | − | − | − |
| methyl-β-d-xylopyranoside | + | − | + | − |
| d-fructose | + | + | − | + |
| d-mannose | + | + | + | − |
| l-rhamnose | + | + | − | − |
| dulcitol | + | − | + | − |
| inositol | + | − | − | − |
| methyl-α-d-mannopyranoside | − | − | + | − |
| N-acetylglucosamine | − | + | + | + |
| amygdalin | − | + | + | + |
| d-trehalose | − | − | + | + |
| inulin | + | + | − | − |
| d-raffinose | + | + | − | + |
| xylitol | + | + | − | − |
| d-tagatose | + | − | + | − |
| d-fucose | − | + | + | − |
| l-fucose | + | + | + | − |
| d-arabitol | + | − | − | − |
| l-arabitol | + | + | − | − |
Differential characteristics between the new strains and reference strains.
Strains: 1, D04T; 2, AATT; 3, Saccharicrinis fermentans DSM 9555T; and 4, Labilibacter aurantiacus HQYD1T. All data are from this study unless indicated otherwise. All strains are positive for alkali phosphatase, esterase (C4), esterase lipase (C8), acid phosphatase, and naphthol-AS-BI-phosphohydrolase, but negative for lipase (C14), valine arylamidase, a-chymotrypsin, α-galactosidase, β-glucuronidase, α-mannosidase and β-fucosidase. All strains are also positive for the utilization of tryptophan deaminase, Voges–Proskauer reaction, and arabinose, but negative for lysine decarboxylase, citrate utilization, H2S production, indole production, rhamnol, and o-nitrophenyl-β-D-galactopyranoside. All strains produced acid from d-arabinose, d-ribose, d-xylose, d-galactose, d-glucose, arbulin, aesculin ferric citrate, salicin, d-cellobiose, d-maltose, d-lactose, d-melibiose, d-saccharose, starch, glycogen, d-gentiobiose, d-turanose, d-lyxose, potassium 2-keto-gluconate, and potassium 5-keto-gluconate, but not from l-sorbose, d-mannitol, d-sorbitol, methyl-α-d-glucopyranoside, d-melezitose and potassium gluconate. +, Positive; −, negative.
*Data taken from: a (Yang et al., 2014); b (Lu et al., 2017).
3.2 Comparison and phylogenetic analysis of the 16S rRNA gene sequences
The nearly complete 16S rRNA gene sequences of strains D04T (1480 bp) and AATT (1525 bp) were obtained. Sequence comparisons showed that strains D04T and AATT had the highest 16S rRNA gene sequence similarity with the type strain of Saccharicrinis fermentans DSM 9555T (94.1% and 94.9%, respectively), followed by Saccharicrinis marinus Y11T (93.3% and 94.5%, respectively). The 16S rRNA gene sequences of strains D04T and AATT were similar to those of Labilibacter aurantiacus HQYD1T type strains (93.3% and 93.4%, respectively). The similarity of the 16S rRNA gene sequence between the two strains was 96.6%. The neighbor-joining, maximum-likelihood, and maximum-parsimony phylogenetic trees formed an independent sub-cluster supported by very high bootstrap values (Figure 1). In the phylogenetic tree, strains D04T and AATT are closely related to the members of the genera Saccharicrinis and Labilibacter. By using 16S rRNA gene sequence similarity and phylogenetic trees, Saccharicrinis fermentans DSM 9555T and Labilibacter aurantiacus HQYD1T were selected as reference strains.
Figure 1

Neighbor-joining phylogenetic tree based on the 16S rRNA gene sequences showing the phylogenetic position of strains D04T and AATT among members of the order Marinilabiliales. Numbers on nodes represent bootstrap values (NJ, ML, and MP) based on 1,000 replications. The sequence of Dysgonomonas gadei 1145589T (AB548675) was used as an outgroup. Bar, 0.02 substitutions per nucleotide position.
3.3 Genomic features
3.3.1 Basic characteristics
The obtained draft genome of D04T is comprised of 187 scaffolds (N50 = 106,391 bp) and is 5,621,283 bp in length. The obtained draft genome of AATT is comprised of 428 scaffolds (N50 = 74,668 bp) and is 6,588,154 bp in length. The G+C content of strains D04T and AATT were 36.6% and 33.4%. The annotated genome of strain D04T contained a total of 4,522 genes, with 4,456 protein-coding genes, 3 rRNAs, and 63 tRNAs. The annotated genome of strain AATT contained a total of 5,207 genes, with 5,130 protein-coding genes, 7 rRNAs, and 70 tRNAs. The range of the AAI values of strains D04T and AATT in the family Marinilabiliaceae genus are 62–72% and 61–71%, respectively, which were below the threshold value (74%) for genus delineation (Nicholson et al., 2020). The AAI value of strains D04T and AATT is 77%, which was higher than the threshold value (74%) for genus delineation (Supplementary Figure 2A). The ANI value of strains D04T and AATT is less than 74%, which is below the threshold (95%) for new species identification (Supplementary Figure 2B) (Yoon et al., 2017b). The dDDH value of strains D04T and AATT is less than 22%, which is below the threshold (70%) for new species identification (Goris et al., 2007). The phylogenomic maximum-likelihood tree based on 120 core genes showed that strains D04T and AATT formed distinct lineages within the family Marinilabiliaceae (Supplementary Figure 3).
