 Ezra Curtis
Ezra Curtis Joanne Moseley1
Joanne Moseley1 Riccardo Racicot
Riccardo Racicot Rachel M. Wright
Rachel M. Wright- 1Department of Biological Sciences, Smith College, Northampton, MA, United States
- 2Department of Biological Sciences, Southern Methodist University, Dallas, TX, United States
Introduction: Exaiptasia diaphana is a popular model organism for exploring the symbiotic relationship observed between cnidarians and their microsymbionts. While physiological roles of algal photosymbionts (Symbiodinaceae) are well studied, the contributions of bacterial communities are less defined in this system.
Methods: We investigated microbial variation between distinct parts of the body and symbiotic state across four genets held in identical environmental conditions using 16s rRNA gene amplicon sequencing.
Results: We found differentially abundant taxa between body part and symbiotic state that highlight the roles these bacteria may play in holobiont heterotrophy and nutrient cycling. Beta-diversity analysis revealed distinct communities between symbiotic states consistent with previous studies; however, we did not observe the presence of previously reported core microbiota. We also found community differences across clonal lines, despite years of identical rearing conditions.
Conclusion: These findings suggest the Exaiptasia bacterial microbiome is greatly influenced by host genetics and unpredictable environmental influences.
Introduction
Coral reefs are estimated to support 25% of all marine species (Fisher et al., 2015) and have immense economic value (Costanza et al., 2014). Due to rising pressures from the global climate crises, coral reefs are declining (Gardner et al., 2003). Increased intensity, duration, and frequency of heatwaves in recent decades have led to regular coral mass bleaching events resulting in high mortality (Hughes et al., 2018a; Hughes et al., 2018b). Rising sea temperatures have also been associated with increased disease susceptibility in corals (Miller et al., 2009; Muller et al., 2018). Researching strategies to mitigate harm to coral reefs is essential. To do so, we must first understand the dynamics contributing to coral health and fitness.
Corals have a well-described relationship with their algal endosymbiont belonging to the family Symbiodineceae, however the contributions of microbiota to coral health and resilience have only recently begun to be explored. Cycling of carbon, nitrogen, sulfur, and host protection through produced antimicrobial compounds and competition with potential opportunistic bacteria are among possible functions of coral-associated bacteria (Daniels et al., 2015; Rädecker et al., 2015; Bourne et al., 2016; Silveira et al., 2017). Performing research with corals has presented scientists with many challenges due to its difficult laboratory maintenance. Such difficulties have slowed progress in understanding the working mechanisms behind coral function. Thus, Exaiptasia diaphana has emerged as a model organism due to its ease of lab maintenance and association with the same algal symbionts as corals. E. diphana’s ability to be kept in symbiotic and aposymbiotic states has also allowed for comparative studies between the two states (Weis et al., 2008). Characterizing the microbiome of E. diaphana opens the potential for use as a model organism for microbial studies.
To date, a limited number of studies have focused on the microbiome of E. diaphana. Previous work has implicated symbiotic state as influential on the anemones’ microbiome (Röthig et al., 2016). The acontia of anemones have also been shown to be associated with a unique microbial compared to the whole anemone (Maire et al., 2021). However, studies focusing on microbiome analysis including anemones from distinct genetic backgrounds are scarce. Comparative studies by Herrera et al. (2017); Brown et al. (2017), and Hartman et al. (2020) identified significant differences between clonal lines. Here, we expand on these findings by analyzing microbiota associated with anemones belonging to four distinct clonal lines reared in identical environmental conditions across symbiotic state and anemone body part. Through this analysis, we uncover possible drivers of variation and identify taxa with potential functional importance.
Methods and materials
Anemone rearing and dissection
Prior to sequencing, stock populations of the anemones were maintained in replicate 10-gallon tanks at 25 degrees in 34 ppt Instant Ocean artificial seawater (ASW). Each tank contained one genet (H2, CC7, VWA, or VWB) in one symbiotic state (symbiont-rich or symbiont-depleted). Symbiont-depleted anemones were subjected to a bleaching protocol as previously described (Lehnert et al., 2012) and maintained in dark conditions for approximately 6–9 months before nucleic acid isolation. Symbiotic states were defined based upon pale appearance to the naked eye, although the symbiont-depleted anemones from all genets retained some algal cells (Figure 1). The H2 and VWB clonal strains originate from Coconut Island, HI (Xiang et al., 2013). VWA is a multi-origin strain (Poole et al., 2016). CC7 was originally obtained from Carolina Biological Supply (Sunagawa et al., 2009). Symbiont-rich anemones were kept on a 12:12 light:dark cycle using white LED lights at about 12µmol photons/m/s. All anemones were fed freshly hatched Artemia shrimp nauplii 2–3x weekly and were subject to regular salinity checks and water changes.
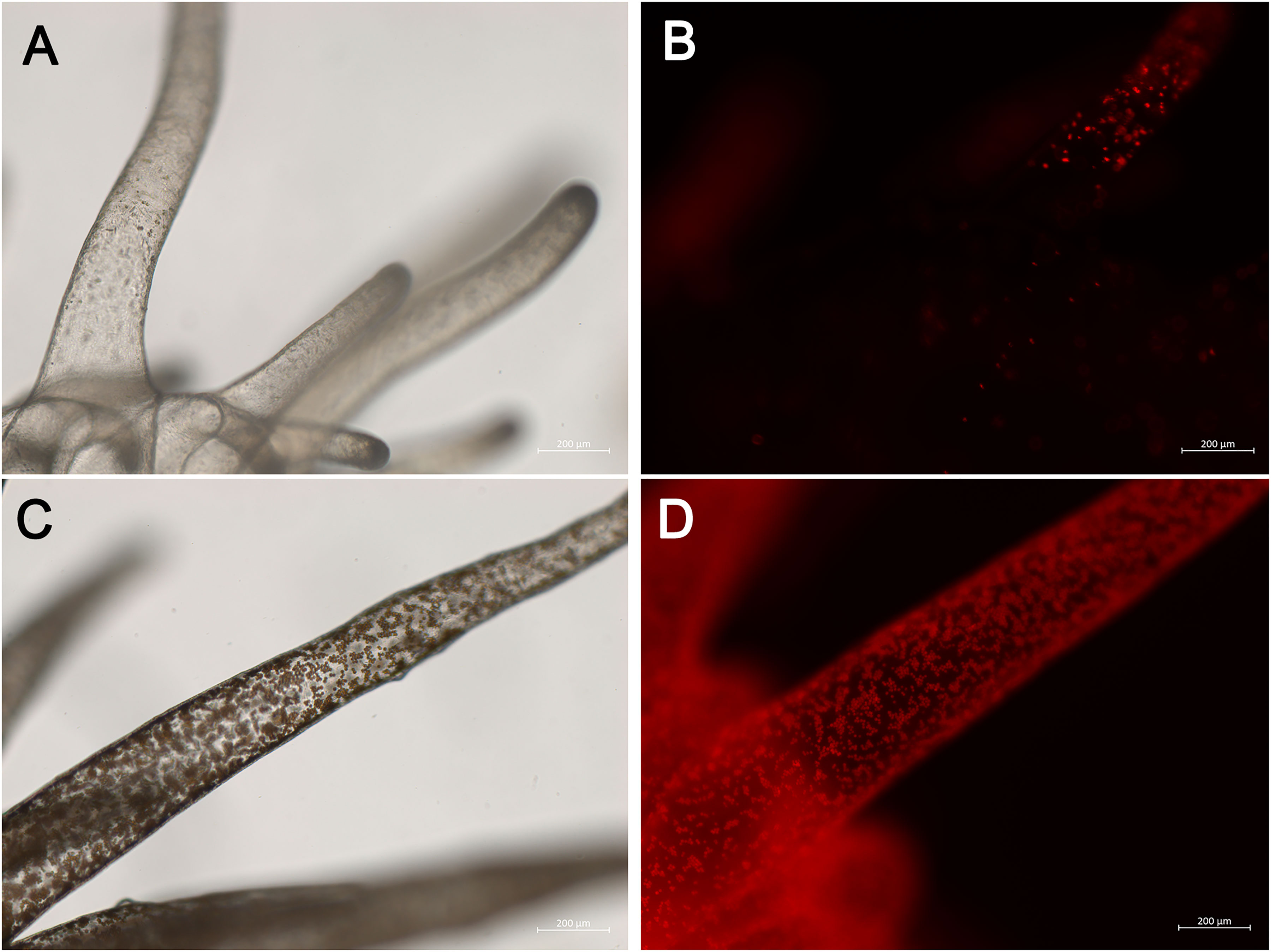
Figure 1 Images of sea anemone E. diaphana demonstrating symbiont-depleted (A, B) and symbiont-rich (C, D) states. Images acquired using the Zeiss Axio Imager M2 Light Microscope under brightfield (A, C) or fluorescent light (B, D) and Zeiss Zen Image Acquisition and Processing software.
