 Neak Muhammad
Neak Muhammad Forbes Avila1,2
Forbes Avila1,2 Ho Le Han
Ho Le Han Kyoung-Ho Kim
Kyoung-Ho Kim Song-Gun Kim
Song-Gun Kim- 1Biological Resource Center/Korean Collection for Type Cultures (KCTC), Korea Research Institute of Bioscience and Biotechnology, Jeongeup, Republic of Korea
- 2Department of Environmental Biotechnology, KRIBB School of Biotechnology, University of Science and Technology (UST), Daejeon, Republic of Korea
- 3The University of Danang, University of Science and Technology, Danang, Vietnam
- 4Department of Microbiology, Pukyong National University, Busan, Republic of Korea
A Gram-stain-negative, long rod, oxidase and catalase-positive strain WSW3-B12T was isolated from red algae on tidal flats in the West Sea, Korea. Phylogenetic analysis based on the 16S rRNA gene sequence revealed that the strain WSW3-B12T had the highest sequence similarity, 92.7%, to Flexithrix dorotheae DSM 6795T, followed by Rapidithrix thailandica TISTR 1750T at 90.8% in the family Flammeovirgaceae of the phylum Bacteroidota. The whole genome sequence determined using both the Nanopore and Illumina platforms revealed that the complete genome consists of 29 contigs, among which contig 1 was a circular chromosome, while the remaining 28 contigs were plasmids. The size of the genome was 10.1 Mbp and the G+C content was 34.1%. The average nucleotide identity (ANI), digital DNA–DNA hybridization (dDDH), average amino acid identity (AAI), and percentage of conserved proteins (POCP), phylogenomic-related indexes between the strain WSW3-B12T and the closest strain Flexithrix dorotheae DSM 6795T, were 76.6%, 19.9%, 57.2%, and 55.6%, respectively, which were all lower than the threshold values to support the creation of a novel genus. A comprehensive genome analysis revealed that the strain WSW3-B12T harbored many of the key genes involved in central metabolism in the main chromosome and also carried important genes for the production of vitamins, quinone, and antimicrobial resistance on the plasmids. The strain also carried genes that are involved in the metabolism of heavy metals such as arsenic, cobalt, copper, and iron on both the chromosome and plasmids. Furthermore, the genome of the strain was highly enriched with carbohydrate-active enzymes (CAZymes), carrying a total of 241 CAZymes. Moreover, a complete CRISPR/Cas system was detected on plasmid 20. The major fatty acids of the strain were iso-C15:0 and C16:1 ω5. The polar lipids contained phosphatidylethanolamine, four unidentified lipids, and four glycolipids. The respiratory quinone was menaquinone 7. Based on the phenotypic, chemotaxonomic, and genomic analyses, the strain WSW3-B12T could be assigned to a novel species and novel genus within the family Flammeovirgaceae, for which the name Chondrinema litorale gen. nov., sp. nov. (type strain WSW3-B12T = KCTC 82707T = GDMCC 1.3198T) is proposed.
1 Introduction
The sea covers around 70% of the surface of our planet, and tidal flats connect the sea and land. Tidal flats are located between marine ecosystems and land environments, providing a habitat for aquatic organisms and an ideal place for isolating unique microbial taxa (Wilms et al., 2006). They are exposed to certain environmental events, such as flooding, habitat destruction, climate changes, and pollution (Duarte et al., 2008), which result in the formation of unique microbial communities (Lv et al., 2016). Microbes in tidal flats play critical roles in regulating ecological processes and biogeochemical cycles (Beam et al., 2020). They are a key player in nutrient cycling by converting organic matter into inorganic forms, which enhance the nutrient supply in marine environments (Lee et al., 2021). Furthermore, these microbes also decompose pollutants flowing into the tidal flats (Cheng et al., 2017; Lee et al., 2019; Kim et al., 2022). The productivity of tidal flat ecosystems is largely due to these microbes. However, the precise relationship between their diversity and functioning is little understood.
The microbes in the tidal flats interact with invertebrates, animals, plants, and seaweed. Seaweed or sea macroalgae, which perform primary production using sunlight and nutrients from the environment, refer to thousands of species of macroscopic, multicellular marine algae. Macroalgae include red, brown, and green algae, which are essential components of the marine food chain (Lobban and Harrison, 1994). Furthermore, macroalgae are biological reservoirs of abundant marine bacteria (Singh and Reddy, 2015; Juhmani et al., 2020). These microbes produce bioactive compounds that can be used to combat drug resistance and cancer (Menaa et al., 2020; Kizhakkekalam and Chakraborty, 2021). A large number of novel bacterial strains have been isolated from marine algae (Karthick and Mohanraju, 2018; Asharaf et al., 2022; Muhammad et al., 2022a).
The phylum Bacteroidota (a heterotypic synonym of Bacteroidetes) was first established by Leadbetter in 1974 (Leadbetter, 1974), subsequently amended by Reichenbach in 1992 (Reichenbach, 1992), and again revised by Ludwig (Ludwig et al., 2010). The phylum was initially described as a monophyletic branch of three genera, Cytophaga, Flavobacterium, and Bacteroides (CFB) (Paster et al., 1994). But it was expanded to include six classes and six orders (https://lpsn.dsmz.de/phylum/bacteroidota). The class Cytophagia is currently represented by the sole order Cytophagales (https://lpsn.dsmz.de/order/cytophagales) comprising 20 families, among which Flammeovirgaceae (https://lpsn.dsmz.de/family/flammeovirgaceae) is the second largest family, comprising nine validly published genera and three genera unassigned to the family. The nine validly published genera are Flammeovirga, Flexithrix, Imperialibacter, Limibacter, Perexilibacter, Rapidithrix, Sediminitox, Tunicatimonas, and Xanthovirga.
Members of the family Flammeovirgaceae are heterotrophic, Gram-stain-negative, non-flagellated, non-spore-forming, rod-shaped cells, and share menaquinone 7 (MK-7) as the major respiratory quinone (Yoon et al., 2011). They are widely distributed in various environments, particularly in marine aquatic systems (Yoon et al., 2012; Wang et al., 2013; Goldberg et al., 2020). Members of the family Flammeovirgaceae and other members of the phylum Bacteroidota are relatively enriched in a large number of glycoside hydrolases (GHs), glycosyl transferases, peptidases, and adhesion proteins that degrade organic biomacromolecules (Fernández-Gómez et al., 2013). They contain a large number of genes encoding carbohydrate-active enzymes, which potentially participate in polysaccharide degradation (Mckee et al., 2021). There are a number of reported strains of the phylum Bacteroidota that can degrade various types of complex polysaccharides (Hehemann et al., 2012; Mann et al., 2013).
During a study on the microbial diversity of the tidal flats in the West Sea, Korea, a number of novel bacterial strains were isolated (Muhammad et al., 2022a; Muhammad et al., 2022b). Strain WSW3-B12T isolated from the red algae of the genus Chondrus was characterized by polyphasic taxonomy and genome analysis. Based on the polyphasic taxonomic analysis, it is proposed that the isolate represents a novel species and a novel genus of the family Flammeovirgaceae in the phylum Bacteroidota. A comparative genomic analysis of the strain WSW3-B12T and its close strains highlighted the uniqueness in terms of multiple plasmids and the presence of various functional genes for possible application. Genome mining emphasized the presence of abundant genes for heavy metal metabolism, for CAZymes of polysaccharide degradation, and for CRISPR candidates that are expected to have potential biotechnological applications. Furthermore, the degradation of polysaccharides (starch, inulin, and cellulose) and the presence of heavy metal metabolism genes (arsenic, cobalt, and copper) highlighted the contribution of strain in the carbon cycle (Fernández-Gómez et al., 2013) and its important role in the remediation of heavy metals (Chen et al., 2018; Dell’Anno et al., 2021) in the tidal flat ecosystem.
2 Materials and methods
2.1 Isolation and cultivation
A red alga belonging to the genus Chondrus was collected in the West Sea, Byeonsan, Republic of Korea (35° 40′ 53.76″ N 126° 31′ 51.96″ E) in late autumn, 2020. The red alga genus Chondrus is abundant across the east and west coasts of the Republic of Korea. The genus can be found on rocks from the middle intertidal zone into the subtidal zone (Lee et al., 2013; Kristyanto et al., 2022). One gram of the alga was cut into small pieces and then placed at the center of agar plates, which was composed of 60% seawater and 1.5% agar. The primary plates were incubated at 15 °C for five days and then routinely observed under a stereoscopic microscope. The colonies from the primary plate were picked up and transferred to marine agar 2216 (MA; BD) at 30°C which is also called Zobell media, a standard plate medium for the cultivation of marine bacteria (ZoBell, 1941). The pure cultures of the novel strain were preserved in 20% glycerol at -80°C. The cultures were deposited in the Korean Collection for Type Cultures (KCTC) and the Guangdong Microbial Culture Collection Center (GDMCC), under the culture depositing numbers KCTC 82707T and GDMCC 1.3198T.
2.2 16S rRNA phylogeny
The genomic DNA of the strain WSW3-B12T was extracted from cells grown in MA at 30°C, by a NucleoSpin Microbial DNA kit (Macherey-nagel, Germany) following the manufacturer’s instructions. The 16S rRNA gene was amplified with bacterial universal primers and sequenced with Sanger’s sequencing method (Biofact, South Korea) (Pheng et al., 2020). Pairwise sequence similarity values of the 16S rRNA gene sequence (1454 bp) were obtained through the EzBioCloud server (http://www.ezbiocloud.net) (Yoon et al., 2017a). All sequences of the related strains retrieved from EzBioCloud were aligned and edited with the BioEdit software (version 7.2.5). Phylogenetic trees were constructed by using neighbor-joining (NJ) (Saitou and Nei, 1987), maximum-likelihood algorithms (ML) (Felsenstein, 1981), and the maximum parsimony (MP) method (Felsenstein, 1981) in the Molecular Evolutionary Genetics Analysis (MEGA X) program (Kumar et al., 2018). For the ML tree, Kimura’s two-parameter model was used for the calculation of the evolutionary distance. For the construction of NJ and MP phylogenetic trees Tajima-Nei model and subtree-pruning-regrafting (SRF) model were used. The bootstrap resampling method of 1000 replicates was applied to evaluate the phylogenetic tree. Bootstrap values >70% are presented in nodes of each branch.
2.3 Morphological, physiological, and biochemical analyses
Gram-staining was performed with the BBL™ Gram-staining kit (BD, USA) according to the manufacturer’s instructions. Cells of the strain WSW3-B12T were incubated for 48 h on MA at 30 °C and examined for morphology through a scanning electron microscope (Regulus 8100, Hitachi) (Jeon et al., 2022). The motility was tested by the hanging drop method (Bowman, 2000). The optimal temperature for growth was determined in MA, for seven days at 4, 10, 15, 20, 25, 30, 37, and 45 °C. The salt tolerance was determined for a novel and reference strains by making a media having the same composition as MA except with the supplementation of salt from 0 to 0.5, and 1.0-8.0% (w/v) (Muhammad et al., 2022a). Growth at pH 5 to 10 (at intervals of 0.5 unit) was determined in MA adjusted with various buffers, at a concentration of 20 mM: acetate for pH 5.0–5.5, phosphate for pH 6.0–8.0, Tris for pH 8.5–9.0, and carbonate for pH 9.5–10.0 (Muhammad et al., 2022b). Catalase activity was tested by the production of oxygen bubbles when 3% (v/v) H2O2 was added to fresh cells (Ueno et al., 2021). The oxidase reaction was carried out using 1% (w/v) tetramethyl-p-phenylenediamine.
