
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 05 April 2023
Sec. Marine Biotechnology and Bioproducts
Volume 10 - 2023 | https://doi.org/10.3389/fmars.2023.1140516
This article is part of the Research Topic Bioactive Terpenoids from Marine Organisms and Their Pharmacological Activities View all 5 articles
 Jiyoon Park1
Jiyoon Park1 Hang Su Cho1
Hang Su Cho1 Dong Hyun Moon1Donghoon Lee2Youngjoo Kal2
Dong Hyun Moon1Donghoon Lee2Youngjoo Kal2 Sangwon Cha2
Sangwon Cha2 Sang Kook Lee1
Sang Kook Lee1 Yeo Joon Yoon1*
Yeo Joon Yoon1* Dong-Chan Oh1*
Dong-Chan Oh1*The xiamycins are bioactive indolosesquiterpenoids that have been isolated from actinobacterial strains belonging to the Streptomyces genus. The overexpression of orf2011, which encodes the LuxR family regulator in a marine Streptomyces strain (HK18) isolated from a hypersaline saltern, significantly increased the production of xiamycin dimers, namely the previously reported dixiamycins A and C (3 and 4), compared to the wild-type strain. In addition, the engineered strain produced new members of the xiamycin family (lipoxiamycins A and B), which possessed a lipophilic chain linked to the indolosesquiterpenoid core structure by a N–O bond. The transcription analysis of the N-hydroxylase-encoding xiaH by semiquantitative reverse transcription polymerase chain reaction (RT-PCR) revealed that the transcription level of xiaH responsible for the formation of a nitroxyl radical was increased by the overexpression of orf2011, which is located outside the xiamycin biosynthetic gene cluster. The structures of these compounds were determined by full spectroscopic analysis, and the connectivity between the lipophilic chain and the indolosesquiterpenoid moiety was confirmed in both lipoxiamycins A and B (1 and 2) by MS/MS analysis. Moreover, the absolute configurations of these compounds were established using quantum mechanics-based electronic circular dichroism and DP4 calculations. Finally, it was demonstrated that lipoxiamycin A (1) displayed inhibitory activity against lipopolysaccharide-induced NO production at an IC50 of 9.89 ± 0.92 µM in RAW 264.7 cells.
Terpenoids are one of the most diverse structural classes of natural products, and as a result, these isoprene-derived natural products have been developed for use in drugs and cosmetics, following their isolation from plants, fungi, and invertebrates (Faulkner, 1984; Bergman et al., 2019; Jiang et al., 2020). For example, paclitaxel, which is isolated from the Pacific yew tree, targets tubulin and is approved as an anticancer drug against various cancers, including ovarian and breast carcinomas (Gotaskie and Andreassi, 1994). This diterpenoid is produced by endophytic fungi, thereby facilitating its biotechnological production from fungal cultures (Xiong et al., 2013). In addition, pseudopterosin A is a marine soft coral (Pseudoptergorgia elisabethae)-derived diterpene glycoside that possesses anti-inflammatory, analgesic, and neuroregulatory effects (Fenical, 1987; Mayer et al., 1998). It is one of the ingredients of a commercially developed anti-wrinkle cosmetic cream that is applied to the skin and is also in clinical trials for use as a wound-healing agent (Alves et al., 2020). Although terpenoids rarely originate from bacteria, increasing genomic information indicates that bacteria can indeed be a valuable source of bioactive terpenoids. For example, the early genomic analysis of Streptomyces coelicolor identified the biosynthetic gene cluster for the production of geosmin, a volatile sesquiterpenoid with an earthy odor (Daum et al., 2009). Indeed, the genomic screening of bacterial DNA libraries has enabled the prioritization of potential terpene producers and the identification of several Streptomyces strains that are capable of producing diterpenoids, such as platensimycin, platencin, viguiepinol, and the oxaloterpins, thereby highlighting the potential of bacteria to biosynthesize terpene metabolites (Motohashi et al., 2007; Smanski et al., 2009; Zhang et al., 2011; Bi and Yu, 2016). In a previous study, we discovered antiviral indolosesquiterpenoids, namely xiamycins C–E, in a marine Streptomyces strain (HK18) isolated from a hypersaline saltern (Kim et al., 2016).
Many previous studies have reported that the manipulation of regulatory elements is a useful strategy for the discovery of new compounds by the activation of silent biosynthetic gene clusters (BGCs) (Olano et al., 2014; Huang et al., 2016; Chen et al., 2017). For instance, overexpression of the LuxR family transcriptional regulator in S. albus led to a further enhancement in the production of 6-epi-alteramide A and a small amount of 6-epi-alteramide B compared to the wild-type strain, which produced only 6-epi-alteramide A (Olano et al., 2014). In addition, the known totopotensamides A and B and a new analog, totopotensamide C, have been produced by a triple mutant strain generated by the inactivation of two negative regulators and by the overexpression of the LuxR family regulator (Chen et al., 2017). Furthermore, violapyrone B has been produced by inactivation of the global regulatory gene wblA in S. somaliensis SCSIO ZH66 (Huang et al., 2016). Taken together, these results indicate that the manipulation of regulatory genes could serve as a powerful tool for the diversification of novel bioactive compounds.
In our efforts to discover novel natural products from Streptomyces sp. HK18, the orf2011 gene encoding the LuxR family regulator was overexpressed in the wild-type HK18 strain. Because the LuxR family resided within a thiopeptide BGC, we expected to activate the thiopeptide BGC primarily. In addition, as the regulatory network involved in secondary metabolism in Streptomyces is a complicated system with only-partially understood mechanisms, the overexpression of the LuxR family regulator could induce the expression of other BGCs secondarily. In our observation, this genetic manipulation did not activate the corresponding thiopeptide BGC, but instead led to a significant change in the production of xiamycin family members possibly by cross-regulation. The mutant strain produced new xiamycin analogs lipoxiamycins A and B (1 and 2), which were not previously reported, as well as dixiamycins A and C (3 and 4), to a greater extent than the wild-type HK18 strain. Here, we report the structural elucidation and biological activities of these lipoxiamycins.
Optical rotations were recorded using a JASCO P-2000 polarimeter with a 1 cm quartz cell at 20°C. Ultraviolet (UV) absorption and circular dichroism (CD) data were recorded on an Applied Photophysics Chirascan Plus Circular Dichroism Detector with a 1 mm quartz cell. Infrared (IR) spectra were obtained using a Thermo N1 COLET iS10 spectrometer. Low-resolution electrospray ionization-mass spectrometric (ESI-MS) data were obtained using an Agilent Technologies 6130 quadrupole mass spectrometer (Agilent, Santa Clara, CA, USA) connected to an Agilent analytical high performance liquid chromatograph (HPLC, 1200 series) using a C18(2) column (Phenomenex Luna, 100 × 4.6 mm). High resolution (HR-ESI-MS) data were recorded using an AB SCIEX Q-TOF 5600 high-resolution mass spectrometer (Framingham, MA, USA) at the National Instrumentation Center for Environmental Management (NICEM, Seoul, Republic of Korea). Nuclear magnetic resonance (NMR) spectra were acquired on a Bruker Avance 800 MHz spectrometer (Bruker, Billerica, MA, USA) at the College of Pharmacy, Seoul National University, and a Bruker Avance II 900 MHz NMR spectrometer at the KBSI (Korea Basic Science Institute at Ochang). For tandem mass spectrometric (MS/MS) analysis and ozonolysis experiments, a Thermo Finnigan LTQ XL linear ion trap mass spectrometer (Thermo Scientific Inc., San Jose, CA, USA) connected to a home-built ionization source, ESI or paper spray ionization (PSI), was utilized.
All strains and plasmids employed in the current study are listed in Table S10. Streptomyces sp. HK18 wild-type strain (Kim et al., 2016) and its mutant were propagated on ISP4 (Difco, Sparks, MD, USA). E. coli DH5α was used for plasmid propagation according to a standard protocol. Non-methylating E. coli ET12567/pUZ8002 (MacNeil et al., 1992) was used as the donor strain for conjugal transfer, and the conjugal transfer of a recombinant plasmid between Streptomyces and E. coli was conducted on Mannitol-Soy flour (MS) medium (Kieser et al., 2000). Isolation of the genomic DNA from the wild-type strain was performed according to the standard protocol using cultures grown in ISP4 medium (Kieser et al., 2000). The DNA fragments used to construct the overexpression plasmid were amplified using the polymerase chain reaction (PCR) of genomic DNA from the wild-type strain and GXL DNA polymerase (Takara, Kyoto, Japan); the corresponding primers are listed in Table S11. The integrative E. coli-Streptomyces shuttle vector pSET152 (Bierman et al., 1992) with a constitutive ermE* promoter (ermEp*) was used for the overexpression of orf2011, which encodes the LuxR family regulator in Streptomyces sp. HK18. To construct the orf2011 overexpression plasmid p2011, the orf2011 fragment from the genomic DNA of the wild-type strain was amplified using the 2011F and 2011R primers (Table S11). The PCR product of the gene was cloned into pSET152 digested with BamHI and XbaI by Gibson assembly using the HiFi DNA assembly master mix (New England Biolabs, Beverly, MA, USA), and the p2011 plasmid was sequenced. The plasmid was then introduced into the wild-type strain by conjugation with E. coli ET12567/pUZ8002 to yield the mutant strain HK18/p2011. The genotype was verified by PCR, and DNA sequencing was performed.
The wild-type strain Streptomyces sp. HK18 and its mutant strain HK18/p2011 strain were individually inoculated in YEME medium (50 mL; prepared from 4 g yeast extract, 10 g malt extract, and 6 g glucose in 1 L artificial seawater) in a 125 mL Erlenmeyer flask and cultivated for 3 d on a rotary shaker at 180 rpm and 30°C. The liquid culture (5 mL) was transferred into YPM + starch medium (250 mL; prepared from 6 g yeast extract, 4 g peptone, 10 g starch, and 4 g mannitol in 1 L artificial seawater) under the same conditions. After 3 d, an aliquot (20 mL) of the culture was transferred to YPM + starch medium (1 L) in a 2.5 L Ultra Yield flask and allowed to incubate for 5 d at 180 rpm and 30°C. The mutant strain HK18/p2011 strain was also cultivated under the same conditions. A 2 L culture of each strain was then extracted using ethyl acetate (EtOAc, 4 L). Subsequently, the extract was concentrated by rotary evaporation and dissolved in methanol (MeOH, 1 mL) for analysis by liquid chromatography–mass spectrometry (LC/MS) using gradient solvent conditions (10–100% aqueous CH3CN over 20 min, 0.1% formic acid, flow rate: 0.7 mL/min, C18(2) column: Phenomenex Luna, 100 × 4.6 mm). Compounds 1–4 eluted at 21.9, 22.5, 15.9, and 16.9 min, respectively. The production yields of 3 and 4 increased by 10 and 11 times respectively in the mutant compared to the wild-type.
