 Umberto Rosani
Umberto Rosani Morgan Gaia
Morgan Gaia Tom O. Delmont
Tom O. Delmont Mart Krupovic
Mart Krupovic- 1Department of Biology, University of Padova, Padova, Italy
- 2Génomique Métabolique, Genoscope, Institut François Jacob, CEA, CNRS, Univ Evry, Université Paris-Saclay, Evry, France
- 3Research Federation for the Study of Global Ocean Systems Ecology and Evolution, FR2022/Tara GOsee, Paris, France
- 4Institut Pasteur, Université Paris Cité, CNRS UMR6047, Archaeal Virology Unit, Paris, France
The family of Malacoherpesviridae is currently represented by only two viruses infecting molluscs, Ostreid herpesvirus 1 (OsHV-1) and Haliotid herpesvirus 1 (HaHV-1), both causing detrimental infections in aquaculture species. Malacoherpesvirus-like sequences were also detected through genome sequencing projects in amphioxus (Branchiostoma species) and annelid worm (Capitella teleta), suggesting the existence of a hidden diversity of malacoherpesviruses in aquatic animals. Here, to extend the knowledge on malacoherpesvirus diversity, we searched for the presence of malacoherpesvirus relatives in genomic, transcriptomic and metagenomic datasets, including from the Tara Oceans expedition, and report 4 novel malacoherpesvirus-like genomes (MalacoHV1-4). Genomic analysis suggested gastropods and bivalves as the most probable hosts for these new malacoherpesviruses. Phylogenetic analysis based on the family B DNA polymerase placed the novel MalacoHV1 and MalacoHV3 as sister lineages of OsHV-1 and HaHV-1, respectively, whereas MalacoHV2 and MalacoHV4 showed higher divergence. The viral genome found associated with amphioxus together with MalacoHV4 formed a sister clade to the mollusc and annelid malacoherpesviruses, suggesting an early divergence of the two virus assemblages. In conclusion, although relatively rare in the available sequence databases, the previously undescribed malacoherpesviruses, MalacoHV1-4, circulate in aquatic ecosystems and should be considered as possible emerging viruses under changing environmental conditions.
1 Introduction
The taxonomic reorganization of the virus world has resulted in formal unification of all double-stranded (ds) DNA viruses with the HK97-fold major capsid proteins (MCPs), namely, prokaryotic viruses of the class Caudoviricetes (tailed bacteriophages and related archaeal viruses) (Dion et al., 2020; Liu et al., 2021) and eukaryotic viruses of the order Herpesvirales (class Herviviricetes), into the realm Duplodnaviria (Koonin et al., 2020). The Herpesvirales comprises three families of herpes-like viruses, Alloherpesviridae, Orthoherpesviridae (formerly, Herpesviridae) and Malacoherpesviridae. Members of the former two families exclusively infect vertebrates, specifically fish, amphibians and mammals, whereas viruses of the Malacoherpesviridae family are primarily associated with invertebrate hosts. Due to their medical importance, members of the Orthoherpesviridae have been extensively sampled and the family currently comprises 118 species. By contrast, Alloherpesviridae and Malacoherpesviridae include only 13 and 2 species, respectively (Walker et al., 2022), limiting our understanding on the diversity, evolution and impact of non-mammalian herpesviruses. For instance, bipartite network analysis of dsDNA viruses based on shared genes placed the family Malacoherpesviridae into a module containing T4-like tailed phages (Iranzo et al., 2016). However, as noted by the authors, this clustering appeared to be artefactual due to the small number of recognizable core genes shared among members of the Herpesvirales and the lack of diversity among known malacoherpesviruses (Iranzo et al., 2016).
Family Malacoherpesviridae currently includes two viruses, Ostreid herpesvirus 1 (OsHV-1) and Haliotid herpesvirus 1 (HaHV-1), infecting bivalves and gastropods, respectively (Davison et al., 2009), both with a major devastating impact on aquaculture (Stentiford et al., 2017). Herpes-like infections have been first reported in bivalves, namely, in Crassostrea virginica in 1972 (Farley et al., 1972), with herpesvirus-like particles being subsequently observed in larvae of the Pacific oyster Crassostrea gigas and associated with significant mortalities (Nicolas et al., 1992). DNA sequencing of the virus isolated from C. gigas larvae in 1995 led to the identification of Ostreid herpesvirus 1 (NCBI ID: GCF_000846065.1), and to its taxonomic classification into a novel viral lineage (Davison et al., 2005; Davison et al., 2009). More recently, an emerging OsHV-1 genotype, called microvar (OsHV-1 μVar), was associated with heavy mortalities in France (Segarra et al., 2010) and worldwide (Jenkins et al., 2013; Bai et al., 2016; Abbadi et al., 2018; Whittington et al., 2018). Acute Viral Necrosis Virus (AVNV) and OsHV-1-SB are two other variants of OsHV-1, respectively detected in the Chinese scallop Chlamys farreri (Wang et al., 2002) and Chinese blood clam Scapharca Anadara broughtonii, where it causes considerable pathogenesis (Bai et al., 2016). The reports of OsHV-1 associated with other marine bivalve species, without disease signs, suggest that these species can act as viral reservoirs, complicating malacoherpesvirus eradication efforts (Prado-Alvarez et al., 2021). The second known representative of the Malacoherpesviridae, HaHV-1, was first associated with high mortality rates in cultured abalone Haliotis diversicolor supertexta in Taiwan (HaHV-1-Taiwan) (Chang et al., 2005). Subsequently, HaHV-1 outbreaks impacting Haliotis rubra and Haliotis laevigata have been reported in Australia, where a similar viral strain, dubbed Victoria, has been sequenced (Corbeil, 2020). The emergence of HaHV-1 infections was back-dated to 1999 in Southern China, by providing electron microscopy detection evidence of the virus in infected H. diversicolor supertexta specimens (Wang et al., 2004).
Despite limited sequence similarity, OsHV-1 and HaHV-1 display a conserved genomic architecture similar to that of other known members of the order Herpesvirales. In particular, OsHV-1 genome contains two unique regions (UL/US) flanked by two pairs of inverted repeats (TRL/IRL and TRS/IRS), separated by a third unique region, called X region. Similar genomic architecture was found in AVNV (Ren et al., 2013) and OsHV-1-SB (Xia et al., 2015). Notably, our results showed that the X region in OsHV-1-SB (strain CN2017) is considerably longer than in OsHV-1 (3.8 versus 1.5 kb, respectively) (Bai et al., 2021). Two other studies, focusing on OsHV-1-μVar genome, suggested the presence of two copies of the X region (Burioli et al., 2017; Abbadi et al., 2018), although it cannot be excluded that this unusual feature results from misassembly of the genome due to the presence of OsHV-1 isomers in the same sample, as reported previously (Davison et al., 2005). Despite these variations, the overall genomic structure of OsHV-1 resembles that of human herpesvirus 1 (HSV-1, subfamily Alphaherpesvirinae) and human herpesvirus 5 (HHV-5, subfamily Betaherpesvirinae) (Lefkowitz et al., 2018). The genome structure of HaHV-1-CN2003, structured as TRL–UL–IRL–IRS–US–TRS, also resembles that of HSV-1. Similar to certain vertebrate orthoherpesviruses (Osterrieder et al., 2014), malacoherpesviruses apparently are capable of occasional integration into the genome of their hosts. Large contiguous fragments of malacoherpesvirus-like genomes were obtained during chromosomal DNA sequencing projects of amphioxi Branchiostoma floridae and Branchiostoma belcheri and the annelid Capitella teleta (Savin et al., 2010; Rosani and Venier, 2017; Mushegian et al., 2018). In marine environments, the presence of herpesvirus-like sequences in protostomes has been claimed in stony corals (Vega Thurber et al., 2008; Correa et al., 2016), Hydra sp. (Grasis et al., 2014) and in the crab Paralithodes platypus (Ryazanova et al., 2015). However, most of these putative viral fragments have doubtful origin, except perhaps for the one in P. platypus (Mushegian et al., 2018), and have never been substantiated by genome sequencing or experimental evidence. Thus, the existence of other invertebrate herpesviruses and the phylogenetic relationship between malacoherpesviruses and other herpesviruses remain unclear. Importantly, high infectivity, rapid spread and persistence in the environment of malacoherpesviruses raised concerns in the aquaculture sector (Pernet et al., 2016; Whittington et al., 2018), calling for a more careful characterization of aquaculture-related viruses. Considering also the current global environmental challenges, the mollusc aquaculture industry could play a pivotal role due to its low carbon footprint (FAO, 2020). Hence, it will be extremely important to trace the presence of other invertebrate herpesviruses and consider their eventual impact on aquaculture.
In the present study, we attempted to expand our knowledge on the genetic diversity of invertebrate herpesviruses by mining host-associated data and global metagenomic datasets to search for malacoherpesvirus-like sequences.
2 Materials and methods
Due to differences in data type and volume of investigated databases, we adopted different bioinformatic approaches to scan them in search for malacoherpesvirus-like sequences, as outlined in Figure 1 and detailed in the following sections.