The degradation of polysaccharides was studied in basal media made of 60% (v/v) seawater, 0.1% (w/v) peptone, 0.6% (w/v) gellan gum as a solidifying agent, and 0.1% (w/v) of each test polysaccharide (cellulose, chitin, ĸ-carrageenan, inulin, sodium alginate, starch, and xylan) (Gao et al., 2017). The degradation of polysaccharides was further determined by using basal broth, which contained 0.2% of the above sugar molecules. Each sugar broth was inoculated with a 48 hours’ old culture and incubated at 30 °C in the shaker. The supernatant of the culture was harvested at 0, 2, and 4 days and then reacted with 3,5-dinitrosalicylic acid (DNS) to detect reduced sugar production (Gao et al., 2017; Deshavath et al., 2020). The hydrolysis of Tweens 20, 40, 80, and casein was determined by the method of Cowan & Steel (Phillips, 1993; Amrina et al., 2021). DNase activity was checked by using DNase agar (BD).
The enzymatic activities and carbon source utilization were determined by using API ZYM, API 20E, API 50CH (bioMérieux), and GEN III Microplates (Biolog), following the manufacturer’s instructions, except that the inoculum suspensions were adjusted to a final concentration of 2% (w/v) NaCl (Jeong et al., 2020). To test for growth under an anaerobic condition, the strain WSW3-B12T was cultivated on MA plates and incubated in an anaerobic jar at 30°C for seven days, following the manufacturer’s instructions (BD GasPak Systems). Susceptibility to antibiotics was tested on MA plates using various antibiotic discs (μg/disc): chloramphenicol (30), erythromycin (15), kanamycin (30), novobiocin (30), neomycin (30), penicillin (10 IU), tetracycline (30), and vancomycin (30) (Jorgensen and Turnidge, 2015).
2.4 Chemotaxonomic analysis
For the fatty acid analysis, cells of the strain WSW3-B12T and the reference strains Flexithrix dorotheae KCTC 82993T, Rapidithrix thailandica KCTC 82795T, and Sediminitomix flava KCTC 12970T were collected from MA agar at the same physiological age. The standard MIDI protocol (version 6.2) was used to extract the fatty acids from the cell (Sasser, 1990). The extracted fatty acid methyl esters were then injected into a gas chromatograph and were analyzed by the Sherlock Microbial Identification System (TSBA; library version 6.0). For respiratory quinones, the compounds were extracted from bacterial biomass using the protocol of Komagata and Suzuki (Komagata and Suzuki, 1988). First, 100 mg of freeze-dried cells was treated with chloroform-methanol (2:1, v/v), and then crude compounds were separated by thin-layer chromatography (TLC, 20cm×20cm, silica gel 60 F254 plate, Merck) using petroleum benzene/diethyl ether (9:1, v/v) as the developing solvent. The quinone band was collected from a TLC plate and dissolved in acetone. Finally, the compound was purified and identified by reverse-phase high-performance liquid chromatography with a UV detector at 270 nm. Polar lipids were extracted from the freeze-dried cells by chloroform/methanol followed by two-dimensional TLC, as described by Minnikin et al. (Komagata and Suzuki, 1988). The two-dimensional TLC (10 cm×10 cm, silica gel 60 F254 plate, Merck) plates were run using chloroform-methanol-water (65:25:4, v/v/v) and chloroform-methanol-acetic acid-water (80:12:15:4, v/v/v/v) as the solvent systems for the first and second dimensions, respectively. Finally, the TLC plates were sprayed with 0.2% ninhydrin for the amino group-containing lipids, α-naphthol for sugar-containing lipids, molybdenum blue for phosphorus-containing lipids, 0.5% phosphomolybdic acid reagent for total lipids, and Dragendorff’s solution for quaternary nitrogen-containing lipids.
2.5 Genome sequencing and analysis
The total genomic DNA of the strain WSW3-B12T was extracted from two-day old culture on MA. The quality and quantity of the DNA samples were measured with a spectrophotometer (Nanodrop 2000/2000c), and the size and length were monitored by electrophoresis using 1% (w/v) agarose gel. The whole genome sequencing was performed by a combination of Illumina Novaseq 6000 and Oxford Nanopore Technologies (ONT) (at HME, Republic of Korea). For Illumina sequencing, the short-length DNA was used to build up a library while for the ONT high-molecular-weight DNA was used to prepare the library using a ligation sequencing kit (SQK-LSK109; ONT) following the manufacturer’s protocol (version RPB_9059_v1_revC_08Mar2018). Sequencing was performed as multiplex runs on a MinION with MinKnow v1.15.1 using FLO-MIN106 R9.4 flow cells. Illumina reads were trimmed and filtered from raw reads using Trimmomatic (Bolger et al., 2014) and Filtlong v0.20 (https://github.com/rrwick/Filtlong) filtering tools. For Nanopore reads, the raw Fast5 files were demultiplexed with Guppy to retain the barcoded reads. Porechop v0.2.4 (https://github.com/rrwick/Porechop) was used to remove the barcode and adapters. The filtered Illumina and Nanopore reads were assembled using four different programs: Spades, CANU (Koren et al., 2017) Unicycler, and Flye (Lin et al., 2016). The quality initial genomes bins were then evaluated using CheckM (Parks et al., 2015).
2.6 Genome annotation and phylogeny
First, the genomes were annotated by NCBI Prokaryotic Genomes Automatic AnnotationPipeline (PGAAP) and Prokka version 1.14.6 (Seemann, 2014). The genome of the strain WSW3-B12T contained 29 contigs. Each contig was predicted for plasmids and then blasted to find the most similar plasmids and their closest host using the PLSDB server v. 2021_06_23_v2 (Schmartz et al., 2022).
To determine the taxonomic position of the strain WSW3-B12T the average nucleotide identity (ANI) and digital DNA-DNA hybridization (dDDH) were calculated using the ANI calculator tool in Kostas Lab Service (http://enve-omics.ce.gatech.edu/ani/) and the Genome to Genome Distance Calculator version 3.0 of DSMZ (https://ggdc.dsmz.de/ggdc.php#) (Meier-Kolthoff et al., 2013). For genus confirmation, amino acid identity (AAI) and percentage of conserved proteins (POCP) values were further calculated using Kostas Lab Service (http://enve-omics.ce.gatech.edu/aai/) and the BLASTP program (https://github.com/hoelzer/pocp), respectively (Qin et al., 2014). The whole-genome-based phylogenetic tree was built based on the alignment of 400 marker genes with FastTree using PhyloPhlAn version 3.0 (Asnicar et al., 2020). A nonmetric multidimensional scaling (nMDS) plot was made using a matrix of functional genes found in the COGs (Clusters of Orthologous Groups) database (Galperin et al., 2021), which was used to find the clustering of the strain WSW3-B12T and the existing strains.
2.7 Genome functional analysis
The functional genes within each genome were annotated using the KEGG database and then translated into pathways using KEGG Decoder (Graham et al., 2018) and KEGG-Expander (https://github.com/bjtully/BioData/tree/master/KEGGDecoder). The prediction and classification of the orthologous groups associated with the protein coding genes were annotated using the COGs database (Galperin et al., 2021). The genomes were further annotated for functional genes using the rapid annotations based on subsystem technology (RAST) database (https://rast.nmpdr.org/rast.cgi). Carbohydrate active enzymes were annotated using the dbCAN2 meta server (Zhang et al., 2018). Biosynthesis gene clusters (BGCs) and metabolic gene clusters (MGCs) possessed by each genome were identified using antiSMASH 6.01 (Blin et al., 2019). The antimicrobial resistance (AMR) genes were predicted using the comprehensive antibiotic resistance database (CARD) database (Konaté et al., 2019; Alcock et al., 2022). The CRISPRCasFinder server (Couvin et al., 2018) and CRISPRcasIdentifier (https://github.com/BackofenLab/CRISPRcasIdentifier) were used to predict the CRISPR-Cas system (Padilha et al., 2020). Finally, the cas enzyme’s sequences were blasted using UniProt for confirmation (The UniProt Consortium, 2023). Phage sequences were identified by the PHAST (phage search tool) (Arndt et al., 2016).
3 Results and discussion
3.1 Isolation and identification
The strain WSW3-B12T was isolated from the red alga genus Chondrus collected from tidal flats at the West Sea, Byeonsan, Republic of Korea. The strain grew well on MA and produced regular pin-pointed, light brown colonies after 48 hours’ incubation. The diameters of the colonies ranged from 325-420 µm. Cells of the strain WSW3-B12T were Gram-stain-negative, non motile and rod-shaped with a length of 5-7 µm and width of 0.46-0.54 µm (Figure S1).
3.2 16S rRNA gene analyses
The analysis of 16S rRNA gene sequences from the EzBioCloud server (http://www.ezbiocloud.net) revealed that the strain WSW3-B12T was phylogenetically affiliated with the family Flammeovirgaceae, and the closest relatives were Flexithrix dorotheae DSM 6795T (92.7%) and Rapidithrix thailandica TISTR 1750T (90.8%). Furthermore, the phylogenetic trees based on 16S rRNA gene sequences showed clustering of the strain WSW3-B12T with Rapidithrix thailandica TISTR 1750T and Flexithrix dorotheae DSM 6795T, (Figure 1). The closest strains Flexithrix dorotheae DSM 6795T, Rapidithrix thailandica TISTR 1750T, and Sediminitomix flava DSM 28229T were isolated from marine silt, algae, and sediment, respectively (Hosoya and Yokota, 2007; Khan et al., 2007; Srisukchayakul et al., 2007). The strains Flexithrix dorotheae KCTC 82993T, Rapidithrix thailandica KCTC 82795T, and Sediminitomix flava KCTC 12970T were used as reference strains. Finally, the 16S rRNA gene sequences of the strain WSW3-B12T were submitted to the NCBI database under the accession number OP823709.
