
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 03 March 2023
Sec. Marine Biotechnology and Bioproducts
Volume 10 - 2023 | https://doi.org/10.3389/fmars.2023.1136926
This article is part of the Research TopicMining, Development and Utilization of Genetic Resources Related to Economic Traits of Aquatic Animals Based on Omics TechnologyView all 5 articles
 Mengyang Chang1†
Mengyang Chang1† Jianlong Ge1,2†
Jianlong Ge1,2† Meijie Liao1,2*
Meijie Liao1,2* Xiaojun Rong1,2Yingeng Wang1,2*Bin Li1,2Xinrong Li1,2
Xiaojun Rong1,2Yingeng Wang1,2*Bin Li1,2Xinrong Li1,2 Jinjin Wang1,2Zheng Zhang1,2
Jinjin Wang1,2Zheng Zhang1,2 Yongxiang Yu1,2Chunyuan Wang1,2
Yongxiang Yu1,2Chunyuan Wang1,2DNA methylation is an important epigenetic modification that regulates many biological processes. The sea cucumber Apostichopus japonicus often suffers from heat stress that affects its growth and leads to significant economic losses. In this study, the mRNA expression patterns and DNA methylation characteristics in the body wall of A. japonicus under heat stress were analyzed by whole-genome bisulfite sequencing (WGBS) and transcriptome sequencing (RNA-seq). We found that CpG was the main DNA methylation type, and heat stress caused a significant increase in the overall methylation level and methylation rate, especially in the intergenic region of the A. japonicus genome. In total, 1,409 differentially expressed genes (DEGs) and 17,927 differentially methylated genes (DMGs) were obtained by RNA-seq and WGBS, respectively. Association analysis between DNA methylation and transcription identified 569 negatively correlated genes in both DMGs and DEGs, which indicated that DNA methylation affects on transcriptional regulation in response to heat stress. These negatively correlated genes were significantly enriched in pathways related to energy metabolism and immunoregulation, such as the thyroid hormone signaling pathway, renin secretion, notch signaling pathway and microRNAs in cancer. In addition, potential key genes, including heat shock protein (hsp70), calcium-activated chloride channel regulator 1(clca1), and tenascin R (tnr), were obtained and their expression and methylation were preliminarily verified. The results provide a new perspective for epigenetic and transcriptomic studies of A. japonicus response to heat stress, and provide a reference for breeding sea cucumbers resistant to high temperatures.
Sea cucumber (Apostichopus japonicus) is a traditional health product with high nutritional and economic value, which has been an important aquaculture species and farmed in many Asian countries, including China, Japan, and Korea (Cao, 2014). As a typical temperate species, the growth of A. japonicus is often hindered by extremely high or low temperatures, but especially high temperatures (Li et al., 2019). Changes in temperature usually reduce the activity and food intake of sea cucumbers, resulting in reduced immunity, which can lead to disease and death (Huo et al., 2021; Li et al., 2021; Huo et al., 2022). Therefore, revealing the molecular mechanism of their sensitivity to temperature and pressure can expand our understanding of how marine animals adapt to environmental challenges.
With the development of high-throughput sequencing technology, RNA-seq has been widely used in aquatic animals. The transcriptome map of the body wall and intestinal tissue of A. japonicus was first constructed in 2011, which provided data for studying the regeneration mechanism of A. japonicus (Sun et al., 2011). Subsequent studies constructed the transcriptome maps of sea cucumbers at different development stages and during summer dormancy stage, and numerous genes related to development, growth, metabolism and immunity were identified (Du et al., 2012; Zhao et al., 2014; Zhou et al., 2016). At present, studies on gene expression patterns of A. japonicus under heat stress have mainly focused on muscle, intestine, and respiratory tree tissues (Xu et al., 2018; Li et al., 2019; Chen, 2020). However, research on the body wall, which is the target organ of skin ulceration syndrome (SUS) is relatively rare.
Epigenetics is a kind of hereditary variation that does not change DNA sequence, and its regulatory mechanism mainly includes DNA methylation modification, histone modification, chromosome remodeling, and non-coding RNA regulation. These modifications interact and jointly regulate genome function (Kulis and Esteller, 2010; Crabtree et al., 2020) and these epigenetic modifications are easily induced by the environment (Feil and Fraga, 2012). DNA methylation is an important part of epigenetics research and one of the modification methods of DNA. It participates in the regulation of gene expression, cell growth and development, and response to adversity and stress by affecting the spatial conformation, stability, and interactions with proteins of nucleic acids (Entrambasaguas et al., 2021).
In aquatic animals, environmental factors influence gene expression by affecting DNA methylation status, so that the biological activities and phenotypes of organisms can be changed to adapt to the environment (Wang et al., 2011). Studies have found that DNA methylation mediates the biological process and then affects sex changes in Dicentrarchus labrax (Kuhl et al., 2011). A study showed that the total methylation rate of genomic DNA in the scallop (Patinopecten yessoensis) was affected by acute temperature stress (Wu et al., 2016). Li et al. (2018) found that after salt stress treatment, the exon methylation level of the growth-related gene igf1 was negatively correlated with its liver expression level in Cynoglossus semilaevis. Studies have also found that DNA methylation plays an important role in regulating the response to adversity in sea cucumbers. Methylation-sensitive amplified polymorphism (MSAP) analysis revealed that methylation levels significantly increased in sea cucumbers with SUS (Gao et al., 2017). Li et al. (2017) found that the expression of the apparent modification-related genes (dnmt1, hdac3, and mll5) of sea cucumbers significantly changed at high temperatures. Zhao et al. (2015) found that sea cucumber methylation levels during summer dormancy were higher than during non-summer dormancy. Although some progress has been achieved in the study of transcription pattern and DNA methylation characteristics of sea cucumber under heat stress, the effect of epigenetic modification on the transcription process is still unclear.
In this study, whole-genome bisulfite sequencing (WGBS) and transcriptome sequencing were used to gain insights into the DNA methylation characteristics and transcription pattern of A. japonicus under heat stress. We analyzed the correlation between gene expression and DNA methylation, and obtained potential key pathways and functional genes. These findings provide the first insight into the epigenetic mechanism of heat stress response in the body wall tissue of A. japonicus.
The experimental sea cucumbers were from Qingdao Ruizi Group Co., Ltd., they had an average weight of 50 ± 2g and were healthy, active, and free from disease or infection. Sea cucumbers were randomly divided into the control (CS) group at 17 °C and the heat stress (HS) group at 32 °C (the median lethal temperature of sea cucumber according to a previous study by our lab; Zhang et al., 2022), with 50 sea cucumbers in each group. The initial water temperature of the HS group was 17°C, and the temperature was increased at a rate of 1°C per day until the water temperature reached 32°C. On the fifth day, when the temperature reached 32°C, three healthy and three SUS sea cucumbers were randomly selected from the CS and HS groups, respectively. The body wall tissues were sectioned, placed in 2 ml EP tubes and put into liquid nitrogen for quick freezing. All samples were stored at -80°C for transcriptome and genome-wide DNA methylation sequencing analyses.
Total RNA was isolated using TRIzol reagent (Qiagen, Carlsbad, CA, USA) following the manufacturer’s procedure. The extracted RNA samples from the CS and HS groups were analyzed using a LabChip GX Touch (Perkin Elmer, CA, USA), and samples with RIN number > 7 were selected for library construction. Finally, the cDNA library was sequenced with PE150 on the Illumina Novaseq 6000 by LC-Bio (Hangzhou, China).
Cutadapt 4.2 (Kechin et al., 2017) was used to remove adaptor reads. High-quality sequences were mapped to the sea cucumber reference genome (assembled by our lab, unpublished data) using HISAT2 (Pertea et al., 2016). The transcriptomes from the six samples were combined and a complete transcriptome was reconstructed using gffcompare (Pertea and Pertea, 2020). The expression levels were estimated for all transcripts using StringTie (Pertea et al., 2016) and analyzed for mRNA expression levels by calculating of Fragments per Kilobase Million (FPKM). The differentially expressed genes (DEGs) were performed with FDR < 0.005 and fold change > 2 by DESeq2 (Love et al., 2014).
Total DNA was isolated using the QIAamp Fast DNA Tissue Kit (Qiagen, 51404). The A260/280 ratio was read by a spectrophotometer and DNA was available when the A260/280 ratio was in the range of 1.8 to 2.0. DNA samples were subjected to bisulfite conversion after sonication using the Bisulfite Conversion Kit (Swift, MI, USA) to construct the WGBS library. The library was then sequenced on an Illumina Hiseq 4000 platform by LC-Bio (Hangzhou, China).
