 Magalí Rey-Campos
Magalí Rey-Campos Luis Daniel González-Vázquez
Luis Daniel González-Vázquez Beatriz Novoa
Beatriz Novoa Antonio Figueras
Antonio Figueras- Institute of Marine Research (IIM), Spanish National Research Council (CSIC), Vigo, Spain
Introduction: Mollusks play a significant role in marine ecosystems and have economic value for aquaculture. Sometimes, unexpected and unexplained mortalities among mollusks have been described. The role of potential pathogens such as viruses remains unknown due to the lack of molluscan cell cultures, which is one of the major drawbacks to determining the viral role in such mortalities. Several oceanographic studies have suggested a high abundance of viruses in the oceans. Virus identification and understanding of viral interaction with organisms in marine ecosystems are in their infancy. Metatranscriptomics could become a useful tool to identify viruses using a shotgun approach and the growing number of viral genomes and sequences deposited in public databases.
Methods: In this work, several bioinformatics approaches were set up to screen Mollusca RNA sequences to find and confirm viral traces in their transcriptomes. This meta-analysis included an extensive search of SRA datasets belonging to mollusks available in the NCBI database, selecting a total of 55 SRA datasets that were further analyzed searching for viral sequences.
Results: Twenty-two bivalves, 19 cephalopods and 16 gastropods from 16 geographical origins and 17 different tissues were considered. The domain search approach was the most productive method to find viral sequences. This virus search showed that Cephalopoda samples (Idiosepius notoides and Amphioctopus fangsiao) exhibited the highest number of virus identifications. Some of the detected viral sequences were similar or identical to others previously identified. However, 33 putative new viruses were identified and analyzed phylogenetically when the RdRp domain was available. Specifically, Cephalopoda samples showed a considerable number of viruses belonging to the Rhabdoviridae family.
1 Introduction
Several events of emergence and spread of infectious diseases have been highly relevant in the course of history (Piret and Boivin, 2021). This context has necessarily changed the view of the importance of identifying and tracking viruses present in the environment and animals, given the possibility of transmission to other animals and potential human susceptibility (Holmes, 2022).
In a marine context, wild animals are constantly exposed to potentially pathogenic viruses that might cause mass mortalities in the case of unfavorable changes in environmental conditions. This is the case for the Pacific oyster (Crassostrea gigas), for which massive mortalities have been detected since 2008 in France. These serious losses were related to the detection of Oyster Herpesvirus type 1 (OsHV-1; Renault et al., 2014). OsHV-1 has spread worldwide, and other countries, such as China and Australia, have reported similar mortality episodes (Bai et al., 2015; Go et al., 2017). Apart from the Pacific oyster, this virus also affects other bivalve species, such as Scapharca broughtonii (ark clam; Bai et al., 2016) and Argopecten irradians (Atlantic bay scallop; Kim et al., 2019). Abalone amyotrophia causes mass mortality of several Haliotis species (Matsuyama et al., 2021) and the abalone ganglioneuritis is responsible for extensive mortalities and significant economic losses in Asia and Australia in the last two decades (Corbeil, 2020). This last disease is listed by the World Organization for Animal Health (WOAH) as a notifiable disease. Other diseases severely affecting crustaceans (i.e., white spot disease, Taura syndrome, yellow head disease, infectious myonecrosis and white tail disease) are also considered notifiable diseases. White tail disease, for instance, is an infection produced by Macrobrachium rosenbergii nodavirus, which has a very negative effect on the aquaculture industry. It belongs to the genus Alphanodavirus, which also affects insects (Dasmahapatra et al., 1985; Warrilow et al., 2018) and other invertebrates, causing major economic losses (Ho et al., 2018; Xu et al., 2020; Xia et al., 2022). The other genus included in this family, Betanodavirus, is characterized by being a neurotropic pathogenic virus affecting a multitude of marine fish species (Munday et al., 2002). Although the advances in sequencing have revealed great diversity and variability within the family Nodaviridae, it has been suggested that the current Alpha-/Betanodavirus taxonomy (Sahul Hameed et al., 2019) is insufficient to classify all the variability (NaveenKumar et al., 2013). Therefore, according to the high abundance of viruses in seawater and the current knowledge about viruses, especially those pathogenic to commercial species, the study of putative viral pathogens present in marine environments or wild animals could facilitate early detection and improve the actions to be taken.
However, virus identification is a challenge. These entities lack conserved ribosomal genes, which excludes the possibility of using amplicon sequencing technologies such as 16S rRNA or 18S rRNA sequencing. Metatranscriptomics is a useful and unbiased method to identify viruses (Razzauti et al., 2015; Durazzi et al., 2021). Nevertheless, to date, only a few studies on the presence of viral sequences in some marine animals have been carried out by identifying viral domains such as RdRp (RNA-dependent RNA polymerase; Rosani and Gerdol, 2017; Rosani et al., 2019) or by building viral databases and searching them by BLAST (Rosani et al., 2019; Rosani, 2022). These works have provided information on known viruses that are part of several marine bivalves and nudibranch gastropods but also viruses that remained unidentified or unclassified and could represent a problem under changing environmental conditions.
Increasing knowledge about viruses is a challenge. Oceans are microorganism reservoirs; hence, marine metazoans must show viral entities naturally constituting their microbiome and the surrounding environment. In the present work, we focused on mollusks, the largest marine phylum, because of their role in both aquatic ecosystems and aquaculture activities. Four different working pipelines were developed to perform a comprehensive meta-analysis in the identification of viruses. Several species of bivalves, cephalopods and gastropods were evaluated, some of them relevant for the seafood industry (Crassostrea gigas, Octopus vulgaris, Haliotis rufescens), others because of immune resistance to pathogens (Mytilus galloprovincialis) and finally others that are known to be particularly susceptible to some viral infections (Crassostrea gigas, Haliotis discus and Haliotis diversicolor). Molluscan transcriptomes were selected considering sequencing attributes, sample/tissue types and geographic origin, and they were analyzed to find viral sequences. The four search methods were merged and compared to obtain a notion of the identification possibilities offered by metatranscriptomics.
2 Materials and methods
2.1 Data retrieval
The SRA archive (https://www.ncbi.nlm.nih.gov/sra) was inspected to retrieve a representative number of molluscan transcriptomic datasets (Supplementary Table 1). The following features were considered: RNA-seq samples with a posting date after 2014, characterized by paired-read layout and sequenced using Illumina technology.
After examining the NCBI-BioProject database, a similar number of bivalves, gastropods and cephalopods were selected, as well as a wide range of tissues and geographical origins. Finally, only control/naïve samples were selected to run the analysis.
2.2 Reference databases
Reference viral databases for the different virus identification pipelines included Clustered-RVDB (873,234 entries that contain viral, virus-related, and virus-like nucleotide sequences; Goodacre et al., 2018), a RefSeq database containing all the available viral genome assemblies and downloaded on 18/05/2021 (10,907 viral assemblies) and a database constituted by InterPro and Pfam IDs. These raw domain databases (InterPro and Pfam) were downloaded on 20/08/2022 (Blum et al., 2021; Mistry et al., 2021) and cleaned of non-viral domains, keeping only those that contained the terms “virus”, “viral”, “RdRp” or “capsid”. These lists of domain IDs were used to identify viral domains after scan processing (1,370 and 1,058 viral codes, respectively; Supplementary Table 2).
2.3 Read processing and assembly
Raw reads in FastQ format were downloaded from the SRA ftp server. After that, raw reads were trimmed using Trimmomatic software (Bolger et al., 2014), removing Illumina adapters and specifying the following instructions: LEADING:3, TRAILING:3, MINLEN:36, and HEADCROP:15. After trimming, read datasets were inspected using FastQC software (Andrews, 2010), assessing the effectiveness of the trimming process and running the process again if necessary. Trimmed reads of each RNA-seq dataset were de novo assembled using Trinity software (Grabherr et al., 2011) and CLC Genomics Workbench 22.0.2 (Qiagen), setting a minimum contig length of 200.
2.4 Identification of putative viral sequences
Four different pipelines were developed for the identification of putative viral sequences. Figure 1 shows a scheme of these approaches:
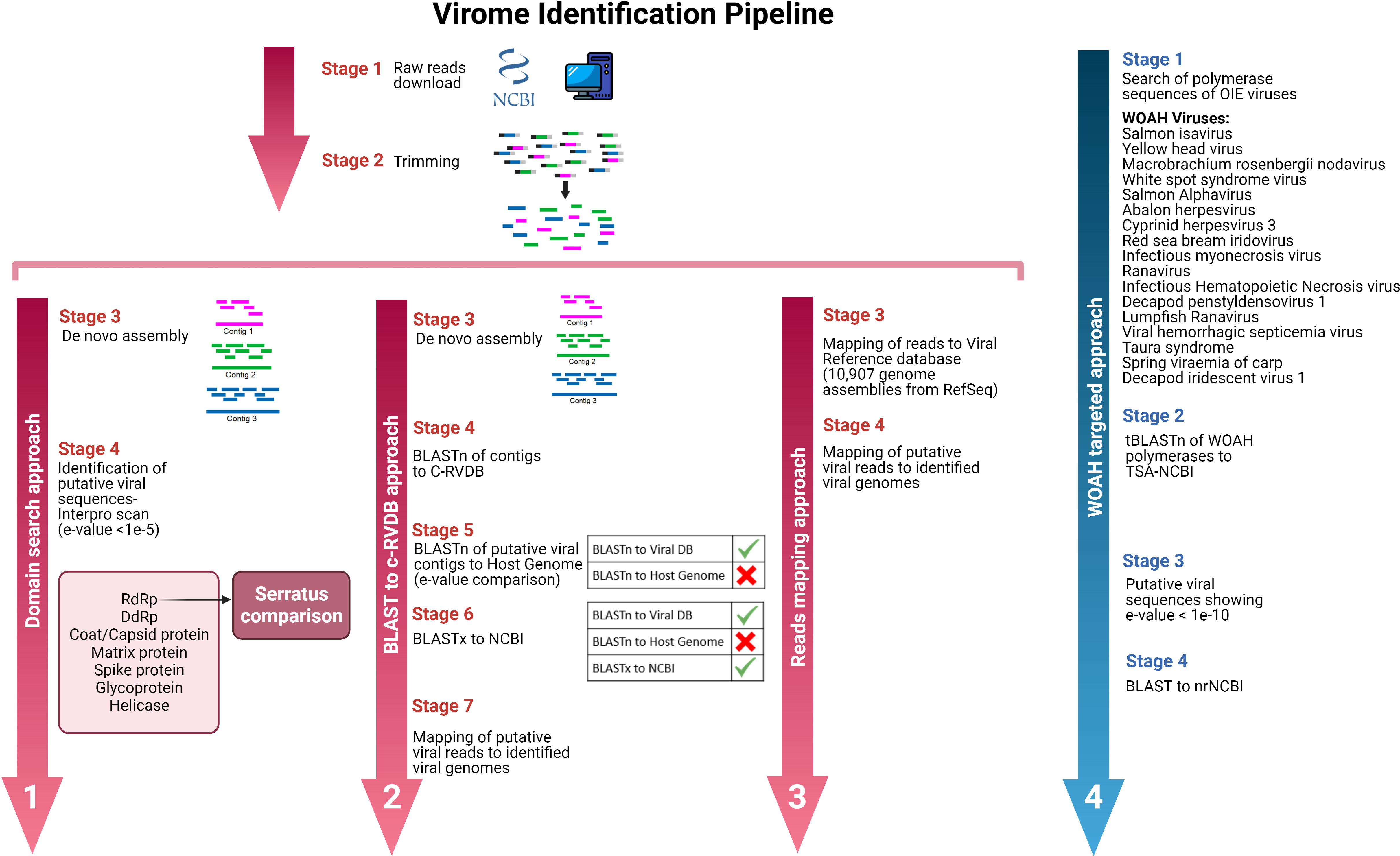
Figure 1 Working plan to find viral sequences in transcriptomic reads/assemblies available in public databases.