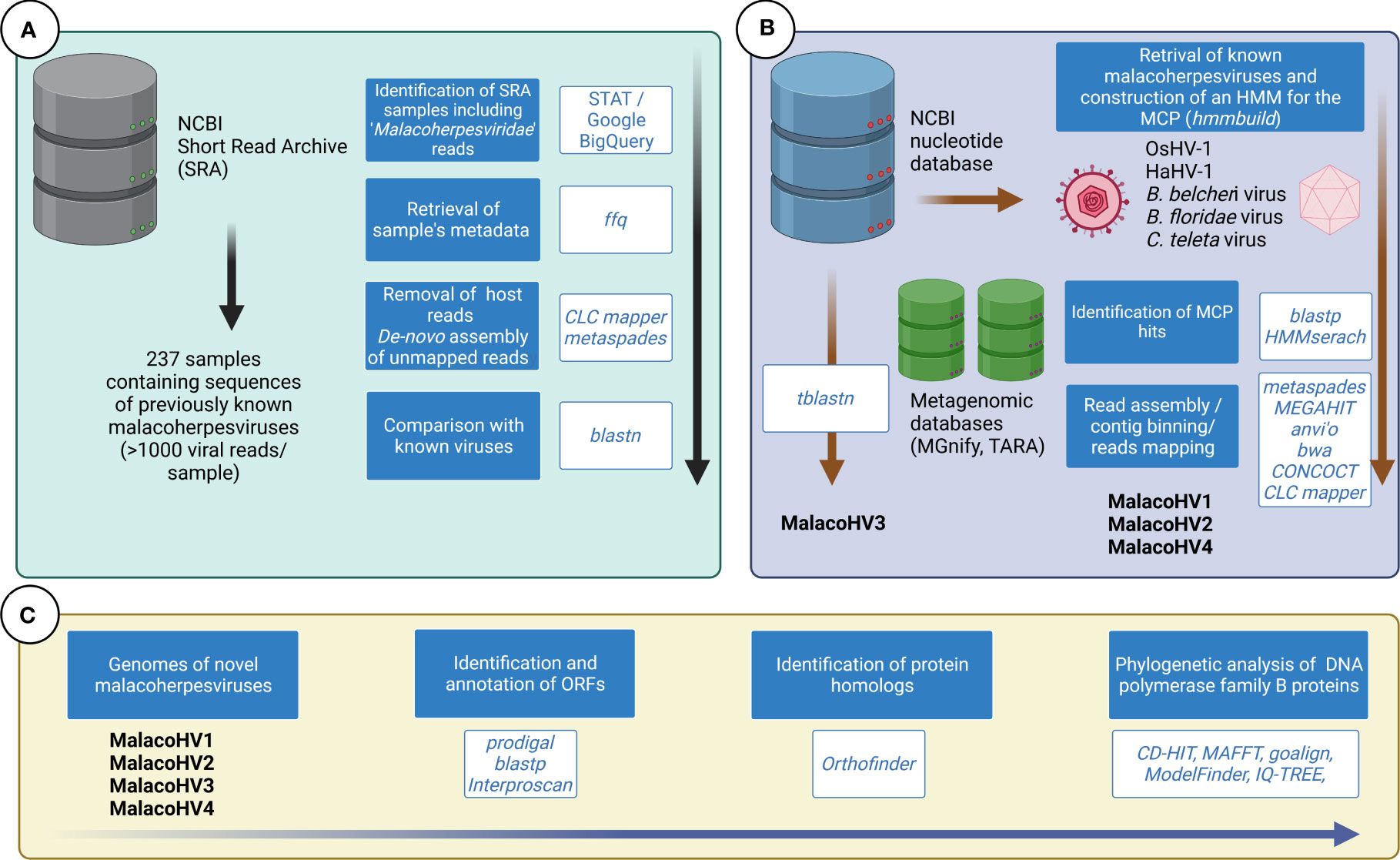
Figure 1 Overview of the bioinformatic approaches used to identify novel malacoherpes-like viruses. (A) The taxonomic classification of the reads from the NCBI SRA database has been investigated to identify samples containing malacoherpesvirus reads. Analysis of selected SRA samples did not reveal novel malacoherpes-like viruses, only the previously characterized ones. (B) The NCBI nucleotide database was scanned for the presence of malacoherpesviruses using tblastn with OsHV-1 and HaHV-1 proteomes used as queries. The major capsid protein (MCP) of known malacoherpesviruses have been used to build an HMM, which was used to scan metagenomic databases. (C) The four novel identified malacoherpesvirus (MalacoHV1-4) genomes were annotated and comprehensively analyzed. DNA polymerase family B proteins were used as markers for phylogenetic analysis.
2.1 Search for Malacoherpesviridae hits in the NCBI SRA database
Taking advantage of the taxonomic classification of the reads deposited in the NCBI SRA database performed with the Taxonomy Analysis Tool (STAT) (Katz et al., 2021), we used Google BigQuery to mine the taxonomic results using the following query syntax: “SELECT *; FROM `nih-sra-datastore.sra.metadata`; WHERE organism = ‘Malacoherpesviridae’”, resulting in a list of SRA accession IDs with the corresponding number of identified reads (SRA accessed the 1st of July 2022). This output table was populated with the related experimental metadata retrieved from the SRA database using the ffq tool (Gálvez-Merchán et al., 2023) (Supplementary Table 1). Manual inspection of the metadata table allowed the detection of datasets associated with projects focusing on malacoherpesviruses (OsHV-1 or HaHV-1) and these were excluded according to the purpose of our analysis. The remaining SRA samples including more than 1,000 Malacoherpesviridae reads were downloaded and further analyzed. In particular, the reads in fastq format were trimmed for quality using fastp v0.23.2 (Chen et al., 2018) with the following parameters: –cut_front –cut_tail –cut_window_size 4 –cut_mean_quality 25 –qualified_quality_phred 25 –unqualified_percent_limit 30 –n_base_limit 5 –length_required 70 –detect_adapter_for_pe –trim_poly_x –trim_poly_g –low_complexity_filter. Trimmed reads were either directly de-novo assembled per sample or, in the case of SRA dataset produced for whole genome sequencing of a given species, the reads were firstly mapped on the reference genome (as declared in the metadata, Supplementary Table 1) using the CLC mapper with similarity and read length set to 0.8 (CLC Genomics, Qiagen, US) and only the unmapped reads were de-novo assembled. Reads were assembled using metaspades v.3.15.5 (Nurk et al., 2017) using the automatically determined kmer sizes based on read lengths and sample sizes. The obtained contigs were screened for the presence of Malacoherpesviridae genes by tblastn using all the OsHV-1 and HaHV-1 predicted proteins as queries and the positive contigs were used as reference for mapping the raw reads from the corresponding samples. For specific cases, the relevant contigs were refined using the iterative assembler PRICE v1.2 (Ruby et al., 2013), run with the following parameters: −rfq 90.95 −icf 1 1 5 −nc 30 −dbmax70 −mol 30 −tol 10 −mpi 80 −target 90 2 1 1. Raw reads were back-mapped on the malacoherpesvirus contigs using the CLC mapper tool, to evaluate their relative coverages. To search for additional malacoherpesvirus contigs in the NCBI databases, we used tblastx searches using the malacoherpesvirus proteomes as queries against the NCBI nucleotide database excluding the taxonomic term “Viruses”. The relevant contigs identified were subjected to Open Reading Frame (ORF) prediction using prodigal v2.6.3 (Hyatt et al., 2010) and the predicted proteins were compared with malacoherpesvirus proteomes obtained from OsHV-1 and HaHV-1 by blastp.
2.2 Genome-resolved meta genomics of Malacoherpesviridae
A Hidden Markov Model (HMM) of the putative Malacoherpesviridae major capsid protein (MCP) was build using hmmbuild based on four aligned proteins: YP_024643.1 (OsHV-1) YP_006908720.1 (HaHV-1) ELT95197.1 (C. teleta) and XP_019628941.1 (B. belcheri). The latter two hits represented viral genes associated with these metazoan genome assemblies because of putative integrations of co-sequencing. The HMM model was further updated with the novel hits (Data Sheet 1). HMMer v3.3.2 (Eddy, 2011) was used in combination with the MCP model to scan metagenomic datasets deposited in the MGnify database (https://www.ebi.ac.uk/metagenomics/). Moreover, the MCP profile was used for guidance to perform a targeted genome-resolved metagenomic survey of Tara Oceans. Briefly, we analyzed 798 metagenomes from Tara Oceans available at the EBI under project PRJEB402 (https://www.ebi.ac.uk/ena/browser/view/PRJEB402) which were organized into 11 ‘metagenomic sets’ based upon their geographic coordinates (Delmont et al., 2022a; Delmont et al., 2022b). Those 0.28 trillion reads were used as inputs for 11 metagenomic co-assemblies using MEGAHIT v1.1.1 (Li et al., 2015). Co-assemblies yielded 11.9 million contigs longer than 2,500 nucleotides (Delmont et al., 2022a; Delmont et al., 2022b). Constrained automatic binning was performed on each co-assembly output. Briefly, (1) anvi’o (Eren et al., 2021) profiled contigs using prodigal v2.6.3 (Hyatt et al., 2010) with default parameters to identify genes, (2) we mapped short reads from the metagenomic set to the contig using BWA v0.7.15 (Li and Durbin, 2009) (minimum identity of 95%) and stored the recruited reads as BAM files using samtools (Li et al., 2009; Eren et al., 2021) profiled each BAM file to estimate the coverage and detection statistics of each contig, and combined mapping profiles into a merged profile database for each metagenomic set. We then clustered contigs with the automatic binning algorithm CONCOCT (Alneberg et al., 2014) by constraining the number of clusters per metagenomic set to a number ranging from 50 to 400 depending on the set (total of 2,550 metabins from ~12 million contigs) (Delmont et al., 2022a; Delmont et al., 2022b). Here, we identified four metabins with a hit for the Malacoherpesviridae MCP HMM, and performed targeted genome-resolved metagenomics to characterize and manually curate metagenome assembled genomes (MAGs) corresponding to Malacoherpesviridae. The metabins are made available to reproduce the findings (see Data availability section).