Cutadapt and perl scripts were used to remove the adapter contamination, low quality, and undetermined bases. Reads that passed quality control were mapped to the reference genome of sea cucumbers using Bismark (Krueger and Andrews, 2011). After alignment, reads were further deduplicated using SAMtools (Li et al., 2009). Plink (V1.90b6.21) software was used for principal component analysis (PCA). PerScript and MethPipe (Song et al., 2013) were used to locate each cytosine in the alignment sequence. The DNA methylation level was determined by the ratio of the number of reads supporting C (methylation) to the total number of reads (methylated and unmethylated). DSS software was used to calculate the differentially methylated regions (DMRs) with 1,000 bp slide windows, 500 bp overlap, P < 0.05 (from TSS to TES) (Akalin et al., 2012). In this study, genes were considered to be differentially methylated genes (DMGs) when at least one DMR was located within 3000 bp upstream of the start codon.
Pearson’s method was used to test the relative expression data of DEGs and DMRs, and to determine the correlation between gene expression and DNA methylation. The selected genes were subjected to Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment anslyses to characterize their function and response pathways.
The body wall of six sea cucumbers were sampled in each group for real-time PCR validation. First-strand cDNA was synthesized using HiScript II Reverse Transcriptase (Vazyme, China). The mRNA expression was examined using Taq Pro Universal SYBR qPCR Master Mix (Vazyme, Takara) and Roche LightCycler96 PCR (Roche, Switzerland). The gene actb was used as a control gene for internal standardization. The nine genes chosen for validation were hsp70 (heat shock protein 70), clca1 (calcium-activated chloride channel regulator 1), tnr (tenascin R), bag3 (recombinant bcl2 associated athanogene 3), traf7 (TNF receptor-associated factor 7), fut4 (fucosyltransferase 4), act-3 (alpha-actinin-3), srpr (larimichthys crocea signal recognition particle receptor), and wdr20 (WD repeat-containing protein 20). The amplification cycling was performed as follows: (1) 30 s at 95°C for 1 cycle, and 5 s at 95°C, 1min at 60°C, and 25 s at 72°C for 40 cycles.
Methylation sites were verified by BSP. The DNA of six cucumbers was modified with bisulfite using an EZ DNA Methylation-Gold™ Kit (Takara, Japan). The modified DNA samples were subjected to BSP amplification using the Takara EpiTaqHS Kit (Takara, Japan). The six genes chosen for validation included hsp70, snrnp25 (small nuclear ribonucleoprotein U11/U12 subunit 25), gpatch2 (G patch domain containing 2), BSL78_04364, BSL78_07932 and BSL78_12691. The amplification cycling was performed as follows: 3 min at 98°C and then 10 s at 98°C, 30 s at 55°C, 30 s at 72°C, and 7 min at 72°C for 35 cycles. After the recombinant plasmid was constructed, positive clones were selected for sequencing analysis
The qRT-PCR and BSP primers are listed in Table S1. The 2-△△CT method was used to analyze the comparative mRNA expression levels. One-way analysis of variance conducted with SPSS 19.0 software (SPSS Inc., Chicago, IL, USA) was used for statistical analysis. The level of significance was set to P < 0.05. All data are presented as mean ± SD (standard deviation of the mean) (N = 3).
In total, 73.33G raw reads were generated from six RNA-seq samples. All raw reads were submitted to the SRA database under the accession number PRJNA901272. In this study, the Q20 ranged from 99.83% to 99.90%. An average of 75.03% of clean sequences could be mapped to the reference genome of A. japonicus, of which 40.14% to 45.70% had unique alignment and 24.99% to 26.75% had multiple alignments to the genome (Table 1).
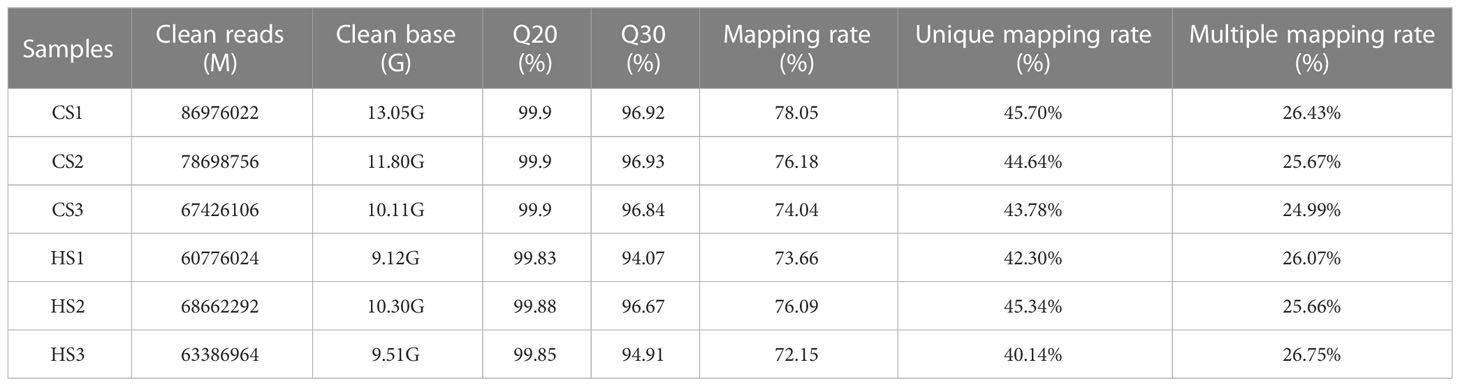
Table 1 Statistical data for RNA-seq.
Data analysis showed that the general distributions of FPKM values for six samples were similar; the CS group and the HS group had similar gene expression levels (Figure S1). In total, 29,290 genes were detected from transcriptome sequencing and 1,409 DEGs were selected, including 528 up-regulated DEGs and 881 down-regulated DEGs (Figure 1). The information for the 1,409 DEGs is shown in Table S2.
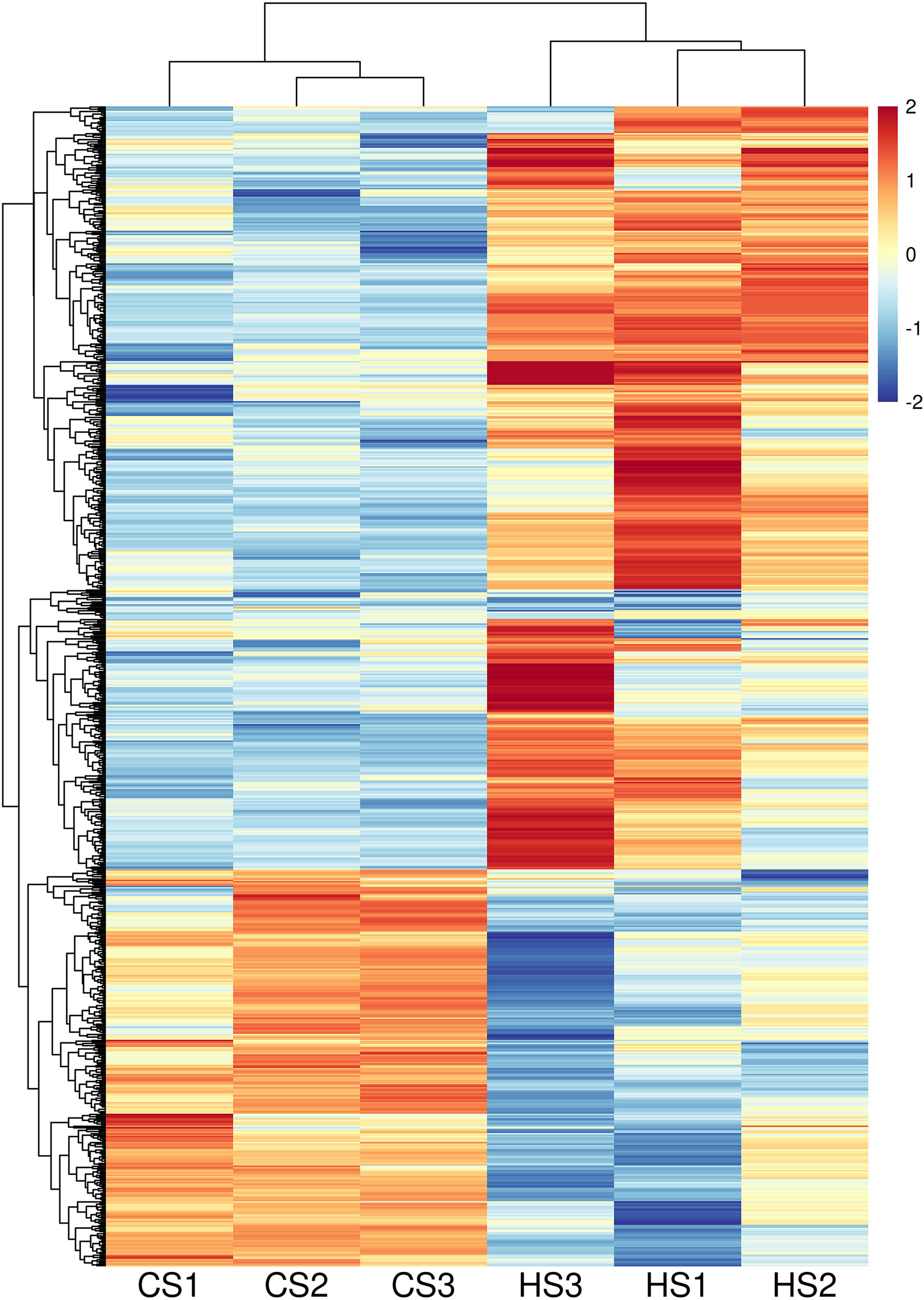
Figure 1 Heatmap of DEGs between CS and HS with FDR < 0.005 and fold change > 2.