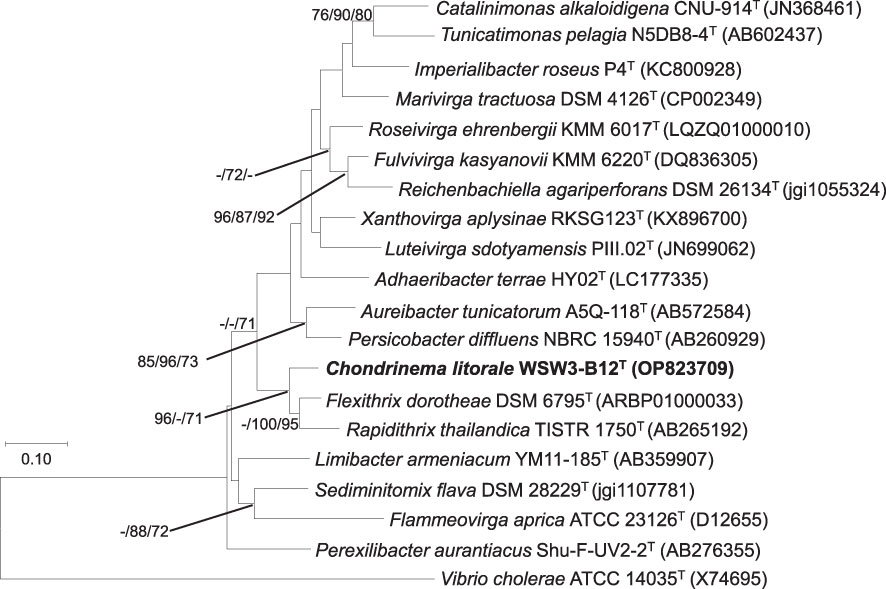
Figure 1 Maximum likelihood tree based on 16S rRNA gene sequences showing the phylogenetic relationship of the strain WSW3-B12T and closely related genus of family Flammeovirgaceae and order Cytophagales. Bootstrap values (>70%) in the order of ML/NJ/MP are shown at the branch points based on 1000 replications. Vibrio cholerae ATCC 14035T were used as outgroups. Bar 0.10 substitutions per nucleotide position.
3.3 Physiological and biochemical characteristics
The strain WSW3-B12T was Gram-stain-negative, oxidase and catalase positive, strictly aerobic and long rod-shaped bacteria. Growth occurred in a temperature range of 15–30°C (optimum, 25–30°C), a pH range of 5.5–9.0 (optimum, 6.5–8.5), and a NaCl range of 1.0–7.0% (optimum, 2.0–5.0%) (w/v). The strain degraded cellulose, inulin, and starch but not chitin, ĸ-carrageenan, sodium alginate, and xylan. Cells were positive for the hydrolysis of Tweens 20, 40, and 80 but negative for the hydrolysis of casein and DNase. The strain was sensitive to chloramphenicol, novobiocin, tetracycline, and vancomycin, but resistant to erythromycin, kanamycin, neomycin, and penicillin. In the API ZYM test, activities of acid phosphatase, alkaline phosphatase, N-acetyl-β-glucosaminidase, cystine arylamidase, α-chymotrypsin, esterase (C4), esterase lipase (C8), α-galactosidase, β-galactosidase, α-glucosidase, β-glucosidase, leucine arylamidase, naphthol-AS-BI-phosphohydrolase, trypsin, and valine arylamidase were present while activities of α-fucosidase, β-glucuronidase, lipase (C14), and α-mannosidase were absent. The strain was only positive for gelatinase in API 20E. In the API 50CH strip, acid is produced from L-arabinose, amidon (starch), arbutin, N- acetylglucosamine, D-cellobiose, gentibiose, D-galactose, glucose, D-lactose, methyl-αD-glucopyranoside, methyl-αD-mannopyranoside, D-maltose, D-melibiose, D-mannose, D-melezitose, D-raffinose, L-rhamnose, D-sucrose, D-trehalose, D-turanose, and D-xylose. In a GEN III MicroPlate (Biolog) test, cells utilize dextrin, L-fucose, D-glucose-6-phosphate, D-glucuronic acid, β-methyl-D-glucoside, L-malic acid, sucrose, D-trehalose, D-turanose, and quinic acid. Details of physiological and biochemical characteristics that differentiate the strain WSW3-B12T from its closest relatives in the family Flammeovirgaceae are listed in (Table 1).
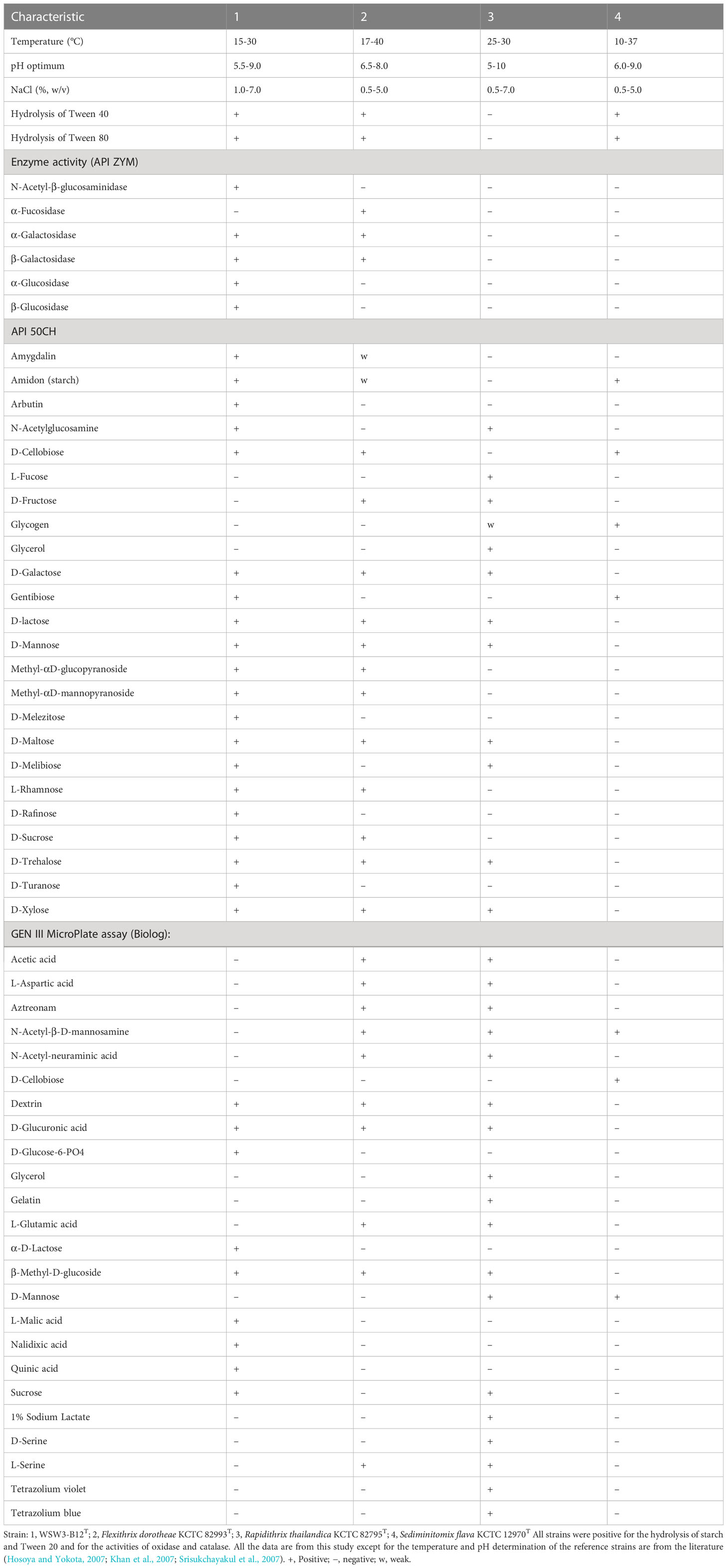
Table 1 Differential characteristics of the strain WSW3-B12T and phylogenetically closely related type species of the genus belongs to Flammeovirgaceae.
3.4 Chemotaxonomic analysis
The major fatty acids (>5.0%) of the strain WSW3-B12T were iso-C15:0 (24.9%), C16:1 ω5c (23.8%), C17:1 ω6c (9.4%), and iso-C16:0 (7.9%). Meanwhile, iso-C17:0 3-OH (3.5%), summed feature 3 (C16:1 ω7c and/or C16:1 ω6c) (3.5%), C15:0 3-OH (3.2%), C16.0 (2.8%), C18:1 ω9c (2.3%), C12:0 (2.0%), C16:0 3-OH (1.5%), and C14:0 (1.4%) were the minor fatty acids (Table 2). The novel strain and all three reference strains showed a higher percentage of C16:1 ω5c (15.3-41.0%) and iso-C15:0 (8.5-24.9%) components of fatty acids. The strain WSW3-B12T had a higher percentage of iso-C16:0 (7.9%) and a lower percentage of C16.0 (2.8%) as compared to the reference strains. Interestingly, among the four strains, only the strain WSW3-B12T had C17:1 ω6c (9.4%) component of fatty acid. The respiratory quinones of the strain WSW3-B12T was menaquinone 7 (MK-7) which was also founded in the reference strains. The polar lipids of the strain WSW3-B12T contained phosphatidylethanolamine, one unidentified aminophospholipid, four unidentified glycolipids, and three unidentified lipids (Figure S2).
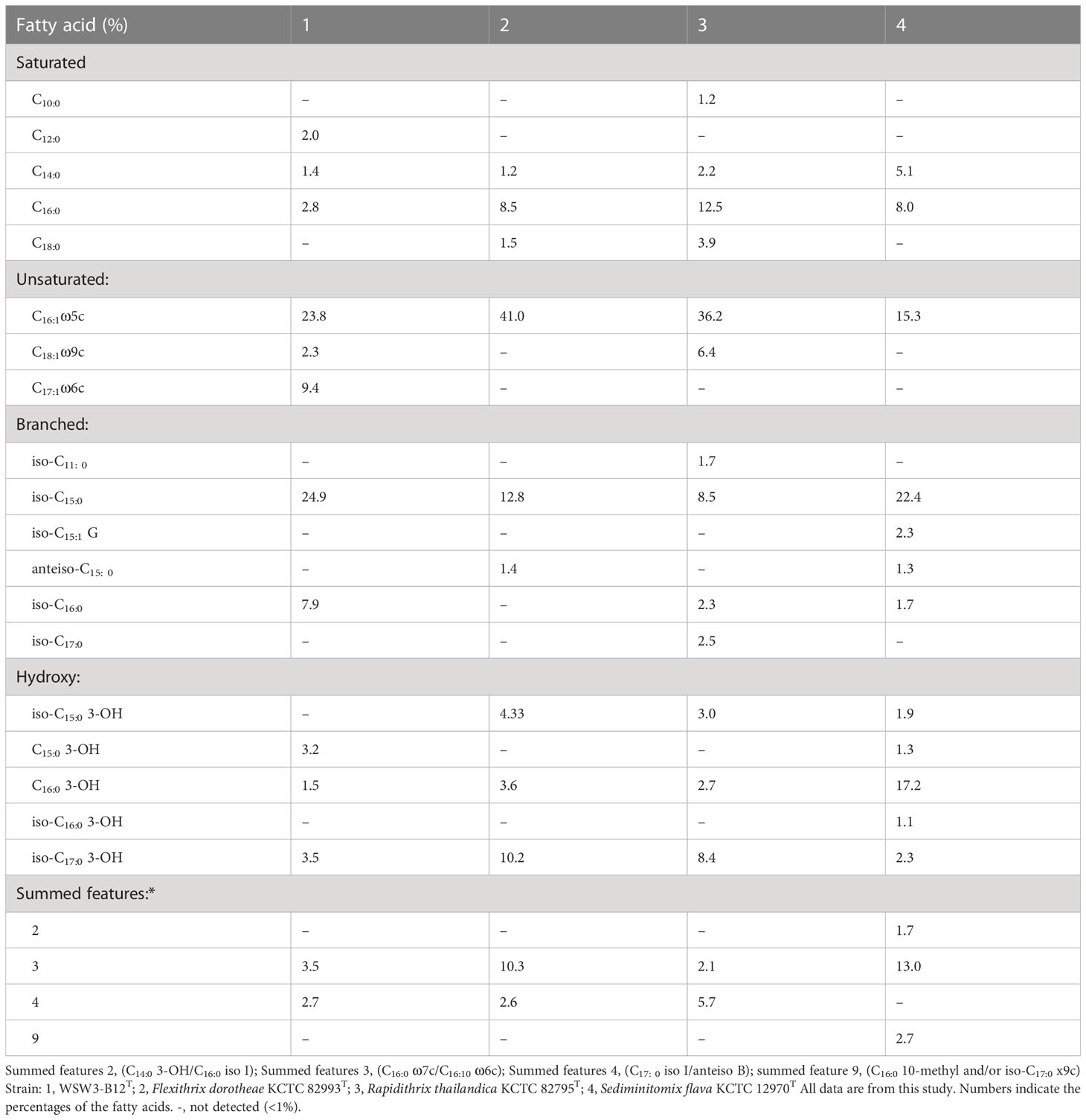
Table 2 Cellular fatty acid compositions (%) of the strain WSW3-B12T and phylogenetically closely related type species of genus belonging to Flammeovirgaceae.