The mutant Streptomyces sp. HK18/p2011 strain was cultivated in YEME liquid medium (50 mL; prepared from 4 g yeast extract, 10 g malt extract, and 4 g glucose in 1 L artificial seawater) in a 125 mL flask at 180 rpm at 30°C on a rotary shaker for 3 d. After this time, an aliquot (5 mL) of the culture was inoculated in YPM + starch medium (250 mL; prepared from 6 g yeast extract, 4 g peptone, 10 g starch, and 4 g mannitol in 1 L artificial seawater). Finally, an aliquot (20 mL) of the bacterial culture medium was transferred to YPM + starch medium (1 L) in a 2.5 L Ultra Yield flask and incubated for 5 d at 30°C and 180 rpm. The whole culture (210 L) was extracted with EtOAc (315 L) to obtain the desired extract (80 g).
The crude extract was adsorbed onto Celite and fractionated using C18 resin (25 g) by the elution of aqueous solutions of MeOH (200 mL each; 20, 40, 60, 80, and 100% MeOH in H2O). After fractionation, lipoxiamycins A and B (1 and 2) were detected in the 100% MeOH fraction, whereas dixiamycins A and C (3 and 4) were eluted in the 80% MeOH/H2O fraction. The 100% MeOH fraction was subjected to reversed-phase HPLC using a YMC column (250 × 10 mm, C18, 5 μm) under gradient solvent conditions (40–70% aqueous CH3CN over 30 min, 100% CH3CN for 20 min, 0.1% formic acid, flow rate: 2 mL/min, detection: UV 254 nm). Lipoxiamycins A and B (1 and 2) eluted at retention times of 45 and 47 min, respectively. These compounds were further purified using a different gradient solvent system (75–85% aqueous CH3CN over 30 min, 0.1% formic acid, flow rate: 2 mL/min, 250 × 10 mm, C18, 5 μm), and pure 1 and 2 eluted at 30 and 34 min. Additionally, to purify dixiamycins A and C (3 and 4), the 80% fraction was injected into a semi-preparative reversed-phase HPLC column (YMC C18: 250 × 10 mm, 5 μm) and eluted using a combination of gradient and isocratic elution (40–70% aqueous CH3CN over 30 min, and 100% CH3CN for 20 min, 0.1% formic acid, flow rate: 2 mL/min, detection: UV 254 nm) and 3 and 4 were isolated at 34 and 36 min.
Lipoxiamycin A (1): yellow, amorphous oil; +33 (c 0.1, MeOH); UV (MeOH) λmax (log ϵ) 239 (2.93), 299 (2.44), and 321 (2.17) nm; IR (neat) νmax 3356, 2954, 2870, 1684, 1461, 1379, 1242, and 1078 cm–1; CD (c 1.52 × 10-4 M, MeOH) (Δϵ) 223 (1.75), 254 (-8.87), 300 (0.46), 341 (3.03); 1H and 13C NMR spectral data, Table 1; HRESIMS m/z [M+H]+ 658.4090 (calcd for 658.4102).
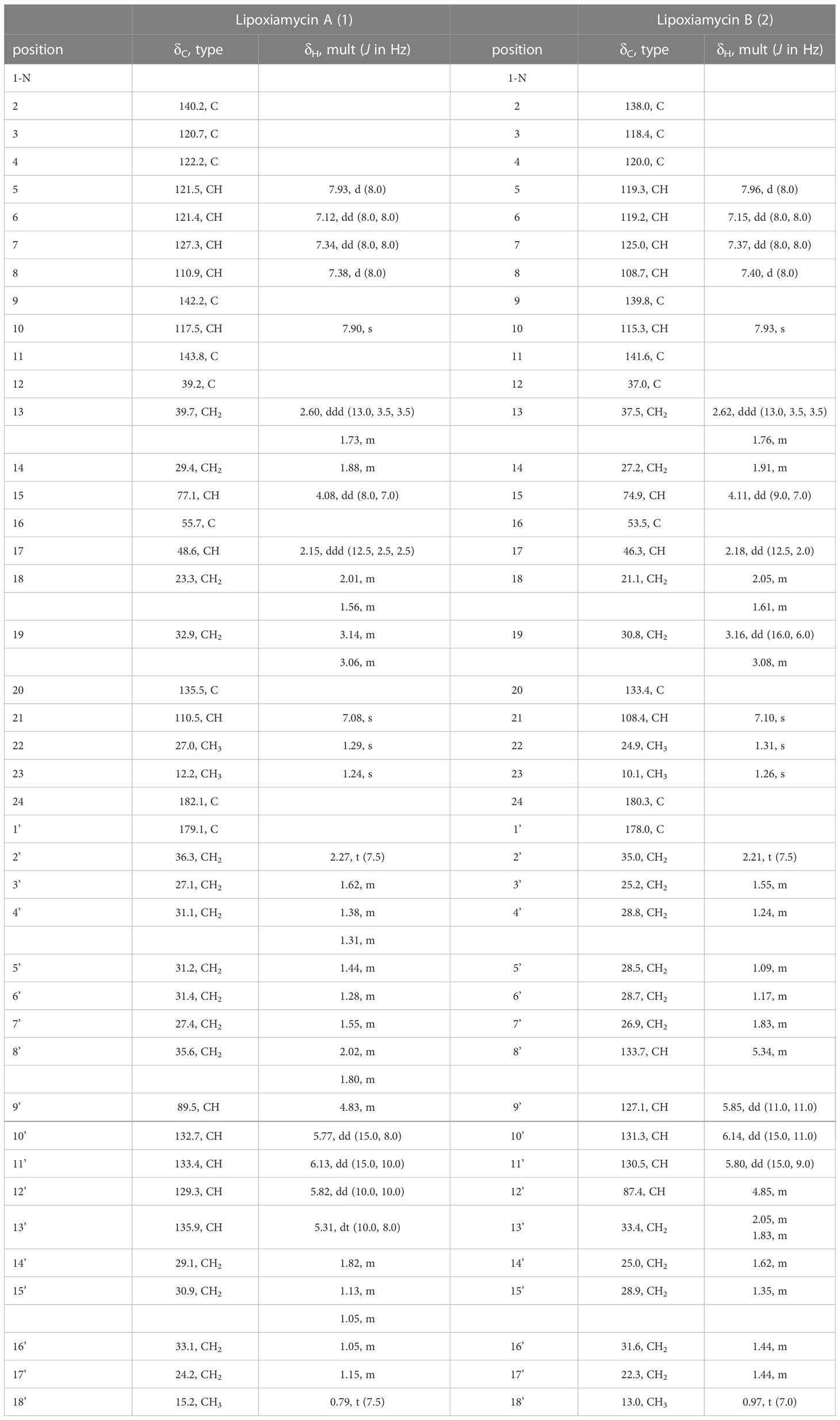
Table 1 1H and 13C NMR data for lipoxiamycins A and B (1 and 2) in CD3OD.
Lipoxiamycin B (2): yellow, amorphous oil; +66 (c 0.1, MeOH); UV (MeOH) λmax (log ϵ) 238 (2.93), 299 (2.44), and 322 (2.17) nm; CD (c 1.52 × 10-4 M, MeOH) (Δϵ) 225 (-0.45), 243 (9.78); IR (neat) νmax 3375, 2926, 2855, 1705, 1461, 1378, and 1235 cm–1; 1H and 13C NMR spectral data, Table 1; HRESIMS m/z [M+H]+ 658.4075 (calcd for 658.4102).
Dixiamycin A (3): yellow, amorphous oil; +29 (c 0.1, MeOH); UV (MeOH) λmax (log ϵ) 216 (2.51), 234 (2.76), 250 (2.39), 296 (2.36), 320 (1.59), and 332 (1.47) nm; CD (c 1.38 × 10-4 M, MeOH) (Δϵ) 215 (3.17), 237 (3.53), 288(2.39), 1H and 13C NMR spectral data, Figures S13–S14; HR-ESI-MS m/z [M+H]+ 725.3572 (calcd for C46H49N2O6 725.3585).
Dixiamycin C (4): yellow, amorphous oil; +130 (c 0.1, MeOH); UV (MeOH) λmax (log ϵ) 216 (2.51), 235 (2.75) 250 (2.39), 287 (2.18), 296 (2.36) nm; CD (c 1.38 × 10-4 M, MeOH) (Δϵ) 216 (2.74), 238 (3.11), 1H and 13C NMR spectral data, Figures S15–S16; HRESIMS m/z [M+H]+ 725.3574 (calcd for C46H49N2O6 725.3585).
For ozonolysis experiment, one microliter of lipoxiamycins A or B (1 or 2) solution (1 mg/mL) was spotted onto a triangular paper tip (Whatman filter paper grade 1 with a width of 5 mm and a height of 10 mm) and dried. Then, the sample-spotted paper tip was exposed to the ozone generating low pressure mercury (LP Hg) lamp (185 nm, Analamp®, BHK, Ontario, CA, USA) for 1 min. After ozonolysis, full mass and MS/MS spectra were recorded in the negative ion mode by PSI MS with MeOH as the spray solvent and a spray voltage of -4.5 kV.
The energy-minimized structures of lipoxiamycins A and B (1 and 2) were generated using the Avogadro 1.2.0 program. A conformational search was conducted using the MacroModel (Version 9.9, Schrödinger LLC, New York, NY, USA) program in Maestro (Version 9.9, Schrödinger LLC) with a mixed torsional/low mode sampling method. Conformers of the diastereomers with energies of < 10 kJ/mol were selected, as calculated using the MMFF force field. Their geometries were calculated by optimization at the B3LYP/6-31++ level of theory, and the shielding tensor values of the optimized conformers were based on equation (1), where is the calculated NMR chemical shift for the x nucleus, and is the shielding tensor for the proton and carbon nuclei in the gas phase. These values were averaged by the Boltzmann population and the associated Gibbs free energy and were used for DP4 analysis. It should be noted that DP4 analysis was facilitated by an Excel sheet provided by the original authors (Smith and Goodman, 2009; Smith and Goodman, 2010; Lodewyk et al., 2012).