2.3 Annotation of viral proteomes and phylogenetic inference of orthologs
The proteomes of the identified viruses were annotated using blastp searches against the NCBI nr database and using InterProScan v.90 (Jones et al., 2014). Orthofinder v2.5.4 (Emms and Kelly, 2019) was used to trace phylogenetic inference of orthologs based on the proteomes of the novel viruses and selected metazoans, namely using the available genomic-predicted datasets of molluscs, crustaceans, annelids and cephalochordates (NCBI Genome database, accessed the 21st of September 2022, Supplementary Table 2).
2.4 Phylogenetic analysis of family B DNA polymerase
A large dataset of family B DNA polymerase sequences (Gaïa et al., 2023) was used to identify the position of Malacoherpesviridae and Alloherpesviridae and their closest branching clades. The dataset consists of eukaryotic sequences from Kazlauskas et al. (Kazlauskas et al., 2020), and sequences identified from metagenome-assembled genomes of environmental viruses (Gaïa et al., 2023). Duplodnaviria and Naldaviricetes sequences, obtained by HMM searches on their genomes collected from the NCBI viral genomic database (accessed April 2022) and from Weinheimer & Aylward (Weinheimer and Aylward, 2022) (for the Jumbo phages), are also included. Redundant sequences (>95% identity) were removed using CD-HIT v4.7 (Fu et al., 2012), and long branches were iteratively removed. The most distant clades to Malacoherpesviridae and Alloherpesviridae were removed, and their remaining related clades were reduced to randomly selected representatives in order to increase the resolution of the resulting phylogenetic tree. The sequences from this restricted dataset are available in fasta format as Data Sheet 2. The alignments were performed using the FFT-NS-2 algorithm of MAFFT v7.490 (Katoh and Standley, 2013) for the large dataset and using the L-INS-i algorithm for the restricted dataset, and sites with more than 70% gap were trimmed using Goalign v0.3.1 (Lemoine and Gascuel, 2021). Tree reconstructions were done with IQ-TREE v1.6.12 (Nguyen et al., 2015) with the VT+F+R6 substitution model, as determined to be most suitable for the analyzed data using ModelFinder (Kalyaanamoorthy et al., 2017). The branch supports were computed using SH-aLRT test (Guindon et al., 2010, 0) and ultrafast bootstrap estimation (Hoang et al., 2018, 2).
3 Results
3.1 Malacoherpesvirus reads in the SRA databases are largely associated with known hosts
To search for new relatives of malacoherpesviruses, we first explored the NCBI Sequence Read Archive (SRA). The occurrence of the term ‘Malacoherpesviridae’ in the taxonomic annotations of the reads of the whole SRA database revealed 237 samples with more than 1,000 matching reads (Supplementary Table 1). A large fraction of these samples (N=168) corresponds to RNA-seq datasets produced in the effort to characterize the transcriptional dynamics during the OsHV-1 and HaHV-1 infections. Fifty-three samples represented DNA sequencing datasets of viral strains linked to the same RNA-seq studies or from samples collected from diseased hosts in different locations. The 16 remaining malacoherpesvirus-positive samples contained reads associated with non-invertebrate hosts and we explored these datasets more closely. One of these samples originated from DNA sequencing of Oncorhynchus nerka (sockeye salmon) collected in the Pitt Lake (British Columbia, Canada) and contained more than 54k malacoherpesvirus spots in addition to the spots corresponding to the salmon DNA. De-novo assembly of the non-salmon reads allowed the detection of 24 contigs highly similar to the OsHV-1 µVar genome (99.3% of average nucleotide identity [ANI] over 46% of covered genome length). Further six DNA sequencing datasets from the turtle Platysternon megacephalum genome project included ~12k malacoherpesvirus spots. Again, assembly of the non-turtle reads revealed fragments covering up to 78.2% of the OsHV-1 genome (99.6% of ANI). Finally, a number of malacoherpesvirus spots was found in 2 DNA sequencing samples of Bos taurus (N=31,625) and in 6 RNA-seq samples of the plant Cleistogenes songorica (N=34,845); the contigs assembled from these datasets also corresponded to OsHV-1 strains and covered >65% of the viral genome length (Data Sheet 3). The presence of malacoherpesvirus reads in these samples is highly unexpected, but the reasons for this are unclear and their investigation goes beyond the scope of this paper.
3.2 Malacoherpesvirus-like sequences are rare among global metagenomic datasets
We next explored an alternative approach for detection of new malacoherpesviruses. In particular, we searched for a hallmark malacoherpesvirus protein, namely, the major capsid protein (MCP), in various public sequence databases. HMMER search queried with the OsHV-1 MCP sequence against the MGnify database (Mitchell et al., 2020) resulted in a significant match to a protein originating from a shotgun metagenome from a study exploring antibiotic resistance genes in an aquaculture facility in Yantai City, China (Wang et al., 2018). Out of the 12 different DNA-seq samples included in this study, only one originating from the wastewater of the aquaculture facility was positive for the malacoherpesvirus MCP. Metagenomic assembly of the reads yielded more than 3 million contigs, with 8 likely representing a new virus, which we tentatively named MalacoHV1/China/2018 (we used a naming convention where the virus name epithet is followed by the geographical region and year of isolation/sequencing; Table 1 and Figure 2). Back-mapping of sequencing reads on the viral scaffolds revealed an average coverage of 9x, with a 15.1 kb-long scaffold showing a coverage of 17x, likely representing the inverted repeat regions characteristic of malacoherpesvirus genomes. With the total length of 174.3 kb, the genome of MalacoHV1 encodes 107 predicted proteins, with 42 showing the highest similarity to HaHV-1 and 2 to OsHV-1 homologs. One protein showed the highest similarity (E-value = 5.76e-35, amino acidic identity = 33%) to a protein of the sea slug gastropod Elysia chlorotica (RUS69626.1, Supplementary Table 3). Overall, these results suggest that the newly identified virus is not closely related to either of the two currently known malacoherpesviruses and is likely to represent a new species in the family.
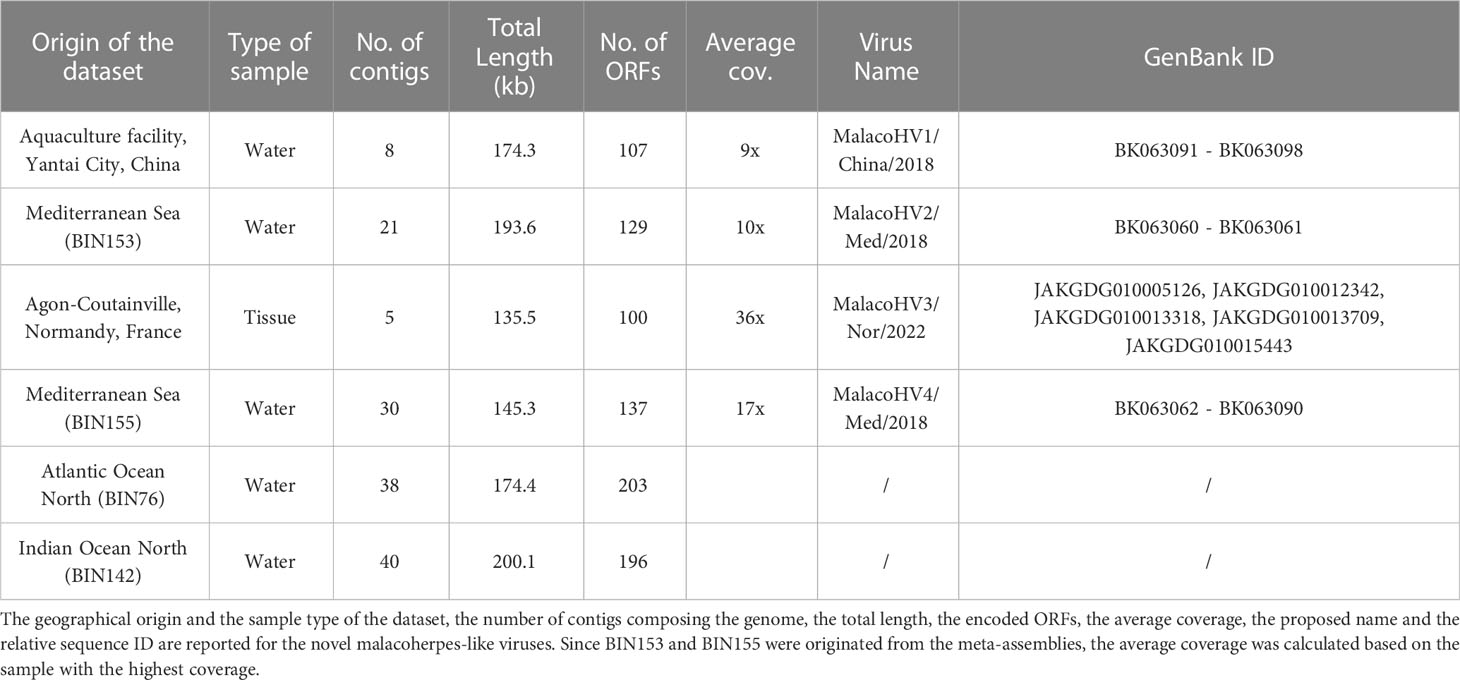
Table 1 Summary of the novel malacoherpes-like viruses.