2.4.1 Domain search approach
The first approach consisted of scanning for viral domains in all the assembled contigs. An InterProScan analysis (Jones et al., 2014) was performed on the assembled contigs using the Pfam database as subject and the nucleotide sequences as input. Once the domains were identified (with e-values lower than 1e-5), filtering was carried out to keep only viral domains (including capsid, polymerase, matrix or any other viral domain available in the databases). This filtering was performed using two viral InterPro and Pfam ID databases (all the searched viral domains can be found in Supplementary Table 2). After identifying contigs showing viral domains, they were BLASTed to nrNCBI (BLASTx) for confirmation. Moreover, all the SRA codes were browsed on the Serratus web page (Edgar et al., 2022) to identify the RdRp domains and compare the results.
2.4.2 BLASTn/C-RVDB search approach
The second approach consisted of a BLASTn search of all the assembled contigs using the nucleotide viral database C-RVDB as a reference. Hits with e-values lower than 1e-50 and query cover higher than 50% were retrieved and BLASTed against the host genome (in the absence of the host genome, we used the genome available from the closest species; information on host genomes can be found in Supplementary Table 3). Host contigs were removed in this protocol step. As a final stage of this approach, putative viral sequences were BLASTed again (BLASTx) against the nrNCBI database to discard misidentified contigs and keep only sequences assigned to viruses. The identified viral sequences were used as a reference, and mapping of the reads putatively attributed to virus was performed (with the restriction mapping conditions of length fraction, 0.5 and similarity fraction, 0.8). The identification of possible viral reads was validated when these reads covered over 50% of the reference.
2.4.3 Reads mapping approach
The third approach to identify putative viruses consisted of mapping raw reads to a RefSeq database containing all the available viral genome assemblies at 18/05/2021 (10,907 viral assemblies). This method was performed using CLC Microbial Genomics Module 21.1 (Qiagen, Hilden, Germany). The mapping parameters used to classify reads as putative viral sequences were as follows: length fraction = 0.5, similarity fraction = 0.8 and minimum seed length = 30. The host genome in which the transcriptomic assay was conducted or, if this was not available, the closest species was used as a reference for filtering host reads (the list of genomes used for this purpose can be accessed in Supplementary Table 3). Samples showing over 30 million reads were randomly subsampled down to this figure before the mapping process due to hardware and computing time limitations. The results were evaluated, and possible viral reads were considered when they covered over 50% of the reference (virus assemblies).
2.4.4 Targeted approach
The last approach to find putative viral sequences was a targeted search. We replicated the search used by Parry and Asgari (2019) to find some crustacean and cephalopod flaviviruses. In this case, 17 viral WOAH listed diseases (cited in Figure 1) affecting aquatic species were considered to find their expression in the molluscan transcriptomes available in public databases. Polymerases or polyproteins of these viral agents were retrieved from the Protein-NCBI database and BLASTed (tBLASTn) to TSA-NCBI (Transcriptome Shotgun Assembly). The subject database was restricted to the “mollusks (taxid:6447)” taxa, and the e-value threshold was set at 1e-10. Putative viral contigs were retrieved to run a new BLAST against nrNCBI to check the alignments and the assignations.
2.5 Phylogenetic analyses
All the RdRp domains detected in this work were identified and taxonomically classified by BLAST homology to nrNCBI. Moreover, the RdRp proteins of the viral families detected in this work were downloaded from the RefSeq database. These sequences (along with those detected by us) were tested to find the best model of evolution (IQ-TREE web server; Kalyaanamoorthy et al., 2017). Moreover, the same IQ-TREE server allowed us to run a maximum likelihood analysis with an ultrafast branch support analysis with 1,000 bootstrap replicates (Nguyen et al., 2015). The resulting trees were edited using the iTOL online tool (Letunic and Bork, 2021).
The ICTV (Walker et al., 2021) and NCBI (Schoch et al., 2020) taxonomy browsers were used for viral taxonomic classification purposes. Moreover, Virus−Host DB was used to check the range of hosts corresponding to identified viruses (Mihara et al., 2016).
3 Results
3.1 Analysed data
In the present work, we searched the SRA archive to retrieve a representative number of Mollusca transcriptomic datasets of interest. The selection process took into account the following features: RNA-seq samples with a posting date after 2014, characterized by paired-read layout and sequenced using Illumina technology. Other information also considered was the geographic origin, which included a wide range of tissues and a representative number of species of each class of mollusks. From 179 NCBI Bioprojects, 55 SRA files were ultimately selected to run the analysis (information on the analyzed datasets is provided in Supplementary Table 1). The geographical distribution of samples analyzed in this virome study is shown in Figure 2A. These datasets included 21 samples from bivalves, 15 from gastropods and 19 from cephalopods. Some of them are relevant for the seafood industry (Crassostrea gigas, Octopus vulgaris, Haliotis rufescens), others show remarkable immune resistance to pathogens (Mytilus galloprovincialis), and others are very susceptible to viral diseases (C. gigas, Haliotis discus and Haliotis diversicolor). Samples included 17 different tissues, each one relevant to several biological functions (gills as the first tissue in contact with microorganisms in filter-feeding bivalves, hemocytes as immune cells, digestive gland as a digestive organ and where toxin accumulation takes place, etc.) from 16 geographical origins, which may have different microbiomes (Figure 2B).
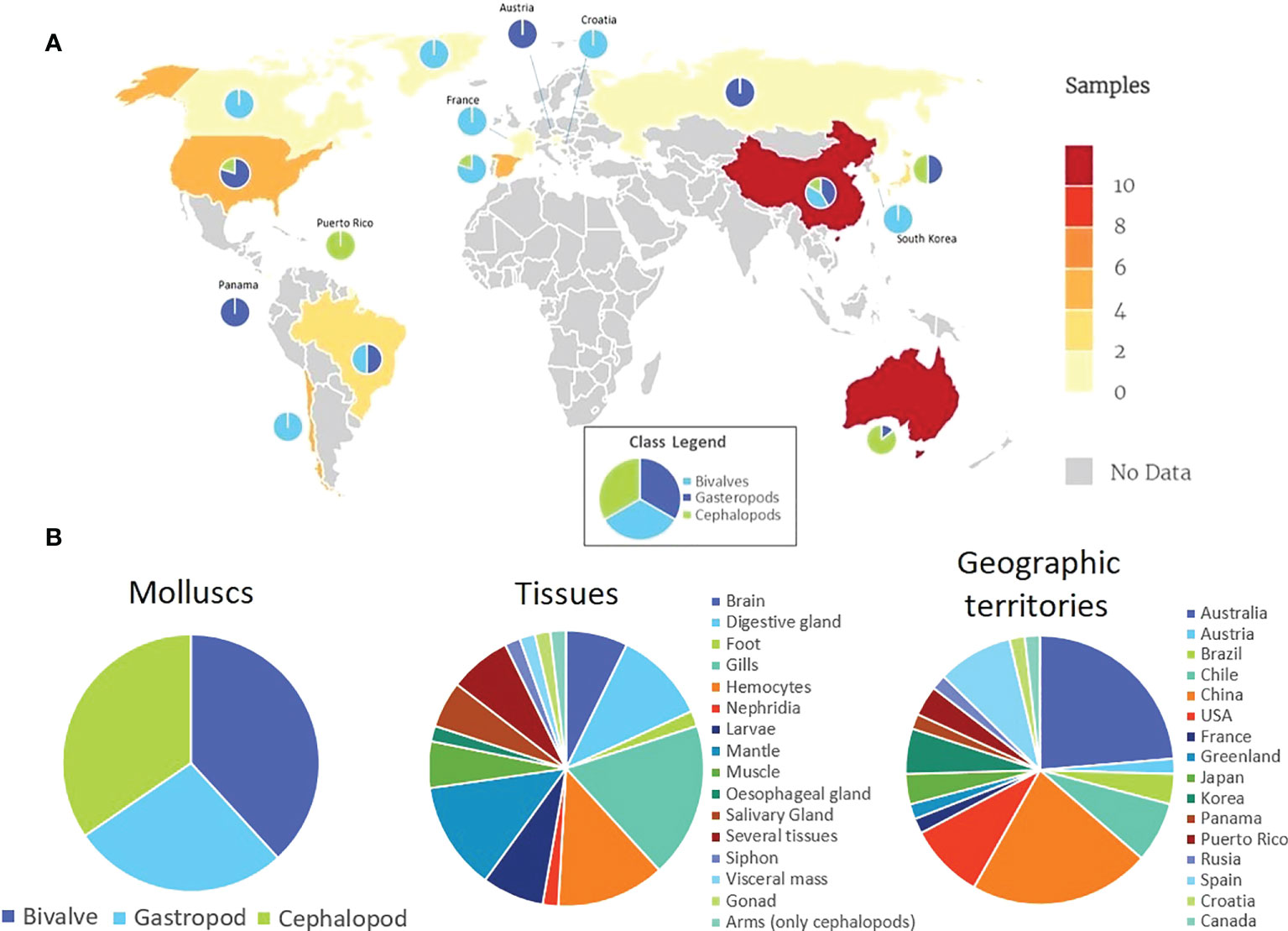
Figure 2 (A) Geographical distribution of samples analyzed in this virome study. The data legend indicates the number of SRA datasets studied. Circular graphs show the number of analyzed gastropods, bivalves and cephalopods from each geographic territory. (B) Summary of analyzed data.