Anemones were transferred to a 6-well plate for each symbiotic state and clonal line (genet), with one or two individuals per well in 3mL ASW. The anemones in the 6-well plates were kept in an incubator (Percival AL30L2) for a week of acclimation under optimal conditions including white light on a 12:12 schedule and twice weekly feedings with freshly hatched Artemia shrimp nauplii.
After the acclimation period, anemones were anesthetized for approximately 10 minutes and rinsed in a solution of sterile 0.5% MgCl2 in ASW in order to relax their bodies for dissection. Once the anemones were anesthetized, they were dissected using a dissecting microscope, fine forceps, and a needle. The anemones were sliced just under the oral disc, resulting in the separation of the oral disc and tentacle region, and the stem and peduncle region. Anemones were kept separate by genet and symbiotic state combination during the dissection process, and the tools were sterilized and wiped clean with ethanol rinses and clean lint-free disposable cloths between uses.
Once the anemones in each group (3–6 replicates each) were dissected into top (oral disc and tentacles) and bottom (stem and peduncle), they were placed into 1.5mL Eppendorf tubes. Each tube was then labeled with a 4-character code indicating the genet, symbiotic state, replicate, and body part, and placed in a -80°C freezer for storage.
Nucleic acid isolation
Nucleic acid isolation was performed using a modified SlimeAway protocol (Vargas et al., 2021). Anemone samples were first homogenized using a motorized pestle inside their Eppendorf tubes on dry ice. Next, a CTAB extraction buffer was prepared by mixing 17µl molecular grade water, 28µl 0.5M EDTA, 56µl 1M TRIS, 224µl 5M NaCl solution, 112µl 0.1M PVP (Polyvinylpyrrolidone K30) solution, and 112µl 0.1M CTAB (Cetyltrimethylammoniumbromide) solution per sample. The buffer was incubated in a water bath at 60°C for 10 minutes, after which 12µl β-Mercaptoethanol per sample was added to the buffer. 550µl CTAB buffer was then added to each sample tube, followed by 5µl Proteinase K, inverting the tube to mix. Samples were incubated at 56°C for between 2 hours to overnight, inverting the tubes regularly to avoid tissue clumping, until there were no visible tissue clumps in solution.
Once the solution was free of clumps, 150µl 5M KoAc solution was added to each tube, and they were incubated again at 56°C for 10 minutes. The samples were then centrifuged for 15 minutes at 4°C at 15000xg, ~600µl supernatant was removed avoiding any pellet or viscous precipitate, and 600µl (1 volume) chloroform was added. After this addition, samples were shaken vigorously for 20 seconds then centrifuged again at the same parameters. Finally, ~400µl of the supernatant was transferred, avoiding the interphase, to a new, labeled 1.5mL Eppendorf tube.
DNA was precipitated using 400µl (1 volume) isopropanol added to each sample, then sample tubes were shaken for 15 seconds before being incubated at room temperature for 10 minutes. They were placed in the -20°C freezer overnight. The next day, the samples were centrifuged at 4°C for 30 minutes at 15000xg. The isopropanol supernatant was discarded without disturbing the pellet, and 62.5µl cold (-20°C) 100% ethanol, 1µl 20mg/mL glycogen, and 2.5µl 3M NaAc solution were added to each sample and mixed. The samples were incubated at -20°C between 2 hours and overnight, then centrifuged at 4°C at 15000xg for 30 minutes. Next the supernatant was carefully removed, the pellet was washed with 500µl cold (-20°C) 75% ethanol and vortexed, and the pellet was centrifuged again with the same settings for 15 minutes. This ethanol removal, wash, and spin step was repeated one more time. The supernatant was once again removed, and the pellet was allowed to dry for 10 minutes, followed by resuspension in 30µl molecular-grade water and stored at -80°C. Samples were treated with Monarch RNase A and subsequently purified with the Monarch PCR & DNA Clean Up Kit (New England Biolabs, Massachusetts). All samples were eluted in 30 µL 1X TE buffer after cleanup. DNA was then quantified using the Qubit HS ssDNA kit and assessed for quality on the Nanodrop 8000 spectrophotometer (ThermoFisher, California).
Library preparation
16S ribosomal RNA libraries were generated from the V3-V4 variable region according to the Illumina 16S Metagenomic Sequencing Library Preparation guide. Samples were normalized to 5 ng/µL in 1X TE buffer. 550 bp amplicons from the V3-V4 region of the rRNA gene were generated for each sample The primers included the V3-V4 locus specific sequences underlined below based on Klindworth et al., 2013, as well as an Illumina overhang adapter sequence.
Forward 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG
Reverse 5’ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC
Amplicons were cleaned using HighPrep beads (MagBio, Maryland) and target size was verified using Fragment Analyzer (Agilent, California). Illumina Nextera DNA CD unique dual indexes were added to the amplicons, then cleaned using HighPrep beads (MagBio, Maryland). Final libraries were quantified using the Qubit HS DNA assay (Thermo Scientific, California) and library size was verified using Fragment Analyzer (Agilent, California).
Sequencing
52 amplicon libraries were normalized to 2 nM, then pooled together in equal volumes. The library pool was then denatured and diluted following the MiSeq dilute and denature guide. Sequencing was performed on the Illumina MiSeq using a paired end 600 cycle v3 sequencing kit (Illumina, California).