3.3.2 Carbohydrate-active enzymes and analysis of SusC and SusD genes
The carbohydrate-active enzyme (CAZy) database builds and breaks down complex carbohydrates and glycoconjugates, covering the following classes of enzyme activities: glycoside hydrolases (GHs), carbohydrate esterases (CEs), glycosyltransferases (GTs), polysaccharide lyases (PLs), auxiliary activities (AA), and carbohydrate-binding modules (CBMs) (Cantarel et al., 2009). Four strains of the family Marinilabiliaceae have been analyzed using the CAZyme database. The strains D04T, AATT, Labilibacter aurantiacus HQYD1T, and Saccharicrinis fermentans DSM 9555T contain 336, 341, 252, and 272 carbohydrate-active enzymes, respectively. Among these carbohydrate-active enzymes, glycoside hydrolases (GHs) are the most abundant CAZymes (more than 70% of the identified enzymes were assigned to the GH family). The strains D04T, AATT, Labilibacter aurantiacus HQYD1T, and Saccharicrinis fermentans DSM 9555T all lack the AA gene (Supplementary Figure 4A). The first starch utilization system (Sus) was reported in human gut bacterium Bacteroides. The Sus operon is proposed to be essential for glycan degradation. SusC (SusC represents the actual TonB-dependent transporter) and SusD (SusD is an associated substrate binding outer membrane lipoprotein) are indispensable (Zheng et al., 2021). Prediction of SusC and SusD genes in strains D04T and AATT was performed using RAST. The strain D04T contains 19 SusC protein genes and 9 SusD protein genes. The strain AATT contains 14 SusC protein genes and 6 SusD protein genes (Supplementary Table 1).
3.3.3 Prediction of secondary metabolites
Based on secondary metabolite analysis predicted by antiSMASH, the strains D04T, AATT, Labilibacter aurantiacus HQYD1T, and Saccharicrinis fermentans DSM 9555T shared gene cluster encoding for RRE-containing (RRE-element containing cluster), NRPS-like (NRPS-like fragment), and terpene cyclase (terpene). Compared with other strains, the two new strains possess more biosynthetic clusters, among which, strains D04T and AATT possess arylpolyene (aryl polyene cluster) and resorcinol (resorcinol cluster) biosynthetic gene clusters. The Labilibacter aurantiacus HQYD1T strain clusters include lanthipeptide-class-IV (class IV lanthipeptide clusters such as venezuelin), NRPS (non-ribosomal peptide synthetase cluster), and T1PKS (type I PKS (polyketide synthase)) biosynthetic clusters (Supplementary Figure 4B).
3.3.4 Analysis of metabolic pathways
Metabolic pathways were analyzed using the KEGG Orthology (KO) database service. All four strains possessed many complete metabolic pathways, including carbon metabolism, energy metabolism, lipid metabolism, nucleic acid metabolism, amino acid metabolism, metabolism of cofactors and vitamins, and biosynthesis of terpenoids and polyketides. The strain D04T possessed the least complete pathways. The strain AATT possessed the most complete pathways. All strains showed a complete PE biosynthesis pathway (M00093), which was consistent with the polar lipid detected in the four strains. It is noteworthy that these strains have complete nitrogen fixation pathways (M00175) (Supplementary Figure 4C).
3.3.5 Analysis of nitrogen fixation
The Marinilabiliaceae, Prolixibacteraceae, and Marinifilaceae of Bacteroidetes was classified as a new order named Marinilabiliales (Wu et al., 2016). At the time of writing, 63 species of Marinilabiliales can be found on LPSN1, of which 12 have no genomic data (Figure 2). Recently, a study was conducted to predict the presence of nitrogen fixation genes in five different strains of the family Prolixibacteraceae (Huang et al., 2020). The nitrogen fixation genes of 53 strains of the order Marinilabiliales were annotated using the genome. Of the 53 strains, 26 were found to contain nitrogen fixation gene clusters. These gene clusters were detected in all three families within the order. The proportion of nitrogen-fixing gene clusters in Marinilabiliaceae, Prolixibacteraceae, and Marinifilaceae genomes was 57%, 50%, and 25%, respectively (Figure 2). Marinilabiliales are mainly found in microbial mats, animal intestines and feces, and various sediments. The Marinilabiliaceae family has more strains with nitrogen fixation potential. This family mainly exists in the deep anoxic zone of the microbial mat, where cyanobacteria are degraded and nourish the surrounding microorganisms as organic matter. Nitrogen-fixing flora provide the cyanobacteria with fixed nitrogen and other factors necessary for growth, forming a reciprocal nitrogen fixation alliance. This alliance is important for the cycling of nitrogen in marine environments and contributes to the productivity and sustainability of marine ecosystems (Steppe et al., 1996; Ben Hania et al., 2017). Nitrogen-fixing enzymes are inactivated when exposed to oxygen. However, the nitrogen-fixing enzyme activity increases significantly when the oxygen concentration in the sediment environment is close to 5% (Smercina et al., 2019) (Supplementary Figure 5). It has been hypothesized that the retention of nitrogen-fixing gene clusters in taxon is related to environmental distribution (Harwood, 2020). The absence of nitrogen fixation-related gene clusters in the genomes of some members of the Marinilabiliales order suggests that the not all Marinilabiliales members contained nif gene clusters. Particularly in the family Marinifilaceae, the nitrogen-fixing gene cluster only exists in the genus Labilibaculum. Analysis of the results showed that the conserved features of nif gene clusters in some lineages of the order Marinilabiliales were lost (Figures 2, 3). Marinilabiliales strains all have a complete set of nitrogen fixation gene clusters, including nifH, nifD, nifK, nifE, nifN, and nifB. It is hypothesized that this taxon has nitrogen fixation potential, providing a basis for studying nitrogen fixation by nitrogen-fixing organisms in ecosystems (Figure 3; Supplementary Table 2). The phylogenetic tree based on the nifH gene is not similar to that based on the 16S rRNA gene; however, the phylogenetic relationship of nifH genes in the novel isolate and reference strains are closely related, similar to that in the phylogenetic tree using the 16S rRNA gene (Figures 4A, B). Nitrogen-fixing bacteria living in the sediment surrounding seagrass play a crucial role in providing bioavailable nitrogen directly to the plant and promoting its growth. This bacterial process helps to maintain the productivity and sustainability of seagrass ecosystems. Similarly, nitrogen-fixing bacteria in deep-sea sediments can use a range of terminal electron acceptors, including oxygen, nitrate, iron, sulfur, sulfate, and organic compounds, to fix nitrogen. The potential coupling of deep-sea diazo nutrition with a variety of biogeochemical cycles can have significant implications for global nutrient cycling. The bacterial strains used in this study were isolated from intertidal sediment, providing a foundation for future research on the ecological functions of microbial communities in intertidal sediments.