3.5 Genomic general features
The combined Nanopore and Illumina sequencing platforms were used to determine the complete genomes of the strains WSW3-B12T. The CheckM analysis showed that the strain had 99.4% completeness and only 1.45% contamination, which indicated the high quality and reliability of the genomes. The complete genome of the strain WSW3-B12T comprises 29 contigs, among which contig 1 was a circular chromosome length of 4.938441 bp while the remaining 28 contigs were plasmids, out of which six were mobile plasmids and the rest were non-mobilizable plasmids. Some genomes of the family Flammeovirgaceae, the phylum Bacteroidota are relatively large (Feng et al., 2021a): the genomes of Flammeovirga yaeyamensis NBRC 100898 (Feng et al., 2021b), Flammeovirga sp. SJP92 (Dong et al., 2017), Flexithrix dorotheae DSM 6795T, and Flammeovirga aprica JL-4T are 7.4, 8.4, 9.5, and 9.3 Mbp, respectively. The details of each plasmid are summarized in Table S1. Interestingly, limited numbers of bacteria have been discovered to have multiple plasmids (Cazares et al., 2020, Feng et al., 2021b). Some marine members of the phylum Bacteroidota carry a single large plasmid (Zhong et al., 2001; Saw et al., 2012). We hypothesize that the strain WSW3-B12T received plasmids from other bacteria in the environment, which explains the presence of the high number of plasmids and the mobile genetic elements in the genome of the strain WSW3-B12T.
Genome annotation by PGAAP pipeline in the NCBI identified a total of 8199 genes, nine ribosomal RNA genes (5S, 3; 16S, 3; 23S, 3), and a total of 51 transfer RNA genes. The G+C content of the strain WSW3-B12T was 34.1%, while for the reference strains Flexithrix dorotheae DSM 6795T and Sediminitomix flava DSM 28229T the content was 36.4 and 35.6%, respectively. The genomic features of the strain WSW3-B12T and the existing strains are presented in (Table 3). The genome of the strain WSW3-B12T was submitted to the NCBI with the GenBank accession number GCA_026250525.
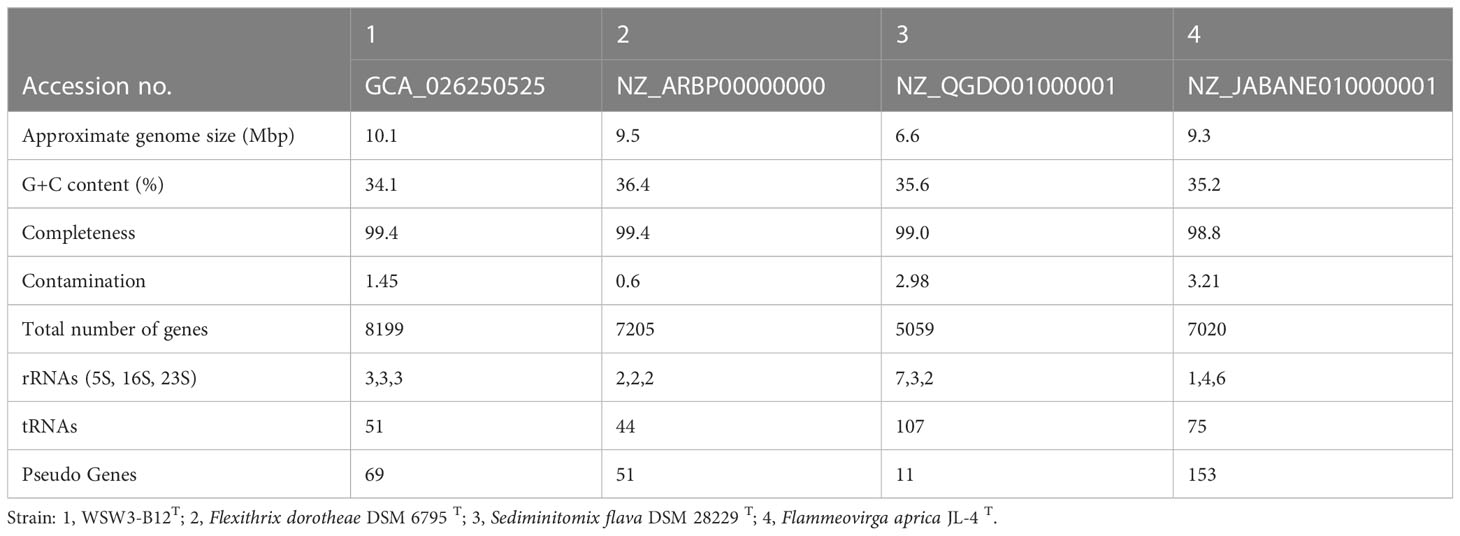
Table 3 Genome features of the strain WSW3-B12T and its closest related members in the family Flammeovirgaceae.
3.6 Whole-genome-based phylogeny
To further confirm the taxonomic position of the strain WSW3-B12T, the ANI and dDDH were calculated among the novel strain and type species of four genus of Flammeovirgaceae Flexithrix dorotheae DSM 6795T, Sediminitomix flava DSM 28229T, Flammeovirga aprica JL-4T, and Xanthovirga aplysinae RKSG123 T. The ANI and dDDH between the strain WSW3-B12T and Flexithrix dorotheae DSM 6795T, Sediminitomix flava DSM 28229T, Flammeovirga aprica JL-4T, and Xanthovirga aplysinae RKSG123T were ranged from 72.3-76.6 and 19.2-26.1%, respectively. The values of ANI and dDDH were below the standard cut-off values for speciation, which are ANI (95-96%) (Yoon et al., 2017b) and dDDH (70%) (Auch et al., 2010). The AAI and POCP values between the strain WSW3-B12T and Flexithrix dorotheae DSM 6795T, Sediminitomix flava DSM 28229T, Flammeovirga aprica JL-4T, and Xanthovirga aplysinae RKSG123 T were ranged from 46.79-57.2 and 28.3-55.6%, respectively. The AAI cut-off value applied for the delineation of genus has a wide range of 60–80% (Park et al., 2022). A POCP value of 50% can be used for genus delineation, with a 55% value as the genus boundary (Qin et al., 2014; Barco et al., 2020). Thus, the strain WSW3-B12T could represent a novel genus because genome-relatedness values are significantly lower than the species and genus boundaries. The phylogenomic tree showed that the strain WSW3-B12T formed a clade with the closest strains, Flexithrix dorotheae DSM 6795T and Sediminitomix flava DSM 28229T, which also belongs to Flammeovirgaceae (Figure 2). The nonmetric multidimensional scaling (nMDS) plots further presented the same cluster between the strains WSW3-B12T and Flexithrix dorotheae DSM 6795T (Figure S3).
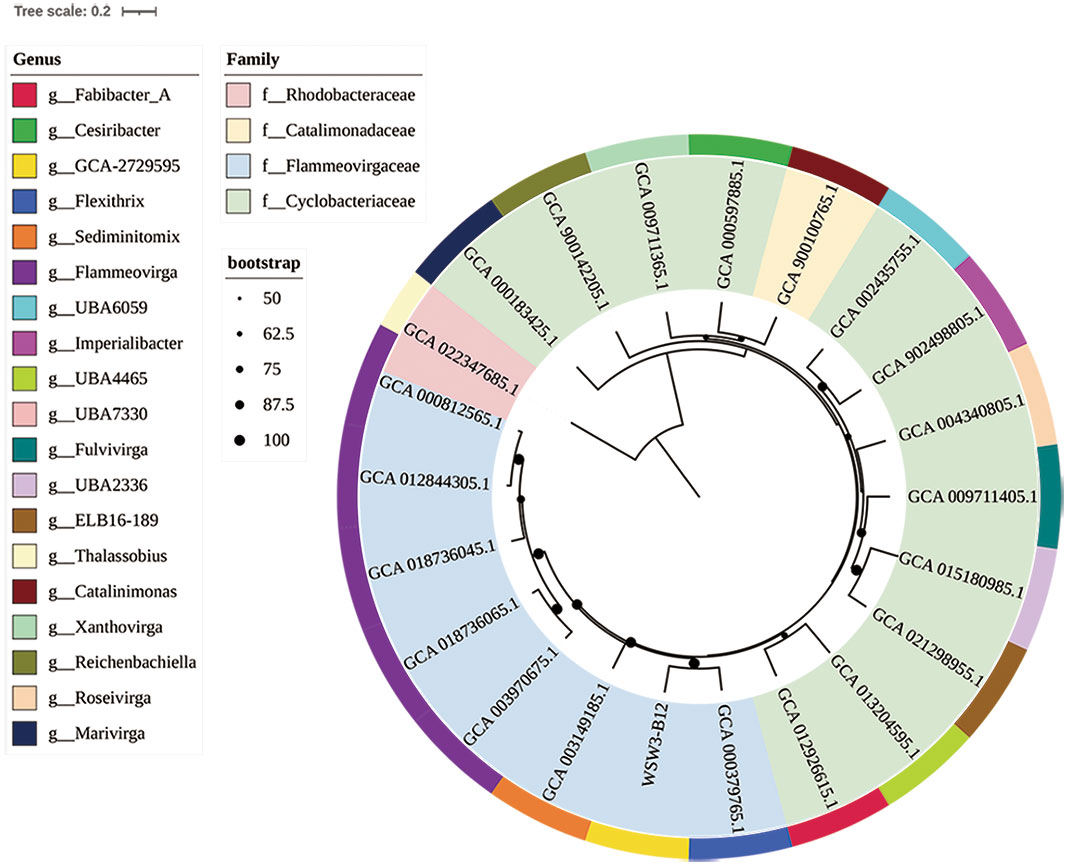
Figure 2 A whole-genome-based phylogenetic tree based on 400 marker genes using PhyloPhlAn version 3.0 shows the position of the strain WSW3-B12T among the members of the families Flammeovirgaceae, Catalimonadaceae, and Cyclobacteriaceae in the order Cytophagales. The strain Thalassobius sp. Cn5-15 (GCA_022347685) of family Rhodobacteraceae was used as an outgroup. Strains: WSW3-B12T, Flexithrix dorotheae DSM 6795T (GCA_000379765), Marinigracilibium pacificum KN852 (GCA_012926615), Flammeovirgaceae sp. bin.302 (GCA_013204595), Flammeovirgaceae sp. CoA8N_M18 (GCA_021298955), Flammeovirgaceae sp. H1_BAC1 (GCA_015180985), Fulvivirga kasyanovii JCM 16186 T (GCA_009711405), Roseivirga ehrenbergii DSM 102268 T (GCA_004340805), Imperialibacter sp. EC-SDR9 T (GCA_902498805), Flammeovirgaceae sp. UBA6059 (GCA_002435755), Catalinimonas alkaloidigena DSM2516T (GCA_900100765), Flammeovirgaceae sp. 311 (GCA_000597885), Xanthovirga aplysinae RKSG123T (GCA_009711365), Reichenbachiella agariperforans DSM 26134T (GCA_900142205), Marivirga tractuosa DSM 4126T (GCA_000183425), Thalassobius sp. Cn5-15 (GCA_022347685), Flammeovirga sp. OC4 (GCA_000812565), Flammeovirga aprica JL-4T (GCA_012844305), Flammeovirga yaeyamensis (GCA_018736045), Flammeovirga kamogawensis (GCA_018736065), F pectinis (GCA_003970675), Sediminitomix flava DSM 28229T GCA_003970675.
