 Lei Zhou1
Lei Zhou1 Xianze Jia1Kaishan Liang1Xiaoling Zuo1Fengqi Huang1Xuzhuo Duan1Zongyang Li1
Xianze Jia1Kaishan Liang1Xiaoling Zuo1Fengqi Huang1Xuzhuo Duan1Zongyang Li1 Shilei Sang2*
Shilei Sang2* Qing Wang1,3*
Qing Wang1,3*- 1Joint Laboratory of Guangdong Province and Hong Kong Region on Marine Bioresource Conservation and Exploitation, College of Marine Sciences, South China Agricultural University, Guangzhou, China
- 2State Environmental Protection Key Laboratory of Environmental Pollution Health Risk Assessment, South China Institute of Environmental Sciences, MEE, Guangzhou, China
- 3Guangdong Laboratory for Lingnan Modern Agriculture, Guangzhou, China
Introduction: Frequent disease outbreaks seriously affect porcupinefish (Diodon hystrix), which is a new aquaculture breed in China. Recently, a novel disease, termed tail fell syndrome (TFS) was increasingly observed in fish stocks, resulting in stalk ulceration, tail loss, ascites and white feces in the intestine. Intestinal microbiota homeostasis is crucial to host health because it influences host and environmental factors, and responds to various internal and external stimuli. However, changes in intestinal microbiota induced by TFS are yet to be elucidated.
Methods: In the present study, we analyzed and compared the intestinal microbiota of normal D. hystrix with TFS fish.
Results: Though microbiota richness and diversity were not affected by TFS, deviations in diversity indices increased and taxa distribution evenness decreased, suggesting TFS lowered microbiota community stability in D. hystrix. Furthermore, at the genus level, Brevibacterium, Mesorhizobium, Ochrobactrum, Ralstonia, Anaerococcus, and Alistipes abundances were significantly increased in TFS D. hystrix, plus, we observed significant decreases in Halomonas, Prevotellaceae_NK3B31_group, and Psychrobacter. Functional comparison predictions between normal and TFS D. hystrix revealed significantly altered pathways were mainly associated with metabolism (biotin metabolism, steroid hormone biosynthesis, flavonoid biosynthesis, biosynthesis of type II polyketide products, and steroid biosynthesis). Overall, the results revealed that TFS impacted intestinal microbiota composition and function in D. hystrix, which should expand our knowledge on diseases associated with porcupine pufferfish aquaculture.
1 Introduction
Diodon hystrix (porcupinefish) is a commercially important fish species mainly distributed in the Indo-West Pacific region (Soto et al., 2013). The fish is a new aquaculture species in China due to its considerable nutritional and medicinal characteristics. With the rapid development of D. hystrix aquaculture in recent years, several diseases have appeared, including the novel tail fell syndrome (TFS), particularly in D. hystrix aquaculture. The disease was first discovered in normal aquaculture environments, with TFS and normal fish emerging from the same environment. The most dominant disease feature is tail stalk festering with entire tail loss, accompanied by ascites and white feces in the intestines, and a high death rate. These observations suggest TFS may be pathogenic in nature.
Under intensive aquaculture conditions, many diseases are caused by pathogens, for example, white spot disease in Penaeus monodon caused by Vibrio alginolyticus infections (Selvin and Lipton, 2003; Zhou et al., 2020; Zhao et al., 2021). Several diseases are caused by a variety of pathogens, including viruses and bacteria (Zhu et al., 2000; Huang et al., 2012). Furthermore, growing evidence suggests that many aquaculture animal diseases induce changes in intestinal bacterial communities (Pérez et al., 2010; Zhou et al., 2020; Zhao et al., 2021). As a complex ecosystem, intestinal microbiota promotes host health by participating in immune responses, nutrient absorption, disease resistance, and the establishment of a balanced community structure (Lin et al., 2014). In Litopenaeus vannamei, infection with white spot syndrome virus altered intestinal microbiota composition and function (Wang et al., 2019). In zebrafish intestines, bacterial invasion affected microbiota composition and induced immune responses (Yang et al., 2017). In grass carp (Ctenopharyngodon idellus), Aeromonas hydrophila infection induced dynamic changes in intestinal microbiota (Zhou et al., 2020). Furthermore, intestinal microbiota also has determinant roles in newly discovered diseases. In sea cucumber (Apostichopus japonicus), different intestinal fungal communities were observed between normal and body vesicular syndrome individuals (Zhao et al., 2021). Our recent study has indicated that TFS, a newly discovered disease, is caused mainly by Vibrio (unpublished data); however, further information on TFS is practically non-existent, however, aberrant intestinal morphology was found, therefore we speculated TFS caused changes in the gut microbiome. However, associations between TFS and the host microbiome remain unclear.
The intestinal microbiota is influenced by host and environmental factors, and responds to various internal and external stimuli, therefore, understanding the composition and structure of intestinal microbial communities is necessary to understand changes and address different situations or stimuli (Clements et al., 2014; Butt and Volkoff, 2019). Over the last decade, clone libraries, culture-based, and denaturing gradient gel electrophoresis approaches have been applied to microbiota investigations (Knapp et al., 2008; Morotomi et al., 2011; Tzuc et al., 2014). While useful, these traditional methods are limited in characterizing global diversity and complex bacterial communities in an integrated environment. In view of this, 16S rRNA sequencing technology is widely used to study the composition of microbial communities in fish intestinal samples (Ghanbari et al., 2015; Wang et al., 2018).
In this study, the intestinal microbiota of TFS and normal D. hystrix was analyzed using high-throughput sequencing technology. This study investigated how TFS induced composition, diversity, and functional changes in gut microbiota in D. hystrix. Our research identifying gut microbiota in diseased D. hystrix, and provides an important clue for TFS management and control.
2 Materials and methods
2.1 Sample collection
D. hystrix were collected from aquaculture ponds in Sanya, China. The average body length of the D. hystrix was 10.54 ± 1.73 cm and the average weight of the D. hystrix was 26.33 ± 4.21g. After collection, eight TFS and eight normal D. hystrix fish were randomly selected and dissected immediately. Fish were anesthetized with MS-222 (Sigma-Aldrich) before sacrifice. Intestinal tracts were aseptically removed from the body cavity, and the contents gently squeezed into sterile containers. These were immediately frozen in liquid nitrogen, and maintained at −80°C until DNA extraction. This work received ethical approval from the Animal Research and Ethics Committees of South China Agriculture University.
2.2 Histopathology
Five TFS and five normal D. hystrix fish were selected, mid-intestinal segments removed, and fixed in Bouin’s fluid. After dehydration, tissue was embedded in paraffin wax, sectioned (5 μm), stained in hematoxylin and eosin, and examined using a Nikon microscope (Nikon Eclipse E100, Japan).
2.3 DNA extraction, amplification and sequencing
Microbial DNA from intestinal contents was extracted using the QIAGEN DNA stool mini kit (QIAGEN, USA) following manufacturer’s instructions. We conducted 1% agarose gel electrophoresis to assess successful DNA extraction. DNA concentrations and purity were then measured using the NanoPhotometer® Classic Launched (IMPLEN, Germany). As previously described, DNA was diluted to 1 ng/μl, and two specific barcoded primers (341F, CCTACGGGNGGCWGCAG and 806R, GGACTACHVGGGTATCTAAT; the barcode was an eight-base sequence unique to each sample) were used to amplify the V3–V4 region of the 16S rRNA gene by PCR (Zhou et al., 2020). PCR product quality was assessed by 2% agarose gel electrophoresis. Purified amplicons were pooled in equimolar amounts, and paired ends sequenced (PE250) on an Illumina platform Hiseq™ 2500 (Gene Denovo Biotechnology Co. Ltd. Guangzhou, China).