Figure 2
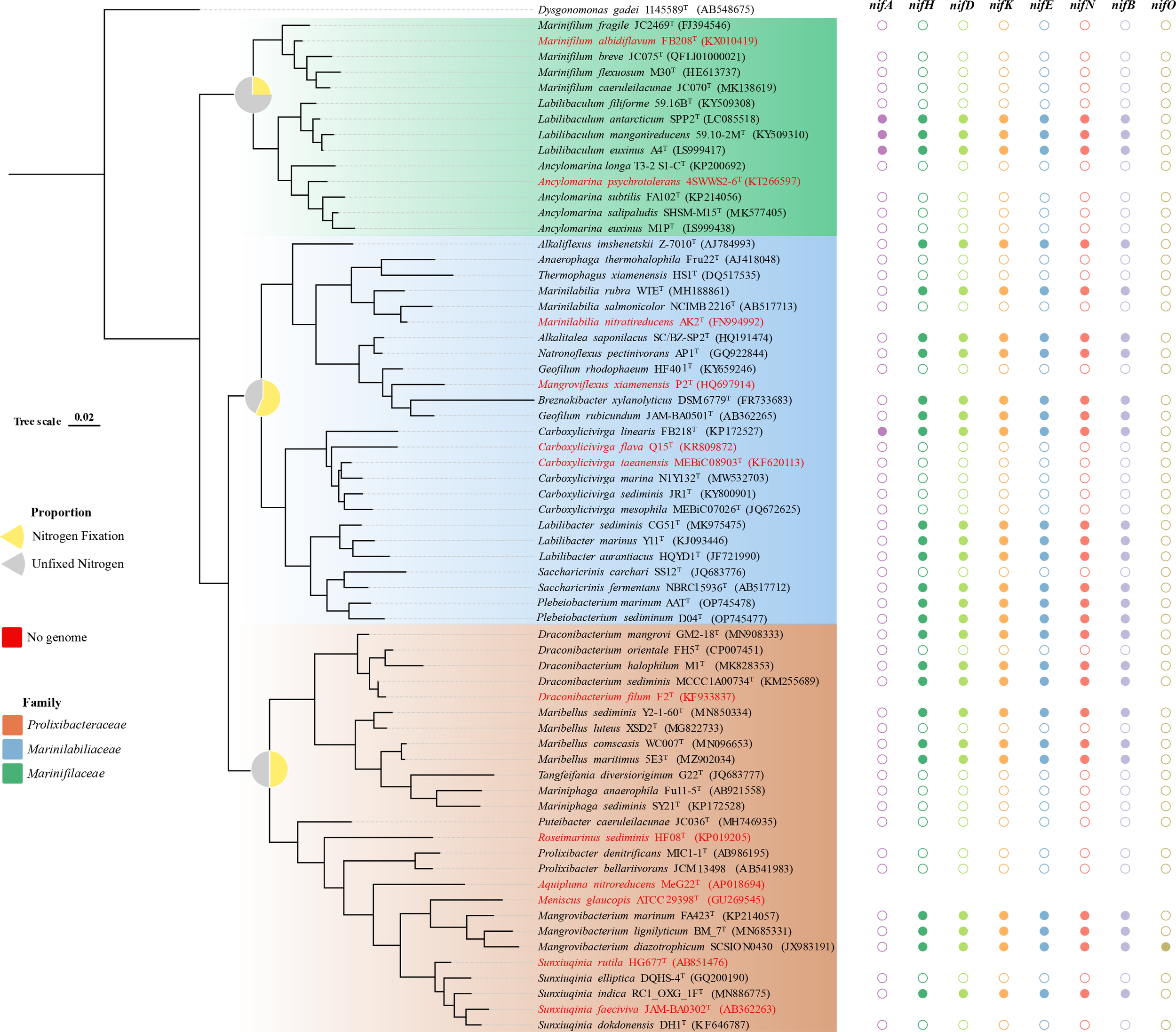
Distribution of the nitrogenase gene cluster among Marinilabiliacea bacteria. The tree was constructed using the maximum-likelihood method employing the 16S rRNA gene sequences. The tree was rooted with Dysgonomonas gadei 1145589T. Different colored shapes (solid: gene cluster exists, hollow: gene cluster absence) represent the presence of genes nifH, nifD, nifK, nifE, nifN, nifB, nifA, and nifO. Pie charts on the nodes show the proportion of nitrogen fixation and nitrogen fixation gene clusters missing in the three families. Marinifilaceae, Marinilabiliaceae, and Prolixibacteraceae are colored in the corresponding leaves of the tree (see figure legend). In the phylogenetic tree, no genome is found when the species name is in red.
Figure 3

Comparative analysis of the nif gene cluster among the genomes of the order Marinilabiliales. Arrows with different colors represent the presence of corresponding nif genes (see figure legend) and the direction of the arrows represents the direction of the gene clusters. The gene numbers indicated by HS and F are homocitrate synthase and ferredoxin (2Fe-2S), respectively. The genes indicated by P are nitrogen regulatory protein P-II. The genes indicated by nifB-DP are nifB-domain protein. Families colored blue indicate the Marinilabiliacea family, orange indicate Prolixibacteraceae, and green indicate Marinifilaceae.
Figure 4

Phylogenetic relationships of the Marinilabiliales species based on 16S rRNA gene sequences (A) and the nifH nucleotide sequence (B). The tree was constructed using the maximum-likelihood method and rooted with Dysgonomonas gadei. Bar, 0.02. Blue, orange, and green indicate Marinilabiliacea, Prolixibacteraceae, and Marinifilaceae, respectively.