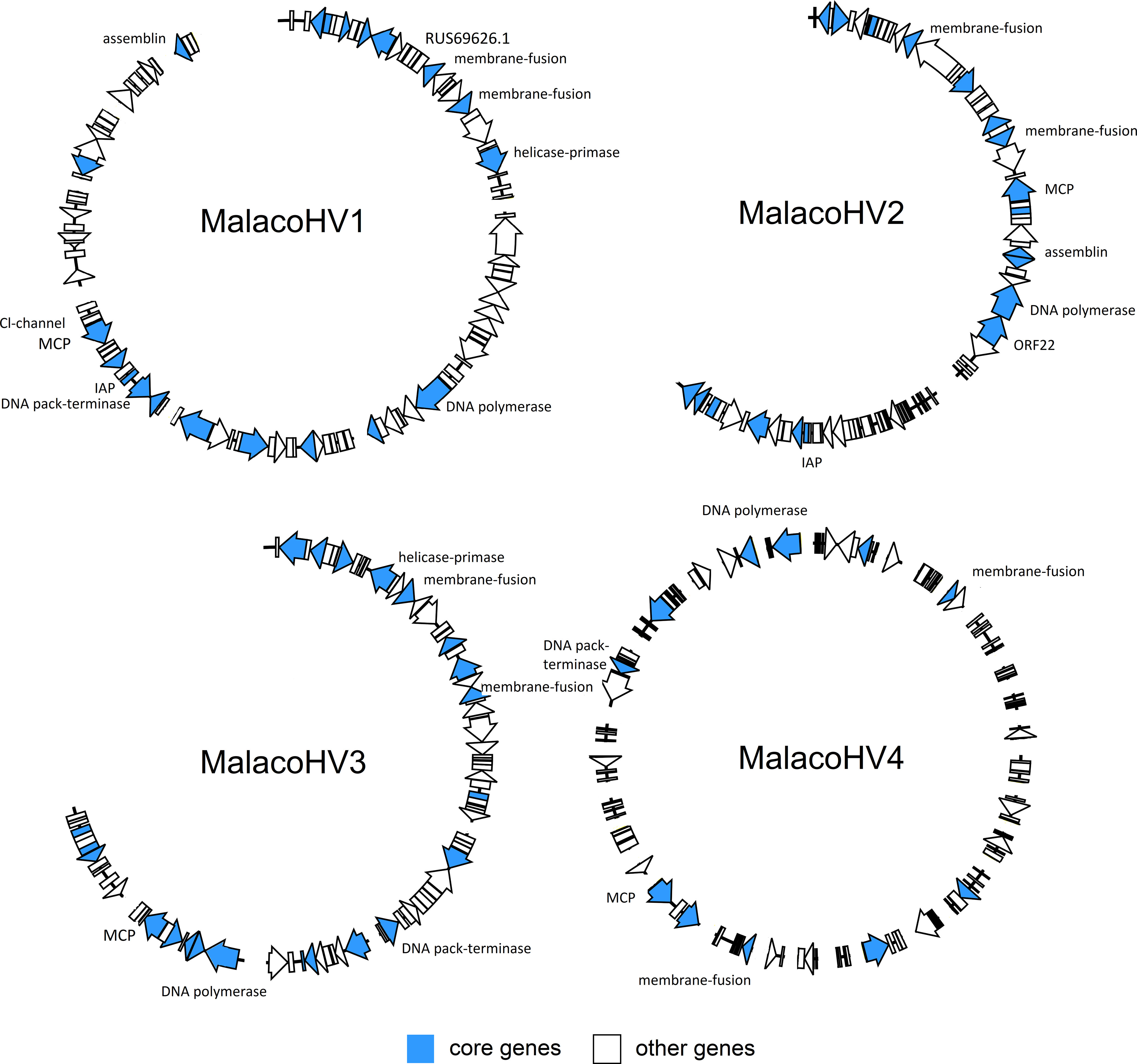
Figure 2 Overview of the novel malacoherpesvirus-like genomic contigs. Along the viral genomes of MalacoHV1, MalacoHV2, MalacoHV3 and MalacoHV4, the predicted ORFs are depicted as arrows, colored ibn light blue if they represent core malacoherpesvirus genes or in white for the other genes. Genes discussed in the paper have been also named. Linear dsDNA genomes have been depicted as open circles for convenient representation, with contigs separated by spaces.
The MCP sequence of MalacoHV1 was used to create a more sensitive MCP profile hidden Markov model (HMM), which was then used to scan the global Tara Oceans database, resulting in the detection of 4 additional MCP homologs. Binning of the datasets including the MCP homologs yielded 4 metagenome-assembled genomes (MAGs), varying in length from 145 to 200 kb and including a total of 129 contigs (Table 1). The first MCP homolog was encoded on a 76.7 kb-long contig (contig_012) carrying 40 predicted genes, with 13 proteins showing significant similarity (E-value <0.0001) with malacoherpesvirus proteins. In particular, 7 proteins had the closest homologs in OsHV-1, 3 in HaHV-1 and 3 in the previously described putative malacoherpesvirus associated with B. belcheri. However, it should be noted that the homologous sequences displayed considerable sequence divergence (22-36% amino acidic identity; Supplementary Table 3). This MCP-encoding contig was binned together with 20 other contigs (BIN153). Notably, the second longest contig (45.6 kb, contig_006) in this bin contained several additional ORFs homologous to those of malacoherpesviruses, whereas the remaining 19 contigs, altogether accounting for 75.1 kb in length, did not include ORFs with similarity to known malacoherpesviruses or other viruses (Supplementary Table 3). As a precaution, we considered only contig_006 and contig_012 as part of a putative new virus, tentatively named MalacoHV2/Med/2018 (Figure 2, Table 1). The second MCP homolog was encoded within a 10 kb contig, which in addition carried 2 other ORFs with homologs in HaHV-1. A total of 16 contigs out of 30 included in the metagenomic bin (BIN155) encoded detectable homologs of viral proteins, suggesting that BIN155 represents a novel viral genomes, called MalacoHV4/Med/2018. Most of the viral proteins had homologs in the malacoherpesviral sequences associated with B. belcheri or B. floridae (Figure 2, Supplementary Table 3). By contrast, in BIN76 and BIN142, the identified MCP homologs were encoded on contigs of a few kilobases, with maximum of 3 ORFs similar to those of known malacoherpesviruses and no other contigs in these bins encoded putative viral proteins. Thus, BIN76 and BIN142 were not considered further.
Finally, to identify additional malacoherpesvirus relatives, we performed tblastn searches queried with the malacoherpesvirus proteomes against the NCBI nucleotide database, which yielded positive hits in 5 scaffolds of the blue mussel Mytilus edulis genome (MEDU_EUS, from an animal collected in Normandy, France; Table 1). These sequences collectively amounted to 135.5 kb of a putative novel malacoherpesvirus genome, called MalacoHV3/Nor/2022, encoding 100 proteins, 61% of which showed moderate similarity (max: 65%, min: 23%, median: 37% amino acidic identity) to OsHV-1 proteins, whereas one was most similar (although with low sequence identity of 25%) to ORF89 of HaHV-1 (Figure 2, Supplementary Table 3). As for MalacoHV1, a scaffold of 15.9 kb of MalacoHV3 had a twice higher coverage (69x) compared to the average coverage, suggesting that it represents repeated genomic regions.
3.3 Homology and phylogenetic analyses differentiate malacoherpesvirus lineages
Comparative genomics of the newly identified and the previously known malacoherpesviruses yielded 126 protein families, 34 of which were present in only one viral genome, whereas 20 were conserved in at least 7 out of the 9 analyzed genomes and were, therefore, considered as malacoherpesvirus core genes (Figure 3). These included the MCP, DNA polymerase, DNA packaging terminase, 2 copies of the class I membrane fusion protein, helicase, helicase-primase and an archaeo-eukaryotic primase, inhibitor of apoptosis (IAPs), YqaJ-like endonuclease and assemblin (capsid maturation protease) as well as 9 uncharacterized protein families (ORF29, ORF102, ORF69, ORF94, ORF40, ORF61, ORF6, ORF91, ORF22, named according to the OsHV-1 nomenclature). A total of 13 protein clusters were specific to the HaHV-1 or OsHV-1 lineages. However, for most of these proteins a function is not known and could not be predicted, with the exception of HTH domain and a zinc-finger proteins in HaHV-1 and a predicted dUTPase in OsHV-1.