2.4 Bioinformatics and statistical analysis
DADA2 was used to perform quality filtering on amplicons, infer exact amplicon sequencing variants (ASVs), and filter and remove chimeras (Callahan et al., 2016). Taxonomic assignment (i.e., kingdom, phylum, class, order, family, genus, and species) by a naive Bayesian model was conducted using the RDP classifier (version 2.2, Wang et al., 2007) based on the SILVA database (version 132, Quast et al., 2013). Singletons across samples and reads which mapped to mitochondria or chloroplasts were discarded.
Alpha diversity comparisons (Sob, Chao1, ACE, Shannon, Simpson, and Pielou indices) between groups were calculated using the Wilcoxon rank test. To compare microbial community differences between groups, non-metric multidimensional scaling (NMDS) ordination and analysis of similarities (ANOSIM) using the Bray-Curtis distance was conducted in the R project Vegan package (Oksanen et al., 2016). Biomarker features in each group were screened using the random forest model in the randomForest R package (Liaw and Wiener, 2002).
Kyoto Encyclopedia of Genes and Genomes pathway analysis of ASVs was inferred using PICRUSt2 (Douglas et al., 2020). Functional differences between groups were analyzed by Welch’s t-test.
3 Results
3.1 Morphological TFS characteristics
D. hystrix with TFS exhibited tail stalk ulceration, resulting in tail loss (Figures 1B, D), whereas normal fish did not exhibit this phenomenon (Figures 1A, C). When we dissected TFS fish abdomen and observed viscera, ascites and white feces in intestines (Figure 1E).
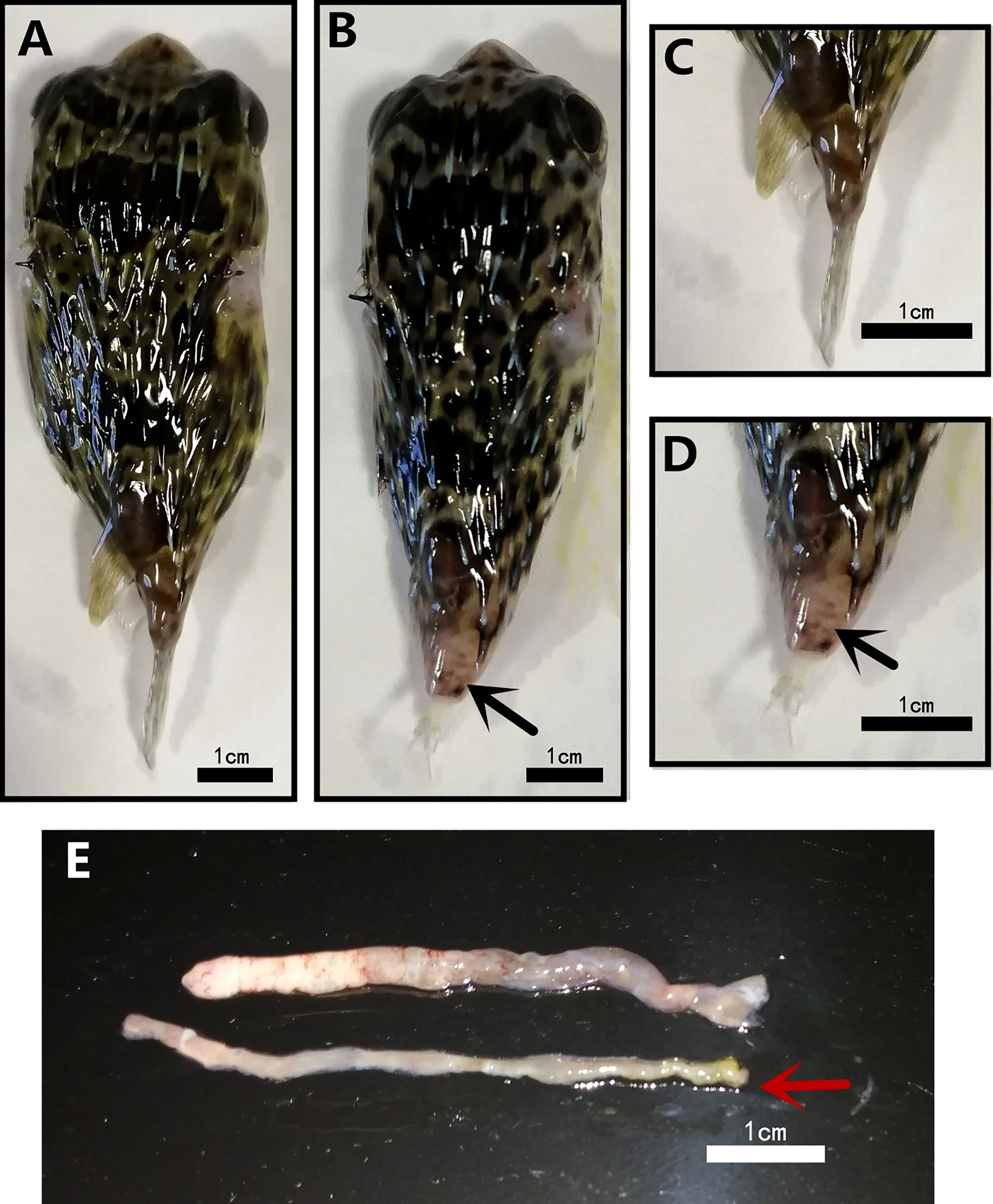
Figure 1 Morphological differences in normal (NOR) and tails fell syndrome (TFS) Diodon hystrix. (A) healthy D. hystrix. (B) TFS D. hystrix. (C) Closeup of the tail of health D. hystrix. (D) Closeup of the tail of TFS D. hystrix, the black arrows indicate the festering and broken tails. (E) The state of the intestines of health and TFS D. hystrix, the red arrows indicate the intestines of TFS D. hystrix.
3.2 Histopathology
TFS and normal fish intestines were sectioned and histologically analyzed. TFS fish intestinal villi had many cavities and gaps (Figures 2A, B). Furthermore, intestinal villi were shorter and distances between villi were larger in TFS fish intestines. Normal fish intestines are shown (Figures 2C, D).