3.4 Chemotaxonomic characteristics
The major fatty acids (>10%) of D04T and AATT were iso-C15:0 and anteiso-C15:0, which was similar with the profiles of the two reference strains, although there were differences in the proportions of some fatty acids. Strain AATT also possesses major fatty acids, including iso-C15:0 3-OH and iso-C17:0 3-OH. Fatty acids (>0.5%) were listed in detail in Table 2. The polar lipid profile of strain D04T consisted of phosphatidylethanolamine (PE), three glycolipid (GL1-3) and one unknown polar lipid. The polar lipid profile of strain AATT consisted of phosphatidylethanolamine (PE), one glycolipid (GL), one aminophosphoglycolipid and one phosphoglycolipid. The presence of phosphatidylethanolamine (PE) was conserved in all strains. Further detailed polar lipid images with different specific strains are given in Supplementary Figure 6. For respiratory quinone analyses, the results indicated that the major respiratory quinone of strains D04T and AATT were identified to be MK-7, which matched other members in the family Marinilabiliaceae.
Table 2
| Fatty acid | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Saturated | ||||
| C12:0 | 0.8 | 1.4 | 2.2 | TR |
| C13:0 | 0.5 | TR | ND | ND |
| C14:0 | 1.3 | 1.5 | 0.6 | 6.6 |
| C16:0 | ND | 5.9 | 1.1 | 5.3 |
| C18:0 | TR | 0.8 | ND | TR |
| Branched saturated | ||||
| iso-C13:0 | 5.0 | 8.8 | 1.3 | 0.6 |
| iso-C14:0 | TR | 0.9 | 1.7 | 1.3 |
| iso-C15:0 | 37.5 | 24.4 | 34.4 | 34.1 |
| iso-C16:0 | 1.0 | 1.1 | 0.5 | 1.2 |
| iso-C15:1 F | ND | ND | 2.2 | 0.9 |
| anteiso-C15:0 | 16.5 | 15.5 | 16.9 | 17.2 |
| Unsaturated | ||||
| C15:1ω6c | ND | 1.1 | ND | TR |
| C18:1ω9c | 0.9 | 0.7 | TR | 0.6 |
| Hydroxy | ||||
| iso-C13:0 3-OH | ND | ND | 0.5 | 4.1 |
| iso-C15:0 3-OH | 9.4 | 11.8 | 6.8 | ND |
| iso-C16:0 3-OH | 1.0 | 0.6 | 1.2 | ND |
| iso-C17:0 3-OH | 7.9 | 10.3 | 7.4 | ND |
| C15:0 2-OH | 0.5 | 1.3 | 1.5 | ND |
| C15:0 3-OH | 2.4 | 3.5 | ND | ND |
| C16:0 3-OH | 1.7 | 1.5 | 0.5 | ND |
| C17:0 2-OH | 0.5 | 1.5 | 0.9 | ND |
| Summed features* | ||||
| 1 | TR | ND | 1.2 | TR |
| 3 | TR | TR | 0.8 | 3.5 |
| 4 | ND | ND | 1.3 | TR |
| 9 | ND | TR | 3.4 | 2.0 |
Major cellular fatty acids comparison of strains D04T and AATT and relative type strains of the family Marinilabiliaceae.
*Summed features are fatty acids that cannot be reliably resolved from another fatty acid using the chosen chromatographic conditions. The MIDI system groups these fatty acids together as one feature with a single percentage of the total. Summed feature1, C15:1 and/or iso-C13:0 3-OH; Summed feature3, C16:1 ω6c and/or C16:1 ω7c; Summed feature4, iso-C17:1 I and/or anteiso-C17:1 B; Summed feature9, iso-C17:1 ω9c and/or C16:1 10-methyl.
Strains: 1, D04T; 2, AATT; 3, Saccharicrinis fermentans DSM 9555T; and 4, Labilibacter aurantiacus HQYD1T. All data are from this study unless indicated otherwise. Fatty acids that represented >10.0% are indicated in bold type. Fatty acids that represented <1.0% in all columns were omitted.TR, traces (<1.0%); ND, Not detected
4 Description of Plebeiobacterium gen. nov.
Plebeiobacterium (Ple.beio.bacterium’ adj. Plebeio, Common, ordinary. n. bakterion a small rod. N.L. fem. n. Plebeiobacterium, Common rod-shaped bacteria)
Cells are Gram-stain-negative, motile, facultatively anaerobic and straight rods. The major cellular fatty acids are dominant cellular fatty acids were identified as iso-C15:0 and anteiso-C15:0. MK-7 is the major menaquinone. The major polar lipid is phosphatidylethanolamine. The type species is Plebeiobacterium marinum.
4.1 Description of Plebeiobacterium marinum sp. nov.
Plebeiobacterium marinum (ma.ri’num. L. fem. marinum, from the sea).