Analysis
The DADA2 (Callahan et al., 2016) pipeline was followed in R v. 4.2.1 (R Core Team, 2022) for filtering, trimming, denoising sequences, merging reads, and performing chimeric checks. All statistical analysis and data visualization was performed using R. Silva ver. 132 was used to assign taxonomy. Non-bacterial sequences were removed from downstream analysis. Sequence data was compiled into a Phyloseq object to perform diversity and abundance analyses (McMurdie and Holmes, 2013). Data were not normalized prior to diversity analyses. Alpha-diversity was measured using the observed number of ASVs, the Shannon index, and Pielou’s evenness. Beta-diversity distances were calculated using the Bray-Curtis method. Vegan (Oksanen et al., 2022) was used to create rarefaction curves. Data was visualized using GGplot2 (Wickham, 2016) and Vegan (Oksanen et al., 2022). DESeq2 (Love et al., 2014) was used for differential abundance analysis. The results of the differential abundance analysis were further filtered using a threshold of ⅔ presence across samples belonging to the differentially abundant category of interest to focus on ASVs that were commonly observed. Core microbiome analysis was performed by making lists of ASVs present in 100% of samples. We also searched for a core set of ASVs present in each category of analysis by sub-setting samples by clonal line, body part, and symbiotic state, similar to Röthig et al. (2016). Results were considered significant at an alpha level of 0.05.
Results
We performed 16S rRNA gene metabarcoding on E. diaphana across symbiotic state and distinct body parts. After merging, denoising, and chimera filtering, 1,106,571 reads remained (Supplementary File S1). Read depth ranged from 538 to 102,169 reads per sample. A total number of 489 ASVs were observed across samples, which is consistent within an order of magnitude with previous studies in E. diaphana (e.g., Hartman et al., 2020; Xiang et al., 2022). The majority of reads belonged to rare ASVs. Rarefaction curves plateaued, indicating sufficient sequencing depth (Supplementary Figure S1).
Diversity metrics across body part, symbiotic state, and genet
Alpha-diversity metrics (observed ASVs, Shannon’s index, Pielou’s evenness) did not indicate any significant differences between symbiotic states and body parts (Figure 2). Pairwise Wilcox rank-sum tests using FDR correction showed significant differences between observed alpha-diversity between four genet pairings, however only one genet pairing showed significant difference using the Shannon metric for alpha-diversity. Anemones from the VWA and VWB clonal lines displayed greater evenness compared to anemones belonging to CC7 and H2 (Figure 2C). Beta-diversity was visualized via principal coordinates analysis (PCoA) based upon the Bray-Curtis dissimilarity index reveals some clustering by symbiotic state and genet (Figure 3). Results of permutational multivariate analysis of variance, where the R2 value represents the proportion of variation in the distance matrix explained by each variable, confirmed substantial variation driven by genet (R2 = 0.31; p = 0.001) and symbiotic state (R2 = 0.03, p = 0.02), but not body part (R2 = 0.02, p = 0.146). Additional pairwise comparisons were performed between each individual genet. Differences between each pairing were significant, further cementing our finding that genets have distinct microbial communities.
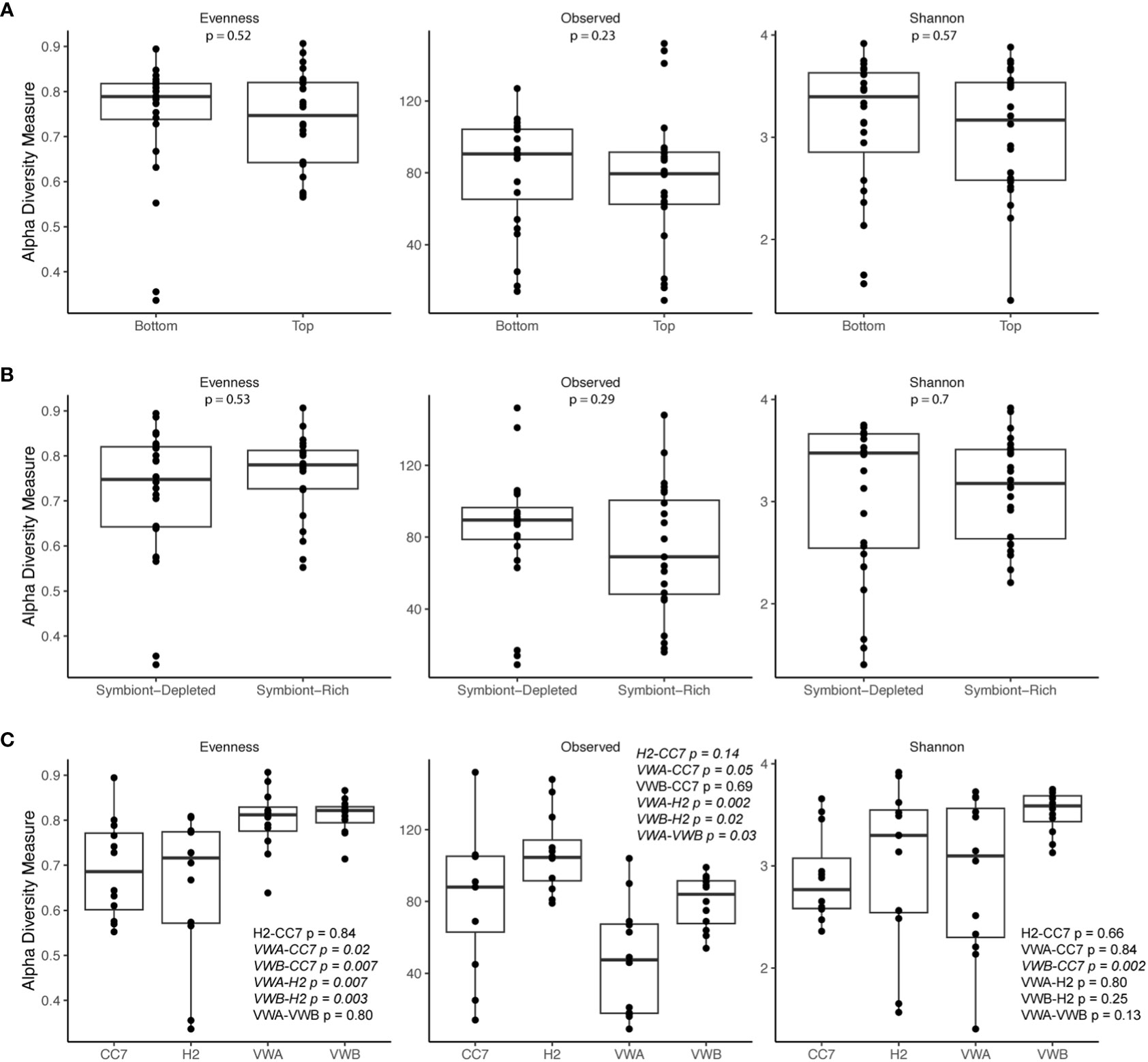
Figure 2 Alpha diversity metrics (Observed, Shannon’s index, Pielou’s evenness) of bacterial communities from body parts (A), symbiotic states (B), and genets (C). The bar represents the median. Outer boundaries of the box represent first and third quartiles. Points represent individual samples. P-values are based on pairwise Wilcox rank-sum tests using FDR correction, with values < 0.05 italicized.