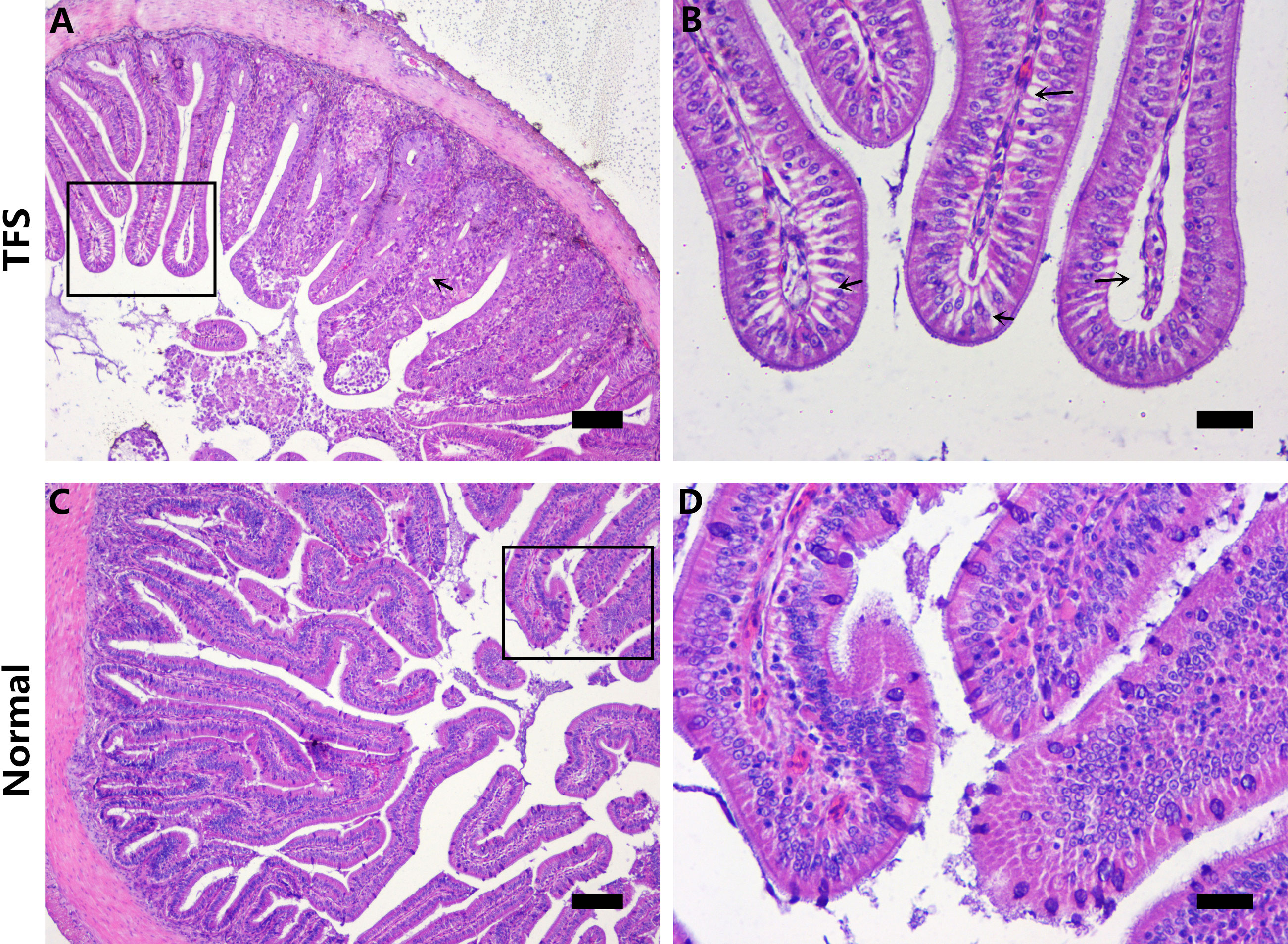
Figure 2 Histological analysis of the intestinal sections of normal (NOR) and TFS D. hystrix. (A) The histological of TFS D. hystrix. (B) High magnification of the boxed areas in A, black arrows indicate the cavitation and voids. (C) The histological of normal D. hystrix. (D) High magnification of the boxed areas in (C) Scale bars (A, C): 100μm, Scale bars (B, D): 25μm.
3.3 Microbiota composition and distribution
After quality control and rarefaction, 512368 high-quality reads were obtained from 16 samples, comprising eight normal D. hystrix and eight TFS D. hystrix. The total number of ASVs was 14434, ranging from 612–2481 ASVs per sample. ASV and taxonomy data from samples are presented (Table S1).
The top 10 taxa with the highest abundance in each group at phylum and genus levels are shown (Figure 3). At the phylum level (Figure 3A), the average abundance of Firmicutes was 37.27%; Bacteroidetes was 25.80%; Proteobacteria was 19.32%, and Actinobacteria was 4.43% in the TFS group. In the normal fish group, the average abundance of Firmicutes was 36.32%; Bacteroidetes was 21.70%; Proteobacteria was 19.04%, and Actinobacteria was 5.95% (Figure 3A). At the genus level, the average abundance of the Lachnospiraceae_NK4A136_group was 5.04% in TFS and 5.64% in normal fish, also the average abundance of Lactobacillus was 4.91% in TFS and 3.39% in normal fish (Figure 3B).
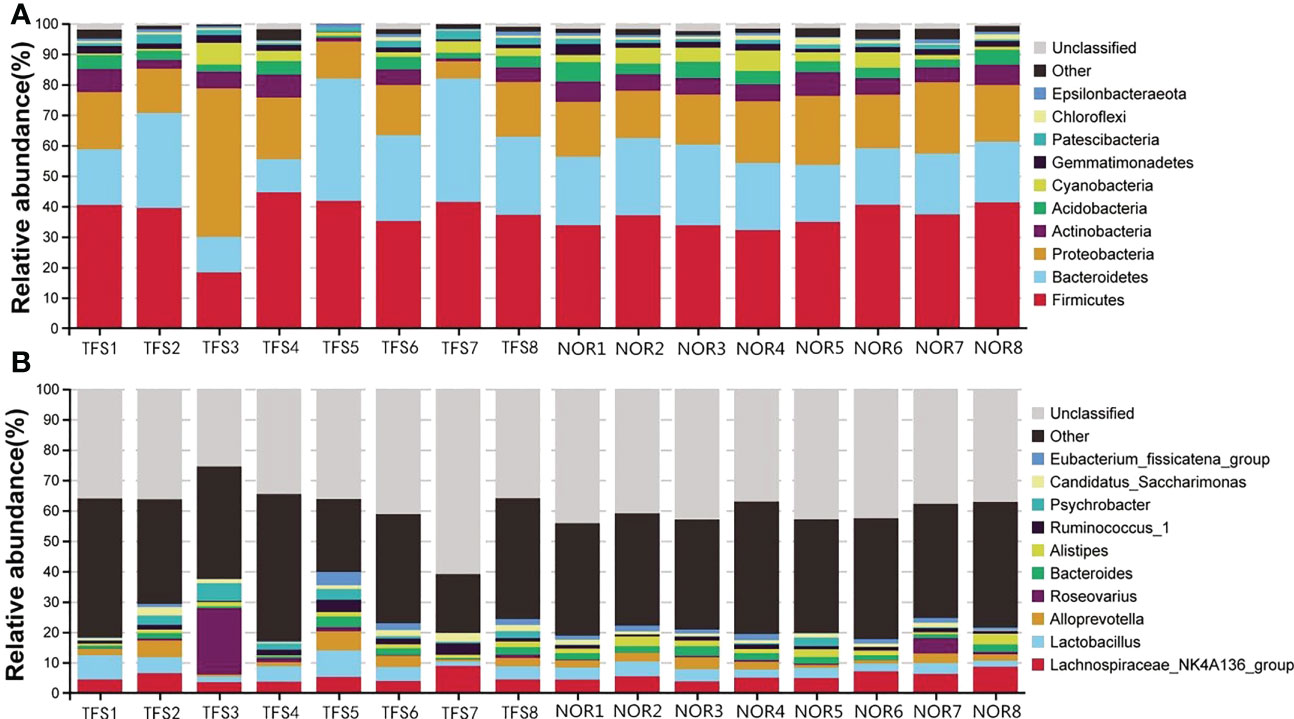
Figure 3 Taxonomy stack distributions of different levels of classification for normal (NOR) and TFS D. hystrix. The top 10 phylum (A) and genus (B) were selected, and the rest were unified to the other category.
Furthermore, we identified nine genus-level indicator taxa associated with TFS by using a random forest model and Wilcoxon test (Figure 4A). Of these, Halomonas, Prevotellaceae_NK3B31_group and Psychrobacter abundances were significantly decreased in TFS fish. In contrast, Brevibacterium, Mesorhizobium, Ochrobactrum, Ralstonia, Anaerococcus, and Alistipes were significantly increased (Figure 4B).