Cells are facultatively anaerobic, Gram-stain-negative, and rod-shaped, roughly 0.3 μm wide and 3.4–4.1 μm long. Gliding motility was not observed. Cells were observed to be yellow colored, flat, smooth, and with circular colonies at 33°C on the modified MA grown after 5 days. Strain was oxidase positive, catalase negative, and nitrate reduced to nitrite. Growth occurs at 20–40°C (optimum 33°C), pH 6.5–7.5 (optimum pH 7.0), and with 0–8% NaCl (optimum 2% w/v). The strain was positive for the hydrolysis of starch but negative for casein, DNA, CM-cellulose, alginate, and Tweens 20, 40, 60, and 80. The major fatty acids are iso-C15:0 and anteiso-C15:0. The predominant respiratory quinone is MK-7. The polar lipid profile consisted of a phosphatidylethanolamine, three glycolipids (GL1-3), and one unknown polar lipid. Cells are positive for alkali phosphatase, esterase (C4), esterase lipase (C8), acid phosphatase, naphthol-AS-BI-phosphohydrolase, β-galactosidase, and β-glucosidase. Cells are also positive for the utilization of tryptophan deaminase, Voges–Proskauer reaction, and arabinose. Acid is produced from d-arabinose, d-ribose, d-xylose, d-galactose, d-glucose, arbulin, aesculin ferric citrate, salicin, d-cellobiose, d-maltose, d-lactose, d-melibiose, d-saccharose, starch, glycogen, d-gentiobiose, d-turanose, d-lyxose, potassium 2-keto-gluconate, potassium 5-keto-gluconate, l-arabinose, d-adonitol, methyl-β-d-xylopyranoside, d-fructose, d-mannose, l-rhamnose, dulcitol, inositol, inulin, d-raffinose, xylitol, d-tagatose, l-fucose, d-arabitol, and l-arabitol. In carbon source oxidation tests, positive results are obtained for dextrin, d-maltose, d-trehalose, d-cellobiose, gentiobiose, sucrose, d-turanose, stachyose, d-raffinose, α-d-lactose, d-melibiose, d-salicin, N-acetyl-β-d-mannosamine, N-acetyl-d-galactosamine, α-d-glucose, d-mannose, d-fructose, d-galactose, 3-methyl glucose, l-fucose, myo-inositol, d-glucose-6-PO4,d-fructose-6-PO4, gelatin, l-alanine, l-histidine, l-pyroglutamic acid, pectin, d-glucuronic acid, glucuronamide, α-keto-glutaric acid, l-malic acid, α-keto-butyric acid, propionic acid, acetoacetic acid, and acetic acid. The genomic DNA G+C content of the type strain is 36.8%.
The type strain D04T (MCCC 1H00493T = KCTC 92026T) was isolated from coastal sediment collected from Xiaoshi Island, Weihai, China. Strain D04T was stored in our own laboratory repository,10 with code SDUM258008. The GenBank accession number for the 16S rRNA gene and the draft genome sequence of D04T are OP745477 and JAPDPI000000000, respectively.
4.2 Description of Plebeiobacterium sediminum sp. nov.
Plebeiobacterium sediminum (se.di.minum. L. gen. pl. n. sediminum of sediments, pertaining to source of isolation).
Cells are facultatively anaerobic, Gram-stain-negative, and rod-shaped, roughly 0.4–0.5 μm wide and 0.6–1.4 μm long. Gliding motility was observed. Cells were observed to be yellow colored, flat, smooth, and with circular colonies at 33°C on the modified MA grown after 5 days. Strain was oxidase positive, catalase negative, and nitrate reduced to nitrite. Growth occurs at 20–40°C (optimum 33°C), pH 6.0–7.5 (optimum pH 6.5), and with 0.5–8% NaCl (optimum 3% w/v). The strain was positive for the hydrolysis of starch, DNA, but negative for casein, CM-cellulose, alginate, and Tweens 20, 40, 60, and 80. The major fatty acids are iso-C15:0, anteiso-C15:0, iso-C15:0 3-OH, and iso-C17:0 3-OH. The predominant respiratory quinone is MK-7. The polar lipid profile consisted of phosphatidylethanolamine, one glycolipid (GL), one aminophosphoglycolipid, and one phosphoglycolipid. Cells are positive for alkali phosphatase, esterase (C4), esterase lipase (C8), acid phosphatase, naphthol-AS-BI-phosphohydrolase, β-galactosidase, leucine arylamidase, cystine arylamidase, trypsin, N-acetyl-β-glucosaminidase, α-glucosidase, and β-glucosidase. Cells are also positive for the utilization of tryptophan deaminase, Voges–Proskauer reaction, and arabinose. Acid is produced from d-arabinose, d-ribose, d-xylose, d-galactose, d-glucose, arbulin, aesculin ferric citrate, salicin, d-cellobiose, d-maltose, d-lactose, d-melibiose, d-saccharose, starch, glycogen, d-gentiobiose, d-turanose, d-lyxose, potassium 2-keto-gluconate, potassium 5-keto-gluconate, erythritol, l-arabinose, d-fructose, d-mannose, l-rhamnose, N-acetylglucosamine, amygdalin, inulin, d-raffinose, xylitol, d-fucose, l-fucose, and l-arabitol. In carbon source oxidation tests, positive results are obtained for dextrin, d-maltose, d-trehalose, d-cellobiose, gentiobiose, sucrose, d-turanose, stachyose, d-raffinose, α-d-lactose, d-melibiose, β-methyl-d-glucoside, d-salicin, α-d-glucose, d-mannose, d-fructose, d-galactose, l-rhamnose, d-glucose-6-PO4, glycyl-l-proline, pectin, d-galacturonic acid, l-galactonic acid lactone, d-glucuronic acid, glucuronamide, methyl pyruvate, α-keto-butyric acid, and acetic acid. The genomic DNA G+C content is 33.4%.
The type strain AATT (MCCC1H00485T = KCTC92028T) was isolated from coastal sediment collected from Xiaoshi Island, Weihai, China. Strain AATT was stored in our own laboratory repository,10 with code SDUM258009. The GenBank accession number for the 16S rRNA gene and the draft genome sequence of AATT are OP745478 and JAPDPJ000000000, respectively.
Statements
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: the accession numbers for the 16S rRNA gene sequence of strains D04T and AATT are OP745477 and OP745478, respectively (Genbank); the accession number for the whole genome shotgun project of strains D04T and AATT are JAPDPI000000000 and JAPDPJ000000000, respectively (GenBank).
Author contributions
W-XY and Q-YL isolated the strains D04T and AATT. W-XY performed experimental operation, collected data, analysis and wrote the manuscript. Z-JD and D-SM offered experiment guidance and critical revision of the article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Science and Technology Fundamental Resources Investigation Program (Grant Nos. 2019FY100700 and 2022FY101100) and the National Natural Science Foundation of China (41876166).
Acknowledgments
The scanning electron microscopy was supported by the Physical-Chemical Materials Analytical and Testing Center of Shandong University at Weihai. A scanning electron microscope (Nova NanoSEM 450, FEI) was used to observe the cell size and morphology.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1213051/full#supplementary-material
Abbreviations
DSM, German Collection of Microorganisms and Cell Cultures GmbH; MCCC, Marine Culture Collection of China; KCTC, Korean Collection for Type Cultures; dDDH, digital DNA-DNA hybridization; AAI, average amino acid identity; MIDI, Microbial Identification System; TLC, thin-layer chromatography; MEGA, Molecular Evolutionary Genetics Analysis; HPLC, High Performance Liquid Chromatography.