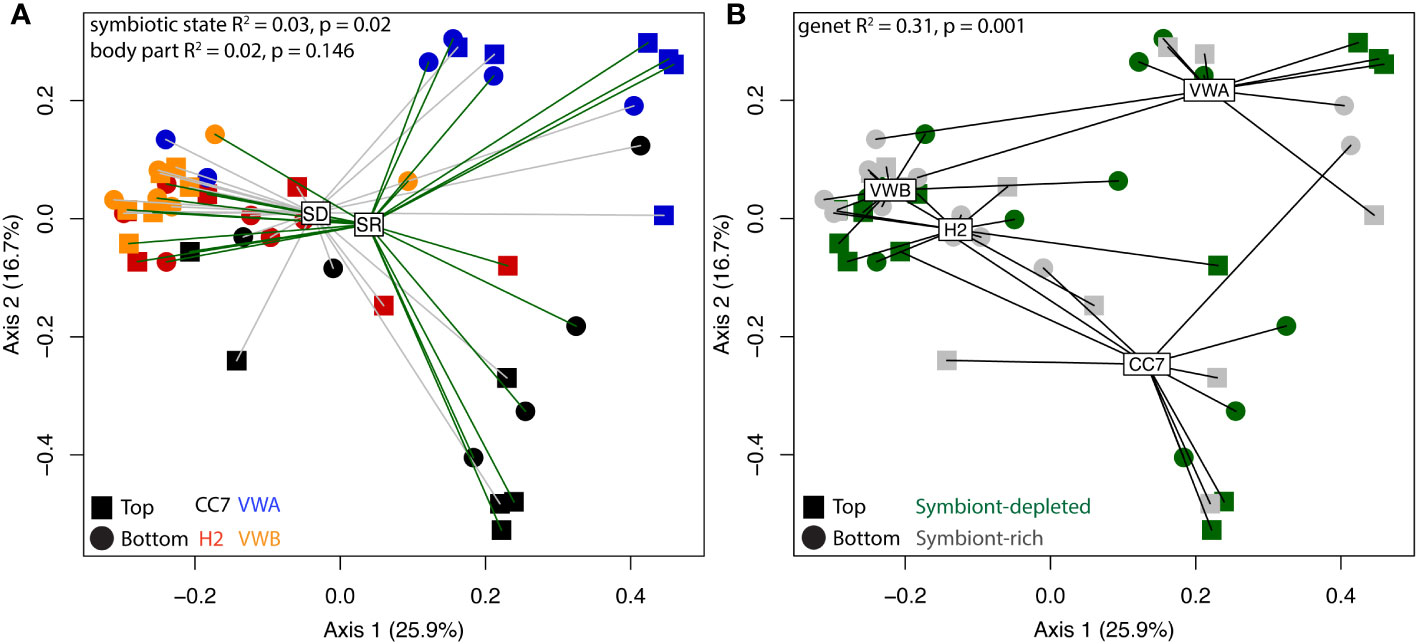
Figure 3 Principal coordinate analysis (PCoA) visualization using Bray-Curtis distances on abundance of ASVs between anemones. Spider-clustering groups samples by symbiont-depleted (SD) and symbiont-rich (SR) anemones (A) and genet (B). Shapes represent body parts and colors represent genets, according to the inset legend. R-square and p-values were generated by permutational multivariate analysis of variance (adonis).
Taxonomic differences between body part, symbiotic state, and genet
The mean relative abundance of bacteria was used to provide an overview of dominant ASVs at multiple taxonomic levels across sample types. ASVs originating from Proteobacteria had the highest relative abundance (74%), followed by Plancomyetes (11%), Actinobacteria (4.5%) and Lentisphaerae (3.5%) (Figure 4). The abundance profile of phyla remained similar between genets, body parts, and symbiotic states.
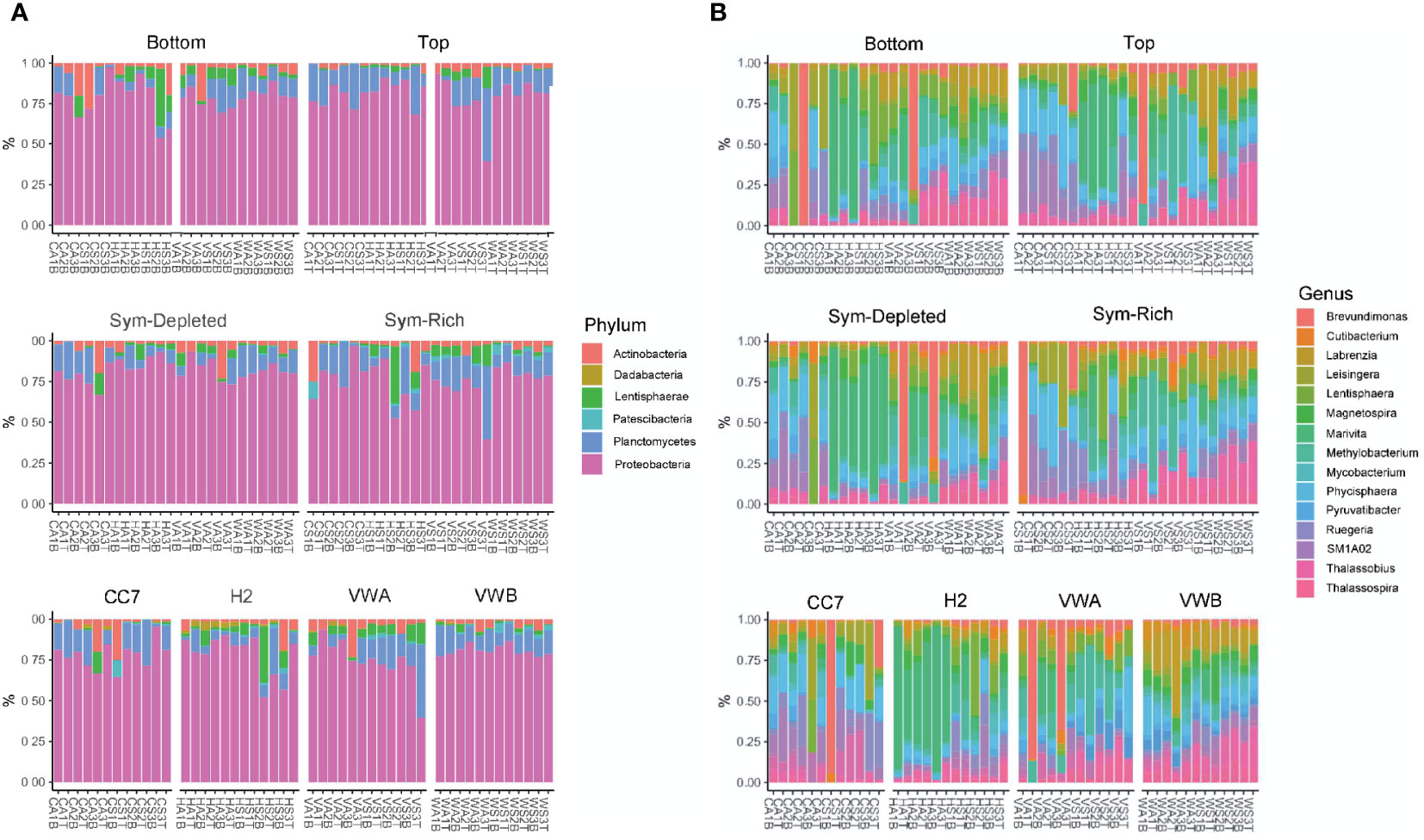
Figure 4 Bacterial community composition at the level of phylum (A) and genus (B). Each color represents a genus or phylum with a relative abundance >1% across all samples.