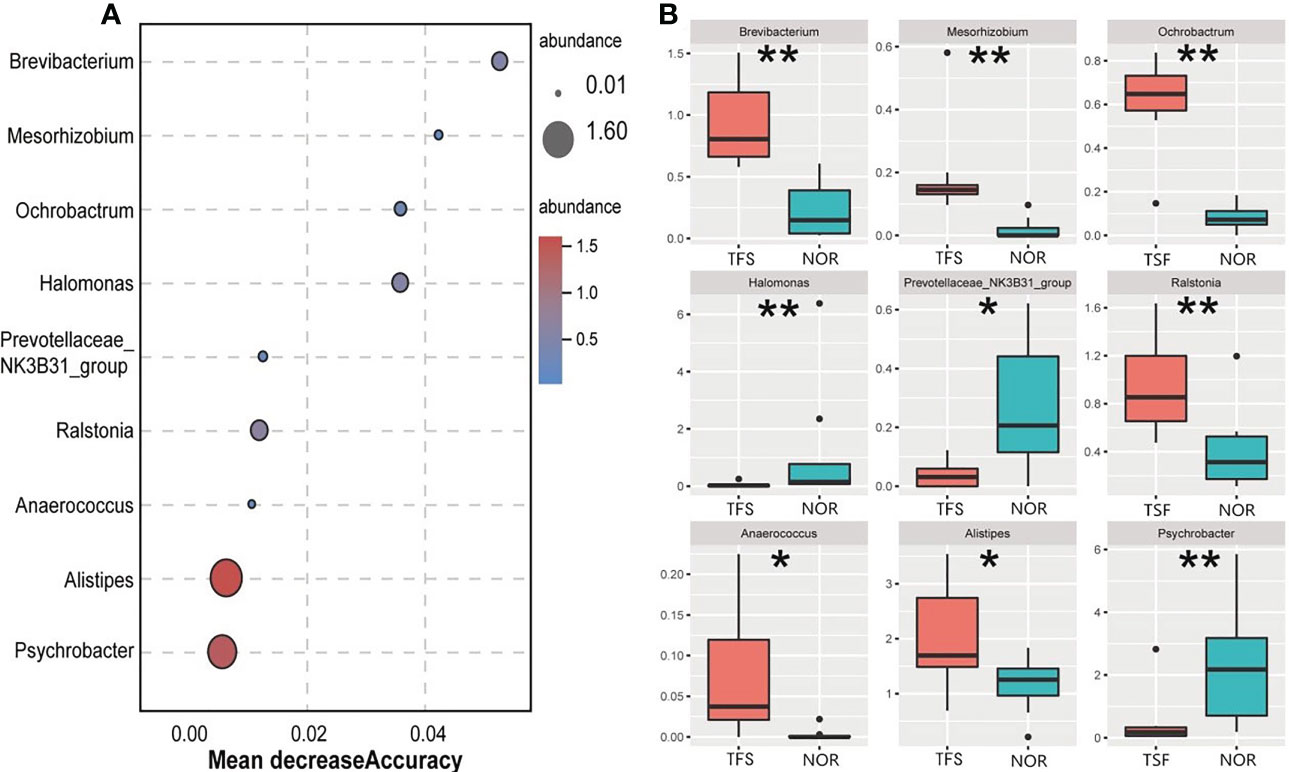
Figure 4 Identified differentially abundant genera between the normal (NOR) and TFS D. hystrix. by random forest model (A) and wilcox test (B). A mean decrease in accuracy measures the increase in misclassification produced on average by removing the given predictor. * P < 0.05, ** P < 0.01.
3.4 Microbiota diversity and structural differences
For alpha diversities, all Sob, Chao1, ACE, Shannon, Shannon, and Pielou indices showed no significant differences between groups. We calculated the coefficient of variation (standard deviation/mean), which indicated larger deviations in alpha diversities in TFS fish (Figure 5; Table 1). In addition, Pielou’s evenness index indicated a decreasing tendency (Wilcoxon rank test: p = 0.089) in the TFS group. These results suggested increased instability in TFS D. hystrix intestinal microbiota.
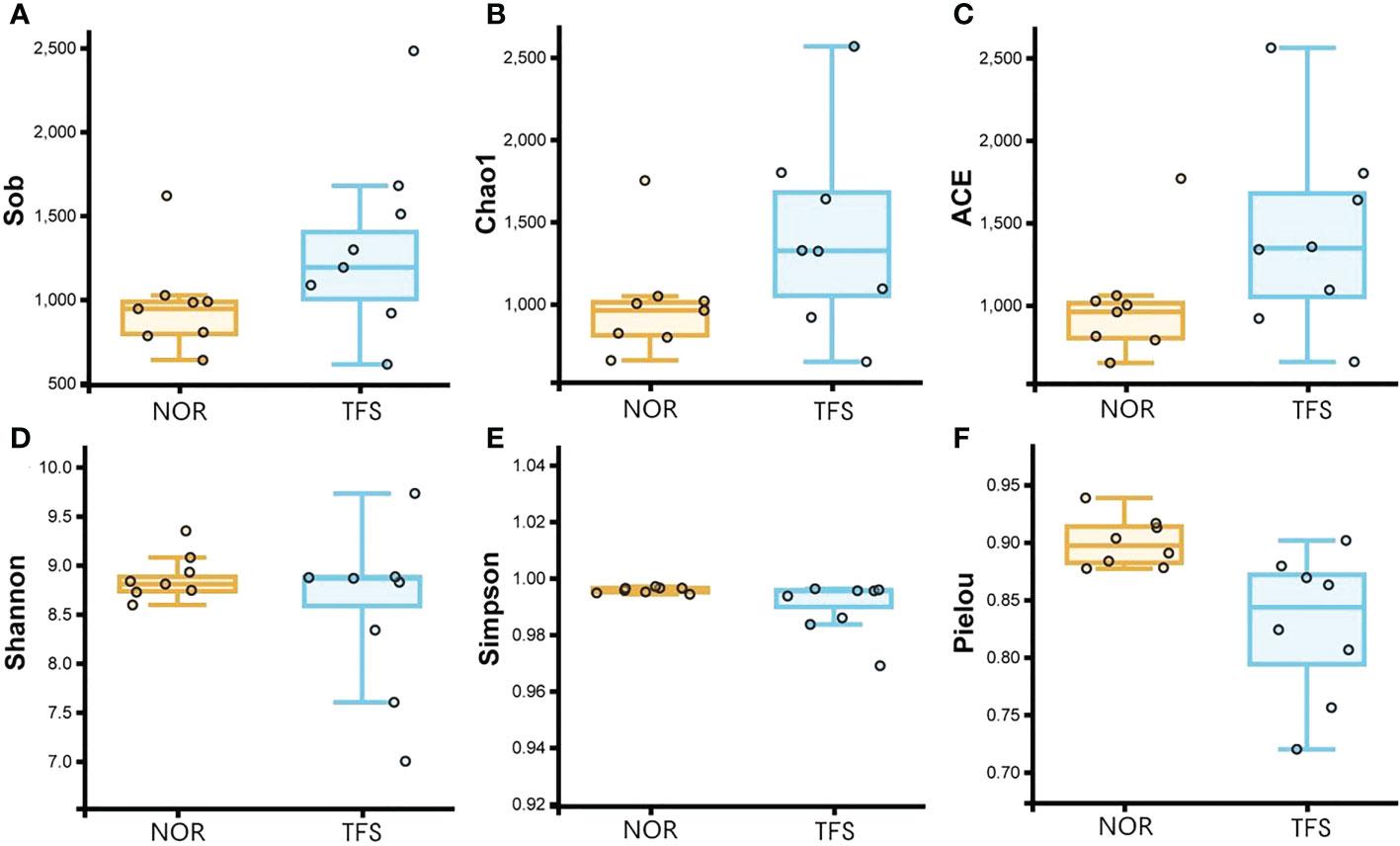
Figure 5 Comparisons of intestinal microbiota alpha diversity between normal (NOR) and TFS D. hystrix. Sob (A), Chao1 (B), ACE (C), Shannon (D), and Simpson (E) and Pielou indexes (F).