3.2 Viral domains detected in RNA-seq files
After a Pfam domain scan of the 55 de novo assemblies, 447 domains related to viral proteins were identified (Supplementary Table 4). Most of these domains were ascribed to a specific viral family in the InterPro database (Blum et al., 2021), which allowed a description of the viral families found in each of the mollusk species under study (Figure 3). This analysis showed several patterns; for example, domains associated with the Reoviridae family were detected only in bivalves (M. chilensis, M. edulis, M. galloprovincialis and P. maximus). Another case is the family Herpesviridae, which was almost absent in cephalopods (detected only in Octopus kaurna) and mostly identified in bivalves and gastropods (Figure 3, Supplementary Table 4). Baculoviridae-, Retroviridae- and Parvoviridae-related domains were detected in almost all the studied mollusks. Finally, domains associated with Rhabdoviridae were found in half of the cephalopod species investigated and were absent in gastropods.
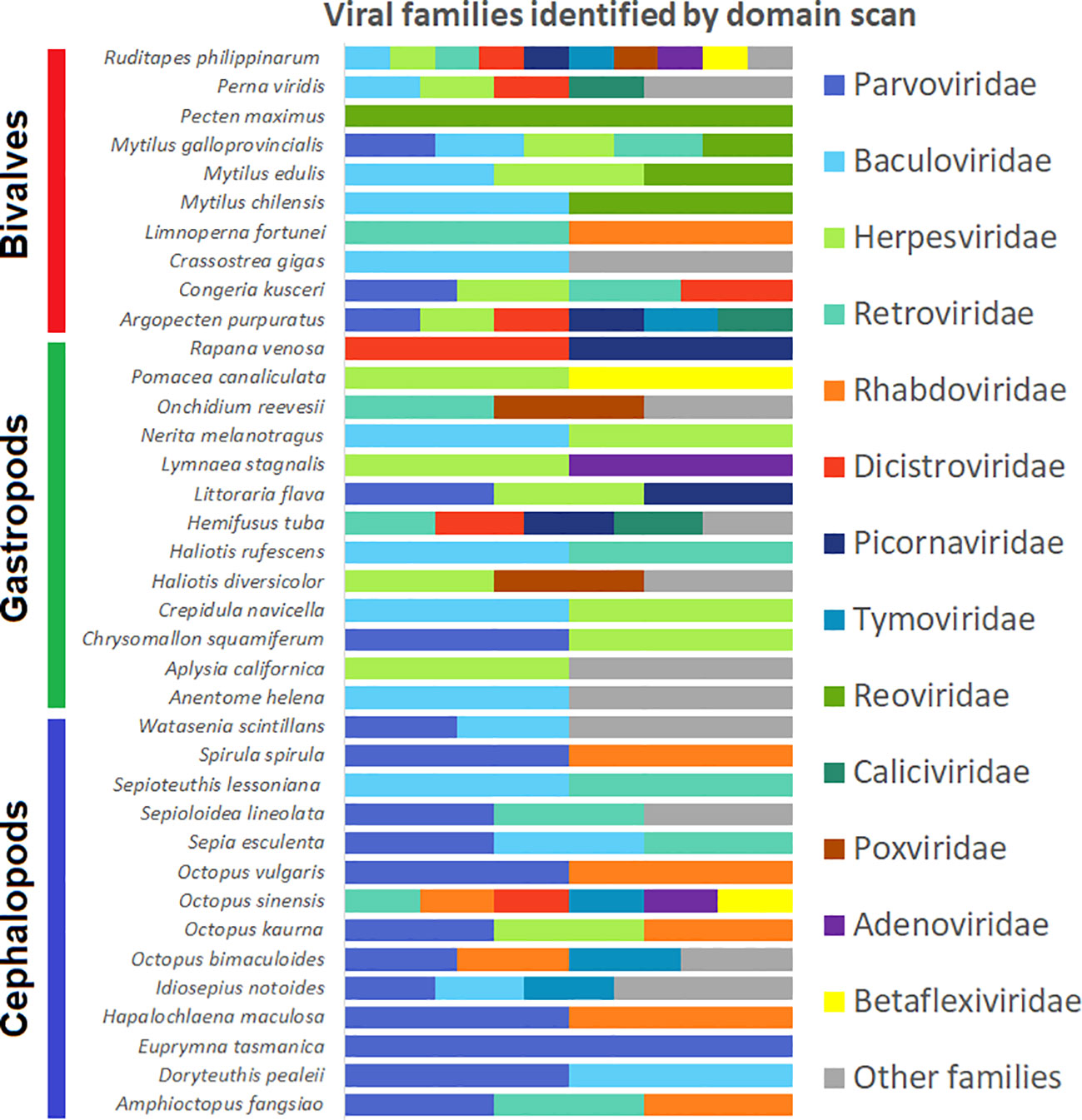
Figure 3 Histogram showing viral families detected in each species based on the specific domains detected by InterProScan.
Focusing on the RNA-dependent RNA polymerase (RdRp) domains, 20 domains were found. The SRA files showing these domains are listed in Table 1. This table also lists the domain ID, the signature description and the e-value of the detection. Among samples, the abundance of RdRp domains detected in cephalopods is highlighted (65% of the detected domains RdRp corresponded to these mollusks). Comparing our result to the Serratus project (Edgar et al., 2022), some detections matched (Flaviviridae in Idiosepius notoides, Rhabdoviridae in Hapalochlaena maculosa, and Marnaviridae in Argopecten purpuratus). However, some RdRp domains identified by the Serratus project were not found in our analysis (Alphaflexiviridae-1 and Marnaviridae-2; Table 1); in contrast, 17 RdRp domains were found in our analysis and were absent in the Serratus database, increasing the information in terms of viral domain detection (details in Table 1).
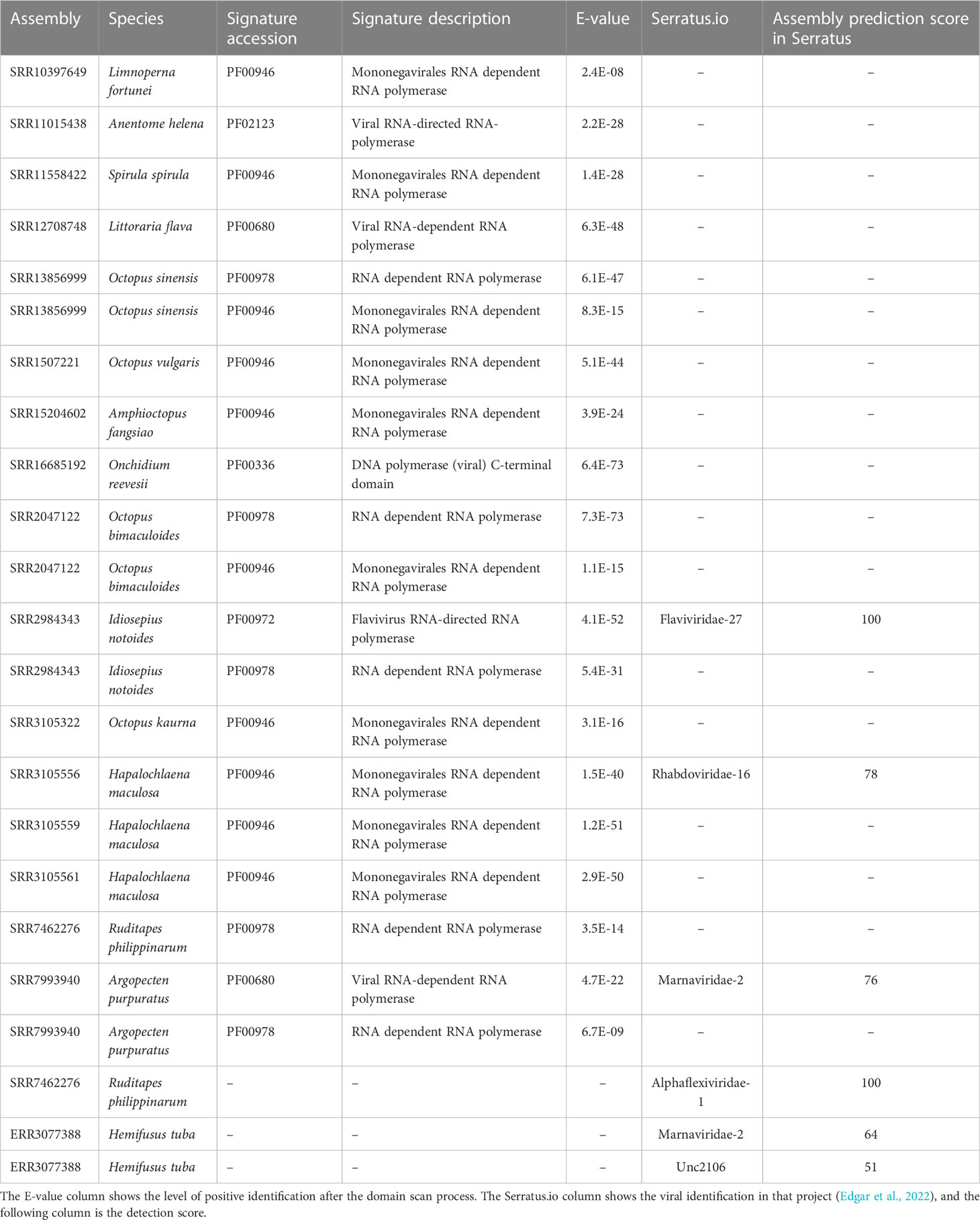
Table 1 RdRp domains detected by InterProScan.
This analysis also helped us to find contigs containing two or more viral domains, allowing us to identify some complete or nearly complete viruses that will be reported in the following sections.
3.3 Viral sequences detected by other approaches
The BLASTn/C-RVDB search approach allowed us to identify sequences with high similarity to previously detected viruses. Similarly, the read mapping approach identified well-conserved viruses that were already found by some of the other methods.
Finally, although the WOAH targeted search did not allow us to find notifiable viruses in mollusks, it served as search bait to identify some other putatively new viruses not found with the other methods. Detailed information on the results of these approaches can be found in Supplementary Table 5.
3.4 Virus identification
All the contigs identified as viral sequences by any of the search methods were retrieved and BLASTed against nrNCBI (BLASTx) to confirm and classify the putative viruses (Supplementary Table 5). A total of 50 viruses were identified in the whole set of transcriptomes studied. Interestingly, the domain search allowed the identification of over 58% of the putative viruses, making the approach more valuable in this sense (Figure 4A). In contrast, the read mapping approach showed to be the most stringent method, according to the characteristics of the bioinformatic technique (mapping versus BLAST). It is also noteworthy that there was a lack of overlap among the methods (Figure 4B), which means that it is necessary to use several strategies to perform a comprehensive analysis.