The up-regulated DEGs were significantly enriched in 340 GO terms, and the down-regulated DEGs were significantly enriched in 308 GO terms (P < 0.05). In the GO terms of up-regulated DEGs, the following terms were frequently enriched in protein folding, response to heat and extracellular exosomes and stress-related terms (Figure 2A). The down-regulated DEGs were mainly enriched in terms related to protein decomposition and metabolism, such as calcium ion binding, cellular protein catabolism, and hydrolase activity (Figure 2B). KEGG enrichment showed that the up-regulated DEGs were significantly enriched in 18 KEGG signaling pathways, including lysine degradation, Toll and IMD signaling pathway, and protein processing in endoplasmic reticulum (Figure 2C). The down-regulated DEGs were significantly enriched in 24 KEGG signaling pathways, including notch signaling pathway, ubiquitin mediated proteolysis, and MAPK signaling pathways (Figure 2D).
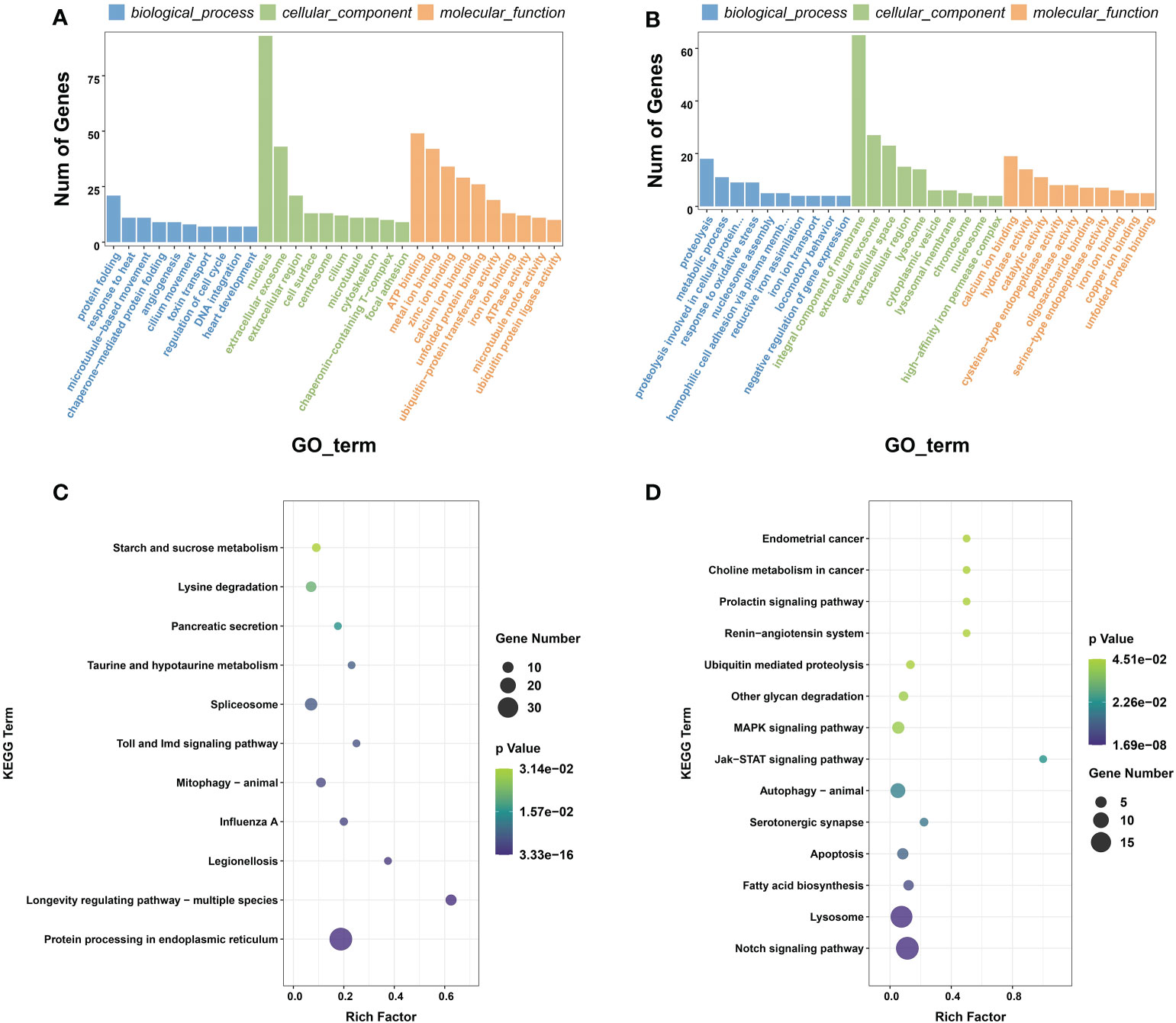
Figure 2 (A) GO functional classification histograms of up-regulated DEGs. (B) GO functional classification histograms of down-regulated DEGs. (C) Scatter plot showing significantly enriched KEGG pathways in up-regulated DEGs. (D) Scatter plot showing significantly enriched KEGG pathways in down-regulated DEGs.
The DNA libraries of six samples were constructed and sequenced on the Hiseq 4000 platform. All raw reads were submitted to the SRA database under the accession number PRJNA901271. First, sequencing adapters and low-quality data were removed, and the average value of clean data was 24.04 Gb for each library. In this study, the Q20 value varied from 96.78% to 97.70%, and the genome mapping rate varied from 41.24% to 42.59% for each sample. The conversion of cytosine under the bisulfite treatment was between 99.35% and 99.66%, with an average of 99.55%, which indicated high efficiency of bisulfite treatment (Table 2). PCA analysis showed that both CS and HS groups had effective repliactions (Figure S2).
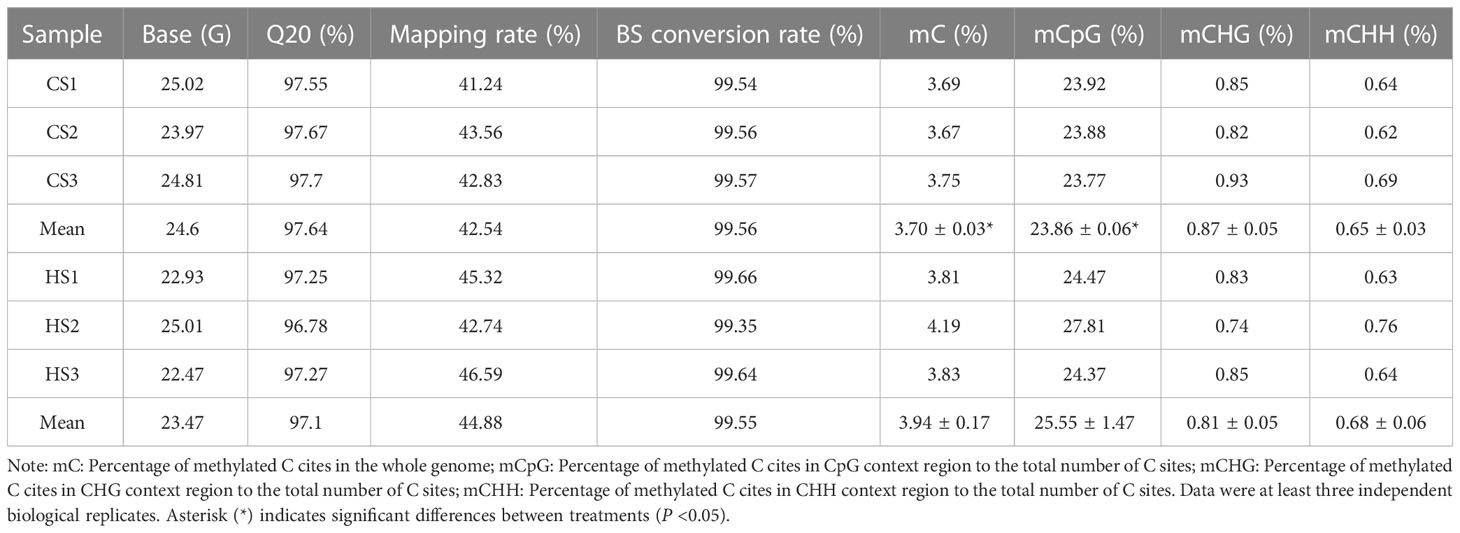
Table 2 Statistical data for whole genome bisulfite sequencing.