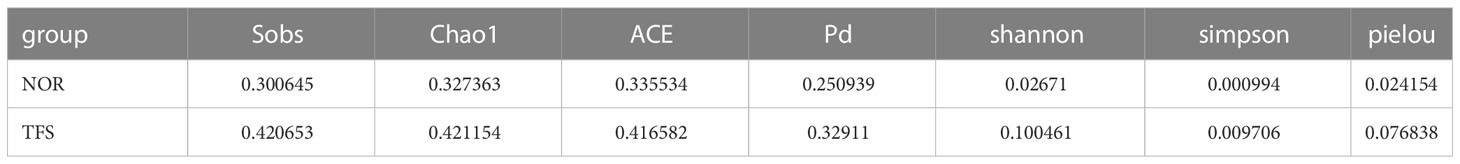
Table 1 Coefficient of Variation in alpha diversities in the intestinal microbiota of normal (NOR) and TFS D. hystrix.
NMDS analysis showed that community structures were significantly altered between groups (Figure 6A). Furthermore, microbial community differences between groups were tested using ANOSIM, and showed statistically significant differences (R = 0.106, P = 0.034) between groups (Figure 6B).
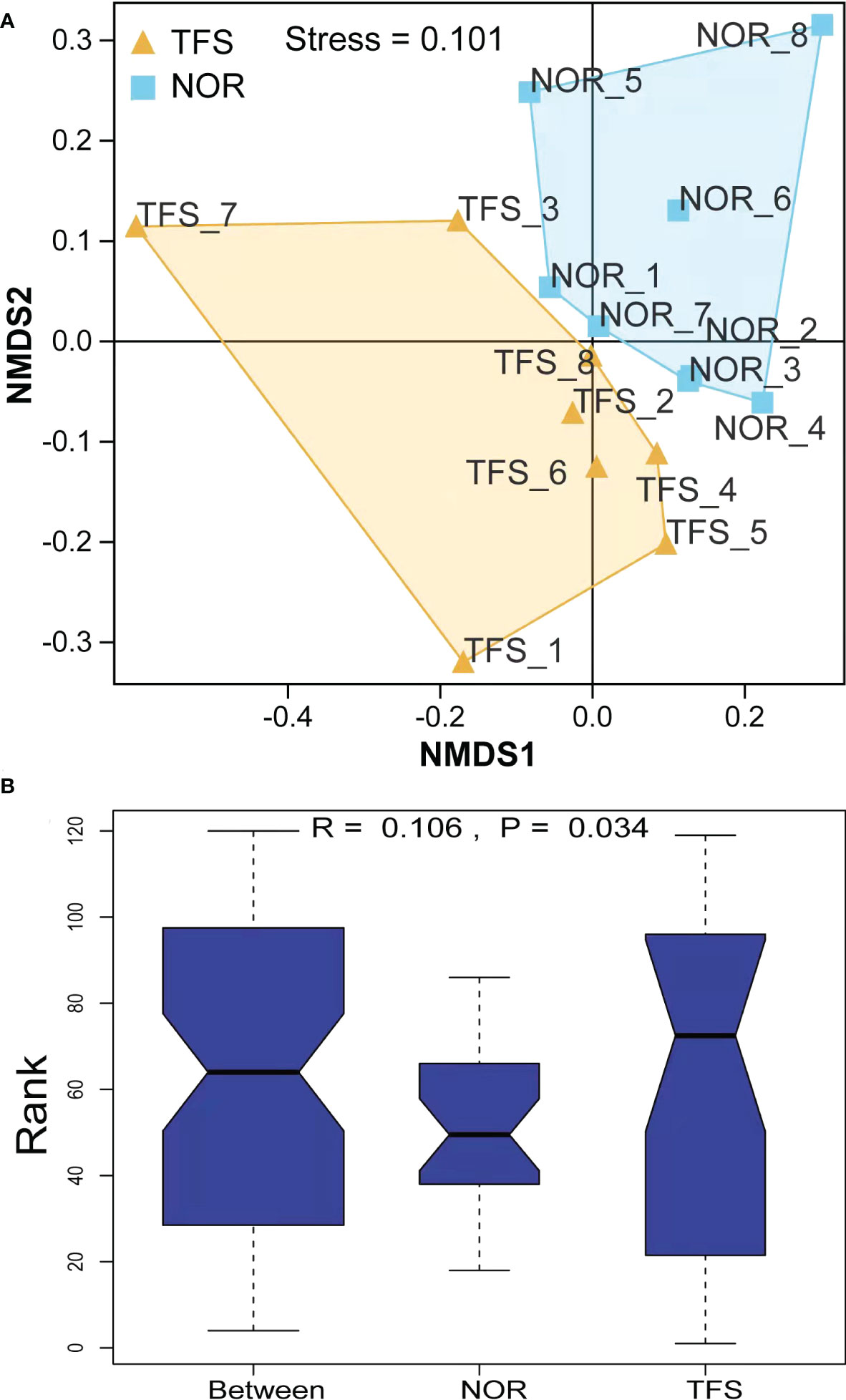
Figure 6 Beta diversity difference in the intestinal microbiota of normal (NOR) and TFS D. hystrix. (A) Nonmetric multidimensional scaling (NMDS) analysis base on Bray-Curtis dissimilarity index. (B) Statistical significance via Analysis of Similarity (ANOSIM) test with 999 permutations.
To identify the most important marker functions correlating with the TFS, we performed Kyoto Encyclopedia of Genes and Genomes pathway analysis of ASVs by using PICRUSt2. As shown in Figure 7, as an example, among the top nine pathways, five (biotin metabolism, steroid hormone biosynthesis, flavonoid biosynthesis, biosynthesis of type II polyketide products, and steroid biosynthesis) were classified as metabolism, one (biofilm formation - Vibrio cholera) was classified as a cellular process, one (non-homologous end-joining) was classified as genetic information processing, and two (V. cholerae infection and systemic lupus erythematosus) were classified as infectious diseases, most of which were metabolism or disease infectious related pathways, thus suggesting that these pathways (especially for “biosynthesis of type II polyketide products” and “flavonoid biosynthesis”) might be the dominant direct or indirect feedback responses to TFS.