At the family level, ASVs belonging to Rhodobacteraceae (21%), Phycisphaeraceae (8.3%), Teraskiellaceae (5.7%), Beijerinckiaceae (5.2%), Caulobacteraceae (4.4%), and Stappiaceae (3.9%) were common. Samples from the VWA clonal line abundances revealed greater evenness between families, with Beijerinckiaceae (12.4%) and Caulobacteraceae (11.2%) being particularly common. In contrast, Stappiaceae (10.2%) was particularly abundant within the VWB genet. Abundances were much more variable across genets at the genus level, however Marivita, Methylobacterium, Phycisphaera, Brevundimonas, Labrenzia, and Thalassobius were common. A core microbiome, defined by the presence of an ASV in 100% of samples, was not identified by our data. Similarly, no ASVs were found to be universal within each body part, symbiotic state, nor clonal line.
Differential abundance analysis
To further investigate taxonomic differences between anemone body part and symbiotic state, differential abundance analysis was performed using DESeq2. We identified a total of 91 differentially abundant ASVs between symbiotic states (Supplementary File S2) and 32 differentially abundant ASVs between anemone body parts (Supplementary File S3; Figure 5). To account for the compositional dissimilarity between samples, we considered ASVs found in at least ⅔ of samples belonging to the category of interest as part of a “common” group. 12 ASVs in this common group were shown to be significantly more abundant in symbiont-depleted anemones than symbiont-rich anemones, including ASVs belonging to Marivita, Magnetospira, Nitrospina, Kordiimonas and Mycobacterium. In contrast, only 2 ASVs, belonging to Mesorhizobium and Maritalea, were found to be significantly more abundant in symbiont-rich anemones with modest L2FC difference. Only 7 common and differentially abundant ASVs between anemone body parts were identified, all of which were more abundant in the bottom half of the anemone. Visualization of the differential abundance analysis results reveal greater consistency in abundance of abundant ASVs in symbiont-depleted anemones compared to symbiont-rich anemones, implicating algal-symbiont identity as a possible driver of variation (Supplementary Figure S2).
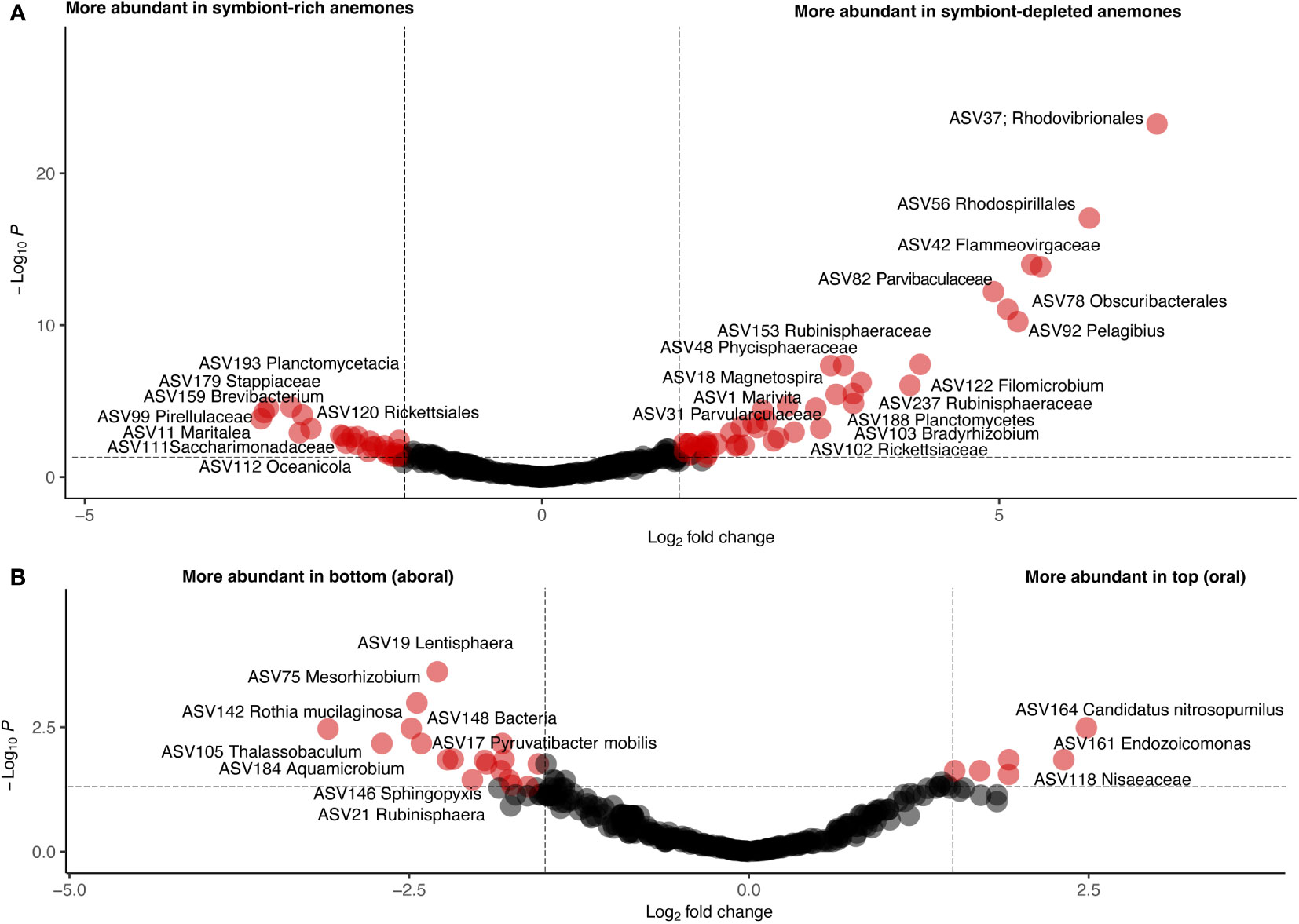
Figure 5 Volcano plots showing differentially abundant ASVs by symbiotic state (A) and body part (B). Each circle represents one ASV. A p-value of 0.05 and a log2-fold change of 2 are indicated by dotted lines. ASVs meeting these thresholds are colored red and labeled with their assigned taxonomy.