The average DNA methylation levels of C sites and different types (CpG, CHG, and CHH) in the body wall tissues of sea cucumbers under heat stress are shown in Table 1. The whole genome methylation level of the 23 chromosomes of A. japonicas is also shown in a Circos plot (Figure 3A). The percentage of methylated C sites to the total number of C sites was 3.94 ± 0.17% in the HS samples and 3.70 ± 0.03% in the CS samples, which indicated a significant increase in the percentage of methylated C sites in the genome under heat stress (P < 0.05). Statistical results of the average methylation level of different types (CpG, CHG, and CHH) showed that the proportions of mCpG, mCHG, and mCHH in the CS group were 24.14%, 0.56%, and 0.47%, respectively. While the mCpG mCHG and mCHH in HS group accounted for 24.22%, 0.98% and 0.76%, respectively. For the sea cucumber genome, the DNA methylation ratio of CpG sites was significantly higher than that of the CHG and CHH sites (Table 2). Overall, DNA methylation of the sea cucumber was mainly observed in CpG sites (84%) and rarely in CHG (13.6%) and CHH sites (3.8%). CpG DNA methylation levels varied to a greater extent across functional regions, whereas CHH and CHG methylation levels varied to a lesser extent (Table S3). The promoter and exon regions had the highest proportion of CpG methylation levels (Figure 3B).
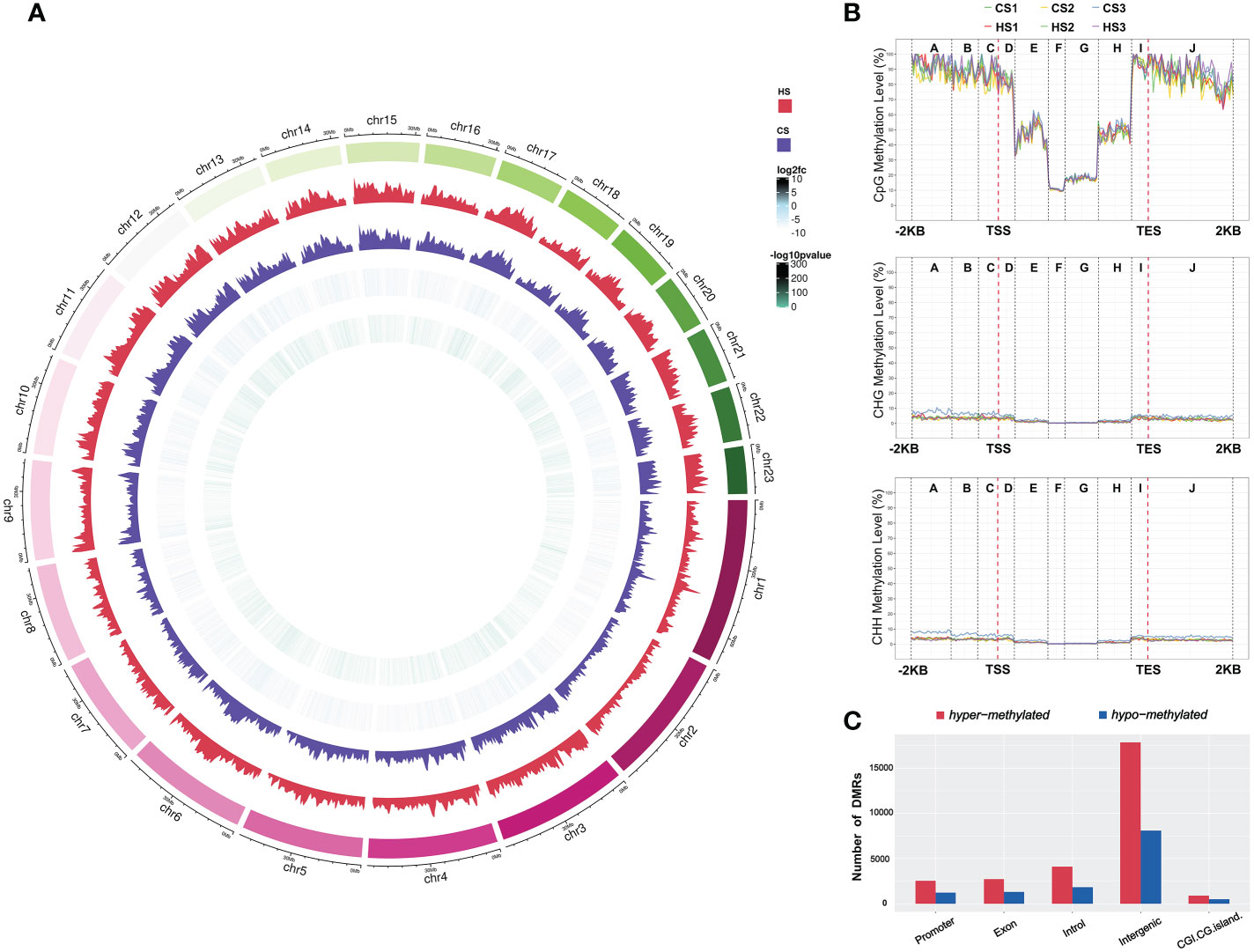
Figure 3 (A) Circos plot showing the distribution of DNA methylation differences at CG context between CS and HS groups across A. japonicus chromosome-scaled genome. The five outer to inner circles represent chromosomes of A. japonicus, methylation levels of the HS group, methylation levels of the CS group, the fold change of methylation level between CS and HS groups, and the significance of methylation levels between CS and HS groups, respectively. (B) CHG, CHH and CpG methylation level distribution of different functional components. A: Distal promoter; (B): Intermediate promoter; (C): Proximal promoter; (D): Fist exon; (E): Fist intron; (F): Internal exon; (G): Internal intron; (H): Last intron; (I): Last exon; (J): Downstream. (C) Number of DMRs in different gene functional regions between the CS and HS groups.
The DSS method was used to identify genome-wide DNA methylation changes between the CS and HS groups. In total, 748,347 DMRs were identified in the sea cucumber genome. Among them, 48,392 DMRs were in promoter, 69,090 DMRs were in exons, 129,779 DMRs were in introns, 481,072 DMRs were in the intergenic region, and 20,014 DMRs were in the CGI.CG island region. The results showed that the percentage of methylated genes in the intergenic region of sea cucumbers was the highest, accounting for 64.02%, whereas the percentage of methylated genes in the CGI.CG island region was the lowest, accounting for 2.72% (Figure 3C).
Overall, 17,927 DMGs were identified in DMRs. The GO enrichment analysis showed that DMGs were significantly enriched in substance transport-related terms, such as protein binding, actin binding, and calcium ion binding (Figure 4A). In addition, the KEGG enrichment analysis showed that these DMGs were significantly enriched in RNA transport, MAPK signaling pathway, ubiquitin mediated proteolysis, and basal transcription factors (Figure 4B).
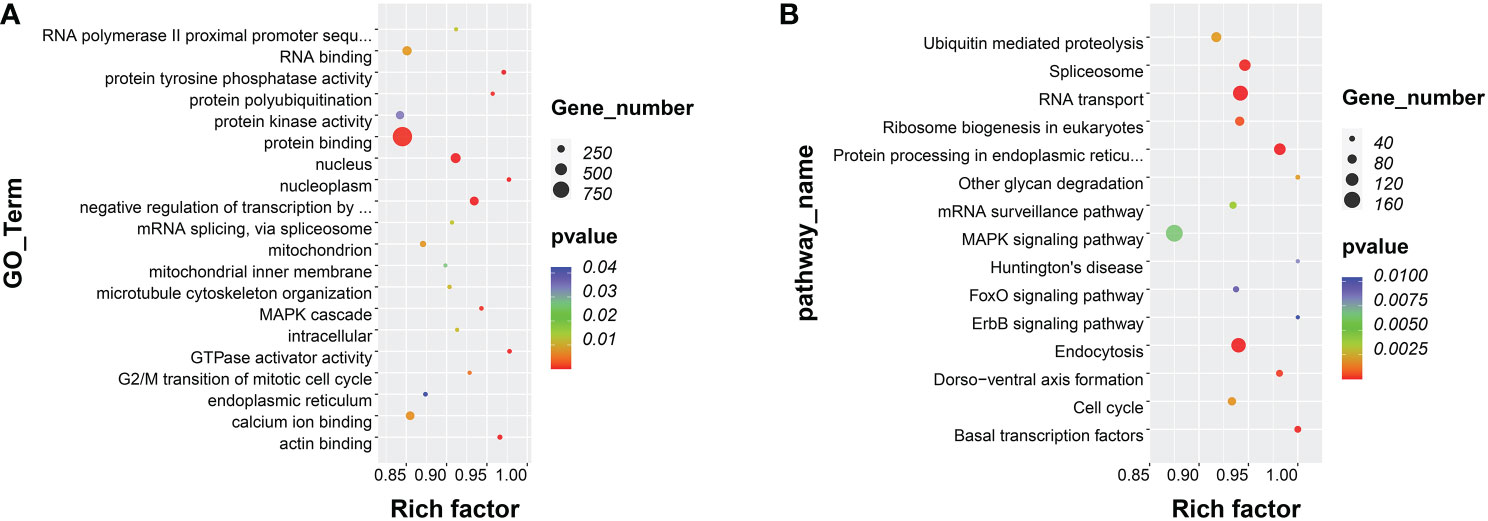
Figure 4 (A) Scatter plot showing the top 20 enriched GO terms, resulting from the DMGs between CS and HS. (B) Scatter plot showing the top 15 enriched KEGG pathways, resulting from the DMGs between CS and HS.