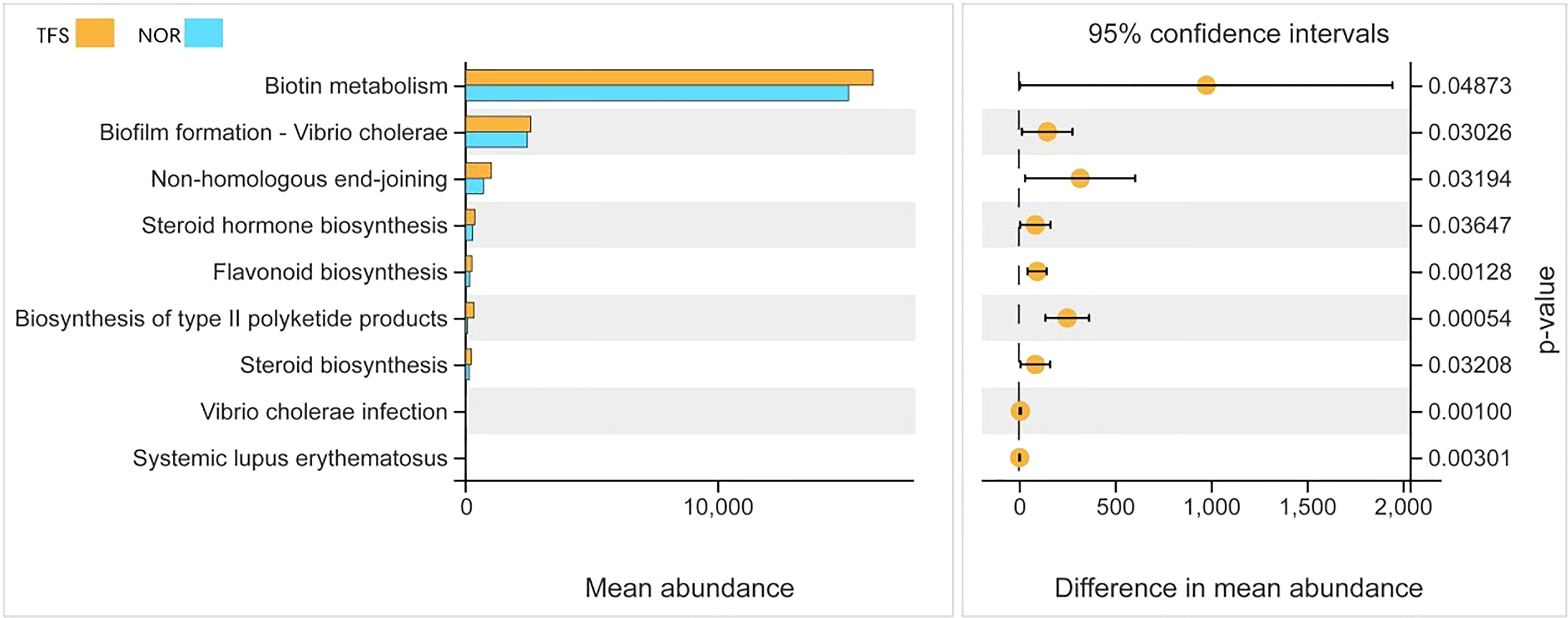
Figure 7 Functional differences in intestinal microbial communities between normal (NOR) and TFS D. hystrix. Functions were predicted by PICRUSt2 analysis of bacterial sequences. Significantly different functions (P < 0.05) of KEGG pathway categories (level 3) are show.
4 Discussion
D. hystrix is a new aquaculture breed currently undergoing large scale cultivation in China, however, with increased aquaculture comes new diseases. TFS is the latest, with a high mortality rate, tail stalk festering, tail-loss, and ascites and white feces in the intestines. We proposed TFS may induce changes in bacteria or fungi in the gut, therefore, we investigated gut microbiota in diseased and normal fish, and observed significant differences in microbial composition and function between groups.
In recent years, an increasing number of studies have identified correlations between immune functions, disease, and intestinal microbiota in fish (Li et al., 2011; Sudheesh et al., 2012; Guijarro et al., 2015; Liu et al., 2016; Qi et al., 2017). Many fish diseases induce intestinal microbiota changes, e.g. the related taxa, Vibrio, Aeromonas, and Shivella were overexpressed in the intestinal flora of Carassius auratus ‘red film’ disease (Li et al., 2017). Similarly, Aeromonas is a biomarker of furuncles in largemouth bass (Li et al., 2016). These phenomena suggested that certain intestinal microbial characteristics were indicative of host health status, independent of disease etiology, as demonstrated by human intestinal diseases (Mancabelli et al., 2017). Thus far, few studies have investigated the relationship between disease severity and gut microbiome disruption during fish disease development; thus, the transition from healthy to diseased gut flora is unclear (Knights et al., 2014). However, intestinal microbiota characteristics may be used as factors in predicting the incidence of fish disease (Xiong et al., 2017; Xiong et al., 2018), therefore, significant compositional differences in intestinal microbial communities observed here may be used to predict TFS occurrence in D. hystrix stocks. Our results help to elucidate differences in intestinal microorganisms between normal and diseased fish.
Bacterial diversity is closely associated with the functional stability of the intestinal microbiota; the more diverse the gut microbial community, the more likely it is to have species with antagonistic properties against invading pathogens; e.g., this is often observed for Coreius guichenoti (Li et al., 2016), C. auratus (Li et al., 2017), and Plecoglossus altivelis (Nie et al., 2017). In this study, we observed no significant differences in intestine microbiota species diversity and richness between groups. However, NMDS and ANOSIM strategies demonstrated differential microbial communities between normal and TFS fish. Also, intestinal microbiota compositional structures were changed in TFS fish. Combined, these data suggested diseased fish intestinal microbial community homeostasis was disrupted.
Previous studies have demonstrated that bacteria are responsible for fish diseases (Senderovich et al., 2010; Rønneseth et al., 2017; Castillo et al., 2019). At the phylum level, we observed Bacteroidetes levels were increased in TFS fish intestines. Many bacteria in the Bacteroidetes phylum cause inflammation, abscesses, and deep abscesses in the abdominal cavity and anus of both animals and humans (Louis and Flint, 2009), thus, TFS appeared to cause intestinal ulceration which may have been associated with these bacteria however, more analyses are required. At the genus level, we observed significantly increased bacteria in diseased fish, including Anaerococcus, Brevibacterium and Alistipes. Anaerococcus are considered enteropathogenic in mammals (Cobo and Navarro-Marí, 2020), Brevibacterium is becoming increasingly pathogenic to humans (Magi et al., 2018), and Alistipes are associated with gut inflammation (Parker et al., 2020). In contrast, Halomonas and Prevotellaceae_NK3B31 groups were decreased in TFS fish guts. The fish digestive tract has a stable ecosystem just like the nature. Normal fish contain huge bacterial numbers and proportions in the gastrointestinal tract; however, different factors can elicit a vast variety of changes. Normally, these changes fluctuate within certain limits, thus maintaining a relatively stable equilibrium. However, once fish get sick, this balance is upset, and intestinal flora becomes imbalanced. Thus, TFS may disrupt intestinal microbiota homeostasis in D. hystrix.
Investigating bacterial functional diversity highlighted the importance of the intestinal microbiome in host immunity. In this study, the gut bacteria of healthy and TFS fish were associated with multiple pathways. The most predominant included steroid biosynthesis and flavonoid biosynthesis. In organisms, steroids regulate innate immune responses and participate in immune regulation. As observed in Drosophila, steroid hormone signaling is essential to regulate innate immune cells and fight against bacterial infection (Regan et al., 2013). The steroid-signaling pathway can also help Caenorhabditis defend against bacteria (Fischer et al., 2013). Moreover, several studies have shown that steroid hormones exert excellent anti-inflammatory properties; e.g., 2-methoxyestradiol and corticosteroids elicit wide ranging anti-inflammatory effects (Shand et al., 2011; Doycheva et al., 2018). Steroid hormones play important roles in enhancing biological immunity; however, our results showed that steroid biosynthesis and flavonoid biosynthesis were down-regulated in the intestinal microbiota in diseased fish, and this downregulation can decrease sterol synthesis, weaken fish immunity, and increase susceptibility to the invasion of pathogens, thus resulting in TFS. However, these functional profiles were predicted by 16S data, therefore, more experimental studies are required.