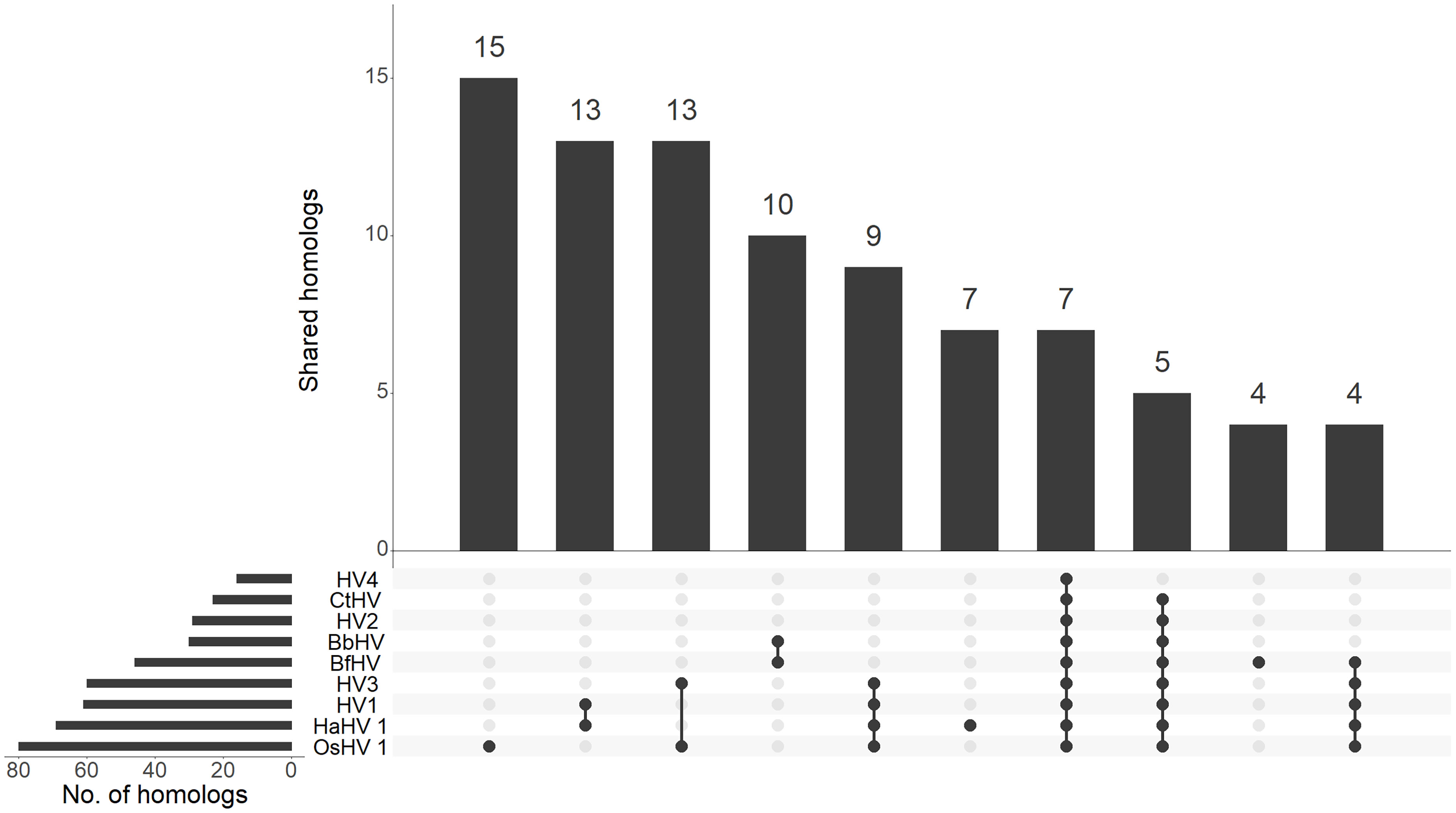
Figure 3 Distribution of malacoherpesvirus homologs. The upset graph depicts the distribution of homologs between malacoherpesviruses. For each species, the number of total and of shared homologs are reported. BbHV, BfHV anc CtHV referred to the viral sequences found associated with B. belcheri, B. floridae and C. teleta chromosomes, respectively.
To further explore the evolutionary relationship among malacoherpesviruses as well as between malacoherpesviruses and other large DNA viruses, we conducted a maximum likelihood phylogenetic analysis based on the conserved family B DNA polymerase. In the phylogeny, the previously described and newly identified malacoherpesviruses formed a well-supported monophyletic clade, which branched as a sister group to alloherpesviruses, albeit with a non-significant branch support (Figure 4). In the malacoherpesvirus clade, DNA polymerases found in the amphioxi genomes clustered with MalacoHV4. Notably, the three sequences formed a sister clade to all other malacoherpesviruses, suggesting an earlier divergence of the viruses associated with amphioxus. In the second subclade, the annelid-associated DNA polymerase appears to be nested among mollusc viruses, suggesting a possible host switch event, whereas MalacoHV2 occupies a basal position in this clade. Overall, these results suggest host-specific divergence of malacoherpesviruses into emerging clades.
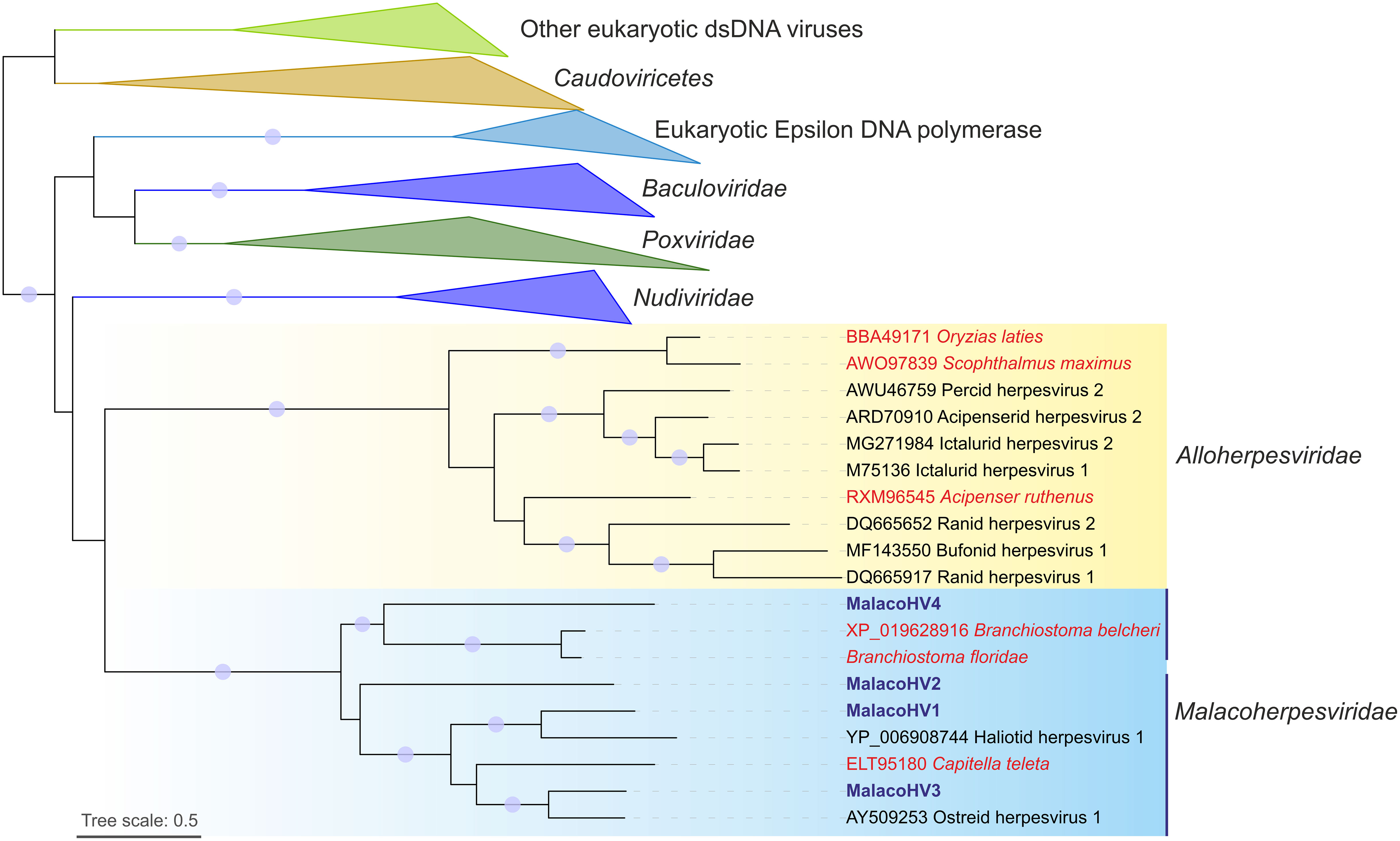
Figure 4 Maximum likelihood phylogenetic tree of family B DNA polymerases from viruses and cellular organisms. The tree is mid-point rooted and the clades corresponding to eukaryotes and viruses other than malacoherpesviruses and alloherpesviruses are collapsed for convenience of representation. Viruses likely integrated in the host genomes are indicated with red font, whereas MalacoHV1-4 are highlighted with blue font. Vertical lines indicate the two subclades of malacoherpesviruses. Circles at the nodes represent the branch support values: SH-aLRT ≥ 80% and UFB (ultrafast bootstrap) ≥ 95%. Tree scale represent the number of substitutions per site.