3.7 Genome functional analysis
3.7.1 Metabolic pathways analysis
First, a comparison of the distribution of genes in the KEGG module pathways for the strain WSW3-B12T and existing stains is presented in a heatmap (Figure 3). The figure shows that the central metabolic pathways that are typically involved in aerobic respiration, sugar metabolism, amino acid synthesis, etc. appeared to be similar among these strains. The strain WSW3-B12T contains genes that are involved in the biosynthesis of vitamins such as cobalamin, thiamin, and retinal. Furthermore, the KEGG pathways showed that the strain harbors genes for various enzymes, among which the genes for oligogalacturonide lyase (OGL) were only found in the genome of WSW3-B12T. Interestingly, OGL genes exists on plasmid 3 instead of the main chromosome. This makes strain WSW3-B12T unique compared to other Flammeovirgaceae genera that do not have the OGL genes. The OGL genes may give the strain WSW3-B12T evolutionary advantages compared to other microorganisms. The OGL genes catalyzed the eliminative removal of unsaturated terminal residues from oligosaccharides of D-galacturonate, which is a key constituent of pectin in plant biomass (Linhardt et al., 1987; Abbott and Boraston, 2008; Abbott et al., 2010). The OGL genes is present in a group of pectinolytic bacteria, such as Erwinia species, that are involved in the degradation of pectin, resulting in characteristic plant cell necrosis and tissue maceration (Collmer and Bateman, 1981).
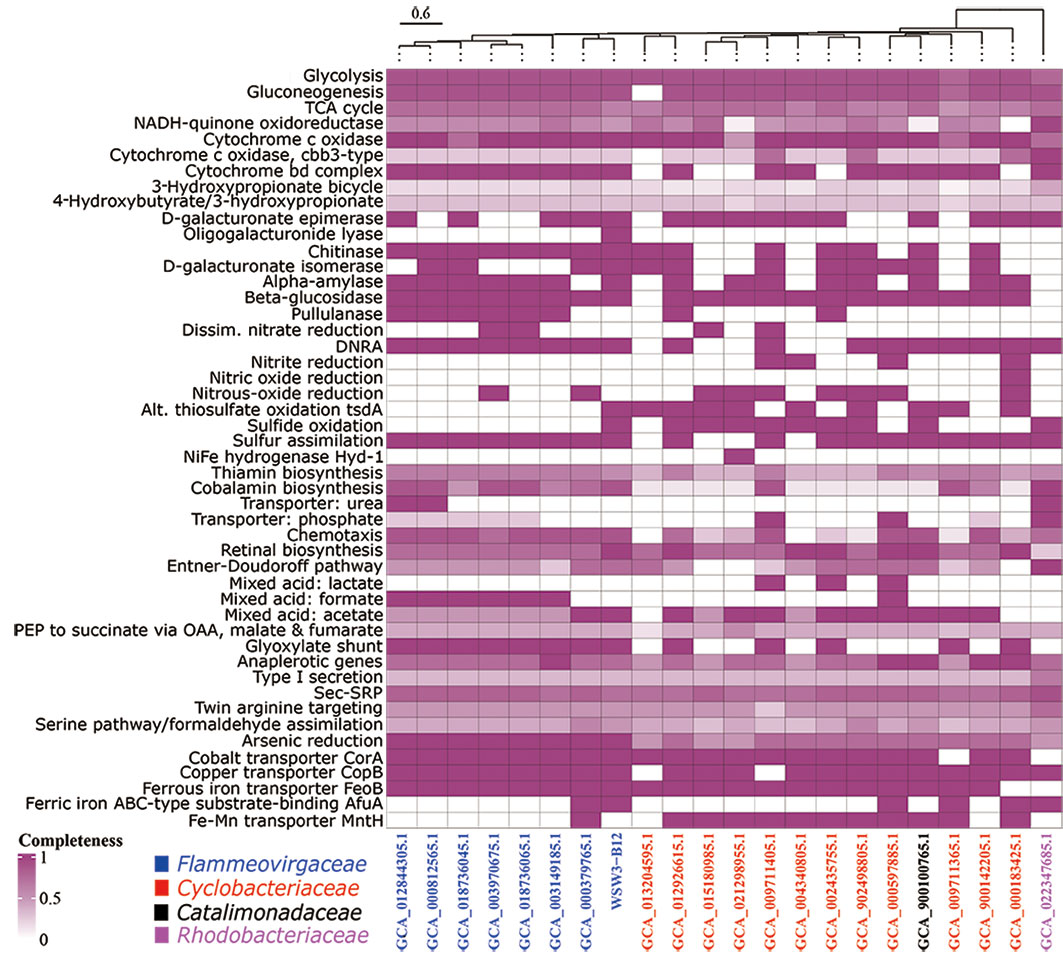
Figure 3 Heatmap of discriminated metabolic pathways within the genome of the strain WSW3-B12T and 21 representative genomes of the existing members of the Flammeovirgaceae, Catalimonadaceae, and Cyclobacteriaceae in the order Cytophagales as used in Figure 2. The cell indicates the completeness of each pathway referring to annotations using KEGG. The heatmap of metabolic pathways was created using the same strains as in Figure 2 for a comparative study.
Second, the COG analysis showed that the major genes were those involved in general function prediction, followed by signal transduction mechanisms, while the minor genes were those involved in cytoskeleton formation (Figure 4). The COG analysis further showed that genes for signal transduction were higher in the strain WSW3-B12T compared to existing strains. Microbes use multiple signal transduction systems to respond to environmental changes and mediate diverse physiological processes and intercellular communication, including antibiotic resistance, virulence, symbiotic bacteria-host interactions, and biofilm assemblage (Wang et al., 2021). In the RAST system, a total of 8525 protein-encoding genes were predicted and assigned to 275 subsystems. In the subsystem category, the highest number of genes were assigned to the metabolism of amino acids and derivatives (303) and carbohydrates (251), followed by cofactors, vitamins, prosthetic groups, and pigment metabolism (171). Although the distribution of genes between novel strains and the closest strains was similar. The numbers of genes involved in regulation/cell signaling and cell wall/capsule synthesis were higher in the strain WSW3-B12T than in the other strains (Table S2). Bacteria use these regulatory and cell signaling pathways for intercellular communication and diverse biological processes including biofilm formation (Wang et al., 2021). Furthermore, the higher number of cell wall and capsule synthesis pathways further helps the strain WSW3-B12T in unfavorable conditions as well as in interaction of the strain with algae where the strain is isolated from.
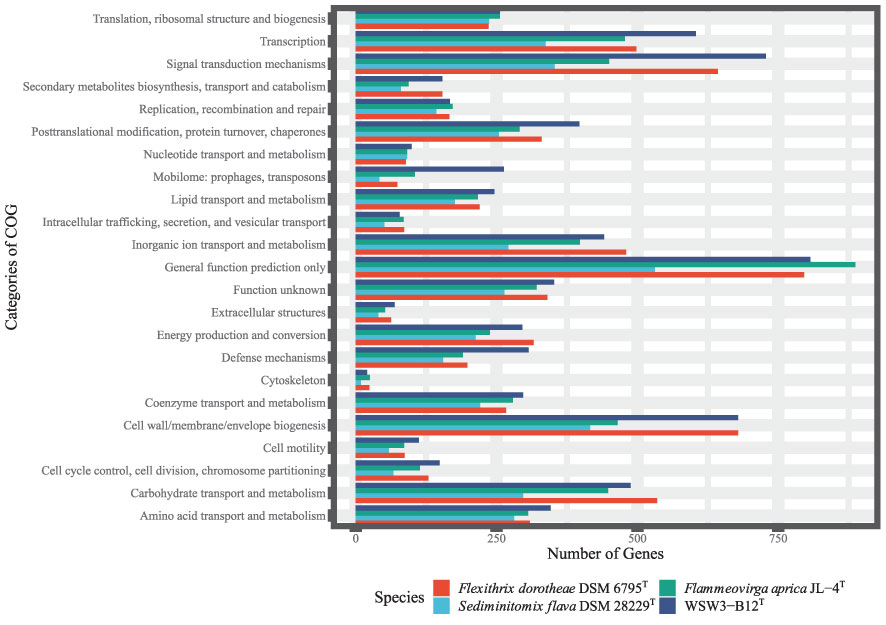
Figure 4 Clusters of Orthologous Group (COG), functional classification of proteins in the genome of the WSW3-B12T and three existing strains of Flammeovirgaceae.
The genes for polysaccharide degradation were identified using dbCAN meta servers, which detected a number of CAZyme gene clusters (CGCs) in the genome of the strain WSW3-B12T and the existing strains. The strain WSW3-B12T carried a total of 241 CAZymes. The distribution of CAZyme gene clusters (CGCs) between the novel and existing strains is summarized in (Table S3). The percentage of CAZyme genes out of the total genes and the ratio of the number of glycoside hydrolases (GHs) per Mbp genome were calculated. The strain WSW3-B12T carried 2.94% CAZym and 13.86 GHs per Mbp of genome (Table S4). GHs and PLs that are expected to participate in degradation of polysaccharide were searched from the dbCAN meta server and compared in terms of the in vitro activities of the strain WSW3-B12T (Table S5). The strain degraded cellulose, inulin, and starch. The strain carried four GH3 and five GH5 for cellulose degradation, one GH32 for inulin degradation, and eight GH13 for starch degradation. These GHs may participate in the degradation of the test polysaccharides. The strain did not carry any genes for the degradation of agar and alginate while it carried two genes for the degradation of κ-carrageenan.