5 Conclusions
In this study, the changes in the intestinal tissue and microbiota of TFS and healthy D. hystrix were explored through histopathological sectioning and 16S rRNA sequencing. Histological analysis indicated that diseased guts were highly disrupted. Analysis of the community diversity of the intestinal microbiota showed that TFS decreased microbiota community stability and induced intestinal microbiota community structural change in D. hystrix. Furthermore, functional comparison predictions between normal and TFS D. hystrix revealed that the significantly altered pathways were associated primarily with metabolism, thus leading to the alteration of a number of pathways related to sterol synthesis. Overall, our data indicate that TFS affects intestinal microbiota composition and function in D. hystrix, thus expanding knowledge of diseases associated with porcupine pufferfish aquaculture.
Data availability statement
The datasets presented in this study can be found in online repositories. The name of the repository and accession number can be found below: NCBI; PRJNA913412.
Ethics statement
The animal study was reviewed and approved by The Animal Research and Ethics Committees of South China Agriculture University. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
LZ, XZ, and QW designed the experiments. XJ, KL, and FH conducted the DNA extraction, amplification and sequencing. FH, XD and ZL extraction, amplification and sequencing of DNA. KL, XD, and ZL conducted the Bioinformatics and statistical analysis. LZ, SS and QW wrote the manuscript with feedback from all authors. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Youth Fund Project of the National Natural Science Foundation of China (42176103,41806151), the Science and Technology Planning Project of Guangzhou (202002030206), The talent team tender grant of Zhanjiang marine equipment and biology (2021E05035), Science and Technology Planning Project of Guangzhou City (PM-ZX913-202104-127\202002030345).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1108737/full#supplementary-material
References
Butt R. L., Volkoff H. (2019). Gut microbiota and energy homeostasis in fish. Front. Microbiol. 10, 9. doi: 10.3389/fendo.2019.00009
Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J., Holmes S. P. (2016). DADA2: High-resolution sample inference from illumina amplicon data. Nat. Methods 13 (7), 581–583. doi: 10.1038/nmeth.3869
Castillo D., Andersen N., Kalatzis P. G., Middelboe M. (2019). Large Phenotypic and genetic diversity of prophages induced from the fish pathogen vibrio anguillarum. Viruses 11, 983. doi: 10.3390/v11110983
Clements K. D., Angert E. R., Montgomery W. L., Choat J. H. (2014). Intestinal microbiota in fishes: what’s known and what’s not. Mol. Ecol. 23, 1891–1898. doi: 10.1111/mec.12699
Cobo F., Navarro-Marí J. M. (2020). First description of anaerococcus octavius as cause of bacteremia. Anaerobe 61, 102130. doi: 10.1016/j.anaerobe.2019.102130
Douglas G. M., Maffei V. J., Zaneveld J. R., Yurgel S. N., Brown J. R., Taylor C. M., et al. (2020). PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688. doi: 10.1038/s41587-020-0548-6
Doycheva D., Deuter C., Grajewski R. (2018). [Topical corticosteroids and non-steroidal anti-inflammatory drugs in the therapy of non-infectious uveitis]. Klin Monbl. Augenheilkd. 235, 586–591. doi: 10.1055/a-0590-4546
Fischer M., Regitz C., Kull R., Boll M., Wenzel U. (2013). Vitellogenins increase stress resistance of caenorhabditis elegans after photorhabdus luminescens infection depending on the steroid-signaling pathway. Microbes Infect. 15, 569–578. doi: 10.1016/j.micinf.2013.05.002
Ghanbari M., Kneifel W., Domig K. J. (2015). A new view of the fish gut microbiome: advances from next-generation sequencing. Aquaculture 448, 464–475. doi: 10.1016/j.aquaculture.2015.06.033
Guijarro J. A., Cascales D., Garcia-Torrico A. I., Garcia-Dominguez M., Mendez J. (2015). Temperature-dependent expression of virulence genes in fish-pathogenic bacteria. Front. Microbiol. 6, 700. doi: 10.3389/fmicb.2015.00700
Huang Z., Tang J., Li M., Fu Y., Dong C., Zhong J. F., et al. (2012). Immunological evaluation of vibrio alginolyticus, vibrio harveyi, vibrio vulnificus and infectious spleen and kidney necrosis virus (ISKNV) combined-vaccine efficacy in Epinephelus coioides. Vet. Immunol. Immunopathol. 150, 61–68. doi: 10.1016/j.vetimm.2012.08.008
Knapp B. A., Seeber J., Podmirseg S. M., Meyer E., Insam H. (2008). Application of denaturing gradient gel electrophoresis for analysing the gut microflora of Lumbricus rubellus hoffmeister under different feeding conditions. Bull. Entomol. Res. 98, 271–279. doi: 10.1017/S0007485308006056
Knights D., Ward T. L., McKinlay C. E., Miller H., Gonzalez A., McDonald D., et al. (2014). Rethinking “enterotypes”. Cell Host Microbe 16, 433–437. doi: 10.1016/j.chom.2014.09.013
Li T., Li H., Gatesoupe F. J., She R., Lin Q., Yan X., et al. (2017). Bacterial signatures of “Red-operculum” disease in the gut of crucian carp (Carassius auratus). Microbial. Ecol. 74, 510–521. doi: 10.1007/s00248-017-0967-1
Li T., Long M., Ji C., Shen Z., Gatesoupe F. J., Zhang X., et al. (2016). Alterations of the gut microbiome of largemouth bronze gudgeon (Coreius guichenoti) suffering from furunculosis. Sci. Rep. 6, 30606. doi: 10.1038/srep30606
Li M. F., Wang C. L., Sun L. (2011). A pathogenic vibrio harveyi lineage causes recurrent disease outbreaks in cultured Japanese flounder (Paralichthys olivaceus) and induces apoptosis in host cells. Aquaculture 319, 30–36. doi: 10.1016/j.aquaculture.2011.06.034
Lin C. S., Chang C. J., Lu C. C., Martel J., Ojcius D. M., Ko Y. F., et al. (2014). Impact of the gut microbiota, prebiotics, and probiotics on human health and disease. BioMed. J. 37, 259–268. doi: 10.4103/2319-4170.138314
Liu T., Wang K. Y., Wang J., Chen D. F., Huang X. L., Ouyang P., et al. (2016). Genome sequence of the fish pathogen yersinia ruckeri SC09 provides insights into niche adaptation and pathogenic mechanism. Int. J. Mol. Sci. 17, 557. doi: 10.3390/ijms17040557
Louis P., Flint H. J. (2009). Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett 294, 1–8.
Magi B., Migliorini L., Sansoni A., Cusi M. G. (2018). Brevibacterium casei bacteraemia in a port-a-cath carrier patient: a case report. Infez. Med. 26, 263–265. Available at: https://www.infezmed.it/media/journal/Vol_26_3_2018_11.pdf.
Mancabelli L., Milani C., Lugli G. A., Turroni F., Cocconi D., van Sinderen D., et al. (2017). Identification of universal gut microbial biomarkers of common human intestinal diseases by meta-analysis. FEMS Microbiol. Ecol. 93, 153. doi: 10.1093/femsec/fix153
Morotomi N., Fukuda K., Nakano M., Ichihara S., Oono T., Yamazaki T., et al. (2011). Evaluation of intestinal microbiotas of healthy Japanese adults and effect of antibiotics using the 16S ribosomal RNA gene based clone library method. Biol. Pharm. Bull. 34, 1011–1020. doi: 10.1248/bpb.34.1011
Nie L., Zhou Q. J., Qiao Y., Chen J. (2017). Interplay between the gut microbiota and immune responses of ayu (Plecoglossus altivelis) during vibrio anguillarum infection. Fish Shellfish Immunol. 68, 479–487. doi: 10.1016/j.fsi.2017.07.054
Oksanen J., Blanchet F. G., Kindt R., Legendre P., Wagner H. (2016). vegan: Community Ecology Package. CRAN-The Comprehensive R Archive Network.