The ground-state geometries were optimized by TmoleX 6.5 using the def-SVP set for all atoms at the B3LYP/DFT functional level (Pescitelli and Bruhn, 2016). The electronic circular dichroism (ECD) spectra were simulated by overlapping each transition, where σ is the width of the band at a 1/e height, ΔEi and Ri are the excitation and rotatory strengths of transition i, respectively, and the value of σ was fixed at 0.10 eV (2):
To confirm the transcriptional changes in orf2011 and xiaH, the total RNA was isolated from the wild-type and HK18/p2011 strains, respectively. The harvested cells were suspended in TRIzol reagent (1 mL; Invitrogen, Carlsbad, CA, USA) and incubated at room temperature for 5 min. The mixture was vortexed with chloroform (200 μL) and subjected to centrifugation at 13,000 × g for 15 min. The supernatant was then collected in a new tube and mixed with one volume of 70% ethanol. The mixture was transferred to an RNeasy Mini spin column (Qiagen, Hilden, Germany) and centrifuged at 8,000 × g for 15 s at room temperature. Subsequently, the spin column was washed with RPE washing buffer (Qiagen, Hilden, Germany), and the resulting total RNA was dissolved in RNase-free water (50 μL). The nucleic acid preparations were treated with DNase I (New England Biolabs, Ipswich, MA, USA), as recommended by the manufacturer. The RT-PCR conditions employed for analysis were as follows: cDNA synthesis, 50°C for 30 min, 95°C for 15 min, followed by 33 cycles of 94°C for 1 min, 55°C for 1 min, and 72°C for 45 s. The cycle number was optimized for each gene to obtain sufficient visibility of the RT-PCR bands, to ensure that amplification was within the linear range, and to obtain semi-quantitative results. Primers (Tm = 50–60°C; 20mer) were designed using the Primer3 software (Rozen and Skaletsky, 2000) (Table S11). With each set of primers, negative controls were carried out using Taq DNA polymerase (New England Biolabs, Ipswich, MA, USA) in the absence of the RT reaction to confirm that the amplified products were not derived from chromosomal DNA. 16S rRNA was used as the positive control due to its known expression in this system. The RT-PCR experiments were performed in triplicate using RNA samples obtained from three independent cultures.
The mouse macrophage RAW 264.7 cell line was obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). The cells were cultured in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1% antibiotics-antimycotics (0.25 mg/mL amphotericin B), and were incubated in a 5% CO2 atmosphere at 37°C. The amount of NO produced by the RAW 264.7 cells in the culture medium was determined using the Griess reagent (0.1% N-(1-naphthyl)ethylenediamine dihydrochloride and 1% sulfanilamide in 5% phosphoric acid). The RAW 264.7 cells were seeded in 24-well plates at a density of 2 × 105 cells/mL and incubated at 37°C in a 5% CO2 atmosphere for 24 h. After this time, the cells were washed with phosphate-buffered saline (PBS), replaced with fresh medium containing 1% FBS, and pre-treated with either the vehicle or with the samples prepared at various concentrations (i.e., 1.25, 2.5, 5, 10, and 20 μM). Following incubation for 1 h at 37°C, the cells were stimulated with lipopolysaccharide (LPS; 1 μg/mL) for 20 h. The control group was subjected to the same conditions but in the absence of LPS. After transferring an aliquot (100 μL) of the supernatant from each well into a 96-well plate, the Griess reagent (180 μL) was added. The absorbance of each plate was measured at 540 nm using a microplate reader. The NO concentration was calculated using a sodium nitrite standard. The cell viability was determined using the colorimetric MTT assay. More specifically, following the NO assessment, the MTT solution was added to each well to give a final concentration of 500 μg/mL. After incubation at 37°C for 4 h, the medium was discarded, and the remaining formazan blue salt was dissolved in dimethyl sulfoxide (1 mL, DMSO). An aliquot (100 μL) of each supernatant was then transferred from each well to a 96-well plate, and the absorbance was measured at 570 nm using a microplate reader. The percentage of cell survival (%) was calculated relative to the LPS-treated group.
Cultivation of the mutant HK18/p2011 strain and subsequent LC/MS-based chemical analysis of the extract identified two new peaks (protonated adducts at m/z 658) with UV spectra (λmax at 239, 299, and 321 nm) similar to those of the xiamycins obtained from the wild-type culture. The UV absorptions at 299 and 321 nm were assumed to originate from the carbazole moiety commonly found in xiamycins, and so the UV peak at 239 nm indicated the presence of structural variations in these compounds, such as additional conjugated double bonds. Based on the MS and UV data, these two compounds, named lipoxiamycins A and B, were considered to be previously unidentified metabolites, and so were subjected to further spectroscopic analysis. Detailed chromatographic analysis confirmed the production of the new derivatives lipoxiamycins A (1) and B (2) in the mutant strain, and the contents of compounds 3 and 4 were significantly increased in the mutant culture extract compared to the wild-type culture. All four of the compounds were spectroscopically characterized, and their corresponding structures are shown in Figure 1.
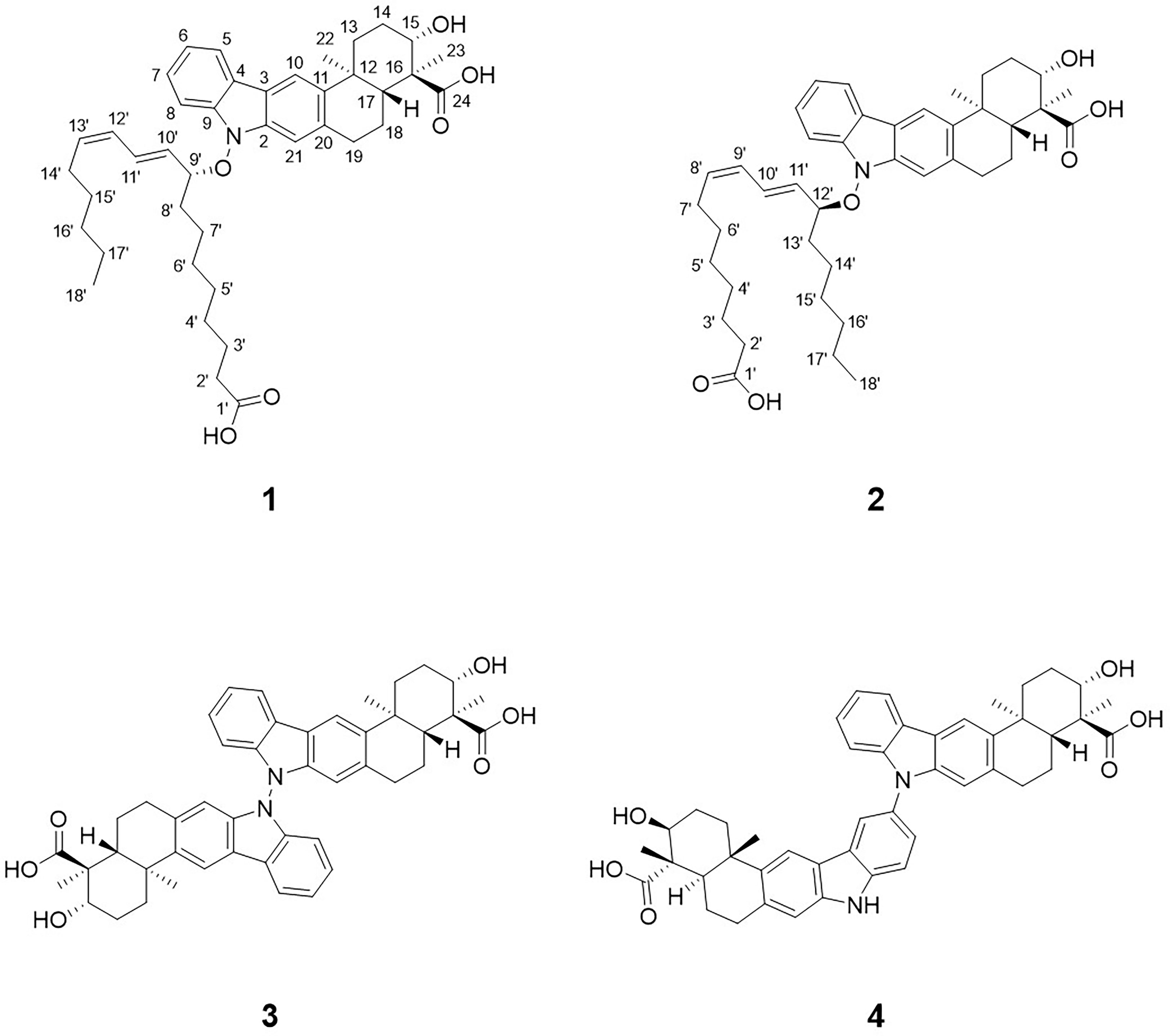
Figure 1 Structures of lipoxiamycin A (1), lipoxiamycin B (2), dixiamycin A (3), and dixiamycin C (4).
Lipoxiamycin A (1) was purified as a yellow, amorphous oil, and its molecular formula was identified as C41H55NO6 using a combination of HR-ESI-MS, 1H NMR, and 13C NMR data (Table 1 and Figure 2). Since the UV spectrum of 1 was virtually identical to that of xiamycin A, the major compound present in the extract, namely lipoxiamycin A (1), was considered to be a new member of the xiamycin family. As expected, its 1H NMR spectrum contained signals originating from the structure of xiamycin A, although additional resonances were also observed. To expedite structural elucidation, the structural motif of xiamycin was identified in lipoxiamycin A (1). The 1H NMR spectrum of 1 contained four hydrogen signals (δH 7.93, 7.38, 7.34, and 7.12) with coupling constants of 8.0 Hz, in addition to two singlet proton signals (δH 7.90 and 7.08); these signals were attributed to the two aromatic ring systems present in the xiamycin motif. In addition, one distinct carbinol proton signal (δH 4.08) and two singlet methyl signals (δH 1.29 and 1.24) were observed, as in the case of xiamycin A. Analysis of the 13C and HSQC NMR spectra revealed that one carbonyl carbon (δC 182.1), 12 aromatic carbon atoms (δC 142.2–110.5), one oxymethine carbon (δC 77.1), four aliphatic methylene carbon atoms (δC 39.7, 32.9, 29.4, and 23.3), one aliphatic carbon corresponding to the methine group (δC 48.6), two quaternary carbon atoms (δC 55.7 and 39.2), and two aliphatic methyl groups (δC 27.0 and 12.2) composed the indolosesquiterpenoid structure of xiamycin A.