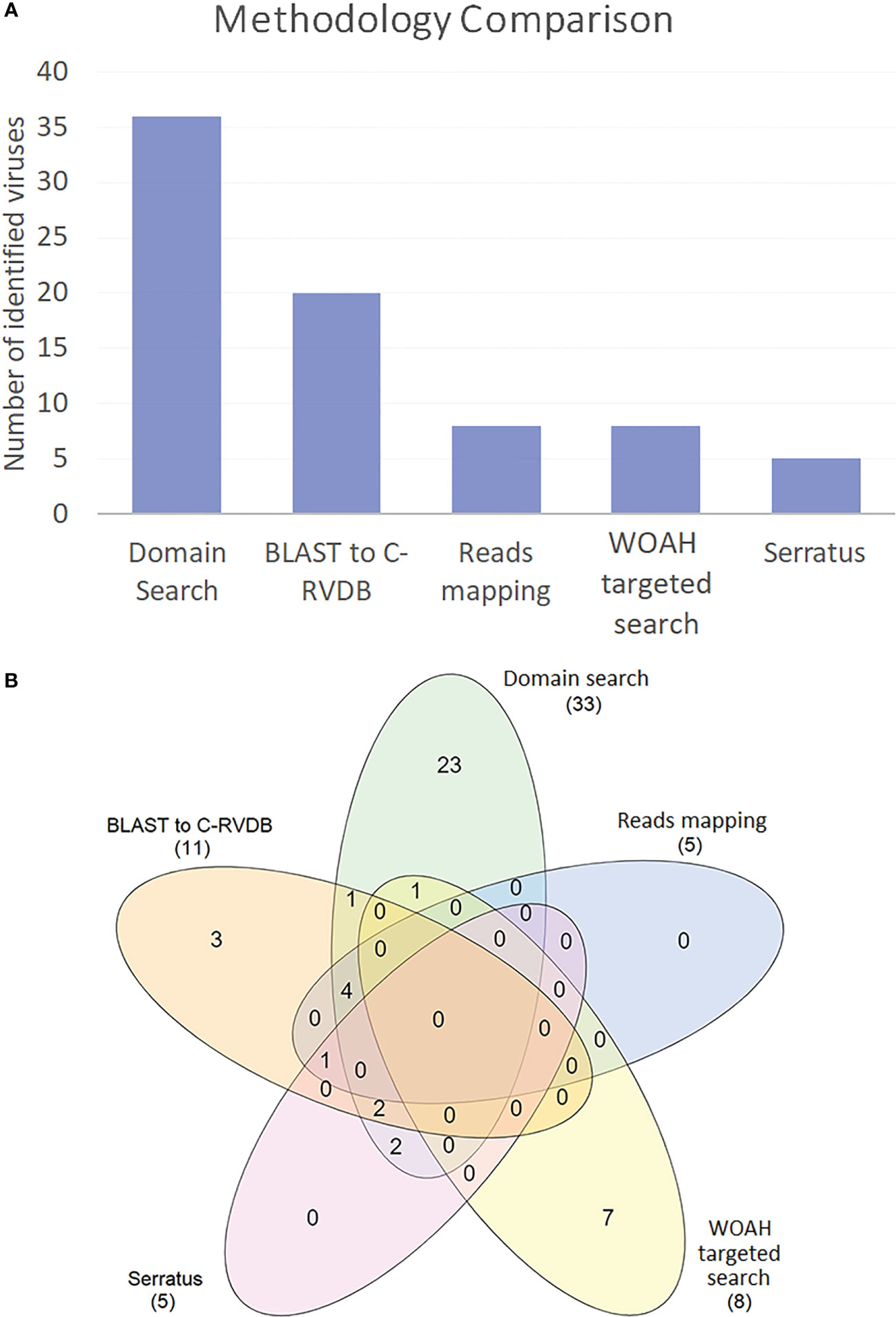
Figure 4 (A) Comparison of all the methods used in this work to find viral sequences in 55 SRA and 7 TSA-NCBI files. (B) Venn diagram showing shared and exclusive viruses identified by each method.
A summary of the results obtained with all the methods is shown in Supplementary Figure 1. The cephalopods Amphioctopus fangsiao and Idiosepius notoides were the species showing the highest number of viral detections (25% out of all the detections; Supplementary Figure 1). The rest of the cephalopods as well as bivalves and gastropods showed similar numbers. In turn, larval samples (mainly belonging to cephalopod samples) revealed the highest number of viruses, followed by visceral mass and brain (the three samples/tissues account for 50% of identifications; Supplementary Figure 1). Finally, in terms of origin, samples from Germany, China and Chile were the most relevant because they showed the most viruses (Supplementary Figure 1).
All of the identified viral sequences could be divided into three groups: i) sequences from putative contaminant viruses, ii) sequences from viruses already detected in mollusks (viruses showing sequence identity higher than 90% to other sequences available in public databases), and iii) new candidate viral sequences detected in this work (identity under 90% to the NCBI database).
In the first group, we found mammalian retroviruses, avian viruses and plant viruses. All of these sequences showed high identities to sequences deposited in public databases (Goose dicistrovirus, Cactus virus X or Murine leukemia virus), possibly constituting environmental or reagent contaminants, as previously reported (Asplund et al., 2019; Cobbin et al., 2021).
In the second group, some viruses previously reported in mollusks were found. This is the case for the Southern pygmy squid flavivirus, found by Parry and Asgari (2019) in the same RNA sample. Moreover, sequences highly similar to Biomphalaria virus 5 (93% identity) were found in our work in Biomphalaria pfeifferi samples. Another virus usually found in gastropods is the Abalone herpesvirus type 2 Taiwan, also detected in our work in Haliotis diversicolor hemocytes. Taxonomically unclassified viruses such as Wenzhou gastropod virus 2, previously detected in clams, were also detected in our study in R. philippinarum samples from China and Spain. Finally, sequences named Adintovirus and Xenomavirus previously found in some bivalves, but for which no details are available, were also found in our work.
In the third group, 33 different putative new viruses belonging to 14 families were found in the mollusk transcriptomes (Table 2) and named according to their putative classification (sequences in the Supplementary Table 6). In bivalves, viral sequences similar to viruses belonging to the families Aliusviridae (Bivalve alius-like virus 1), Dicistroviridae (Bivalve dicistro-like virus 1), Nodaviridae (Bivalve noda-like virus 1-2) and Picornaviridae (Bivalve picorna-like virus 1-2) were identified. Moreover, 4 yet unclassified viruses (Bivalve RNA virus 1-4) were detected in different species.
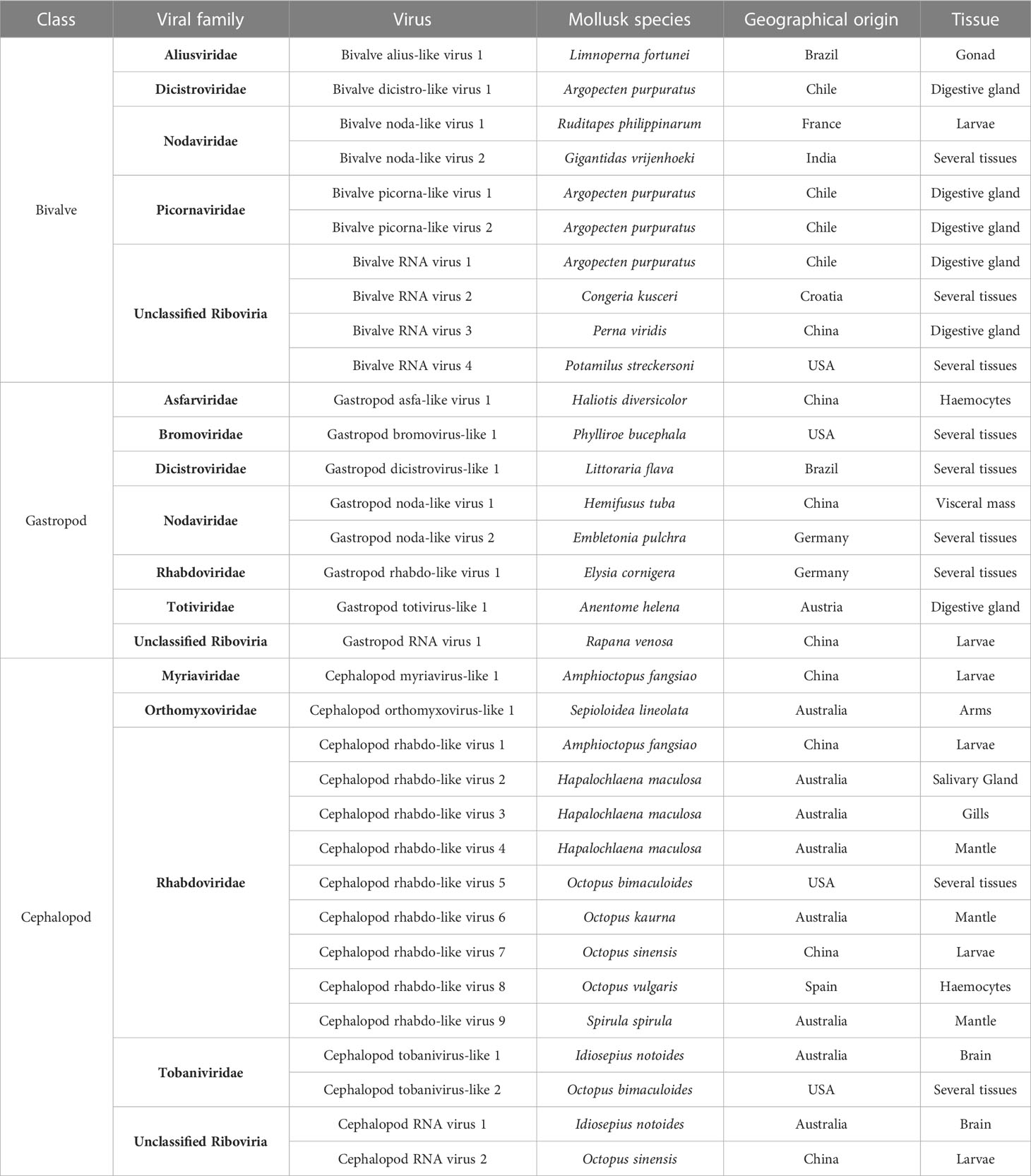
Table 2 New putative viruses detected in RNA-seq datasets from mollusks.
In gastropods, viruses included in the families Asfarviridae (Gastropod asfa-like virus 1), Bromoviridae (Gastropod bromovirus-like 1), Dicistroviridae (Gastropod dicistrovirus-like 1), Nodaviridae (Gastropod noda-like virus 1-2), Rhabdoviridae (Gastropod rhabdo-like virus 1) and Totiviridae (Gastropod totivirus-like 1) were identified. One unclassified virus (Gastropod RNA virus 1) was also detected.
In cephalopods, viruses putatively belonging to the families Myriaviridae (Cephalopod myriavirus-like 1), Orthomyxoviridae (Cephalopod orthomyxovirus-like 1), Rhabdoviridae (Cephalopod rhabdo-like virus 1-9), Tobaniviridae (Cephalopod tobanivirus-like 1-2) and some unclassified viruses (Cephalopod RNA virus 1-2) were identified.