3.7.2 Metabolism of heavy metals
The KEGG pathways showed that the strain WSW3-B12T and the existing strain of Flammeovirgaceae had complete pathways for arsenic reduction (Figure 3) and also carried transporter genes such as CorA, CopA, FeoB, and AfuA for cobalt, copper, ferrous, and ferric iron, respectively. The CorA proteins are generally associated with the transport of magnesium ions, but some members of the CorA family can also transport other ions such as cobalt (Co) and nickel (Ni) (Zhang et al., 2009). The strain WSW3-B12T carried CorA in both chromosome and plasmid 11. CopA is one of the most well-known microbial metal transporters. The monomer of CopA binds to two Cu2+ ions and subsequently transfers them to the periplasm using the Cu chaperone (CusF) and ATP energy (Li et al., 2022). For the transportation of ferrous and ferric iron, the strain carries the FeoB protein, a cytoplasmic membrane transporter protein, and AfuA, an ABC-type transporter protein. The ABC transporters help the microbes in membrane translocation of HMs ions (Hantke, 2003; Lau et al., 2016). Interestingly, FeoB genes exist on both chromosome and plasmid 6. Plasmid 6 even has two FeoB genes, compared to only one on the chromosome (Figure S5).
More HMs metabolism genes were discovered on the chromosome and in the plasmids as a result of the Prokka annotation. Many iron and copper metabolic genes were distributed in both the chromosome and the plasmids proportionally. Meanwhile, most of the arsenic and cobalt metabolic genes come from the plasmids. Thus, the plasmid has an interesting relationship with heavy metal metabolism. Details of these genes are listed in (Table S6).
3.7.3 Prediction of biosynthetic gene clusters and antibiotic resistance genes
The strain possessed six types of putative BGCs (terpene, type I polyketide synthases, type III polyketide synthases, betalactone, acyl amino acid, and lanthipeptide class I). Compared to the novel strain, only two BGCs in Flexithrix dorotheae DSM 6795T, three BGCs in Sediminitomix flava DSM 28229T, and five BGCs in Flammeovirga aprica JL-4T were detected. (Table S7). Among the BGCs, the polyketide synthases are a family of multi-domain enzymes that produce polyketides, a large class of antimicrobial secondary metabolites (Hochmuth and Piel, 2009), while lanthipeptide class I are ribosomally synthesized antimicrobial compounds that are also called lantibiotics. Lanthipeptide can be used as an alternative to antibiotics in the food industry and veterinary medicine, and against multi-drug resistant strains methicillin-resistant Staphylococcus aureus.(MRSA) and vancomycin-resistant enterococcus (VRE) (Barbosa et al., 2015).
Based on CARD database prediction, there are a total of four antibiotic resistance genes, one on the chromosome and three on the plasmids (plasmid 3, 5, and 9). The chromosome contains a glycopeptide antibiotic resistance gene cluster and the plasmids contain fluoroquinolone and tetracycline antibiotic resistance.
3.7.4 CRISPR-Cas system and prophage analysis
CRISPR-Cas system consists of CRISPR array (spacers and repeats) and Cas system (collection of Cas enzymes). First, the CRISPRCasFinder was used to detect the CRISPR array. CRISPRcasIdentifier was then used to identify CRISPR related enzymes in the genome. The sequences were then blasted using UniProt for confirmation (Table S8). The CRISPRCasFinder server detected seven CRISPR arrays candidates in the genome of the strain WSW3-B12T. However, only one CRISPR array, located in plasmid 20, that are long enough to be consider CRISPR array. The CRISPRcasIndetifier and Uniprot also only detects Cas enzymes in the plasmid 20 upstream of the CRISPR array. There were eight Cas enzymes, Cas1 to 7 and an unknown Cas enzyme (Figure S4). Thus, the CRISPR/Cas system could be classified as an incomplete Class I type I-B that is missing the Cas8 enzyme (Makarova et al., 2020). Based on this analysis we confirmed that the strain WSW3-B12T have the CRISPR-Cas system in the plasmid 20.
When we compared the genome of the strain WSW3-B12T with that of other reference strains, only the genome of Sediminitomix flava DSM 28229T contained two CRISPR arrays. The other reference strains did not have a CRISPR array. When we searched similar sequences from Uniprot, the highest sequence identity of Cas3, 5, 6, and 7 of the strain WSW3-B12T were found from the Cas genes of Sediminitomix flava DSM 28229T (Table S8). Sediminitomix flava DSM 28229T contains only Cas 3, 5, 6, and 7 on its chromosome. Thus, it also has an incomplete Class I type I-B CRISPR/Cas system. It is very likely that the Cas enzymes (3, 5, 6, and 7) of strains WSW3-B12T and Sediminitomix flava DSM 28229T came from the same lineage. Based on their isolation sites, this is also probable because strain WSW3-B12T was isolated from the West Sea, Korea and Sediminitomix flava DSM 28229T was isolated from Okinawa, Japan: both locations are connected by a sea current.
The PHAST search tool identified two prophage regions, one of which is incomplete and the other is questionable. The high number of CRISPRs candidates per strain described as the prokaryotic defense mechanism against external attacks and the presence of bacteriophages in their genomes suggest that the strain is also prone to phage infections. The CRISPR-based technologies are also useful for either sequence-specific killing of pathogenic bacteria or for the removal of accessory genes (antimicrobial resistance, virulence, etc.) (Palacios Araya et al., 2021).
3.7.5 Plasmid functional analysis
The function of the chromosome and each plasmid were analyzed using the KEGG database. Based on this analysis we classified the pathways into three categories based on their locations: found exclusively in the chromosome (Figure S5A); found both in the chromosome and plasmids (Figure S5B); and found exclusively in the plasmids (Figure S5C). The existence of plasmids in addition to the chromosome could help the strain WSW3-B12T to have more robust and highly expressed metabolic pathways. The addition of plasmids may increase the competitive edge of the strain WSW3-B12T in terms of nutrition, growth, and defense in the marine environments. For example, the chromosome of the strain WSW3-B12T lacked cysteine pathways but plasmids 3 and 11 carried genes for the cysteine pathway that may supplement cysteine metabolism (Figure S5C).
3.8 Description of Chondrinema gen. nov.
Chondrinema (Chon.dri.ne’ma. N.L. masc. n. Chondrus, a genus of red algae; Gr. neut. n. nema, a thread; N.L. neut. n. Chondrinema, a thread associated with the algal genus Chondrus).
This genus falls within the phylum Bacteroidota, class Cytophagia, and order Cytophagales. Cells are Gram-stain negative, oxidase and catalase-positive, and aerobic. The major fatty acids of the strain are iso-C15:0 and C16:1 ω5. The major respiratory quinone and polar lipid are menaquinone 7 (MK-7) and phosphatidylethanolamine.
3.9 Description of Chondrinema litorale sp. nov.
Chondrinema litorale (li. to. ra’le. L. neut. adj. litorale, of or belonging to the seashore)
Cells grow well on MA and produce light brown colonies with a diameter in a range from 325-420 µm. Cells of the strain are rod-shaped with a length 5-7 µm and width 0.46-0.54 µm.
Growth occurs at 15-30°C (optimum, 20-30°C), at pH 5.5-9.0 (optimum, pH 6.5-8.0), and with 1-7.0% NaCl (w/v) (optimum, 2-5%). The cell can degrade cellulose, inulin, and starch. Cells are positive for the hydrolysis of Tweens 20, 40, and 80. They are also positive for the activities of acid phosphatase, alkaline phosphatase, N-acetyl-β-glucosaminidase, cystine arylamidase, α-chymotrypsin, esterase (C4), esterase lipase (C8), α-galactosidase, β-galactosidase, α-glucosidase, β-glucosidase, leucine arylamidase, naphthol-AS-BI-phosphohydrolase, trypsin, and valine arylamidase. The cell can produce acid from L-arabinose, amidon (starch), arbutin, N-acetylglucosamine, D-cellobiose, gentibiose, D-galactose, glucose, D-lactose, methyl-αD-glucopyranoside, methyl-αD-mannopyranoside, D-maltose, D-melibiose, D-mannose, D-melezitose, D-raffinose, L-rhamnose, D-sucrose, D-trehalose, D-turanose, and D-xylose.
The type strain WSW3-B12T (= KCTC 82707T = GDMCC 1.3198T) was isolated from a red algae genus (Chondrus). The size of the genome is 10.1 Mbp and the G+C content is 34.1%. The genome contains one circular chromosome and 28 plasmids.
Repositories: The GenBank/EMBL/DDBJ accession number for the 16S rRNA gene sequence of strain WSW3-B12T is OP823709 and that for the whole genome sequence is GCA_026250525.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: GenBank, OP823709.1 and GCA_026250525.1.
Author contributions
NM and S-GK designed the study. NM performed the isolation and the morphological, physiological, and biochemical characterization. FA and Y-JL performed bioinformatic analyses with help from K-HK and HLH. NM, FA and S-GK wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The Korea Research Institute of Bioscience and Biotechnology (KRIBB) Research Initiative Program (KGM5232322) and a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. NRF-2021M3H9A1030164) supported this research.