Footnotes
1.^ https://lpsn.dsmz.de/family/Marinifilaceae
2.^ https://www.ezbiocloud.net/pa
4.^ https://www.genome.jp/tools/kofamkoala/
6.^ https://antismash.secondarymetabolites.org/
7.^ http://enve-omics.ce.gatech.edu/aai/
References
1
Aziz R. K. Bartels D. Best A. A. DeJongh M. Disz T. Edwards R. A. et al . (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics9, 75. doi: 10.1186/1471-2164-9-75
2
Ben Hania W. Joseph M. Bunk B. Spröer C. Klenk H. P. Fardeau M. L. et al . (2017). Characterization of the first cultured representative of a bacteroidetes clade specialized on the scavenging of cyanobacteria. Environ. Microbiol.19 (3), 1134–1148. doi: 10.1111/1462-2920.13639
3
Bernardet J. F. Nakagawa Y. Holmes B. Subcommittee On The Taxonomy Of, F Cytophaga-Like Bacteria Of The International Committee On Systematics Of, P (2002). Proposed minimal standards for describing new taxa of the family flavobacteriaceae and emended description of the family. Int. J. Syst. Evol. Microbiol.52 (Pt 3), 1049–1070. doi: 10.1099/00207713-52-3-1049
4
Blin K. Shaw S. Kloosterman A. M. Charlop-Powers Z. van Wezel G. P. Medema M. H. et al . (2021). antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res.49 (W1), W29–w35. doi: 10.1093/nar/gkab335
5
Cantarel B. L. Coutinho P. M. Rancurel C. Bernard T. Lombard V. Henrissat B. (2009). The carbohydrate-active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res.37 (Database issue), D233–D238. doi: 10.1093/nar/gkn663
6
Chakraborty S. Andersen K. H. Visser A. W. Inomura K. Follows M. J. Riemann L. (2021). Quantifying nitrogen fixation by heterotrophic bacteria in sinking marine particles. Nat. Commun.12 (1), 4085. doi: 10.1038/s41467-021-23875-6
7
CLSI . (2021). Performance standards for antimicrobial susceptibility testing, M100, 31st ed. (Wayne, PA: Clinical and Laboratory Standards Institute).
8
Denger K. Warthmann R. Ludwig W. Schink B. (2002). Anaerophaga thermohalophila gen. nov., sp. nov., a moderately thermohalophilic, strictly anaerobic fermentative bacterium. Int. J. Syst. Evol. Microbiol.52 (Pt 1), 173–178. doi: 10.1099/00207713-52-1-173
9
Du Z. J. Wang Y. Dunlap C. Rooney A. P. Chen G. J. (2014). Draconibacterium orientale gen. nov., sp. nov., isolated from two distinct marine environments, and proposal of draconibacteriaceae fam. nov. Int. J. Syst. Evol. Microbiol.64 (Pt 5), 1690–1696. doi: 10.1099/ijs.0.056812-0
10
Fang D. B. Han J. R. Liu Y. Du Z. J. (2017). Seonamhaeicola marinus sp. nov., isolated from marine algae. Int. J. Syst. Evol. Microbiol.67 (11), 4857–4861. doi: 10.1099/ijsem.0.002396
11
Felsenstein J. (1981). Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol.17 (6), 368–376. doi: 10.1007/bf01734359
12
Felsenstein J. (1985). Confidence limits on phylogenies: an approach using the bootsrap. Evolution39 (4), 783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x
13
Gaby J. C. Buckley D. H. (2011). A global census of nitrogenase diversity. Environ. Microbiol.13 (7), 1790–1799. doi: 10.1111/j.1462-2920.2011.02488.x
14
Gcrhardt P. Murray R. G. E. Wood W. A. Krieg N. R. (1994). Methods for general and molecular bacteriology. (Washington, DC: American Society for Microbiology). 607 p.
15
Goris J. Konstantinidis K. T. Klappenbach J. A. Coenye T. Vandamme P. Tiedje J. M. (2007). DNA-DNA Hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol.57 (Pt 1), 81–91. doi: 10.1099/ijs.0.64483-0
16
Gutierrez J. C. Santero E. Tortolero M. (1997). Ammonium repression of the nitrite-nitrate (nasAB) assimilatory operon of azotobacter vinelandii is enhanced in mutants expressing the nifO gene at high levels. Mol. Gen. Genet.255 (2), 172–179. doi: 10.1007/s004380050486
17
Halm H. Lam P. Ferdelman T. G. Lavik G. Dittmar T. LaRoche J. et al . (2012). Heterotrophic organisms dominate nitrogen fixation in the south pacific gyre. Isme J.6 (6), 1238–1249. doi: 10.1038/ismej.2011.182
18
Harwood C. S. (2020). Iron-only and vanadium nitrogenases: fail-safe enzymes or something more? Annu. Rev. Microbiol.74, 247–266. doi: 10.1146/annurev-micro-022620-014338
19
Herbert R. A. (1999). Nitrogen cycling in coastal marine ecosystems. FEMS Microbiol. Rev.23 (5), 563–590. doi: 10.1111/j.1574-6976.1999.tb00414.x
20
Huang Z. Hu Y. Lai Q. Guo Y. (2020). Description of maribellus sediminis sp. nov., a marine nitrogen-fixing bacterium isolated from sediment of cordgrass and mangrove. Syst. Appl. Microbiol.43 (4), 126099. doi: 10.1016/j.syapm.2020.126099
21
Inoue J. Oshima K. Suda W. Sakamoto M. Iino T. Noda S. et al . (2015). Distribution and evolution of nitrogen fixation genes in the phylum bacteroidetes. Microbes Environ.30 (1), 44–50. doi: 10.1264/jsme2.ME14142
22
Isobe K. Ohte N. (2014). Ecological perspectives on microbes involved in n-cycling. Microbes Environ.29 (1), 4–16. doi: 10.1264/jsme2.me13159