Among these viruses, some Asfarviridae viruses have been previously identified in gastropods, such as the Abalone asfa-like virus. Additionally, totivirus has been found in Biomphalaria spp., and viruses belonging to the family Dicistroviridae in bivalves (Bivalve RNA virus G1-G5). Moreover, sequences fitting to viral families usually detected in insects such as Aliusviridae and Myriaviridae were also identified in our mollusk dataset (Bivalve alius-like virus 1 and Cephalopod myriavirus-like 1).
The family Nodaviridae constitutes another relevant viral family detected in our search. We detected some noda-like viruses in 4 mollusk transcriptomes (Table 2) from Embletonia pulchra, Ruditapes philippinarum, Gigantidas vrijenhoeki and Hemifusus tuba. Among these viruses, the one found in E. pulchra was the most similar to Macrobrachium rosenbergii nodavirus, showing 45% sequence homology.
The most striking case was the detection of sequences belonging to the family Rhabdoviridae. Ten different rhabdoviruses could be related to the sequences identified in our work. Most of them were found on cephalopod samples (Cephalopod rhabdo-like viruses 1-9), and only one member of this family was found in the gastropod Elysia cornigera (Gastropod rhabdo-like 1) (Table 2).
Some of these putative newly detected viruses were complete or nearly complete, showing a well-defined domain structure (Figure 5). Six of them contained the RdRp domain used for further phylogenetic analyses.
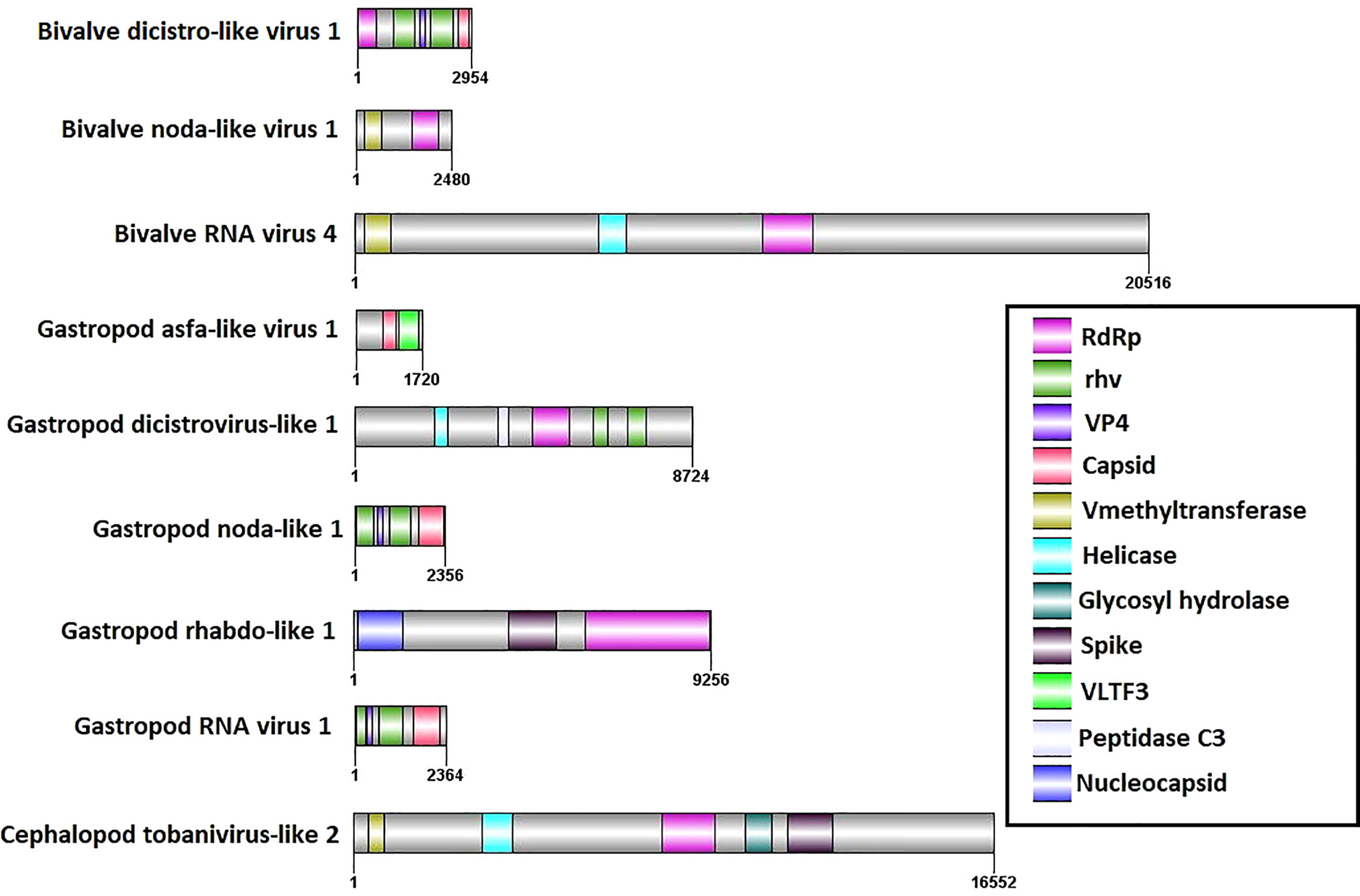
Figure 5 Domain organization of the 9 full or almost complete viral sequences detected in this work.
3.5 RdRp domain and phylogenetic analyses
According to the relevance of RdRp as an essential protein encoded in the genomes of most RNA viruses, the sequences detected in this work were identified and taxonomically classified by BLAST homology to nrNCBI. These RdRp sequences belonged to the order Picornavirales and the families Rhabdoviridae, Bromoviridae, Totiviridae, Aliusviridae and Nodaviridae. After screening the public databases, some phylogenetic analyses, including sequences found in mollusks and reference sequences available in the RefSeq database, were performed.
All the sequence groups were evaluated to determine the best model of evolution. After finding the best models, maximum likelihood phylogenies were run, and Figure 6 shows the results of the analysis. Notably, some of the sequences studied showed a high number of changes with respect to the available sequences (Totiviridae, Bromoviridae and Aliusviridae sequences). In contrast, in the case of the Nodaviridae-like sequences found in mollusks, they grouped in branches between the Alpha- and Betanodavirus sequences, following the great divergence between vertebrate and invertebrate hosts. Finally, it is noteworthy that cephalopod Rhabdoviridae viruses, one of the most abundant viral families found in the analysis, provide relevant new information about the Rhabdovirus evolution.
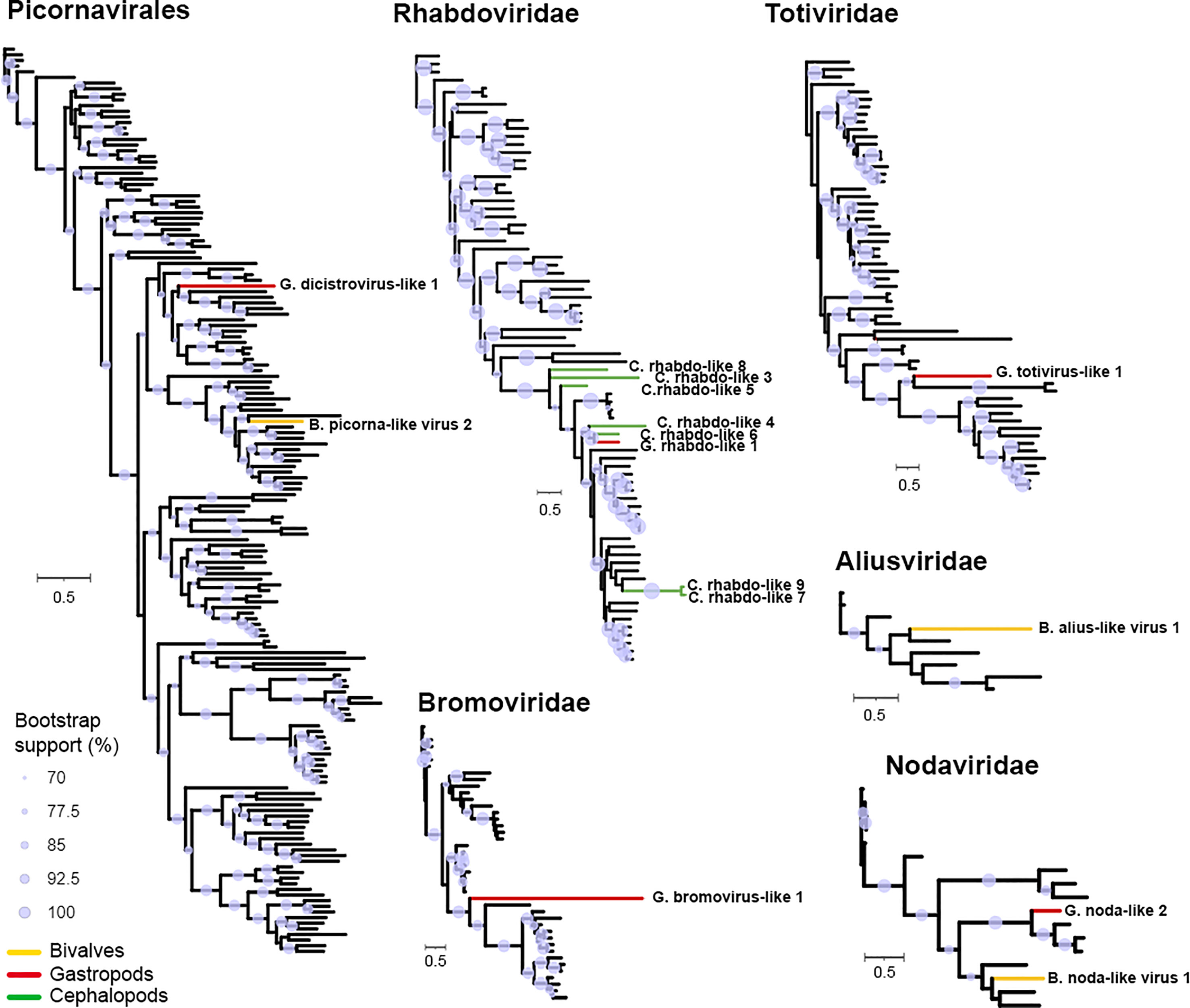
Figure 6 RdRp maximum likelihood analysis. Sequences identified in this work are highlighted in color (gastropods: red, cephalopods: green and bivalves: yellow). Purple circles indicate branches with over 70% support by bootstrapping.