Acknowledgments
The authors thank Prof. Aharon Oren from the Hebrew University of Jerusalem for his help on the nomenclature of the novel strain.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1186809/full#supplementary-material
References
Abbott D. W., Boraston A. B. (2008). Structural biology of pectin degradation by Enterobacteriaceae. Microbiol. Mol. Biol. Rev. 72, 301–316. doi: 10.1128/mmbr.00038-07
Abbott D. W., Gilbert H. J., Boraston A. B. (2010). The active site of oligogalacturonate lyase provides unique insights into cytoplasmic oligogalacturonate beta-elimination. J. Biol. Chem. 285, 39029–39038. doi: 10.1074/jbc.M110.153981
Alcock B. P., Huynh W., Chalil R., Smith K. W., Raphenya A. R., Wlodarski M. A., et al. (2022). CARD 2023: expanded curation, support for machine learning, and resistome prediction at the comprehensive antibiotic resistance database. Nucleic Acids Res. 6:51, D690–D699. doi: 10.1093/nar/gkac920
Amrina R. A., Furusawa G., Lau N.-S. (2021). Saccharobesus litoralis gen. nov., sp. nov., a novel alginate-degrading bacterium isolated from the surface of intertidal algal turf. Int. J. Syst. Evol. Microbiol. 71, 5087. doi: 10.1099/ijsem.0.005087
Arndt D., Grant J. R., Marcu A., Sajed T., Pon A., Liang Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Asharaf S., Chakraborty K., Chakraborty R. D. (2022). Seaweed-associated heterotrophic bacteria: are they future novel sources of antimicrobial agents against drug-resistant pathogens? Arch. Microbiol. 204, 232. doi: 10.1007/s00203-022-02835-8
Asnicar F., Thomas A. M., Beghini F., Mengoni C., Manara S., Manghi P., et al. (2020). Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat. Commun. 11, 2500. doi: 10.1038/s41467-020-16366-7
Auch A. F., Von Jan M., Klenk H. P., Göker M. (2010). Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand Genomic Sci. 2, 117–134. doi: 10.4056/sigs.531120
Barbosa J., Caetano T., Mendo S. (2015). Class I and class II lanthipeptides produced by Bacillus spp. J. Nat. Prod. 78, 2850–2866. doi: 10.1021/np500424y
Barco R. A., Garrity G. M., Scott J. J., Amend J. P., Nealson K. H., Emerson D. (2020). A genus definition for bacteria and archaea based on a standard genome relatedness index. mBio. 11, e02475–e02419. doi: 10.1128/mBio.02475-19
Beam J. P., George S., Record N. R., Countway P. D., Johnston D. T., Girguis P. R., et al. (2020). Mud, microbes, and macrofauna: seasonal dynamics of the iron biogeochemical cycle in an intertidal mudflat. Front. Mar. Sci. 7. doi: 10.3389/fmars.2020.562617
Blin K., Shaw S., Steinke K., Villebro R., Ziemert N., Lee S. Y., et al. (2019). antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 47, W81–W87. doi: 10.1093/nar/gkz310
Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bowman J. P. (2000). Description of cellulophaga algicola sp. nov., isolated from the surfaces of Antarctic algae, and reclassification of Cytophaga uliginosa (ZoBell and upham 1944) reichenbach 1989 as Cellulophaga uliginosa comb. nov. Int. J. Syst. Evol. Microbiol. 50, 1861–1868. doi: 10.1099/00207713-50-5-1861
Cazares A., Moore M. P., Hall J. P. J., Wright L. L., Grimes M., Emond-Rhéault J. G., et al. (2020). A megaplasmid family driving dissemination of multidrug resistance in Pseudomonas. Nat. Commun. 11, 1370. doi: 10.1038/s41467-020-15081-7
Chen Y., Jiang Y., Huang H., Mou L., Ru J., Zhao J., et al. (2018). Long-term and high-concentration heavy-metal contamination strongly influences the microbiome and functional genes in yellow river sediments. Sci. Total Environ. 637, 1400–1412. doi: 10.1016/j.scitotenv.2018.05.109
Cheng H., Wu Y. H., Huo Y. Y., Zhou P., Liu Q., Wang C. S., et al. (2017). Complete genome sequence of Altererythrobacter dongtanensis KCTC 22672T, isolated from a tidal flat. Mar. Genomics 34, 11–14. doi: 10.1016/j.margen.2017.02.003
Collmer A., Bateman D. F. (1981). Impaired induction and self-catabolite repression of extracellular pectate lyase in Erwinia chrysanthemi mutants deficient in oligogalacturonide lyase. Proc. Natl. Acad. Sci. U S A. 78, 3920–3924. doi: 10.1073/pnas.78.6.3920
Couvin D., Bernheim A., Toffano-Nioche C., Touchon M., Michalik J., Néron B., et al. (2018). CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for cas proteins. Nucleic Acids Res. 46, W246–W251. doi: 10.1093/nar/gky425
Dell’Anno F., Rastelli E., Tangherlini M., Corinaldesi C., Sansone C., Brunet C., et al. (2021). Highly contaminated marine sediments can host rare bacterial taxa potentially useful for bioremediation. Front. Microbiolo 12. doi: 10.3389/fmicb.2021.584850
Deshavath N. N., Mukherjee G., Goud V. V., Veeranki V. D., Sastri C. V. (2020). Pitfalls in the 3, 5-dinitrosalicylic acid (DNS) assay for the reducing sugars: interference of furfural and 5-hydroxymethylfurfural. Int. J. Biol. Macromol. 156, 180–185. doi: 10.1016/j.ijbiomac.2020.04.045
Dong Q., Ruan L., Shi H. (2017). Genome sequence of a high agarase-producing strain Flammeovirga sp. SJP92. Stand Genom Sci. 12, 1–6. doi: 10.1186/s40793-017-0221-y
Duarte C. M., Dennison W. C., Orth R. J., Carruthers T. J. (2008). The charisma of coastal ecosystems: addressing the imbalance. Estuaries coasts. 31, 233–238. doi: 10.1007/s12237-008-9038-7
Felsenstein J. (1981). Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17, 368–376. doi: 10.1007/bf01734359
Feng Y., Wang Y., Zhu B., Gao G. F., Guo Y., Hu Y. (2021a). Metagenome-assembled genomes and gene catalog from the chicken gut microbiome aid in deciphering antibiotic resistomes. Commun. Biol. 4, 1305. doi: 10.1038/s42003-021-02827-2
Feng Z., Zhang Z., Liu Y., Gu J., Cheng Y., Hu W., et al. (2021b). The second chromosome promotes the adaptation of the genus Flammeovirga to complex environments. Microbiol. Spectr. 9, e0098021. doi: 10.1128/Spectrum.00980-21
Fernández-Gómez B., Richter M., Schüler M., Pinhassi J., Acinas S. G., González J. M., et al. (2013). Ecology of marine Bacteroidetes: a comparative genomics approach. ISME J. 7, 1026–1037. doi: 10.1038/ismej.2012.169
Galperin M. Y., Wolf Y. I., Makarova K. S., Vera Alvarez R., Landsman D., Koonin E. V. (2021). COG database update: focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 49, D274–D281. doi: 10.1093/nar/gkaa1018
Gao B., Jin M., Li L., Qu W., Zeng R. (2017). Genome sequencing reveals the complex polysaccharide-degrading ability of novel deep-sea bacterium Flammeovirga pacifica WPAGA1. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.00600
Goldberg S. R., Correa H., Haltli B. A., Kerr R. G. (2020). Fulvivirga aurantia sp. nov. and Xanthovirga aplysinae gen. nov., sp. nov., marine bacteria isolated from the sponge Aplysina fistularis, and emended description of the genus Fulvivirga. Int. J. Syst. Evol. Microbiol. 70, 2766–2781. doi: 10.1099/ijsem.0.004108
Graham E. D., Heidelberg J. F., Tully B. J. (2018). Potential for primary productivity in a globally-distributed bacterial phototroph. ISME J. 12, 1861–1866. doi: 10.1038/s41396-018-0091-3
Hantke K. (2003). Is the bacterial ferrous iron transporter FeoB a living fossil? Trends Microbiol. 11, 192–195. doi: 10.1016/s0966-842x(03)00100-8
Hehemann J. H., Correc G., Thomas F., Bernard T., Barbeyron T., Jam M., et al. (2012). Biochemical and structural characterization of the complex agarolytic enzyme system from the marine bacterium Zobellia galactanivorans. J. Biol. Chem. 287, 30571–30584. doi: 10.1074/jbc.M112.377184
Hochmuth T., Piel J. (2009). Polyketide synthases of bacterial symbionts in sponges–evolution-based applications in natural products research. Phytochemistry. 70, 1841–1849. doi: 10.1016/j.phytochem.2009.04.010
Hosoya S., Yokota A. (2007). Reclassification of Flexibacter aggregans (Lewin 1969) leadbetter 1974 as a later heterotypic synonym of Flexithrix dorotheae lewin 1970. Int. J. Syst. Evol. Microbiol. 57, 1086–1088. doi: 10.1099/ijs.0.64798-0
Jeon D., Jiang L., Peng Y., Seo J., Li Z., Park S. H., et al. (2022). Sphingomonas cannabina sp. nov., isolated from cannabis sativa l. 'Cheungsam'. Int. J. Syst. Evol. Microbiol. 72, 005566. doi: 10.1099/ijsem.0.005566
Jeong Y. S., Kang W., Sung H., Lee J. Y., Yun J. H., Shin N. R., et al. (2020). Flammeovirga pectinis sp. nov., isolated from the gut of the Korean scallop, Patinopecten yessoensis. Int. J. Syst. Evol. Microbiol. 70, 499–504. doi: 10.1099/ijsem.0.003783
Jorgensen J. H., Turnidge J. D. (2015). “Susceptibility test methods: dilution and disk diffusion methods,” in Manual of clinical microbiology. Eds. H.Jorgensen J., Carroll K. C., Funke G., Pfaller M. A., Landry M. L., Richter S. S., Warnock D. W. (New York: NY: Wiley), 1253–1273.
Juhmani A. S., Vezzi A., Wahsha M., Buosi A., Pascale F., Schiavon R., et al. (2020). Diversity and dynamics of seaweed associated microbial communities inhabiting the lagoon of venice. Microorganisms. 8, 1657. doi: 10.3390/microorganisms8111657
Karthick P., Mohanraju R. (2018). Antimicrobial potential of epiphytic bacteria associated with seaweeds of little andaman, India. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.00611
Khan S. T., Nakagawa Y., Harayama S. (2007). Sediminitomix flava gen. nov., sp. nov., of the phylum Bacteroidetes, isolated from marine sediment. Int. J. Syst. Evol. Microbiol. 57, 1689–1693. doi: 10.1099/ijs.0.64854-0
Kim J., Cha I. T., Lee K. E., Son Y. K., Yu J., Seol D. (2022). Characteristics and adaptability of Flavobacterium panici BSSL-CR3 in tidal flat revealed by comparative genomic and enzymatic analysis. Arch. Microbiol. 205, 22. doi: 10.1007/s00203-022-03359-x
Kizhakkekalam V. K., Chakraborty K. (2021). Seaweed-associated heterotrophic bacteria: new paradigm of prospective anti-infective and anticancer agents. Arch. Microbiol. 203, 1241–1250. doi: 10.1007/s00203-020-02106-4
Komagata K., Suzuki K. (1988). Lipid and cell-wall analysis in bacterial systematics. Methods Microbiol. 19, 161–207. doi: 10.1016/S0580-9517(08)70410-0
Konaté M. M., Plata G., Park J., Usmanova D. R., Wang H., Vitkup D. (2019). Molecular function limits divergent protein evolution on planetary timescales. Elife. 8, e39705. doi: 10.7554/eLife.39705
Koren S., Walenz B. P., Berlin K., Miller J. R., Bergman N. H., Phillippy A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kristyanto S., Kim K. R., Jung J., Kim H. M., Kim K., Jeon C. O. (2022). Tenacibaculum aquimarinum sp. nov., isolated from a marine alga and seawater. Int. J. Syst. Evol. Microbiol. 72, 005477. doi: 10.1099/ijsem.0.005477
Kumar S., Stecher G., Li M., Knyaz C., Tamura K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lau C. K., Krewulak K. D., Vogel H. J. (2016). Bacterial ferrous iron transport: the feo system. FEMS Microbiol. Rev. 40, 273–298. doi: 10.1093/femsre/fuv049
Leadbetter E. (1974). Order II. cytophagales nomen novum. bergey's manual of determinative bacteriology. 8th ed Vol. 99 (Baltimore: The Williams & Wilkins Co.).
Lee H., Heo Y. M., Kwon S. L., Yoo Y., Kim D., Lee J., et al. (2021). Environmental drivers affecting the bacterial community of intertidal sediments in the yellow Sea. Sci. Total Environ. 755, 142726. doi: 10.1016/j.scitotenv.2020.142726
Lee H., Lee D. W., Kwon S. L., Heo Y. M., Jang S., Kwon B. O., et al. (2019). Importance of functional diversity in assessing the recovery of the microbial community after the hebei spirit oil spill in Korea. Environ. Int. 128, 89–94. doi: 10.1016/j.envint.2019.04.039
Lee S. J., Park M., Ogandaga M C. A., Park S. K., Kim H., Kim Y., et al. (2013). A study on the growth and disease of Chondrus ocellatus in Korea. J. fish Pathol. 26, 265–274. doi: 10.7847/jfp.2013.26.3.265
Li W., Wang L., Li X., Zheng X., Cohen M. F., Liu Y. X. (2022). Sequence-based functional metagenomics reveals novel natural diversity of functioning CopA in environmental microbiomes. Genom Proteom Bioinf. S1672-0229, 00101–00112. doi: 10.1016/j.gpb.2022.08.006
Lin Y., Yuan J., Kolmogorov M., Shen M. W., Chaisson M., Pevzner P. A. (2016). Assembly of long error-prone reads using de bruijn graphs. Proc. Natl. Acad. Sci. U S A. 113, E8396–e8405. doi: 10.1073/pnas.1604560113
Linhardt R., Galliher P., Cooney C. (1987). Polysaccharide lyases. Appl. Biochem. Biotechnol. 12, 135–176.