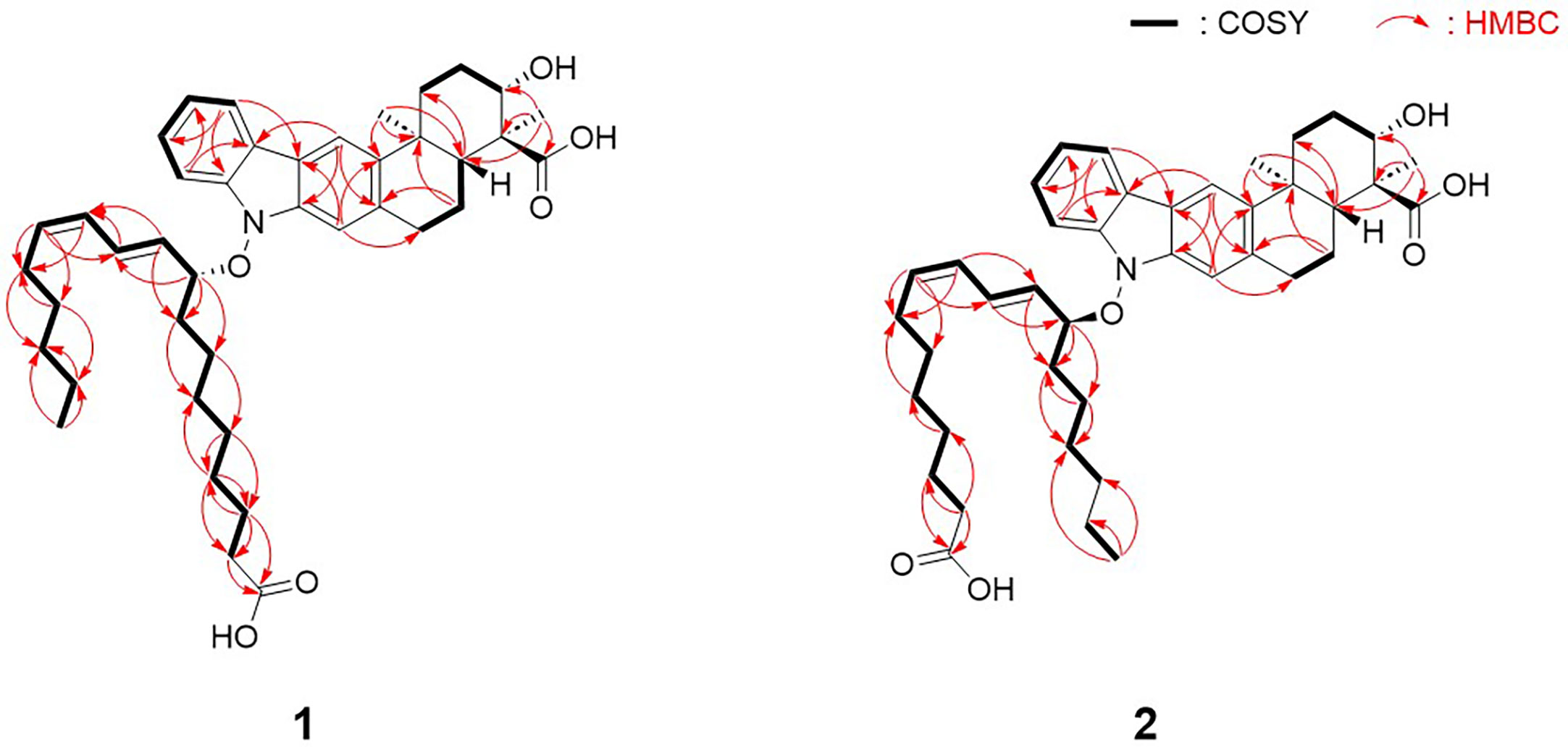
Figure 2 Key COSY and HMBC correlations of lipoxiamycins A (1) and B (2).
Analysis of the one-dimensional (1D) and two-dimensional (2D) NMR spectra and a comparison of the NMR data with those of xiamycin A enabled the assignment of the indolosesquiterpenoid core of compound 1. The four downfield proton signals [H-5–H-8 (δH 7.93, 7.38, 7.34, and 7.12)] of the first aromatic ring were assigned based on their COSY correlations and coupling constants (8.0 Hz). In addition, the HMBC correlations between H-6/H-8 (δH 7.12 and 7.38) and C-4 (δC 122.2) and those between H-5/H-7 (δH 7.93 and 7.34) and C-9 (δC 142.2) constructed the six-membered aromatic ring. The second aromatic ring system was elucidated by analysis of the HMBC correlations between H-10 (δH 7.90) and C-2/C-20 (δC 140.2 and 135.5), and between H-21 (δH 7.08) and C-3/C-11 (δC 120.7 and 143.8). Furthermore, the 3-bond HMBC correlations between H-5 (δH 7.93) and C-3 (δC 120.7) and between H-10 (δH 7.90) and C-4 (δC 122.2) indicated the connection of these two aromatic rings. Moreover, the COSY correlations from H2-13 (δH 2.60 and 1.73) to H-15 (δH 4.08) and from H-17 (δH 2.15) to H2-19 (δH 3.14 and 3.06) represent two spin systems. These two partial structures were connected based on the HMBC correlations between H3-22 (δH 1.29) and C-11/C-13/C-17 (δC 143.8, 39.7, and 48.6) and between H3-23 (δH 1.24) and C-15/C-16/C-17 (δC 77.1, 55.7, and 48.6). The correlation between H-15 (δH 4.08) and C-15 (δC 77.1) indicated the presence of a hydroxy group, while the presence of a carboxylic group at the C-16 position (δC 55.7) was supported by the 3JCH HMBC correlation between the methyl group (H3-23, δH 1.24) and C-24 (δC 182.1) in the downfield region. HMBC correlations between H2-18 (δH 2.01 and 1.56) and C-20 (δC 135.5), and between H2-19 (δH 3.14 and 3.06) and C-21 (δC 110.5) confirmed the C-19–C-20 connectivity. Analogous to xiamycin A, a carbazole moiety was assembled by connecting the two aromatic rings through a nitrogen atom, thereby completing the indolosesquiterpenoid motif of the xiamycin family.
Due to the fact that lipoxiamycin A (1) was revealed to contain an indolosesquiterpenoid structure as a new xiamycin member, the unassigned 1H and 13C peaks were further analyzed in detail. In addition to the above-described signals, the 1H NMR spectrum contained four olefinic proton signals (δH 6.13, 5.82, 5.77, and 5.31), 22 aliphatic proton signals (δH 2.27–1.05), one oxymethine proton signal (δH 4.83), and one methyl group signal (δH 0.79). Consistently, one carbonyl carbon (δC 179.1), four olefinic carbon atoms (δC 135.9, 133.4, 132.7, and 129.3), one oxymethine carbon (δC 89.5), 11 aliphatic methylene groups (δC 36.3–24.2), and one methyl group (δC 15.2) were observed in the 13C and HSQC NMR spectra. Analysis of the COSY correlations allowed the assignment of another partial structure as a linear lipophilic chain. In particular, an array of COSY correlations from H2-2’ (δH 2.27) to H3-18’ (δH 0.79) and HMBC correlations between H-2’/H-3’ (δH 2.27 and 1.62) and C-1’ (δC 179.1) indicated that this chain was 9-hydroxyoctadeca-10-12-dienoic acid.
The fatty acid possesses a C5 aliphatic chain connected to the conjugated diene. This part was confirmed by detailed analysis of 2D NMR data. The connectivity of C-18’ (δC 15.2)–C-17’ (δC 24.2)–C-16’ (δC 33.1) in the terminus of the chain was assigned by H3-18’ (δH 0.79)/H2-17’ (δH 1.15) and H2-17’/H2-16’ (δH 1.05) COSY correlations along with H3-18’/C-17’ and H3-18’/C-16’ HMBC couplings. The double bond proton at H-13’ (δH 5.31) correlated with H2-14’ (δH 1.82) in the COSY spectrum, assigning C-14’ (δC 29.1) adjacent to C-13’ (δC 135.9). C-14’ (δC 29.1) was connected to C-15’ (δC 30.9) by H2-14’/H2-15’ (δH 1.13 and 1.05) COSY correlations. This connectivity was clearly supported by HMBC correlations from H-13’ to C-15’. Even though H2-15’ and H2-16’ are overlapped in the 1H NMR spectrum, H2-14’/C-16’ HMBC correlations enabled to extend the chain from C-15’ to C-16’, finally confirming that the C5 aliphatic chain from the double bond (Figure 2).
To reveal the double bond position in the lipophilic chain unequivocally, ozonolysis of lipoxiamycin A (1) was performed with an ozone generating LP Hg lamp, followed by PSI MS and MS/MS analyses (Thomas et al., 2006; Stinson et al., 2018). As a result of ozonolysis of lipoxiamycin A (1), ions corresponding to the ozonolysis product (m/z 588) at carbon 12’ as well as the molozonide intermediate (m/z 705) were observed in the full MS spectrum, along with the ion corresponding to [M – H]– of lipoxiamycin A (1) at m/z 657 (Figure S33A). In the MS/MS analysis of the molozonide intermediate (m/z 705), the fragment ions corresponding to the possible ozonolysis products (m/zs 560, 588, and 604) from the proposed structure of lipoxiamycin A (1) were clearly observed (Figure S34A). Other major fragment ions in the region of m/z 200-400 were mainly originated from the cleavage of the N–O bond (Figure 3).
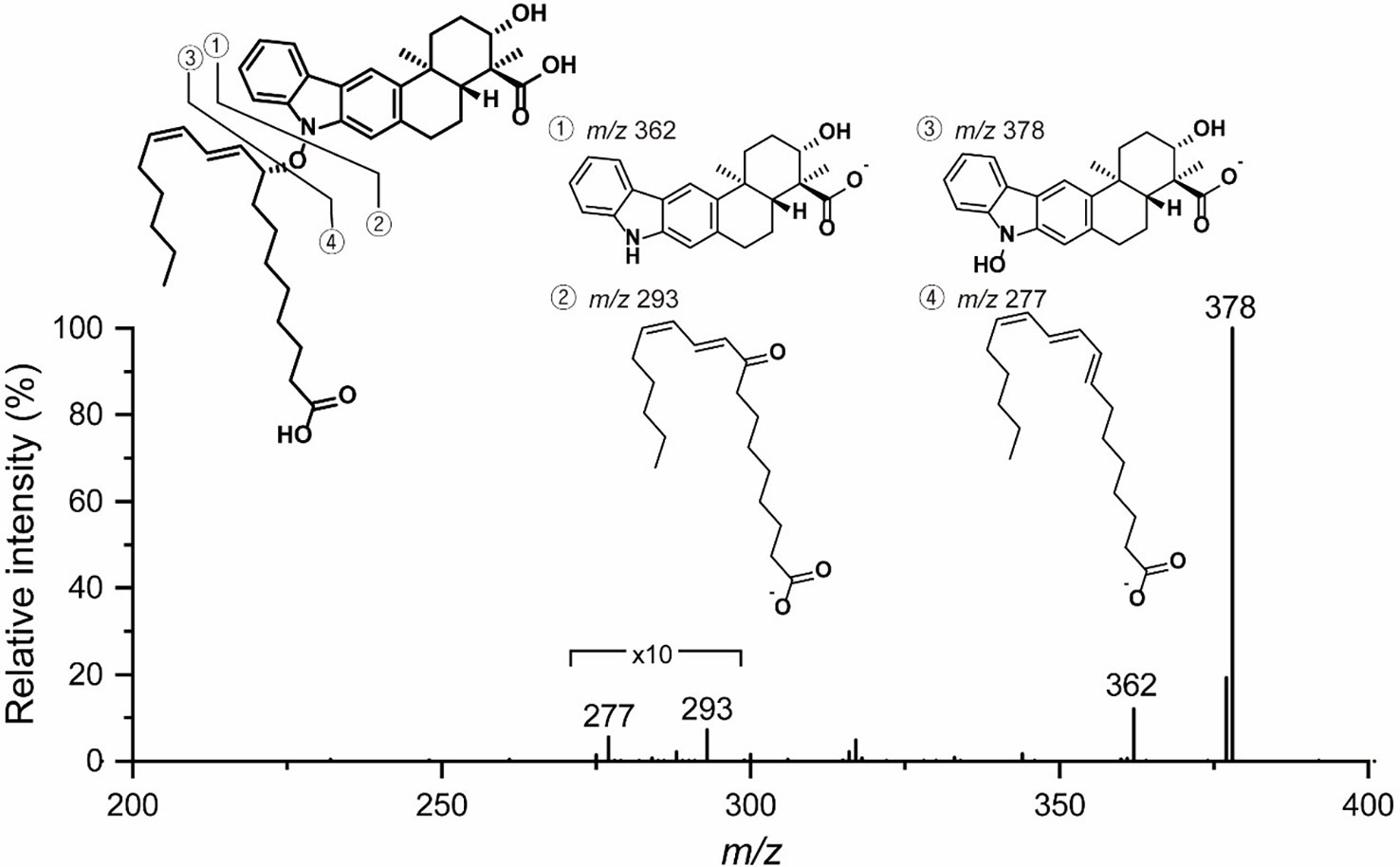
Figure 3 MS/MS analysis of lipoxiamycin A (1).