To investigate the effect of DNA methylation on transcription, 808 shared genes were identified in both DMGs and DEGs (Figure 5A). The results of association analysis of DMRs and DEGs are shown in Table S4. In total, 569 negatively correlated genes were identified, which accounted for 70.42% of shared genes, including 293 hyper-methylated and down-regulated genes, and 276 hypo-methylated and up-regulated genes. In addition, 223 negatively correlated genes with DMRs located in the promoter region and 276 negatively correlated genes with DMRs located in the gene body region were identified. The results showed that more genes had a negative correlation between transcription patterns and DNA methylation characteristics (Figure 5B). The gene expression and methylation levels of these genes in the CS and HS groups are shown in Figure 5C. Enrichment analysis of these negatively correlated genes indicated that genes were mainly enriched in GO terms such as transmembrane transport, integrated component of membrane, and protein binding (Figure 5D). KEGG enrichment results showed that genes were mainly enriched in the thyroid hormone signaling pathway, renin secretion, cGMP-PKG signaling pathway, notch signaling pathway, antigen processing and presentation, intestinal immune network for IgA production, and microRNAs in cancer. Some key functional genes were screened, including hsp70, clca1, tnr, bag3, traf7, and fut4 (Figure 5E).
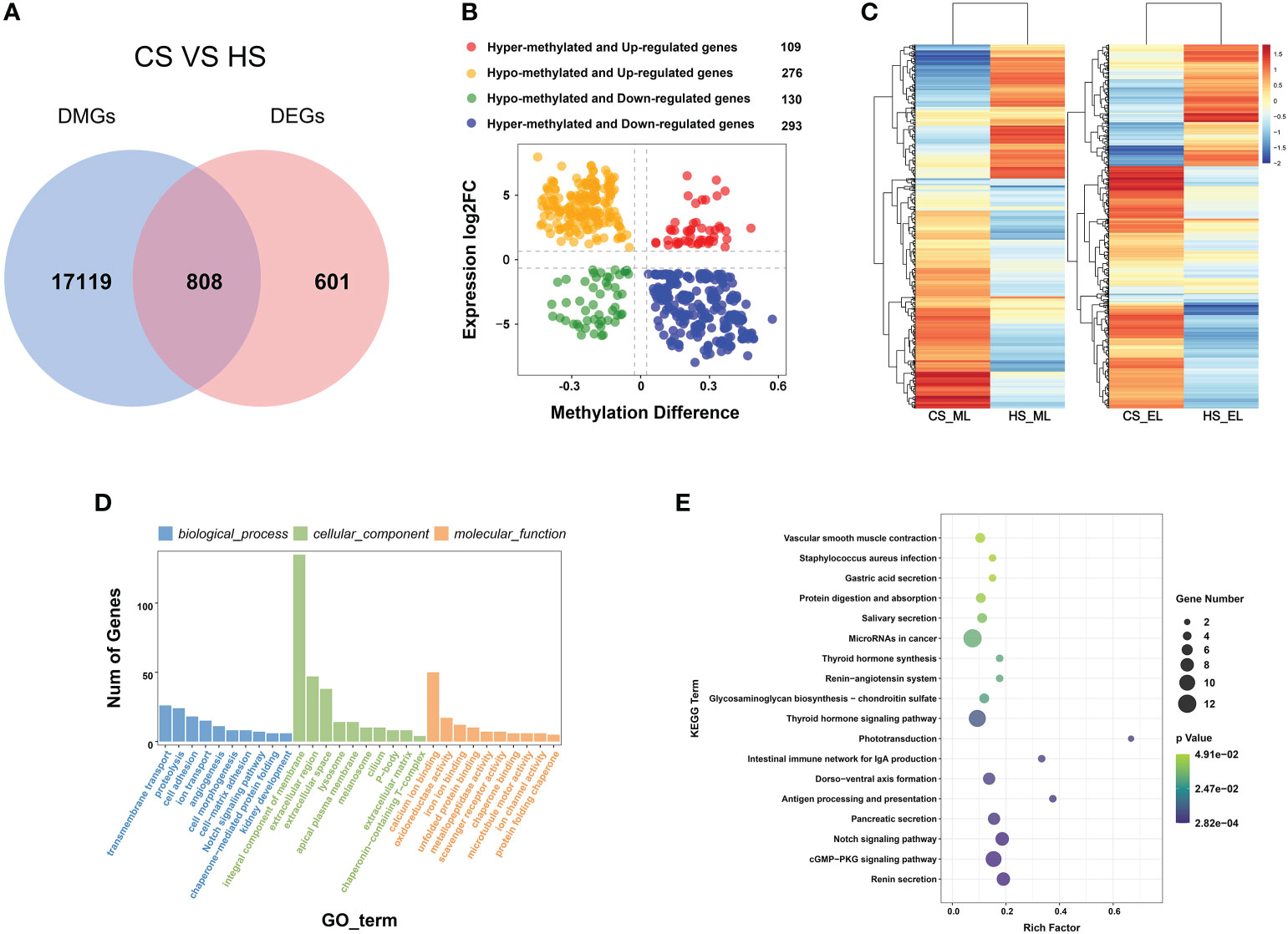
Figure 5 (A) Venn plot of DMGs and DEGs between CS and HS. (B) Scatter plot of the correlation between expression levels of DEGs and methylation difference of DMGs in 808 shared genes. (C) Heatmap of methylation levels (ML) and expression levels (EL) of 808 shared genes in CS and HS. (D) Bar chart of GO enrichment of 569 negatively correlated genes between DEGs and DMGs. (E) Scatter plot of KEGG enrichment of 569 negatively correlated genes between DEGs and DMGs.
The results showed that the expression levels of tnr, clca1, hsp70, act-3, bag3 and traf7 were up-regulated in sea cucumber under heat stress, and the expression levels of srpr, wdr20 and fut4 were down-regulated in sea cucumber under heat stress. The qRT-PCR results showed that the expression of DEGs was consistent with the transcriptome sequencing results (Figure 6A). In this study, six DMGs were randomly selected for BSP, and the WGBS results were verified. According to a standard regression analysis, the results were consistent between BSP and WGBS (Figure 6B).
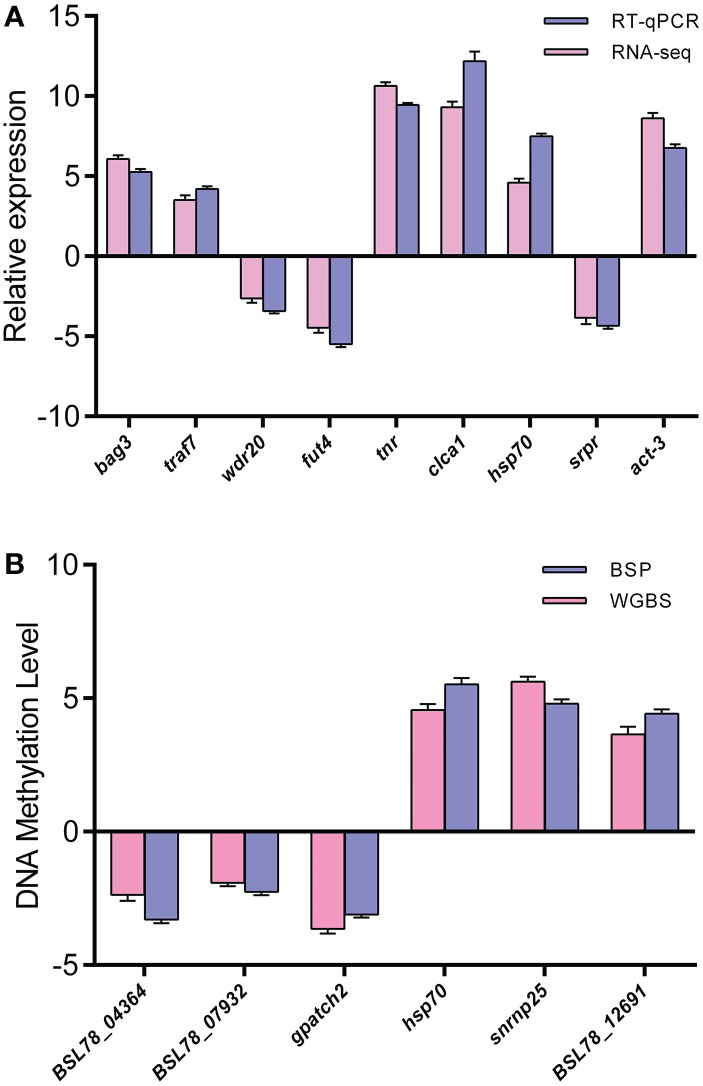
Figure 6 (A) Comparison of mRNA expression levels among the nine DEGs obtained using qRT-PCR validation. Log2Fold Change are expressed as the ratio of gene expression after normalization to Actb. (B) Comparison of DNA methylation levels among the six DMGs obtained using BSP validation.