23
Jordan E. M. Thompson F. L. Zhang X. H. Li Y. Vancanneyt M. Kroppenstedt R. M. et al . (2007). Sneathiella chinensis gen. nov., sp. nov., a novel marine alphaproteobacterium isolated from coastal sediment in qingdao, China. Int. J. Syst. Evol. Microbiol.57 (Pt 1), 114–121. doi: 10.1099/ijs.0.64478-0
24
Kanehisa M. Sato Y. Kawashima M. Furumichi M. Tanabe M. (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res.44 (D1), D457–D462. doi: 10.1093/nar/gkv1070
25
Kapili B. J. Barnett S. E. Buckley D. H. Dekas A. E. (2020). Evidence for phylogenetically and catabolically diverse active diazotrophs in deep-sea sediment. Isme J.14 (4), 971–983. doi: 10.1038/s41396-019-0584-8
26
Kroppenstedt R. M. (1982). Separation of bacterial menaquinones by HPLC using reverse phase (RP18) and a silver loaded ion exchanger as stationary phases. J. Liquid Chromatogr. \& Related Technol.5 (12), 2359–2367. doi: 10.1080/01483918208067640
27
Kumar S. (1996). A stepwise algorithm for finding minimum evolution trees. Mol. Biol. Evol.13 (4), 584–593. doi: 10.1093/oxfordjournals.molbev.a025618
28
Kuypers M. M. M. Marchant H. K. Kartal B. (2018). The microbial nitrogen-cycling network. Nat. Rev. Microbiol.16 (5), 263–276. doi: 10.1038/nrmicro.2018.9
29
Lee I. Ouk Kim Y. Park S. C. Chun J. (2016). OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol.66 (2), 1100–1103. doi: 10.1099/ijsem.0.000760
30
Lehnen N. Marchant H. K. Schwedt A. Milucka J. Lott C. Weber M. et al . (2016). High rates of microbial dinitrogen fixation and sulfate reduction associated with the Mediterranean seagrass posidonia oceanica. Syst. Appl. Microbiol.39 (7), 476–483. doi: 10.1016/j.syapm.2016.08.004
31
López-Torrejón G. Jiménez-Vicente E. Buesa J. M. Hernandez J. A. Verma H. K. Rubio L. M. (2016). Expression of a functional oxygen-labile nitrogenase component in the mitochondrial matrix of aerobically grown yeast. Nat. Commun.7, 11426. doi: 10.1038/ncomms11426
32
Lu D.-C. Zhao J.-X. Wang F.-Q. Xie Z.-H. Du Z.-J. (2017). Labilibacter aurantiacus gen. nov., sp. nov., isolated from sea squirt (Styela clava) and reclassification of saccharicrinis marinus as labilibacter marinus comb. nov. Int. J. System. Evolution. Microbiol.67 (2), 441–446. doi: 10.1099/ijsem.0.001649
33
Luo R. Liu B. Xie Y. Li Z. Huang W. Yuan J. et al . (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience1 (1), 18. doi: 10.1186/2047-217x-1-18
34
Luo Z. Zhong Q. Han X. Hu R. Liu X. Xu W. et al . (2021). Depth-dependent variability of biological nitrogen fixation and diazotrophic communities in mangrove sediments. Microbiome9 (1), 212. doi: 10.1186/s40168-021-01164-0
35
Meier-Kolthoff J. P. Auch A. F. Klenk H.-P. Göker M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinf.14, 60. doi: 10.1186/1471-2105-14-60
36
Minnikin D. E. O’Donnell A. G. Goodfellow M. Alderson G. Athalye M. Schaal A. et al . (1984). An integrated procedure for the extraction of bacterial isoprenoid quinones and polar lipids. Microbiol. Methods2 (5), 233–241. doi: 10.1016/0167-7012(84)90018-6
37
Mohr W. Lehnen N. Ahmerkamp S. Marchant H. K. Graf J. S. Tschitschko B. et al . (2021). Terrestrial-type nitrogen-fixing symbiosis between seagrass and a marine bacterium. Nature600 (7887), 105–109. doi: 10.1038/s41586-021-04063-4
38
Mu D. S. Liang Q. Y. Wang X. M. Lu D. C. Shi M. J. Chen G. J. et al . (2018). Metatranscriptomic and comparative genomic insights into resuscitation mechanisms during enrichment culturing. Microbiome6 (1), 230. doi: 10.1186/s40168-018-0613-2
39
Nicholson A. C. Gulvik C. A. Whitney A. M. Humrighouse B. W. Bell M. E. Holmes B. et al . (2020). Division of the genus chryseobacterium: observation of discontinuities in amino acid identity values, a possible consequence of major extinction events, guides transfer of nine species to the genus epilithonimonas, eleven species to the genus kaistella, and three species to the genus halpernia gen. nov., with description of kaistella daneshvariae sp. nov. and epilithonimonas vandammei sp. nov. derived from clinical specimens. Int. J. Syst. Evol. Microbiol.70 (8), 4432–4450. doi: 10.1099/ijsem.0.003935
40
Pi H. W. Lin J. J. Chen C. A. Wang P. H. Chiang Y. R. Huang C. C. et al . (2022). Origin and evolution of nitrogen fixation in prokaryotes. Mol. Biol. Evol.39 (9), msac181. doi: 10.1093/molbev/msac181
41
Raymond J. Siefert J. L. Staples C. R. Blankenship R. E. (2004). The natural history of nitrogen fixation. Mol. Biol. Evol.21 (3), 541–554. doi: 10.1093/molbev/msh047
42
Saitou N. Nei M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol.4 (4), 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
43
Sarkar A. Reinhold-Hurek B. (2014). Transcriptional profiling of nitrogen fixation and the role of NifA in the diazotrophic endophyte azoarcus sp. strain BH72. PloS One9 (2), e86527. doi: 10.1371/journal.pone.0086527