The configurations of the conjugated double bond were determined to be 10’E, and 12’Z based on the 15 Hz 1H–1H coupling constant between H-10’ (δH 5.77) and H-11’ (δH 6.13) and the 10 Hz coupling constant between H-12’ (δH 5.82) and H-13’ (δH 5.31). This geometry was also supported by H-10’ (δH 5.77)/H-12’, H-12’ (δH 5.82)/H-13’ (δH 5.31), H-13’/H-14’ (δH 1.82), and H-11’ (δH 6.13)/H-8’b (δH 1.80) ROESY correlations. Subsequently, the 9-hydroxyoctadeca-10E-12Z-dienoic acid moiety was connected to the carbazole nitrogen atom of the indolosesquiterpenoid core through a N–O bond based on the previously determined molecular formula and chemical shifts of the C-9’ (δC 89.5) and H-9’ (δH 4.83) atoms.
The presence of N–O bonds in the lipoxiamycins were unequivocally confirmed by the appearance of two sets of characteristic fragment ions in the MS/MS analysis. Particularly, as illustrated in Figure 3, the MS/MS analysis of lipoxiamycin A (1) in the negative ion mode produced two pairs of fragment ions. Upon cleavage of the N–O bond, two fragment ions were obtained, i.e., at m/zs 362 and 293. In contrast, two other fragment ions were produced at m/zs 378 and 277 when the C–O bond was cleaved during a collision-induced dissociation. Importantly, MS/MS analysis of lipoxiamycin B (2) showed almost identical results. In addition, H-8 (δH 7.38)/H-9’ (δH 4.83), H-8/H-11’ (δH 6.13), and H-21 (δH 7.08)/H-9’ (δH 4.83) ROESY correlations supported this N–O coupling (Figure 4A).
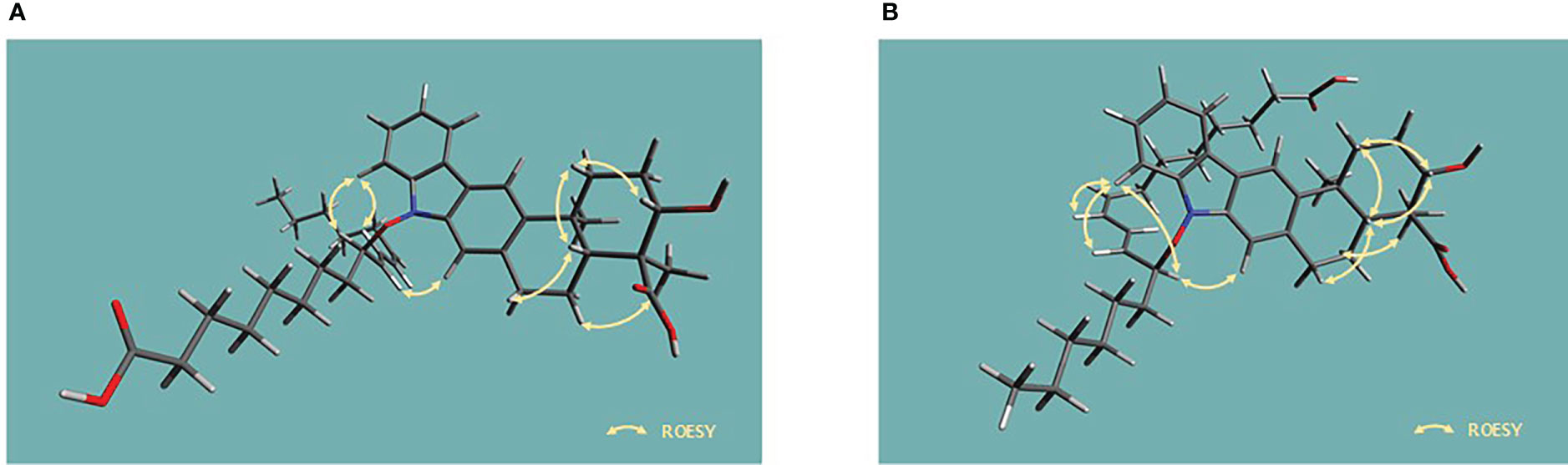
Figure 4 Energy-minimized structures for the key ROESY correlations present in (A) lipoxiamycin A (1) and (B) lipoxiamycin B (2).
Lipoxiamycin B (2) was obtained as a yellow, amorphous oil, and its HR-ESI mass data showed that 1 and 2 have the same molecular formula, i.e., C41H55NO6. Overall, the 1H, 13C, and HSQC NMR data for 2 showed spectroscopic features similar to those of 1. A detailed comparison of the NMR data revealed that both compounds bear the same indolosesquiterpenoid core, although a structural difference was found in their lipophilic chains. More specifically, the chain of 2 was determined to be 12-hydroxyoctadeca-8,10-dienoic acid by the observation of consecutive COSY correlations between H-2’ (δH 2.21) and H-18’ (δH 0.97), in addition to HMBC correlations between H-2’/H-3’ (δH 2.21 and 1.55) and carbonyl carbon C-1’ (δC 178.0). This fatty acid has a C6 aliphatic chain from the oxygenated carbon C-12’ (δC 87.4), which is different from that of 1 bearing a C5 chain from the double bond carbon. Because of the overlapped 1H and 13C signals, both COSY and HMBC correlations were complementarily utilized to confirm the structure of the fatty acid chain in 2. HMBC correlations from the terminal methyl group protons (H3-18’) to C-17’ and C-16’. Furthermore, C-15’ was connected to C-16’ based on the 4-bond 1H-13C correlations from H3-18 to C-16’. H-12’/H-13’ COSY correlation connected C-12’ and C-13’. This connectivity was further supported by the HMBC correlations from H-12’ to C-13’. H-12’/C-14’ heteronuclear coupling assembled C-14’ next to C-13’. One of the protons (1.83 ppm) at C-13’ displayed an HMBC correlation to C-15’, thus completing the C6 aliphatic chain from the oxygenated carbon C-12’ (Figure 2).
The double bond positions in 12-hydroxyoctadeca-8,10-dienoic acid were further analyzed by MS/MS analysis. After ozonolysis of lipoxiamycin B (2), the molozonide intermediate (m/z 705) was observed, but no distinctive ozonolysis product was found in the full mass spectrum (Figure S34B). MS/MS analysis of the molozonide intermediate (m/z 705) of lipoxiamycin B (2) (Figure S35B) produced a very different fragmentation pattern in the region of m/z 520 – 620 compared to that of the molozonide intermediate of lipoxiamycin A (1) (Figure S35A). The major fragment ion observed from the molozonide intermediate of lipoxiamycin B (2) was the ion at m/z 532, corresponding to the alcohol form of the ozonolysis product. Given that the ozonolysis of lipoxiamycin B (2) resulted in the loss of a carboxyl group in the lipophilic chain, it is reasonable to observe fewer ions from lipoxiamycin B (2) compared to lipoxiamycin A (1) in the negative ion mode. Ozonolysis experiments of lipoxiamycins A and B (1 and 2) clearly suggest that their double bond positions in the lipophilic chain are distinct from each other and support the proposed locations of the double bond in each structure.
The geometry of the C-8’–C-9’ double bond was determined to be 8’Z based on the H-8’–H-9’ coupling constant (11.0 Hz). In addition, the large coupling constant of the H-10’–H-11’ double bond (15.0 Hz) indicated that this double bond adopted the 10’Z geometry. Analogous to 1, the chain and the xiamycin core were coupled via a N–O bond, as determined from the molecular formula and the presence of ROESY correlations between H-8 (δH 7.40) and H-11’ (δH 5.80), H-8 (δH 7.40) and H-12’ (δH 4.85), and H-21 (δH 7.10) and H-12’ (δH 4.85), as shown in Figure 4B.
Since lipoxiamycins A and B (1 and 2) are produced via the same biosynthetic pathway in xiamycin-producing Streptomyces sp. HK18 cells, as previously reported, it was reasonable to expect that the indolosesquiterpenoid skeleton would have the same absolute configuration. Indeed, this was also supported by the ROESY correlations commonly observed for these lipoxiamycins and for xiamycin A, as presented in Figure 4A. In particular, the presence of ROESY correlations between H-15 (δH 4.08) and H-13b (δH 1.73), H-17 (δH 2.15) and H-13b (δH 1.73), and H-17 (δH 2.15) and H-19a (δH 3.14) indicated that these protons were assigned to axial positions on the same side of the structure, whereas the ROESY couplings between H3-23 (δH 1.24) and H-22/H-18a (δH 1.29 and 2.01) revealed that these protons were on the opposite side. Furthermore, the stereogenic centers at the C-9’ (δC 89.5) position of 1 and the C-12’ (δC 129.3) position of 2 were configurationally analyzed using quantum mechanics-based DP4 calculations to give the 9’R configuration in 1 and the 12’S configuration in 2, with probabilities of 100.0 and 99.9%, respectively (Tables S2 and S3). These configurations were further supported by a comparison of the experimental and calculated CD spectra, which showed opposite Cotton effects at ~250 nm (Figure 5).