RNA-seq has been widely used in studies of gene expression patterns of A. japonicus under heat stress on muscle, intestine, and respiratory tree tissues (Xu et al., 2018; Li et al., 2019; Chen, 2020). Li et al. (2019) found that 46 DEGs were involved in the innate and adaptive immunity of A. japonicus muscle tissue under heat stress, which contained many genes of heat shock family. Interestingly, genes of heat shock families such as hsp26, hsp70, hsc70 and hsp90 were selected in intestine tissue under heat stress (Xu et al., 2018). Chen (2020) also revealed that DEGs in the intestine were significantly enriched in immune-related GO terms after heat stress of A. japonicus, such as toll-like receptor 3, carboxy-terminal kinesin 2-like, and complement component C3. Studies have also shown that A. japonicus can respond to heat stress by regulating the expression of immune-related genes with some non-coding RNAs such as miRNA, circRNA and lncRNA (Huo et al., 2020; Huo et al., 2021; Chang et al., 2022). In the present study, the down-regulated DEGs were significantly enriched in immune-related pathways such as notch signaling pathway, ubiquitin mediated proteolysis, and MAPK signaling pathway. It was reported that these pathways can regulate the production of cytokines and participate in immune response (Meurette and Mehlen, 2018; Holdsworth et al., 2020; Liu et al., 2021). These results indicated that heat stress may induce a significant decrease in the immune regulation ability of sea cucumbers and provided an insight into the occurrence of SUS of A. japonicus under heat stress.
Research on aquatic organisms has shown that DNA methylation can be altered in response to temperature shift, diet composition, ploidy, and heterosis (Zhuo et al., 2019; Han et al., 2021; Yuan et al., 2021). Our results showed that methylation levels in the body wall of sea cucumbers increased after heat stress. Interestingly, a similar methylation pattern was also observed in the Yesso scallop (Patinopecten yessoensis), Pacific oyster (Crassostrea gigas) and Pacific abalone (Haliotis discus hannai), whose DNA methylation levels increased under heat stress (Huang et al., 2021; Wang et al., 2021; Yuan et al., 2021). In addition, previous studies showed that the DNA methylation levels in body wall with SUS were markedly higher than those of healthy body wall in A. japonicus (Gao et al., 2017; Sun et al., 2020). These results demonstrate that aquatic animals generally respond to environmental stress by increasing the DNA methylation level. However, a different result was obtained in a previous study in which the DNA methylation level of the A. japonicus digestive tractdecreased after heat stress at 32°C (Wen et al., 2021). We speculate that this divergence may be due to the difference in tissue type and selection of reference genome. Our results indicated that the body wall DNA methylated cytosines of sea cucumbers under heat stress were mainly located on CpG dinucleotides. Previous studies have also shown that DNA methylation in the body wall and intestinal tissues of A. japonicus mainly occurred at the CpG sites during Vibrio splendens infestation, aestivation, and heat stress (Sun et al., 2020; Yang et al., 2020; Wen et al., 2021). In addition, the DNA methylation pattern in A. japonicus was consistent with research results on the Pacific abalone, Yesso scallop, Pacific oyster (Huang et al., 2021; Yuan et al., 2021; Venkataraman et al., 2022), and other metazoans (Lechner et al., 2013). Although CpG was the main type of DNA methylation, there were some differences in the characteristics of CpG in different regions of the genome. The DNA methylation level of the exon region in A. japonicus intestine during aestivation (Yang et al., 2020) was lower than that in our study. We speculate that the difference may be related to temperature selection, because temperature can affect the methylation level of exons (Iamjan et al., 2021). The DNA methylation characteristics of different tissues after heat stress might be different, and WGBS data are needed for more tissue types in the future.
The relationship between methylation and gene expression was explored by analyzing the correlation between the expression level of DEGs and the methylation characteristics of DMRs. As a result, 808 shared genes, including 569 negative regulatory genes and 239 positive regulatory genes, were screened by DNA methylation and transcription association analysis; this indicated that DNA methylation mainly regulates gene expression through negativeregulation. This was consistent with previous studies on DNA methylation of Pacific abalone and European sea bass (Anastasiadi et al., 2017; Huang et al., 2021). Both negative and positive correlations between methylation levels and gene expression levels have also been found in the body wall and intestine of A. japonicus (Chen, 2020; Sun et al., 2020).The effect of methylation on gene expression often depends on the location in the genome. Generally, methylation of the gene body promotes gene expression, whereas methylation of the promoter inhibits gene expression (Palou-Márquez et al., 2021). However, our study showed that most of the negative regulatory genes were located in the gene body, and there were few negative regulatory genes in the promoter. The same result was found in a study on intestinal DNA methylation in A. japonicus under heat stress (Chen, 2020). This condition may be caused by re-methylation of CpG islands within the gene or inhibition of antisense transcription factors (Lou et al., 2014).
KEGG enrichment of 569 negative regulatory genes showed that they were mainly enriched in energy metabolism pathways, such as thyroid hormone signaling pathway, renin secretion and cGMP-PKG signaling pathway, as well as notch signaling pathway, antigen processing and presentation, intestinal immune network for IgA production, and microRNAs in cancer, which are related to immunoregulation. The cGMP-PKG signaling pathway can generally mediate the expression of cytokine PDE5 and affect the proliferation and apoptosis of cancer cells (Xiao et al., 2021). The thyroid hormone signaling pathway is an essential signaling pathways for regulating growth, development and energy metabolism, and the downstream molecules can participate in the development and metamorphosis of marine invertebrates (Gilleron et al., 2006). The notch signaling pathway, as a classical immune-related signaling pathway, has a wide and close relationship with the occurrence and development of tumors. The signal transduction between adjacent cells through the Notch receptor can regulate cell differentiation, proliferation, and apoptosis (Sajadimajd et al., 2022). Therefore, these pathways may be important adaptation strategies of sea cucumbers under heat stress.
The DNA methylation levels and expression levels of negative regulatory genes in these pathways were randomly verified by qRT-PCR and BSP. These negatively regulated genes are ideal targets in the future to reveal the epigenetic regulation of heat stress response in A. japonicus. For example, hsp70 can reduce the effects of toxic substance accumulation during stress (Radons, 2016). Furthermore, hsp70 may play a role in both innate and acquired immune systems (Treweek et al., 2015). Studies have found that the expression level of hsp70 significantly increased significantly with the increase of heat stress time. The rapid and persistent response of hsp70 indicates its critical role in the heat stress response of A. japonicus (Xu et al., 2015; Xu et al., 2016). The up-regulated expression of hsp70 in sea cucumbers under heat stress has also been detected in our previous study (Chang et al., 2022). Additionally, clca1 was found to be a key gene in IL-13 (Pro-inflammatory interleukin-13) dependent mucosal metaplasia (Alevy et al., 2012). In Cynoglossus semilaevis, clca1 acts as an activator of hepatic macrophages and regulates the expression of cytokines to inhibit pathogenic infection (Li and Sun, 2016). Tnr plays an important role in the development of the nervous system, the regeneration of tissues and organs after trauma and it can regulate the morphology of extracellular matrix (Meloty-Kapella et al., 2006). Another study showed that tnr could act as an important ECM component involved in neurodevelopmental regeneration and participates in the intestinal regeneration of A. japonicus (Yamazaki et al., 2020). We speculated that A. japonicus could repair tissue damage during heat stress and resist environmental changes by up-regulating the expression of tnr.
This study was the first attempt to investigate the gene transcription pattern and genome-wide DNA methylation characteristics in the body wall tissue of sea cucumbers in response to heat stress. We found significant changes in overall DNA methylation levels of sea cucumbers under heat stress, and obtained a list of DEGs and DMGs. In particular, many negatively correlated genes in both DMGs and DEGs were identified, which indicated that DNA methylation affects transcriptional regulation during heat stress. Further analysis showed that these negatively correlated genes were significantly enriched in pathways related to energy metabolism and immunoregulation. These results provide important targets for further revealing the response or adaptation mechanism of sea cucumbers to heat stress, and deepened our understanding of the epigenetic regulatory mechanism of heat stress response in sea cucumbers.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA901271 https://www.ncbi.nlm.nih.gov/, PRJNA901272.
ML and YW conceived and designed the experiments. MC, JG, XR, BL, XL, JW, ZZ, YY and CW performed the experiments. MC and JG analyzed the data. MC and JG wrote the paper. All authors read and approved the manuscript. All authors contributed to the article and approved the submitted version.