Discussion
The bacterial microbiome is increasingly appreciated as a component of holobiont health in systems ranging from honey bees (Raymann and Moran, 2018) to humans (Cho and Blaser, 2012). Reef-building corals also depend on fragile associations with healthy microbes (Gilbert et al., 2012; Glasl et al., 2016), and microbe manipulation is currently being considered as a tool to help corals resist devastating effects of climate change (Epstein et al., 2019; Rosado et al., 2019; Doering et al., 2021; Li et al., 2022). In this study, we characterized microbial variation in a lab-tractable organism often used to investigate basic cnidarian biology, Exaiptasia diaphana. Though other studies have previously examined the bacterial microbiome in E. diaphana (Röthig et al., 2016; Dungan et al., 2020; Costa et al., 2021; Maire et al., 2021), this study compares across clonal lines and body part, in addition to algal symbiotic state. Comparing microbiota across genets, which were reared in independent tanks but otherwise identical conditions for years, helps elucidate the role that host genetics may play in structuring bacterial communities. We did not identify a core microbiome consisting of taxa present across all samples, consistent with previous findings of either no or few shared taxa across clonal lines (Brown et al., 2017; Herrera et al., 2017). The absence of shared taxa between anemones raised in identical rearing conditions, including water and Artemia from a shared bulk supply, suggests possible genetic drivers of microbial variation. Diversity analyses further confirmed microbiome clustering by genotype. While core microbiomes have been identified across genets previously in E. diaphana (Hartman et al., 2020; Maire et al., 2021), these studies sourced genets from similar geographic regions and therefore are likely less genetically distinct than the clonal lines used here, such as CC7 sourced from off the southeastern coast of the United States and H2 and VWB sourced from Hawaii (Sunagawa et al., 2009; Xiang et al., 2013). Previous work in another anemone, Nematostella vectensis, also found differences in bacterial communities across different source populations of animals despite years of laboratory rearing under identical conditions (Mortzfeld et al., 2016). We also note that we used ethanol wipes to sterilize equipment between samples. Ethanol would not remove DNA from dissecting equipment, but our repeated wiping and rinsing has sufficed to prevent nucleic acid contamination in the past. However, we did not treat our equipment with DNase between samples. Cross-contamination may have reduced our ability to detect differentially abundant ASVs between symbiotic states. However, the stark differences in bacterial communities between genets support a hypothesis that cross-contamination was minimal.
Community composition at the phylum level for previously characterized CC7 and H2 clonal lines is consistent with previous findings, however highly inconsistent at lower levels of classification (Röthig et al., 2016; Herrera et al., 2017). For example, Herrera et al. (2017) and Röthig et al. (2016) identified Kordia and Glaciecola as the most abundant genera in H2 and CC7 anemones respectively, but these genera were absent from these anemones. Similarity at higher orders of classification may suggest that some functional roles are being filled by alternate taxa. For example, Rhodobacteraceae, a family highly abundant in E. diaphana, is known for carbon and sulfur cycling capabilities (Pujalte et al., 2014). Similarly, Beijerinckiaceae are known for their nitrogen fixation capabilities (Marín and Arahal, 2014). While these anemones are treated as clonal lines in labs across the globe, live culture through clonal division allows unique mutations to accumulate. Thus, genetic differences between “clones” in different facilities may explain some incongruence of genet-specific microbiota across studies.
Comparing the results described here to other studies using the same lines (e.g., CC7 in Röthig et al., 2016), reveals how strong environmental effects may limit how confidently we can determine the roles of specific taxa in general holobiont biology. One source of this variation could be symbiont identity, as Röthig et al. (2016) inoculated the symbiotic anemones with a specific algal culture. Röthig et al. also contrasts from our study starved prior to sampling. Diet has been implicated as a driver of microbiome variation in other systems, including cold water corals, and thus may account for some of the observed differences within the genet (Kartzinel et al., 2019; Galand et al., 2020; Delaroque et al., 2022). Further investigations of the environmental factors that initially shape distinct bacterial communities, and the molecular factors that allow decades-long persistence in new environments, could help optimize microbiome engineering efforts. Additionally, future studies should continue to investigate genetic and environmental drivers of bacterial microbiome variation to further assess the amount of variation one can expect between systems in this model organism. We should also focus on what can be learned about host health without the requirement of identifying specific taxa that may have only brief or regional associations with reef organisms. For example, functional characterization of microbiota can reveal similarities in general metabolic capability and thus functional capability between communities each composed of unique taxa (Zhang et al., 2015). This concept of functional redundancy has been demonstrated within the human microbiome and has been suggested in studies focusing on coral microbiomes (Hernandez-Agreda et al., 2018; Tian et al., 2020).
No large-scale differences between anemone body parts were observed, contrasting from a previous study comparing acontia to whole anemone microbiomes (Maire et al., 2021). However, the acontia in our samples were not expelled prior to dissection and therefore may be represented in the bottom (i.e., lower stalk and pedal disk) of our samples. Future studies may identify more distinctions in microbial communities by finer dissection, e.g., pedal disk vs. stalk vs. mouth, in Exaiptasia. For example, trisection analysis performed in Nematostella revealed an abundance of Spirochaetaceae in the capitulum relative to two aboral sections of these anemones (Bonacolta et al., 2021). We identified 32 differentially abundant ASVs between body parts, seven of which were present after filtering for consistency between samples. Each of those seven ASVs were more abundant in the lower body region than in the upper mouth and tentacles. One ASV belongs to Terasakiellaceae, which has been implicated in nitrogen cycling in a cold-water coral (Weiler et al., 2018).
Symbiont-depleted anemones displayed higher structural consistency compared to symbiont-rich anemones. This variation could be explained by differing symbiont identity between the anemones. Characterizing algal symbionts was beyond the scope of this work, but previous work has demonstrated stable associations between algal symbiont types and Exaiptasia genets. For example, H2 is associated with Breviolum pseudominutum (Xiang et al., 2013) and CC7 hosts Symbiodinium linuchae (Sunagawa et al., 2009). VWA and VWB have been characterized as hosting Breviolum minutum (B1) in other labs. Only Mesorhizobium and Maritalea of the 14 differentially abundant ASVs identified after filtering for sample consistency were abundant in symbiotic anemones. Mesorhizobium is suggested to have a nitrogen-fixing role (Carlos et al., 2013) potentially increasing nitrogen availability and thus facilitating growth and high density of the anemone’s algal symbiont (Rädecker et al., 2015). Future functional analysis of these identified taxa is necessary to provide confirmation of the proposed contributions to cnidarian fitness.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: SRA data: PRJNA907389.
Author contributions
JM, EC, and RW designed the experiment. JM isolated genetic material. RR prepared sequencing libraries and carried out preliminary sequence analysis. EC and RW completed data analysis. EC wrote the manuscript, with contributions from JM, RR, and RW. All authors contributed to the article and approved the submitted version.
Funding
Funding was provided by the Nancy Kershaw Tomlinson Fund for Undergraduate Honors Research to EC and JM and Smith College to RW.