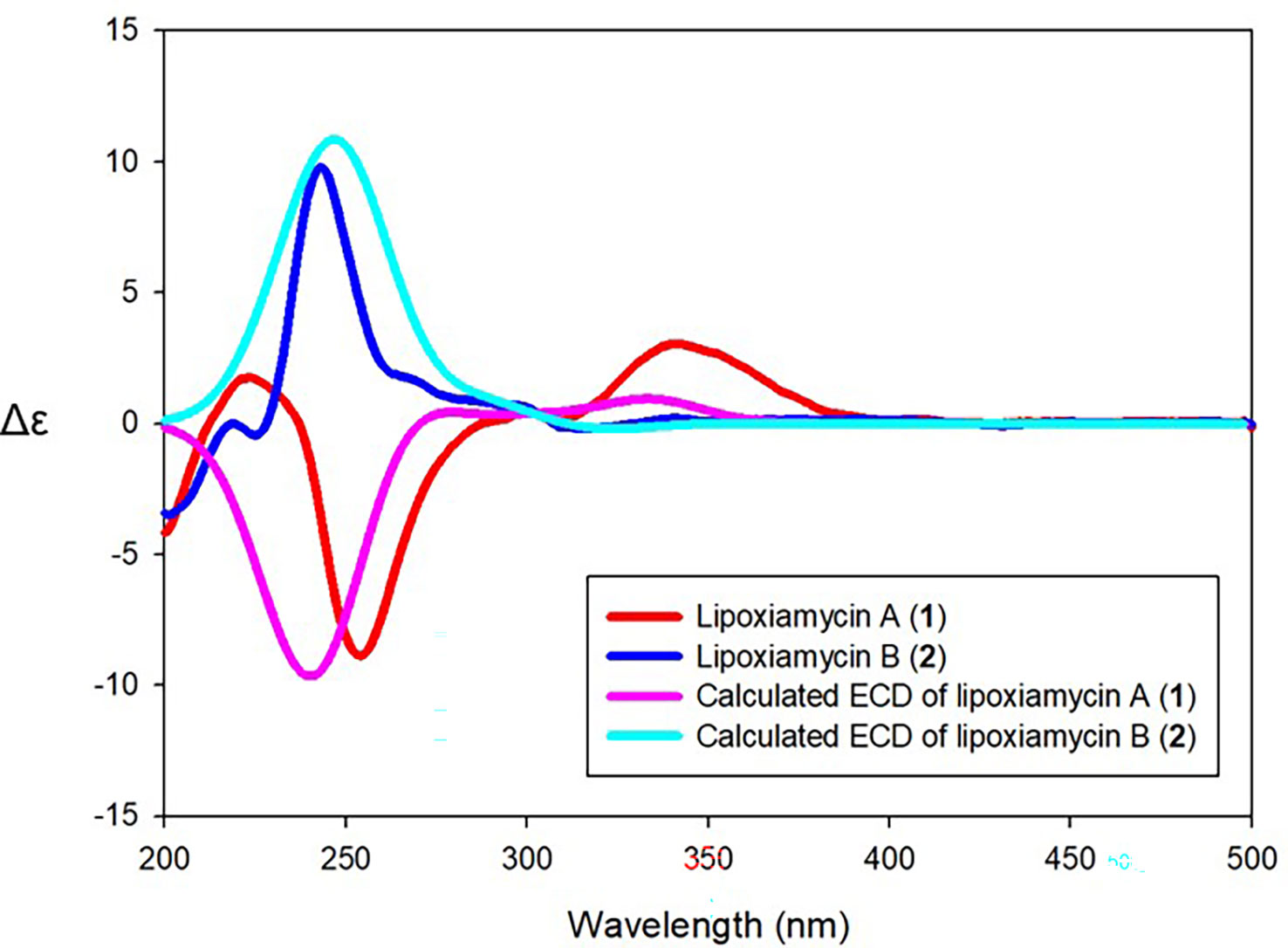
Figure 5 Comparison of the experimental and calculated ECD spectra of compounds 1 and 2.
Moreover, HR-ESI-MS analysis confirmed that compounds 3 and 4 possessed the same molecular formula (i.e., C46H48N2O6). Due to the fact that xiamycin A possesses the molecular formula C23H25NO3, compounds 3 and 4 were considered to be dimers of xiamycin A. It was found that the 1H and 13C NMR data of 3 were comparable to those of dixiamycins A and B, thereby indicating that compound 3 is a symmetrical dimer of xiamycin A (Ding et al, 2010). Dixiamycins A and B have the same planar structure but are atropdiastereomers formed by three-dimensional rotational disturbances at the N–N bond. Thus, upon comparison of the CD spectrum of 3 with those of dixiamycins A and B, it was possible to identify compound 3 as dixiamycin A, which possesses the aS configuration (Baunach et al., 2013). A detailed comparison of the 1H and 13C NMR spectra of 4 with previously reported xiamycin A dimers identified 4 as dixiamycin C, which is an asymmetrical dimer (Meng et al., 2015).
Previous studies have suggested that the dimerization of xiamycin occurs via radical N–C and N–N aryl–aryl coupling reactions promoted by XiaH, which is a flavin-dependent aromatic hydroxylase, and is essential for the conversion of the indolosesquiterpene xiamycin into oxiamycin and dixiamycins (Ding et al., 2010; Baunach et al., 2013; Teufel et al., 2016). Indeed, in a feeding experiment, it was not possible to detect oxiamycin and the dixiamycins in the XiaH deletion strain, whereas all other mutants produced xiamycin analogs (Baunach et al., 2013). Another previous report suggested that XiaK, a homolog of XiaH in S. pactum SCSIO 02999, is an FAD-dependent N-hydroxylase that performs diverse chemical couplings (Zhang et al., 2017). A feeding experiment employing E. coli BL21(DE3) that expressed XiaK from S. pactum SCSIO 02999 revealed that this enzyme catalyzes the N-hydroxylation of xiamycin A (Zhang et al., 2017). Additionally, the XiaK-overexpressing mutant S. coelicolor YF11 successfully converted Xiamycin A to chloroxiamycin in a study by Zhang et al. (2017). Taken together, although direct evidence for the catalytic mechanism of XiaH has not been previously presented, the above studies indicate that this enzyme catalyzes both aryl couplings and ether formation at the N1 position of xiamycin A.
To validate the role of XiaH in the production of xiamycin analogs in this study, transcription analysis of XiaH and orf2011 was conducted by semiquantitative RT-PCR of the wild-type and HK18/p2011 strains, respectively (Figure 6). Transcription analysis revealed that the expression level of the xiaH gene, which is essential for the formation of a nitroxyl radical, was increased by the overexpression of orf2011 (Figure 6), which was located outside of the xiamycin BGC. Secondary metabolite BGCs are generally regulated by pathway-specific transcription factors located within the target BGC (Chen et al., 2010). However, the cluster-situated regulator FscRI located in the candicidin BGC also binds to the promoter of the antimycin BGC in S. albus S4 (McLean et al., 2016), which represents the first report of the cross-regulation of individual BGCs. In addition, it is known that some transcription factors that control secondary metabolism are located outside the secondary metabolite BGCs. For example, a previous study demonstrated that CagR of the two-component CagRS system in S. clavuligerus F613-1 possessed 162 target sites that are involved in the biosynthesis and primary metabolism of clavulanic acid (Fu et al., 2019). Another study reported that the nucleoid-associated protein Lsr2 in S. venezuelae ATCC 10712 broadly repressed the transcription of several secondary metabolite BGCs (Gehrke et al., 2019), thereby demonstrating that controlling the expression of Lsr2 is a promising engineering strategy for the activation of silent secondary metabolite BGCs. Similarly, it is plausible that xiaH is regulated by the overexpression of orf2011 outside of the xiamycin BGC. However, the regulatory network involved in the production of secondary metabolites in Streptomyces is a complicated system and the detailed regulatory mechanism of orf2011 is currently unclear. Further studies are required to elucidate the complex regulatory cascade linking orf2011 to xiaH.
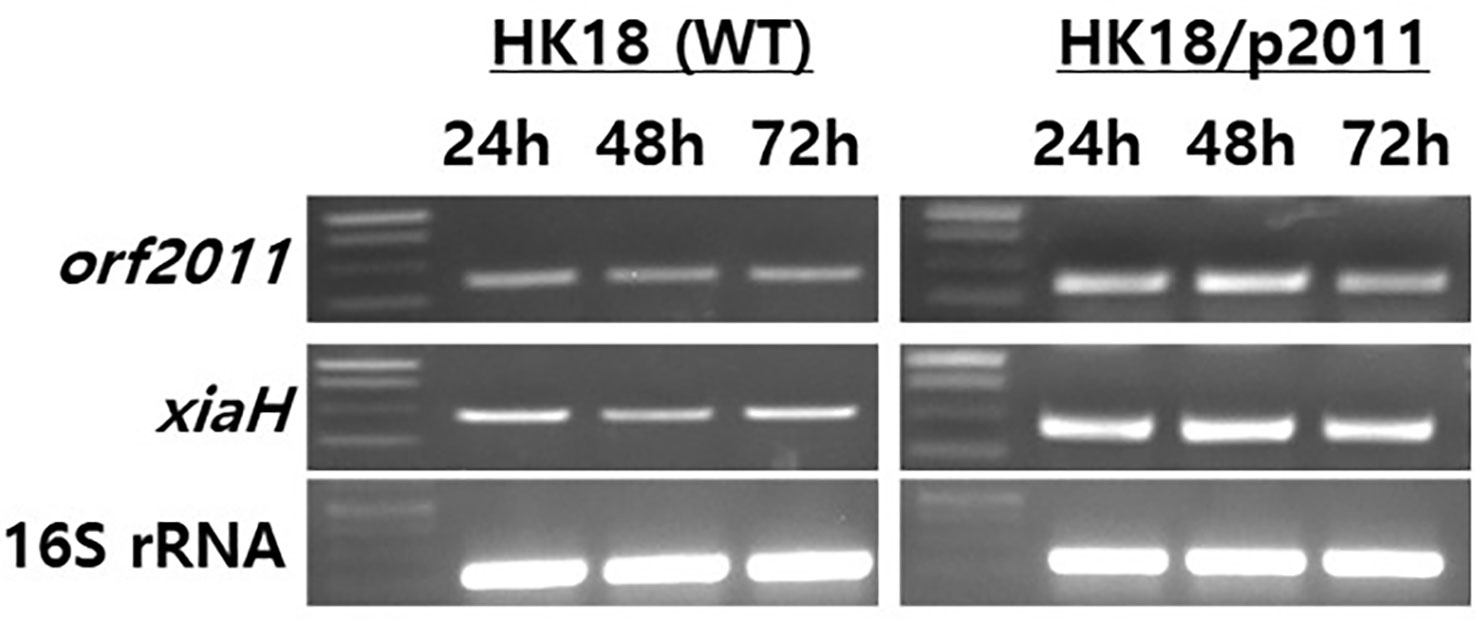
Figure 6 Gene expression analysis of orf2011 and xiaH by RT-PCR. The total RNAs were isolated from cultures of HK18 and HK18/p2011 at the times indicated. The 16S rRNA gene was used as a control.