Lobban C. S., Harrison P. J. (1994). Seaweed ecology and physiology (New York: Cambridge University Press). doi: 10.1007/BF02798420
Ludwig W., Euzéby J., Whitman W. B. (2010). “Road map of the phyla bacteroidetes, spirochaetes, tenericutes (Mollicutes), acidobacteria, fibrobacteres, fusobacteria, dictyoglomi, gemmatimonadetes, lentisphaerae, verrucomicrobia, chlamydiae, and planctomycetes,” in Bergey’s manual® of systematic bacteriology (New York: Springer), 1–19.
Lv X., Ma B., Yu J., Chang S. X., Xu J., Li Y., et al. (2016). Bacterial community structure and function shift along a successional series of tidal flats in the yellow river delta. Sci. Rep. 6, 36550. doi: 10.1038/srep36550
Makarova K. S., Wolf Y. I., Iranzo J., Shmakov S. A., Alkhnbashi O. S., Brouns S. J. J., et al. (2020). Evolutionary classification of CRISPR–cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol. 18, 67–83. doi: 10.1038/s41579-019-0299-x
Mann A. J., Hahnke R. L., Huang S., Werner J., Xing P., Barbeyron T., et al. (2013). The genome of the alga-associated marine flavobacterium Formosa agariphila KMM 3901T reveals a broad potential for degradation of algal polysaccharides. Appl. Environ. Microbiol. 79, 6813–6822. doi: 10.1128/aem.01937-13
Mckee L. S., La Rosa S. L., Westereng B., Eijsink V. G., Pope P. B., Larsbrink J. (2021). Polysaccharide degradation by the Bacteroidetes: mechanisms and nomenclature. Environ. Microbiol. Rep. 13, 559–581. doi: 10.1111/1758-2229.12980
Meier-Kolthoff J. P., Auch A. F., Klenk H. P., Göker M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinf. 14, 60. doi: 10.1186/1471-2105-14-60
Menaa F., Wijesinghe P., Thiripuranathar G., Uzair B., Iqbal H., Khan B. A., et al. (2020). Ecological and industrial implications of dynamic seaweed-associated microbiota interactions. Mar. Drugs 18, 641. doi: 10.3390/md18120641
Muhammad N., Le Han H., Lee Y. J., Ko J., Nguyen T. T. H., Kim S. G. (2022a). Flavobacterium litorale sp. nov., isolated from red alga. Int. J. Syst. Evol. Microbiol. 72, 005458. doi: 10.1099/ijsem.0.005458
Muhammad N., Nguyen T. T. H., Lee Y. J., Ko J., Avila F., Kim S. G. (2022b). Vibrio ostreae sp. nov., a novel gut bacterium isolated from a yellow sea oyster. Int. J. Syst. Evol. Microbiol. 72, 005586. doi: 10.1099/ijsem.0.005586
Padilha V. A., Alkhnbashi O. S., Shah S. A., De Carvalho A., Backofen R. (2020). CRISPRcasIdentifier: machine learning for accurate identification and classification of CRISPR-cas systems. Gigascience. 9, giaa062. doi: 10.1093/gigascience/giaa062
Palacios Araya D., Palmer K. L., Duerkop B. A. (2021). CRISPR-based antimicrobials to obstruct antibiotic-resistant and pathogenic bacteria. PloS Pathog. 17, e1009672. doi: 10.1371/journal.ppat.1009672
Park M. J., Kim Y. J., Park M., Yu J., Namirimu T., Roh Y. R., et al. (2022). Establishment of genome based criteria for cclassification of the family Desulfovibrionaceae and proposal of two novel genera, Alkalidesulfovibrio gen. nov. and Salidesulfovibrio gen. nov. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.738205
Parks D. H., Imelfort M., Skennerton C. T., Hugenholtz P., Tyson G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Paster B. J., Dewhirst F. E., Olsen I., Fraser G. J. (1994). Phylogeny of Bacteroides, prevotella, and Porphyromonas spp. and related bacteria. J. Bacteriol. 176, 725–732. doi: 10.1128/jb.176.3.725-732.1994
Pheng S., Han H. L., Park D. S., Chung C. H., Kim S. G. (2020). Lactococcus kimchii sp. nov., a new lactic acid bacterium isolated from kimchi. Int. J. Syst. Evol. Microbiol. 70, 505–510. doi: 10.1099/ijsem.0.003782
Phillips I. (1993). Cowan and steel's manual for the identification of medical bacteria. J. Clin. Pathol. 46, 975. doi: 10.1017/CBO9780511527104
Qin Q. L., Xie B. B., Zhang X. Y., Chen X. L., Zhou B. C., Zhou J., et al. (2014). A proposed genus boundary for the prokaryotes based on genomic insights. J. Bacteriol. 196, 2210–2215. doi: 10.1128/jb.01688-14
Reichenbach H. (1992). “The order cytophagales,” in The prokaryotes: a handbook on the biology of bacteria: ecophysiology, isolation, identification, applications. Eds. Balows A., Trüper H. G., Dworkin M., Schleifer K. H., Harder W. (New York: Springer), 3631–3675.
Saitou N., Nei M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Sasser M. (1990). “Identification of bacteria by gas chromatography of cellular fatty acids,” in MIDI technical note 101 (Newark: DE: MIDI Inc).
Saw J. H. W., Yuryev A., Kanbe M., Hou S., Young A. G., Aizawa S., et al. (2012). Complete genome sequencing and analysis of Saprospira grandis str. lewin, a predatory marine bacterium. Stand Genom Sci. 6, 84–93. doi: 10.4056/sigs.2445005
Schmartz G. P., Hartung A., Hirsch P., Kern F., Fehlmann T., Müller R., et al. (2022). PLSDB: advancing a comprehensive database of bacterial plasmids. Nucleic Acids Res. 50, D273–D278. doi: 10.1093/nar/gkab1111
Seemann T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics. 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Singh R. P., Reddy C. R. (2015). Seaweed-microbial interactions: key functions of seaweed-associated bacteria. FEMS Microbiol. Ecol. 88, 213–230. doi: 10.1111/1574-6941.12297
Srisukchayakul P., Suwanachart C., Sangnoi Y., Kanjana-Opas A., Hosoya S., Yokota A., et al. (2007). Rapidithrix thailandica gen. nov., sp. nov., a marine gliding bacterium isolated from samples collected from the andaman sea, along the southern coastline of Thailand. Int. J. Syst. Evol. Microbiol. 57, 2275–2279. doi: 10.1099/ijs.0.65087-0
The UniProt Consortium (2023). UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531. doi: 10.1093/nar/gkac1052
Ueno A., Tamazawa S., Tamamura S., Murakami T., Kiyama T., Inomata H., et al. (2021). Desulfovibrio subterraneus sp. nov., a mesophilic sulfate-reducing deltaproteobacterium isolated from a deep siliceous mudstone formation. Int. J. Syst. Evol. Microbiol. 71, 004683. doi: 10.1099/ijsem.0.004683
Wang H., Li J., Zheng T., Hill R. T., Hu X. (2013). Imperialibacter roseus gen. nov., sp. nov., a novel bacterium of the family Flammeovirgaceae isolated from permian groundwater. Int. J. Syst. Evol. Microbiol. 63, 4136–4140. doi: 10.1099/ijs.0.052662-0
Wang R., Zhang W., Ding W., Liang Z., Long L., Wong W. C., et al. (2021). Profiling signal transduction in global marine biofilms. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.768926
Wilms R., Sass H., Köpke B., Köster J., Cypionka H., Engelen B. (2006). Specific bacterial, archaeal, and eukaryotic communities in tidal-flat sediments along a vertical profile of several meters. Appl. Environ. Microbiol. 72, 2756–2764. doi: 10.1128/aem.72.4.2756-2764.2006
Yoon J., Adachi K., Park S., Kasai H., Yokota A. (2011). Aureibacter tunicatorum gen. nov., sp. nov., a marine bacterium isolated from a coral reef sea squirt, and description of Flammeovirgaceae fam. nov. Int. J. Syst. Evol. Microbiol. 61, 2342–2347. doi: 10.1099/ijs.0.027573-0
Yoon S. H., Ha S. M., Kwon S., Lim J., Kim Y., Seo H., et al. (2017a). Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 67, 1613–1617. doi: 10.1099/ijsem.0.001755
Yoon S. H., Ha S. M., Lim J., Kwon S., Chun J. (2017b). A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek. 110, 1281–1286. doi: 10.1007/s10482-017-0844-4
Yoon J., Oku N., Park S., Katsuta A., Kasai H. (2012). Tunicatimonas pelagia gen. nov., sp. nov., a novel representative of the family Flammeovirgaceae isolated from a sea anemone by the differential growth screening method. Antonie Van Leeuwenhoek. 101, 133–140. doi: 10.1007/s10482-011-9626-6
Zhang Y., Rodionov D. A., Gelfand M. S., Gladyshev V. N. (2009). Comparative genomic analyses of nickel, cobalt and vitamin B12 utilization. BMC Genomics 10, 78. doi: 10.1186/1471-2164-10-78
Zhang H., Yohe T., Huang L., Entwistle S., Wu P., Yang Z., et al. (2018). dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46, W95–W101. doi: 10.1093/nar/gky418
Zhong Z., Toukdarian A., Helinski D., Knauf. V., Sykes S., Wilkinson J. E., et al. (2001). Sequence analysis of a 101-kilobase plasmid required for agar degradation by a Microscilla isolate. Appl. Environ. Microbiol. 67, 5771–5779. doi: 10.1186/1471-2164-10-78
Keywords: red algae, microbial diversity, polyphasic taxonomy, comparative genomics, carbohydrate-active enzymes
Citation: Muhammad N, Avila F, Lee Y-J, Han HL, Kim K-H and Kim S-G (2023) Chondrinema litorale gen. nov., sp. nov., of the phylum Bacteroidota, carrying multiple megaplasmids isolated from a tidal flat in the West Sea, Korea. Front. Mar. Sci. 10:1186809. doi: 10.3389/fmars.2023.1186809
Received: 15 March 2023; Accepted: 09 May 2023;
Published: 23 May 2023.
Edited by:
Xue-Wei Xu, Ministry of Natural Resources, ChinaReviewed by:
Heng-Lin Cui, Jiangsu University, ChinaGuohong Liu, Fujian Academy of Agricultural Sciences, China
Copyright © 2023 Muhammad, Avila, Lee, Han, Kim and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Song-Gun Kim, c2draW1Aa3JpYmIucmUua3I=