Acknowledgments
The authors are grateful for anemones provided by Virginia Weis at Oregon State University, and for microscopic equipment and expertise provided by Judith Woperis and Michael Barresi, microscopic images generated by Nicole Capozzi, sequencing services from the Center for Molecular Biology, and computational resources through the Clark Science Center Computing and Technical Services at Smith College.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1113043/full#supplementary-material
References
Bonacolta A. M., Connelly M. T., Rosales S. M., Del Campo J., Traylor-Knowles N. (2021). The starlet sea anemone, Nematostella vectensis, possesses body region-specific bacterial associations with spirochetes dominating the capitulum. FEMS Microbiol. Lett. 368 (3). doi: 10.1093/femsle/fnab002
Bourne D. G., Morrow K. M., Webster N. S. (2016). Insights into the coral microbiome: Underpinning the health and resilience of reef ecosystems. Annu. Rev. Microbiol. 70, 317–340. doi: 10.1146/annurev-micro-102215-095440
Brown T., Otero C., Grajales A., Rodriguez E., Rodriguez-Lanetty M. (2017). Worldwide exploration of the microbiome harbored by the cnidarian model, Exaiptasia pallida (Agassiz in verrill 1864) indicates a lack of bacterial association specificity at a lower taxonomic rank. PeerJ 5 (e3235), e3235. doi: 10.7717/peerj.3235
Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J. A., Holmes S. P. (2016). DADA2: High-resolution sample inference from illumina amplicon data. Nat. Methods 13 (7), 581–583. doi: 10.1038/nmeth.3869
Carlos C., Torres T. T., Ottoboni L. M. M. (2013). Bacterial communities and species-specific associations with the mucus of Brazilian coral species. Sci. Rep. 3, 1624. doi: 10.1038/srep01624
Cho I., Blaser M. J. (2012). The human microbiome: At the interface of health and disease. Nat. Rev. Genet. 13 (4), 260–270. doi: 10.1038/nrg3182
Costa R. M., Cárdenas A., Loussert-Fonta C., Toullec G., Meibom A., Voolstra C. R. (2021). Surface topography, bacterial carrying capacity, and the prospect of microbiome manipulation in the Sea anemone coral model aiptasia. Front. Microbiol. 12, 637834. doi: 10.3389/fmicb.2021.637834
Costanza R., de Groot R., Sutton P., van der Ploeg S., Anderson S. J., Kubiszewski I., et al. (2014). Changes in the global value of ecosystem services. Global Environ. Change: Hum. Policy Dimen. 26, 152–158. doi: 10.1016/j.gloenvcha.2014.04.002
Daniels C., Baumgarten S., Yum L., MIchell C., Bayer T., Arif C., et al. (2015). Metatranscriptome analysis of the reef-building coral Orbicella faveolata indicates holobiont response to coral disease. Front. Mar. Sci. 2. doi: 10.3389/fmars.2015.00062
Delaroque C., Wu G. D., Compher C., Ni J., Albenberg L., Liu Q., et al. (2022). Diet standardization reduces intra-individual microbiome variation. Gut Microbes 14 (1), 2149047. doi: 10.1080/19490976.2022.2149047
Doering T., Wall M., Putchim L., Rattanawongwan T., Schroeder R., Hentschel U., et al. (2021). Towards enhancing coral heat tolerance: A “microbiome transplantation” treatment using inoculations of homogenized coral tissues. Microbiome 9 (1), 102. doi: 10.1186/s40168-021-01053-6
Dungan A. M., Hartman L. M., Tortorelli G., Belderok R., Lamb A. M., Pisan L., et al. (2020). Exaiptasia diaphana from the great barrier reef: A valuable resource for coral symbiosis research. Symbiosis 80 (2), 195–206. doi: 10.1007/s13199-020-00665-0
Epstein H. E., Smith H. A., Torda G., Oppen M. J. H. (2019). Microbiome engineering: enhancing climate resilience in corals. Front. Ecol. Environ. 17 (2), 100–108. doi: 10.1002/fee.2001
Fisher R., O’Leary R. A., Low-Choy S., Mengersen K., Knowlton N., Brainard R. E., et al. (2015). Species richness on coral reefs and the pursuit of convergent global estimates. Curr. Biol.: CB 25 (4), 500–505. doi: 10.1016/j.cub.2014.12.022
Galand P. E., Remize M., Meistertzheim A.-L., Pruski A. M., Peru E., Suhrhoff T. J., et al. (2020). Diet shapes cold-water corals bacterial communities. Environ. Microbiol. 22 (1), 354–368. doi: 10.1111/1462-2920.14852
Gardner T. A., Côté I. M., Gill J. A., Grant A., Watkinson A. R. (2003). Long-term region wide declines in Caribbean corals. Science 301 (5635), 958–960. doi: 10.1126/science.1086050
Gilbert J. A., Hill R., Doblin M. A., Ralph P. J. (2012). Microbial consortia increase thermal tolerance of corals. Mar. Biol. 159 (8), 1763–1771. doi: 10.1007/s00227-012-1967-9
Glasl B., Herndl G. J., Frade P. R. (2016). The microbiome of coral surface mucus has a key role in mediating holobiont health and survival upon disturbance. ISME J. 10 (9), 2280–2292. doi: 10.1038/ismej.2016.9
Hartman L. M., van Oppen M. J. H., Blackall L. L. (2020). Microbiota characterization of Exaiptasia diaphana from the great barrier reef. Anim. Microb. 2 (1), 10. doi: 10.1186/s42523-020-00029-5
Hernandez-Agreda A., Leggat W., Bongaerts P., Herrera C., Ainsworth T. D. (2018). Rethinking the coral microbiome: Simplicity exists within a diverse microbial biosphere. mBio 9 (5). doi: 10.1128/mBio.00812-18
Herrera M., Ziegler M., Voolstra C. R., Aranda M. (2017). Laboratory-cultured strains of the Sea anemone Exaiptasia reveal distinct bacterial communities. Front. Mar. Sci. 4. doi: 10.3389/fmars.2017.00115
Hughes T. P., Anderson K. D., Connolly S. R., Heron S. F., Kerry J. T., Lough J. M., et al. (2018b). Spatial and temporal patterns of mass bleaching of corals in the anthropocene. Science 359 (6371), 80–83. doi: 10.1126/science.aan8048
Hughes T. P., Kerry J. T., Baird A. H., Connolly S. R., Dietzel A., Eakin C. M., et al. (2018a). Global warming transforms coral reef assemblages. Nature 556 (7702), 492–496. doi: 10.1038/s41586-018-0041-2
Kartzinel T. R., Hsing J. C., Musili P. M., Brown B. R. P., Pringle R. M. (2019). Covariation of diet and gut microbiome in African megafauna. Proc. Natl. Acad. Sci. United States America 116 (47), 23588–23593. doi: 10.1073/pnas.1905666116
Klindworth A., Pruesse E., Schweer T., Peplies J., Quast C., Horn M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41 (1), e1. doi: 10.1093/nar/gks808
Lehnert E. M., Burriesci M. S., Pringle J. R. (2012). Developing the anemone Aiptasia as a tractable model for cnidarian-dinoflagellate symbiosis: The transcriptome of aposymbiotic A. pallida. BMC Genomics 13, 271. doi: 10.1186/1471-2164-13271
Li J., Yang Q., Dong J., Sweet M., Zhang Y., Liu C., et al. (2022). Microbiome engineering: A promising approach to improve coral health. Proc. Estonian Acad. Sci.: Eng. doi: 10.1016/j.eng.2022.07.010
Love M. I., Huber W., Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 550. doi: 10.1186/s13059-014-0550-8
Maire J., Blackall L. L., van Oppen M. J. H. (2021). Microbiome characterization of defensive tissues in the model anemone exaiptasia diaphana. BMC Microbiol. 21 (1), 152. doi: 10.1186/s12866-021-02211-4
Marín I., Arahal D. R. (2014). “The family beijerinckiaceae,” in The prokaryotes: Alphaproteobacteria and betaproteobacteria. Eds. Rosenberg E., DeLong E. F., Lory S., Stackebrandt E., Thompson F. (Berlin Heidelberg: Springer), 115–133.