44
Sayavedra L. Li T. Bueno Batista M. Seah B. K. B. Booth C. Zhai Q. et al . (2021). Desulfovibrio diazotrophicus sp. nov., a sulfate-reducing bacterium from the human gut capable of nitrogen fixation. Environ. Microbiol.23 (6), 3164–3181. doi: 10.1111/1462-2920.15538
45
Smercina D. N. Evans S. E. Friesen M. L. Tiemann L. K. (2019). To fix or not to fix: controls on free-living nitrogen fixation in the rhizosphere. Appl. Environ. Microbiol.85 (6), e02546–e02518. doi: 10.1128/aem.02546-18
46
Sohm J. A. Webb E. A. Capone D. G. (2011). Emerging patterns of marine nitrogen fixation. Nat. Rev. Microbiol.9 (7), 499–508. doi: 10.1038/nrmicro2594
47
Sorokin D. Y. Panteleeva A. N. Tourova T. P. Kaparullina E. N. Muyzer G. (2011). Natronoflexus pectinivorans gen. nov. sp. nov., an obligately anaerobic and alkaliphilic fermentative member of bacteroidetes from soda lakes. Extremophiles15 (6), 691–696. doi: 10.1007/s00792-011-0399-7
48
Steppe T. F. Olson J. B. Paerl H. W. Litaker R. W. Belnap J. (1996). Consortial N2 fixation: a strategy for meeting nitrogen requirements of marine and terrestrial cyanobacterial mats. FEMS Microbiol. Ecol.21 (3), 149–156. doi: 10.1111/j.1574-6941.1996.tb00342.x
49
Weisburg W. G. Barns S. M. Pelletier D. A. Lane D. J. (1991). 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol.173 (2), 697–703. doi: 10.1128/jb.173.2.697-703.1991
50
Wu W. J. Zhao J. X. Chen G. J. Du Z. J. (2016). Description of ancylomarina subtilis gen. nov., sp. nov., isolated from coastal sediment, proposal of marinilabiliales ord. nov. and transfer of marinilabiliaceae, prolixibacteraceae and marinifilaceae to the order marinilabiliales. Int. J. Syst. Evol. Microbiol.66 (10), 4243–4249. doi: 10.1099/ijsem.0.001342
51
Yang S.-H. Seo H.-S. Woo J.-H. Oh H.-M. Jang H. Lee J.-H. et al . (2014). Carboxylicivirga gen. nov. in the family marinilabiliaceae with two novel species, carboxylicivirga mesophila sp. nov. and carboxylicivirga taeanensis sp. nov., and reclassification of cytophaga fermentans as saccharicrinis fermentans gen. nov., comb. nov. Int. J. System. Evolution. Microbiol.64 (Pt_4), 1351–1358. doi: 10.1099/ijs.0.053462-0
52
Yoon S. H. Ha S. M. Kwon S. Lim J. Kim Y. Seo H. et al . (2017a). Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol.67 (5), 1613–1617. doi: 10.1099/ijsem.0.001755
53
Yoon S. H. Ha S. M. Lim J. Kwon S. Chun J. (2017b). A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek110 (10), 1281–1286. doi: 10.1007/s10482-017-0844-4
54
Zehr J. P. (2011). Nitrogen fixation by marine cyanobacteria. Trends Microbiol.19 (4), 162–173. doi: 10.1016/j.tim.2010.12.004
55
Zehr J. P. Jenkins B. D. Short S. M. Steward G. F. (2003). Nitrogenase gene diversity and microbial community structure: a cross-system comparison. Environ. Microbiol.5 (7), 539–554. doi: 10.1046/j.1462-2920.2003.00451.x
56
Zhang H. Yohe T. Huang L. Entwistle S. Wu P. Yang Z. et al . (2018). dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res.46 (W1), W95–w101. doi: 10.1093/nar/gky418
57
Zhao B. Chen S. (2012). Alkalitalea saponilacus gen. nov., sp. nov., an obligately anaerobic, alkaliphilic, xylanolytic bacterium from a meromictic soda lake. Int. J. Syst. Evol. Microbiol.62 (Pt 11), 2618–2623. doi: 10.1099/ijs.0.038315-0
58
Zheng R. Cai R. Liu R. Liu G. Sun C. (2021). Maribellus comscasis sp. nov., a novel deep-sea bacteroidetes bacterium, possessing a prominent capability of degrading cellulose. Environ. Microbiol.23 (8), 4561–4575. doi: 10.1111/1462-2920.15650
59
Zhilina T. N. Appel R. Probian C. Brossa E. L. Harder J. Widdel F. et al . (2004). Alkaliflexus imshenetskii gen. nov. sp. nov., a new alkaliphilic gliding carbohydrate-fermenting bacterium with propionate formation from a soda lake. Arch. Microbiol.182 (2-3), 244–253. doi: 10.1007/s00203-004-0722-0
Summary
Keywords
Plebeiobacterium , analysis of genome, metabolic pathways, nitrogen fixation, polyphasic taxonomy
Citation
Yu W-X, Liang Q-Y, Du Z-J and Mu D-S (2023) Characterization of Plebeiobacterium marinum gen. nov., sp. nov. and Plebeiobacterium sediminum sp. nov., revealing the potential nitrogen fixation capacity of the order Marinilabiliales. Front. Mar. Sci. 10:1213051. doi: 10.3389/fmars.2023.1213051
Received
27 April 2023
Accepted
14 June 2023
Published
30 June 2023
Volume
10 - 2023
Edited by
Xue-Wei Xu, Ministry of Natural Resources, China
Reviewed by
Heng-Lin Cui, Jiangsu University, China; Guohong Liu, Fujian Academy of Agricultural Sciences, China
Updates
Copyright
© 2023 Yu, Liang, Du and Mu.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Da-Shuai Mu, dashuai.mu@sdu.edu.cn
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.