This work was funded by the National Key R&D Program of China (2018YFD0901604), Key R&D Program of Qingdao (22-3-3-hygg-1-hy), Agriculture Seed Improvement Project of Shandong Province (2020LZGC015), Key Laboratory of Healthy Mariculture for the East Sea, Ministry of Agriculture and Rural Affairs, P.R.China (2022ESHML03) and Central Public-interest Scientific Institution Basal Research Fund, CAFS (2020TD40).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1136926/full#supplementary-material
Supplementary Figure 1 | Boxplot of gene expression level in CS and HS groups.
Supplementary Figure 2 | PCA analysis of the correlation of CS and HS groups.
Akalin A., Kormaksson M., Li S., Garrett-Bakelman F. E., Figueroa M. E., Melnick A., et al. (2012). MethylKit: A comprehensive r package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 13, R87. doi: 10.1186/gb-2012-13-10-r87
Alevy Y. G., Patel A. C., Romero A. G., Patel D. A., Tucker J., Roswit W. T., et al. (2012). IL-13-induced airway mucus production is attenuated by MAPK13 inhibition. J. Clin. Invest. 122, 4555–4568. doi: 10.1172/JCI64896
Anastasiadi D., Díaz N., Piferrer F. (2017). Small ocean temperature increases elicit stage-dependent changes in DNA methylation and gene expression in a fish, the European sea bass. Sci. Rep. 7, 12401. doi: 10.1038/s41598-017-10861-6
Cao X. (2014). Study on the effect of temperature on the growth, physiology and nutritional components of apostichopus japonicus. [master’s thesis] ([Dalian]: Dalian Ocean University).
Chang M., Li B., Liao M., Rong X., Wang Y., Wang J., et al. (2022). Differential expression of miRNAs in the body wall of the sea cucumber Apostichopus japonicus under heat stress. Front. Physiol. 13. doi: 10.3389/fphys.2022.929094
Chen M. (2020). Comparative analysis of genome-wide methylation and RNA-seq in Apostichopus japonicus at different temperature. [master’s thesis] (Shanghai: Shanghai Ocean University).
Crabtree J. R., Macagno A. L. M., Moczek A. P., Rohner P. T., Hu Y. (2020). Notch signaling patterns head horn shape in the bull-headed dung beetle Onthophagus taurus. Dev. Genes Evol. 230, 213–225. doi: 10.1007/s00427-020-00645-w
Du H., Bao Z., Hou R., Wang S., Su H., Yan J., et al. (2012). Transcriptome sequencing and characterization for the sea cucumber Apostichopus japonicus (Selenka 1867). PloS One 7, e33311. doi: 10.1371/journal.pone.0033311
Entrambasaguas L., Ruocco M., Verhoeven K. J. F., Procaccini G., Marín-Guirao L. (2021). Gene body DNA methylation in sea grasses: Inter- and intraspecific differences and interaction with transcriptome plasticity under heat stress. Sci. Rep. 11, 14343. doi: 10.1038/s41598-021-93606-w
Feil R., Fraga M. F. (2012). Epigenetics and the environment: emerging patterns and implications. Nat. Rev. Genet. 13, 97–109. doi: 10.1038/nrg3142
Gao L., Yuan Z., Yu S., Yang Y., Li Y., He C. (2017). Genome-wide identification of HSP70/110 genes in sea cucumber Apostichopus japonicus and comparative analysis of their involvement in aestivation. Comparative biochemistry and physiology. Part D, Genomics & proteomics. 28, 162–171. doi: 10.1016/j.cbd.2018.07.005
Gilleron J., Nebout M., Scarabelli L., Senegas-Balas F., Palmero S., Segretain D., et al. (2006). A potential novel mechanism involving connexin 43 gap junction for control of sertoli cell proliferation by thyroid hormones. J. Cell. Physiol. 209, 153–161. doi: 10.1002/jcp.20716
Han L., Sun Y., Cao Y., Gao P., Quan Z., Chang Y., et al. (2021). Analysis of the gene transcription patterns and DNA methylation characteristics of triploid sea cucumbers (Apostichopus japonicus). Sci. Rep. 11, 7564. doi: 10.1038/s41598-021-87278-9
Holdsworth M. J., Vicente J., Sharma G., Abbas M., Zubrycka A. (2020). The plant n-degron pathways of ubiquitin-mediated proteolysis. J. Integr. Plant Biol. 62, 70–89. doi: 10.1111/jipb.12882
Huang Z., Xiao Q., Yu F., Gan Y., Lu C., Peng W., et al. (2021). Comparative transcriptome and DNA methylation analysis of phenotypic plasticity in the pacific abalone (Haliotis discus hannai). Front. Physiol. 12. doi: 10.3389/fphys.2021.683499
Huo D., Su F., Cui W., Liu S., Zhang L., Yang H., et al. (2022). Heat stress and evisceration caused lipid metabolism and neural transduction changes in sea cucumber: Evidence from metabolomics. Mar. pollut. Bull. 182, 113993. doi: 10.1016/j.marpolbul.2022.113993
Huo D., Sun L., Storey K. B., Zhang L., Liu S., Sun J., et al. (2020). The regulation mechanism of lncRNAs and mRNAs in sea cucumbers under global climate changes: Defense against thermal and hypoxic stresses. Sci. Total. Environ. 709, 136045. doi: 10.1016/j.scitotenv.2019.136045
Huo D., Sun L., Sun J., Lin C., Liu S., Zhang L., et al. (2021). Emerging roles of circRNAs in regulating thermal and hypoxic stresses in Apostichopus japonicus (Echinodermata: Holothuroidea). Ecotoxicol. Environ. Safety. 228, 112994. doi: 10.1016/j.ecoenv.2021.112994
Iamjan S. A., Thanoi S., Watiktinkorn P., Fachim H., Dalton C. F., Nudmamud-Thanoi S., et al. (2021). Changes of BDNF exon IV DNA methylation are associated with methamphetamine dependence. Epigenomics 13, 953–965. doi: 10.2217/epi-2020-0463
Kechin A., Boyarskikh U., Kel A., Filipenko M. (2017). CutPrimers: A new tool for accurate cutting of primers from reads of targeted next generation sequencing. J. Comput. Biol.: J. Comput. Mol. Cell Biol. 24, 1138–1143. doi: 10.1089/cmb.2017.0096
Krueger F., Andrews S. R. (2011). Bismark: A flexible aligner and methylation caller for bisulfite-seq applications. Bioinf. (Oxford England). 27, 1571–1572. doi: 10.1093/bioinformatics/btr167
Kuhl H., Tine M., Beck A., Timmermann B., Kodira C., Reinhardt R. (2011). Directed sequencing and annotation of three Dicentrarchus labrax l. chromosomes by applying Sanger- and pyrosequencing technologies on pooled DNA of comparatively mapped BAC clones. Genomics 98, 202–212. doi: 10.1016/j.ygeno.2011.06.004
Kulis M., Esteller M. (2010). DNA Methylation and cancer - ScienceDirect. Adv. Genet. 70, 27–56. doi: 10.1016/B978-0-12-380866-0.60002-2
Lechner M., Marz M., Ihling C., Sinz A., Stadler P. F., Krauss V. (2013). The correlation of genome size and DNA methylation rate in metazoans. Theory Biosci. 132, 47–60. doi: 10.1007/s12064-012-0167-y
Li C., Fang H., Xu D. (2019). Effect of seasonal high temperature on the immune response in Apostichopus japonicus by transcriptome analysis. Fish. Shellfish. Immunol. 92, 765–771. doi: 10.1016/j.fsi.2019.07.012
Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinf. (Oxford England). 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li X., Sun L. (2016). Toll-like receptor 2 of tongue sole Cynoglossus semilaevis: Signaling pathway and involvement in bacterial infection. Fish. Shellfish. Immunol. 51, 321–328. doi: 10.1016/j.fsi.2016.03.001
Li S., Sun G., Li X., Li G., Yang J., Yang S. (2017). Characteristics of epigenetic regulation of related genes under high temperature stress in sea cucumber Apostichopus japonicus. J. Fishery. Sci. China. 24, 470–476. doi: 10.3724/SP.J.1118.2017.16275
Li X., Wang C., Li N., Gao Y., Ju Z., Liao G., et al. (2021). Combined effects of elevated temperature and crude oil pollution on oxidative stress and apoptosis in sea cucumber (Apostichopus japonicus, selenka). Int. J. Environ. Res. Public Health 18, 801. doi: 10.3390/ijerph18020801
Li Y., Wang R., Xun X., Wang J., Bao L., Thimmappa R., et al. (2018). Sea Cucumber genome provides insights into saponin biosynthesis and aestivation regulation. Cell Discov. 4, 29. doi: 10.1038/s41421-018-0030-5
Liu S., Xu X., Fang Z., Ning Y., Deng B., Pan X., et al. (2021). Piezo1 impairs hepatocellular tumor growth via deregulation of the MAPK-mediated YAP signaling pathway. Cell Calcium. 95, 102367. doi: 10.1016/j.ceca.2021.102367