4 Discussion
Viruses constitute a relevant part of the oceans’ microbiome (Suttle, 2007), affecting the composition of other microorganisms, as well as the biogeochemical cycles, and directly or indirectly influencing the health of plants and metazoans (Marx, 2022). Over the last decade, several studies have tried to define the diversity and abundance of viruses in marine environments.
With the main objective of defining abundance and identifying viral diversity, sequencing methods have allowed several extensive studies involving oceanic expeditions and testing samples distributed throughout the oceans, including even the Arctic region (Brum et al., 2015; Roux et al., 2016; Gregory et al., 2019; Dominguez-Huerta et al., 2022). This enormous amount of data uncovered the viral biodiversity in oceans around the globe.
Mollusks, the taxa under study in this work, constitute a widespread group of invertebrates inhabiting a wide variety of habitats (freshwater, marine and terrestrial environments). Their study has become relevant because they are valuable as bioindicators (Chaudhary et al., 2022; De Silva et al., 2022; Gonçalves et al., 2022; Jong et al., 2022; Lemos et al., 2022; Pokhrel et al., 2022) and because many mollusk species are important in the aquaculture industry, as is the case for mussels, oysters, clams and abalones, representing 23% of total aquaculture production (FAO, 2022). It is important to highlight the habit of consuming some uncooked food, being a potential risk for human health (Prato et al., 2004; Guyader et al., 2008; Lattos et al., 2021). Well-demonstrated cases of norovirus, causing gastroenteritis and hepatitis A virus, highlight the importance of detection in samples of commercial interest.
Marine mollusks exposed to the aforementioned diversity of viruses could be a good model to study invertebrate immunity (Qiao et al., 2021b; Qiao et al., 2021a).
Characterizing mollusk viruses without the availability of cell lines is a challenge (Renault and Novoa, 2004). Arzul et al. (2017) reported molecular techniques and histologic approaches to study some of the most relevant viruses affecting some mollusks (oyster, abalone and scallop), but they highlighted the great difficulty resulting from the lack of tools. Despite this obstacle, there are some known viral pathologies that have caused serious problems in the aquaculture industry (Renault et al., 2014; Bai et al., 2015; Bai et al., 2016; Kim et al., 2019).
Massive sequencing studies are increasingly enabling the identification of pathogens. Frequently, the screening of data available in public databases allows the identification of putative new viruses and already known and well-classified viruses. This is the case for several pioneering studies performed in bivalves (Rosani and Gerdol, 2017; Rosani et al., 2019) and nudibranchs (Rosani, 2022). Some of these identified viruses were the Wenzhou gastropod virus 2 in Ruditapes philippinarum samples, a virus that we also found in several samples analyzed in our study. We detected this virus in Hemifusus tuba, a gastropod sampled in China, a different location from the two previous clam detections (Spain and USA).
After exploring different genomic tools and comparing our results to those of other studies (i.e., the Serratus project, Edgar et al., 2022), it is apparent that the different approaches and search algorithms are complementary. A domain scan approach seems to be a successful method since the high level of molecular variation of viruses, especially RNA viruses (Peck and Lauring, 2018; Sanjuán and Domingo-Calap, 2021), and their low representation in databases is an obstacle added to the lack of tools such as molluscan cell lines. Moreover, strategies based on domain searches result in some identifications of DNA viruses inserted in host genomes. Recently, the widespread genomic distribution of eukaryotic transposons showing hallmarks typical of some dsDNA viruses was assessed (Starrett et al., 2021). These transposable elements, easily identified as viral domains, are common in eukaryotic genomes. This could also be the case for Mytilus Mediterranean mussel adintovirus, found in Mytilus spp. in our work and considered a genomic transposon in the work of Starrett et al. (2021). This type of genomic element remains poorly studied in mollusks, and further research is necessary.
Another caution in this type of study should be the detection of contaminant viruses. For instance, depending on the biology of the host, it might be common to find viruses associated with food such as plants or algae in the case of some mollusks. Other common contaminants are those associated with laboratory reagents. Asplund et al. (2019) and Cobbin et al. (2021) reported a list of viruses found in specific reagents, including some retroviruses and parvoviruses. We detected some of these viruses, being Parvovirus domains extensively found in our mollusk dataset (30% of analyzed transcriptomes showed sequences identified as Parvovirus); hence, this detection could be associated with the use of certain reagents (e.g., library preparation kits, culture media) during the development of the experiments.
After considering all the results obtained with the different approaches, differences were found among species in terms of viral number and diversity, but no predominance of detection was observed when comparing the three classes of mollusks. Larvae, visceral mass and brain showed the majority of viral domains and sequence detection. Most viruses detected in gills were from bivalve samples, which was somewhat expected due to the filter-feeding biology of these animals (Musella et al., 2020; Li et al., 2022). In regard to the environment, 10% of the samples were from freshwater locations. We detected an Aliusviridae-like virus in Limnoperna fortunei and a Totiviridae-like virus in Anentome helena. There is little information on these viruses infecting mollusks, thus further analyses including more samples are needed.
Picorna-like viruses (Picornavirales), nodaviruses (Nodamuvirales) and rhabdoviruses (Mononegavirales) (Figure 7) were the most detected viruses in our mollusk dataset. Picorna-like viruses constitute a highly diverse and poorly defined group but are widely found in association with eukaryotes and in the marine environment (Culley et al., 2003; Culley et al., 2006). Nervous necrosis virus (NNV-Betanodavirus) is responsible for mass mortalities in the aquaculture industry worldwide, affecting approximately 30 fish species, with great economic and environmental impacts (Munday et al., 2002; Costa and Thompson, 2016). Moreover, Alphanodavirus also affects insects (Dasmahapatra et al., 1985; Warrilow et al., 2018) and other invertebrates, such as crustaceans, causing major economic losses (Ho et al., 2018; Xu et al., 2020; Xia et al., 2022). Previous works have referred to the detection of this viral family in several mollusks, indicating ubiquity in the marine environment (Gomez et al., 2008; Volpe et al., 2018; Bitchava et al., 2019). In some cases, infectivity of NNV taken from mollusk reservoirs has been demonstrated, indicating a serious risk for outbreaks in susceptible cultured fish (Gomez et al., 2010). We detected nodavirus sequences in several mollusks: Hemifusus tuba (Gastropod noda-like virus 1), Embletonia pulchra (Gastropod noda-like virus 2), Ruditapes philippinarum (Bivalve noda-like virus 1) and Gigantidas vrijenhoeki (Bivalve noda-like virus 2). This last species is a vent mussel, showing the great diversity of locations and species that can be in contact with these viruses (Ryu et al., 2021).
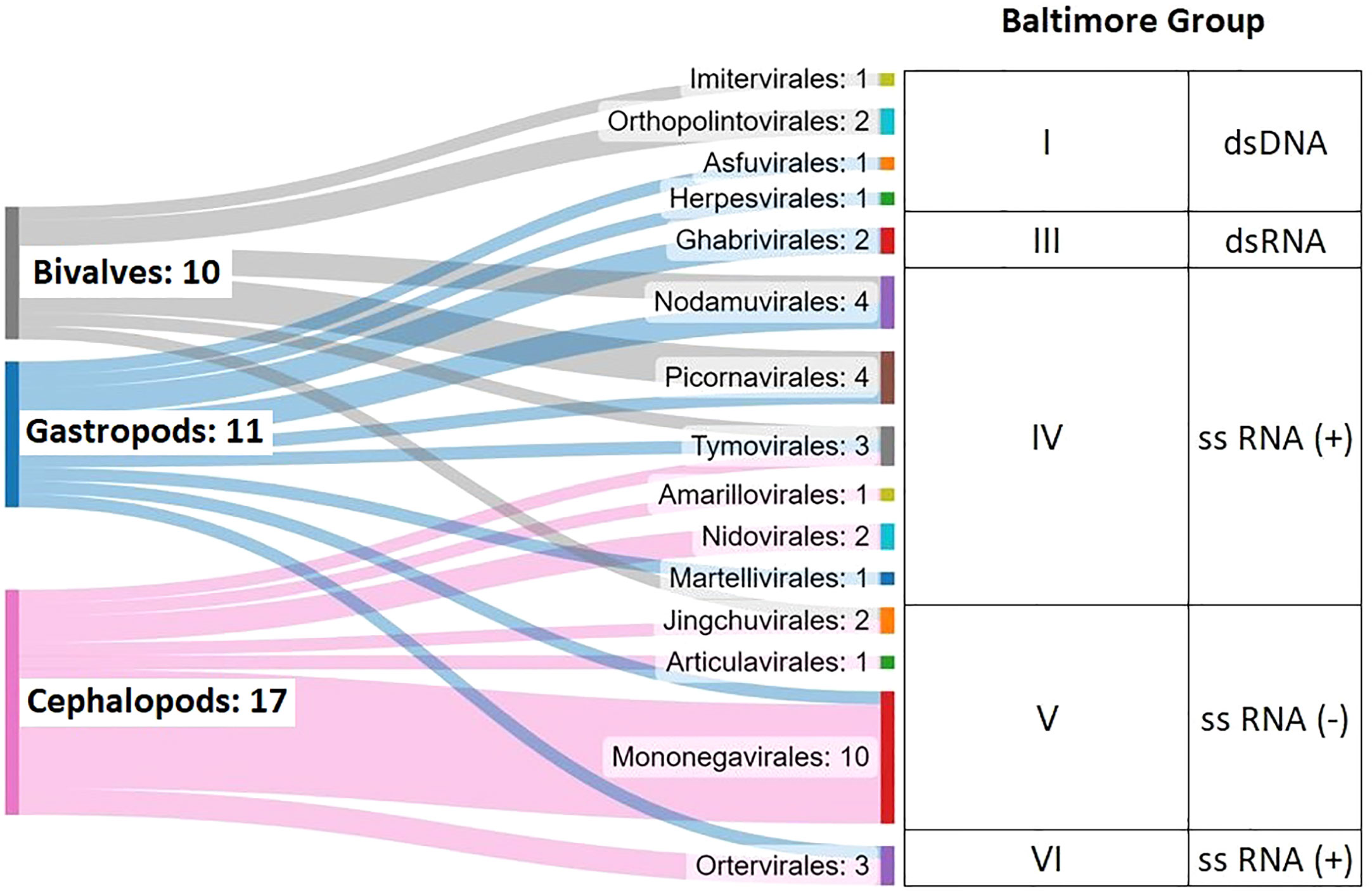
Figure 7 Summary of the viruses identified in this work, considering 62 Mollusca transcriptome datasets.
Finally, cephalopod samples showed a remarkable number of rhabdoviruses (named Cephalopod rhabdo-like virus 1-9 in the present work).Three aquatic rhabdoviruses, spring viremia of carp virus (SVCV), infectious hematopoietic necrosis virus (IHNV) and micropterus salmoides rhabdovirus (MSRV), cause severe diseases among farmed fish species. Scarce information on viruses infecting and causing pathologies in cephalopods has been reported (Gestal et al., 2019). These animals are of great interest because of their economic value (4 million tons of catches per year worldwide on average; FAO). Among the studied cephalopods, several Octopus spp. with commercial interest showed the presence of rhabdoviruses (Cephalopod rhabdo-like virus 5-8), which would imply an economic risk and an explanation for possible drops in population counts.