3.4 The distribution of malacoherpesvirus orthologs suggests their host spectrum
To gain insight into the potential malacoherpesvirus host spectrum, we searched for homologs of the core malacoherpesvirus proteins in 54 metazoan species, namely, bivalves, crustaceans, annelids and cephalochordates for which genome sequences are available. With exception of B. belcheri and C. teleta genomes which carry extended genomic regions similar to malacoherpesviruses (potential proviruses), in all other metazoans homologs of malacoherpesvirus genes are encoded sporadically. In particular, we identified 6 protein clusters present in two or more malacoherpesvirus genomes and shared with more than 3 of the selected metazoans. Four of these protein families, namely, IAPs, DNA polymerase, the class I membrane fusion protein and the uncharacterized ORF22, are encoded by the previously described core malacoherpesvirus genes. In addition, the ribonucleoside-diphosphate reductase small subunit (RDR2) and a chloride channel protein were also present (Table 2). From one to six IAP copies are present in the malacoherpesvirus genomes. By contrast, homologs of putative class I membrane fusion proteins, a core gene conserved in all malacoherpesviruses and always present in two copies, could be found in 16 out of the 54 selected metazoan species, including C. gigas, Mytilus galloprovincialis, Mercenaria mercenaria (hard clam) and Lepeophtheirus salmonis (copepod) (Table 2).
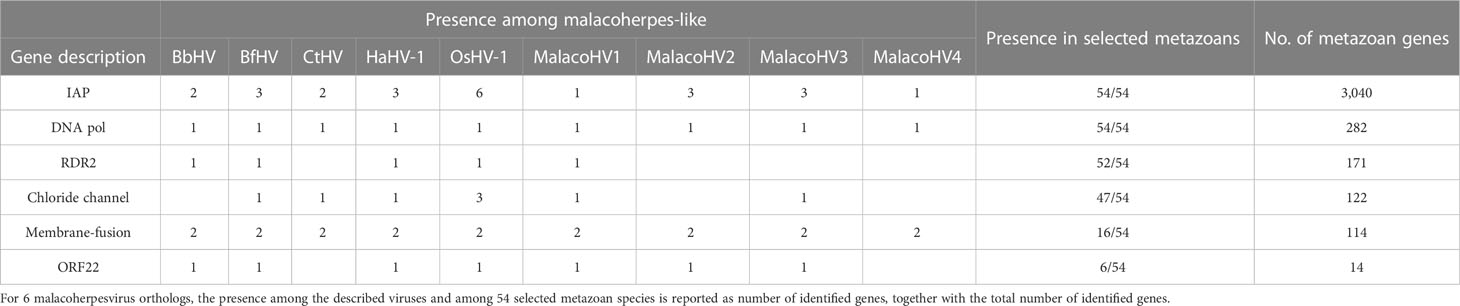
Table 2 Distribution of malacoherpesvirus core genes in selected metazoan species.
4 Discussion
The small number of described malacoherpesviruses raised a question of whether these viruses are rare in marine ecosystems or whether they have been systematically overlooked in previous analyses (Iranzo et al., 2016; Mushegian et al., 2018). Somewhat unexpectedly, extensive searches of malacoherpesvirus-like MCPs in global sequencing datasets, including marine metagenomic datasets, performed in this study did not uncover the suspected hidden malacoherpesvirus diversity in marine ecosystems. Two non-mutually exclusive possibilities could explain the scarcity of these viruses. First, malacoherpesviruses could be genuinely rare, present in low titres in marine environments. Second, the analyzed datasets are biased toward sequences from the plankton fraction, i.e., unicellular organisms and extracellular virus particles; hence, viruses of multicellular eukaryotes could be underrepresented, highlighting the need to expand the exploration of the ocean viromes to animals and other multicellular eukaryotes. Nevertheless, our analysis led to the identification of 4 novel malacoherpes-like viruses that share limited protein sequence similarities to the previously known viruses. Phylogenetic analysis based on the family B DNA polymerase placed the novel MalacoHV1 and MalacoHV3 as sister lineages to OsHV-1 and HaHV-1, respectively, likely representing new species, whereas MalacoHV2 showed higher divergence and occupied a basal position in the subclade including mollusc herpesviruses. Notably, viral sequences integrated or co-sequenced with two amphioxus genomes, together with MalacoHV4, form a sister clade to mollusc and annelid herpesviruses. The considerable nucleotide similarity between the viral genomic regions of the two amphioxi suggests that the two elements belong to the same virus species, although the presence of a viral genome rearrangement as well as different flanking genes between B. belcheri and B. floridae loci can suggest independent integration events. The conservation of these possibly integrated viruses in the amphioxus genomes poses questions regarding a possible advantage in maintaining them, although it should be noted that no conclusive experimental evidence exists to support their integration. It is tempting to speculate that the two endogenous virus elements function in antivirus defense, as has been demonstrated for many other endogenized viruses (Koonin and Krupovic, 2018). Although the malacoherpesvirus-like genes in C. teleta are present on a separate genomic scaffold, we hypothesize that this contig also represents an integrated virus. Consistent with this possibility, many of the viral genes on this contig carry multiple introns, possibly underpin a progressive domestication and pseudogenization through intron invasion, as reported for basal eukaryotes (Filée, 2014). This would also suggest that the putative provirus lost the ability to replicate and proliferate as an autonomous. Further research will be needed to understand whether potential proviruses detected in the genomes of B. belcheri, B. floridae and C. teleta represent endogenized relics of malacoherpesviruses or transiently integrated viral genomes capable of excision and reactivation as it has been demonstrated for their vertebrate relatives in the family Orthoherpesviridae (Cohen, 2020).
The phylogenetic relationship between malacoherpesviruses based on the family B DNA polymerase is corroborated by the comparative genomics analysis, with MalacoHV1 and MalacoHV3 sharing higher fractions of orthologous genes with HaHV-1 and OsHV-1, respectively. Although lineage-specific proteins were identified, the absence of functional information for most of them hampered further conclusions regarding specific virus-host interactions. Notably, however, all malacoherpesviruses encode copies of IAPs, revealing a common strategy to overcome host apoptosis, as has been also shown for some other viruses (Clem, 2015). Notably, IAPs showed characteristic expansions among bivalves, particularly in oyster and hard clam (Song et al., 2021; Witkop et al., 2022). It is worth noting that the HaHV-1-like lineage shares several genes with Elysia chlorotica, further suggesting that MalacoHV1, similar to HaHV-1, can infect gastropod species. One of these proteins is predicted to be a putative transposon-associated RNA-guided endonuclease TnpB, known as Fanzor in eukaryotes (Bao and Jurka, 2013), and similar proteins are found also in two bivalves (M. mercenaria and Dreissena polymorpha) as well as in some giant viruses (Kaumoebavirus and Yasminevirus).
Similarly, based on the fact that contigs representing the MalacoHV3 genome originate from the blue mussel M. edulis sequencing dataset, MalacoHV3 likely infects a bivalve host. Consistently, MalacoHV3 genome has low GC% content, possibly revealing a host adaptation. Indeed, bivalve genomes have lower GC% content compared to gastropods (Adachi et al., 2021). The presence of malacoherpesvirus core proteins in several mollusc species further pointed to possible host associations, but additional genomic data is needed to substantiate these findings. Notably, the membrane fusion protein that we found in several metazoan genomes is homologous to the fusogens of not only other large dsDNA viruses, such as baculoviruses, and diverse RNA viruses, but also occasionally encoded by endogenous viruses of the Metaviridae family (also known as Gypsy/Ty3 retrotransposons), distant relatives of vertebrate retroviruses which are widespread in eukaryotic genomes (Krupovic et al., 2018; Llorens et al., 2020). Thus, it is likely that the fusogen homologs identified in the metazoan species represent integrated metaviruses rather than true orthologs of malacoherpesvirus proteins.
Overall, we demonstrated that although rare, the diversity of circulating mollusc herpesviruses is greater than currently appreciated. Our data extends the recent reports on the overlooked diversity of malacoherpesviruses, where one meta-transcriptomic study noted two divergent DNA-packaging terminases putatively associated with sandworm and peanut worm herpesviruses (Porter et al., 2019) and the other study focusing on RNA viruses described divergent malacoherpesvirus-like transcripts in nudibranchs (gastropods) (Rosani, 2022). It is conceivable that malacoherpesviruses described in this study have evaded detection until now due to the fact that they infect economically less-relevant species or due to limited pathogenicity, possibly resulting from the long-lasting co-evolution with their hosts (Little et al., 2010; Geoghegan and Holmes, 2018).
Finally, we would like to draw the attention to the geographic origins of the novel malacoherpesviruses: MalacoHV1 was detected in Yantai, a coastal city in Shandong province (China), MalacoHV2 and MalacoHV4 in the Mediterranean Sea, and MalacoHV3 in Normandy, France, all three regions devoted to aquaculture. This raises some concerns linked to the potential pathogenicity of the new malacoherpesviruses, further substantiated by the fact that the two other known malacoherpesviruses, HaHV-1 and OsHV-1, display broad host spectrum and persistence (Arzul et al., 2001; Chang et al., 2005; Bai et al., 2015; Corbeil et al., 2016; Fuhrmann et al., 2021). Therefore, more focused analyses to characterize the virome of marine animals are required in order to anticipate and increase the detection capability of possible future viral outbreaks impacting aquaculture. In particular, given that the routine PCR-based detection of malacoherpesviruses with HaHV-1 and OsHV-1 specific primers will not yield signal for MalacoHV1-4, new sets of PCR primers should be designed for screening of aquaculture.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author. Nucleotide sequence data reported are available in the Third Party Annotation Section of the DDBJ/ENA/GenBank databases under the accession numbers TPA: BK063060-BK063098.