Under normal conditions, nitric oxide (NO) plays an important role in the physiological and pathological functions of many organs (Hobbs et al., 1999). However, the overproduction of NO results in the pathogenesis of inflammatory disorders and the development of various diseases (Bogdan, 2001). Thus, inhibiting the overproduction of NO via inhibition of the expression and/or activity of the inducible isoform of nitric oxide synthase (iNOS) is an attractive therapeutic target. Thus, the inhibitory activities of 1–4 toward the production of NO determined using the iNOS assay with RAW 264.7 cells, using AMT (2-amino-5,6-dihydro-6-methyl-4H-1,3-thiazine hydrochloride, 200 nM) as a positive control. It was found that lipoxiamycin A (1) displayed a moderate inhibitory activity against LPS-induced NO production (IC50 = 9.89 ± 0.92 µM; Figure 7), whereas dixiamycin A (3) significantly inhibited NO production (IC50 = 4.12 ± 0.22 µM; Figure 8). Based on the MTT assay, it was therefore apparent that both dixiamycin A and lipoxiamycin A exhibited mild cytotoxicities at an average cell viability percentage of 75–83% at the tested concentrations. Lipoxiamycin B (2) and dixiamycin C (4) displayed weak activity in this assay (Figures S32 and S33)
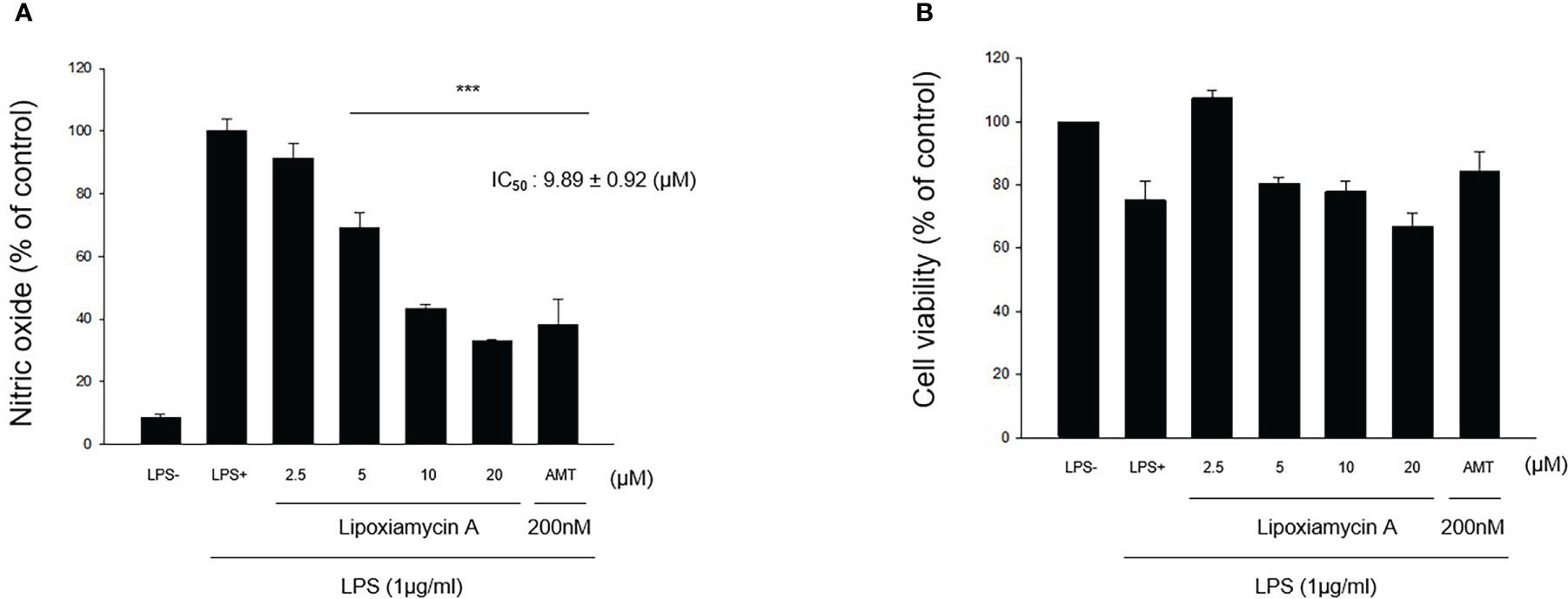
Figure 7 Effect of lipoxiamycin A (1) on LPS-stimulated NO production in macrophage cells. (A) RAW 264.7 cells were pre-treated with lipoxiamycin A (2.5, 5, 10, and 20 µM) for 1 h and then stimulated with LPS (1 µg/mL) for 20 (h) The supernatant from each well was collected to determine the degree of NO production using the Griess reaction. (B) The cell viability was determined using the MTT assay. Data are expressed as mean ± SD (N=3) based on data recorded from three independent experiments. ***p<.001 vs. the LPS-treated control group.
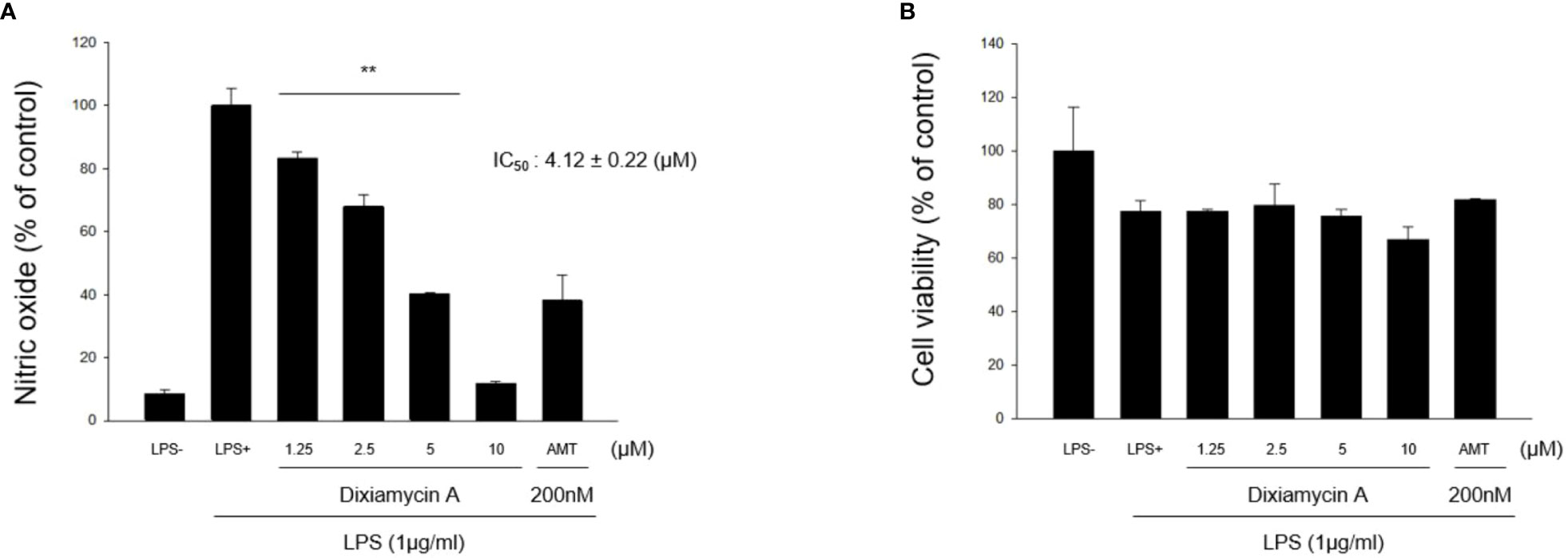
Figure 8 Effect of dixiamycin A (3) on LPS-stimulated NO production in macrophage cells. (A) RAW 264.7 cells were pre-treated with dixiamycin A (1.25, 2.5, 5, and 10 µM) for 1 h and then stimulated with LPS (1 µg/mL) for 20 (h) The supernatant from each well was collected to determine the degree of NO production using the Griess reaction. (B) The cell viability was determined using the MTT assay. Data are expressed as mean ± SD (N=3) based on data recorded from three independent experiments. **p<.01 vs. the LPS-treated control group.
Genetic engineering of the marine Streptomyces sp. HK18 and overexpression of the orf2011 gene that encodes a LuxR family regulator was found to induce the production of lipoxiamycins A and B (1 and 2), new members of the xiamycin family, along with previously reported xiamycin dimers, dixiamycins A (3), and C (4). Importantly, the novel compounds, lipoxiamycins A (1) and B (2), were produced only from mutant strains that were obtained by genetic modification. The structures of lipoxiamycins A (1) and B (2) were found to be unique due to the presence of a lipophilic chain linked to the indolosesquiterpenoid xiamycin core through a N–O bond. Reverse transcription polymerase chain reaction (RT-PCR) analysis of the xiaH gene, which encodes the corresponding N-hydroxylase, revealed that the transcription level of xiaH, which catalyzes the formation of a nitroxyl radical, was increased in the mutant, thereby indicating the crucial role of XiaH in the biosynthesis of the induced compounds. It should be noted here that compounds bearing N–O linkages have not been frequently reported in nature, although a macrolide-type nocardamin glucuronide has been found to possess a N–O linkage between its N-hydroxy-N’-succinylcadaverine unit and glucuronide acid (Lopez et al., 2019). In addition, asperidine C, a fungal piperidine-derived alkaloid, has been reported to contain piperidine and benzene substructures that are linked through their nitrogen and oxygen atoms (Phainuphong et al., 2018). Furthermore, maximiscin, a polyketide-shikimate-nonribosomal peptide hybrid, exhibits connectivity through a N–O bond (Robles et al., 2016). Despite these previous reports, lipoxiamycins A and B are the first members of the indolosesquiterpenoid xiamycin family that possess a N–O bond. It was also demonstrated in the current study that lipoxiamycin A (1) inhibits lipopolysaccharide-induced NO production, indicating its anti-inflammatory potential. Our discovery of rare N–O bond-bearing terpenoids via the genetic engineering of a marine Streptomyces strain therefore highlights marine bacteria to act as potential sources of bioactive terpenoids, wherein it is expected that their structural diversity could be enhanced by further genomic analysis and manipulation. Although the overexpression of regulators in Streptomyces strains led to unexpected changes in secondary metabolite production, this approach in other strains will lead to the facile and efficient discovery of novel compounds for the further diversification of secondary metabolites.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Conceptualization JP, HC, YY, D-CO. Experiments JP, HC, DM, DL, YK. Data curation JP, HC, DL, YK, SC, SL, YY, D-CO. Writing–original draft preparation JP, HC, YK, SC, SL, YY, D-CO. Writing–review and editing JP, HC, YY, D-CO. Project administration and funding acquisition YY, D-CO. All authors contributed to the article and approved the submitted version.