Thus, these analysis tools might have to be incorporated into fishery resource exploitation works. Additionally, the novel rhabdovirus group found in cephalopods needs to be considered in future research, either as part of the large reservoir that constitutes the group of mollusks or as a cause of possible unknown pathologies of wild populations. This is especially interesting given the aforementioned economic relevance of cephalopods and their use as experimental models due to their high neuronal complexity.
In summary, a meta-analysis was conducted to find viral sequences to continue describing the Mollusca virome. Domain search allowed us to find the majority of viruses, but several search methods needed to be explored. The mollusks in which more viruses were found were cephalopods.
Among the 50 viruses detected, 33 were considered putative new viruses according to their divergence with respect to those deposited in the databases. The most relevant were the single-stranded RNA viruses: picorna-like virus, noda-like virus and rhabdo-like virus, being the rhabdo-like viruses clearly associated with cephalopod samples.
This work support massive sequencing techniques as a great tool to monitor and diagnose pathogens to foresee and explain potential massive mortalities. Moreover, the great diversity of viruses in the oceans is also reflected here, as well as the requirement of using several computer-based analysis tools to obtain more comprehensive results.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository or repositories and accession number(s) can be found in the article or Supplementary Material.
Author contributions
BN and AF conceived and designed the project. MR-C, LG, and AF conducted the experimental work and the bioinformatics analyses. AF, MR-C, and BN analyzed the generated data. MR-C wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Our research was funded by the Ministerio de Ciencia e Innovación (PID2021-124955OB-I00) and Xunta de Galicia (IN607B 2022/13).
Acknowledgments
We want to thank Judit Castro for her technical assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1209103/full#supplementary-material
Supplementary Table 1 | General information on the molluscan transcriptomes under study.
Supplementary Table 2 | List of InterPro and Pfam viral domains.
Supplementary Table 3 | Genomes used to discard host contigs.
Supplementary Table 4 | List of viral domains detected in the 55 SRA datasets.
Supplementary Table 5 | Summary of all the viruses identified in this work.
Supplementary Table 6 | Sequences of putative new viruses discovered in this work.
Supplementary Figure 1 | (A) Pie chart showing the number of viruses detected in each mollusk species. (B) Pie chart showing the number of viruses detected in each type of sample/tissue. (C) Pie chart showing the number of viruses detected in each geographic territory country.
References
Andrews S. (2010) FastQC: a quality control tool for high throughput sequence data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
Arzul I., Corbeil S., Morga B., Renault T. (2017). Viruses infecting marine mollusks. J. Invertebr. Pathol. 147, 118–135. doi: 10.1016/j.jip.2017.01.009
Asplund M., Kjartansdóttir K. R., Mollerup S., Vinner L., Fridholm H., Herrera J. A. R., et al. (2019). Contaminating viral sequences in high-throughput sequencing viromics: a linkage study of 700 sequencing libraries. Clin. Microbiol. Infect. 25 (10), 1277–1285. doi: 10.1016/j.cmi.2019.04.028
Bai C., Gao W., Wang C., Yu T., Zhang T., Qiu Z., et al. (2016). Identification and characterization of ostreid herpesvirus 1 associated with massive mortalities of Scapharca broughtonii broodstocks in China. Dis. Aquat. Organ. 118 (1), 65–75. doi: 10.3354/dao02958
Bai C., Wang C., Xia J., Sun H., Zhang S., Huang J. (2015). Emerging and endemic types of ostreid herpesvirus 1 were detected in bivalves in China. J. Invertebr. Pathol. 124, 98–106. doi: 10.1016/j.jip.2014.11.007
Bitchava K., Chassalevris T., Lampou E., Athanassopoulou F., Economou V., Dovas C. I. (2019). Occurrence and molecular characterization of betanodaviruses in fish and invertebrates of the Greek territorial waters. J. Fish. Dis. 42 (12), 1773–1783. doi: 10.1111/jfd.13098
Blum M., Chang H. Y., Chuguransky S., Grego T., Kandasaamy S., Mitchell A., et al. (2021). The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 49 (D1), D344–D354. doi: 10.1093/nar/gkaa977
Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Brum J. R., Ignacio-Espinoza J. C., Roux S., Doulcier G., Acinas S. G., Alberti A., et al. (2015). Ocean plankton. patterns and ecological drivers of ocean viral communities. Science 348 (6237), 1261498. doi: 10.1126/science.1261498
Chaudhary D. K., Kim H., Reible D., Lee M., Kim S., Kim L. H., et al. (2022). Seasonal trends of mercury bioaccumulation and assessment of toxic effects in Asian clams and microbial community from field study of estuarine sediment. Environ. Res. 212, 113439. doi: 10.1016/j.envres.2022.113439
Cobbin J. C., Charon J., Harvey E., Holmes E. C., Mahar J. E. (2021). Current challenges to virus discovery by meta-transcriptomics. Curr. Opin. Virol. 51, 48–55. doi: 10.1016/j.coviro.2021.09.007
Corbeil S. (2020). Abalone viral ganglioneuritis. Pathogens 9 (9), 720. doi: 10.3390/pathogens9090720
Costa J. Z., Thompson K. D. (2016). Understanding the interaction between betanodavirus and its host for the development of prophylactic measures for viral encephalopathy and retinopathy. Fish Shellfish Immunol. 53, 35–49. doi: 10.1016/j.fsi.2016.03.033
Culley A. I., Lang A. S., Suttle C. A. (2003). High diversity of unknown picorna-like viruses in the sea. Nature 424 (6952), 1054–1057. doi: 10.1038/nature01886
Culley A. I., Lang A. S., Suttle C. A. (2006). Metagenomic analysis of coastal RNA virus communities. Science 312 (5781), 1795–1798. doi: 10.1126/science.1127404
Dasmahapatra B., Dasgupta R., Ghosh A., Kaesberg P. (1985). Structure of the black beetle virus genome and its functional implications. J. Mol. Biol. 182 (2), 183–189. doi: 10.1016/0022-2836(85)90337-7
De Silva N. A. L., Marsden I. D., Gaw S., Glover C. N. (2022). The relationship between population attributes of the mud snail Amphibola crenata and sediment contamination: a multi-estuary assessment. Mar. Pollut. Bull. 180, 113762. doi: 10.1016/j.marpolbul.2022.113762
Dominguez-Huerta G., Zayed A. A., Wainaina J. M., Guo J., Tian F., Pratama A. A., et al. (2022). Diversity and ecological footprint of global ocean RNA viruses. Science 376 (6598), 1202–1208. doi: 10.1126/science.abn6358
Durazzi F., Sala C., Castellani G., Manfreda G., Remondini D., De Cesare A. (2021). Comparison between 16S rRNA and shotgun sequencing data for the taxonomic characterization of the gut microbiota. Sci. Rep. 11 (1), 3030. doi: 10.1038/s41598-021-82726-y
Edgar R. C., Taylor J., Lin V., Altman T., Barbera P., Meleshko D., et al. (2022). Petabase-scale sequence alignment catalyses viral discovery. Nature 602 (7895), 142–147. doi: 10.1038/s41586-021-04332-2
FAO (2022). The state of world fisheries and aquaculture 2022. towards blue transformation (Rome: FAO). doi: 10.4060/cc0461en
Gestal C., Pascual S., Guerra Á., Fiorito G., Vieites J. (2019). Handbook of pathogens and diseases in cephalopods (Cham: Springer). doi: 10.1007/978-3-030-11330-8_7
Go J., Deutscher A. T., Spiers Z. B., Dahle K., Kirkland P. D., Jenkins C. (2017). Mass mortalities of unknown aetiology in pacific oysters Crassostrea gigas in port stephens, new south Wales, Australia. Dis. Aquat. Organ. 125 (3), 227–242. doi: 10.3354/dao03146
Gomez D. K., Baeck G. W., Kim J. H., Choresca C. H. Jr., Park S. C. (2008). Molecular detection of betanodaviruses from apparently healthy wild marine invertebrates. J. Invertebr. Pathol. 97 (3), 197–202. doi: 10.1016/j.jip.2007.10.012
Gomez D. K., Mori K., Okinaka Y., Nakai T., Park S. C. (2010). Trash fish can be a source of betanodaviruses for cultured marine fish. Aquaculture 302, 158–163. doi: 10.1016/j.aquaculture.2010.02.033
Gonçalves J. M., Beckmann C., Bebianno M. J. (2022). Assessing the effects of the cytostatic drug 5-fluorouracil alone and in a mixture of emerging contaminants on the mussel Mytilus galloprovincialis. . Chemosphere 305, 135462. doi: 10.1016/j.chemosphere.2022.135462
Goodacre N., Aljanahi A., Nandakumar S., Mikailov M., Khan A. S. (2018). A reference viral database (RVDB) to enhance bioinformatics analysis of high-throughput sequencing for novel virus detection. mSphere 3 (2), e00069–e00018. doi: 10.1128/mSphereDirect.00069-18
Grabherr M. G., Haas B. J., Yassour M., Levin J. Z., Thompson D. A., Amit I., et al. (2011). Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29 (7), 644–652. doi: 10.1038/nbt.1883
Gregory A. C., Zayed A. A., Conceição-Neto N., Temperton B., Bolduc B., Alberti A., et al. (2019). Marine DNA viral macro- and microdiversity from pole to pole. Cell 177 (5), 1109–1123.e14. doi: 10.1016/j.cell.2019.03.040
Guyader F. S. L., Saux J. C. L., Ambert-Balay K., Krol J., Serais O., Parnaudeau S., et al. (2008). Aichi virus, norovirus, astrovirus, enterovirus, and rotavirus involved in clinical cases from a French oyster-related gastroenteritis outbreak. J. Clin. Microbiol. 46 (12), 4011–4017. doi: 10.1128/jcm.01044-08
Ho K. L., Gabrielsen M., Beh P. L., Kueh C. L., Thong Q. X., Streetley J., et al. (2018). Structure of the macrobrachium rosenbergii nodavirus: a new genus within the nodaviridae? PloS Biol. 16 (10), e3000038. doi: 10.1371/journal.pbio.3000038