Author contributions
UR and MK conceived the study. UR, TD, MG, and MK performed the analyses, UR and MK drafted the first version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
UR was financially supported by the Italian PRIN 2017 project VIRIDE (2017EKFA98).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer LX is currently organizing a Research Topic with the author UR.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1159754/full#supplementary-material
References
Abbadi M., Zamperin G., Gastaldelli M., Pascoli F., Rosani U., Milani A., et al. (2018). Identification of a newly described OsHV-1 µvar from the North Adriatic Sea (Italy). J. Gen. Virol. 99, 693–703. doi: 10.1099/jgv.0.001042
Adachi K., Yoshizumi A., Kuramochi T., Kado R., Okumura S.-I. (2021). Novel insights into the evolution of genome size and AT content in mollusks. Mar. Biol. 168, 25. doi: 10.1007/s00227-021-03826-x
Alneberg J., Bjarnason B. S., de Bruijn I., Schirmer M., Quick J., Ijaz U. Z., et al. (2014). Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146. doi: 10.1038/nmeth.3103
Arzul I., Nicolas J. L., Davison A. J., Renault T. (2001). French Scallops: a new host for ostreid herpesvirus-1. Virology 290, 342–349. doi: 10.1006/viro.2001.1186
Bai C., Gao W., Wang C., Yu T., Zhang T., Qiu Z., et al. (2016). Identification and characterization of ostreid herpesvirus 1 associated with massive mortalities of Scapharca broughtonii broodstocks in China. Dis. Aquat. Organ. 118, 65–75. doi: 10.3354/dao02958
Bai C.-M., Rosani U., Zhang X., Xin L.-S., Bortoletto E., Wegner K. M., et al. (2021). Viral decoys: the only two herpesviruses infecting invertebrates evolved different transcriptional strategies to deflect post-transcriptional editing. Viruses 13, 1971. doi: 10.3390/v13101971
Bai C., Wang C., Xia J., Sun H., Zhang S., Huang J. (2015). Emerging and endemic types of ostreid herpesvirus 1 were detected in bivalves in China. J. Invertebr. Pathol. 124, 98–106. doi: 10.1016/j.jip.2014.11.007
Bao W., Jurka J. (2013). Homologues of bacterial TnpB_IS605 are widespread in diverse eukaryotic transposable elements. Mob. DNA 4, 12. doi: 10.1186/1759-8753-4-12
Burioli E., Prearo M., Houssin M. (2017). Complete genome sequence of ostreid herpesvirus type 1 µVar isolated during mortality events in the pacific oyster Crassostrea gigas in France and Ireland. Virology 509, 239–251. doi: 10.1016/j.virol.2017.06.027
Chang P. H., Kuo S. T., Lai S. H., Yang H. S., Ting Y. Y., Hsu C. L., et al. (2005). Herpes-like virus infection causing mortality of cultured abalone Haliotis diversicolor supertexta in Taiwan. Dis. Aquat. Organ. 65, 23–27. doi: 10.3354/dao065023
Chen S., Zhou Y., Chen Y., Gu J. (2018). Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinforma. Oxf. Engl. 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Clem R. J. (2015). Viral IAPs, then and now. Semin. Cell Dev. Biol. 39, 72–79. doi: 10.1016/j.semcdb.2015.01.011
Corbeil S., Williams L. M., McColl K. A., Crane M. S. J. (2016). Australian Abalone (Haliotis laevigata, h. rubra and h. conicopora) are susceptible to infection by multiple abalone herpesvirus genotypes. Dis. Aquat. Organ. 119, 101–106. doi: 10.3354/dao02989
Correa A. M. S., Ainsworth T. D., Rosales S. M., Thurber A. R., Butler C. R., Vega Thurber R. L. (2016). Viral outbreak in corals associated with an In situ bleaching event: atypical herpes-like viruses and a new megavirus infecting symbiodinium. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.00127
Davison A. J., Eberle R., Ehlers B., Hayward G. S., McGeoch D. J., Minson A. C., et al. (2009). The order herpesvirales. Arch. Virol. 154, 171–177. doi: 10.1007/s00705-008-0278-4
Davison A. J., Trus B. L., Cheng N., Steven A. C., Watson M. S., Cunningham C., et al. (2005). A novel class of herpesvirus with bivalve hosts. J. Gen. Virol. 86, 41–53. doi: 10.1099/vir.0.80382-0
Delmont T. O., Gaia M., Hinsinger D. D., Frémont P., Vanni C., Fernandez-Guerra A., et al. (2022a). Functional repertoire convergence of distantly related eukaryotic plankton lineages abundant in the sunlit ocean. Cell Genomics 2, 100123. doi: 10.1016/j.xgen.2022.100123
Delmont T. O., Pierella Karlusich J. J., Veseli I., Fuessel J., Eren A. M., Foster R. A., et al. (2022b). Heterotrophic bacterial diazotrophs are more abundant than their cyanobacterial counterparts in metagenomes covering most of the sunlit ocean. ISME J. 16, 927–936. doi: 10.1038/s41396-021-01135-1
Dion M. B., Oechslin F., Moineau S. (2020). Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 18, 125–138. doi: 10.1038/s41579-019-0311-5
Eddy S. R. (2011). Accelerated profile HMM searches. PloS Comput. Biol. 7, e1002195. doi: 10.1371/journal.pcbi.1002195
Emms D. M., Kelly S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238. doi: 10.1186/s13059-019-1832-y
Eren A. M., Kiefl E., Shaiber A., Veseli I., Miller S. E., Schechter M. S., et al. (2021). Community-led, integrated, reproducible multi-omics with anvi’o. Nat. Microbiol. 6, 3–6. doi: 10.1038/s41564-020-00834-3
FAO (2020). The state of world fisheries and aquaculture 2020: sustainability in action (Rome, Italy: FAO). doi: 10.4060/ca9229en
Farley C. A., Banfield W. G., Kasnic G., Foster W. S. (1972). Oyster herpes-type virus. Science 178, 759–760. doi: 10.1126/science.178.4062.759
Filée J. (2014). Multiple occurrences of giant virus core genes acquired by eukaryotic genomes: the visible part of the iceberg? Virology 466–467, 53–59. doi: 10.1016/j.virol.2014.06.004
Fu L., Niu B., Zhu Z., Wu S., Li W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinforma. Oxf. Engl. 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Fuhrmann M., Georgiades E., Cattell G., Brosnahan C., Lane H. S., Hick P. M. (2021). Aquatic pathogens and biofouling: pilot study of ostreid herpesvirus 1 translocation by bivalves. Biofouling 1–15, 949–963. doi: 10.1080/08927014.2021.1985474
Gaïa M., Meng L., Pelletier E., Forterre P., Vanni C., Fernandez-Guerra A., et al. (2023). Mirusviruses link herpesviruses to giant viruses. Nature 616, 783–789. doi: 10.1038/s41586-023-05962-4
Gálvez-Merchán Á., Min K. H., Pachter L., Booeshaghi A. S. (2023). Metadata retrieval from sequence databases with ffq. Bioinformatics 39, btac667. doi: 10.1093/bioinformatics/btac667
Geoghegan J. L., Holmes E. C. (2018). The phylogenomics of evolving virus virulence. Nat. Rev. Genet. 19, 756–769. doi: 10.1038/s41576-018-0055-5
Grasis J. A., Lachnit T., Anton-Erxleben F., Lim Y. W., Schmieder R., Fraune S., et al. (2014). Species-specific viromes in the ancestral holobiont Hydra. PloS One 9, e109952. doi: 10.1371/journal.pone.0109952
Guindon S., Dufayard J.-F., Lefort V., Anisimova M., Hordijk W., Gascuel O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Hoang D. T., Chernomor O., von Haeseler A., Minh B. Q., Vinh L. S. (2018). UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522. doi: 10.1093/molbev/msx281
Hyatt D., Chen G.-L., LoCascio P. F., Land M. L., Larimer F. W., Hauser L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 11, 119. doi: 10.1186/1471-2105-11-119
Iranzo J., Krupovic M., Koonin E. V. (2016). The double-stranded DNA virosphere as a modular hierarchical network of gene sharing. mBio 7, e00978–e00916. doi: 10.1128/mBio.00978-16
Jenkins C., Hick P., Gabor M., Spiers Z., Fell S. A., Gu X., et al. (2013). Identification and characterisation of an ostreid herpesvirus-1 microvariant (OsHV-1 µ-var) in Crassostrea gigas (Pacific oysters) in Australia. Dis. Aquat. Organ. 105, 109–126. doi: 10.3354/dao02623
Jones P., Binns D., Chang H.-Y., Fraser M., Li W., McAnulla C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kalyaanamoorthy S., Minh B. Q., Wong T. K. F., von Haeseler A., Jermiin L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Katoh K., Standley D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Katz K. S., Shutov O., Lapoint R., Kimelman M., Brister J. R., O’Sullivan C. (2021). STAT: a fast, scalable, MinHash-based k-mer tool to assess sequence read archive next-generation sequence submissions. Genome Biol. 22, 270. doi: 10.1186/s13059-021-02490-0
Kazlauskas D., Krupovic M., Guglielmini J., Forterre P., Venclovas Č. (2020). Diversity and evolution of b-family DNA polymerases. Nucleic Acids Res. 48, 10142–10156. doi: 10.1093/nar/gkaa760
Koonin E. V., Dolja V. V., Krupovic M., Varsani A., Wolf Y. I., Yutin N., et al. (2020). Global organization and proposed megataxonomy of the virus world. Microbiol. Mol. Biol. Rev. MMBR 84, e00061–e00019. doi: 10.1128/MMBR.00061-19
Koonin E. V., Krupovic M. (2018). The depths of virus exaptation. Curr. Opin. Virol. 31, 1–8. doi: 10.1016/j.coviro.2018.07.011
Krupovic M., Blomberg J., Coffin J. M., Dasgupta I., Fan H., Geering A. D., et al. (2018). Ortervirales: new virus order unifying five families of reverse-transcribing viruses. J. Virol. 92, e00515–e00518. doi: 10.1128/JVI.00515-18
Lefkowitz E. J., Dempsey D. M., Hendrickson R. C., Orton R. J., Siddell S. G., Smith D. B. (2018). Virus taxonomy: the database of the international committee on taxonomy of viruses (ICTV). Nucleic Acids Res. 46, D708–D717. doi: 10.1093/nar/gkx932
Lemoine F., Gascuel O. (2021). Gotree/Goalign: toolkit and go API to facilitate the development of phylogenetic workflows. NAR Genomics Bioinforma. 3, lqab075. doi: 10.1093/nargab/lqab075
Li H., Durbin R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., et al. (2009). The sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li D., Liu C.-M., Luo R., Sadakane K., Lam T.-W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de bruijn graph. Bioinforma. Oxf. Engl. 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Little T. J., Shuker D. M., Colegrave N., Day T., Graham A. L. (2010). The coevolution of virulence: tolerance in perspective. PloS Pathog. 6, e1001006. doi: 10.1371/journal.ppat.1001006
Liu Y., Demina T. A., Roux S., Aiewsakun P., Kazlauskas D., Simmonds P., et al. (2021). Diversity, taxonomy, and evolution of archaeal viruses of the class Caudoviricetes. PloS Biol. 19, e3001442. doi: 10.1371/journal.pbio.3001442
Llorens C., Soriano B., Krupovic M., Ictv Report Consortium (2020). ICTV virus taxonomy profile: metaviridae. J. Gen. Virol. 101, 1131–1132. doi: 10.1099/jgv.0.001509
Mitchell A. L., Almeida A., Beracochea M., Boland M., Burgin J., Cochrane G., et al. (2020). MGnify: the microbiome analysis resource in 2020. Nucleic Acids Res. 48, D570–D578. doi: 10.1093/nar/gkz1035
Mushegian A., Karin E. L., Pupko T. (2018). Sequence analysis of malacoherpesvirus proteins: pan-herpesvirus capsid module and replication enzymes with an ancient connection to “Megavirales”. Virology 513, 114–128. doi: 10.1016/j.virol.2017.10.009
Nguyen L.-T., Schmidt H. A., von Haeseler A., Minh B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nicolas J. L., Comps M., Cochennec N. (1992). Herpes-like virus infecting pacific-oyster larvae, Crassostrea gigas. Bull. Eur. Assoc. Fish Pathol. U. K. 12 (1), 11.