This research was supported by the National Research Foundation of Korea (NRF) grants funded by the Korean Government (Ministry of Science and ICT) (2020R1A2C2003518, 2021R1A4A2001251, and 2022R1A2C3004621).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1140516/full#supplementary-material
Alves A., Sousa E., Kijjoa A., Pinto M. (2020). Marine-derived compounds with potential use as cosmeceuticals and nutricosmetics. Molecules 25, 2536. doi: 10.3390/molecules25112536
Baunach M., Ding L., Bruhn T., Bringmann G., Hertweck C. (2013). Regiodivergent N–C and N–N aryl coupling reactions of indoloterpenes and cycloether formation mediated by a single bacterial flavoenzyme. Angew. Chem. Int. Ed. 52, 9040–9043. doi: 10.1002/anie.201303733
Bergman M. E., Davis B., Phillips M. A. (2019). Medically useful plant terpenoids: biosynthesis, occurrence, and mechanism of action. Molecules 24, 3961. doi: 10.3390/molecules24213961
Bi Y., Yu Z. (2016). Diterpenoids from streptomyces sp. SN194 and their antifungal activity against Botrytis cinerea. J. Agric. Food Chem. 64, 8525–8529. doi: 10.1021/acs.jafc.6b03645
Bierman M., Logan R., O’Brien K., Seno E. T., Rao R. N., Schoner B. E. (1992). Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to streptomyces spp. Gene 116, 43–49. doi: 10.1016/0378-1119(92)90627-2
Bogdan C. (2001). Nitric oxide and the immune response. Nat. Immunol. 2, 907–916. doi: 10.1038/ni1001-907
Chen Y., Smanski M. J., Shen B. (2010). Improvement of secondary metabolite production in Streptomyces by manipulating pathway regulation. Appl. Microbiol. Biotechnol. 86, 19–25. doi: 10.1007/s00253-009-2428-3
Chen R., Zhang Q., Tan B., Zheng L., Li H., Zhu Y., et al. (2017). Genome mining and activation of a silent PKS/NRPS gene cluster direct the production of totopotensamides. Org. Lett. 19, 5697–5700. doi: 10.1021/acs.orglett.7b02878
Daum M., Herrmann S., Wilkinson B., Bechthold A. (2009). Genes and enzymes involved in bacterial isoprenoid biosynthesis. Curr. Opin. Chem. Biol. 13, 180–188. doi: 10.1016/j.cbpa.2009.02.029
Ding L., Münch J., Goerls H., Maier A., Fiebig H.-H., Lin W.-H., et al. (2010). Xiamycin, a pentacyclic indolosesquiterpene with selective anti-HIV activity from a bacterial mangrove endophyte. Bioorg. Med. Chem. Lett. 20, 6685–6687. doi: 10.1016/j.bmcl.2010.09.010
Faulkner D. J. (1984). Marine natural products: metabolites of marine invertebrates. Nat. Prod. Rep. 1, 551–598. doi: 10.1039/NP9840100551
Fenical W. (1987). Marine soft corals of the genus Pseudoterogorgia: a resource for novel anti-inflammatory diterpenoids. J. Nat. Prod. 50, 1001–1008. doi: 10.1021/np50054a001
Fu J., Qin R., Zong G., Liu C., Kang N., Zhong C., et al. (2019). The CagRS two-component system regulates clavulanic acid metabolism via multiple pathways in Streptomyces clavuligerus F613-1. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.00244
Gehrke E. J., Zhang X., Pimentel-Elardo S. M., Johnson A. R., Rees C. A., Jones S. E., et al. (2019). Silencing cryptic specialized metabolism in Streptomyces by the nucleoid-associated protein Lsr2. Elife 8, e47691. doi: 10.7554/eLife.47691
Gotaskie G. E., Andreassi B. F. (1994). Paclitaxel: a new antimitotic chemotherapeutic agent. Cancer Pract. 2, 27–33.
Hobbs A. J., Higgs A., Moncada S. (1999). Inhibition of nitric oxide synthase as a potential therapeutic target. Annu. Rev. Pharmacol. Toxicol. 39, 191–220. doi: 10.1146/annurev.pharmtox.39.1.191
Huang H., Hou L., Li H., Qiu Y., Ju J., Li W. (2016). Activation of a plasmid-situated type III PKS gene cluster by deletion of a wbl gene in deep sea-derived Streptomyces somaliensis SCSIO ZH66. Microb. Cell Fact. 15, 1–9. doi: 10.1186/s12934-016-0515-6
Jiang M., Wu Z., Guo H., Liu L., Chen S. (2020). A review of terpenes from marine-derived fungi: 2015–2019. Mar. Drugs 18, 321. doi: 10.3390/md18060321
Kieser T., Bibb M. J., Buttner M. J., Chater K. F., Hopwood D. A. (2000). Practical Streptomyces genetics. Soc. Gen. Microbiol.
Kim S.-H., Ha T.-K.-Q., Oh W. K., Shin J., Oh D.-C. (2016). Antiviral indolosesquiterpenoid xiamycins c–e from a halophilic actinomycete. J. Nat. Prod. 79, 51–58. doi: 10.1021/acs.jnatprod.5b00634
Lodewyk M. W., Siebert M. R., Tantillo D. J. (2012). Computational prediction of 1H and 13C chemical shifts: a useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem. Rev. 112, 1839–1862. doi: 10.1021/cr200106v
Lopez J. A. V., Nogawa T., Futamura Y., Shimizu T., Osada H. (2019). Nocardamin glucuronide, a new member of the ferrioxamine siderophores isolated from the ascamycin-producing strain Streptomyces sp. 80H647. J. Antibiot. 72, 991–995. doi: 10.1038/s41429-019-0217-5
MacNeil D. J., Gewain K. M., Ruby C. L., Dezeny G., Gibbons P. H., MacNeil T. (1992). Analysis of Streptomyces avermitilis genes required for avermectin biosynthesis utilizing a novel integration vector. Gene 111, 61–68. doi: 10.1016/0378-1119(92)90603-m
Mayer A. M., Jacobson P. B., Fenical W., Jacobs R. S., Glaser K. B. (1998). Pharmacological characterization of the pseudopterosins: novel anti-inflammatory natural products isolated from the Caribbean soft coral, Pseudopterogorgia elisabethae. Life Sci. 62, PL401–PL407. doi: 10.1016/s0024-3205(98)00229-x
McLean T. C., Hoskisson P. A., Seipke R. F. (2016). Coordinate regulation of antimycin and candicidin biosynthesis. mSphere 1, e00305–e00316. doi: 10.1128/mSphere.00305-16
Meng Z., Yu H., Li L., Tao W., Chen H., Wan M., et al. (2015). Total synthesis and antiviral activity of indolosesquiterpenoids from the xiamycin and oridamycin families. Nat. Commun. 6, 1–8. doi: 10.1038/ncomms7096
Motohashi K., Ueno R., Sue M., Furihata K., Matsumoto T., Dairi T., et al. (2007). Studies on terpenoids produced by actinomycetes: oxaloterpins A, B, C, D, and E, diterpenes from Streptomyces sp. KO-3988. J. Nat. Prod. 70, 1712–1717. doi: 10.1021/np070326m
Olano C., García I., González A., Rodriguez M., Rozas D., Rubio J., et al. (2014). Activation and identification of five clusters for secondary metabolites in Streptomyces albus J1074. Microb. Biotechnol. 7, 242–256. doi: 10.1111/1751-7915.12116
Pescitelli G., Bruhn T. (2016). Good computational practice in the assignment of absolute configurations by TDDFT calculations of ECD spectra. Chirality 28, 466–474. doi: 10.1002/chir.22600
Phainuphong P., Rukachaisirikul V., Saithong S., Phongpaichit S., Sakayaroj J., Srimaroeng C., et al. (2018). Pyrrolidine and piperidine derivatives from the soil-derived fungus Aspergillus sclerotiorum PSU-RSPG178. Bioorg. Med. Chem. 26, 4502–4508. doi: 10.1016/j.bmc.2018.07.036
Robles A. J., Du L., Cichewicz R. H., Mooberry S. L. (2016). Maximiscin induces DNA damage, activates DNA damage response pathways, and has selective cytotoxic activity against a subtype of triple-negative breast cancer. J. Nat. Prod. 79, 1822–1827. doi: 10.1021/acs.jnatprod.6b00290
Rozen S., Skaletsky H. (2000). Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 132, 365–386. doi: 10.1385/1-59259-192-2:365
Smanski M. J., Peterson R. M., Rajski S. R., Shen B. (2009). Engineered Streptomyces platensis strains that overproduce antibiotics platensimycin and platencin. Antimicrob. Agents Chemother. 53, 1299–1304. doi: 10.1128/AAC.01358-08
Smith S. G., Goodman J. M. (2009). Assigning the stereochemistry of pairs of diastereoisomers using GIAO NMR shift calculation. J. Org. Chem. 74, 4597–4607. doi: 10.1021/jo900408d
Smith S. G., Goodman J. M. (2010). Assigning stereochemistry to single diastereoisomers by GIAO NMR calculation: the DP4 probability. J. Am. Chem. Soc. 132, 12946–12959. doi: 10.1021/ja105035r
Stinson C. A., Zhang W., Xia Y. (2018). UV Lamp as a facile ozone source for structural analysis of unsaturated lipids via electrospray ionization-mass spectrometry. J. Am. Soc. Mass Spectrom. 29, 481–489. doi: 10.1007/s13361-017-1861-2
Teufel R., Agarwal V., Moore B. S. (2016). Unusual flavoenzyme catalysis in marine bacteria. Curr. Opin. Chem. Biol. 31, 31–39. doi: 10.1016/j.cbpa.2016.01.001
Thomas M. C., Mitchell T. W., Blanksby S. J. (2006). Ozonolysis of phospholipid double bonds during electrospray ionization: a new tool for structure determination. J. Am. Chem. Soc. 128, 58–59. doi: 10.1021/ja056797h
Xiong Z.-Q., Yang Y.-Y., Zhao N., Wang Y. (2013). Diversity of endophytic fungi and screening of fungal paclitaxel producer from anglojap yew, taxus x media. BMC Microbiol. 13, 1–10. doi: 10.1186/1471-2180-13-71
Zhang Q., Li H., Yu L., Sun Y., Zhu Y., Zhu H., et al. (2017). Characterization of the flavoenzyme XiaK as an N-hydroxylase and implications in indolosesquiterpene diversification. Chem. Sci. 8, 5067–5077. doi: 10.1039/c7sc01182b
Keywords: indolosesquiterpenoid, bacteria, overexpression, N-O bond, structure elucidation
Citation: Park J, Cho HS, Moon DH, Lee D, Kal Y, Cha S, Lee SK, Yoon YJ and Oh D-C (2023) Discovery of new indolosesquiterpenoids bearing a N-O linkage by overexpression of LuxR regulator in a marine bacterium Streptomyces sp.. Front. Mar. Sci. 10:1140516. doi: 10.3389/fmars.2023.1140516
Received: 09 January 2023; Accepted: 22 March 2023;
Published: 05 April 2023.
Edited by:
Hui Cui, Guangzhou University of Chinese Medicine, ChinaReviewed by:
Valeria Costantino, University of Naples Federico II, ItalyCopyright © 2023 Park, Cho, Moon, Lee, Kal, Cha, Lee, Yoon and Oh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yeo Joon Yoon, eWVvam9vbnlvb25Ac251LmFjLmty; Dong-Chan Oh, ZG9uZ2NoYW5vaEBzbnUuYWMua3I=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.