Lou S., Lee H. M., Qin H., Li J. W., Gao Z., Liu X., et al. (2014). Whole-genome bisulfite sequencing of multiple individuals reveals complementary roles of promoter and gene body methylation in transcriptional regulation. Genome Biol. 15, 408. doi: 10.1186/s13059-014-0408-0
Love M. I., Huber W., Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/s13059-014-0550-8
Meloty-Kapella C. V., Degen M., Chiquet-Ehrismann R., Tucker R. P. (2006). Avian tenascin-W: expression in smooth muscle and bone, and effects on calvarial cell spreading and adhesion in vitro. Dev. Dynamics. 235, 1532–1542. doi: 10.1002/dvdy.20731
Meurette O., Mehlen P. (2018). Notch signaling in the tumor microenvironment. Cancer Cell. 34, 536–548. doi: 10.1016/j.ccell.2018.07.009
Palou-Márquez G., Subirana I., Nonell L., Fernández-Sanlés A., Elosua R. (2021). DNA Methylation and gene expression integration in cardiovascular disease. Clin. Epigenet. 13, 75. doi: 10.1186/s13148-021-01064-y
Pertea M., Kim D., Pertea G. M., Leek J. T., Salzberg S. L. (2016). Transcript level expression analysis of RNA-seq experiments with HISAT, StringTie and ballgown. Nat. Protoc. 11, 1650–1667. doi: 10.1038/nprot.2016.095
Pertea G., Pertea M. (2020). GFF utilities: GffRead and GffCompare. F1000Research 9, J–304. doi: 10.12688/f1000research.23297.2
Radons J. (2016). The human HSP70 family of chaperones: Where do we stand. Cell Stress Chaperones. 21, 379–404. doi: 10.1007/s12192-016-0676-6
Sajadimajd S., Mohammadi B., Bahrami G., Madani S. H., Hatami R., Miraghaee S. S. (2022). Modulation of notch signaling and angiogenesis via an isolated polysaccharide from momordica charantia in diabetic rats. J. Food Biochem. 46, e14033. doi: 10.1111/jfbc.14033
Song Q., Decato B., Hong E. E., Zhou M., Fang F., Qu J., et al. (2013). A reference methylome database and analysis pipeline to facilitate integrative and comparative epigenomics. PloS One 8, e81148. doi: 10.1371/journal.pone.0081148
Sun L., Chen M., Yang H., Wang T., Liu B., Shu C., et al. (2011). Large Scale gene expression profiling during intestine and body wall regeneration in the sea cucumber apostichopus japonicus. comparative biochemistry and physiology. Part D. Genomics Proteomics. 6, 195–205. doi: 10.1016/j.cbd.2011.03.002
Sun H., Zhou Z., Dong Y., Yang A., Jiang J. (2020). Insights into the DNA methylation of sea cucumber Apostichopus japonicus in response to skin ulceration syndrome infection. Fish. Shellfish. Immunol. 104, 155–164. doi: 10.1016/j.fsi.2020.05.005
Treweek T. M., Meehan S., Ecroyd H., Carver J. A. (2015). Small heat-shock proteins: Important players in regulating cellular proteostasis. Cell. Mol. Life Sci. 72, 429–451. doi: 10.1007/s00018-014-1754-5
Venkataraman Y. R., White S. J., Roberts S. B. (2022). Differential DNA methylation in pacific oyster reproductive tissue in response to ocean acidification. BMC Genomics 23, 556. doi: 10.1186/s12864-022-08781-5
Wang X., Li A., Wang W., Zhang G., Li L. (2021). Direct and heritable effects of natural tidal environments on DNA methylation in pacific oysters (Crassostrea gigas). Environ. Res. 197, 111058. doi: 10.1016/j.envres.2021.111058
Wang T., Yang H., Zhao H., Chen M., Wang. B. (2011). Transcriptional changes in epigenetic modifiers associated with gene silencing in the intestine of the sea cucumber, Apostichopus japonicus (Selenka), during aestivation. Chin. J. Oceanol. Limnol. 29, 1267–1274. doi: 10.1007/s00343-011-0143-2
Wen Z., Zuo S., Chen M., Zhou H., Sun G., Feng Y., et al. (2021). DNA Methylation level of genomic DNA of Apostichopus japonicus. J. Fisheries. China. 42, 46–54. doi: 10.19663/j.issn2095-9869.20201204002
Wu B., Yang A., Sun X., Liu Z., Zhou L., Gong Y. (2016). Effects of acute temperature stress on genome-wide DNA methylation profiles in Patinopecten yessoensis. Prog. Fishery. Sci. 37, 140–146. doi: 10.11758/yykxjz.20150826001
Xiao H., Jiang Q., Qiu H., Wu K., Ma X., Yang J., et al. (2021). Gastrodin promotes hippocampal neurogenesis via PDE9-cGMP-PKG pathway in mice following cerebral ischemia. Neurochem. Int. 150, 105171. doi: 10.1016/j.neuint.2021.105171
Xu D., Sun L., Liu S., Zhang L., Yang H. (2015). Histological, ultrastructural and heat shock protein 70 (HSP70) responses to heat stress in the sea cucumber Apostichopus japonicus. Fish. Shellfish. Immunol. 45, 321–326. doi: 10.1016/j.fsi.2015.04.015
Xu D., Sun L., Liu S., Zhang L., Yang H. (2016). Molecular cloning of hsf1 and hsbp1 cDNAs, and the expression of hsf1, hsbp1 and hsp70 under heat stress in the sea cucumber Apostichopus japonicus. Comparative biochemistry and physiology. Part B. Biochem. Mol. Biol. 198, 1–9. doi: 10.1016/j.cbpb.2016.03.001
Xu D., Zhou S., Sun L. (2018). RNA-Seq based transcriptional analysis reveals dynamic genes expression profiles and immune-associated regulation under heat stress in Apostichopus japonicus. Fish. Shellfish. Immunol. 78, 169–176. doi: 10.1016/j.fsi.2018.04.037
Yamazaki Y., Sakai Y., Yu J., Mino S., Sawabe T. (2020). Tracking the dynamics of individual gut microbiome of sea cucumber Apostichopus japonicus during gut regeneration. PeerJ 8, e10260. doi: 10.7717/peerj.10260
Yang Y., Zheng Y., Sun L., Chen M. (2020). Genome-wide DNA methylation signatures of sea cucumber Apostichopus japonicus during environmental induced aestivation. Genes 11, 1020. doi: 10.3390/genes11091020
Yuan C., Mao J., Sun H., Wang Y., Guo M., Wang X., et al. (2021). Genome-wide DNA methylation profile changes associated with shell colouration in the yesso scallop (Patinopecten yessoensis) as measured by whole-genome bisulfite sequencing. BMC Genomics 22, 740. doi: 10.1186/s12864-021-08055-6
Zhang Y., Yu S., Liao M., Dong Y. (2022). Evaluation and prediction of extreme high temperature on sea cucumber (Apostichopus japonicus) pond aquaculture in China. Journal of Fishery Sciences of China. 29, 408–420. doi: 11.3446. S.20211108.1003.002
Zhao Y., Chen M., Storey K. B., Sun L., Yang H. (2015). DNA Methylation levels analysis in four tissues of sea cucumber Apostichopus japonicus based on fluorescence-labeled methylation-sensitive amplified polymorphism (F-MSAP) during aestivation. Comp. Biochem. Physiol. Part B. Biochem. Mol. Biol. 181, 26–32. doi: 10.1016/j.cbpb.2014.11.001
Zhao Y., Yang H., Storey K. B., Chen M. (2014). RNA-Seq dependent transcriptional analysis unveils gene expression profile in the intestine of sea cucumber Apostichopus japonicus during aestivation. Comp. Biochem. Physiol. Part D. Genomics Proteomics. 10, 30–43. doi: 10.1016/j.cbd.2014.02.002
Zhou X., Cui J., Liu S., Kong D., Sun H., Gu C., et al. (2016). Comparative transcriptome analysis of papilla and skin in the sea cucumber, Apostichopus japonicus. PeerJ 4, e1779. doi: 10.7717/peerj.1779
Keywords: Apostichopus japonicus, DNA Methylation, transcription patterns, heat stress, regulation mechanism
Citation: Chang M, Ge J, Liao M, Rong X, Wang Y, Li B, Li X, Wang J, Zhang Z, Yu Y and Wang C (2023) Genome-wide DNA methylation and transcription analysis reveal the potential epigenetic mechanism of heat stress response in the sea cucumber Apostichopus japonicus. Front. Mar. Sci. 10:1136926. doi: 10.3389/fmars.2023.1136926
Received: 03 January 2023; Accepted: 14 February 2023;
Published: 03 March 2023.
Edited by:
Gen Hua Yue, Temasek Life Sciences Laboratory, SingaporeReviewed by:
Guohua Sun, Ludong University, ChinaCopyright © 2023 Chang, Ge, Liao, Rong, Wang, Li, Li, Wang, Zhang, Yu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meijie Liao, bGlhb21qQHlzZnJpLmFjLmNu; Yingeng Wang, d2FuZ3lnQHlzZnJpLmFjLmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.