Nurk S., Meleshko D., Korobeynikov A., Pevzner P. A. (2017). metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/gr.213959.116
Osterrieder N., Wallaschek N., Kaufer B. B. (2014). Herpesvirus genome integration into telomeric repeats of host cell chromosomes. Annu. Rev. Virol. 1, 215–235. doi: 10.1146/annurev-virology-031413-085422
Pernet F., Lupo C., Bacher C., Whittington R. J. (2016). Infectious diseases in oyster aquaculture require a new integrated approach. Philos. Trans. R. Soc Lond. B. Biol. Sci. 371 (B371), 20150213. doi: 10.1098/rstb.2015.0213
Porter A. F., Shi M., Eden J.-S., Zhang Y.-Z., Holmes E. C. (2019). Diversity and evolution of novel invertebrate DNA viruses revealed by meta-transcriptomics. Viruses 11. doi: 10.3390/v11121092
Prado-Alvarez M., García-Fernández P., Faury N., Azevedo C., Morga B., Gestal C. (2021). First detection of OsHV-1 in the cephalopod octopus vulgaris. is the octopus a dead-end for OsHV-1? J. Invertebr. Pathol. 183, 107553. doi: 10.1016/j.jip.2021.107553
Ren W., Chen H., Renault T., Cai Y., Bai C., Wang C., et al. (2013). Complete genome sequence of acute viral necrosis virus associated with massive mortality outbreaks in the Chinese scallop, Chlamys farreri. Virol. J. 10, 110. doi: 10.1186/1743-422X-10-110
Rosani U. (2022). Tracing RNA viruses associated with nudibranchia gastropods. PeerJ 10, e13410. doi: 10.7717/peerj.13410
Rosani U., Venier P. (2017). Oyster RNA-seq data support the development of Malacoherpesviridae genomics. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.01515
Ruby J. G., Bellare P., Derisi J. L. (2013). PRICE: software for the targeted assembly of components of (Meta) genomic sequence data. G3 Bethesda Md 3, 865–880. doi: 10.1534/g3.113.005967
Ryazanova T. V., Eliseikina M. G., Kalabekov I. M., Odintsova N. A. (2015). A herpes-like virus in king crabs: characterization and transmission under laboratory conditions. J. Invertebr. Pathol. 127, 21–31. doi: 10.1016/j.jip.2015.02.003
Savin K. W., Cocks B. G., Wong F., Sawbridge T., Cogan N., Savage D., et al. (2010). A neurotropic herpesvirus infecting the gastropod, abalone, shares ancestry with oyster herpesvirus and a herpesvirus associated with the amphioxus genome. Virol. J. 7, 308. doi: 10.1186/1743-422X-7-308
Segarra A., Pépin J. F., Arzul I., Morga B., Faury N., Renault T. (2010). Detection and description of a particular ostreid herpesvirus 1 genotype associated with massive mortality outbreaks of pacific oysters, Crassostrea gigas, in France in 2008. Virus Res. 153, 92–99. doi: 10.1016/j.virusres.2010.07.011
Song H., Guo X., Sun L., Wang Q., Han F., Wang H., et al. (2021). The hard clam genome reveals massive expansion and diversification of inhibitors of apoptosis in Bivalvia. BMC Biol. 19, 15. doi: 10.1186/s12915-020-00943-9
Stentiford G. D., Sritunyalucksana K., Flegel T. W., Williams B. A. P., Withyachumnarnkul B., Itsathitphaisarn O., et al. (2017). New paradigms to help solve the global aquaculture disease crisis. PloS Pathog. 13, e1006160. doi: 10.1371/journal.ppat.1006160
Vega Thurber R. L., Barott K. L., Hall D., Liu H., Rodriguez-Mueller B., Desnues C., et al. (2008). Metagenomic analysis indicates that stressors induce production of herpes-like viruses in the coral Porites compressa. Proc. Natl. Acad. Sci. U. S. A. 105, 18413–18418. doi: 10.1073/pnas.0808985105
Walker P. J., Siddell S. G., Lefkowitz E. J., Mushegian A. R., Adriaenssens E. M., Alfenas-Zerbini P., et al. (2022). Recent changes to virus taxonomy ratified by the international committee on taxonomy of viruses, (2022). Arch. Virol. 167, 2429–2440. doi: 10.1007/s00705-022-05516-5
Wang J., Guo Z., Feng J., Liu G., Xu L., Chen B., et al. (2004). Virus infection in cultured abalone, Haliotis diversicolor reeve in Guangdong Province, China. J. Shellfish Res. 23, 1163–1169.
Wang J.-H., Lu J., Zhang Y.-X., Wu J., Luo Y., Liu H. (2018). Metagenomic analysis of antibiotic resistance genes in coastal industrial mariculture systems. Bioresour. Technol. 253, 235–243. doi: 10.1016/j.biortech.2018.01.035
Wang C., Wang X., Song X., Huang J., Song W. (2002). Purification and ultrastructure of a spherical virus in cultured scallop Chlamys farrer. J. Fishery China 26 (2), 2002.
Weinheimer A. R., Aylward F. O. (2022). Infection strategy and biogeography distinguish cosmopolitan groups of marine jumbo bacteriophages. ISME J. 16, 1657–1667. doi: 10.1038/s41396-022-01214-x
Whittington R. J., Paul-Pont I., Evans O., Hick P., Dhand N. K. (2018). Counting the dead to determine the source and transmission of the marine herpesvirus OsHV-1 in Crassostrea gigas. Vet. Res. 49, 34. doi: 10.1186/s13567-018-0529-7
Witkop E. M., Proestou D. A., Gomez-Chiarri M. (2022). The expanded inhibitor of apoptosis gene family in oysters possesses novel domain architectures and may play diverse roles in apoptosis following immune challenge. BMC Genomics 23, 201. doi: 10.1186/s12864-021-08233-6
Keywords: Malacoherpesviridae, Herpesvirales, OsHV-1, HaHV-1, metagenomic, herpesviruses, HK97-fold major capsid proteins
Citation: Rosani U, Gaia M, Delmont TO and Krupovic M (2023) Tracing the invertebrate herpesviruses in the global sequence datasets. Front. Mar. Sci. 10:1159754. doi: 10.3389/fmars.2023.1159754
Received: 06 February 2023; Accepted: 29 June 2023;
Published: 14 July 2023.
Edited by:
Eric Pante, Center for the National Scientific Research (CNRS), FranceReviewed by:
Germain Chevignon, Laboratoire de Génétique et Pathologie des Mollusques Marins (LGPMM), FranceLusheng Xin, Chinese Academy of Fishery Sciences (CAFS), China
Copyright © 2023 Rosani, Gaia, Delmont and Krupovic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Umberto Rosani, dW1iZXJ0by5yb3NhbmlAdW5pcGQuaXQ=