Holmes E. C. (2022). The ecology of viral emergence. Annu. Rev. Virol. 9 (1), 173–192. doi: 10.1146/annurev-virology-100120-015057
Jones P., Binns D., Chang H. Y., Fraser M., Li W., McAnulla C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinf. (Ox. Eng.) 30 (9), 1236–1240. doi: 10.1093/bioinformatics/btu031
Jong M. C., Li J., Noor H. M., He Y., Gin K. Y. (2022). Impacts of size-fractionation on toxicity of marine microplastics: enhanced integrated biomarker assessment in the tropical mussels, Perna viridis. Sci. Total Environ. 835, 155459. doi: 10.1016/j.scitotenv.2022.155459
Kalyaanamoorthy S., Minh B. Q., Wong T. K. F., von Haeseler A., Jermiin L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14 (6), 587–589. doi: 10.1038/nmeth.4285
Kim H. J., Jun J. W., Giri S. S., Yun S., Kim S. G., Kim S. W., et al. (2019). Mass mortality in Korean bay scallop (Argopecten irradians) associated with ostreid herpesvirus-1 μVar. Transbound Emerg. Dis. 66 (4), 1442–1448. doi: 10.1111/tbed.13200
Lattos A., Chaligiannis I., Papadopoulos D., Giantsis I. A., Petridou E. I., Vafeas G., et al. (2021). How safe to eat are raw bivalves? host pathogenic and public health concern microbes within mussels, oysters, and clams in Greek markets. Foods 10, 2793. doi: 10.3390/foods10112793
Lemos L., Gantiva L., Kaylor C., Sanchez A., Quinete N. (2022). American Oysters as bioindicators of emerging organic contaminants in Florida, united states. Sci. Total Environ. 835, 155316. doi: 10.1016/j.scitotenv.2022.155316
Letunic I., Bork P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49 (W1), W293–W296. doi: 10.1093/nar/gkab301
Li S., Young T., Archer S., Lee K., Sharma S., Alfaro A. C. (2022). Mapping the green-lipped mussel (Perna canaliculus) microbiome: a multi-tissue analysis of bacterial and fungal diversity. Curr. Microbiol. 79 (3), 76. doi: 10.1007/s00284-021-02758-5
Marx V. (2022). Why the ocean virome matters. Nat. Methods 19 (8), 924–927. doi: 10.1038/s41592-022-01567-3
Matsuyama T., Kiryu I., Inada M., Takano T., Matsuura Y., Kamaishi T. (2021). Susceptibility of four abalone species, Haliotis gigantea, Haliotis discus discus, Haliotis discus hannai and Haliotis diversicolor, to abalone asfa-like virus. Viruses 13 (11), 2315. doi: 10.3390/v13112315
Mihara T., Nishimura Y., Shimizu Y., Nishiyama H., Yoshikawa G., Uehara H., et al. (2016). Linking virus genomes with host taxonomy. Viruses 8, 66. doi: 10.3390/v8030066
Mistry J., Chuguransky S., Williams L., Qureshi M., Salazar G. A., Sonnhammer E. L. L., et al. (2021). Pfam: the protein families database in 2021. Nucleic Acids Res. 49 (D1), D412–D419. doi: 10.1093/nar/gkaa913
Munday B. L., Kwang J., Moody N. (2002). Betanodavirus infections of teleost fish: a review. J. Fish Dis. 25, 127–142. doi: 10.1046/j.1365-2761.2002.00350.x
Musella M., Wathsala R., Tavella T., Rampelli S., Barone M., Palladino G., et al. (2020). Tissue-scale microbiota of the Mediterranean mussel (Mytilus galloprovincialis) and its relationship with the environment. Sci. Total Environ. 717, 137209. doi: 10.1016/j.scitotenv.2020.137209
NaveenKumar S., Shekar M., Karunasagar I., Karunasagar I. (2013). Genetic analysis of RNA1 and RNA2 of macrobrachium rosenbergii nodavirus (MrNV) isolated from India. Virus Res. 173 (2), 377–385. doi: 10.1016/j.virusres.2013.01.003
Nguyen L. T., Schmidt H. A., von Haeseler A., Minh B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32 (1), 268–274. doi: 10.1093/molbev/msu300
Parry R., Asgari S. (2019). Discovery of novel crustacean and cephalopod flaviviruses: insights into the evolution and circulation of flaviviruses between marine invertebrate and vertebrate hosts. J. Virol. 93 (14), e00432–e00419. doi: 10.1128/JVI.00432-19
Peck K. M., Lauring A. S. (2018). Complexities of viral mutation rates. J. Virol. 92 (14), e01031–e01017. doi: 10.1128/JVI.01031-17
Piret J., Boivin G. (2021). Pandemics throughout history. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.631736
Pokhrel P., Mashiko S., Akther S., Suzuki J., Fujita M. (2022). Antioxidant capacity and carbon-based scope for growth of brackish water clams Corbicula japonica under the combined effects of natural and anthropogenic factors. Environ. pollut. 308, 119676. doi: 10.1016/j.envpol.2022.119676
Prato R., Lopalco P. L., Chironna M., Barbuti G., Germinario C., Quarto M. (2004). Norovirus gastroenteritis general outbreak associated with raw shellfish consumption in south Italy. BMC Infect. Dis. 4, 37. doi: 10.1186/1471-2334-4-37
Qiao X., Wang L., Song L. (2021a). The primitive interferon-like system and its antiviral function in mollusks. Dev. Comp. Immunol. 118, 103997. doi: 10.1016/j.dci.2021.103997
Qiao X., Zong Y., Liu Z., Wu Z., Li Y., Wang L., et al. (2021b). The cGAS/STING-TBK1-IRF regulatory axis orchestrates a primitive interferon-like antiviral mechanism in oyster. Front. Immunol. 12. doi: 10.3389/fimmu.2021.689783
Razzauti M., Galan M., Bernard M., Maman S., Klopp C., Charbonnel N., et al. (2015). A comparison between transcriptome sequencing and 16S metagenomics for detection of bacterial pathogens in wildlife. PloS Negl. Trop. Dis. 9 (8), e0003929. doi: 10.1371/journal.pntd.0003929
Renault T., Bouquet A. L., Maurice J. T., Lupo C., Blachier P. (2014). Ostreid herpesvirus 1 infection among pacific oyster (Crassostrea gigas) spat: relevance of water temperature to virus replication and circulation prior to the onset of mortality. Appl. Environ. Microbiol. 80 (17), 5419–5426. doi: 10.1128/AEM.00484-14
Renault T., Novoa B. (2004). Viruses infecting bivalve mollusks. Aquat. Living Resour. 17 (4), 397–409. doi: 10.1051/alr:2004049
Rosani U. (2022). Tracing RNA viruses associated with nudibranchia gastropods. PeerJ 10, e13410. doi: 10.7717/peerj.13410
Rosani U., Gerdol M. (2017). A bioinformatics approach reveals seven nearly-complete RNA-virus genomes in bivalve RNA-seq data. Virus Res. 239, 33–42. doi: 10.1016/j.virusres.2016.10.009
Rosani U., Shapiro M., Venier P., Allam B. (2019). A needle in a haystack: tracing bivalve-associated viruses in high-throughput transcriptomic data. Viruses 11 (3), 205. doi: 10.3390/v11030205
Roux S., Brum J. R., Dutilh B. E., Sunagawa S., Duhaime M. B., Loy A., et al. (2016). Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 537 (7622), 689–693. doi: 10.1038/nature19366
Ryu T., Kim J. G., Lee J., Yu O. H., Yum S., Kim D., et al. (2021). First transcriptome assembly of a newly discovered vent mussel, Gigantidas vrijenhoeki, at onnuri vent field on the northern central Indian ridge. Mar. Genomics 57, 100819. doi: 10.1016/j.margen.2020.100819
Sahul Hameed A. S., Ninawe A. S., Nakai T., Chi S. C., Johnson K. L., Ictv Report Consortium (2019). ICTV virus taxonomy profile: nodaviridae. J. Gen. Virol. 100 (1), 3–4. doi: 10.1099/jgv.0.001170
Sanjuán R., Domingo-Calap P. (2021). Genetic diversity and evolution of viral populations. Encyclopedia Virol. 1, 53–61. doi: 10.1016/B978-0-12-809633-8.20958-8
Schoch C. L., Ciufo S., Domrachev M., Hotton C. L., Kannan S., Khovanskaya R., et al. (2020). NCBI taxonomy: a comprehensive update on curation, resources and tools. Database (Oxford) 2020, baaa062. doi: 10.1093/database/baaa062
Starrett G. J., Tisza M. J., Welch N. L., Belford A. K., Peretti A., Pastrana D. V., et al. (2021). Adintoviruses: a proposed animal-tropic family of midsize eukaryotic linear dsDNA (MELD) viruses. Virus Evol. 7 (1), veaa055. doi: 10.1093/ve/veaa055
Suttle C. A. (2007). Marine viruses–major players in the global ecosystem. Nat. Rev. Microbiol. 5 (10), 801–812. doi: 10.1038/nrmicro1750
Volpe E., Grodzki M., Panzarin V., Guercio A., Purpari G., Serratore P., et al. (2018). Detection and molecular characterization of betanodaviruses retrieved from bivalve mollusks. J. Fish. Dis. 41 (4), 603–611. doi: 10.1111/jfd.12759
Walker P. J., Siddell S. G., Lefkowitz E. J., Mushegian A. R., Adriaenssens E. M., Alfenas-Zerbini P., et al. (2021). Changes to virus taxonomy and to the international code of virus classification and nomenclature ratified by the international committee on taxonomy of viruse, (2021). Arch. Virol. 166 (9), 2633–2648. doi: 10.1007/s00705-021-05156-1
Warrilow D., Huang B., Newton N. D., Harrison J. J., Johnson K. N., Chow W. K., et al. (2018). The taxonomy of an Australian nodavirus isolated from mosquitoes. PloS One 13 (12), e0210029. doi: 10.1371/journal.pone.0210029
Xia J., Wang C., Yao L., Wang W., Zhao W., Jia T., et al. (2022). Investigation on natural infection of covert mortality nodavirus in farmed giant freshwater prawn (Macrobrachium rosenbergii). Anim. (Basel). 12 (11), 1370. doi: 10.3390/ani12111370
Keywords: mollusks, virome, abundance, diversity, metatranscriptomics, RNA-Seq, genomics
Citation: Rey-Campos M, González-Vázquez LD, Novoa B and Figueras A (2023) Metatranscriptomics unmasks Mollusca virome with a remarkable presence of rhabdovirus in cephalopods. Front. Mar. Sci. 10:1209103. doi: 10.3389/fmars.2023.1209103
Received: 20 April 2023; Accepted: 08 June 2023;
Published: 22 June 2023.
Edited by:
Danny Ionescu, Leibniz-Institute of Freshwater Ecology and Inland Fisheries (IGB), GermanyReviewed by:
Ioannis A. Giantsis, University of Western Macedonia, GreeceEric James Schott, University of Maryland, United States
Copyright © 2023 Rey-Campos, González-Vázquez, Novoa and Figueras. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio Figueras, YW50b25pb2ZpZ3VlcmFzQGlpbS5jc2ljLmVz