Parker B. J., Wearsch P. A., Veloo A. C. M., Rodriguez-Palacios A. (2020). The genus alistipes: Gut bacteria with emerging implications to inflammation, cancer, and mental health. Front. Immunol. 11, 906. doi: 10.3389/fimmu.2020.00906
Pérez T., Balcázar J. L., Ruiz-Zarzuela I., Halaihel N., Vendrell D., de Blas I., et al. (2010). Host-microbiota interactions within the fish intestinal ecosystem. Mucosal Immunol. 3, 355–360. doi: 10.1038/mi.2010.12
Qi X. Z., Xue M. Y., Yang S. B., Zha J. W., Wang G. X., Ling F. (2017). Ammonia exposure alters the expression of immune-related and antioxidant enzymes-related genes and the gut microbial community of crucian carp (Carassius auratus). Fish Shellfish Immunol. 70, 485–492. doi: 10.1016/j.fsi.2017.09.043
Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, 590–596. doi: 10.1093/nar/gks1219
Rønneseth A., Castillo D., D’Alvise P., Tønnesen Ø., Haugland G., Grotkjaer T., et al. (2017). Comparative assessment of vibrio virulence in marine fish larvae. J. Fish Dis. 40, 1373–1385. doi: 10.1111/jfd.12612
Regan J. C., Brandão A. S., Leitão A. B., Mantas Dias A. R., Sucena E., Jacinto A., et al. (2013). Steroid hormone signaling is essential to regulate innate immune cells and fight bacterial infection in drosophila. PloS Pathog. 9, e1003720. doi: 10.1371/journal.ppat.1003720
Selvin J., Lipton A. P. (2003). Vibrio alginolyticus associated with white spot disease of penaeus monodon. Dis. Aquat. Org. 57, 147–150. doi: 10.3354/dao057147
Senderovich Y., Izhaki I., Halpern M. (2010). Fish as reservoirs and vectors of Vibrio cholerae. PloS One 5, e8607. doi: 10.1371/journal.pone.0008607
Shand F. H., Langenbach S. Y., Keenan C. R., Ma S. P., Wheaton B. J., Schuliga M. J., et al. (2011). In vitro and in vivo evidence for anti-inflammatory properties of 2-methoxyestradiol. J. Pharmacol. Exp. Ther. 336, 962–972. doi: 10.1124/jpet.110.174854
Soto E., Bogdanovic L., Krecek R. C., Jovonovich J. A., Arauz M., Overstreet R. M. (2013). Parasitosis of metabronema sp (Nematoda: Cystidicolidae) in Caribbean porcupinefish, diodon hystrix (L.). J. Fish Dis. 36, 1031–1034. doi: 10.1111/jfd.12109
Sudheesh P. S., Al-Ghabshi A., Al-Mazrooei N., Al-Habsi S. (2012). Comparative pathogenomics of bacteria causing infectious diseases in fish. Int. J. Evol. Biol. 2012, 457264. doi: 10.1155/2012/457264
Tzuc J. T., Escalante D. R., Rojas Herrera R., Gaxiola Cortes G., Ortiz M. L. (2014). Microbiota from litopenaeus vannamei: digestive tract microbial community of pacific white shrimp (Litopenaeus vannamei). SpringerPlus 3, 280. doi: 10.1186/2193-1801-3-280
Wang Q., Garrity G. M., Tiedje J. M., Cole J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Wang J., Huang Y., Xu K., Zhang X., Sun H., Fan L., et al. (2019). White spot syndrome virus (WSSV) infection impacts intestinal microbiota composition and function in Litopenaeus vannamei. Fish Shellfish Immunol. 84, 130–137. doi: 10.1016/j.fsi.2018.09.076
Wang A. R., Ran C., Ringo E., Zhou Z. G. (2018). Progress in fish gastrointestinal microbiota research. Rev. Aquac. 10, 626–640. doi: 10.1111/raq.12191
Xiong J., Yu W., Dai W., Zhang J., Qiu Q., Ou C. (2018). Quantitative prediction of shrimp disease incidence via the profiles of gut eukaryotic microbiota. Appl. Microbiol. Biotechnol. 102, 3315–3326. doi: 10.1007/s00253-018-8874-z
Xiong J., Zhu J., Dai W., Dong C., Qiu Q., Li C. (2017). Integrating gut microbiota immaturity and disease-discriminatory taxa to diagnose the initiation and severity of shrimp disease. Environ. Microbiol. 19, 1490C1501. doi: 10.1111/1462-2920.13701
Yang H. T., Zou S. S., Zhai L. J., Wang Y., Zhang F. M., An L. G., et al. (2017). Pathogen invasion changes the intestinal microbiota composition and induces innate immune responses in the zebrafish intestine. Fish Shellfish Immunol. 71, 35–42. doi: 10.1016/j.fsi.2017.09.075
Zhao Z. L., Zhou Z. C., Dong Y., Pan Y. J., Jiang J. W., Jiang Y., et al. (2021). Association of intestinal fungal communities with the body vesicular syndrome: An emerging disease of sea cucumber (Apostichopus japonicus). Aquaculture 530, 735–758. doi: 10.1016/j.aquaculture.2020.735758
Zhou L., Wei J. F., Lin K. T., Gan L., Wang J. J., Sun J. J., et al. (2020). Intestinal microbial profiling of grass carp (Ctenopharyngodon idellus) challenged with aeromonas hydrophila. Aquaculture 524, 735292. doi: 10.1016/j.aquaculture.2020.735292
Zhu C. H., He J. G., Huang Z. J. (2000). Separation. identification and pathogenicity of ulceration disease of Epinephelus in cage acta. Sci. Nat. Univ. Sunyatseni 39, 278–282. Available at: https://kns.cnki.net/kcms2/article/abstract?v=iJQOcxL8JgzDYLcI8m8karp4aucUf8ZxnYI4k6ANlhcjoHLkluW9gce5ug7eBrbBLWvAW6peNqUQUM8oQQntHSZk-6kAVIxvLbhzo4tRm0s7gRwe_8-zGA==&uniplatform=NZKPT&language=gb.
Keywords: aquaculture, high-throughput sequencing, Diodon hystrix, intestinal microbiota, tail fell syndrome
Citation: Zhou L, Jia X, Liang K, Zuo X, Huang F, Duan X, Li Z, Sang S and Wang Q (2023) Tail fell syndrome impacts intestinal microbiota in porcupinefish (Diodon hystrix). Front. Mar. Sci. 10:1108737. doi: 10.3389/fmars.2023.1108737
Received: 26 November 2022; Accepted: 06 February 2023;
Published: 23 February 2023.
Edited by:
Jin Zhou, Tsinghua University, ChinaReviewed by:
Chao Ran, Feed Research Institute (CAAS), ChinaCheng Peng, Guangdong Academy of Science (CAS), China
Yong Zhang, Sun Yat-sen University, China
Copyright © 2023 Zhou, Jia, Liang, Zuo, Huang, Duan, Li, Sang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shilei Sang, c2FuZ3NoaWxlaUBzY2llcy5vcmc=; Qing Wang, d2FuZ3FpbmdAc2NhdS5lZHUuY24=