McMurdie P. J., Holmes S. (2013). Phyloseq: An r package for reproducible interactive analysis and graphics of microbiome census data. PloS One 8 (4), e61217. doi: 10.1371/journal.pone.0061217
Miller J., Muller E., Rogers C., Waara R., Atkinson A., Whelan K. R. T., et al. (2009). Coral disease following massive bleaching in 2005 causes 60% decline in coral cover on reefs in the US virgin islands. In Coral Reefs 28 (4), 925–937). doi: 10.1007/s00338-009-0531-7
Mortzfeld B. M., Urbanski S., Reitzel A. M., Künzel S., Technau U., Fraune S. (2016). Response of bacterial colonization in Nematostella vectensis to development, environment and biogeography. Environ. Microbiol. 18 (6), 1764–1781. doi: 10.1111/1462-2920.12926
Muller E. M., Bartels E., Baums I. B. (2018). Bleaching causes loss of disease resistance within the threatened coral species Acropora cervicornis. eLife 7. doi: 10.7554/eLife.35066
Oksanen J., Blanchet F. G., Friendly M., Kindt R., Legendre P., McGlinn D., et al. (2022). Vegan: Community ecology package. r package version 2. 6–4. Available at: https://CRAN.R-project.org/package=vegan
Poole A. Z., Kitchen S. A., Weis V. M. (2016). The role of complement in cnidarian-dinoflagellate symbiosis and immune challenge in the Sea anemone aiptasia pallida. Front. Microbiol. 7, 519 . doi: 10.3389/fmicb.2016.00519
Pujalte M. J., Lucena T., Ruvira M. A., Arahal D. R., Macián M. C. (2014). “The family rhodobacteraceae,” in The prokaryotes: Alphaproteobacteria and betaproteobacteria, vol. 439–512 . Eds. Rosenberg E., DeLong E. F., Lory S., Stackebrandt E., Thompson F. (Berlin Heidelberg: Springer).
Rädecker N., Pogoreutz C., Voolstra C. R., Wiedenmann J., Wild C. (2015). Nitrogen cycling in corals: The key to understanding holobiont functioning? Trends Microbiol. 23 (8), 490–497. doi: 10.1016/j.tim.2015.03.008
Raymann K., Moran N. A. (2018). The role of the gut microbiome in health and disease of adult honey bee workers. Curr. Opin. Insect Sci. 26, 97–104. doi: 10.1016/j.cois.2018.02.012
R Core Team (2022). R: A language and environment for statistical computing (Vienna, Austria: R Foundation for Statistical Computing). Available at: https://www.R-project.org/.
Rosado P. M., Leite D. C. A., Duarte G. A. S., Chaloub R. M., Jospin G., Nunes da Rocha U., et al. (2019). Marine probiotics: Increasing coral resistance to bleaching through microbiome manipulation. ISME J. 13 (4), 921–936. doi: 10.1038/s41396-018-0323-6
Röthig T., Costa R. M., Simona F., Baumgarten S., Torres A. F., Radhakrishnan A., et al. (2016). Distinct bacterial communities associated with the coral model aiptasia in aposymbiotic and symbiotic states with symbiodinium. Front. Mar. Sci. 3. doi: 10.3389/fmars.2016.00234
Silveira C. B., Cavalcanti G. S., Walter J. M., Silva-Lima A. W., Dinsdale E. A., Bourne D. G., et al. (2017). Microbial processes driving coral reef organic carbon flow. FEMS Microbiol. Rev. 41 (4), 575–595. doi: 10.1093/femsre/fux018
Sunagawa S., Wilson E. C., Thaler M., Smith M. L., Caruso C., Pringle J. R., et al. (2009). Generation and analysis of transcriptomic resources for a model system on the rise: the sea anemone aiptasia pallida and its dinoflagellate endosymbiont. BMC Genomics 10, 258. doi: 10.1186/1471-2164-10-258
Tian L., Wang X.-W., Wu A.-K., Fan Y., Friedman J., Dahlin A., et al. (2020). Deciphering functional redundancy in the human microbiome. Nat. Commun. 11 (1), 6217. doi: 10.1038/s41467-020-19940-1
Vargas S., Caglar C., Büttner G., Schätzle S., Deister F., Woerheide G. (2021) Slime away: a simple CTAB-based high molecular weight DNA and RNA extraction protocol for “difficult” invertebrates (rev20200518) v1. doi: 10.17504/protocols.io.bwcwpaxe.
Weiler B. A., Verhoeven J. T. P., Dufour S. C. (2018). Bacterial communities in tissues and surficial mucus of the cold-water coral paragorgia arborea. Front. Mar. Sci. 5. doi: 10.3389/fmars.2018.00378
Weis V. M., Davy S. K., Hoegh-Guldberg O., Rodriguez-Lanetty M., Pringle J. R. (2008). Cell biology in model systems as the key to understanding corals. Trends Ecol. Evol. 23 (7), 369–376. doi: 10.1016/j.tree.2008.03.004
Wickham H. (2016). Package “ggplot2”: elegant graphics for data analysis (New York: Springer-Verlag).
Xiang T., Hambleton E. A., DeNofrio J. C., Pringle J. R., Grossman A. R. (2013). Isolation of clonal axenic strains of the symbiotic dinoflagellate symbiodinium and their growth and host specificity (1). J. Phycol. 49 (3), 447–458. doi: 10.1111/jpy.12055
Xiang N., Rädecker N., Pogoreutz C., Cárdenas A., Meibom A., Wild C., et al. (2022). Presence of algal symbionts affects denitrifying bacterial communities in the sea anemone Aiptasia coral model. ISME Commun. 2 (1), 1–10. doi: 10.1038/s43705-022-00190-9
Keywords: Aiptasia, Exaiptasia, symbiosis, microbial community, coral reef
Citation: Curtis E, Moseley J, Racicot R and Wright RM (2023) Bacterial microbiome variation across symbiotic states and clonal lines in a cnidarian model. Front. Mar. Sci. 10:1113043. doi: 10.3389/fmars.2023.1113043
Received: 01 December 2022; Accepted: 31 January 2023;
Published: 10 February 2023.
Edited by:
Virginia M Weis, Oregon State University, United StatesReviewed by:
Sebastian Fraune, Heinrich Heine University of Düsseldorf, GermanyAnna Roik, Helmholtz Institute for Functional Marine Biodiversity (HIFMB), Germany
Copyright © 2023 Curtis, Moseley, Racicot and Wright. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rachel M. Wright, cmFjaGVsd3JpZ2h0OEBnbWFpbC5jb20=